

Abstract
TetR/AcrR‐like transcription regulators enable bacteria to sense a wide variety of chemical compounds and to dynamically adapt the expression levels of specific genes in response to changing growth conditions. Here, we describe the structural characterisation of SCO3201, an atypical TetR/AcrR family member from Streptomyces coelicolor that strongly represses antibiotic production and morphological development under conditions of overexpression. We present crystal structures of SCO3201 in its ligand‐free state as well as in complex with an unknown inducer, potentially a polyamine. In the ligand‐free state, the DNA‐binding domains of the SCO3201 dimer are held together in an unusually compact conformation and, as a result, the regulator cannot span the distance between the two half‐sites of its operator. Interaction with the ligand coincides with a major structural rearrangement and partial conversion of the so‐called hinge helix (α4) to a 310‐conformation, markedly increasing the distance between the DNA‐binding domains. In sharp contrast to what was observed for other TetR/AcrR‐like regulators, the increased interdomain distance might facilitate rather than abrogate interaction of the dimer with the operator. Such a ‘reverse’ induction mechanism could expand the regulatory repertoire of the TetR/AcrR family and may explain the dramatic impact of SCO3201 overexpression on the ability of S. coelicolor to generate antibiotics and sporulate.
Keywords: gene regulation, polyamine, SAXS, transcription, X‐ray crystallography
Crystallographic analysis of SCO3201 reveals an unusually compact ligand‐free state, incompatible with DNA‐binding, which is also observed in solution by small‐angle X‐ray scattering. An alternative conformation seen in a second crystal structure coincides with the presence of an unidentified ligand, a potential polyamine. The allosteric transition linking the two states is characterised by a remarkable α‐to‐310 transition within the hinge helix that physically connects the DNA‐binding and ligand‐binding domains.
Abbreviations
- HTH
helix‐turn‐helix motif
- SAXS
small‐angle X‐ray scattering
- RMSD
root‐mean‐square deviation
Introduction
Streptomyces are a genus of gram‐positive, soil‐dwelling bacteria, capable of producing a wide variety of secondary metabolites of immediate biomedical interest. These include an estimated 80% of currently available antibiotics, as well as numerous antitumour agents, antifungals and antivirals [1, 2, 3]. Certain species of Streptomyces have in addition gained popularity as hosts for heterologous protein expression [4]. A characteristic feature of streptomycetes is their complex differentiation cycle, which involves profound changes in metabolism as well as in morphology [5, 6, 7]. The differentiation process is triggered by variations in the intra‐ and extracellular concentrations of specific compounds, particularly nutrients that constitute vital sources of phosphorus and nitrogen [8]. Concentrations of these substances are often gauged by one‐component systems [9], such as the TetR/AcrR [10] family of transcription regulators. Members of this family act as homodimeric repressors or activators, which bind to palindromic sequences in the promoters of target genes and are released by a cognate small molecule, the inducer. TetR/AcrR‐like regulators are bipartite and consist of an N‐terminal DNA‐binding domain that comprises a helix‐turn‐helix (HTH) motif, followed by a C‐terminal domain required for dimerisation and inducer recognition [11]. In all cases where crystal structures of DNA‐bound as well as inducer‐bound forms are available, interaction with the small molecule is found to allosterically increase the distance between the DNA‐binding domains, which then exceeds the spacing between the half‐sites in the operator [10, 12]. As a consequence, DNA‐binding is abrogated and transcription enabled [13].
SCO3201, a TetR/AcrR‐like regulator from Streptomyces coelicolor, was originally described by Xu et al. [14], who screened a genomic library for factors that influence antibiotic production and development. Overexpression of SCO3201 was found to strongly reduce levels of actinorhodin (ACT), a polyketide antibiotic, as well as to inhibit morphological differentiation and sporulation of S. coelicolor [14]. The protein was shown to physically interact with the promoters of at least 16 genes, including some that encode downstream regulators, thereby directly and indirectly affecting numerous genes that play a role in the differentiation process [15]. An initial crystallographic analysis of SCO3201 at 2.1 Å resolution revealed a highly asymmetric homodimer, with an unidentified small molecule bound to one of its subunits [15]. In this study, we describe a new crystal form that lacks the unknown ligand, leading to an essentially symmetric and unusually compact conformation of the dimer. This compact conformation is also observed in solution by means of small‐angle X‐ray scattering (SAXS). Furthermore, we have repeated the crystal structure determination of the asymmetric, ligand‐bound dimer at a higher resolution (1.89 Å), providing more detailed insight into the nature of the small molecule, a possible polyamine. Taken together, our data suggest that a novel allosteric mechanism underlies the ligand response of SCO3201.
Results
The crystal structure of the ligand‐free form of SCO3201 reveals an unusually compact ‘closed’ state
The asymmetric unit of the ligand‐free P21 crystal form that we obtained contains four polypeptide chains (A–D), which form two homodimers (AB and CD). The overall fold of SCO3201 (Fig. 1) is typical of the TetR/AcrR family of transcription factors [10] and comprises an all‐helical regulatory domain that also mediates dimerisation, preceded by a DNA‐binding three‐helix‐bundle (α1–α3). The second and third helix of this DNA‐binding domain correspond to a classical helix‐turn‐helix (HTH) motif. A distinguishing feature of SCO3201 is the additional α‐helix (α8) within a solvent‐exposed loop of the canonical TetR/AcrR fold, which juts out from the core of the ligand‐binding domain (Fig. 1A). However, the most striking aspect of the structure is arguably the exceptionally short distance separating the HTH motifs of the homodimer. The recognition helices (α3), which are crucial for sequence‐specific DNA binding [10], are 22.6 Å apart in dimer AB and 24.5 Å in dimer CD (this is measured as the distance between the centres of mass of the helical residues, amino acid residues 66–72, considering main‐chain atoms only). In most structures of ligand‐free and DNA‐bound TetR/AcrR‐like regulators the corresponding distance is within the range 35–41 Å, while even longer distances have been observed for inducer‐bound repressors [16]. The positioning of the DNA‐binding domains of SCO3201, which gives the dimer a remarkably compact (‘closed’) appearance compared to other members of the TetR/AcrR family, mainly arises because of a pronounced bend in the so‐called hinge helix (α4). This helix plays a key role in the allosteric induction mechanism of TetR/AcrR‐like repressors, as it physically links the HTH motif to the ligand‐binding domain. Interestingly, the equivalent of α4 in SCO3201 is a composite helix, as its N‐terminal part in direct contact with the DNA‐binding domain adopts a 310‐conformation. This results in a bend of approximately 30° at the junction with the α‐helical section, in good agreement with the average bend angle for 310‐α transitions (37° ± 19°) observed by Pal et al. [17]. Each of the four independent protein chains in the crystal features a similarly bent composite hinge helix and the AB and CD dimers can be superimposed with an RMSD of 1.6 Å for a total of 358 aligned Cα‐atoms (Fig. 1B).
Fig. 1.
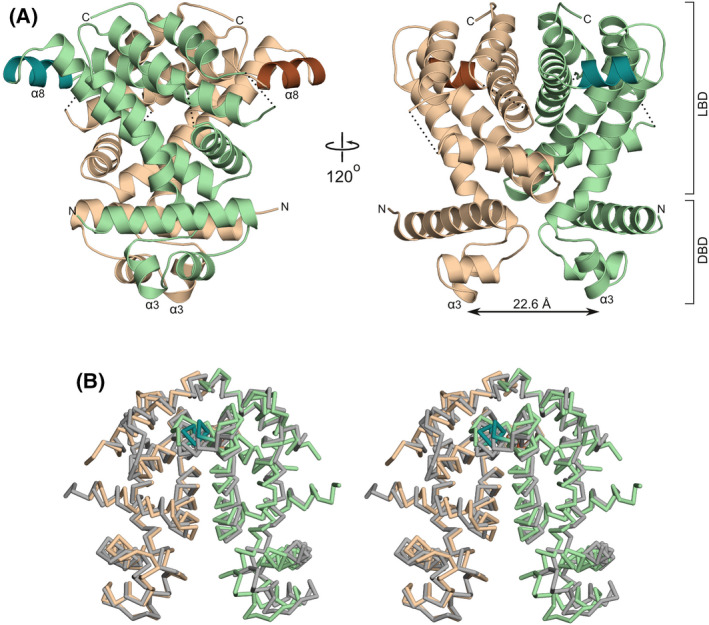
The structure of the ligand‐free SCO3201 homodimer. (A) Ribbon representation of the dimer in two orientations. Chains A and B are shown in beige and green, respectively. The exposed helix α8, which has no equivalent in other TetR/AcrR regulators, is highlighted in brown (chain A) and dark green (chain B). Designations LBD and DBD indicate the ligand‐binding and DNA‐binding domains, respectively. The distance separating the recognition helices (α3) of the two HTH‐motifs is indicated by an arrow. (B) Stereo view of the superposed AB (colours as in panel A) and CD (grey) homodimers, shown as Cα‐traces.
Ligand binding coincides with major conformational changes
A second crystal form, belonging to space group P212121, turned out to be indistinguishable from the one that we had already analysed in earlier work [15]. However, crystals diffracted to higher resolution this time (1.86 Å versus 2.1 Å). As in the earlier structure, the asymmetric unit comprises a single homodimer, whose subunits adopt markedly different conformations. While chain B is virtually identical to the four molecules in the ligand‐free structure (with Cα‐RMSD values ranging between 0.79 and 1.0 Å), the DNA‐binding domain of chain A has moved in the outward direction by approximately 10 Å, resulting in an increased α3–α3 distance of 34.8 Å (Fig. 2). This change coincides with the binding of an unknown ligand to a cavity within the A‐subunit (Figs 2 and 3). The movement of the DNA‐binding domain is caused by a repositioning of the composite hinge helix, which undergoes a 10° outward rotation (away from the ligand binding pocket) as well as a 3 Å shift in the upward direction in Fig. 2A. This movement is accompanied by a rearrangement of the adjacent helices α5, α6 and α7. All of these changes result from direct contacts between the helices and the ligand, which force the binding pocket to expand. Interestingly, the upward shift of the hinge helix would cause its outer 310‐segment to collide with the adjacent α7 helix, while the DNA‐binding domain would be drawn into the ligand‐binding domain. Such clashes are avoided by an extension of the 310‐helix (Fig. 2B), which now includes two additional residues belonging to the α‐helical region in the ligand‐free structure (Ala81 and Pro82). The result is not only an appreciable elongation of the hinge helix, but also a reduction of the bend angle to approximately 15°. This straightening is a direct consequence of the fact that the remodelled 310‐α junction does not obey the sequence requirements for composite helices that were formulated by Pal et al. [17]. In particular, Pro82 no longer occupies the favourable second position in the α‐helix, but instead acts as the final residue of the 310‐helix. This precludes proper capping of the α‐helix and formation of a larger bend. Instead, Pro82 is rigidly integrated into the structural framework of the 310‐helix by hydrogen bonds between its side chain (Cδ‐H) and the carbonyl moieties of Glu78 and Ala79 (with Cδ‐O distances of 3.1 and 3.4 Å, respectively).
Fig. 2.
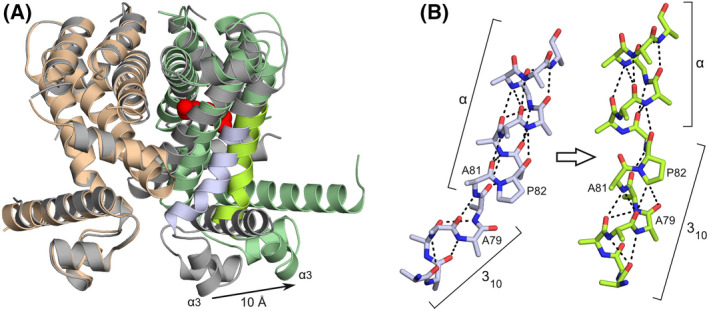
Comparison of the ligand‐bound and ligand‐free SCO3201 structures. (A) Superposition of liganded (protein chains shown as beige and green ribbons with the unknown small molecule, tentatively modelled as spermidine, represented by red spheres) and free SCO3201 (dimer AB, grey ribbons). The outward movement of one of the HTH motifs by 10 Å, which coincides with ligand binding to the same protein chain, is indicated by an arrow. The portion of the hinge helix that undergoes substantial conformational changes (bright green and blue in the respective structures) is shown in more detail in panel (B). (B) Conformational changes within the composite hinge helix upon ligand binding. The N‐terminal 310‐segment is extended by two residues previously part of the α‐helical region (Ala81 and Pro82), leading to stretching and straightening of the hinge helix. All side‐chain atoms beyond Cβ have been removed to improve clarity, except those of Pro82. Hydrogen bonds are shown as dashed lines.
Fig. 3.
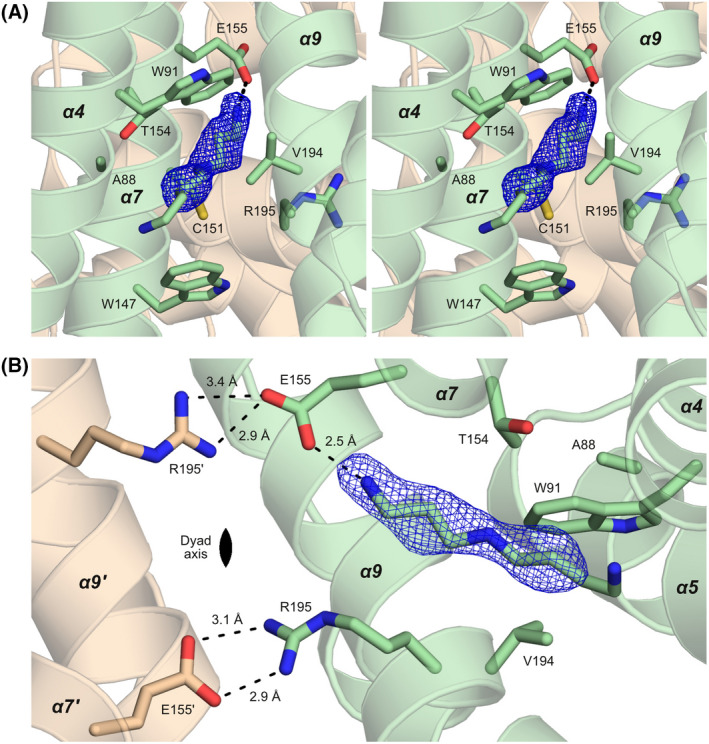
The interaction of SCO3201 with the inducer. (A) Stereo image of the liganded cavity. The view is from the opening of the cavity at the protein surface towards the interior of the dimer, with the dyad axis oriented vertically in the plane of the image. Difference density (mFo‐DFc) corresponding to the inducer, contoured at 3.0 σ, is shown in blue. Nearby side chains are represented as stick models, as is the spermidine molecule that was modelled into the density. The short‐distance interaction between the head group of the inducer and the side chain of Glu155 is represented by a dashed line. (B) The dimerisation interface of ligand‐bound SCO3201, viewed along the dyad axis. Hydrogen bonds involving the inducer, Glu155 and Arg195 are shown as dashed lines, together with the corresponding interatomic distances. As can be seen here, the local non‐crystallographic symmetry of the dimerisation interface (the Glu155‐Arg195′ and Glu155′‐Arg195 salt bridges in particular) is broken by the interaction of Glu155 with the ligand.
In contrast to the extensive changes to helices α4–α7, the main dimerisation interface consisting of helices α9 and α10 remains essentially unaffected by the presence of the ligand. These helices can be superimposed onto the corresponding part of the ligand‐free AB dimer with a Cα‐RMSD of 0.38 Å (68 aligned residues). In the ligand‐free dimer, however, two highly similar salt bridges related by non‐crystallographic symmetry (Fig. 3B) connect the carboxylate of Glu155 in helix α7 to the guanidinium moiety of Arg195 in helix α9 of the dimerisation partner. In the liganded structure, the head group of the unknown small molecule is at 2.5 Å, that is well within hydrogen bonding distance, from one of the carboxylate oxygen atoms of Glu155. This interaction abrogates one of the hydrogen bonds between Glu155 and the side chain of Arg195′, thereby breaking the electrostatic symmetry of the two salt bridges.
The elusive ligand may be an aliphatic polyamine
Unfortunately, mass spectrometry analysis of dissolved crystals failed to identify the unknown ligand. However, owing to the markedly improved resolution of the data, the contours of the small molecule are now clearly defined in the electron density map (Fig. 3). Although the identity of the ligand cannot be unambiguously deduced from the density alone, the properties of the binding site and the way it interacts with the ligand necessitate a linear aliphatic chain of at least 8 heavy (non‐hydrogen) atoms, including a positively charged (or at least polar) head group. In view of these requirements, aliphatic polyamines [18] seem to be likely candidates. Well‐balanced levels of these ubiquitous compounds, the most abundant of which are spermidine, spermine and putrescine [18, 19], are considered essential for cell growth, proliferation and survival in all organisms [20]. In Streptomyces, polyamines have been shown to play key roles in gene regulation and metabolism [21]. On the basis of these considerations, we have tentatively modelled spermidine into the electron density map. The observed Van der Waals contacts to hydrophobic residues lining the binding pocket and the short‐distance interaction of the head group with the carboxylate moiety of the Glu155 side chain are consistent with this proposition. However, other polyamines (including the closely related compounds putrescine, cadaverine and spermine) are equally plausible candidates and further biochemical studies will be required to unambiguously identify the natural ligand. As the binding site widens towards the protein surface, the outer region of the bound molecule may well be disordered and, as a result, remain invisible in the density map. Consequently, the unknown molecule could be considerably longer than the part that is actually observed.
SCO3201 adopts the closed conformation in solution
The markedly different conformations observed in our crystal structures prompted us to characterise SCO3201 in solution by means of SAXS (Fig. 4). Comparison of the extrapolated forward scattering (I 0 ) to that of a bovine serum albumin (BSA) reference sample suggests a particle mass of 46.4 kDa, while the concentration‐independent Bayesian method of Hajizadeh et al. [22], yields a value of 46.7 kDa. These results are in excellent agreement with the predicted mass of the SCO3201 homodimer (46.7 kDa). A more detailed analysis of the data using crysol [23] (Fig. 4A) indicates that the scattering from SCO3201 is explained almost to within experimental error (χ2 = 1.2) by the CD dimer from the ligand‐free crystal structure. In contrast, using the asymmetric liganded structure as a model (either with or without the small molecule) leads to a considerable lack of fit (χ2 = 2.6). The same is true for a symmetrical ‘doubly liganded’ model, constructed by superimposing a copy of the A chain of the liganded structure onto the dimerisation interface of the B chain and combining that with the original A chain (χ2 = 2.5). An ab initio model calculated from the experimental data using the program dammin [24] closely matches the dimer from 5EFY (Fig. 4B). We therefore conclude that, in our SAXS experiments, SCO3201 predominantly adopts a closed conformation similar to the one seen in the ligand‐free crystal structure. Several things may explain why the ligand‐induced conformation is not observed here. In the first place, the basic conditions (pH 8.5) might favour dissociation of the complex, as the pH of the solution could have a considerable impact on the protonation state of both the ligand and the glutamate residue it is in contact with. Indeed, crystals of the complex were obtained at pH 5.0, whereas the ligand‐free protein crystallised at pH 8.5. Compared to the crystallisation assays, the SAXS experiments were also carried out at much lower protein concentrations, which would be expected to shift the binding equilibrium towards the dissociated state. Furthermore, only a small fraction of the purified protein may have been liganded in the first place. While this might not prevent crystallisation of the liganded species, the unliganded form would in this case dominate the SAXS signal. Finally, we cannot exclude the possibility that the unknown ligand was not co‐purified from the expression host, but contained in the chemicals used during crystal structure determination, in particular the paraffin oil (a complex and poorly defined mixture of organic compounds) that we used for cryoprotection. However, mass spectrometry analysis of a protein solution extensively equilibrated with paraffin oil also failed to identify the unknown ligand.
Fig. 4.
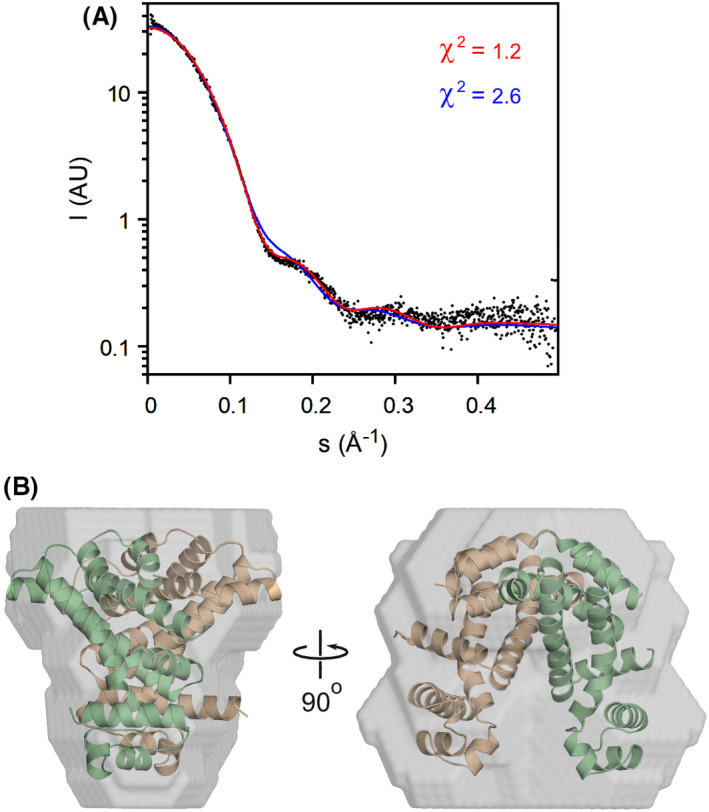
SAXS analysis of SCO3201 in solution. (A) Experimental SAXS data (black dots) and theoretical curves calculated for the ligand‐free (red) and ligand‐bound (blue) crystallographic structures. Theoretical curves were fitted to the experimental data using CRYSOL, with 7PT0 and the CD dimer of 5EFY as the models, respectively. (B) Ab initio model (grey surface) with the superposed 5EFY CD dimer shown in ribbon representation (beige and green).
Discussion
The crystallographic structure of the ligand‐free form of SCO3201 reported in the present study reveals a domain arrangement that, to our knowledge, has not been encountered in other members of the TetR/AcrR family. The DNA‐binding domains of the protein dimer are unusually close to one another, mainly as a result of a pronounced inward bend in the hinge helices. We have several reasons to believe that this arrangement is not merely the result of crystal packing effects, but corresponds to the native state of the ligand‐free regulator. First, a transition from a 310‐helix to an α‐helix is fully expected to produce the kind of kinked junction that is observed here [17]. Second, all four of the independent polypeptide chains in the crystal feature a composite hinge helix with a comparable bend, despite the fact that each chain experiences a different lattice environment. And finally, our SAXS data corroborate the existence of the closed conformation in solution.
Intriguingly, the compact arrangement of the two HTH‐motifs, with their recognition helices at less than 25 Å from each other, would preclude binding to consecutive instances of the major groove on one face of the promoter. This DNA‐binding mode, which is a hallmark of the TetR/AcrR family [10], requires an α3–α3 distance very close to 35 Å [16]. This implies that the SCO3201 dimer has to undergo a major conformational change if it is to interact with the operator. Although DNA binding by members of the TetR/AcrR family is typically accompanied by appreciable structural changes in the regulatory domain [10], all currently known cases involve DNA‐binding domains that are far apart in the free repressor and need to be brought into closer proximity to enable promoter binding. Inducers can then act simply by preventing such rearrangements [25]. Strikingly, this is the exact opposite of what we seem to observe for SCO3201.
In our second crystal form, interaction with a small‐molecule ligand causes a large (~ 10 Å) increase in the distance between the DNA‐binding domains, which would now be compatible with DNA binding. This observation suggests that SCO3201 could possibly function by means of a ‘reverse’ induction mechanism, in the sense that ligand binding might increase, rather than decrease, the affinity of the regulator for DNA. To our knowledge, such a mechanism has not yet been suggested for any naturally occurring TetR/AcrR family members. Mutagenesis studies of TetR(D), however, have given rise to artificial variants of this repressor (referred to as ‘revTetR’) that, instead of being released from DNA by tetracycline like the wild‐type protein, show enhanced promoter binding in the presence of the antibiotic [26, 27, 28]. For the related LacI/GalR family of transcription factors, cases of increased as well as decreased DNA‐binding in the presence of cognate small‐molecule ligands have been reported [29].
It is currently unclear whether or not the SCO3201 dimer is able to interact with a second inducer molecule, which might further enlarge the physical distance between the HTH motifs and lead to a complex transcriptional response to increasing ligand concentrations. It should be noted, however, that many TetR/AcrR‐like repressors only bind a single ligand molecule per homodimer [10], either in a central cavity around the symmetry axis of the dimer, as in FrrA [25], or in one of several available pockets within the individual subunits, for example in QacR [30] and TtgR [31].
The conformational changes that we observe in SCO3201 are ultimately caused by a slight expansion of the binding pocket, a direct consequence of the bulkiness of the inducer. This effect should be virtually independent of the precise binding specificity of the cavity, a phenomenon that we have also observed for the induction mechanisms of TetR(D) [32] and FrrA [25]. As noted before, modularity of ligand recognition and allostery is expected to result in a considerable degree of evolutionary flexibility, allowing existing transcriptional regulators to mutate and adapt to new inducers and regulatory circuits while their induction mechanism remains intact [32].
Loop‐to‐helix transitions within helix α4, sometimes linked to the presence of helix‐destabilising residues (Pro, Gly) and generally triggered by direct interactions of the helix with the inducer, have been reported for several TetR/AcrR‐like repressors [33, 34]. The allosteric mechanism of SCO3201, in contrast, relies on a composite hinge helix and the elongation of its 310‐segment in response to the expansion of the ligand‐binding cavity. Interconversion of 310‐ and α‐helices is believed to be an important regulatory mechanism in biomolecular signalling, in particular in voltage‐gated channels and other membrane‐associated switches [35]. However, we are not aware of such mechanisms having been described for transcription factors. A search of the PDB using DALI [36] indicates that SCO3201 most strongly resembles entry 2RAE (Z‐score: 16.4), which corresponds to the 2.2 Å crystal structure of a repressor from Rhodococcus sp. RHA1. Although this protein lacks the additional α‐helix within loop α7‐α8, the relative positioning of most of its helices is roughly comparable to that of counterparts in SCO3201 (Fig. 5A). Like SCO3201, 2RAE possesses a composite hinge helix, with a helix‐destabilising residue (Gly67) at the transition between the α‐ and 310‐segments (Fig. 5B). As in SCO3201, the 310‐segment also contains a proline residue (Pro65), whose side chain contributes to the 310 hydrogen‐bonding network. Although no ligands are present in the deposited model, inspection of the 2mFo‐DFc map reveals an area of poorly explained electron density within the putative binding cavity, presumably arising from the presence of a planar organic molecule (Fig. 5B). This observation strongly suggests that 2RAE corresponds to the induced form of the repressor. Unlike the ligand‐bound form of SCO3201, both subunits of the 2RAE homodimer are liganded (they are related by crystallographic symmetry), leading to an α3‐α3 distance that appears to be too large for DNA binding (41.8 Å). Despite this intriguing difference, SCO3201 and 2RAE together constitute a novel category of regulators whose allosteric mechanisms exploit the special properties of 310‐α junctions in composite helices [17]. A more detailed analysis of these molecular switches awaits the structural characterisation of complexes of the proteins with promoter DNA, as well as the unambiguous identification of their natural inducers.
Fig. 5.
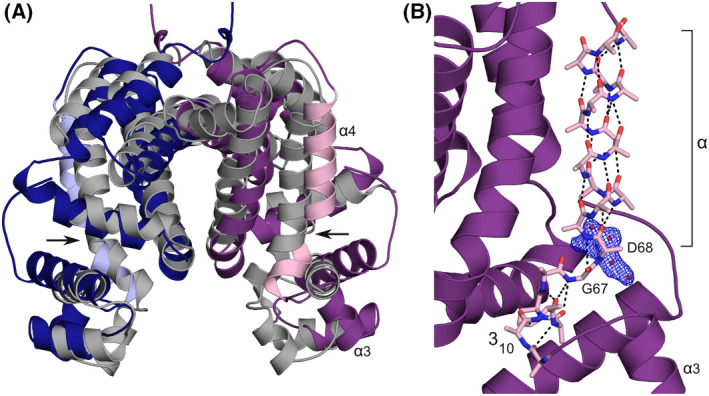
Comparison of SCO3201 to 2RAE, the most similar PDB entry identified by DALI [36]. (A) Superposition of 2RAE (chains related by crystallographic symmetry in dark blue and purple) onto the ligand‐free SCO3201 dimer (chains C and D, grey). The hinge helices (α4) of 2RAE have been highlighted in light blue and pink. The kink in the hinge helix of 2RAE, around residue Gly67, is indicated by arrows. (B) Detailed view of the composite hinge helix of 2RAE (pink stick model, side‐chain atoms beyond Cβ removed except in Pro65) with the rest of the protein chain in ribbon representation (purple). Hydrogen bonds are shown as dashed lines. Electron density (2mFo‐DFc) likely to correspond to an unidentified ligand, contoured at 1.2 σ, is shown in blue. The authors of 2RAE interpreted this density as four water molecules at less than 2.2 Å from each other (red spheres).
Involvement of SCO3201 in polyamine sensing, possibly by means of a reverse induction mechanism, may explain the dramatic effect of overexpression of the regulator on morphological differentiation and antibiotic production in S. coelicolor [14]. A recent study has demonstrated that Streptomyces are able to detoxify and assimilate polyamines very efficiently, enabling growth even under conditions where these molecules constitute the sole nitrogen source [37]. Moreover, polyamines are naturally abundant in the environments where Streptomyces are typically found [21]. In S. coelicolor, de novo biosynthesis of putrescine and spermidine has been reported in the late‐stationary phase in rich medium, while cadaverine synthesis occurred under conditions of iron limitation [38]. These observations strongly suggest that polyamines play key roles in cell homeostasis, with depletion likely to act as a nutritional stress signal. As soon as the polyamine concentration falls below a certain threshold, genes involved in morphological development and antibiotic production would need to be derepressed, triggering differentiation and antibiotic production. Overexpression of SCO3201 would amplify polyamine‐dependent repression (and potentially prolong it) as a result of the law of mass action. Moreover, sequestering of polyamine by the overexpressed regulator might protect the former from being metabolised, allowing maintenance of repression throughout growth [14]. However, the fact that deletion of SCO3201 has no detectable impact on morphological differentiation and antibiotic production [14] suggests that control of the relevant genes involves additional factors besides SCO3201. Two regulators of polyamine catabolism have been described in S. coelicolor: EpuRII (SCO5656), which controls the glnA3 gene encoding a γ‐glutamylpolyamine synthetase, and the global regulator of nitrogen metabolism, GlnR, which has been shown to interact with the glnA2 gene, encoding a second γ‐glutamylpolyamine synthetase [37]. Interestingly, the expression of glnR can only be induced by cadaverine [37], whereas the expression of EpuRII can be induced by putrescine, cadaverine and spermidine [39]. It is therefore conceivable that several dedicated factors sense different polyamines or groups of polyamines in the context of an intricate regulatory network that mediates cross‐talk between a variety of nutritional signals. Apart from sensors of nitrogen sources [40, 41, 42, 43], such a network is likely to include factors that respond to the presence of inorganic phosphate [44, 45, 46] and polyphosphates [47, 48, 49].
Materials and methods
Protein expression and purification
In all experiments, a SCO3201 deletion mutant was used that lacks the 20‐residue region of low complexity at the N‐terminus of the full‐length sequence, which we found interfered with crystallisation. The construct, corresponding to amino acids 21–216 of NCBI reference sequence WP_011028825.1, was expressed and purified essentially as described earlier [15]. In brief, the His6‐tagged protein was overproduced in Escherichia coli strain C41 using a modified pET‐28b vector that instead of the thrombin recognition sequence encodes a TEV‐protease site. Cultures in LB medium containing 50 μg·mL−1 kanamycin were grown shaking at 37 °C until they reached an OD (600 nm) of 0.8, followed by a further 4 h incubation in the presence of 1 mm IPTG. Upon centrifugation (15 min at 10 000 g , 4 °C), cells from 1 L of culture were resuspended in 30 mL lysis buffer (20 mm Tris/HCl pH 7.0, 400 mm NaCl, 20 mm of imidazole and 1 mm of DTT) and disrupted by means of sonication for 8 min on ice using a Sonopuls HD 2070 (Bandelin Electronic GmbH, Berlin, Germany). Sonication was repeated twice, with 2 min of cooling between cycles. Cell debris was removed by centrifugation for 1 h at 40 000 g and the cleared lysate applied to a Ni2+‐charged Poros 20 MC column (Thermo Fisher Scientific GmbH, Dreieich, Germany). The protein was eluted using lysis buffer containing 250 mm of imidazole. TEV protease was added directly to the eluate to a final concentration of 0.1 mg·mL−1. The solution was then incubated for 3 days at 4 °C, leading to complete removal of the His6‐tag. SCO3201 was further purified by gel filtration (Superdex 75, GE Healthcare GmbH, Munich, Germany) in a buffer containing 20 mm Tris/HCl pH 8.5, 400 mm NaCl, 50 mm imidazole and 1 mm DTT. The protein was concentrated to 30 mg·mL−1 using a Vivaspin 15 concentrator (Sartorius Lab Instruments GmbH, Göttingen, Germany) with a 10 kDa molecular weight cut‐off. The concentrate was flash‐frozen in 50 μL of aliquots using liquid nitrogen and stored at −80 °C until further use. Total protein yield was approximately 100 mg from 1 L of culture. The protein appeared as a single band on Coomassie‐stained SDS/PAGE gels.
X‐ray crystallography
Crystals of ligand‐free SCO3201 belonging to space group P21 were obtained using the hanging drop vapour diffusion method at 20 °C, with a reservoir solution containing 12% ethanol and 0.1 m Tris/HCl pH 8.5. For cryoprotection, crystals were covered in paraffin oil (alkane chain length ≥ 20, Carl Roth GmbH, Karlsruhe, Germany) prior to flash freezing in liquid nitrogen. Diffraction data to a resolution of 2.7 Å were collected at beamline BL14.2, operated by the Joint Berlin MX‐Laboratory at the BESSY II electron storage ring (Berlin‐Adlershof, Germany) [50].
Crystals of the liganded form of SCO3201, which belonged to space group P212121, were also obtained via hanging drop vapour diffusion at 20 °C. Ahead of crystallisation assays, 1 mL protein solution (2 mg·mL−1) was incubated with 10 μL of paraffin oil. The rationale behind this approach was that the oil, which was used as a cryoprotectant in the determination of the earlier structure (4CGR), might contain the unknown ligand, in which case pre‐equilibration might increase occupancy and improve crystal quality. The solution, without the organic phase, was subsequently concentrated to 50 μL (40 mg·mL−1) and used for crystallisation. The reservoir solution in these experiments contained 1 m of (NH4)H2PO4 and 0.1 m of Tris/HCl pH 5.0. Again, paraffin oil was used for cryoprotection. A dataset with a resolution of 1.86 Å was collected at beamline P13, operated by EMBL Hamburg at the PETRA III storage ring (DESY, Hamburg, Germany) [51].
All diffraction data were processed with xds [52, 53]. Both crystal structures were solved using phaser [54], in combination with the ccp4 suite [55]. For the ligand‐free form, chain B of protein data bank (PDB) entry 4CGR was used as the molecular replacement search model. Searches with chain A or the complete homodimer (AB) did not yield meaningful solutions. For the liganded form, the dimer was used as the search model. The structures were refined using the ccp4 suite [55] and refmac5 [56, 57]. All molecular graphics illustrations were produced with pymol 0.99rc6 (DeLano Scientific LLC, Palo Alto, CA, USA). Centres of mass and angles between helices were determined using calcom [58] and qhelix [59], respectively. Data collection and refinement statistics can be found in Table 1.
Table 1.
Data collection and structure refinement statistics. Values in parentheses correspond to the highest resolution shell.
| Protein or complex | SCO3201 | SCO3201/spermidine |
|---|---|---|
| PDB ID | 5EFY | 7PT0 |
| Data collection | ||
| Radiation source, beamline | BESSY, BL14.2 | PETRA III, EMBL c/o DESY, P13 |
| Detector | RAYONIX MX‐225 | Pilatus 6M |
| Wavelength (Å) | 0.9184 | 0.9763 |
| Temperature (K) | 100 | 100 |
| Resolution range (Å) | 94.6–2.70 (2.85–2.70) | 61.6–1.89 (2.00–1.89) |
|
Unit cell parameters a, b, c (Å) α, β, γ (°) |
51.79, 98.43, 89.61 90, 100.78, 90 |
55.13, 79.88, 96.87 90, 90, 90 |
| Space group | P21 | P212121 |
| Unique reflections | 23 495 (3916) | 34 934 (5288) |
| Redundancy | 3.0 (2.9) | 6.3 (5.5) |
| Mean I/σ (I) | 7.7 (1.4) | 20.7 (1.1) |
| Completeness (%) | 96.6 (97.7) | 99 (94.1) |
| Rmeas (%) | 6.1 (101.3) | 4.0 (148) |
| Wilson B‐factor (Å2) | 79.1 | 59.5 |
| CC 1/2 | 0.998 (0.279) | 1 (0.53) |
| Refinement | ||
| Rcryst/number of reflections | 0.258/22 236 | 0.204/33 204 |
| Rfree/number of reflections | 0.297/1227 | 0.220/1727 |
| Number of non‐hydrogen atoms, protein/water/ligand | 5309/23/– | 3002/87/10 |
| RMSD bond lengths (Å) | 0.016 | 0.010 |
| RMSD bond angles (°) | 1.58 | 1.60 |
| RMSD torsion angles (°) | 4.68 | 5.29 |
| Ramachandran parameters (%), favoured/allowed/outlier | 99.5/0.5/– | 96.6/2.8/0.6 |
| Average B‐factors (Å2) | 77.6 | 64.6 |
SAXS
SAXS data were collected at beamline BM29 [60] of the EMBL Outstation at ESRF, Grenoble, using a PILATUS 1 m pixel detector. Measurements covered the momentum transfer range 0.002–0.60 Å−1 (s = 4π·sin(θ)/λ, where 2θ is the scattering angle and λ is the X‐ray wavelength, 0.992 Å). All experiments took place at a temperature of 293 K. A capillary flow cell was used to avoid radiation damage. Several SCO3201 dilutions (0.19–5.5 mg·mL−1) were analysed in a buffer containing 20 mm Tris/HCl pH 8.5, 400 mm NaCl, 50 mm imidazole and 1 mm DTT. All data were normalised to the intensity of the transmitted beam and radially averaged. Scattering of the buffer was subtracted and the resulting curves were scaled to unity protein concentration (1 mg·mL−1). For further data analysis, version 3.0.3 of the atsas package was used [61]. Theoretical scattering patterns based on structural models were fitted to the experimental data using the program crysol, which accounts for protein surface hydration [23]. Ab initio models were calculated using the program dammin [24].
Conflict of interest
The authors declare no conflict of interest.
Author contributions
SW, M‐JV and WH conceived, initiated and supervised this study; PW expressed, purified and crystallised SCO3201; PW and GJP collected X‐ray diffraction data; PW and WH solved and refined the crystal structures; SW analysed the SAXS data; SW interpreted the experimental results, analysed the induction mechanism and produced the artwork; SW and WH wrote the manuscript.
Peer review
The peer review history for this article is available at https://publons.com/publon/10.1111/febs.16606.
Acknowledgements
We would like to thank Martha E. Brennich (EMBL Grenoble) for SAXS data collection.
Contributor Information
Sebastiaan Werten, Email: sebastiaan.werten@i-med.ac.at.
Winfried Hinrichs, Email: winfried.hinrichs@uni-greifswald.de.
Data availability statement
Atomic coordinates and X‐ray diffraction data for the crystal structures of ligand‐free and induced SCO3201 have been deposited in the Protein Data Bank (PDB) with accession codes 5EFY and 7PT0. SAXS data and models have been deposited in the Small‐Angle Scattering Biological Data Bank (SASBDB) with accession code SASDN75.
References
- 1. Procópio REL, da Silva IR, Martins MK, de Azevedo JL, de Araújo JM. Antibiotics produced by Streptomyces. Braz J Infect Dis. 2012;16:466–71. [DOI] [PubMed] [Google Scholar]
- 2. Antoraz S, Santamaría RI, Díaz M, Sanz D, Rodríguez H. Toward a new focus in antibiotic and drug discovery from the Streptomyces arsenal. Front Microbiol. 2015;6:461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kemung HM, Tan LTH, Khan TM, Chan KG, Pusparajah P, Goh BH, et al. Streptomyces as a prominent resource of future anti‐MRSA drugs. Front Microbiol. 2018;9:2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berini F, Marinelli F, Binda E. Streptomycetes: attractive hosts for recombinant protein production. Front Microbiol. 2020;11:1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Claessen D, de Jong W, Dijkhuizen L, Wösten HAB. Regulation of Streptomyces development: reach for the sky! Trends Microbiol. 2006;14:313–9. [DOI] [PubMed] [Google Scholar]
- 6. Flärdh K, Buttner MJ. Streptomyces morphogenetics: dissecting differentiation in a filamentous bacterium. Nat Rev Microbiol. 2009;7:36–49. [DOI] [PubMed] [Google Scholar]
- 7. Flärdh K, Richards DM, Hempel AM, Howard M, Buttner MJ. Regulation of apical growth and hyphal branching in Streptomyces. Curr Opin Microbiol. 2012;15:737–43. [DOI] [PubMed] [Google Scholar]
- 8. McCormick JR, Flärdh K. Signals and regulators that govern Streptomyces development. FEMS Microbiol Rev. 2012;36:206–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Romero‐Rodríguez A, Robledo‐Casados I, Sánchez S. An overview on transcriptional regulators in Streptomyces. Biochim Biophys Acta BBA. 2015;1849:1017–39. [DOI] [PubMed] [Google Scholar]
- 10. Cuthbertson L, Nodwell JR. The TetR family of regulators. Microbiol Mol Biol Rev. 2013;77:440–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hinrichs W, Kisker C, Düvel M, Müller A, Tovar K, Hillen W, et al. Structure of the Tet repressor‐tetracycline complex and regulation of antibiotic resistance. Science. 1994;264:418–20. [DOI] [PubMed] [Google Scholar]
- 12. Orth P, Schnappinger D, Hillen W, Saenger W, Hinrichs W. Structural basis of gene regulation by the tetracycline inducible Tet repressor‐operator system. Nat Struct Biol. 2000;7:215–9. [DOI] [PubMed] [Google Scholar]
- 13. Aleksandrov A, Schuldt L, Hinrichs W, Simonson T. Tet repressor induction by tetracycline: a molecular dynamics, continuum electrostatics, and crystallographic study. J Mol Biol. 2008;378:898–912. [DOI] [PubMed] [Google Scholar]
- 14. Xu D, Seghezzi N, Esnault C, Virolle M‐J. Repression of antibiotic production and sporulation in Streptomyces coelicolor by overexpression of a TetR family transcriptional regulator. Appl Environ Microbiol. 2010;76:7741–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xu D, Waack P, Zhang Q, Werten S, Hinrichs W, Virolle MJ. Structure and regulatory targets of SCO3201, a highly promiscuous TetR‐like regulator of Streptomyces coelicolor M145. Biochem Biophys Res Commun. 2014;450:513–8. [DOI] [PubMed] [Google Scholar]
- 16. Haberl F, Lanig H, Clark T. Induction of the tetracycline repressor: characterization by molecular‐dynamics simulations. Proteins. 2009;77:857–66. [DOI] [PubMed] [Google Scholar]
- 17. Pal L, Dasgupta B, Chakrabarti P. 3(10)‐helix adjoining alpha‐helix and beta‐strand: sequence and structural features and their conservation. Biopolymers. 2005;78:147–62. [DOI] [PubMed] [Google Scholar]
- 18. Michael AJ. Polyamines in eukaryotes, bacteria, and archaea. J Biol Chem. 2016;291:14896–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Michael AJ. Polyamine function in archaea and bacteria. J Biol Chem. 2018;293:18693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wallace HM. The polyamines: past, present and future. Essays Biochem. 2009;46:1–9. [DOI] [PubMed] [Google Scholar]
- 21. Krysenko S, Matthews A, Busche T, Bera A, Wohlleben W. Poly‐ and monoamine metabolism in Streptomyces coelicolor: the new role of glutamine synthetase‐like enzymes in the survival under environmental stress. Microb Physiol. 2021;31:233–47. [DOI] [PubMed] [Google Scholar]
- 22. Hajizadeh NR, Franke D, Jeffries CM, Svergun DI. Consensus Bayesian assessment of protein molecular mass from solution X‐ray scattering data. Sci Rep. 2018;8:7204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Svergun D, Barberato C, Koch MHJ. CRYSOL– a program to evaluate X‐ray solution scattering of biological macromolecules from atomic coordinates. J Appl Cryst. 1995;28:768–73. [Google Scholar]
- 24. Svergun DI. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys J. 1999;76:2879–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Werner N, Werten S, Hoppen J, Palm GJ, Göttfert M, Hinrichs W. The induction mechanism of the flavonoid‐responsive regulator FrrA. FEBS J. 2021;289:507–18. [DOI] [PubMed] [Google Scholar]
- 26. Kamionka A, Bogdanska‐Urbaniak J, Scholz O, Hillen W. Two mutations in the tetracycline repressor change the inducer anhydrotetracycline to a corepressor. Nucleic Acids Res. 2004;32:842–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Scholz O, Henßler EM, Bail J, Schubert P, Bogdanska‐Urbaniak J, Sopp S, et al. Activity reversal of Tet repressor caused by single amino acid exchanges. Mol Microbiol. 2004;53:777–89. [DOI] [PubMed] [Google Scholar]
- 28. Resch M, Striegl H, Henssler EM, Sevvana M, Egerer‐Sieber C, Schiltz E, et al. A protein functional leap: how a single mutation reverses the function of the transcription regulator TetR. Nucleic Acids Res. 2008;36:4390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Swint‐Kruse L, Matthews KS. Allostery in the LacI/GalR family: variations on a theme. Curr Opin Microbiol. 2009;12:129–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schumacher MA, Miller MC, Grkovic S, Brown MH, Skurray RA, Brennan RG. Structural mechanisms of QacR induction and multidrug recognition. Science. 2001;294:2158–63. [DOI] [PubMed] [Google Scholar]
- 31. Alguel Y, Meng C, Terán W, Krell T, Ramos JL, Gallegos MT, et al. Crystal structures of multidrug binding protein TtgR in complex with antibiotics and plant antimicrobials. J Mol Biol. 2007;369:829–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Werten S, Schneider J, Palm GJ, Hinrichs W. Modular organisation of inducer recognition and allostery in the tetracycline repressor. FEBS J. 2016;283:2102–14. [DOI] [PubMed] [Google Scholar]
- 33. Manjasetty BA, Halavaty AS, Luan CH, Osipiuk J, Mulligan R, Kwon K, et al. Loop‐to‐helix transition in the structure of multidrug regulator AcrR at the entrance of the drug‐binding cavity. J Struct Biol. 2016;194:18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sawai H, Yamanaka M, Sugimoto H, Shiro Y, Aono S. Structural basis for the transcriptional regulation of heme homeostasis in Lactococcus lactis . J Biol Chem. 2012;287:30755–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vieira‐Pires RS, Morais‐Cabral JH. 310 helices in channels and other membrane proteins. J Gen Physiol. 2010;136:585–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Holm L. Using Dali for protein structure comparison. Methods Mol Biol. 2020;2112:29–42. [DOI] [PubMed] [Google Scholar]
- 37. Krysenko S, Okoniewski N, Nentwich M, Matthews A, Bäuerle M, Zinser A, et al. A second gamma‐Glutamylpolyamine synthetase, GlnA2, is involved in polyamine catabolism in Streptomyces coelicolor . Int J Mol Sci. 2022;23:3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Burrell M, Hanfrey CC, Kinch LN, Elliott KA, Michael AJ. Evolution of a novel lysine decarboxylase in siderophore biosynthesis. Mol Microbiol. 2012;86:485–99. [DOI] [PubMed] [Google Scholar]
- 39. Krysenko S, Okoniewski N, Kulik A, Matthews A, Grimpo J, Wohlleben W, et al. Gamma‐Glutamylpolyamine synthetase GlnA3 is involved in the first step of polyamine degradation pathway in Streptomyces coelicolor M145. Front Microbiol. 2017;8:726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Feng W‐H, Mao X‐M, Liu Z‐H, Li Y‐Q. The ECF sigma factor SigT regulates actinorhodin production in response to nitrogen stress in Streptomyces coelicolor . Appl Microbiol Biotechnol. 2011;92:1009–21. [DOI] [PubMed] [Google Scholar]
- 41. Lewis RA, Shahi SK, Laing E, Bucca G, Efthimiou G, Bushell M, et al. Genome‐wide transcriptomic analysis of the response to nitrogen limitation in Streptomyces coelicolor A3(2). BMC Res Notes. 2011;4:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tiffert Y, Franz‐Wachtel M, Fladerer C, Nordheim A, Reuther J, Wohlleben W, et al. Proteomic analysis of the GlnR‐mediated response to nitrogen limitation in Streptomyces coelicolor M145. Appl Microbiol Biotechnol. 2011;89:1149–59. [DOI] [PubMed] [Google Scholar]
- 43. Zhu Y, Zhang P, Zhang J, Xu W, Wang X, Wu L, et al. The developmental regulator MtrA binds GlnR boxes and represses nitrogen metabolism genes in Streptomyces coelicolor . Mol Microbiol. 2019;112:29–46. [DOI] [PubMed] [Google Scholar]
- 44. Martín JF, Rodríguez‐García A, Liras P. The master regulator PhoP coordinates phosphate and nitrogen metabolism, respiration, cell differentiation and antibiotic biosynthesis: comparison in Streptomyces coelicolor and Streptomyces avermitilis . J Antibiot (Tokyo). 2017;70:534–41. [DOI] [PubMed] [Google Scholar]
- 45. Yang R, Liu X, Wen Y, Song Y, Chen Z, Li J. The PhoP transcription factor negatively regulates avermectin biosynthesis in Streptomyces avermitilis . Appl Microbiol Biotechnol. 2015;99:10547–57. [DOI] [PubMed] [Google Scholar]
- 46. Smirnov A, Esnault C, Prigent M, Holland IB, Virolle M‐J. Phosphate homeostasis in conditions of phosphate proficiency and limitation in the wild type and the phoP mutant of Streptomyces lividans . PLoS ONE. 2015;10:e0126221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chouayekh H, Virolle M‐J. The polyphosphate kinase plays a negative role in the control of antibiotic production in Streptomyces lividans . Mol Microbiol. 2002;43:919–30. [DOI] [PubMed] [Google Scholar]
- 48. Werten S, Rustmeier NH, Gemmer M, Virolle M‐J, Hinrichs W. Structural and biochemical analysis of a phosin from Streptomyces chartreusis reveals a combined polyphosphate‐ and metal‐binding fold. FEBS Lett. 2019;593:2019–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shikura N, Darbon E, Esnault C, Deniset‐Besseau A, Xu D, Lejeune C, et al. The Phosin PptA plays a negative role in the regulation of antibiotic production in Streptomyces lividans . Antibiotics (Basel). 2021;10:325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mueller U, Darowski N, Fuchs MR, Förster R, Hellmig M, Paithankar KS, et al. Facilities for macromolecular crystallography at the Helmholtz‐Zentrum Berlin. J Synchrotron Radiat. 2012;19:442–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cianci M, Bourenkov G, Pompidor G, Karpics I, Kallio J, Bento I, et al. P13, the EMBL macromolecular crystallography beamline at the low‐emittance PETRA III ring for high‐ and low‐energy phasing with variable beam focusing. J Synchrotron Radiat. 2017;24:323–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kabsch W. Integration, scaling, space‐group assignment and post‐refinement. Acta Crystallogr D Biol Crystallogr. 2010;66:133–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Winn MD, Murshudov GN, Papiz MZ. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 2003;374:300–21. [DOI] [PubMed] [Google Scholar]
- 57. Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum‐likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–55. [DOI] [PubMed] [Google Scholar]
- 58. Costantini S, Paladino A, Facchiano AM. CALCOM: a software for calculating the center of mass of proteins. Bioinformation. 2008;2:271–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lee HS, Choi J, Yoon S. QHELIX: a computational tool for the improved measurement of inter‐helical angles in proteins. Protein J. 2007;26:556–61. [DOI] [PubMed] [Google Scholar]
- 60. Pernot P, Round A, Barrett R, de Maria Antolinos A, Gobbo A, Gordon E, et al. Upgraded ESRF BM29 beamline for SAXS on macromolecules in solution. J Synchrotron Radiat. 2013;20:660–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Manalastas‐Cantos K, Konarev PV, Hajizadeh NR, Kikhney AG, Petoukhov MV, Molodenskiy DS, et al. ATSAS 3.0: expanded functionality and new tools for small‐angle scattering data analysis. J Appl Cryst. 2021;54:343–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Atomic coordinates and X‐ray diffraction data for the crystal structures of ligand‐free and induced SCO3201 have been deposited in the Protein Data Bank (PDB) with accession codes 5EFY and 7PT0. SAXS data and models have been deposited in the Small‐Angle Scattering Biological Data Bank (SASBDB) with accession code SASDN75.