

Abstract
Sex chromosomes have evolved repeatedly across the tree of life. As they are present in different copy numbers in males and females, they are expected to experience different selection pressures than the autosomes, with consequences including a faster rate of evolution, increased accumulation of sexually antagonistic alleles and the evolution of dosage compensation. Whether these consequences are general or linked to idiosyncrasies of specific taxa is not clear as relatively few taxa have been studied thus far. Here, we use whole‐genome sequencing to identify and characterize the evolution of the X chromosome in five species of Timema stick insects with XX:X0 sex determination. The X chromosome had a similar size (approximately 12% of the genome) and gene content across all five species, suggesting that the X chromosome originated prior to the diversification of the genus. Genes on the X showed evidence of relaxed selection (elevated dN/dS) and a slower evolutionary rate (dN + dS) than genes on the autosomes, likely due to sex‐biased mutation rates. Genes on the X also showed almost complete dosage compensation in somatic tissues (heads and legs), but dosage compensation was absent in the reproductive tracts. Contrary to prediction, sex‐biased genes showed little enrichment on the X, suggesting that the advantage X‐linkage provides to the accumulation of sexually antagonistic alleles is weak. Overall, we found the consequences of X‐linkage on gene sequences and expression to be similar across Timema species, showing the characteristics of the X chromosome are surprisingly consistent over 30 million years of evolution.
Keywords: insects, population genetics, sex chromosomes, sexual selection & conflicts
1. INTRODUCTION
One of the most common forms of genomic variation between individuals within species stems from sex chromosomes. Sex chromosomes differ in copy number between males and females, which has a large effect on the evolutionary forces acting on genes located on them. Specifically, when males are the heterogametic sex (i.e. in XY or X0 systems), the genes on the X chromosome are present in only a single copy in males, whereas genes on the autosomes are present in two copies. This fundamental difference is expected to have a large effect on the evolutionary forces acting on genes located on the X chromosome, with consequences including a faster rate of sequence evolution, increased accumulation of sexually antagonistic alleles and the evolution of dosage compensation mechanisms (Bachtrog et al., 2011; Lenormand & Roze, 2021; Wright et al., 2016). Such effects should apply across taxa, meaning we should observe common patterns of X chromosome evolution in diverse species. Despite this, studies of sex chromosome evolution have shown a great deal of variation among study systems, including differences in the influence selection and drift have on the content of the X and in the extent of dosage compensation (Bachtrog et al., 2011; Gu & Walters, 2017). Currently, it is difficult to understand the factors that govern this variation as only few taxa (typically with only a single or few representative species) have been studied. To elucidate these factors, studies of X chromosome evolution are thus needed from multiple species from a wide range of taxa (Palmer et al., 2019). Such studies will allow us to disentangling general sex chromosome‐linked patterns from species‐specific patterns, and allow us to develop a fuller understanding of sex chromosome evolution.
The rate of sequence evolution is expected to differ between the X chromosome and autosomes for two main reasons. Firstly, the X is hemizygous in males, meaning that recessive or partially recessive mutations in X‐linked genes will be more exposed to selection than mutations in genes on the autosomes, allowing for beneficial mutations to be fixed and deleterious mutations to be purged more effectively (Charlesworth et al., 1987). On the other hand, as the X is only present in a single copy in males, its effective population size is expected to be smaller than that of the autosomes. This means that the effects of drift will also be stronger for genes on the X chromosome than on the autosomes, which will tend to reduce the fixation rate for advantageous mutations but increase it for deleterious mutations (Vicoso & Charlesworth, 2009; Wright, 1931). Together, these effects have been used as an explanation for the overall faster evolution of the X chromosome (the faster‐X effect) seen in many species (Charlesworth et al., 2018; Mank et al., 2010; Meisel & Connallon, 2013; Parsch & Ellegren, 2013). In addition, the difference in X chromosome copy number between males and females is expected to facilitate the fixation of sexually antagonistic alleles (Gibson et al., 2002; Mullon et al., 2012; Rice, 1984). This is because the X chromosome spends two‐thirds of its time in females, giving an advantage to dominant female‐beneficial alleles on the X. In addition, recessive alleles on the X will be exposed to selection on the X in males, giving an advantage to male‐beneficial alleles. These complex forces have the potential to shape how sexually antagonistic variation is distributed across the genome, which can in turn influence broad evolutionary processes such as speciation (Coyne & Orr, 2004; Payseur et al., 2018) and sexual conflict (Bachtrog et al., 2011; Mank et al., 2014; Wilkinson et al., 2015).
The fact that genes on the X are present in different copy numbers in males and females can also create a problem for gene expression, as for many genes, expression is proportional to their copy number (Birchler, 2016; Birchler & Veitia, 2012). As such, species with differentiated sex chromosomes should have evolved dosage compensation mechanisms to equalize expression of the X chromosome in males and females (Charlesworth, 1978, 1996; Ohno, 1967). Note that such dosage compensation mechanisms can evolve either in response to sex chromosomes differentiation or alongside it (Lenormand et al., 2020). Dosage compensation has been demonstrated across a wide range of taxa (Disteche, 2012; Gu & Walters, 2017; Mank, 2013), including model species such as Drosophila melanogaster (Conrad & Akhtar, 2012) and Caenorhabditis elegans (Meyer, 2000) where this phenomenon has been studied in detail (Conrad & Akhtar, 2012; Lucchesi, 1998; Meyer, 2000; Parkhurst & Meneely, 1994; Straub & Becker, 2011). Despite this commonality, it has become increasingly clear that the extent to which genes on the X are dosage compensated varies among species. Several studied species show only partial or no dosage compensation (Mank, 2013). The extent of dosage compensation may also differ by tissue type, with reduced dosage compensation observed in the reproductive tracts of for example, C. elegans (Kelly et al., 2002; Pirrotta, 2002) or D. melanogaster (Mahadevaraju et al., 2021; Meiklejohn et al., 2011; Oliver, 2002). However, it is not clear how widespread tissue‐specific dosage compensation is, as work in non‐model species often use whole‐body samples for examining expression (Gu & Walters, 2017).
Here, we expand our knowledge of the evolutionary characteristics of sex chromosomes by identifying and studying the X chromosome in Timema stick insects. Aspects of X chromosome evolution have been previously studied in several insect orders (Odonata (Chauhan et al., 2021), Hemiptera (Pal & Vicoso, 2015; Richard et al., 2017), Orthoptera (Rayner et al., 2021), Strepsiptera (Mahajan & Bachtrog, 2015), Coleoptera (Mahajan & Bachtrog, 2015; Prince et al., 2010) and Diptera (Bone & Kuroda, 1996; Deng et al., 2011; Jiang et al., 2015; Marín et al., 1996; Nozawa et al., 2014; Rose et al., 2016; Vicoso & Bachtrog, 2015)). However, to date no studies have examined X chromosome evolution in stick insects (Phasmatodea), an order which originated approximately 130 mya (Simon et al., 2019) and contains around 3100 extant species (Bradler & Buckley, 2018). First, we identified the X chromosome in five species of Timema that diverged approximately 30 mya (Riesch et al., 2017). Timema have an XX/X0 sex determination system (Schwander & Crespi, 2009). To determine if genes on the X chromosome show the predicted faster rate of sequence evolution than genes on the autosomes, we examined sequence evolution rates in each species. In addition, we tested if the X chromosome is enriched for sexually antagonistic alleles. This was done by examining if genes with sex‐biased expression are enriched on the X chromosome, as the evolution of sex‐biased expression is thought to be driven primarily by sexually antagonistic selection (Ellegren & Parsch, 2007; Griffin et al., 2013; Innocenti & Morrow, 2010). Finally, we examined if the genes on the X chromosome are dosage compensated by comparing male and female gene expression in three composite tissues (heads, legs and reproductive tracts). Our study thus provides a detailed study of several key aspects of X chromosome evolution in a previously unstudied group, revealing that the characteristics of these X chromosomes are conserved over at least 30 million years of evolution.
2. MATERIALS AND METHODS
2.1. Sample collection and sequencing
We used a combination of available genomic data from females in addition to newly collected data for males for five sexually reproducing species of Timema (T. bartmani, T. cristinae, T. californicum, T. podura and T. poppensis). Reads from five females per species were downloaded from NCBI (Bioproject accession number: PRJNA670663). We collected four males from each of the five species from natural populations in California, from the same (or a geographically very close) population as the available females (Table S8). DNA extractions were done on whole‐body adult males using the Qiagen Mag Attract HMW DNA kit following the manufacturer instructions. Sequencing libraries were generated for each male using a TruSeq DNA nano prep kit (550 bp insert size). Libraries were then sequenced using Illumina HiSeq 2500 at the Lausanne Genomic Technologies Facility.
2.2. Using coverage to identify X‐linked scaffolds
Reads were trimmed before mapping using Trimmomatic (v. 0.36) (Bolger et al., 2014) to remove adapter and low‐quality sequences (options: ILLUMINACLIP:3:25:6 LEADING:9 TRAILING:9 SLIDINGWINDOW:4:15 MINLEN:90). Reads from each individual were mapped to their species' reference genome (Jaron et al., 2022) (Bioproject accession number: PRJEB31411) using BWA‐MEM (v. 0.7.15) (Li, 2013). Multi‐mapping and poor quality alignments were filtered (removing reads with XA:Z or SA:Z tags or a mapq <30). PCR duplicates were removed with Picard (v. 2.9.0) (http://broadinstitute.github.io/picard/). Coverage was then estimated for all scaffolds at least 1000 bp in length using BEDTools (v. 2.26.0) (Quinlan & Hall, 2010). Per base coverage distributions were inspected visually for each individual and libraries with extremely non‐normal coverage distributions were excluded from further analysis (Figures S1–S5).
To compare coverage between males and females, coverage was first summed for all male and all female libraries per scaffold (for scaffolds at least 1000 bp in length). Male and female coverage was then normalized by modal coverage to adjust for differences in overall coverage. X‐linked scaffolds were then identified using the log2 ratio of male to female coverage. Autosomal scaffolds should have equal coverage in males and females (log2 ratio of male to female coverage ≈ 0) and X‐linked scaffolds should have half the coverage in males as in females (log2 ratio of male to female coverage ≈ −1). Each species showed frequency peaks near these values (Figure S6). X‐linked scaffolds were classified in two ways: a ‘liberal’ classification (following [Vicoso & Bachtrog, 2015]) whereby scaffolds were classified as X‐linked if the log2 ratio of male to female coverage < autosomal peak – 0.5, and a ‘stringent’ classification (following (Pal & Vicoso, 2015)) whereby scaffolds were classified as X‐linked if the log2 ratio of male to female coverage was within 0.1 of the value of the X‐linked peak.
To compare X‐linked scaffolds between species, we examined the overlap of one‐to‐one orthologs (previously identified in (Jaron et al., 2022)) on the X‐linked scaffolds. To determine if the overlap of X‐linked orthologs between species was greater than expected, we used the SuperExactTest package (v. 0.99.4) (Wang et al., 2015) in R (R Core Team, 2017). Additionally, we used a tblastn approach to identify which regions of the Bacillus rossius genome correspond to the 210 shared X‐linked orthologs in Timema (maximum e‐value = 1 × 10−20, minimum query coverage = 50%).
2.3. Heterozygosity, nucleotide diversity, effective population size
To calculate heterozygosity, reads were mapped to the reference genomes as described above. We then additionally performed indel realignment with GATK (v. 3.7) (Van der Auwera et al., 2013). A maximum‐likelihood estimate of the number of heterozygous sites per scaffold was then calculated using AngsD (v. 0.921) (Korneliussen et al., 2014) (options: ‐doSaf 1 ‐gl 1 ‐minQ 20 ‐minMapQ 40 ‐fold 1 ‐doCounts 1, with a minimum depth of 5 and a maximum depth of twice the median genome coverage once sites with 0 coverage were excluded) for each sample (for all scaffolds at least 1000 bp in length). The proportion of heterozygous sites was then calculated for each scaffold and the median proportion of heterozygous sites was weighted by the number of covered sites on a scaffold. We used a weighted median for scaffolds as shorter contigs are typically enriched for repeats and therefore less likely to give reliable estimates of heterozygosity. A similar approach was used to calculate pairwise nucleotide diversity for each species, but using only female samples (options: ‐doSaf 1 ‐gl 1 ‐minQ 20 ‐minMapQ 40 ‐fold 1 ‐doCounts 1 with a minimum depth of 5 and a maximum depth of twice the median genome coverage once sites with 0 coverage were excluded). The number of individuals (−nind) and the minimum number of individuals a site must be present in (−minind) were both set to the number of samples analysed. Effective population size (Ne) was estimated for the X and autosomes using the estimates of nucleotide diversity, assuming that nucleotide diversity is equal to 4Neμ (μ = mutation rate). Estimates of the mutation rate were taken from Drosophila melanogaster (2.8 × 10−09) (Keightley et al., 2014) and Heliconius melpomene (2.9 × 10−09) (Keightley et al., 2015).
2.4. Excluding male achiasmy
An absence of recombination in males could influence the genetic diversity of the X and autosomes. Previous work in different phasmid species has shown that males do recombine (Marescalchi et al., 1986; White, 1976), however, this has not been shown in Timema. To examine this, we re‐surveyed images of Giemsa‐stained chromosome preparations from Schwander and Crespi (2009)) which were originally generated for karyotyping purposes. Specifically, we looked for the presence and position of chiasmata on pre‐metaphase plates obtained from male testes.
2.5. Selection analyses
We took values of dN/dS (number of non‐synonymous substitutions per non‐synonymous site/number of synonymous substitutions per synonymous site) for each species branch from Jaron et al. (2022). Briefly, branch‐site models with rate variation at the DNA level (Davydov et al., 2019) were run using the Godon software (https://bitbucket.org/Davydov/godon/, version 2020‐02‐17, option BSG ‐‐ncat 4) for each gene with an ortholog found in at least three of the species of Timema used here. Godon estimates the proportion of sites evolving under purifying selection (p0), neutrality (p1) and positive selection. We used only sites evolving under purifying or neutrality to calculate dN/dS. To test for differences in dN/dS between the X and autosomes, we used Wilcoxon tests with p‐values adjusted for multiple comparisons using Benjamini and Hochberg's algorithm (Benjamini & Hochberg, 1995). GC was calculated using the seqinR package (v.4.2–8) (Charif & Lobry, 2007) in R. We examined the occurrence of positive selection with two approaches. First, as in Jaron et al. (2022), we used the branch‐site models as described above to identify branches with evidence for positively selected sites. However, such branch‐site models may lack power to detect positive selection so in addition we also used GODON to perform site‐based model comparisons. Specifically, we compared the fit of model M8 (which allows for positively selected sites) and M8a (which does not allow for any positively selected sites) (Yang et al., 2000), also with rate variation at the DNA level (Davydov et al., 2019). The two models were compared using a log‐likelihood ratio test for each gene. p‐values were corrected for multiple tests using Benjamini and Hochberg's algorithm. We then used a Fisher's exact test to determine if positively selected genes were overrepresented on the X.
2.6. Gene expression analyses
RNA‐seq reads from three composite tissues (heads, legs and reproductive tracts) for males and females (three replicates per sex) for each of the five species are publically available (Parker et al., 2019a, 2019b) (Bioproject accession number: PRJNA392384). Adapter sequences were removed using Cutadapt (v. 2.3) (Martin, 2011) before quality trimming reads with Trimmomatic (v. 0.36) (Bolger et al., 2014) (options: LEADING:10 TRAILING:10 SLIDINGWINDOW:4:20 MINLEN:80). Trimmed reads were then mapped to reference genomes using STAR (v. 2.6.0c, default options). HTSeq v.0.9.1 (Anders et al., 2015) was used to count the number of reads uniquely mapped to the exons of each gene, with the following options (htseq‐count ‐‐order = name ‐‐type = exon ‐‐idattr = gene_id ‐‐stranded = reverse). Expression analyses were performed using the Bioconductor package EdgeR (v 3.32.1) (Robinson et al., 2010), and done separately for each species and tissue. Normalization factors for each library were computed using the TMM method and were used to calculate normalized expression levels (either FPKM (Fragments Per Kilobase of transcript per Million mapped reads) or TPM (Transcripts Per Million mapped reads)). For the main analyses, genes with low expression (less than 2 FPKM [or TPM] in 2 or more libraries per sex) were excluded. This filtering step was used to exclude any sex‐specifically expressed genes as our goal was to examine how the expression of genes differs in males and females. This decision could influence our results if sex‐limited gene expression was extensive on the X chromosome. To investigate this, we repeated our analysis with the inclusion of sex‐limited genes and found similar results to the main analyses (Figures S7 and S8). To examine dosage compensation, we used the log2 ratio of mean male expression level to female expression level and used Wilcoxon tests (adjusted for multiple testing using Benjamini and Hochberg's algorithm (1995)) to determine if this ratio differed between genes on the autosomal and X‐linked scaffolds.
To determine the significance of sex on gene expression, we fit a generalized linear model (GLM) with a negative binomial distribution with sex as an explanatory variable and used a GLM likelihood ratio test to determine the significance for each gene. p‐values were then corrected for multiple tests using Benjamini and Hochberg's algorithm (1995). In the main analysis, sex‐biased genes were then defined as genes that showed difference in expression between males and females with a false‐discovery rate (FDR) < 0.05 (we also repeated analyses with the additional condition that genes must show a greater than two‐fold difference in expression to ensure our results are robust to the effects of sex‐biased allometry (Montgomery & Mank, 2016)). Note that all genes not classified as sex‐biased were classified as unbiased genes. We then examined if male‐ or female‐ biased genes were under‐ or over‐ represented on autosomal or X‐linked scaffolds using Fisher's exact tests (adjusted for multiple‐testing using Benjamini and Hochberg's algorithm (Benjamini & Hochberg, 1995)). To examine species by sex interactions in gene expression, we used a similar GLM approach as above, with sex, species and species by sex interaction as explanatory variables for genes with an ortholog in each species. Genes with an FDR < 0.1 for the interaction term were considered to have significant species by sex interaction. To determine if sex‐biased genes on the X were underrepresented for species by sex interactions, we used a Fisher's exact test.
It is possible that dosage compensation may vary along the X chromosome. Unfortunately, our current reference genomes are too fragmented to investigate this question. Previous work by Nosil et al. (2018) however produced a more contiguous genome assembly of one of our study species, T. cristinae, which has been further constructed into a linkage map. To use this synteny information for each of our species, we anchored the scaffolds from each of our genome assemblies to the Nosil et al. (2018) reference genome (BioProject Accession PRJNA417530) using MUMmer (version 4.0.0beta2) (Marçais et al., 2018) with parameter – mum. The alignments were processed by other tools within the package: show‐coords with parameters – THrcl to generate tab‐delimited alignment files and dnadiff to generate 1‐to‐1 alignments. We used only uniquely anchored scaffolds for which we were able to map at least 10 k nucleotides to the Nosil et al. (2018) reference genome. Nosil et al. (2018) indicated that linkage group 13 was the X chromosome (Nosil et al., 2018). To determine if this was correct, we repeated our coverage analyses on the Nosil et al. (2018) assembly. From this, we found that most scaffolds that make up linkage group 13 did not show reduced coverage in males (Figure S9). In addition, several scaffolds from other linkage groups did show reduced coverage in males (Figure S9). In order to use as much of the synteny information as possible we ‘cleaned’ the Nosil et al. (2018) assembly by removing X‐linked scaffolds from autosomal linkage groups 1–12. These scaffolds were then assigned to a new, unordered collection of scaffolds from the X chromosome, together with X‐linked scaffolds from linkage group 13 and from those not assigned to any linkage group in Nosil et al. (2018). Scaffolds from linkage group 13 that were not X‐linked were assigned to linkage group NA. We classified scaffolds in the Nosil et al. (2018) assembly as X‐linked if most of a scaffold was covered by aligned scaffolds from our assembly assigned to the X rather than autosomes (i.e. if aligned scaffolds assigned as X covered more than twice as many bases as those assigned as autosomal for a particular scaffold, it was classed as X‐linked).
3. RESULTS
3.1. Identifying X‐linked scaffolds
We used a coverage approach to identify X‐linked scaffolds in our previous genome assemblies (Jaron et al., 2022). Genomic reads from four males and five females from each species were mapped onto the corresponding reference genome. After filtering low‐quality alignments and non‐uniquely mapping reads (see Methods) the median coverage per sample ranged from 11x to 31x (18.5x on average, see Table S1). Visual inspection of coverage distributions (Figures S1–S5) found that while most libraries had either one (in females) or two (in males) coverage peaks, three libraries (T. podura (H56, Figure S4), T. poppensis (ReSeq Ps08 and Reseq_Ps12, Figure S5) did not show a clear coverage peak, and were excluded from all further analyses.
To identify X‐linked scaffolds, we used the log2 ratio of (normalized) coverage of males to females. As males have only a single copy of the X and females have two, X‐linked scaffolds should have twice as much coverage in females as males (log2 male:female coverage ≈ −1). Autosomes are expected to have the same coverage in both sexes (log2 male:female coverage ≈ 0). Considering all scaffolds with a log2 ratio of male to female coverage < autosomal peak −0.5 to be X‐linked, we classified between 12 and 14% of each genome as X‐linked (Table 1, Figure S6), which fits well with the X chromosome size observed in karyotypes (Schwander & Crespi, 2009). This approach may mean some autosomal contigs may be misclassified as X‐linked, thus we also repeated all our analyses with a more stringent classification scheme (scaffolds with a log2 ratio of male to female coverage within 0.1 of the X‐linked peak) (Table S2, Figure S6). Using the more stringent classification scheme produced very similar results (not shown). Of note, most differences between the classification schemes are for short scaffolds (1000–4999 bp), which represent ~20% of the genome assemblies. When these are excluded, the two classification schemes classify almost the same set of scaffolds as X‐linked (Figure S10, Table S3). Finally, we also examined the heterozygosity of the X in males. As expected, the heterozygosity of the X is close to zero and much lower than on the autosomes, corroborating our X‐linked scaffold assignments (Figure S11).
TABLE 1.
Number of genes and scaffolds classified as X‐linked or autosomal
| Species | Min scaffold length | % genome X‐linked | N Autosomal genes | N X‐linked genes | N genes not classified |
|---|---|---|---|---|---|
| T. bartmani | 1000 | 12.11 | 12 804 | 1244 | 18 |
| T. cristinae | 1000 | 12.21 | 12 542 | 1316 | 24 |
| T. californicum | 1000 | 12.22 | 13 207 | 1344 | 12 |
| T. podura | 1000 | 13.75 | 15 069 | 1457 | 3 |
| T. poppensis | 1000 | 12.40 | 14 115 | 1454 | 36 |
3.2. The X chromosome is conserved across Timema
Comparing orthologs across the five different species shows X chromosome gene content is conserved, with >90% of X‐linked orthologs shared between the 5 species (Figure 1, S12). This overlap is much greater than expected by chance (FDR < 6.863 × 10−316). This suggests that the X chromosome is homologous in all five species, which last shared a common ancestor approximately 30 million years ago (Riesch et al., 2017). Additionally, we used species genome alignments to an independent T. cristinae genome assembly (Nosil et al., 2018) to assign scaffolds to linkage groups based on a single reference. By applying coverage analyses, we were able to identify and correct the X‐linked scaffolds in the Nosil et al. (2018) assembly (see Methods). Using this corrected reference, we found that contigs aligned to X‐linked scaffolds showed reduced coverage in males but not females in each species (Figures S13–S17), again indicating the X chromosome is the same in all species. Finally, using BLAST we found that the majority (72%) of the shared X‐linked genes in Timema (for which we were able to obtain a significant hit) were also present on the X chromosome of Bacillus rossius (Figure S18). The split between Timema and all other extant phasmids (the Euphasmatodea, which includes Bacillus) occurred approximately 120 mya (Simon et al., 2019), suggesting that the X chromosome in phasmids predates this split.
FIGURE 1.
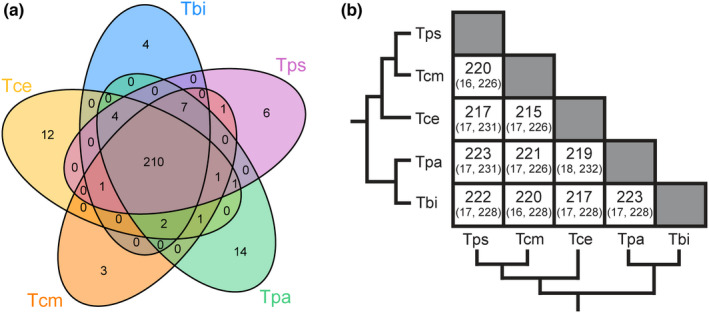
The X chromosome is conserved between Timema species. (a) Venn‐diagrams showing the number of shared X‐linked orthologs between species. (b) Number of shared orthologs (expected, maximum possible). The observed amount of overlap was much greater than expected in all comparisons (FDR <6.863 × 10−316). Species names are abbreviated as Tbi = T. bartmani, Tce = T. cristinae, Tcm = T. californicum, Tps = T. poppensis and Tpa = T. podura. [Correction added on 12 August 2022, after first online publication: Figure 1 has been replaced.]
3.3. The X chromosome has reduced genetic variation
We used female samples to examine two related measures of genetic variation: heterozygosity and nucleotide diversity (π). Only female samples were used, as males are hemizygous for the X, which would affect the estimation of heterozygosity on the X directly. It would also affect the estimation of nucleotide diversity indirectly, as X haplotypes containing recessive lethals on the X will be absent in male samples. We found that both heterozygosity and nucleotide diversity (π) were lower on X‐linked than on autosomal scaffolds (Figures S11, S19), as expected. In all species, the ratios of X to autosomes for both of these measures (heterozygosity = 0.16 to 0.62 (Figure 2), π = 0.19 to 0.48 (Figure 2)) were lower than the 0.75 that would be expected from the reduced effective population size of the X relative to the autosomes. This pattern was also seen when comparing the X to each of the autosomal linkage groups individually (Figures S20, S21). Nucleotide diversity estimates were then used to estimate the effective population size of each species. From this, we estimated the autosomal effective population size in Timema to range from ~150 000 (T. poppensis) to ~2 000 000 (T. podura) (Table S4). Finally, we corroborated that male meiosis was chiasmate, which was revealed by the presence of at least one chiasma per autosome on pre‐metaphase plates (Figure S22).
FIGURE 2.
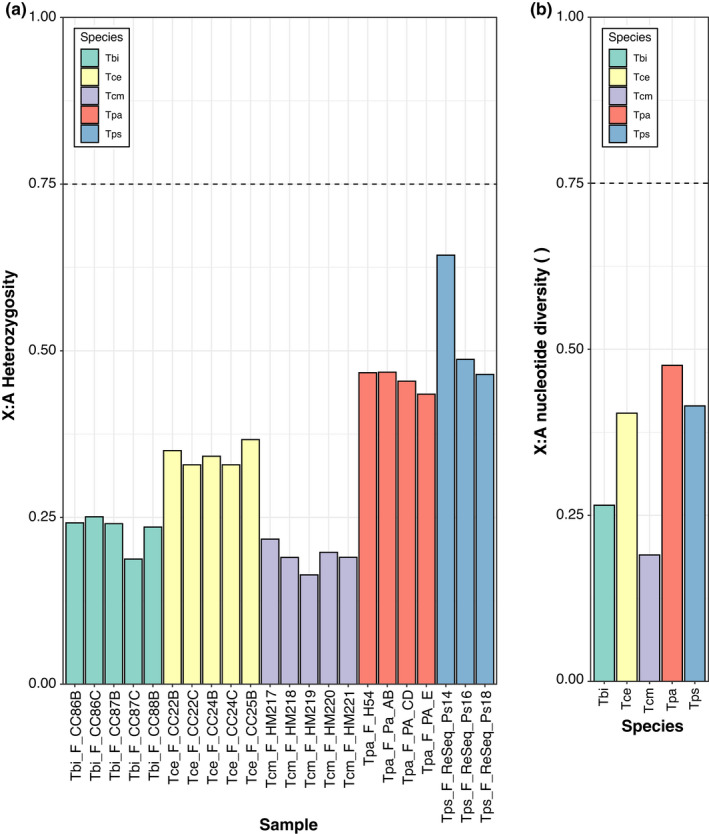
Genetic variation on the X chromosome is lower than expected. (a) Ratio of heterozygosity on the X and autosomes in 3–5 females per species. (b) Ratio of pairwise nucleotide diversity (π) on the X and autosomes. Species names are abbreviated as Tbi = T. bartmani, Tce = T. cristinae, Tcm = T. californicum, Tps = T. poppensis, and Tpa = T. podura. The dotted lines indicate the neutral expectation in a population with a balanced sex ratio.
3.4. The X chromosome shows evidence for reduced purifying selection but no differences in positive selection
Genes on the X chromosome show an elevated dN/dS relative to genes on the autosomes in each species (Figure 3A). This effect remains when accounting for differences in GC and mean expression level (permutation ANOVA p < 0.05 for all species). This increased ratio appears to be driven by reduced purifying selection, as X‐linked genes are not enriched for positively selected genes (Figure 3B, Table S9).
FIGURE 3.
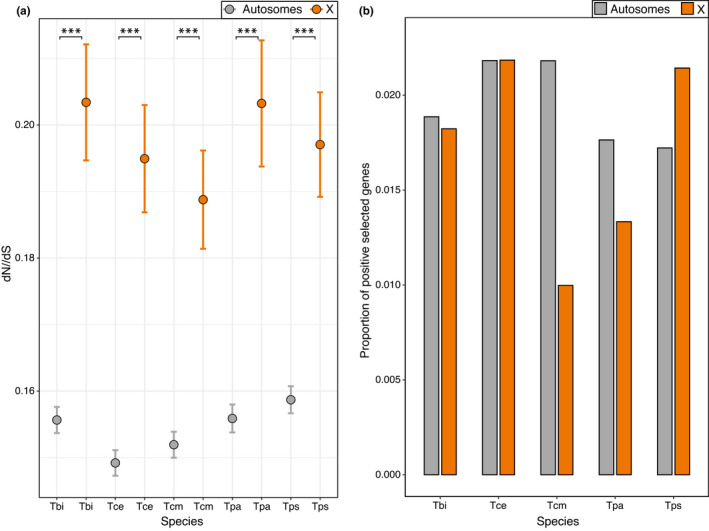
Sequence evolution on the X and autosomes. (a) Average dN/dS across genes. Error bars indicate standard error. Asterisks indicate the significance (FDR) of Wilcoxon tests (***<0.001, **<0.01, *<0.05). (b) Proportion of positively selected genes. Positively selected genes were not enriched on the X chromosome (Fisher's exact test p value = 0.50). Species names are abbreviated as Tbi = T. bartmani, Tce = T. cristinae, Tcm = T. californicum, Tps = T. poppensis and Tpa = T. podura.
Although dN/dS is elevated for genes on the X, overall divergence (branch length, dN + dS) is lower for genes on the X than genes on the autosomes (Figure S23A). More specifically, dS is lower on the X for all species (Figure S23B) and values of dN are only higher on the X for two species (T. podura and T. poppensis, Figure S23C). The reduction in dS on the X is likely due to an overall lower mutation rate in the female than male germline, resulting in fewer mutations on the X due to the X's more frequent transmission through females (Ellegren, 2007; Kirkpatrick & Hall, 2004). Taken together, this suggests that while the genes on the X are subject to reduced purifying selection, the overall divergence of X‐linked genes is smaller.
3.5. Timema show complete dosage compensation in heads and legs, but no dosage compensation in reproductive tracts
We examined if the X chromosome is dosage compensated in Timema by comparing gene expression in males and females in three different composite tissues (heads, legs and reproductive tracts) for each of our five species. While for the main analysis we calculated expression values as FPKM, we also repeated our analyses using TPM. Analyses based on TPM showed very similar results and are provided as supplemental figures and tables. We examined the log2 ratio of male to female expression on the X and the autosomes. For the autosomes and a dosage‐compensated X, the log2 value should be approximately 0, whereas a non‐dosage‐compensated X would have a value of approximately −1. We found that in heads and legs the ratio was close to 0 with only small differences between the X and the autosomes (Figures 4, S24, S25), indicating almost‐complete complete dosage compensation in these tissues. By contrast, in the reproductive tracts the ratio of male to female expression for genes on the X is close to −1 (Figures 4, S24, S25), indicating an absence of dosage compensation in this tissue. This observation was also seen when comparing the X to each of the autosomal linkage groups individually (Figures S26, S27). An alternative, mutually non‐exclusive possibility is that the greatly reduced expression observed in genes on the X in male reproductive tracts is due to a large enrichment of female‐biased genes and depletion of male‐biased genes. Although we cannot formally exclude this possibility, three lines of evidence indicate that lack of dosage compensation in reproductive tracts is the best explanation for our findings. Firstly, a lack of dosage compensation is expected to result in a two‐fold reduction of expression, meaning X linked genes in males should show a major peak with a two‐fold reduction in expression (Mank & Ellegren, 2009; Pal & Vicoso, 2015; Vicoso et al., 2013), which is what we observe (Figure 5). Secondly, if the X chromosomes facilitates the accumulation of sex‐biased genes, we should be able to observe this effect in all tissues (Jaquiéry et al., 2021). While we find a large enrichment of female‐biased genes and a depletion of male‐biased genes on the X in the reproductive tracts, this is not found in the other tissues, with only a slight enrichment of female‐biased genes in the heads of two species and a depletion of male‐biased genes in one species (Figures 6, S28 and Tables S5, S6). This suggests that selection for the enrichment of female‐biased genes and the depletion of male‐biased genes on the X is weak, and thus unlikely to generate the large effect sizes we see in the reproductive tracts. Finally, a reduction in expression of the male X due to a lack of dosage compensation is expected to be consistent across species, since they share the same X chromosome (see above). By contrast, sex‐biased expression independently of dosage is more likely to be species‐specific given the very fast turnover of sex‐biased genes observed between closely related species in several taxa (Harrison et al., 2015; Zhang et al., 2007) including Timema (Parker et al., 2019b). To distinguish between these patterns, we tested if sex‐biased genes on the X in the reproductive tracts are underrepresented for genes with species by sex interactions. We found that genes on the X are underrepresented for genes showing species by sex interactions in the reproductive tracts (p = 0.010), and that this is not the case for the heads (p = 0.938) or legs (p = 0.795). This shows that sex‐differences in expression on the X are more consistent between species in the reproductive tracts than in the heads and legs (also see Figure S29), and further supports a lack of dosage compensation in the reproductive tracts rather than a large enrichment of female‐biased genes on the X.
FIGURE 4.
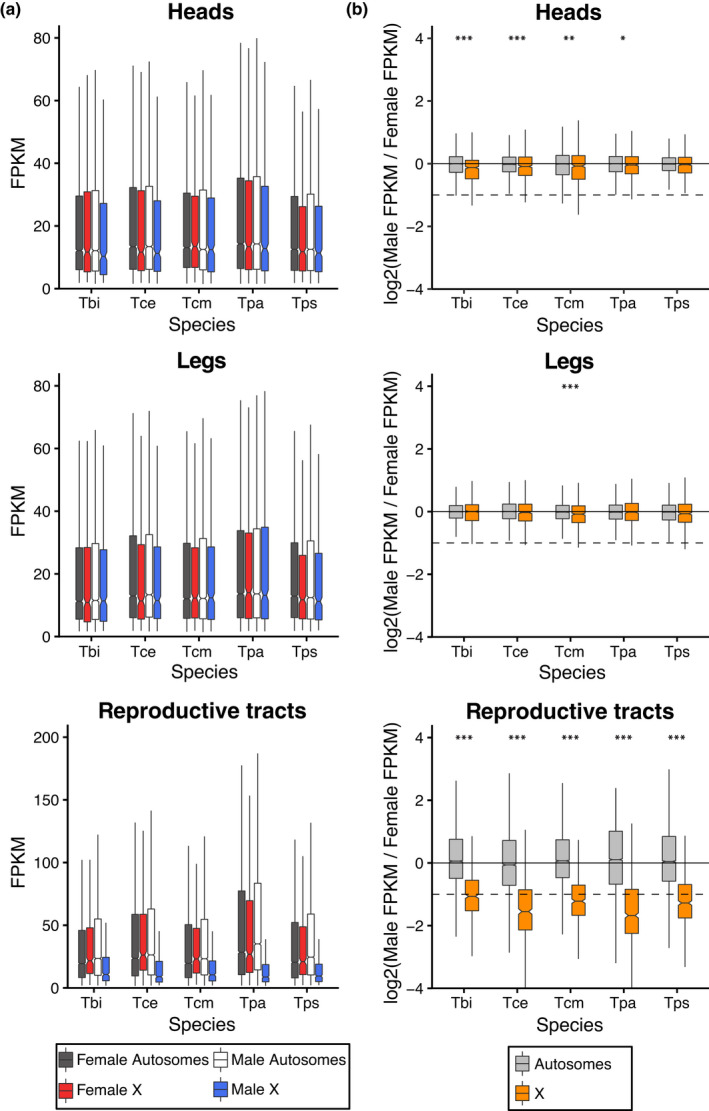
Gene expression on the X and autosomes in heads, legs and reproductive tracts. (a) Average expression levels in males and females on the X and autosomes (b) Log2 of male to female expression ratio for the X and autosomes. Dashed lines represent a two‐fold reduction in expression in males (as expected if there was no dosage compensation). Species names are abbreviated as Tbi = T. bartmani, Tce = T. cristinae, Tcm = T. californicum, Tps = T. poppensis and Tpa = T. podura.
FIGURE 5.
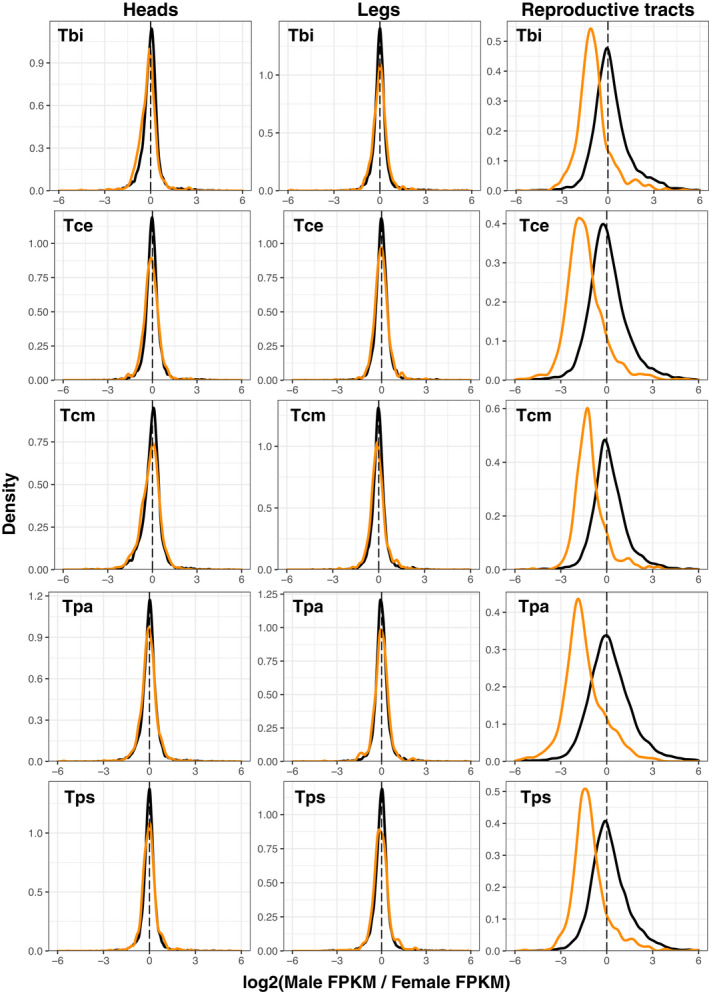
Ratio of male and female gene expression on the X (orange) and autosomes (black) in heads, legs and reproductive tracts. Species names are abbreviated as Tbi = T. bartmani, Tce = T. cristinae, Tcm = T. californicum, Tps = T. poppensis and Tpa = T. podura.
FIGURE 6.
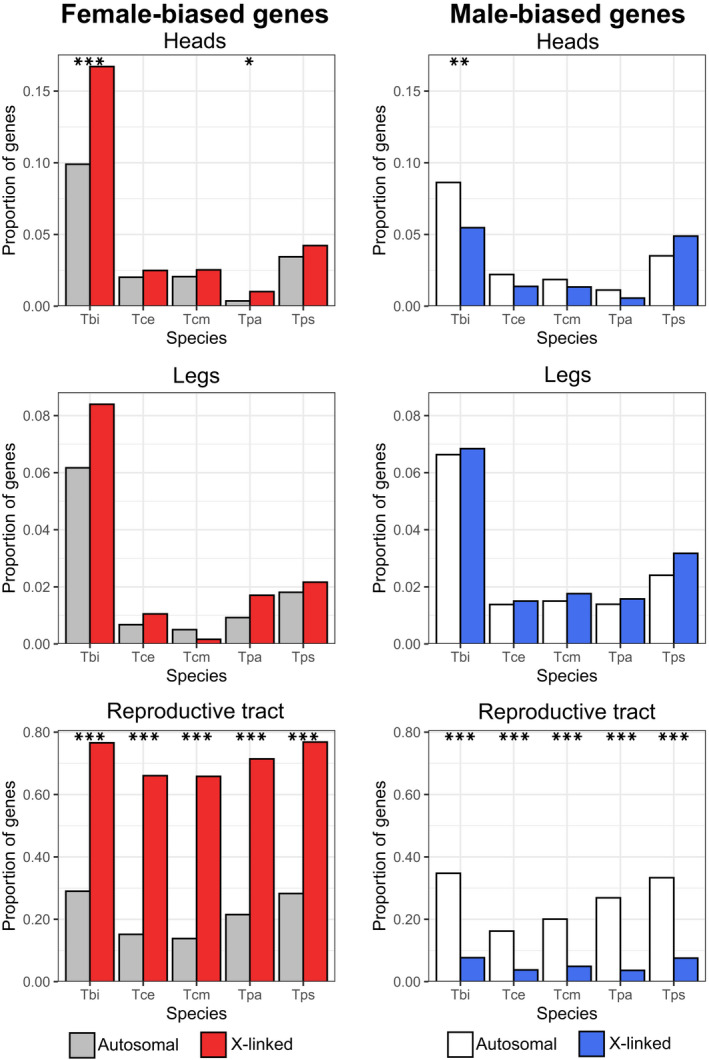
Proportion of female‐ and male‐ biased genes on the X and autosomes in reproductive tract, head and leg samples. Note the scale changes between tissue‐types. Asterisks indicate the significance level (FDR) of Fisher's exact tests (***<0.001, **<0.01, *<0.05). Species names are abbreviated as Tbi = T. bartmani, Tce = T. cristinae, Tcm = T. californicum, Tps = T. poppensis and Tpa = T. podura.
By examining the expression of the X and autosomes in the different tissues, we can infer the type of dosage compensation. In the dosage‐compensated tissues (heads and legs), genes on the autosomes and X have similar overall expression levels in both males and females, and the overall expression of the X is similar to that of the autosomes (Figures 4, S24 and Table S7). By contrast, in the reproductive tracts, where dosage compensation is lacking, expression of genes on the X is much lower in males than in females. This difference seems to be driven by changes in male expression, as X‐linked gene expression in females remains similar to the autosomes (Figures 4, S24 and Table S7). This supports a mechanism of dosage compensation by hyper‐transcription of the X in males, a mechanism common among other insects (Gu & Walters, 2017).
Although we find almost complete dosage compensation, it is possible that its extent could vary along the X chromosome (Mullon et al., 2015). To address this, we examined the extent of dosage along the longest X‐linked scaffolds in the genome, and found no evidence of variation along the X chromosome (Figures S30–S34, see also S35–S46 for expression variation along the autosomes).
3.6. Sex‐biased genes are not enriched on the X chromosome
Sexually antagonistic mutations are expected to fix more easily on the X chromosome than the autosomes. As the evolution of sex‐biased gene expression is thought to be primarily driven by sexually antagonistic selection, it is expected that the X chromosome will be a hotspot for sex‐biased gene expression (Ellegren & Parsch, 2007; Gibson et al., 2002; Griffin et al., 2013; Innocenti & Morrow, 2010; Rice, 1984). Contrary to this expectation, we find most sex‐biased genes on the autosomes (% of sex‐biased genes on the X ranges from 8.3 to 14.3, Table S5), and we find little evidence for enrichment of sex‐biased genes on the X in the head and leg tissues (Figure 6 and Tables S5, S6). Note that almost all genes on the X appear to be sex‐biased in the reproductive tracts, but this effect is largely due to a lack of dosage compensation in this tissue (see above). In addition, while we do find an enrichment of female‐biased genes in the heads of two species (T. bartmani and T. podura, Figure 6) and a depletion of male‐biased genes in T. bartmani, the effect sizes are small and the effect becomes insignificant when considering only sex‐biased genes with at least a twofold difference in expression between males and females (Figure S28). These findings suggest that the selective pressures to accumulate female‐biased genes and reduce male‐biased genes on the X are weak and/or that there are constraints to the gene content or expression levels on the Timema X chromosome.
4. DISCUSSION
The difference in copy numbers of the X chromosome in males and females is expected to have profound effects on its evolution. In particular, the X chromosome is predicted to evolve at a faster rate due to a combination of hemizygous selection and increased drift (Charlesworth et al., 1987; Vicoso & Charlesworth, 2009; Wright, 1931), to accumulate sexually antagonistic alleles (Gibson et al., 2002; Rice, 1984), and to evolve dosage compensation mechanisms (Disteche, 2012; Gu & Walters, 2017; Lenormand et al., 2020; Mank, 2013). Support for these predictions has been mixed from the taxa examined so far, yet the factors responsible for the variation among different taxa are poorly understood (Bachtrog et al., 2011; Gu & Walters, 2017). In this study, we examine these predictions across five species of Timema stick insects. Overall, we find evidence for relaxed selection and complete dosage compensation in somatic tissues, but little evidence for the accumulation of sexually antagonistic alleles (in the form of sex‐biased genes) on the X. Patterns of X chromosome evolution were generally consistent across Timema species, suggesting that the factors influencing sex chromosome evolution in this group are also largely the same.
Sex chromosome conservation is highly variable between taxa, with extensive turnover between species in some groups, for example beetles (Coleoptera) (Blackmon & Demuth, 2014), flies (Diptera) (Vicoso & Bachtrog, 2015) or frogs (Ranidae) (Jeffries et al., 2018), and conservation for over a hundred million years in others, for example Eutherian mammals (Cortez et al., 2014; Lahn & Page, 1999; Marshall Graves, 2015), moths and butterflies (Lepidoptera) (Fraïsse et al., 2017), or birds (Shetty et al., 1999; Xu & Zhou, 2020). The factors influencing turnover rate are complex and interacting (Vicoso, 2019), however, one key factor is the level of differentiation between the X and Y chromosomes, with more differentiated chromosomes less likely to turnover (Pokorná & Kratochvíl, 2009; Vicoso, 2019). Our finding that the X chromosome in Timema is old (likely conserved for over 120 million years) supports this idea. XX/X0 sex determination systems as found in Timema (Schwander & Crespi, 2009) are thought to derive from XX/XY systems with highly differentiated X and Y chromosomes and represent the end point of the gradual loss of gene content from the Y (Bergero & Charlesworth, 2009). As such, XX/X0 systems could be considered the most extreme example of sex chromosome differentiation possible, which may mean that XX/X0 systems are particularly unlikely to turnover (but see (Blackmon & Demuth, 2014)).
Gene sequence evolution on the X chromosome is expected to be faster than that on the autosomes due to a combination of increased drift and hemizygous selection (Charlesworth et al., 1987; Vicoso & Charlesworth, 2009). While this prediction should apply universally, empirical support is mixed (Charlesworth et al., 2018; Mank et al., 2010; Meisel & Connallon, 2013; Pinharanda et al., 2019; Whittle et al., 2020). The cause of this variation is unclear, in particular, because typically only single lineages are examined. In single lineages it is difficult to disentangle the influence of X‐linkage from lineage‐specific effects such as recent bottlenecks, differences in operational sex ratio or population size. By examining the influence of X‐linkage in multiple Timema species covering a span of 30 million years of divergence, we can assess how consistent the effects of X linkage are, allowing for a comprehensive assessment of X chromosome evolution in this genus. Using this approach, we found consistent evidence for a relaxed selection (as measured by dN/dS), known as the ‘faster X effect’, in all species, and that this effect is primarily driven by reduced purifying selection on the X. Interestingly, while dN/dS was higher for genes on the X, the overall amount of divergence was lower. This result is likely driven by a lower net mutation rate on the X relative to autosomes as a result of a lower mutation rate in the female than male germline (Ellegren, 2007; Kirkpatrick & Hall, 2004).
The reduction of purifying selection on the X relative to the autosomes is expected to be largest when the effective population size of the X (NeX) is much smaller than that of the autosomes (NeA) (Vicoso & Charlesworth, 2009). The neutral expectation in a population with a balanced sex ratio is that the ratio of NeX to NeA will be approximately 0.75 (Hartl & Clark, 2006; Wright, 1969), however, demographic and selective processes can have a large effect on this ratio (Caballero, 1995; Charlesworth, 2001; Nunney, 1993). In Timema, NeX / NeA values are much smaller than 0.75 (0.19–0.48, estimated from nucleotide diversity), meaning the effective population size of the X is much smaller than that of the autosomes. Departures from the expected 0.75 nucleotide diversity ratio are common across animals and are typically thought to be a consequence of sex‐biased demography (Mank et al., 2010). One common cause for such departures is that in many systems reproductive skew is stronger for males than females, meaning fewer males contribute to the next generation than females. In Timema, females mate multiply and skew offspring towards particular males (Arbuthnott et al., 2015), suggesting that in Timema reproductive skew is indeed greater in males than females. In male heterogametic species (such as Timema), such a reproductive skew should disproportionately reduce diversity on the autosomes, and result in an X to autosome nucleotide diversity ratio >0.75. Here however, we observe the opposite pattern, a reduction in the X to autosome nucleotide diversity ratio. As such, the reduced diversity on the X we observe is unlikely to be due to sex‐specific variation in reproductive success. There are many potential alternative demographic and selective explanations for this finding, however the most likely (discussed below) are that the X has a lower recombination rate than the autosomes, and/or that Timema undergo frequent population bottlenecks. Since we show that crossing over occurs in both sexes, the effective recombination rate for the X is likely lower than for the autosomes (as recombination between different copies of the X can only occur in females). Low recombination rates intensify the consequences of selective sweeps and background selection on genetic diversity and could thus contribute to the reduced genetic diversity on the X we observe (Betancourt et al., 2004; Charlesworth, 2012, 2013; Wilson Sayres, 2018). Population bottlenecks could also contribute to the reduced genetic diversity on the X. Although bottlenecks will reduce genetic diversity for both the X and the autosomes, it is expected that they will disproportionately reduce genetic diversity on the X (Pool & Nielsen, 2007). This factor may be particularly relevant as Timema live in fire‐prone habitats where fires may lead to frequent population bottlenecks.
Independently of the mechanisms responsible for the strongly reduced effective population size of the X in Timema, it supports our interpretation that the faster‐X effect is driven by less effective purifying selection. Most previous studies of the faster‐X effect have focused on species with NeX/NeA values ≥0.75 (Mank et al., 2010), making direct comparisons with our study difficult. The exceptions to this are studies on birds and Heliconius butterflies which have values of Nez / NeA in a similar range as Timema. Note that both these taxa have heterogametic females (ZW), where low Nez/NeA ratios are believed to stem from strong reproductive skew among males (Mank et al., 2010; Vicoso & Charlesworth, 2009). In birds, a faster‐Z (faster‐X) effect is commonly observed and appears to be driven primarily by less effective purifying selection (Mank et al., 2010). In contrast, in Heliconius evidence for a faster‐Z effect is weaker and is thought to be driven by increased levels of adaptive evolution on the Z, with no evidence for reduced purifying selection on the Z (Pinharanda et al., 2019). The cause of this difference is unclear. However, it has been suggested that it may be due to the overall higher effective population size in Heliconius (Ne ≈ 2 000 000 (Keightley et al., 2015)) compared to birds (Ne = 200 000–600 000 (Primmer et al., 2002; Axelsson et al., 2004; Jennings & Edwards, 2005; Backström et al., 2008)), meaning that selection can be efficient in Heliconius even with the relatively reduced effective population size of the sex chromosome (Mank et al., 2010; Pinharanda et al., 2019). This interpretation conflicts with our findings in Timema, as the (autosomal) effective population size varies greatly between species (from ~150 000 in T. poppensis to ~2 000 000 in T. podura), yet we observe a similar reduction in purifying selection on the X in all species. The reason for this variation between study systems is thus unclear, and highlights the need for future studies across diverse taxa. It should be noted that the values of Ne we estimate for Timema should be considered as rough approximations as our calculations of Ne assume the mutation rate in Timema is similar to that in D. melanogaster and H. melpomene. Since these are distantly related species the mutation rate in Timema could differ markedly, which would mean our estimates of Ne would be systematically under‐ or over‐ estimated. While this complicates direct comparisons with other taxa, it is unlikely to influence our comparisons among Timema species for which only the relative differences in Ne are important.
The X chromosome has long been predicted to be a hotspot for sexually antagonistic variation (Gibson et al., 2002; Rice, 1984). The evolution of sex‐biased gene expression is thought to be driven by sexually antagonistic selection, and thus sex‐biased gene expression should be overrepresented on the X chromosome (Ellegren & Parsch, 2007; Griffin et al., 2013; Innocenti & Morrow, 2010) but see (Hitchcock & Gardner, 2020; Ruzicka & Connallon, 2020). In Timema, we find very little support for this prediction with only a small enrichment of sex‐biased genes on the X in one of our five species. In combination with studies in Drosophila melanogaster (Ruzicka et al., 2019), Callosobruchus maculatus (Sayadi et al., 2019) and Ischnura elegans (Chauhan et al., 2021) which also found little or no enrichment of sexually antagonistic alleles or sex‐biased gene expression on the sex chromosomes, our study suggests that the advantage of accumulating sexually antagonistic alleles on the X be may be smaller than often assumed. The reasons for this are unclear. However, it is likely that the advantage X‐linkage gives to sexually antagonistic alleles is balanced by other forces such as epistatic interactions (Arnqvist et al., 2014) or sex‐specific dominance (Fry, 2010). Both forces favour the accumulation of sexually antagonistic alleles on the autosomes. Additionally, it should be noted that our approach to use sex‐biased genes as a proxy for sexually antagonistic variation may be underpowered as it will miss any genes that are evolving under sexual conflict but have not (yet) evolved sex‐biased gene expression. Despite this, several studies do show the expected enrichment of sexually antagonistic alleles or sex‐biased gene expression on the X: in Tribolium castaneum (Whittle et al., 2020), Diptera (Innocenti & Morrow, 2010; Vicoso & Bachtrog, 2015), Hemiptera (Pal & Vicoso, 2015) and nematodes (Albritton et al., 2014). A key challenge for future work will thus be to integrate studies that quantify multiple factors that influence the accumulation of sexually antagonistic alleles (e.g. epistatic interactions, sex‐specific dominance, reproductive skew) to understand how the variation between studies and taxa is produced.
In many species dosage compensation is thought to be important for ameliorating the costs of misexpression of genes on the X chromosome (Marín et al., 2000). Although common, there is a great deal of variation in the extent to which genes are dosage compensated. Here, we find almost complete dosage compensation in the somatic tissues of all five Timema species, as reported for other species with X0 systems (e.g. Nematodes (Meyer, 2000) and crickets (Rayner et al., 2021)). Dosage compensation is expected for X0 systems as they are thought to arise from XY systems where the Y chromosome has degraded to the point it can be lost without a large decrease in fitness. By this stage, most genes on the X should already be haplo‐sufficient in males. In addition, the evolution of dosage compensation could itself hasten the loss of the Y chromosome, as genes that have functional copies on both the X and the Y will be misexpressed if chromosome‐wide dosage compensation evolves (Lenormand & Roze, 2021; Vicoso & Bachtrog, 2009).
In contrast to the somatic tissues, male reproductive tracts displayed a lack of dosage compensation. Reduced expression of X chromosome in male reproductive tissue has been observed in a number of species including mammals (Disteche, 2012; Khil et al., 2004; Sangrithi & Turner, 2018), C. elegans (Kelly et al., 2002; Pirrotta, 2002) and insects such as D. melanogaster (Mahadevaraju et al., 2021; Meiklejohn et al., 2011; Oliver, 2002) or Teleogryllus oceanicus (Rayner et al., 2021). Whether reduced X expression in male reproductive tracts generally stems from a lack of dosage compensation is, however, not clear. Indeed, reduced X expression can be caused by several non‐mutually exclusive mechanisms, including the accumulation of female‐biased genes on the X (Mank, 2009), the movement of male‐beneficial genes to the autosomes (Vibranovski et al., 2009) or the inactivation of X chromosomes in the germ line (Lee, 2005; Vibranovski, 2014). In Timema, we suggest an absence of dosage compensation is the most likely explanation as expression of X‐linked genes in the male reproductive tracts show a major peak of genes with expression approximately half of that observed in females, similar to that observed in species that lack dosage compensation (Mank & Ellegren, 2009; Vicoso et al., 2013). It should be noted, however, that the expression reduction we see on the male X in Timema is actually slightly less than the half we would expect from a lack of dosage compensation alone. This suggests that other factors, described above, may also have an influence. Disentangling the contribution of each of these factors will require further work. Our work, however, clearly shows that expression in reproductive tissues behaves differently than in somatic tissues, highlighting the importance of studying these tissues separately particularly when assessing the extent of dosage compensation (Gu & Walters, 2017).
Dosage compensation in Timema somatic tissues appears to be achieved by the upregulation of the X in males (type I, (Gu & Walters, 2017)) as indicated by similar expression levels of genes on the X and the autosomes in both sexes. This interpretation assumes that the ancestral state of X chromosome expression (before it was a sex chromosome) was similar to the other autosomes. This seems likely as expression across all Timema autosomes is similar. However, assessing the ancestral state of X expression is largely impossible as the Timema X chromosome appears to be very old (~120 million years). As a consequence, there are likely no species available for comparison with conserved genome organization but with X chromosomes that are homologous to different Timema autosomes. Similar to Timema, upregulation of the X chromosome in males also appears to be the mechanism for dosage compensation in all other XX/XY or XX/X0 insect systems yet studied, even when dosage compensation is incomplete (Gu & Walters, 2017). With our study, this amounts to information from seven different insect orders (Odonata (Chauhan et al., 2021), Phasmatodea (this study), Hemiptera (Pal & Vicoso, 2015; Richard et al., 2017), Orthoptera (Rayner et al., 2021), Strepsiptera (Mahajan & Bachtrog, 2015), Coleoptera (Mahajan & Bachtrog, 2015; Prince et al., 2010) and Diptera (Bone & Kuroda, 1996; Deng et al., 2011; Jiang et al., 2015; Marín et al., 1996; Nozawa et al., 2014; Rose et al., 2016; Vicoso & Bachtrog, 2015)) suggesting that this mechanism may be universal for insect species with heterogametic males.
5. CONCLUSIONS
How consistent the consequences of sex linkage are for gene sequence and expression evolution across taxa remains an open question. Here, we examine several key aspects of sex chromosome evolution in a previously neglected group, phasmids. Overall, we find evidence for several predicted consequences, including complete dosage compensation of the X (in somatic tissues) and a faster rate of evolution of X‐linked than autosomal genes. By contrast, we find little evidence that sex linkage facilitates the accumulation of sexually antagonistic alleles. While our results were consistent across different Timema species, they also show key differences from studies in other taxa, highlighting the importance of studying sex chromosome evolution across a diverse set of species to distinguish general patterns caused by sex linkage from species‐specific idiosyncrasies.
AUTHOR CONTRIBUTIONS
D.J.P. and T.S. designed the study. Z.D. performed molecular work. K.S.J., and D.J.P. analysed the data with input from T.S. and M.R.R.. D.J.P. and T.S wrote the paper with input from all authors.
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/jeb.14075.
Supporting information
Appendix S1
ACKNOWLEDGEMENTS
The authors thank Chloe Larose, Kirsten Jalvingh and Bart Zijlstra for their assistance in the field. They thank Sébastien Moretti for his assistance with running Godon. The authors also thank Matthew Hahn for useful comments. This project was supported by several grants from the Swiss Science Foundation: PP00P3_170627 and 31003A_182495 to TS as well as CRSII3_160723 to TS, MRR and Nicolas Galtier. The authors thank the former Vital‐IT platform of the SIB Swiss Institute of Bioinformatics (SIB) and the DCSR for maintaining the computer infrastructure at the University of Lausanne. Open access funding provided by Universite de Lausanne.
Parker, D. J. , Jaron, K. S. , Dumas, Z. , Robinson‐Rechavi, M. , & Schwander, T. (2022). X chromosomes show relaxed selection and complete somatic dosage compensation across Timema stick insect species. Journal of Evolutionary Biology, 35, 1734–1750. 10.1111/jeb.14075
Contributor Information
Darren J. Parker, Email: d.parker@bangor.ac.uk.
Tanja Schwander, Email: tanja.schwander@unil.ch.
DATA AVAILABILITY STATEMENTS
Raw sequence reads have been deposited in NCBI's sequence read archive under the bioproject: PRJNA725673 (Table S8). Scripts for the analyses in this paper are available at: https://github.com/DarrenJParker/Timema_sex_chr_evol_code and are archived at Zenodo https://doi.org/10.5281/zenodo.6876189. Data was processed to generate plots and statistics using R v4.0.3 and Python v.3.7.3 unless otherwise stated.
REFERENCES
- Albritton, S. E. , Kranz, A.‐L. , Rao, P. , Kramer, M. , Dieterich, C. , & Ercan, S. (2014). Sex‐biased gene expression and evolution of the x chromosome in nematodes. Genetics, 197, 865–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders, S. , Pyl, P. T. , & Huber, W. (2015). HTSeq–a python framework to work with high‐throughput sequencing data. Bioinformatics, 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbuthnott, D. , Crespi, B. J. , & Schwander, T. (2015). Female stick insects mate multiply to find compatible mates. The American Naturalist, 186, 519–530. [DOI] [PubMed] [Google Scholar]
- Arnqvist, G. , Vellnow, N. , & Rowe, L. (2014). The effect of epistasis on sexually antagonistic genetic variation. Proceedings of the Royal Society B: Biological Sciences, 281, 20140489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelsson, E. , Smith, N. G. C. , Sundström, H. , Berlin, S. , & Ellegren, H. (2004). Male‐biased mutation rate and divergence in autosomal, Z‐linked and W‐linked introns of chicken and Turkey. Molecular Biology and Evolution, 21, 1538–1547. [DOI] [PubMed] [Google Scholar]
- Bachtrog, D. , Kirkpatrick, M. , Mank, J. E. , McDaniel, S. F. , Pires, J. C. , Rice, W. , & Valenzuela, N. (2011). Are all sex chromosomes created equal? Trends in Genetics, 27, 350–357. [DOI] [PubMed] [Google Scholar]
- Backström, N. , Fagerberg, S. , & Ellegren, H. (2008). Genomics of natural bird populations: A gene‐based set of reference markers evenly spread across the avian genome. Molecular Ecology, 17, 964–980. [DOI] [PubMed] [Google Scholar]
- Benjamini, Y. , & Hochberg, Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society: Series B: Statistical Methodology, 57, 289–300. [Google Scholar]
- Bergero, R. , & Charlesworth, D. (2009). The evolution of restricted recombination in sex chromosomes. Trends in Ecology & Evolution, 24, 94–102. [DOI] [PubMed] [Google Scholar]
- Betancourt, A. J. , Kim, Y. , & Orr, H. A. (2004). A pseudohitchhiking model of X vs. autosomal diversity. Genetics, 168, 2261–2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler, J. A. (2016). Parallel universes for models of X chromosome dosage compensation in drosophila: A review. Cytogenetic and Genome Research, 148, 52–67. [DOI] [PubMed] [Google Scholar]
- Birchler, J. A. , & Veitia, R. A. (2012). Gene balance hypothesis: Connecting issues of dosage sensitivity across biological disciplines. Proceedings of the National Academy of Sciences of the United States of America, 109, 14746–14753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackmon, H. , & Demuth, J. P. (2014). Estimating tempo and mode of Y chromosome turnover: Explaining Y chromosome loss with the fragile Y hypothesis. Genetics, 197, 561–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A. M. , Lohse, M. , & Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bone, J. R. , & Kuroda, M. I. (1996). Dosage compensation regulatory proteins and the evolution of sex chromosomes in drosophila . Genetics, 144, 705–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradler, S. , & Buckley, T. R. (2018). Biodiversity of Phasmatodea. In Insect biodiversity (pp. 281–313). John Wiley & Sons, Ltd. [Google Scholar]
- Caballero, A. (1995). On the effective size of populations with separate sexes, with particular reference to sex‐linked genes. Genetics, 139, 1007–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charif, D. , & Lobry, J. R. (2007). SeqinR 1.0–2: A contributed package to the R project for statistical computing devoted to biological sequences retrieval and analysis. Springer Verlag. [Google Scholar]
- Charlesworth, B. (1978). Model for evolution of Y chromosomes and dosage compensation. Proceedings of the National Academy of Sciences of the United States of America, 75, 5618–5622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth, B. (1996). The evolution of chromosomal sex determination and dosage compensation. Current Biology, 6, 149–162. [DOI] [PubMed] [Google Scholar]
- Charlesworth, B. (2001). The effect of life‐history and mode of inheritance on neutral genetic variability. Genetical Research, 77, 153–166. [DOI] [PubMed] [Google Scholar]
- Charlesworth, B. (2012). The role of background selection in shaping patterns of molecular evolution and variation: Evidence from variability on the drosophila X chromosome. Genetics, 191, 233–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth, B. (2013). Background selection 20 years on: The Wilhelmine E. key 2012 invitational lecture. The Journal of Heredity, 104, 161–171. [DOI] [PubMed] [Google Scholar]
- Charlesworth, B. , Campos, J. L. , & Jackson, B. C. (2018). Faster‐X evolution: Theory and evidence from drosophila . Molecular Ecology, 27, 3753–3771. [DOI] [PubMed] [Google Scholar]
- Charlesworth, B. , Coyne, J. A. , & Barton, N. H. (1987). The relative rates of evolution of sex chromosomes and autosomes. The American Naturalist, 130, 113–146. [Google Scholar]
- Chauhan, P. , Swaegers, J. , Guillen, R. A. S. , Svensson, E. I. , Wellenreuther, M. , & Hansson, B. (2021). Genome assembly, sex‐biased gene expression and dosage compensation in the damselfly Ischnura elegans . Genomics, 113, 1828–1837. [DOI] [PubMed] [Google Scholar]
- Conrad, T. , & Akhtar, A. (2012). Dosage compensation in Drosophila melanogaster: Epigenetic fine‐tuning of chromosome‐wide transcription. Nature Reviews. Genetics, 13, 123–134. [DOI] [PubMed] [Google Scholar]
- Cortez, D. , Marin, R. , Toledo‐Flores, D. , Froidevaux, L. , Liechti, A. , Waters, P. D. , Grützner, F. , & Kaessmann, H. (2014). Origins and functional evolution of Y chromosomes across mammals. Nature, 508, 488–493. [DOI] [PubMed] [Google Scholar]
- Coyne, J. A. , & Orr, H. A. (2004). Speciation. Sinauer Associates. [Google Scholar]
- Davydov, I. I. , Salamin, N. , & Robinson‐Rechavi, M. (2019). Large‐scale comparative analysis of codon models accounting for protein and nucleotide selection. Molecular Biology and Evolution, 36, 1316–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, X. , Hiatt, J. B. , Nguyen, D. K. , Ercan, S. , Sturgill, D. , Hillier, L. W. , Schlesinger, F. , Davis, C. A. , Reinke, V. J. , Gingeras, T. R. , Shendure, J. , Waterston, R. H. , Oliver, B. , Lieb, J. D. , & Disteche, C. M. (2011). Evidence for compensatory upregulation of expressed X‐linked genes in mammals, Caenorhabditis elegans and Drosophila melanogaster . Nature Genetics, 43, 1179–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disteche, C. M. (2012). Dosage compensation of the sex chromosomes. Annual Review of Genetics, 46, 537–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren, H. (2007). Characteristics, causes and evolutionary consequences of male‐biased mutation. Proceedings of the Biological Sciences, 274, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren, H. , & Parsch, J. (2007). The evolution of sex‐biased genes and sex‐biased gene expression. Nature Reviews. Genetics, 8, 689–698. [DOI] [PubMed] [Google Scholar]
- Fraïsse, C. , Picard, M. A. L. , & Vicoso, B. (2017). The deep conservation of the lepidoptera Z chromosome suggests a non‐canonical origin of the W. Nature Communications, 8, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry, J. D. (2010). The genomic location of sexually antagonistic variation: Some cautionary comments. Evolution, 64, 1510–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson, J. R. , Chippindale, A. K. , & Rice, W. R. (2002). The X chromosome is a hot spot for sexually antagonistic fitness variation. Proceedings of the Biological Sciences, 269, 499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin, R. M. , Dean, R. , Grace, J. L. , Rydén, P. , & Friberg, U. (2013). The shared genome is a pervasive constraint on the evolution of sex‐biased gene expression. Molecular Biology and Evolution, 30, 2168–2176. [DOI] [PubMed] [Google Scholar]
- Gu, L. , & Walters, J. R. (2017). Evolution of sex chromosome dosage compensation in animals: A beautiful theory, undermined by facts and bedeviled by details. Genome Biology and Evolution, 9, 2461–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison, P. W. , Wright, A. E. , Zimmer, F. , Dean, R. , Montgomery, S. H. , Pointer, M. A. , & Mank, J. E. (2015). Sexual selection drives evolution and rapid turnover of male gene expression. Proceedings of the National Academy of Sciences of the United States of America, 112, 4393–4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl, D. L. , & Clark, A. G. (2006). Principles of population genetics (4th ed.). Sinauer Associates. [Google Scholar]
- Hitchcock, T. J. , & Gardner, A. (2020). A gene's‐eye view of sexual antagonism. Proceedings of the Biological Sciences, 287, 20201633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innocenti, P. , & Morrow, E. H. (2010). The sexually antagonistic genes of Drosophila melanogaster . PLoS Biology, 8, e1000335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaquiéry, J. , Simon, J.‐C. , Robin, S. , Richard, G. , Peccoud, J. , Boulain, H. , Legeai, F. , Tanguy, S. , Prunier‐Leterme, N. , & Le Trionnaire, G. (2021). Masculinization of the X‐chromosome in aphid soma and gonads . bioRxiv 2021.08.13.453080.
- Jaron, K. S. , Parker, D. J. , Anselmetti, Y. , Van, P. T. , Bast, J. , Dumas, Z. , Figuet, E. , François, C. M. , Hayward, K. , Rossier, V. , Simion, P. , Robinson‐Rechavi, M. , Galtier, N. , & Schwander, T. (2022). Convergent consequences of parthenogenesis on stick insect genomes. Science Advances, 8, eabg3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffries, D. L. , Lavanchy, G. , Sermier, R. , Sredl, M. J. , Miura, I. , Borzée, A. , Barrow, L. N. , Canestrelli, D. , Crochet, P.‐A. , Dufresnes, C. , Fu, J. , Ma, W.‐J. , Garcia, C. M. , Ghali, K. , Nicieza, A. G. , O’Donnell, R. P. , Rodrigues, N. , Romano, A. , Martínez‐Solano, Í. , … Perrin, N. (2018). A rapid rate of sex‐chromosome turnover and non‐random transitions in true frogs. Nature Communications, 9, 4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings, W. B. , & Edwards, S. V. (2005). Speciational history of Australian grass finches (Poephila) inferred from thirty gene trees. Evolution, 59, 2033–2047. [PubMed] [Google Scholar]
- Jiang, X. , Biedler, J. K. , Qi, Y. , Hall, A. B. , & Tu, Z. (2015). Complete dosage compensation in Anopheles stephensi and the evolution of sex‐biased genes in mosquitoes. Genome Biology and Evolution, 7, 1914–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley, P. D. , Ness, R. W. , Halligan, D. L. , & Haddrill, P. R. (2014). Estimation of the spontaneous mutation rate per nucleotide site in a Drosophila melanogaster full‐sib family. Genetics, 196, 313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley, P. D. , Pinharanda, A. , Ness, R. W. , Simpson, F. , Dasmahapatra, K. K. , Mallet, J. , Davey, J. W. , & Jiggins, C. D. (2015). Estimation of the spontaneous mutation rate in Heliconius melpomene . Molecular Biology and Evolution, 32, 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly, W. G. , Schaner, C. E. , Dernburg, A. F. , Lee, M.‐H. , Kim, S. K. , Villeneuve, A. M. , & Reinke, V. (2002). X‐chromosome silencing in the germline of C. elegans . Development, 129, 479–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khil, P. P. , Smirnova, N. A. , Romanienko, P. J. , & Camerini‐Otero, R. D. (2004). The mouse X chromosome is enriched for sex‐biased genes not subject to selection by meiotic sex chromosome inactivation. Nature Genetics, 36, 642–646. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick, M. , & Hall, D. W. (2004). Male‐biased mutation, sex linkage, and the rate of adaptive evolution. Evolution, 58, 437–440. [PubMed] [Google Scholar]
- Korneliussen, T. S. , Albrechtsen, A. , & Nielsen, R. (2014). ANGSD: Analysis of next generation sequencing data. BMC Bioinformatics, 15, 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahn, B. T. , & Page, D. C. (1999). Four evolutionary strata on the human X chromosome. Science, 286, 964–967. [DOI] [PubMed] [Google Scholar]
- Lee, J. T. (2005). Sex chromosome inactivation: The importance of pairing. Current Biology, 15, R249–R252. [DOI] [PubMed] [Google Scholar]
- Lenormand, T. , Fyon, F. , Sun, E. , & Roze, D. (2020). Sex chromosome degeneration by regulatory evolution. Current Biology, 30, 3001–3006.e5. [DOI] [PubMed] [Google Scholar]
- Lenormand, T. , & Roze, D. (2021). Y recombination arrest and degeneration in the absence of sexual dimorphism . bioRxiv 2021.05.18.444606. [DOI] [PubMed]
- Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM . arXiv 1303.3997. arxiv.org.
- Lucchesi, J. C. (1998). Dosage compensation in flies and worms: The ups and downs of X‐chromosome regulation. Current Opinion in Genetics & Development, 8, 179–184. [DOI] [PubMed] [Google Scholar]
- Mahadevaraju, S. , Fear, J. M. , Akeju, M. , Galletta, B. J. , Pinheiro, M. M. L. S. , Avelino, C. C. , Cabral‐de‐Mello, D. C. , Conlon, K. , Dell’Orso, S. , Demere, Z. , Mansuria, K. , Mendonça, C. A. , Palacios‐Gimenez, O. M. , Ross, E. , Savery, M. , Yu, K. , Smith, H. E. , Sartorelli, V. , Yang, H. , … Oliver, B. (2021). Dynamic sex chromosome expression in drosophila male germ cells. Nature Communications, 12, 892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan, S. , & Bachtrog, D. (2015). Partial dosage compensation in Strepsiptera, a sister group of beetles. Genome Biology and Evolution, 7, 591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mank, J. E. (2009). The W, X, Y and Z of sex‐chromosome dosage compensation. Trends in Genetics, 25, 226–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mank, J. E. (2013). Sex chromosome dosage compensation: Definitely not for everyone. Trends in Genetics, 29, 677–683. [DOI] [PubMed] [Google Scholar]
- Mank, J. E. , & Ellegren, H. (2009). All dosage compensation is local: Gene‐by‐gene regulation of sex‐biased expression on the chicken Z chromosome. Heredity, 102, 312–320. [DOI] [PubMed] [Google Scholar]
- Mank, J. E. , Hosken, D. J. , & Wedell, N. (2014). Conflict on the sex chromosomes: Cause, effect, and complexity. Cold Spring Harbor Perspectives in Biology, 6, a017715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mank, J. E. , Vicoso, B. , Berlin, S. , & Charlesworth, B. (2010). Effective population size and the faster‐X effect: Empirical results and their interpretation. Evolution, 64, 663–674. [DOI] [PubMed] [Google Scholar]
- Marçais, G. , Delcher, A. L. , Phillippy, A. M. , Coston, R. , Salzberg, S. L. , & Zimin, A. (2018). MUMmer4: A fast and versatile genome alignment system. PLoS Computational Biology, 14, e1005944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marescalchi, O. , Masetti, M. , & Scali, V. (1986). C‐banding karyotype and male meiosis of Acrophylla wuelfingi (redtenbacher) (Phasmatodea: Phasmatidae). Australian Journal of Entomology, 25, 223–227. [Google Scholar]
- Marín, I. , Franke, A. , Bashaw, G. J. , & Baker, B. S. (1996). The dosage compensation system of drosophila is co‐opted by newly evolved X chromosomes. Nature, 383, 160–163. [DOI] [PubMed] [Google Scholar]
- Marín, I. , Siegal, M. L. , & Baker, B. S. (2000). The evolution of dosage‐compensation mechanisms. BioEssays, 22, 1106–1114. [DOI] [PubMed] [Google Scholar]
- Marshall Graves, J. A. (2015). Evolution of vertebrate sex chromosomes and dosage compensation. Nature Reviews. Genetics, 17, 33–46. [DOI] [PubMed] [Google Scholar]
- Martin, M. (2011). Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet J, 17, 10–12. [Google Scholar]
- Meiklejohn, C. D. , Landeen, E. L. , Cook, J. M. , Kingan, S. B. , & Presgraves, D. C. (2011). Sex chromosome‐specific regulation in the drosophila male germline but little evidence for chromosomal dosage compensation or meiotic inactivation. PLoS Biology, 9, e1001126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisel, R. P. , & Connallon, T. (2013). The faster‐X effect: Integrating theory and data. Trends in Genetics, 29, 537–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, B. J. (2000). Sex in the worm: Counting and compensating X‐chromosome dose. Trends in Genetics, 16, 247–253. [DOI] [PubMed] [Google Scholar]
- Montgomery, S. H. , & Mank, J. E. (2016). Inferring regulatory change from gene expression: The confounding effects of tissue scaling. Molecular Ecology, 25, 5114–5128. [DOI] [PubMed] [Google Scholar]
- Mullon, C. , Pomiankowski, A. , & Reuter, M. (2012). The effects of selection and genetic drift on the genomic distribution of sexually antagonistic alleles. Evolution, 66, 3743–3753. [DOI] [PubMed] [Google Scholar]
- Mullon, C. , Wright, A. E. , Reuter, M. , Pomiankowski, A. , & Mank, J. E. (2015). Evolution of dosage compensation under sexual selection differs between X and Z chromosomes. Nature Communications, 6, 7720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosil, P. , Villoutreix, R. , de Carvalho, C. F. , Farkas, T. E. , Soria‐Carrasco, V. , Feder, J. L. , Crespi, B. J. , & Gompert, Z. (2018). Natural selection and the predictability of evolution in Timema stick insects. Science, 359, 765–770. [DOI] [PubMed] [Google Scholar]
- Nozawa, M. , Fukuda, N. , Ikeo, K. , & Gojobori, T. (2014). Tissue‐ and stage‐dependent dosage compensation on the neo‐X chromosome in Drosophila pseudoobscura . Molecular Biology and Evolution, 31, 614–624. [DOI] [PubMed] [Google Scholar]
- Nunney, L. (1993). The influence of mating system and overlapping generations on effective population size. Evolution, 47, 1329–1341. [DOI] [PubMed] [Google Scholar]
- Ohno, S. (1967). Sex chromosomes and sex‐linked genes. Springer Verlag. [Google Scholar]
- Oliver, B. (2002). Genetic control of germline sexual dimorphism in drosophila . International Review of Cytology, 219, 1–60. [DOI] [PubMed] [Google Scholar]
- Pal, A. , & Vicoso, B. (2015). The X chromosome of hemipteran insects: Conservation, dosage compensation and sex‐biased expression. Genome Biology and Evolution, 7, 3259–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer, D. H. , Rogers, T. F. , Dean, R. , & Wright, A. E. (2019). How to identify sex chromosomes and their turnover. Molecular Ecology, 28, 4709–4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker, D. J. , Bast, J. , Jalvingh, K. , Dumas, Z. , Robinson‐Rechavi, M. , & Schwander, T. (2019a). Repeated evolution of asexuality involves convergent gene expression changes. Molecular Biology and Evolution, 36, 350–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker, D. J. , Bast, J. , Jalvingh, K. , Dumas, Z. , Robinson‐Rechavi, M. , & Schwander, T. (2019b). Sex‐biased gene expression is repeatedly masculinized in asexual females. Nature Communications, 10, 4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhurst, S. M. , & Meneely, P. M. (1994). Sex determination and dosage compensation: Lessons from flies and worms. Science, 264, 924–932. [DOI] [PubMed] [Google Scholar]
- Parsch, J. , & Ellegren, H. (2013). The evolutionary causes and consequences of sex‐biased gene expression. Nature Reviews. Genetics, 14, 83–87. [DOI] [PubMed] [Google Scholar]
- Payseur, B. A. , Presgraves, D. C. , & Filatov, D. A. (2018). Introduction: Sex chromosomes and speciation. Molecular Ecology, 27, 3745–3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinharanda, A. , Rousselle, M. , Martin, S. H. , Hanly, J. J. , Davey, J. W. , Kumar, S. , Galtier, N. , & Jiggins, C. D. (2019). Sexually dimorphic gene expression and transcriptome evolution provide mixed evidence for a fast‐Z effect in Heliconius . Journal of Evolutionary Biology, 32, 194–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirrotta, V. (2002). Silence in the germ. Cell, 110, 661–664. [DOI] [PubMed] [Google Scholar]
- Pokorná, M. , & Kratochvíl, L. (2009). Phylogeny of sex‐determining mechanisms in squamate reptiles: Are sex chromosomes an evolutionary trap? Zoological Journal of the Linnean Society, 156, 168–183. [Google Scholar]
- Pool, J. E. , & Nielsen, R. (2007). Population size changes reshape genomic patterns of diversity. Evolution, 61, 3001–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Primmer, C. R. , Borge, T. , Lindell, J. , & Saetre, G.‐P. (2002). Single‐nucleotide polymorphism characterization in species with limited available sequence information: High nucleotide diversity revealed in the avian genome. Molecular Ecology, 11, 603–612. [DOI] [PubMed] [Google Scholar]
- Prince, E. G. , Kirkland, D. , & Demuth, J. P. (2010). Hyperexpression of the X chromosome in both sexes results in extensive female bias of X‐linked genes in the flour beetle. Genome Biology and Evolution, 2, 336–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan, A. R. , & Hall, I. M. (2010). BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics, 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . (2017). R: A language and environment for statistical computing. R Foundation for Statistical Computing. [Google Scholar]
- Rayner, J. G. , Hitchcock, T. J. , & Bailey, N. W. (2021). Variable dosage compensation is associated with female consequences of an X‐linked, male‐beneficial mutation. Proceedings of the Biological Sciences, 288, 20210355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice, W. R. (1984). Sex chromosomes and the evolution of sexual dimorphism. Evolution, 38, 735–742. [DOI] [PubMed] [Google Scholar]
- Richard, G. , Legeai, F. , Prunier‐Leterme, N. , Bretaudeau, A. , Tagu, D. , Jaquiéry, J. , & Le Trionnaire, G. (2017). Dosage compensation and sex‐specific epigenetic landscape of the X chromosome in the pea aphid. Epigenetics & Chromatin, 10, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riesch, R. , Muschick, M. , Lindtke, D. , Villoutreix, R. , Comeault, A. A. , Farkas, T. E. , Timothy, E. , Lucek, K. , Hellen, E. , Soria‐Carrasco, V. , Dennis, S. R. , de Carvalho, C. F. , Safran, R. J. , Sandoval, C. P. , Feder, J. , Gries, R. , Crespi, B. J. , Gries, G. , Gompert, Z. , & Nosil, P. (2017). Transitions between phases of genomic differentiation during stick‐insect speciation. Nature Ecology & Evolution, 1, Article number: 0082. [DOI] [PubMed] [Google Scholar]
- Robinson, M. D. , McCarthy, D. J. , & Smyth, G. K. (2010). edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose, G. , Krzywinska, E. , Kim, J. , Revuelta, L. , Ferretti, L. , & Krzywinski, J. (2016). Dosage compensation in the African malaria mosquito Anopheles gambiae . Genome Biology and Evolution, 8, 411–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzicka, F. , & Connallon, T. (2020). Is the X chromosome a hot spot for sexually antagonistic polymorphisms? Biases in current empirical tests of classical theory. Proceedings of the Royal Society B: Biological Sciences, 287, 20201869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzicka, F. , Hill, M. S. , Pennell, T. M. , Flis, I. , Ingleby, F. C. , Mott, R. , Fowler, K. , Morrow, E. H. , & Reuter, M. (2019). Genome‐wide sexually antagonistic variants reveal long‐standing constraints on sexual dimorphism in fruit flies. PLoS Biology, 17, e3000244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangrithi, M. N. , & Turner, J. M. A. (2018). Mammalian X chromosome dosage compensation: Perspectives from the germ line. BioEssays, 40, e1800024. [DOI] [PubMed] [Google Scholar]
- Sayadi, A. , Martinez Barrio, A. , Immonen, E. , Dainat, J. , Berger, D. , Tellgren‐Roth, C. , Nystedt, B. , & Arnqvist, G. (2019). The genomic footprint of sexual conflict. Nature Ecology and Evolution, 3, 1725–1730. [DOI] [PubMed] [Google Scholar]
- Schwander, T. , & Crespi, B. J. (2009). Multiple direct transitions from sexual reproduction to apomictic parthenogenesis in Timema stick insects. Evolution, 63, 84–103. [DOI] [PubMed] [Google Scholar]
- Shetty, S. , Griffin, D. K. , & Graves, J. A. (1999). Comparative painting reveals strong chromosome homology over 80 million years of bird evolution. Chromosome Research, 7, 289–295. [DOI] [PubMed] [Google Scholar]
- Simon, S. , Letsch, H. , Bank, S. , Buckley, T. R. , Donath, A. , Liu, S. , Machida, R. , Meusemann, K. , Misof, B. , Podsiadlowski, L. , Zhou, X. , Wipfler, B. , & Bradler, S. (2019). Old World and New World Phasmatodea: Phylogenomics resolve the evolutionary history of stick and leaf insects. Frontiers in Ecology and Evolution, 7, 345. [Google Scholar]
- Straub, T. , & Becker, P. B. (2011). Transcription modulation chromosome‐wide: Universal features and principles of dosage compensation in worms and flies. Current Opinion in Genetics & Development, 21, 147–153. [DOI] [PubMed] [Google Scholar]
- Van der Auwera, G. A. , Carneiro, M. O. , Hartl, C. , Poplin, R. , Del Angel, G. , Levy‐Moonshine, A. , Jordan, T. , Shakir, K. , Roazen, D. , Thibault, J. , Banks, E. , Garimella, K. V. , Altshuler, D. , Gabriel, S. , & DePristo, M. A. (2013). From FastQ data to high confidence variant calls: The genome analysis toolkit best practices pipeline. Current Protocols in Bioinformatics, 43, 11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vibranovski, M. D. (2014). Meiotic sex chromosome inactivation in drosophila . Journal of Genomics, 2, 104–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vibranovski, M. D. , Zhang, Y. , & Long, M. (2009). General gene movement off the X chromosome in the drosophila genus. Genome Research, 19, 897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicoso, B. (2019). Molecular and evolutionary dynamics of animal sex‐chromosome turnover. Nature Ecology and Evolution, 3, 1632–1641. [DOI] [PubMed] [Google Scholar]
- Vicoso, B. , & Bachtrog, D. (2009). Progress and prospects toward our understanding of the evolution of dosage compensation. Chromosome Research, 17, 585–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicoso, B. , & Bachtrog, D. (2015). Numerous transitions of sex chromosomes in Diptera. PLoS Biology, 13, e1002078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicoso, B. , & Charlesworth, B. (2009). Effective population size and the faster‐X effect: An extended model. Evolution, 63, 2413–2426. [DOI] [PubMed] [Google Scholar]
- Vicoso, B. , Emerson, J. J. , Zektser, Y. , Mahajan, S. , & Bachtrog, D. (2013). Comparative sex chromosome genomics in snakes: Differentiation, evolutionary strata, and lack of global dosage compensation. PLoS Biology, 11, e1001643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, M. , Zhao, Y. , & Zhang, B. (2015). Efficient test and visualization of multi‐set intersections. Scientific Reports, 5, 16923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, M. J. D. (1976). Blattodea, Mantodea, Isoptera, Grylloblattodea, Pasmatodea, Dermaptera and Embioptera. Animal cytogenetics vol. 3: Insecta 2. Gebruder Borntraeger. [Google Scholar]
- Whittle, C. A. , Kulkarni, A. , & Extavour, C. G. (2020). Absence of a faster‐X effect in beetles (Tribolium, coleoptera). G3: Genes, genomes, Genetics, 10, 1125–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson, G. S. , Breden, F. , Mank, J. E. , Ritchie, M. G. , Higginson, A. D. , Radwan, J. , Jaquiery, J. , Salzburger, W. , Arriero, E. , Barribeau, S. M. , Phillips, P. C. , Renn, S. C. , & Rowe, L. (2015). The locus of sexual selection: Moving sexual selection studies into the post‐genomics era. Journal of Evolutionary Biology, 28, 739–755. [DOI] [PubMed] [Google Scholar]
- Wilson Sayres, M. A. (2018). Genetic diversity on the sex chromosomes. Genome Biology and Evolution, 10, 1064–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, A. E. , Dean, R. , Zimmer, F. , & Mank, J. E. (2016). How to make a sex chromosome. Nature Communications, 7, 12087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S. (1931). Evolution in mendelian populations. Genetics, 16, 97–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S. (1969). Evolution and the genetics of populations, volume 2: Theory of gene frequencies. University of Chicago Press. [Google Scholar]
- Xu, L. , & Zhou, Q. (2020). The female‐specific W chromosomes of birds have conserved gene contents but are not feminized. Genes, 11, 1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z. , Nielsen, R. , Goldman, N. , & Pedersen, A. M. (2000). Codon‐substitution models for heterogeneous selection pressure at amino acid sites. Genetics, 155, 431–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Sturgill, D. , Parisi, M. , Kumar, S. , & Oliver, B. (2007). Constraint and turnover in sex‐biased gene expression in the genus drosophila . Nature, 450, 233–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Data Availability Statement
Raw sequence reads have been deposited in NCBI's sequence read archive under the bioproject: PRJNA725673 (Table S8). Scripts for the analyses in this paper are available at: https://github.com/DarrenJParker/Timema_sex_chr_evol_code and are archived at Zenodo https://doi.org/10.5281/zenodo.6876189. Data was processed to generate plots and statistics using R v4.0.3 and Python v.3.7.3 unless otherwise stated.