

Abstract
Aims
The aim was to comprehensively investigate the effects of genetic variability on the pharmacokinetics of rosuvastatin.
Methods
We conducted a genome‐wide association study and candidate gene analyses of single dose rosuvastatin pharmacokinetics in a prospective study (n = 159) and a cohort of previously published studies (n = 88).
Results
In a genome‐wide association meta‐analysis of the prospective study and the cohort of previously published studies, the SLCO1B1 c.521 T > C (rs4149056) single nucleotide variation (SNV) associated with increased area under the plasma concentration–time curve (AUC) and peak plasma concentration of rosuvastatin (P = 1.8 × 10−12 and P = 3.2 × 10−15). The candidate gene analysis suggested that the ABCG2 c.421C > A (rs2231142) SNV associates with increased rosuvastatin AUC (P = .0079), while the SLCO1B1 c.388A > G (rs2306283) and SLCO2B1 c.1457C > T (rs2306168) SNVs associate with decreased rosuvastatin AUC (P = .0041 and P = .0076). Based on SLCO1B1 genotypes, we stratified the participants into poor, decreased, normal, increased and highly increased organic anion transporting polypeptide (OATP) 1B1 function groups. The OATP1B1 poor function phenotype associated with 2.1‐fold (90% confidence interval 1.6–2.8, P = 4.69 × 10−5) increased AUC of rosuvastatin, whereas the OATP1B1 highly increased function phenotype associated with a 44% (16–62%; P = .019) decreased rosuvastatin AUC. The ABCG2 c.421A/A genotype associated with 2.2‐fold (1.5–3.0; P = 2.6 × 10−4) increased AUC of rosuvastatin. The SLCO2B1 c.1457C/T genotype associated with 28% decreased rosuvastatin AUC (11–42%; P = .01).
Conclusion
These data suggest roles for SLCO1B1, ABCG2 and SLCO2B1 in rosuvastatin pharmacokinetics. Poor SLCO1B1 or ABCG2 function genotypes may increase the risk of rosuvastatin‐induced myotoxicity. Reduced doses of rosuvastatin are advisable for patients with these genotypes.
Keywords: ABCG2, pharmacogenomics, rosuvastatin, SLCO1B1, SLCO2B1
What is already known about this subject
Rosuvastatin pharmacogenetics has been studied in targeted candidate gene analyses.
Due to the targeted nature and limited sample sizes of previous studies, relevant genetic variants could have been left unidentified.
This is the largest pharmacogenetic study on rosuvastatin pharmacokinetics in healthy volunteers thus far, and the first comprehensive study.
What this study adds
The SLCO1B1 genotype predicting poor organic anion transporting polypeptide 1B1 function and the ABCG2 genotype predicting poor breast cancer resistance protein function double rosuvastatin exposure.
The SLCO2B1 c.1457C > T variant associates with decreased rosuvastatin exposure.
Lower rosuvastatin starting and maximum doses are advised for patients with poor organic anion transporting polypeptide 1B1 or breast cancer resistance protein function.
1. INTRODUCTION
Rosuvastatin is commonly used as a cholesterol‐lowering drug in the primary and secondary prevention of cardiovascular diseases. 1 , 2 It inhibits 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase, thus limiting the rate of cholesterol synthesis and lowering plasma cholesterol levels. 3 Rosuvastatin therapy is generally well tolerated. Among its well‐known adverse effects, however, are muscle symptoms varying in severity from mild myalgia to potentially fatal rhabdomyolysis. 4 , 5 , 6 , 7 , 8 The risk of rosuvastatin‐induced myotoxicity is dose‐dependent, 5 and increased rosuvastatin concentrations due to drug–drug interactions or variability in genes related to rosuvastatin pharmacokinetics may predispose patients to myotoxicity. Understanding the factors that may affect rosuvastatin exposure is therefore important.
Being a hydrophilic compound, rosuvastatin needs active transport by uptake and efflux transporters to effectively cross cell membranes. 6 , 9 , 10 The oral bioavailability of rosuvastatin is approximately 20%. 11 , 12 Absorption of rosuvastatin is limited by the breast cancer resistance protein (BCRP, official name ABCG2, encoded by ABCG2), expressed on the apical membrane of enterocytes. 5 , 13 BCRP mediates the efflux of rosuvastatin back into the small intestinal lumen, and decreased BCRP function may lead to increased rosuvastatin exposure. For example, the area under the plasma rosuvastatin concentration–time curve (AUC) was 144% higher in healthy volunteers homozygous for the ABCG2 c.421C > A (rs2231142, p.Gln141Lys) decreased function variant, compared with the c.421C/C homozygotes. 14 Concomitant use of BCRP‐inhibiting drugs such as cyclosporine, eltrombopag or febuxostat markedly increases rosuvastatin concentrations. 15 , 16 , 17 , 18
Rosuvastatin is administered in the active acid form and is subject to only limited metabolism. 3 Its main metabolites are active N‐desmethylrosuvastatin, formed by CYP2C9, and inactive rosuvastatin lactone. Approximately 90% of orally administered rosuvastatin is excreted unchanged into bile and urine. 5 , 12 The uptake of rosuvastatin into hepatocytes is primarily mediated by organic anion transporting polypeptide (OATP) 1B1 (encoded by SLCO1B1). In addition, other members of the solute carrier superfamily, OATP1B3, OATP2B1 and, to a lesser extent, the sodium‐dependent taurocholate cotransporting polypeptide NTCP (encoded by SLCO1B3, SLCO2B1 and SLC10A1, respectively), transport rosuvastatin into hepatocytes. 9 , 19 , 20 Changes in the function of these uptake transporters may affect systemic and liver exposures to rosuvastatin. For example, homozygosity for the reduced function c.521 T > C (rs4149056, p.Val174Ala) single nucleotide variation (SNV) in SLCO1B1 is associated with a 1.8–2.7‐fold increase in rosuvastatin peak plasma concentration (C max) and a 1.6–2.2‐fold increase in rosuvastatin AUC. 21 , 22 The c.521 T > C SNV is also associated with a potentially increased risk of rosuvastatin‐associated myotoxicity. 23 , 24
Understanding of genetic variability in rosuvastatin pharmacokinetics has so far been based on targeted candidate gene studies, and genetic variants affecting rosuvastatin pharmacokinetics may therefore still be unidentified. Therefore, we considered it important to conduct a comprehensive pharmacogenetic study on rosuvastatin pharmacokinetics. To that end, we carried out a genome‐wide association study of rosuvastatin pharmacokinetics in 247 healthy volunteers, and a candidate gene analysis of 16 genes previously found to be involved in rosuvastatin pharmacokinetics.
2. METHODS
2.1. Subjects and study design
The study sample consists of a prospective rosuvastatin pharmacokinetic study and a cohort of previously published studies with rosuvastatin pharmacokinetic data. Following written informed consent, a total of 159 unrelated, healthy white Finnish volunteers (79 men and 80 women, mean ± standard deviation: age 25 ± 4 y, height 174 ± 9 cm, weight 70 ± 13 kg and body mass index 23 ± 3 kg/m2) participated in the prospective study. The participants' health was confirmed by medical history, physical examination and routine laboratory tests before they entered the study. None used continuous medication, including hormonal contraception, and all were nonsmokers.
The study protocol was approved by the Coordinating Ethics Committee of the Helsinki and Uusimaa Hospital District (record number 86/13/03/00/2015) and the Finnish Medicines Agency Fimea (EudraCT number 2015–000540‐41). Following an overnight fast, the volunteers ingested a single oral dose of 40 mg rosuvastatin (Crestor; AstraZeneca UK Ltd., Cheshire, UK). A standardized warm meal was served 4 hours, and light meals 7 and 10 hours after rosuvastatin administration. Timed venous blood samples were drawn prior to and 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 10 and 24 hours after the ingestion of rosuvastatin. The samples were 4 or 9 mL each and they were collected in EDTA‐containing tubes that were placed on ice immediately after sampling. Plasma was separated within 30 minutes and aliquots were stored at −70°C until analysis. Use of any other drugs was prohibited from 1 week before to 3 days after rosuvastatin administration. The participants were not allowed to use alcohol from 1 day before to 2 days after the day of rosuvastatin administration, and grapefruit products from 2 days before to 2 days after rosuvastatin administration.
The cohort of previously published studies consisted of 88 unrelated, healthy white Finnish volunteers (38 women and 50 men, mean ± standard deviation age 23 ± 3 y, height 175 ± 9 cm, weight 69 ± 11 kg and body mass index 22 ± 2 kg/m2) who had participated in previously published single‐dose pharmacokinetic studies on either 10 mg (n = 37) or 20 mg (n = 51) rosuvastatin. 14 , 18 , 25 , 26 In the studies, the rosuvastatin plasma concentrations were determined either prior to and 0.5, 1, 2, 3, 4, 5, 7, 9, 12, 24, 34 and 48 hours after rosuvastatin ingestion, 14 , 25 , 26 or prior to and 0.5, 1, 1.5, 2, 3, 4, 5, 6, 7, 8, 10, 23 and 47 hours after rosuvastatin ingestion. 18 The participants in the cohort of previously published studies were unrelated to the prospective study participants. The study protocols, including the present genetic analyses, were approved by competent ethics committees and the Finnish Medicines Agency, Fimea. All participants gave written informed consent.
2.2. Analysis of drug concentrations in plasma samples
Rosuvastatin reference standard and deuterated internal standard (rosuvastatin‐D6) were purchased from Toronto Research Chemicals (North York, Ontario, Canada). Prior to analysis, plasma (150 μL) proteins were precipitated with acetonitrile (450 μL) containing the internal standard, and the sample mixture was drawn through the Phree Phospholipid Removal 96‐well extraction plate (Phenomenex, Torrance, CA, USA) according to the manufacturer's protocol. The supernatant was then evaporated and the residue was reconstituted in 100 μL of 0.1% formic acid: acetonitrile (80:20, v:v).
Rosuvastatin concentrations were determined using a Shimadzu Nexera liquid chromatography system (Shimadzu Corporation, Kyoto, Japan) coupled to an API 3000 tandem mass spectrometer interfaced with electrospray ion source (AB Sciex, Toronto, Ontario, Canada). The mobile phase consisted of 0.1% formic acid in water (A) and acetonitrile (B) (gradient separation, 15–95% B, total run time 7.5 min). The mobile phase flow was set at 300 μL/min and the injection volume was 10 μL. The quantification was based on positive multiple reactions monitoring of the mass‐to‐charge transitions 482–258 and 488–264 for rosuvastatin and rosuvastatin‐D6. The lower and upper limits of quantification in plasma were 0.1 and 50 ng/mL. Samples in which rosuvastatin concentration exceeded 50 ng/mL were diluted with blank plasma. The day‐to‐day coefficient of variation was below 15% at relevant rosuvastatin concentrations.
2.3. Pharmacokinetics
The pharmacokinetic variables (C max; time to C max, Tmax; AUC from zero to infinity, AUC0–∞; elimination half‐life, t ½) of rosuvastatin, were calculated using standard noncompartmental methods using Phoenix WinNonlin, version 8.2 (Certara, Princeton, NJ, USA).
2.4. Genotyping
Genomic DNA was extracted from EDTA whole blood samples using the Maxwell 16 LEV Blood DNA Kit on a Maxwell 16 Research automated nucleic acid extraction system (Promega, Madison, WI, USA). Whole genome genotyping was carried out with the Infinium Core Exome (prospective study, n = 159) or Global Screening Array (the cohort of previously published studies, n = 88) microchips (Illumina, San Diego, CA, USA) at the Technology Centre of the Institute for Molecular Medicine, Finland (University of Helsinki, Finland). Hardy–Weinberg equilibrium P > 10−5 and proportion missing ≤0.03 were used as quality thresholds for including data in the genome‐wide association study.
To supplement missing data and to verify genotype calls, the participants were genotyped for 28 relevant SNVs with TaqMan genotyping assays on a QuantStudio 12 K Flex Real‐Time PCR System (Thermo Fisher Scientific, Waltham, MA, USA; Table S1). Haplotype computations for SLCO1B1*1 (previously known as *1A, c.388A‐c.463C‐c.521 T‐c.1929A), *5 (c.388A‐c.463C‐c.521C‐c.1929A), *14 (c.388G‐c.463A‐c.521 T‐c.1929A), *15 (c.388G‐c.463C‐c.521C‐c.1929A), *20 (previously also known as *35, c.388G‐c.463C‐c.521 T‐c.1929C) and *37 (previously known as *1B, c.388G‐c.463C‐c.521 T‐c.1929A) were performed with PHASE v2.1.1. 27 , 28
2.5. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, and are permanently archived in the Concise Guide to PHARMACOLOGY 2021/22. 29
2.6. Statistical analysis
Data are presented as geometric means with geometric coefficient of variation or 90% confidence intervals (CI), or estimated marginal means with 90% CI. Statistical analyses were carried out using the statistical programs JMP Genomics 8.0 (SAS Institute, Inc., Cary, NC, USA) and IBM SPSS Statistics for Windows, version 27 (Armonk, NY, USA). Before analysis, the pharmacokinetic variables analysed (AUC0–∞, C max, t ½) were logarithmically transformed. For the cohort of previously published studies, the AUC0–∞ and C max values were adjusted linearly to a 40 mg rosuvastatin dose. Sex and logarithmically transformed bodyweight were tested as demographic covariates in the prospective study using stepwise linear regression analysis, with P value thresholds of .05 for entry and .10 for removal.
For the genome‐wide association and candidate gene analyses, a stepwise linear regression analysis fixed for significant demographic covariates was used to investigate possible associations of genetic variants with rosuvastatin pharmacokinetic variables. Genome‐wide meta‐analysis of the prospective study and the cohort of previously published studies was carried out by weighting the regression coefficients from each study by the inverse variance. Additive coding was employed for genetic variants, and multiallelic variants were expanded. Only SNVs included in both the prospective study and the cohort of previously published studies were included in the meta‐analysis and the candidate gene analysis. SNVs with minor allele frequencies ≤0.05 (genome wide association analysis) or ≤0.01 (candidate gene analysis) were excluded from the analyses. A P value below 5 × 10−8 was considered genome‐wide significant. Thresholds of .05 for entry and .10 for removal were used in the candidate gene analysis.
Further gene‐based analyses were carried out using analysis of variance, adjusting for covariates, with pairwise comparisons with the Fisher's least significant difference method. Study (prospective study or cohort of previously published studies) was set as a random factor in the analysis. Since it is possible to achieve greater statistical power by combining variants with similar effects, SLCO1B1 was analysed as 5 genotype‐predicted OATP1B1 function classes: poor function (two decreased function alleles); decrease function (one decreased and 1 normal or increased function allele); normal function (two normal function alleles); increased function (one increased and 1 normal function allele); or highly increased function (two increased function alleles). 30 , 31 SLCO1B1*1 and *37 were considered normal function alleles, *5 and *15 as decreased function alleles, and *14 and *20 as increased function alleles. A P value <.05 was considered statistically significant.
3. RESULTS
The AUC0–∞ of rosuvastatin varied 10‐fold and C max 25‐fold among the participants of the prospective study (Table 1). Body weight was associated with decreased rosuvastatin C max and AUC0–∞ (−4.5% per 10% increase in body weight; P = .016 and P = .003) in the prospective study and was used as a covariate for these variables in all further analyses. Among the total group of 247 participants, rosuvastatin AUC0–∞ varied 13‐fold and C max 37‐fold after adjusting the values to a 40‐mg rosuvastatin dose.
TABLE 1.
Pharmacokinetic variables of rosuvastatin in 247 healthy volunteers
| Variable | Prospective study (n = 159) | Cohort of previously published studies (n = 88) | Total (n = 247) | |||
|---|---|---|---|---|---|---|
| Geometric mean (geometric CV) | Range | Geometric mean (geometric CV) | Range | Geometric mean (geometric CV) | Range | |
| Dose‐adjusted C max (ng/mL) | 17.8 (66%) | 4.1–103.8 | 16.0 (61%) | 2.8–56.4 | 17.1 (64%) | 2.8–103.8 |
| T max (h) a | 4 | 0.5–8 | 5 | 0.5–7 | 5 | 0.5–8 |
| Dose‐adjusted AUC0‐∞ (ng × h/mL) | 159.6 (52%) | 53.6–535.8 | 142.2 (53%) | 40.2–417.2 | 153.2 (53%) | 40.2–535.8 |
| t ½ (h) | 10.4 (31%) | 4.1–37.8 | 11.9 (48%) | 1.7–45.5 | 10.9 (38%) | 1.7–45.5 |
Rosuvastatin dose in the prospective study was 40 mg, and 10–20 mg in the previously published studies. The AUC0–∞ and C max values from the cohort of previously published studies were adjusted to 40‐mg dose. The observed AUC covered the AUC0–∞ well, except for 2 outliers. The geometric mean (geometric CV) % extrapolated AUC was 3.6% (79%). AUC0–∞, area under the plasma concentration–time curve from 0 hour to infinity; CI, confidence interval; CV, coefficient of variation; C max, peak plasma concentration; T max time to C max; t ½, elimination half‐life.
T max data given as median.
3.1. Genome‐wide association study
In the genome‐wide association analysis of the prospective study, no SNV showed a genome‐wide significant association with rosuvastatin pharmacokinetics. In the meta‐analysis with the cohort of previously published studies with rosuvastatin pharmacokinetic data, the SLCO1B1 c.521 T > C SNV associated with rosuvastatin AUC0–∞ and C max at the genome‐wide significance level (Figure 1). The AUC0–∞ of rosuvastatin was 42% (P = 1.8 × 10−12) and the C max 59% (P = 3.2 × 10−15) larger per copy of the c.521C allele. No other variants showed genome‐wide significant associations with rosuvastatin exposure, even after adjusting for the SLCO1B1 c.521 T > C variant. No associations were observed with rosuvastatin t ½.
FIGURE 1.
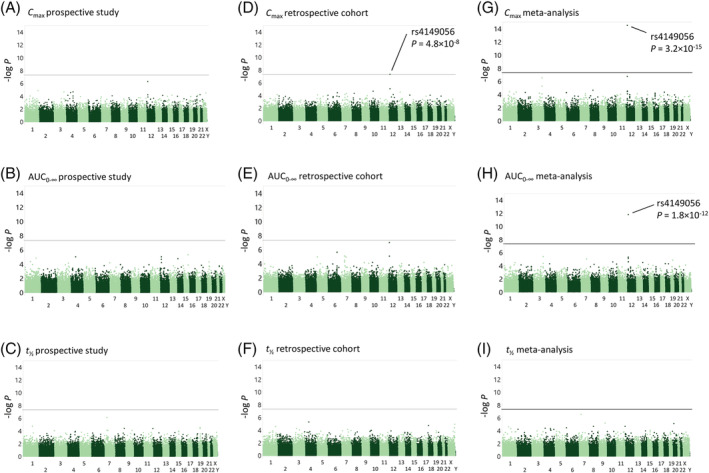
Manhattan plots of pharmacokinetic variables of rosuvastatin. The results of the genome‐wide association analysis of the prospective study for peak plasma concentration (C max) (a), area under the plasma concentration–time curve from zero to infinity (AUC0–∞) (b), and elimination half‐life (t ½) (c) are shown on the left, the results of the cohort of previously published studies for peak plasma concentration (C max) (d), AUC0–∞ (e), and elimination half‐life (t ½) (f) are shown in the middle, and the results of the meta‐analysis of the prospective study and the cohort of previously published studies for C max (g), AUC0–∞ (h), and t ½ (i) on the right. Horizontal lines indicate the genome‐wide significance level of 5 × 10−8
3.2. Candidate gene analysis
To identify which of the 16 candidate genes associate with rosuvastatin pharmacokinetics and to mitigate the risk of false negatives due to the conservative multiple testing correction in the genome‐wide analyses, a linear regression analysis of 42 missense and functional variants was conducted. In the analysis, the SLCO1B1 c.521 T > C and the ABCG2 c.421C > A (tentatively named ABCG2*2) SNVs associated with increased rosuvastatin AUC0–∞ (Table 2). In contrast, the SLCO1B1 c.388A > G (rs2306283, p.Asn130Asp), the ABCG2 c.34G > A (rs2231137, p.Val12Met) and the SLCO2B1 1457C > T (rs2306168, p.Ser486Phe) SNVs associated with decreased AUC0–∞. As the ABCG2 c.34G > A SNV showed only a borderline significant association without correction for multiple testing and has shown an opposite association with rosuvastatin pharmacokinetics in a previous study, 32 it was excluded from further analyses.
TABLE 2.
Results of the candidate gene analyses on rosuvastatin AUC0–∞ in 247 healthy volunteers
| Covariate/SNV | Effect a | ||||
|---|---|---|---|---|---|
| Average (%) | 90% CI | P‐value | Bonferroni adjusted P‐value | Adjusted R 2 for each step | |
| Weight | −6.1 | −8.5, −3.6 | 9.7 × 10−5 | 0.031 | |
| SLCO1B1 c.521 T > C | 52.4 | 38.6, 67.7 | 4.1 × 10−12 | 1.8 × 10−10 | 0.181 |
| SLCO1B1 c.388A > G | −12.4 | −18.8, −5.5 | .0041 | .18 | 0.214 |
| ABCG2 c.421C > A | 18.8 | 6.8, 32.1 | .0079 | .34 | 0.236 |
| SLCO2B1 c.1457C > T | −25.3 | −37.6, −10.7 | .0076 | .33 | 0.258 |
| ABCG2 c.34G > A | −11.9 | −20.7, −2.1 | .048 | >.99 | 0.267 |
AUC0–∞, area under the plasma concentration–time curve from zero to infinity; CI, confidence interval; SNV, single nucleotide variation.
Per minor allele copy or 10% increase in body weight.
3.3. Gene‐based analyses
We next performed an analysis of variance to estimate the effect sizes of different SLCO1B1, ABCG2 and SLCO2B1 genotypes on rosuvastatin pharmacokinetics. For this purpose, the participants were divided into different classes based on their SLCO1B1 genotype‐predicted OATP1B1 phenotype, ABCG2 c.421C > A (ABCG2*2) genotype and SLCO2B1 c.1457C > T genotype. The largest AUC values were seen in individuals with poor OATP1B1 function phenotype as well as those with the ABCG2*2 allele (Figure 2). In contrast, AUC was smallest in the groups with increased or highly increased OATP1B1 function. The SLCO2B1 c.1457C/T genotype also seemed to be more common in individuals with smaller AUC values.
FIGURE 2.
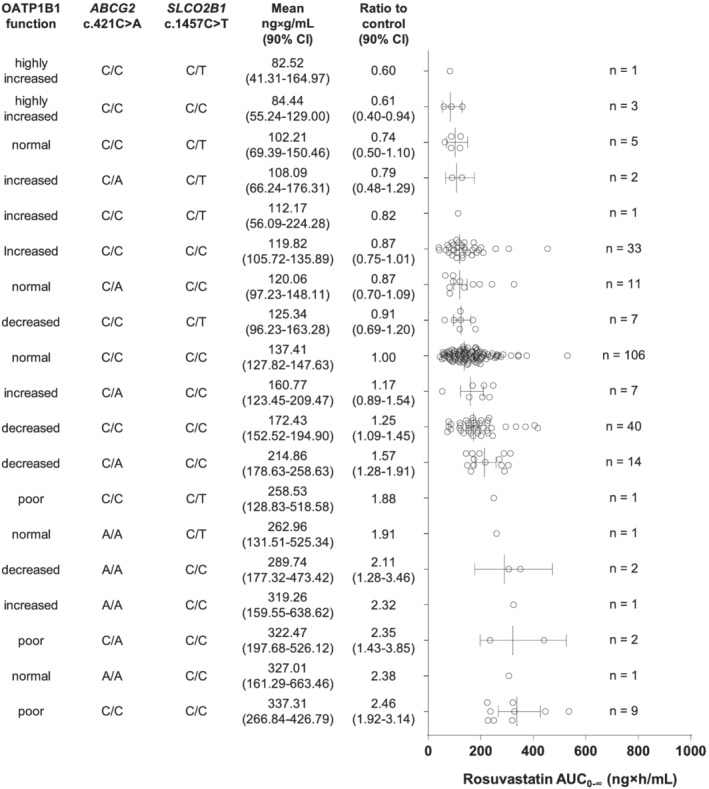
Rosuvastatin area under the plasma concentration–time curve from zero to infinity (AUC0–∞) values in individuals divided into different classes based on the SLCO1B1 phenotype, ABCG2 diplotype and SLCO2B1 c.1457C > T genotype. Data are estimated marginal means with 90% confidence interval (CI). HIF, highly increased function; IF, increased function; NF, normal function; DF, decreased function; PF, poor function
In a gene‐based analysis adjusted for the ABCG2 and SLCO2B1 genotypes, OATP1B1 poor function phenotype associated with increased rosuvastatin AUC0–∞ and C max, whereas the OATP1B1 highly increased function phenotype associated with decreased rosuvastatin AUC0–∞ (Table 3). The mean AUC0–∞ and C max were increased 2.1‐fold (90% CI 1.6, 2.8; P = 4.69 × 10−5) and 2.8‐fold (90% CI 2.0, 3.9; P = 1.75 × 10−6) in the poor function group, compared with the normal function group. The highly increased function group had 44% (90% CI 16%, 62%; P = .019) decreased mean AUC0–∞ compared with the normal function group.
TABLE 3.
Pharmacokinetics of rosuvastatin in individuals with different SLCO1B1 genotype‐predicted OATP1B1 functions
| Variable | Highly increased (n = 4) | Increased (n = 44) | Normal (n = 124) | Decreased (n = 63) | Poor (n = 12) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Mean (90% CI) | Ratio to normal (90% CI) P‐value | Mean (90% CI) | Ratio to normal (90% CI) P‐value | Mean (90% CI) | Mean (90% CI) | Ratio to normal (90% CI) P‐value | Mean (90% CI) | Ratio to normal (90% CI) P‐value | |
| C max (ng/mL) | 10.4 (6.8–15.9) | 0.64 (0.40–1.02) P = .12 | 14.8 (11.8–18.4) | 0.91 (0.68–1.22) P = .59 | 16.2 (13.4–19.7) | 19.6 (17.1–22.6) | 1.21 (0.95–1.54) P = .19 | 45.0 (33.9–59.9) | 2.77 (1.97–3.91) P = 1.75 × 10−6 |
| AUC0‐∞ (ng × h/mL) | 83.8 (58.2–120.7) | 0.56 (0.38–0.84) P = .019 | 140.9 (116.7–170.1) | 0.95 (0.73–1.22) P = .71 | 149.0 (126.3–176.0) | 180.5 (160.0–203.8) | 1.21 (0.99–1.49) P = .13 | 312.0 (244.4–398.6) | 2.09 (1.56–2.81) P = 4.69 × 10−5 |
| t ½ (h) | 12.1 (8.9–16.5) | 1.11 (0.79–1.56) P = .62 | 11.7 (10.0–13.8) | 1.07 (0.87–1.32) P = .60 | 11.0 (9.5–12.6) | 11.1 (10.0–12.3) | 1.00 (0.85–1.20) P = .93 | 11.2 (9.1–13.7) | 1.02 (0.79–1.30) P = .91 |
Data are estimated marginal means adjusted for weight (AUC0‐∞ and C max), SLCO2B1 c.1457C > T genotype and ABCG2 c.421C/A genotype. AUC0–∞, area under the plasma concentration–time curve from zero to infinity; CI, confidence interval; C max, peak plasma concentration; t ½, elimination half‐life.
The ABCG2*2 allele associated with increased concentrations of rosuvastatin in an analysis adjusted for OATP1B1 function and SLCO2B1 genotype (Table 4). Individuals who were ABCG2*2 homozygotes, predicting poor BCRP function, showed 116% larger AUC0–∞ (90% CI 53%, 203%; P = 2.6 × 10−4) and 104% higher C max (90% CI 37%, 203%; P = .0036) compared with the control, *1/*1 group.
TABLE 4.
Pharmacokinetics of rosuvastatin in individuals with different ABCG2 c.421C > A (ACBG2*2) genotypes
| Variable | ABCG2 c.421C/C (n = 206) | ABCG2 c.421C/A (n = 36) | ABCG2 c.421A/A (n = 5) | ||
|---|---|---|---|---|---|
| Mean (90% CI) | Mean (90% CI) | Ratio to c.421C/C (90% CI); P‐value | Mean (90% CI) | Ratio to c.421C/C (90% CI); P‐value | |
| C max (ng/mL) | 16.4 (14.4–18.6) | 16.8 (14.1–20.0) | 1.03 (0.83–1.27); P = .85 | 33.3 (22.8–48.6) | 2.04 (1.37–3.03); P = .0036 |
| AUC0‐∞ (ng × h/mL) | 138.7 (124.4–154.5) | 159.0 (137.0–184.5) | 1.15 (0.95–1.38); P = .22 | 298.6 (215.9–413.1) | 2.16 (1.53–3.03); P = 2.6 × 10 −4 |
| t ½ (h) | 11.5 (10.5–12.6) | 11.4 (10.1–12.9) | 0.99 (0.85–1.16); P = .94 | 10.6 (8.0–13.9) | 0.92 (0.69–1.23); P = .64 |
Data are estimated marginal means adjusted for weight (AUC0‐∞ and C max), SLCO2B1 c.1457C > T genotype and SLCO1B1 genotype‐predicted OATP1B1 function. AUC0–∞, area under the plasma concentration–time curve from zero to infinity; CI, confidence interval; C max, peak plasma concentration; t ½, elimination half‐life.
In addition, the SLCO2B1 c.1457C > T SNV associated with lower concentrations and prolonged t ½ of rosuvastatin (Table 5). The analysis was adjusted for OATP1B1 function and ABCG2 genotype. The mean AUC0–∞ was 28% (90% CI 11%, 42%; P = .011) and the C max 33% (90% CI 14%, 48%; P = .008) lower, and the t ½ 29% longer (90% CI 8%, 54%; P = .02) in C/T heterozygotes than in noncarriers. No participant was homozygous for the SLCO2B1 c.1457C > T variant.
TABLE 5.
Pharmacokinetics of rosuvastatin in individuals with different SLCO2B1 c.1457C > T genotypes
| Variable | SLCO2B1 c.1457C/T (n = 18) | SLCO2B1 c.1457C/C (n = 229) | |
|---|---|---|---|
| Mean (90% CI) | Ratio to c.1457C/C (90% CI); P‐value | Mean (90% CI) | |
| C max (ng/mL) | 13.9 (11.1–17.4) | 0.67 (0.52–0.86); P = .0080 | 20.7 (18.7–23.0) |
| AUC0‐∞ (ng × h/mL) | 128.3 (105.7–155.7) | 0.72 (0.58–0.89); P = .011 | 178.6 (163.6–195.1) |
| t ½ (h) | 13.4 (11.4–15.8) | 1.29 (1.08–1.54); P = .022 | 10.4 (9.7–11.2) |
Data are estimated marginal means adjusted for weight (AUC0‐∞ and C max), SLCO1B1 genotype‐predicted OATP1B1 function and ABCG2 c.421C > A genotype. None of the study participants was homozygous for the SLCO2B1 c.1457C > T variant. AUC0–∞, area under the plasma concentration–time curve from zero to infinity; CI, confidence interval; C max, peak plasma concentration; t ½, elimination half‐life.
4. DISCUSSION
The aim of this study was to investigate the effects of genetic variability on the pharmacokinetics of the cholesterol‐lowering drug rosuvastatin using a genome‐wide approach. The results give new information on how functional variants in SLCO1B1, ABCG2 and SLCO2B1 affect rosuvastatin pharmacokinetics in humans. In the genome‐wide meta‐analysis, the SLCO1B1 c.521 T > C SNV associated with increased rosuvastatin AUC0–∞ and C max. A candidate gene analysis revealed 4 more SNVs in the SLCO1B1, ABCG2 and SLCO2B1 genes associating with rosuvastatin AUC0–∞. In gene‐based analyses of these 3 genes, the SLCO1B1 genotypes that predict poor OATP1B1 function and the ABCG2 c.421A/A genotype that predicts poor BCRP function showed the strongest associations with rosuvastatin exposure, with mean AUC0–∞ and C max values being more than doubled in these genotype groups. In addition, the SLCO2B1 c.1457C/T genotype associated with decreased rosuvastatin concentrations and a prolonged rosuvastatin t ½.
The strongest associations with rosuvastatin pharmacokinetics were observed with SLCO1B1 c.521 T > C SNV. SLCO1B1 encodes the uptake transporter OATP1B1 expressed on the basolateral membrane of human hepatocytes. 20 , 33 OATP1B1 is the primary transporter to mediate rosuvastatin uptake into hepatocytes. The c.521 T > C SNV has been shown to lead to reduced OATP1B1 function and has been associated with raised plasma concentrations of rosuvastatin and a potentially higher risk of rosuvastatin‐induced myotoxicity. 24 In the present study, which is the largest pharmacogenetic study on rosuvastatin pharmacokinetics so far, the c.521 T > C SNV associated genome‐wide significantly with increased AUC0–∞ and C max of rosuvastatin.
In the candidate gene analysis, besides the c.521 T > C, the SLCO1B1 c.388A > G SNV showed a significant association with rosuvastatin AUC. To evaluate the combined effects of SLCO1B1 functional variants, SLCO1B1 was included in the gene‐based analysis as 5 genotype‐predicted OATP1B1 function phenotype classes. 30 , 31 In this analysis, the poor function phenotype associated with 2.1‐fold increased AUC and 2.8‐fold increased C max values of rosuvastatin as compared with normal function OATP1B1. This falls within the range of estimates from previous studies, which have indicated a 1.6–2.2‐fold increase in rosuvastatin AUC in individuals with the c.521C/C genotype. 21 , 22 , 25 Interestingly, rosuvastatin AUC was lower in individuals with highly increased OATP1B1 function than in those with normal OATP1B1 function. A similar association was recently observed with simvastatin acid AUC. 31
As in previous studies, 25 the SLCO1B1 genotype was not associated with the t ½ of rosuvastatin in our study. Because OATP1B1 transports rosuvastatin from the blood into the hepatocytes, poor OATP1B1 function may be assumed to reduce the hepatic extraction ratio of rosuvastatin. For the AUC to double, as was seen in our study, the hepatic extraction ratio of rosuvastatin should be reduced from the normal 63% 11 to 46%. Such a change in the extraction ratio would increase oral bioavailability 1.46‐fold and decrease clearance (Cl) by 27%. As the t ½ depends on both the distribution volume (V d) and Cl (t ½ = ln 2 × V d/Cl), the results suggest that poor OATP1B1 function reduces both the V d and Cl of rosuvastatin.
In the candidate gene analyses, 2 SNVs in the ABCG2 gene associated with changes in rosuvastatin exposure. We tentatively named the c.421A allele *2, similarly to the star allele names commonly used with cytochrome P450 and SLCO1B1 variants. Using simpler names when referring to this allele would be reasonable since the ABCG2*2 has been widely studied and found to be functionally relevant.
In this study, the gene‐based analyses adjusting for SLCO1B1 and SLCO2B1 genotypes showed significant associations for the *2/*2 genotype. The ABCG2*2/*2 genotype predicting poor BCRP function associated with increased AUC and C max. The t ½ of rosuvastatin remained unchanged over the ABCG2 genotype groups. These findings indicate that changes in rosuvastatin concentrations are likely to be the result of altered rosuvastatin bioavailability, following changes in the BCRP‐mediated efflux of rosuvastatin in the small intestine. Homozygosity for the decreased function *2 allele associated with 2.2‐fold increase in rosuvastatin AUC. This effect size lies in the vicinity of what has been seen in previous pharmacogenetic studies. 14 , 32 , 34 A similar 2.1‐fold increase in rosuvastatin AUC without changes in t ½ has also been seen when rosuvastatin was administered after pretreatment with the BCRP‐inhibiting drug febuxostat. 18
The evidence on the function of the other ABCG2 allele, c.34G > A, is controversial. In some studies, no effect on BCRP transporter activity or expression was observed in vitro. 35 , 36 , 37 By contrast, the c.34G > A allele has been shown to disturb the cell membrane localization of BCRP. 38 In a previously published pharmacokinetic study in 62 healthy Chinese men, the AUC and C max of rosuvastatin were increased in patients with the c.34A/A genotype. 32 In our candidate gene analysis, the c.34A/A genotype associated with a decreased rosuvastatin AUC and C max. Therefore, and since the association was only marginally below the level of statistical significance without correction for multiple testing, we excluded the c.34G > A SNV from further analyses.
Another candidate gene with significant association with rosuvastatin pharmacokinetics was SLCO2B1, encoding the OATP2B1 transporter. In our study, the SLCO2B1 c.1457C/T genotype associated with decreased rosuvastatin AUC and C max. Similarly, the c.1457C > T SNV has associated with reduced AUC and C max of the OATP2B1 substrates fexofenadine and celiprolol. 39 , 40 Previous studies have shown that OATP2B1 is expressed, e.g., in the small intestine, on the sinusoidal membrane of hepatocytes, and in heart and skeletal muscle. 41 , 42 Interestingly, the OATP2B1‐inhibiting drug ronacaleret decreased rosuvastatin exposure by approximately half in healthy volunteers, 43 suggesting that decreased function of OATP2B1 may limit the oral bioavailability of rosuvastatin. Taken together, the data suggest that the SLCO2B1 c.1457C > T SNV reduces the oral bioavailability of rosuvastatin by impairing its transport from the gut lumen into enterocytes.
In our study, rosuvastatin t ½ was 29% longer in the SLCO2B1 c.1457C/T heterozygotes than in the C/C homozygotes. A similar difference has also been seen for 3S,5R‐fluvastatin. 44 Interestingly, a previous study showed that apple juice reduces the AUC and C max and prolongs the t ½ of the OATP2B1 substrate fexofenadine. 39 The authors suggested that the changes in fexofenadine AUC and C max indicate decreased fexofenadine bioavailability through OATP2B1 inhibition, and that the prolonged t ½ might be due to flip‐flop pharmacokinetics caused by sustained absorption of fexofenadine. A similar mechanism might explain the prolongation of the rosuvastatin t ½ in the carriers of the SLCO2B1 c.1457C > T variant in our study. More studies are needed to further elucidate the role of SLCO2B1 in rosuvastatin pharmacokinetics.
The normal dose range of rosuvastatin is 5–40 mg/d. Given that the risk of statin‐induced muscle toxicity increases with systemic statin concentrations, taking the genetic variability in rosuvastatin pharmacokinetics into consideration could help prevent cases of myotoxicity. 6 In patients with poor OATP1B1 or BCRP function genotypes, rosuvastatin concentrations may be twice as high as in noncarriers of these variants. It could therefore be advisable to use a lower‐than‐normal starting dose of rosuvastatin, and to avoid the usual maximum dose of 40 mg in these patients. These recommendations are consistent with the current dosing recommendations for patients of Asian ethnicity, whose rosuvastatin exposure is known to be approximately double that of Caucasian patients. 21 , 45 In the Justification for the Use of Statins in Primary Prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER) trial, there was no association between SLCO1B1 genotype and the rate of clinical myalgia in patients receiving 20 mg rosuvastatin daily. 46 A maximum dose of 20 mg rosuvastatin could therefore be a safe choice for patients with poor OATP1B1 or BCRP function genotypes. This corroborates the recent recommendation by the Clinical Pharmacogenetics Implementation Consortium, 47 which was based on previously published data from targeted candidate gene pharmacokinetic studies with relatively small numbers of participants.
In the JUPITER trial, an intronic SNV in ABCG2 associated with a larger reduction in low‐density lipoprotein (LDL) cholesterol levels in patients receiving 20 mg rosuvastatin. 48 This SNV is in a strong linkage disequilibrium with the ABCG2 c.421C > A SNV. In contrast, the SLCO1B1 c.521 T > C SNV has associated either with no change or a slightly impaired LDL cholesterol‐lowering efficacy of statins. 48 , 49 , 50 Reduced OATP1B1 function may therefore increase the risk of myotoxicity of rosuvastatin without an increase in LDL cholesterol‐lowering efficacy, whereas reduced BCRP function may increase both the risk of myotoxicity and the efficacy of rosuvastatin.
In this study, there were no participants homozygous for both the SLCO1B1 c.521 T > C and ABCG2 c.421C > A SNVs. The combined effects of these 2 variants cannot be directly extrapolated from the present results. However, according to pharmacokinetic principles, their effects can be assumed to be additive. Systemic rosuvastatin exposure may therefore be increased more than 4‐fold in these patients compared with noncarriers of the variant alleles. Cyclosporine, which inhibits both BCRP and OATP1B1 as well as OATP1B3 and NCTP, increases rosuvastatin AUC up to 7.1‐fold. 15 The patients with both OATP1B1 and BCRP poor function genotypes could therefore be predisposed to myotoxicity even when using lower rosuvastatin doses.
The allele frequencies in the study population may affect the results from this study. In our prospective study, the minor allele frequencies of the SNVs with significant associations were 17% for SLCO1B1 c.521 T > C, 41% for SLCO1B1 c.388A > G, 7% for ABCG2 c.421C > A, 10% for ABCG2 c.34G > A and 4% for SLCO2B1 c.1457C > T. It may be possible that the effects of some SNVs or genotypes could have remained undetected due to their low frequencies in the Finnish population.
This was a single‐dose study carried out in healthy participants. Since rosuvastatin shows linear pharmacokinetics, 51 , 52 the results can be extrapolated to continuous dosing. Variability in rosuvastatin pharmacokinetics in patients with hypercholesterolaemia and/or previous cardiovascular events, who often have multiple medications, may be greater than in the group of young, healthy volunteers in our study. In addition to genetic variation, drug–drug interactions, increased age and impaired renal function, for example, may predispose patients to statin‐induced myotoxicity. 4 , 53 , 54 Therefore, it is even more important to be able to anticipate higher than average rosuvastatin concentrations in these patients.
In conclusion, this study suggests that the SLCO1B1, ABCG2 and SLCO2B1 genotypes affect rosuvastatin pharmacokinetics. The mechanisms underlying the effects are likely changes in rosuvastatin bioavailability (ABCG2, SLCO2B1) and hepatic uptake of rosuvastatin (SLCO1B1). Taking these genotypes into consideration when prescribing rosuvastatin could help to reduce dose‐dependent adverse effects such as myotoxicity and ensure the efficacy of rosuvastatin therapy. In particular, patients with the ABCG2*2/*2 genotype or with SLCO1B1 genotypes predicting poor OATP1B1 function should avoid high doses of rosuvastatin, as their rosuvastatin exposure may be more than doubled.
COMPETING INTERESTS
The authors declare no conflicts of interest.
CLINICAL TRIAL REGISTRATION
European Union Drug Regulating Authorities Clinical Trials Database (EudraCT) registration number 2015–000540‐41.
CONTRIBUTORS
M.L. and M.Ni. wrote the manuscript; S. T, A.T., J.T.B. and M.Ni. designed the research; E.K.T., S.T., M.Ne., J.V., M.P‐H., T.O.L., T.T., J.T.B., A.T. and M.Ni. performed the research; M.L. and M.Ni. analysed the data.
Supporting information
TABLE S1 SNVs included in the candidate gene analysis.
ACKNOWLEDGEMENTS
The authors thank Eija Mäkinen‐Pulli and Lisbet Partanen for their skilful technical assistance, and Anssi Mykkänen and Päivi Hirvensalo for their valuable insight in designing the statistical analyses.
This study was supported by grants from the European Research Council (Grant agreement 282 109), Sigrid Jusélius Foundation, Orion Research Foundation sr, the Finnish Medical Foundation, and State funding for university‐level health research (Helsinki, Finland).
Lehtisalo M, Taskinen S, Tarkiainen EK, et al. A comprehensive pharmacogenomic study indicates roles for SLCO1B1, ABCG2 and SLCO2B1 in rosuvastatin pharmacokinetics. Br J Clin Pharmacol. 2023;89(1):242‐252. doi: 10.1111/bcp.15485
The authors confirm that the Principal Investigator for this paper is Mikko Niemi and that he had direct clinical responsibility for study participants.
Funding information FP7 Ideas: European Research Council, Grant/Award Number: 282106; Orionin Tutkimussäätiö; Sigrid Juséliuksen Säätiö; Suomen Lääketieteen Säätiö
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available to the extent allowed by the EU General Data Protection Regulation, other applicable regulations and participant consent from the corresponding author upon reasonable request. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- 1. Schachter M. Chemical, pharmacokinetic and pharmacodynamic properties of statins: an update. Fundam Clin Pharmacol. 2005;19(1):117‐125. doi: 10.1111/j.1472-8206.2004.00299.x [DOI] [PubMed] [Google Scholar]
- 2. Wenner MM. The search beyond statins. Nat Med. 2010;16:150‐153. [DOI] [PubMed] [Google Scholar]
- 3. Filppula AM, Hirvensalo P, Parviainen H, et al. Comparative hepatic and intestinal metabolism and pharmacodynamics of statins. Drug Metab Dispos. 2021;49(8):658‐667. doi: 10.1124/dmd.121.000406 [DOI] [PubMed] [Google Scholar]
- 4. Thompson PD, Clarkson P, Karas RH. Statin‐associated myopathy. Jama. 2003;289(13):1681‐1690. doi: 10.1001/jama.289.13.1681 [DOI] [PubMed] [Google Scholar]
- 5. Neuvonen PJ, Niemi M, Backman JT. Drug interactions with lipid‐lowering drugs: mechanisms and clinical relevance. Clin Pharmacol Ther. 2006;80(6):565‐581. doi: 10.1016/j.clpt.2006.09.003 [DOI] [PubMed] [Google Scholar]
- 6. Niemi M. Transporter pharmacogenetics and statin toxicity. Clin Pharmacol Ther. 2010;87(1):130‐133. doi: 10.1038/clpt.2009.197 [DOI] [PubMed] [Google Scholar]
- 7. Egan A, Colman E. Weighing the benefits of high‐dose simvastatin against the risk of myopathy. N Engl J Med. 2011;365(4):285‐287. doi: 10.1056/NEJMp1106689 [DOI] [PubMed] [Google Scholar]
- 8. Alfirevic A, Neely D, Armitage J, et al. Phenotype standardization for statin‐induced myotoxicity. Clin Pharmacol Ther. 2014;96(4):470‐476. doi: 10.1038/clpt.2014.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ho RH, Tirona RG, Leake BF, et al. Drug and bile acid transporters in rosuvastatin hepatic uptake: function, expression, and pharmacogenetics. Gastroenterology. 2006;130(6):1793‐1806. [DOI] [PubMed] [Google Scholar]
- 10. Elsby R, Hilgendorf C, Fenner K. Understanding the critical disposition pathways of statins to assess drug‐drug interaction risk during drug development: it's not just about OATP1B1. Clin Pharmacol Ther. 2012;92(5):584‐598. doi: 10.1038/clpt.2012.163 [DOI] [PubMed] [Google Scholar]
- 11. Martin PD, Warwick MJ, Dane AL, Brindley C, Short T. Absolute oral bioavailability of rosuvastatin in healthy white adult male volunteers. Clin Ther. 2003;25(10):2553‐2563. doi: 10.1016/S0149-2918(03)80316-8 [DOI] [PubMed] [Google Scholar]
- 12. Kanukula R, Salam A, Rodgers A, Kamel B. Pharmacokinetics of rosuvastatin: a systematic review of randomised controlled trials in healthy adults. Clin Pharmacokinet. 2021;60(2):165‐175. doi: 10.1007/s40262-020-00978-9 [DOI] [PubMed] [Google Scholar]
- 13. Deng F, Tuomi SK, Neuvonen M, et al. Comparative hepatic and intestinal efflux transport of statins. Drug Metab Dispos. 2021;49(9):750‐759. doi: 10.1124/dmd.121.000430 [DOI] [PubMed] [Google Scholar]
- 14. Keskitalo JE, Zolk O, Fromm MF, Kurkinen KJ, Neuvonen PJ, Niemi M. ABCG2 polymorphism markedly affects the pharmacokinetics of atorvastatin and rosuvastatin. Clin Pharmacol Ther. 2009;86(2):197‐203. doi: 10.1038/clpt.2009.79 [DOI] [PubMed] [Google Scholar]
- 15. Simonson SG, Raza A, Martin PD, et al. Rosuvastatin pharmacokinetics in heart transplant recipients administered an antirejection regimen including cyclosporine. Clin Pharmacol Ther. 2004;76(2):167‐177. doi: 10.1016/j.clpt.2004.03.010 [DOI] [PubMed] [Google Scholar]
- 16. Allred AJ, Bowen CJ, Park JW, et al. Eltrombopag increases plasma rosuvastatin exposure in healthy volunteers. Br J Clin Pharmacol. 2011;72(2):321‐329. doi: 10.1111/j.1365-2125.2011.03972.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Takeuchi K, Sugiura T, Matsubara K, et al. Interaction of novel platelet‐increasing agent eltrombopag with rosuvastatin via breast cancer resistance protein in humans. Drug Metab Dispos. 2014;42(4):726‐734. doi: 10.1124/dmd.113.054767 [DOI] [PubMed] [Google Scholar]
- 18. Lehtisalo M, Keskitalo JE, Tornio A, et al. Febuxostat, but not allopurinol, markedly raises the plasma concentrations of the breast cancer resistance protein substrate rosuvastatin. Clin Transl Sci. 2020;13(6):1236‐1243. doi: 10.1111/cts.12809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kitamura S, Maeda K, Wang Y, Sugiyama Y. Involvement of multiple transporters in the hepatobiliary transport of rosuvastatin. Drug Metab Dispos. 2008;36(10):2014‐2023. doi: 10.1124/dmd.108.021410 [DOI] [PubMed] [Google Scholar]
- 20. Niemi M, Pasanen MK, Neuvonen PJ. Organic anion transporting polypeptide 1B1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol Rev. 2011;63(1):157‐181. doi: 10.1124/pr.110.002857 [DOI] [PubMed] [Google Scholar]
- 21. Lee E, Ryan S, Birmingham B, et al. Rosuvastatin pharmacokinetics and pharmacogenetics in white and Asian subjects residing in the same environment. Clin Pharmacol Ther. 2005;78(4):330‐341. doi: 10.1016/j.clpt.2005.06.013 [DOI] [PubMed] [Google Scholar]
- 22. Choi JH, Lee MG, Cho JY, Lee JE, Kim KH, Park K. Influence of OATP1B1 genotype on the pharmacokinetics of rosuvastatin in Koreans. Clin Pharmacol Ther. 2008;83(2):251‐257. doi: 10.1038/sj.clpt.6100267 [DOI] [PubMed] [Google Scholar]
- 23. Liu JE, Liu XY, Chen S, et al. SLCO1B1 521T > C polymorphism associated with rosuvastatin‐induced myotoxicity in Chinese coronary artery disease patients: a nested case‐control study. Eur J Clin Pharmacol. 2017;73(11):1409‐1416. doi: 10.1007/s00228-017-2318-z [DOI] [PubMed] [Google Scholar]
- 24. Merćep I, Radman I, Trkulja V, et al. Loss of function polymorphisms in SLCO1B1 (c.521T>C, rs4149056) and ABCG2 (c.421C>A, rs2231142) genes are associated with adverse events of rosuvastatin: a case‐control study. Eur J Clin Pharmacol. 2022;78:227‐236. [DOI] [PubMed] [Google Scholar]
- 25. Pasanen MK, Fredrikson H, Neuvonen PJ, Niemi M. Different effects of SLCO1B1 polymorphism on the pharmacokinetics of atorvastatin and rosuvastatin. Clin Pharmacol Ther. 2007;82(6):726‐733. doi: 10.1038/sj.clpt.6100220 [DOI] [PubMed] [Google Scholar]
- 26. Keskitalo JE, Kurkinen KJ, Neuvonen M, Backman JT, Neuvonen PJ, Niemi M. No significant effect of ABCB1 haplotypes on the pharmacokinetics of fluvastatin, pravastatin, lovastatin, and rosuvastatin. Br J Clin Pharmacol. 2009;68(2):207‐213. doi: 10.1111/j.1365-2125.2009.03440.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978‐989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stephens M, Donnelly P. A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 2003;73:1162‐1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Alexander SP, Kelly E, Mathie A, et al. THE CONCISE GUIDE TO PHARMACOLOGY 2021/22: Transporters. Br J Pharmacol. 2021;178(Suppl 1):S412‐S513. [DOI] [PubMed] [Google Scholar]
- 30. Neuvonen M, Tornio A, Hirvensalo P, Backman JT, Niemi M. Performance of Plasma Coproporphyrin I and III as OATP1B1 Biomarkers in Humans. Clin Pharmacol Ther. 2021;110(6):1622‐1632. doi: 10.1002/cpt.2429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mykkänen AJH, Taskinen S, Neuvonen M, et al. Genome‐wide association study of simvastatin pharmacokinetics. Clin Pharmacol Ther. 2022;112(3):676‐686. doi: 10.1002/cpt.2674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wan Z, Wang G, Li T, et al. Marked alteration of rosuvastatin pharmacokinetics in healthy Chinese with ABCG2 34G>A and 421C>A homozygote or compound heterozygote. J Pharmacol Exp Ther. 2015;354(3):310‐315. doi: 10.1124/jpet.115.225045 [DOI] [PubMed] [Google Scholar]
- 33. König J, Cui Y, Nies AT, Keppler D. A novel human organic anion transporting polypeptide localized to the basolateral hepatocyte membrane. Am J Physiol Gastrointest Liver Physiol. 2000;278(1):G156‐G164. doi: 10.1152/ajpgi.2000.278.1.G156 [DOI] [PubMed] [Google Scholar]
- 34. Liu M, Wu XJ, Zhao GL, et al. Effects of polymorphisms in NR1H4, NR1I2, SLCO1B1, and ABCG2 on the pharmacokinetics of rosuvastatin in healthy Chinese volunteers. J Cardiovasc Pharmacol. 2016;68(5):383‐390. doi: 10.1097/FJC.0000000000000426 [DOI] [PubMed] [Google Scholar]
- 35. Kondo C, Suzuki H, Itoda M, et al. Functional analysis of SNPs variants of BCRP/ABCG2. Pharm Res. 2004;21(10):1895‐1903. doi: 10.1023/B:PHAM.0000045245.21637.d4 [DOI] [PubMed] [Google Scholar]
- 36. Morisaki K, Robey RW, Ozvegy‐Laczka C, et al. Single nucleotide polymorphisms modify the transporter activity of ABCG2. Cancer Chemother Pharmacol. 2005;56(2):161‐172. doi: 10.1007/s00280-004-0931-x [DOI] [PubMed] [Google Scholar]
- 37. Tamura A, Wakabayashi K, Onishi Y, et al. Re‐evaluation and functional classification of non‐synonymous single nucleotide polymorphisms of the human ATP‐binding cassette transporter ABCG2 . Cancer Sci. 2007;98(2):231‐239. doi: 10.1111/j.1349-7006.2006.00371.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mizuarai S, Aozasa N, Kotani H. Single nucleotide polymorphisms result in impaired membrane localization and reduced atpase activity in multidrug transporter ABCG2. Int J Cancer. 2004;109(2):238‐246. doi: 10.1002/ijc.11669 [DOI] [PubMed] [Google Scholar]
- 39. Imanaga J, Kotegawa T, Imai H, et al. The effects of the SLCO2B1 c.1457C > T polymorphism and apple juice on the pharmacokinetics of fexofenadine and midazolam in humans. Pharmacogenet . Genomics. 2011;21:84‐93. [DOI] [PubMed] [Google Scholar]
- 40. Ieiri I, Doi Y, Maeda K, et al. Microdosing clinical study: pharmacokinetic, pharmacogenomic (SLCO2B1), and interaction (grapefruit juice) profiles of celiprolol following the oral microdose and therapeutic dose. J Clin Pharmacol. 2012;52(7):1078‐1089. doi: 10.1177/0091270011408612 [DOI] [PubMed] [Google Scholar]
- 41. Kalliokoski A, Niemi M. Impact of OATP transporters on pharmacokinetics. Br J Pharmacol. 2009;158(3):693‐705. doi: 10.1111/j.1476-5381.2009.00430.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Knauer MJ, Urquhart BL, Meyer zu Schwabedissen HE, et al. Human skeletal muscle drug transporters determine local exposure and toxicity of statins. Circ Res. 2010;106:297‐306. [DOI] [PubMed] [Google Scholar]
- 43. Johnson M, Patel D, Matheny C, Ho M, Chen L, Ellens H. Inhibition of intestinal OATP2B1 by the calcium receptor antagonist ronacaleret results in a significant drug‐drug interaction by causing a 2‐fold decrease in exposure of rosuvastatin. Drug Metab Dispos. 2017;45(1):27‐34. doi: 10.1124/dmd.116.072397 [DOI] [PubMed] [Google Scholar]
- 44. Hirvensalo P, Tornio A, Neuvonen M, et al. Enantiospecific pharmacogenomics of fluvastatin. Clin Pharmacol Ther. 2019;106(3):668‐680. doi: 10.1002/cpt.1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Birmingham BK, Bujac SR, Elsby R, et al. Rosuvastatin pharmacokinetics and pharmacogenetics in Caucasian and Asian subjects residing in the United States. Eur J Clin Pharmacol. 2015;71(3):329‐340. doi: 10.1007/s00228-014-1800-0 [DOI] [PubMed] [Google Scholar]
- 46. Danik JS, Chasman DI, MacFadyen JG, Nyberg F, Barratt BJ, Ridker PM. Lack of association between SLCO1B1 polymorphisms and clinical myalgia following rosuvastatin therapy. Am Heart J. 2013;165(6):1008‐1014. doi: 10.1016/j.ahj.2013.01.025 [DOI] [PubMed] [Google Scholar]
- 47. Cooper‐DeHoff RM, Niemi M, Ramsey LB, et al. The Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for SLCO1B1, ABCG2, and CYP2C9 and statin‐associated musculoskeletal symptoms. Clin Pharmacol Ther. 2022;111(5):1007‐1021. doi: 10.1002/cpt.2557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chasman DI, Giulianini F, MacFadyen J, Barratt BJ, Nyberg F, Ridker PM. Genetic determinants of statin‐induced low‐density lipoprotein cholesterol reduction: the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) trial. Circ Cardiovasc Genet. 2012;5(2):257‐264. doi: 10.1161/CIRCGENETICS.111.961144 [DOI] [PubMed] [Google Scholar]
- 49. SEARCH Collaborative Group , Link E, Parish S, et al. SLCO1B1 variants and statin‐induced myopathy‐‐a genomewide study. N Engl J Med. 2008;359:789‐799. [DOI] [PubMed] [Google Scholar]
- 50. Postmus I, Trompet S, Deshmukh HA, et al. Pharmacogenetic meta‐analysis of genome‐wide association studies of LDL cholesterol response to statins. Nat Commun. 2014;5(1):5068. doi: 10.1038/ncomms6068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. McTaggart F, Buckett L, Davidson R, et al. Preclinical and clinical pharmacology of Rosuvastatin, a new 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase inhibitor. Am J Cardiol. 2001;87(5A):28B‐32B. doi: 10.1016/S0002-9149(01)01454-0 [DOI] [PubMed] [Google Scholar]
- 52. US Food and Drug Administration . Crestor Clinical Pharmacology and Biopharmaceutics Review. 2003.
- 53. Schech S, Graham D, Staffa J, et al. Risk factors for statin‐associated rhabdomyolysis. Pharmacoepidemiol Drug Saf. 2007;16(3):352‐358. doi: 10.1002/pds.1287 [DOI] [PubMed] [Google Scholar]
- 54. Thompson PD, Panza G, Zaleski A, Taylor B. Statin‐associated side effects. J Am Coll Cardiol. 2016;67(20):2395‐2410. doi: 10.1016/j.jacc.2016.02.071 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1 SNVs included in the candidate gene analysis.
Data Availability Statement
The data that support the findings of this study are available to the extent allowed by the EU General Data Protection Regulation, other applicable regulations and participant consent from the corresponding author upon reasonable request. The data are not publicly available due to privacy or ethical restrictions.