

Abstract
Biological ageing is connected to life history variation across ecological scales and informs a basic understanding of age‐related declines in organismal function. Altered DNA methylation dynamics are a conserved aspect of biological ageing and have recently been modelled to predict chronological age among vertebrate species. In addition to their utility in estimating individual age, differences between chronological and predicted ages arise due to acceleration or deceleration of epigenetic ageing, and these discrepancies are linked to disease risk and multiple life history traits. Although evidence suggests that patterns of DNA methylation can describe ageing in plants, predictions with epigenetic clocks have yet to be performed. Here, we resolve the DNA methylome across CpG, CHG, and CHH‐methylation contexts in the loblolly pine tree (Pinus taeda) and construct epigenetic clocks capable of predicting ages in this species within 6% of its maximum lifespan. Although patterns of CHH‐methylation showed little association with age, both CpG and CHG‐methylation contexts were strongly associated with ageing, largely becoming hypomethylated with age. Among age‐associated loci were those in close proximity to malate dehydrogenase, NADH dehydrogenase, and 18S and 26S ribosomal RNA genes. This study reports one of the first epigenetic clocks in plants and demonstrates the universality of age‐associated DNA methylation dynamics which can inform conservation and management practices, as well as our ecological and evolutionary understanding of biological ageing in plants.
Keywords: biological age, chronological age, DNA methylation, epigenetic clock, Pinus taeda
1. INTRODUCTION
Alterations to the epigenome are a conserved hallmark of biological ageing, and recent findings have demonstrated that age‐associated DNA methylation patterns can be modelled to generate epigenetic age predictors capable of estimating chronological and biological age with unprecedented accuracy (Berdyshev et al., 1967; Christensen et al., 2009; Hannum et al., 2013; Richardson, 2003). In one of the first DNA methylation‐based age predictors or “epigenetic clocks” developed by Horvath (2013), the methylation status of 353 cytosines predicts human chronological age with an error of ±3.6 years. Epigenetic clocks have subsequently been developed in a variety of other mammalian (Horvath, 2013; Weidner et al., 2014), avian (De Paoli‐Iseppi et al., 2019; Raddatz et al., 2021), and fish species (Anastasiadi & Piferrer, 2020; Bertucci et al., 2021; Mayne et al., 2020), and are currently being applied to biomedical and conservation problems, as well to questions regarding their relationship to the underlying biology of ageing and senescence (Bertucci & Parrott, 2020; Kabacik et al., 2018). However, whereas the phenomenon of epigenetic ageing appears to be a conserved aspect of biological ageing in vertebrates, age‐associated changes to DNA methylation and their ability to predict chronological age in plants is relatively unexplored (Parrott & Bertucci, 2019; Yao et al., 2021).
DNA methylation refers to the covalent addition of a methyl group to the 5′ carbon of cytosine nucleotides (Jung & Pfeifer, 2015; Ng & Adrian, 1999; Suzuki & Bird, 2008), and although the functional consequences of these modifications vary across genomic context and taxonomic groups, DNA methylation is broadly associated with repressed transcriptional activity of genes and transposable elements through direct silencing and promotion of repressive chromatin states (Ng & Adrian, 1999; Zilberman, 2008). Similar to vertebrates, changes in DNA methylation levels in plants are observed with exposure to stress, age, and development (Dubrovina & Kiselev, 2016; Jiang et al., 2014; Law & Jacobsen, 2010; Probst & Mittelsten Scheid, 2015). However, compared to the distribution of cytosine methylation within vertebrate genomes, which almost exclusively occurs in CpG dinucleotides, DNA methylation in plant genomes is frequently observed within CpG, CHG and CHH contexts (where H = A, T, or C). Methylated cytosines located within gene bodies in plants primarily occurs in CpG contexts (Takuno et al., 2016), whereas CpG, CHG, and CHH‐methylation is typically found within highly repetitive genomic regions, potentially functioning to silence transposable element activity (Ausin et al., 2016; Slotkin & Martienssen, 2007). How these different sequence contexts might relate to age‐associated DNA methylation patterning is not resolved.
Despite the ability of current epigenetic clocks to predict chronological age, discrepancies between epigenetic age and chronological age are observed and reflect variation in biological age (Horvath & Raj, 2018; Parrott & Bertucci, 2019; Xiao et al., 2019). Accelerated epigenetic ageing is associated with age related losses in organismal function and in humans, and predicts risk for age‐associated disease and mortality (Levine et al., 2018; Perna et al., 2016; Zheng et al., 2016). In addition, epigenetic‐to‐chronological age discordance mirrors variation in life history traits (Anderson et al., 2021; Hamlat et al., 2021). For example, birth weight, age and size at maturity, as well as the timing of reproductive senescence are all correlated to epigenetic age in humans, demonstrating intriguing links between ageing processes and life history variation (Binder et al., 2018; Ryan et al., 2018; Simpkin et al., 2015). However, the links between the rate of epigenetic ageing and variation in organismal function and life history traits (including those with potential commercial implications) are largely unexplored in plants.
Loblolly pine (Pinus taeda) is a large tree with a broad geographic range spanning from southern New Jersey to eastern Texas, including parts of northern Florida (Baker & Langdon, 1990) and is capable of living for up to 275 years (Baker & Langdon, 1990). The loblolly pine is often commercially harvested for timber and is also used to diversify forest habitats, control erosion, and improve water quality (Baker & Langdon, 1990). Research and management practices aimed at improving growth and yield from loblolly pine stands often manipulate resource availability (Albaugh et al., 2004; Coyle et al., 2016; Fox et al., 2007). However, the underlying biological mechanisms promoting optimal growth rates across variable environmental conditions remain unclear (Albaugh et al., 2004). As plant growth has been linked to DNA methylation processes (Genger et al., 2003; Horvath et al., 2003), resolving and utilizing age‐associated methylation patterns in trees may aid in evaluating strategies for increasing stand yield (i.e., increasing leaf area and other traits that promote growth efficiency [Dubrovina & Kiselev, 2016; Fox et al., 2007; Medlyn et al., 2003; Samuelson et al., 2001]).
Here, we investigate age‐associated DNA methylation patterns in P. taeda and evaluate their genomic distributions among differing methylation contexts. We then test differing modelling strategies to develop a novel epigenetic clock for P. taeda, which can be used to investigate the factors most important to growth, development, and tree ageing. This study demonstrates the utility of epigenetic clocks in nonvertebrate models and indicates that age‐associated DNA methylation, both within CpG contexts and beyond, may be a universal aspect of organismal ageing.
2. MATERIALS AND METHODS
2.1. Sample collection
Between 31 December 2019 and 14 January 2020, cambium samples from standard coring procedures were obtained at 1.37 m of stem height from 24 P. taeda individuals ranging from 1 to 119 years of age. Cambium tissues have been reliably sampled for differential DNA methylation analyses in other species (Wang et al., 2016). Tree cores spanned the radius of the tree stem, and the cambium was separated from the bark and xylem for DNA extractions. Cambium samples were utilized instead of leaves as it is currently unknown whether age‐related methylation patterns differ among these organs. This study was conducted at the United States Department of Energy's Savannah River Site (Aiken, SC, USA), a National Environmental Research Park. The United States Department of Agriculture (USDA) Forest Service manages the natural resources of the Savannah River Site (Kilgo & Blake, 2005). Using records from the Forest Service, we identified stands which were planted between 1 and 55 years prior and sampled three trees from each stand (1, 10, 19, 28, 37, 46, and 55 years old). Three additional trees of advanced age (approximately 82, 97, and 119 years old) were identified by counting rings within core samples as planting records for older stands were unavailable. Core samples are often used for estimating tree ages (Mahoney et al., 1991); however, coring estimates might contain bias for age underestimation as it can take several years for a tree to reach the height of 1.37 m where coring was performed. Cambium samples from adult trees (>1‐year) were taken at breast height (1.37 m) using a metal hole punch. Saplings (1‐year old) were sampled at the base of the primary stem with a metal blade. All samples were immediately stored in RNAlater at −20°C until DNA extraction. Diameter at breast height was measured using a diameter tape and height was measured using a TruPulse 200x Rangefinder (Laser Technology, Inc.).
2.2. DNA extraction
DNA was extracted from cambium tissue using Qiagen's DNeasy Plant Pro Kit (catalogue no. 69204, Qiagen) following the manufacturer's protocol with the addition of 100 μl of the solution PS per sample due to high amounts of phenolic compounds in pine species. Briefly, samples were cut into small pieces using a sterile blade and then homogenized using a Mini‐Beadbeater (BioSpec) for 4–8 min at 2000 oscillations/min. DNA was eluted in 50 μl of the supplied elution buffer, and the concentration and purity of DNA samples were assessed using a Qubit fluorometer 2.0 (Invitrogen) and Nanodrop spectrometer (Thermo‐Scientific), respectively.
2.3. Reduced representation bisulphite sequencing library preparation
Reduced representation bisulphite sequencing (RRBS) libraries were prepared using Diagenode's Premium RRBS Kit (catalogue no. C02030032, Diagenode). Due to the occurance of CHG‐methylation in plant genomes, we adapted the protocol by digesting genomic DNA with the BsaWI restriction enzyme (catalogue no. R0567S, New England BioLabs) instead of MspI, as BsaWI cuts the recognition site W^CCGGW. Digestion efficiency of BsaWI in our samples was confirmed by visualizing digested genomic DNA from test samples using standard gel electrophoresis. For library preparations, 200 ng of genomic DNA from each sample was digested with 5 units/ng of BsaWI for 12 h at 60°C, after which samples underwent a 20 min heat inactivation of the restriction enzyme at 80°C. To obtain sufficient library concentrations, two RRBS libraries were prepared for each sample. In the second library preparation, the protocol was further altered to add additional extension time in the final amplification (72°C for 1 min during cycling and 10 min during final extension). Libraries were eluted in 22 μl of the supplied elution buffer and stored at −80°C until sequencing. Other than these alterations, the manufacturer's protocol was followed exactly.
2.4. RRBS sequencing, quality control, and alignment
RRBS libraries were assessed for concentration and fragment size distribution on a Fragment analyser (Advanced Analytical Technologies, Inc.) at the Georgia Genomics and Bioinformatics Core at the University of Georgia. Libraries were then pooled and sequenced single‐end for 100 cycles on the Illumina NextSeq 2000 with 20% PhiX control added. Two library preparations for each sample were sequenced across five high‐output flow cells and approximately 400 million reads from each flow cell were generated. The quality of the resulting reads was assessed using FastQC (version 0.11.5; Andrews, 2010). Reads were trimmed using TrimGalore! (version 0.4.5) to remove adapter sequences and low‐quality reads (Phred score < 25), using the –rrbs option for RRBS data. Trimmed reads were concatenated into a single file and aligned to a bisulphite converted index of the loblolly genome (Ptaeda2.0) using Bismark (version 0.20.0) (Krueger & Andrews, 2011) allowing for one mismatch (option n‐1). The subsequent alignments were sorted and indexed using SAMtools (version 0.1.19) (Li et al., 2009).
2.5. File processing
The resulting Bam files were analysed using the methylKit package (Akalin et al., 2012) in R (version 4.0.5) (2021). An average of 18,177,435 (± 1,342,074) reads comprised each Bam file, and cytosines from each individual were divided into CpG, CHG, or CHH contexts using the read. context parameter in the processBismarkAln function of methylKit. To broadly assess age‐associated DNA‐methylation, while also developing an epigenetic clock, we filtered our data using two approaches. In the first approach, we filtered out cytosines covered by <5x reads and not represented in <80% of samples. To ensure comparability across CpG, CHG, and CHH contexts, cytosines on opposite strands were not merged (destrand = FALSE). This data set is considered our “exploratory” data set, in which general age‐associated patterns could be resolved. In the second approach, we increased coverage requirements (≥10x) to yield a data set that could be used to construct epigenetic age predictors.
2.6. Exploratory data set
The resulting exploratory data set was analysed using the files generated for CpG, CHG, and CHH cytosine‐methylation contexts. We filtered out cytosines displaying zero or near‐zero variance in methylation status across samples using the nearZeroVar function from the Classification And Regression Training (caret) package (Kuhn, 2015) in R. Following filtering of invariant cytosines, we performed Spearman correlations between the methylation status of each cytosine and chronological age using the corr.test function from the psych package (Revelle, 2019) in R. Given the exploratory nature of the downstream analyses, we considered cytosines with an absolute correlation coefficient greater than 0.5 (R > |.5|) as being “age‐associated”, and those with p‐values less than .05 (p < .05) following an FDR correction for multiple comparisons as “significantly correlated” with age.
To assess genomic characteristics of age‐associated and significantly correlated CpG, CHG, and CHH cytosines, we classified the genomic locations of all covered cytosines according to genomic context within the P. taeda genome using the GenomicRanges package (Lawrence et al., 2013). We first used CpG plot (Madeira et al., 2019) to identify CpG islands (CGIs; parameters: widow = 100, min len = 200, minoe = 0.6, minpc = 50). We then used CGI coordinates to generate coordinates for shore regions (± 2000 bp from CGIs), and shelf regions (± 2000–4000 bp from CGIs) using Bedtools and Samtools (Li et al., 2009; Quinlan & Hall, 2010). All remaining sites not falling in island, shore, or shelf regions were characterized as open sea regions. Enrichment above background within each methylation context was performed with binomial tests using all cytosines following invariant filtering within each context (21,566, 25,501, and 33,151 CpG, CHG, and CHH cytosines, respectively) as the background levels. To identify genes in close proximity to cytosines according to age‐associated methylation patterns, we performed BLAST searches on 400 bp regions (200 bp upstream and 200 bp downstream) centered around each of the 35 cytosines from CpG and CHG contexts with the greatest correlations with age.
2.7. Clock data set
Cytosine methylation in CHH contexts is highly variable among plants (Bartels et al., 2018) and is subject to elevated measurement variation when compared to CHG and CpG‐methylation (How‐Kit et al., 2017). Thus, whereas much larger sample sizes are probably required to thoroughly assess the associations of CHH‐methylation with ageing, our exploratory analyses indicated that the methylation status of CHH cytosines was not associated with age and we only generated epigenetic clocks using CpG and CHG cytosines. We first removed any invariant sites using the nearZeroVar function from the caret package (Kuhn, 2015) and performed imputation on missing data using a K‐nearest neighbour (KNN) approach in the impute package (Hastie et al., 2021) in R. We set k = 2 as each age group among our samples was only represented by a maximum of three samples.
2.8. Elastic net clocks
We trained elastic net models for CpG and CHG‐methylated cytosines using the glmnet package (Friedman et al., 2010) in R. We used an elastic net penalized regression model (alpha = 0.5, family = gaussian) to select CpG‐ and CHG‐methylated cytosines and assign penalties to individual model coefficients using the subset of CpG‐loci and CHG‐loci. We constructed elastic net models utilizing a leave one out cross validation (LOOCV) approach to evaluate age‐related methylation patterns in P. taeda. This approach constructs epigenetic clocks for all samples, while leaving out one sample and predicting its age. This process is repeated until each tree has been left out and predicted ages for each tree are then assessed. To further assess the influence of the proportion and combinations of individual samples represented in the training and test sets on clock performance, we performed k‐fold cross validations (k = 100) using training sets of randomly chosen trees (12–18 trees per training set) and applied attendant models to remaining trees, which comprised the test set (Appendix S1 and Figure S1). As results from k‐fold validation steps indicated that models tended to become overfit to training sets as the proportion of samples used decreased (p < 2e−16), we split our data by randomly choosing three individuals for our test set (ages: 10, 19, and 55), and used the remaining individuals (n = 18) to construct elastic net regularized clocks.
When constructing our elastic net clocks, we used a LOOCV approach (nfold = 18) to select the optimal lambda value (resulting in minimum mean error) for the training set model (n = 18 samples). We used the resulting model trained on trees from our training set to then predict ages of the trees in our test set. Following the generation of our elastic net epigenetic clocks, we performed BLAST searches of 400 bp regions centered around each clock cytosine and used GenomicRanges to determine the genomic contexts for these sites (genes and CpG islands, shores, shelves, or open seas).
2.9. Pearson clocks
Bertucci et al. (2021) demonstrated that epigenetic clocks constructed using elastic net approaches on small sample sizes may be overfit, and linear models constructed using cytosines for which the relationship between methylation status and age is greatest (assessed with Pearson correlation tests) have the potential to outperform elastic net models. Therefore, we performed Pearson correlations on all cytosines represented across samples and identified the top five and 10 cytosines with the greatest Pearson correlation with age (R > |.5|) from our training set using the corr.test function (using FDR correction, alpha = .05) from the psych package. We generated our linear models using the lm function in R, and then used them to predict the ages of the individuals in our test data set. Following the generation of our linear model epigenetic clocks, we performed BLAST searches of 400 bp regions centered around clock cytosines and used GenomicRanges to determine genomic context (genes and CpG islands, shores, shelves, or open seas).
To evaluate performance of our elastic net and Pearson clocks and to compare whether methylation context (CpG, CHG, or CpG + CHG) had an effect on age predictions, we used multiple regression to compare the mean absolute errors (MAE) generated when using each clock to predict ages of trees from our test set. We used clock type (elastic net, Pearson (5), or Pearson (10)) and methylation type (CpG, CHG, or CpG + CHG) as factors and chronological age as a covariate in our model and corrected resulting p‐values using Bonferroni correction for multiple comparisons. Overfit of each clock to our training set was evaluated by comparing the MAE for training and test sets from each clock using t‐tests. Additionally, we also evaluated how often specific cytosines were selected in our models through further k‐fold cross validation methods. Setting k = 100, we constructed additional separate elastic net models (n = 100) trained on randomly chosen trees from our sample set (n = 18 trees per each training set), which generated additional unique lists (n = 100) of sites used to predict age in P. taeda. This frequency measure is referred to as robustness through the remainder of the manuscript. The robustness of each of the sites used in models reported in this manuscript was evaluated by obtaining the frequency at which each of our model sites were found among each of our additional k = 100 clock site lists.
3. RESULTS
3.1. Characterizing age‐related cytosine methylation
Among the CpGs analysed, 533 (2.47%) displayed age‐associated methylation, with 207 CpGs having positive correlations (max R = .84) and 326 having negative correlations (min R = −.79) (Figure 1). Following FDR (alpha = .05) correction for multiple comparisons, 10 CpGs (9 negative and 1 positive) showed significant correlations with age. When examining locations of our age‐associated cytosines with respect to genomic contexts, CpG‐methylated cytosines were enriched in island (p = 4.85e−10) and shore regions (p = .038), while being depleted in shelf (p = 9e−04) and open‐sea regions (p = 2.20e−06) (Figure 2). Among the age‐associated CpG‐methylated sites found in islands, many became hypomethylated with age (Table 1). Whereas this pattern was also observed for CpGs in shore and open‐seas, CpGs in shelf regions were more evenly split between hypo‐ and hypermethylation with increasing age. Malate dehydrogenase (mdh), as well as 18S and 26S ribosomal genes of related pine species, were among the genes our methylated CpG‐containing 400 bp regions mapped to (Table S1).
FIGURE 1.
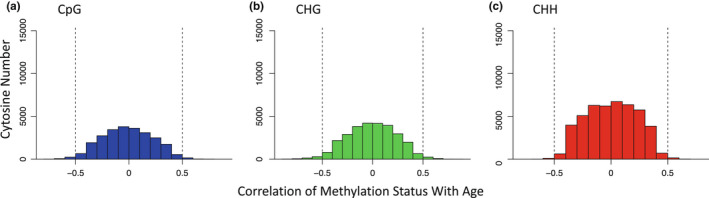
Histograms displaying the distribution of spearman correlation coefficients of cytosine methylation status with age following filtering of invariant sites across CpG, CHG, and CHH‐methylation contexts from 24 loblolly pine trees of differing ages. (a) of 21,567 CpGs analysed, 533 (2.5%) showed correlations between methylation status and age of R > |.5|. (b) of 25,501 CHGs analysed, 869 (3.4%) displayed correlations between methylation status and age of R > |.5|. (c) of 33,151 CHHs analysed, 308 (0.93%) showed correlations between age and methylation status of R > |.5|.
FIGURE 2.
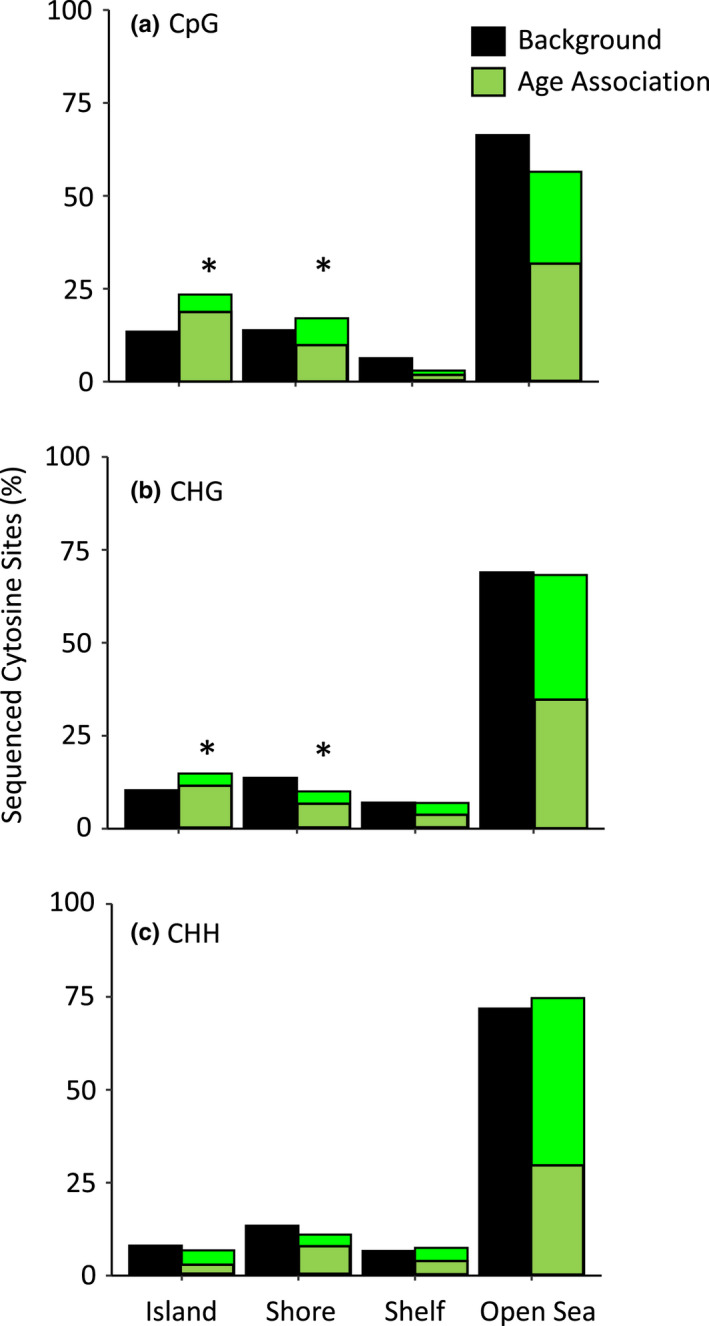
Enrichment and depletion of age‐related DNA methylation within genomic regions with varying CpG densities. Cytosines for which methylation status was associated with age (R > |0.5|) were compared against all RBSS‐captured cytosines (background). Darker green shading indicates proportion of cytosines for which methylation decreased with age and lighter green shading indicates cytosines for which methylation increased with age. (a) Age‐associated CpG‐methylation was enriched in CpG island and shore regions and were depleted in CpG shelf and open‐sea regions. (b) Age‐associated CHG‐methylation was enriched in CpG islands and depleted in shore regions. (c) There were no differences in the distributions of age‐associated CHH‐methylated cytosines compared to background CHH‐methylated cytosine levels (asterisks indicate p < .05).
TABLE 1.
Distributions of hypo‐ (−) and hypermethylated (+) cytosines with age among genomic regions
| Context | Cytosine subset | Island cytosines | Shore cytosines | Shelf cytosines | Open Sea cytosines | ||||
|---|---|---|---|---|---|---|---|---|---|
| − | + | − | + | − | + | − | + | ||
| CpG | Age‐associated | 111 | 14 | 56 | 35 | 7 | 9 | 152 | 149 |
| Significantly correlated | 2 | 0 | 2 | 1 | 0 | 0 | 4 | 1 | |
| CHG | Age‐associated | 105 | 24 | 57 | 30 | 33 | 27 | 325 | 269 |
| Significantly correlated | 21 | 0 | 5 | 1 | 1 | 0 | 33 | 9 | |
| CHH | Age‐associated | 7 | 14 | 20 | 14 | 11 | 12 | 84 | 147 |
For CHG‐methylation, 869 cytosines (3.41%) were age associated, with 350 displaying positive correlations (max R = .87) and 519 negatively correlated with age (min R = −.89; Figure 1). Following FDR corrections, 70 of these 869 age‐associated sites (60 hypomethylated and 10 hypermethylated) showed significant (p < .05) correlations with age. Compared to background levels, age‐associated CHG cytosines were enriched in island regions (p = 3.62e−05) and were depleted in shores (p = 1.3e−03) (Figure 2). The majority of CHG‐methylated cytosines were hypomethylated with age, regardless of genomic context (Table 1). One of the top 35 CHG age‐associated cytosine sites mapped to an apparent homologue of mdh and became hypermethylated with age (Table S2).
Within CHH‐methylation contexts, 308 (0.093%) cytosines were found to be age‐associated, with 187 displaying a positive correlation with age (max R = 76) and 121 negatively correlated to age (min R = −.71; Figure 1). However, following FDR corrections, no cytosines were significantly (p < .05) correlated with ageing, and compared to background levels, there were no differences in the distributions of age‐associated CHH‐methylated cytosines based on genomic context (p > .05). CHH‐methylated cytosines found in island regions showed a trend of being mostly hypermethylated (67%) with age (Table 1), while CHHs found in shore regions were mostly hypomethylated (59%) with age. Shelf‐region CHHs were more evenly split between hypo‐ and hypermethylation with age (48% vs. 52%), while most of the open‐sea region CHH cytosines showed hypermethylation with age (63%) (Figure 2).
3.2. Construction of epigenetic clocks capable of predicting age
LOOCV approaches incorporating all individuals resulted in a large MAE (17.44 ± 3.77 years) of predicted ages and poor correlations to chronological ages (R 2 = .23), with age estimates of the oldest trees in our sampling set (ages 82, 97, and 119) displaying the largest discordance between chronological and epigenetic age (Figure S2). We then reran the LOOCV without the three oldest individuals, which yielded a MAE of 6.77 (± 1.20), and predicted ages showed a higher correlation with chronological age (R 2 = 0.85; Figure 3). Subsequently, epigenetic age predictors were constructed using elastic net regularized regression approaches using either CpG‐methylation, CHG‐methylation, or a combination of CpG and CHG‐methylation (Figure 4). All three elastic net clocks predicted ages of trees from our training set within 0.48 years, and predicted ages were highly correlated with chronological ages for these trees (R 2 = 0.99) (Table 2). However, the ability of these models to predict chronological ages of trees in the test set was slightly lower as R 2 values ranged from 0.75 to 0.97, with the CpG model (Table S3) being outperformed by the CHG (Table S4) and the combined CpG and CHG model (Table S5).
FIGURE 3.
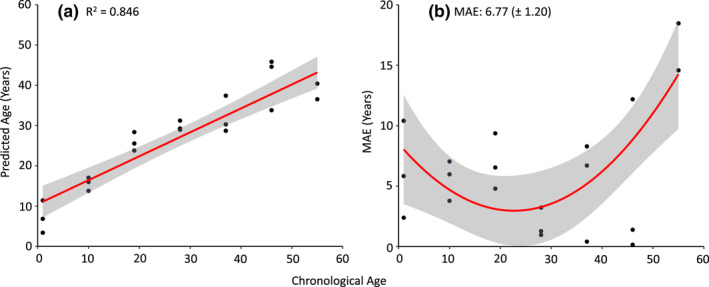
Accuracy and precision of age predictions using elastic net models to predict chronological age for n = 21 Pinus taeda trees (ages 1–55 years) using a leave‐one‐out‐cross‐validation approach. (a) Predicted ages were highly correlated with chronological ages (R 2 = .85, obtained from a regression of predicted age with chronological age). (b) Precision of predicted ages was assessed by comparing estimated age for each individual to their chronological age and is expressed as mean absolute error (MAE) in years.
FIGURE 4.
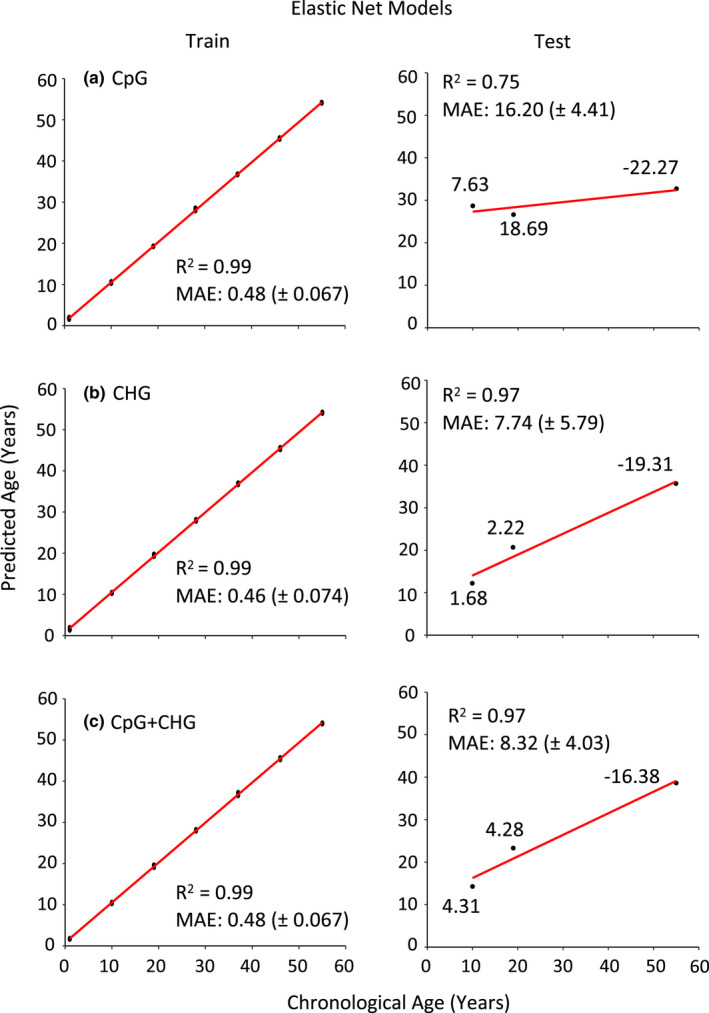
Elastic net models used to predict chronological age in Pinus taeda for both training and test subsets. Clock accuracy is measured by the Pearson's correlation coefficient and precision by the mean absolute error (MAE) (± standard error). (a) a total of 34 CpGs out of 18,844 CpGs following invariant filtering were selected in our elastic net model. (b) a total of 35 CHGs out of 22,263 CHGs were selected and incorporated into the clock. (c) Combining CpG and CHG data sets, there were 41,107 cytosines following invariant filtering, with 38 incorporated into the clock. Values of individual data points represent the error between predicted ages from our models compared to chronological ages among test individuals, with positive values indicating overestimation and negative values indicating underestimation of chronological age.
TABLE 2.
Performance of elastic net models when predicting ages of trees from training and test sets
| Elastic net models | ||||||||
|---|---|---|---|---|---|---|---|---|
| Context | Number of initial sites | Number of filtered sites | Number of clock sites | Train R 2 | Train MAE (years) | Test R 2 | Test MAE (years) | Overfit t‐test p‐value |
| CpG | 26,520 | 18,844 | 34 | .999 | 0.483 (± 0.067) | .75 | 16.20 (± 4.41) | .95 |
| CHG | 30,728 | 22,263 | 35 | .999 | 0.460 (± 0.074) | .97 | 7.74 (± 5.79) | .34 |
| CpG + CHG | 52,748 | 41,107 | 38 | .999 | 0.477 (± 0.067) | .97 | 8.32 (± 4.03) | .19 |
In addition to our elastic net models, we constructed epigenetic clocks for each methylation context (CpG, CHG, and CpG + CHG) separately using cytosines with the greatest Pearson correlation coefficients to chronological age (Figure 5). When the top five Pearson age‐correlated cytosines were selected, training set ages were predicted within 5.89 years and were highly correlated to chronological age (R 2 = 0.98 to 0.99) (Table 3, Figure 5). As expected, the performance on test sets was somewhat lower, with MAE ranging from 12.37 years for the CpG clock and 4.06 years for the CHG clock (Figure 5). We then constructed models using the top 10 Pearson age‐corelated cytosines. Relative to the models using the top five correlated cytosines, these models performed better on the training set (R 2 = 0.89 to 0.99); however, performance suffered on the test set, with R 2 values ranging from 0.53 when using only CHG‐methylated cytosines to 0.75 when using CpG‐methylated cytosines (Table 3). For annotations of CpG, CHG, and combined CpG and CHG cytosines from Pearson correlation models, see Tables S6–S8.
FIGURE 5.
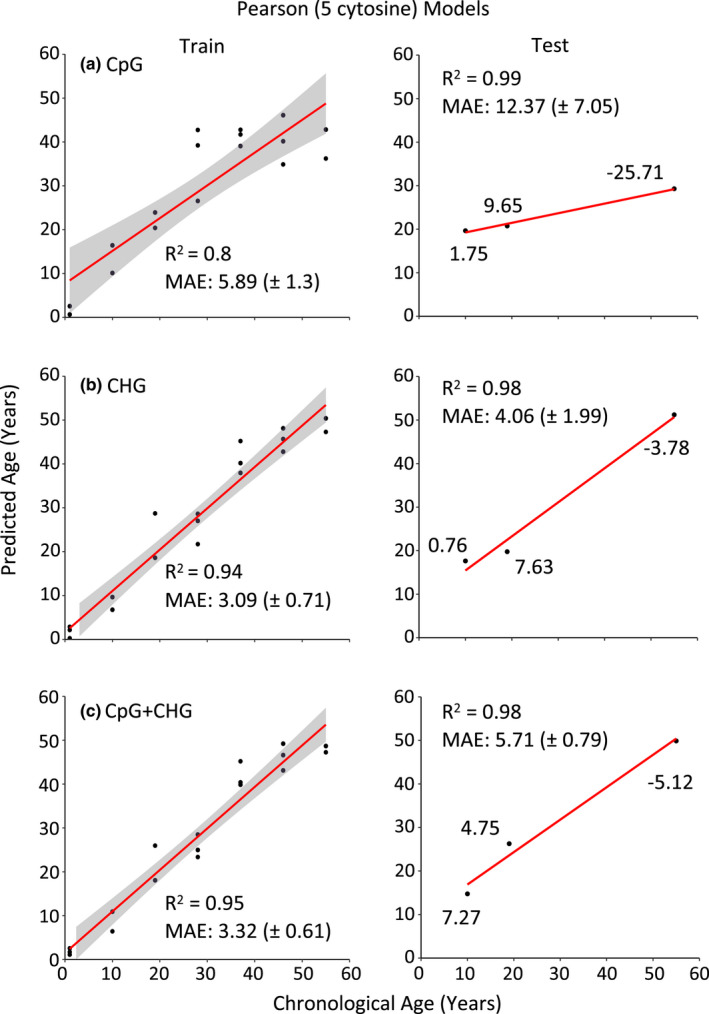
Pearson models incorporating the five cytosines with the strongest age‐associated methylation patterns predict chronological ages in Pinus taeda for both training and test subsets for CpG, CHG, and combined (CpG + CHG) models. Clock accuracy is measured by the Pearson's correlation coefficient and precision by the mean absolute error (MAE) (± standard error). Values of individual datapoints represent the error between predicted ages from our models compared to chronological ages among our test individuals, with positive values indicating overestimation and negative values indicating underestimation of chronological age.
TABLE 3.
Performance of Pearson models when predicting ages of trees from training and test sets
| Context | Number of initial sites | Number of filtered sites | Train R 2 | Train MAE (years) | Test R 2 | Test MAE (years) | Overfit t‐test p‐value |
|---|---|---|---|---|---|---|---|
| Pearson model (5 cytosines) | |||||||
| CpG | 26,520 | 18,844 | .8 | 5.89 (± 1.30) | .99 | 12.37 (± 7.05) | .46 |
| CHG | 30,728 | 22,263 | .94 | 3.09 (± 0.71) | .98 | 4.06 (± 1.99) | .68 |
| CpG + CHG | 52,748 | 41,107 | .95 | 3.23 (± 0.61) | .98 | 5.71 (± 0.79) | .06 |
| Pearson model (10 cytosines) | |||||||
| CpG | 26,520 | 18,844 | .89 | 4.49 (± 0.93) | .75 | 36.44 (± 9.43) | .08 |
| CHG | 30,728 | 22,263 | .99 | 1.72 (± 0.32) | .53 | 12.33 (± 6.65) | .25 |
| CpG + CHG | 52,748 | 41,107 | .97 | 1.93 (± 0.53) | .69 | 8.76 (± 6.37) | .4 |
When comparing predictive performance among our elastic net and Pearson clocks, MAE of predicted ages for trees from our test set ranged from 4.06 to 36.44 years (1.47%–13.25% of the total lifespan for P. taeda). There was no effect of clock type (elastic net or Pearson) on MAE values of predicted ages for trees from our test set (p > .05). To assess the potential overfit of our models, we compared the MAE of the training sets to those of test sets. Although performance was generally lower for test sets, our statistical comparison indicated that they were not significantly overfit to our training set (p > .05) (Tables 2 and 3). When comparing how robust specific cytosines used among our models were at predicting age in P. taeda, the average robustness of sites used in our models was greater for our Pearson models that included the 5 highest age‐correlated sites compared to the 10 highest age‐correlated sites (p = 4.380e−06), which was also the case when comparing these models to our elastic net models (p = 8.580e−06). Clock type (CpG, CHG, or CpG combined with CHG) had no effect on robustness (p > .05).
We next used BLAST to assess if clock cytosines were proximal to specific genes. Among the known genes a few of our loci mapped to were mdh, NADH dehydrogenase, and rRNA (18S and 26S). Loci that mapped to mdh and rRNA genes were found in both CpG and CHG contexts, while loci mapping to NADH dehydrogenase were found only within the CpG context. The loci mapping to these genes were distributed among varying genomic contexts (islands, shores, shelves, and open seas). Both mdh and NADH dehydrogenase sites became hypermethylated with age, and sites mapping to rRNA became hypomethylated with age (similar to patterns we observed in our exploratory data set). Within our combined CpG + CHG elastic net clock six of 38 cytosines returned hits based on homology to mdh and rRNA, all of which were CpG‐methylated (Table S7). The CpG cytosine that mapped to mdh was found in a shore region, while cytosines in regions mapping to rRNA were found in shore and open sea regions. For annotations and robustness of cytosines at predicting age from CpG and CHG clocks, see Tables S3–S8.
4. DISCUSSION
We demonstrate the use of methylation patterns to generate epigenetic clocks for P. taeda. These clocks are capable of predicting chronological ages within ~6% of the maximum lifespan (275 years) of this species, although predicted ages from clocks utilizing CHG‐methylated cytosines were more slightly more correlated with chronological age compared to those using CpG‐methylated cytosines only. Our study shares similarities to that reported by Shahryary et al. (2020) in that both make use of age‐related methylation patterns to predict chronological ages of trees; however; as described in the review by Yao et al. (2021), there are key differences in the methodological approaches and interpretations of our studies. Shahryary et al. (2020) leveraged a novel software program (AlphaBeta) that generated a model capable of accurately predicting whole tree chronological age by estimating divergence times of epimutations among loci fron leaf methylomes collected from differing branches of known chronlogocial age from an individual Poplar tree (Populus trichocarpa). The approach requires methylome divergence along with calibrated epimutation rates as input, and has so far only be applied to a single tree. By contrast, the epigenetic clocks described in our study were generated utilizing either elastic net penalized regression or correlation tests to select relatively fewer numbers of sites whose methylation status is highly correlated with age. The resulting models from this approach can then be utilized to perform age predictions for many individuals and potentially other species, as has been the case for other similarly constructed methylation clocks (Mammalian Methylation Consortium et al., 2021).
Although there was no effect of clock type on the MAE values of predicted ages for our test set trees, we found that models constructed with five sites for which methylation status was highly correlated with age (assessed by Pearson correlation coefficients) were more accurate when predicting age in P. taeda compared to models constructed with elastic net methods. Regarding methylation context, although age‐associated CpG and CHG‐methylation patterns were both distributed across multiple genomic regions, we observed an enrichment of age‐associated CpG‐methylation within island and shore regions, and CHG‐methylation in island regions. Within both methylation contexts, age‐associated methylation mostly resulted in hypomethylation, regardless of genomic (island, shore, shelf, or open sea) context. In regard to potential functional implications, only CpG‐methylation has been previously associated with gene expression in P. taeda (Takuno et al., 2016). As increased CpG‐methylation at promoter sequences (commonly associated with CpG island regions) is associated with reduced expression of downstream genes (Gehring & Henikoff, 2007), hypomethylation at many island‐region sites may indicate that expression of specific genes increases with age in P. taeda. In contrast to CpG‐methylation, CHG‐methylation is associated with gene splicing (Chaudhary et al., 2021; Zhang et al., 2018). Alterations in CHG‐methylation status can affect the proper splicing of genes (Chaudhary et al., 2021; Zhang et al., 2018) which can have deleterious consequences (Ong‐Abdullah et al., 2015). When comparing age‐related changes in methylation status among cytosines from both CpG and CHG contexts mapping to known genes in our BLAST searches, gains and losses in methylation were consistent for sites that mapped to similar genes for both contexts. Compared to CpG and CHG‐methylation, CHH‐methylation in plants has largely been associated with transposable element silencing (Dubin et al., 2015), and rates of epimutations in transposable element regions are generally low (van der Graaf et al., 2015). Thus, the stability of CHH‐methylation through development could explain the poor associations between CHH‐methylation and ageing observed in the current study.
Although the majority of cytosines in our analyses did not map to annotated genes, the expression of critical genes involved in conserved molecular pathways that regulate life‐history traits has been observed to influence multiple physiological processes (Flatt & Partridge, 2018; Partridge & Gems, 2002; Perls et al., 2002). Among the genes proximal to several of our age‐associated CpG and CHG‐methylated sites in the exploratory and clock data sets were malate dehydrogenase (mdh), NADH dehydrogenase, and 18S and 26S rRNA. The expression of mdh, a multisubunit metabolic enzyme in many organisms including plants (Longo & Scandalios, 1969; Yudina, 2012), has been linked to respiration and CO2 assimilation rates, and normal plant growth and development (Tomaz et al., 2010). Expression has been shown to increase with age, which is postulated to maintain aerobic metabolism (Sharma & Patnaik, 1982). NADH dehydrogenase, another important metabolic enzyme involved in the electron transport chain (Weiss et al., 1992), is also associated with plant growth and development (Sweetman et al., 2019). Proper subunit splicing of this enzyme is essential for its function (Bonen, 2008; Malek & Knoop, 1998), with defective splicing leading to decreased activity of the electron transport chain, slower growth, and delayed germination and phenotypic development (Hsieh et al., 2015). Expression and activity of rRNA have been shown to increase with increased metabolic demands (Russell & Zomerdijk, 2005), also affecting growth, cell adaptation, stress responses, and cell proliferation (Russell & Zomerdijk, 2005). Increased methylation near rRNA promoter regions greatly reduces expression (Ghoshal et al., 2004; Zatsepina et al., 1993); therefore, the loss of methylation we observed for cytosines proximal to 18S and 26S rRNA genes in island and shore regions coupled with the methylation gains we observed for CpG‐methylated cytosines near mdh and NADH dehydrogenase in shore and open sea regions may indicate expression of these genes increases to match increasing metabolic demands as P. taeda trees age. If so, an increased requirement for proper splicing of NADH and mdh would be needed (Gendrel et al., 2002; Jeddeloh et al., 1999), which might explain the increased CHG‐methylation near these genes with age. Thus, taken together with current functional understanding of the roles of methylation patterns in plants, our findings may indicate a possible link between epigenetic ageing and increases in gene expression and splicing variation with age, although these predictions require further evaluation.
Beyond their ability to predict chronological age, the epigenetic clocks developed here are likely to find utility in forest management applications and addressing questions surrounding the basic biology of ageing. Currently, stem coring is the most common and reliable method for determination of tree chronological age (Villalba & Veblen, 1997). While not entirely destructive, stem coring does introduce a wound into a tree stem that could potentially allow pest or pathogen access (Tsen et al., 2016). Thus, the chronological age estimation methods described in our study offer a potentially less invasive approach for tree age determination. For example, cambium can be collected by removing bark and not actually penetrating the tree xylem, which would allow for faster repair. If methylation patterns are similar between cambium and leaves, then this approach would offer an even less invasive approach for predicting their chronological age. Unlike the other trees in our data set for which chronological age was determined using planting records, age estimates for the three oldest trees were estimated based solely on coring techniques. Ultimately, these trees showed the highest discordance between their epigenetic age and that estimated using coring techniques. As tree ring formation varies with growth rates and fluctuating environmental conditions (Gonzalez‐Benecke et al., 2015; Jayawickrama et al., 1997; Wong & Lertzman, 2001), erroneous age estimations of up to several decades using these methods may occur (Wong & Lertzman, 2001). However, it is also possible that the epigenetic clocks developed in this study are not able to accurately age older trees.
The directionality and magnitude of epigenetic‐to‐chronological age discordance in humans and other vertebrates correlates to variation in life history traits (Anderson et al., 2021; Hamlat et al., 2021; Ryan, 2021). Variation in life history traits can have consequences for stand productivity (Kellner & Swihart, 2016; Schulze, 2003). If epigenetic‐to‐chronological age discordance, especially in early life, is connected to variation in tree growth, productivity, wood quality, and/or responses to disturbances, epigenetic clocks might inform breeding programs and could be used in evaluating efficiencies of experimental manipulations to increase early‐life growth and subsequent wood yield from managed P. taeda stands (Baker & Langdon, 1990; Clason, 1989). Age‐associated variation in DNA methylation patterns is also associated with species‐specific lifespans in vertebrates (Mammalian Methylation Consortium et al., 2021). The findings presented here raise the possibility that age‐related DNA methylation patterns might be used to estimate lifespan and yield insights into the underlying genetic and epigenetic determinants across different plant species.
A limitation of this study was the relatively small number of individual trees analysed. The P. taeda genome, like that of many tree species, is quite large (20 billion base pairs) (Neale et al., 2014). As a result, resolving patterns of genomic methylation are sequencing intensive, and more cost‐efficient technical approaches (e.g., amplicon sequencing, bait capture techniques) are probably needed if measures of epigenetic age are to be scaled to stand and population applications (Meek & Larson, 2019). Despite a low sample size, age predictions from our methylation clocks were highly accurate, predicting similar MAE values as those for many vertebrate species (Bertucci et al., 2021; Bors et al., 2021; Lemaître et al., 2020; Meer et al., 2018; Stubbs et al., 2017). Additionally, our Pearson clocks utilizing five highly age‐correlated sites showed high robustness when predicting age in P. taeda, which may allow for a more targeted approach in further studies seeking to quantify tree age through epigenetic signatures. Regardless of these limitations, our study clearly demonstrates the relationship between DNA methylation and chronological age in a long‐lived tree species and indicates that alterations in DNA methylation may be a universal aspect of ageing across the tree of life.
AUTHOR CONTRIBUTIONS
Emily M. Bertucci, Benjamin B. Parrott, Doug P. Aubrey, and Andy Horcher developed the concept for the manuscript. Emily M. Bertucci and Randall Sutton collected the samples. Emily M. Bertucci prepared the libraries and assisted Steven T. Gardner with data analysis. Steven T. Gardner analysed the data and wrote the manuscript while being assisted by Emily M. Bertucci and Benjamin B. Parrott.
CONFILCT OF INTEREST
The authors have no conflicts of interest to declare.
OPEN RESEARCH BADGES
This article has earned an Open Data Badge for making publicly available in the NCBI Sequence Read Archive (SRA) under BioProjects PRJNA862919. Scripts used to analyze data are publicly available at https://github.com/stg0015/Loblolly‐scripts.
DISCLAIMER
This report was prepared as an account of work sponsored by an agency of the United States Government. Neither the United States Government nor any agency thereof, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by tradename, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favouring by the United States Government or any agency thereof. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof.
Supporting information
Figures S1–S2
Tables S1–S8
Appendix S1
ACKNOWLEDGEMENTS
We would like to thank Samantha Bock for her assistance with bioinformatic analyses, as well as the members of the Parrott laboratory at SREL for their feedback pertaining to the results of this study. We would also like to thank Dr Sibbett, the Subject Editor, and two anonymous reviewers for their efforts, which went above and beyond and substantially improved the manuscript. This study was supported in part by the USDA Forest Service‐Savannah River, under Interagency DE‐EM0003622 with the U.S. Department of Energy, the National Science Foundation (Award no. 2026210, BBP), and the U.S. Department of Energy Office of Environmental Management under award number DE‐EM0004391 to the University of Georgia Research Foundation.
Gardner, S. T. , Bertucci, E. M. , Sutton, R. , Horcher, A. , Aubrey, D. , & Parrott, B. B. (2023). Development of DNA methylation‐based epigenetic age predictors in loblolly pine (Pinus taeda). Molecular Ecology Resources, 23, 131–144. 10.1111/1755-0998.13698
Handling Editor: Luke Browne
DATA AVAILABILITY STATEMENT
This article has earned an Open Data Badge for making publicly available in the NCBI Sequence Read Archive (SRA) under BioProjects PRJNA862919. Scripts used to analyse data have been made publicly available at https://github.com/stg0015/Loblolly‐scripts.
REFERENCES
- Akalin, A. , Kormaksson, M. , Li, S. , Garrett‐Bakelman, F. E. , Figueroa, M. E. , Melnick, A. , & Mason, C. E. (2012). methylKit: A comprehensive R package for the analysis of genome‐wide DNA methylation profiles. Genome Biology, 13(10), R87. 10.1186/gb-2012-13-10-r87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albaugh, T. J. , Lee Allen, H. , Dougherty, P. M. , & Johnsen, K. H. (2004). Long term growth responses of loblolly pine to optimal nutrient and water resource availability. Forest Ecology and Management, 192(1), 3–19. 10.1016/j.foreco.2004.01.002 [DOI] [Google Scholar]
- Anastasiadi, D. , & Piferrer, F. (2020). A clockwork fish: Age prediction using DNA methylation‐based biomarkers in the European seabass. Molecular Ecology Resources, 20(2), 387–397. 10.1111/1755-0998.13111 [DOI] [PubMed] [Google Scholar]
- Anderson, J. A. , Johnston, R. A. , Lea, A. J. , Campos, F. A. , Voyles, T. N. , Akinyi, M. Y. , Alberts, S. C. , Archie, E. A. , & Tung, J. (2021). High social status males experience accelerated epigenetic aging in wild baboons. eLife, 10, e66128. 10.7554/eLife.66128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data [Online]. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- Ausin, I. , Feng, S. , Yu, C. , Liu, W. , Kuo, H. Y. , Jacobsen, E. L. , Zhai, J. , Gallego‐Bartolome, J. , Wang, L. , Egertsdotter, U. , Street, N. R. , Jacobsen, S. E. , & Wang, H. (2016). DNA methylome of the 20‐gigabase Norway spruce genome. Proceedings of the National Academy of Sciences of the United States of America, 113(50), E8106–E8113. 10.1073/pnas.1618019113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, J. B. , & Langdon, O. G. (1990). Pinus taeda L., Loblolly Pine. In Burns R. M. & Honkala B. H. (Eds.), Silvics of North America, Volume 1, Conifers (pp. 497–512). U.S. Department of Agriculture, Forest Service, Agriculture Handbook 654. [Google Scholar]
- Bartels, A. , Han, Q. , Nair, P. , Stacey, L. , Gaynier, H. , Mosley, M. , Huang, Q. Q. , Pearson, J. K. , Hsieh, T. F. , An, Y. C. , & Xiao, W. (2018). Dynamic DNA methylation in plant growth and development. International Journal of Molecular Sciences, 19(7), 2144. 10.3390/ijms19072144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdyshev, G. D. , Korotaev, G. K. , Boiarskikh, G. V. , & Vaniushin, B. F. (1967). Nucleotide composition of DNA and RNA from somatic tissues of humpback and its changes during spawning. Biokhimiia (Moscow, Russia), 32(5), 988–993. [PubMed] [Google Scholar]
- Bertucci, E. M. , Mason, M. W. , Rhodes, O. E. , & Parrott, B. B. (2021). Exposure to ionizing radiation disrupts normal epigenetic aging in Japanese medaka. Aging, 13(19), 22752–22771. 10.18632/aging.203624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertucci, E. M. , & Parrott, B. B. (2020). Is CpG density the link between epigenetic aging and lifespan? Trends in Genetics, 36(10), 725–727. 10.1016/j.tig.2020.06.003 [DOI] [PubMed] [Google Scholar]
- Binder, A. M. , Corvalan, C. , Mericq, V. , Pereira, A. , Santos, J. L. , Horvath, S. , Shepherd, J. , & Michels, K. B. (2018). Faster ticking rate of the epigenetic clock is associated with faster pubertal development in girls. Epigenetics, 13(1), 85–94. 10.1080/15592294.2017.1414127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonen, L. (2008). Cis‐ and trans‐splicing of group II introns in plant mitochondria. Mitochondrion, 8(1), 26–34. 10.1016/j.mito.2007.09.005 [DOI] [PubMed] [Google Scholar]
- Bors, E. K. , Baker, C. S. , Wade, P. R. , O'Neill, K. B. , Shelden, K. E. W. , Thompson, M. J. , Fei, Z. , Jarman, S. , & Horvath, S. (2021). An epigenetic clock to estimate the age of living beluga whales. Evolutionary Applications, 14(5), 1263–1273. 10.1111/eva.13195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary, S. , Jabre, I. , & Syed, N. H. (2021). Epigenetic differences in an identical genetic background modulate alternative splicing in A. thaliana . Genomics, 113(6), 3476–3486. 10.1016/j.ygeno.2021.08.006 [DOI] [PubMed] [Google Scholar]
- Christensen, B. C. , Houseman, E. A. , Marsit, C. J. , Zheng, S. , Wrensch, M. R. , Wiemels, J. L. , Nelson, H. H. , Karagas, M. R. , Padbury, J. F. , Bueno, R. , Sugarbaker, D. J. , Yeh, R. F. , Wiencke, J. K. , & Kelsey, K. T. (2009). Aging and environmental exposures Alter tissue‐specific DNA methylation dependent upon CpG Island context. PLoS Genetics, 5(8), e1000602. 10.1371/journal.pgen.1000602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clason, T. R. (1989). Early growth enhancement increases loblolly pine rotation yields. Southern Journal of Applied Forestry, 13(2), 94–99. 10.1093/sjaf/13.2.94 [DOI] [Google Scholar]
- Mammalian Methylation Consortium , Lu, A. T. , Fei, Z. , Haghani, A. , Robeck, T. R. , Zoller, J. A. , Li, C. Z. , Zhang, J. , Ablaeva, J. , Adams, D. M. , Almunia, J. , Ardehali, R. , Arneson, A. , Baker, C. S. , Belov, K. , Black, P. , Blumstein, D. T. , Bors, E. K. , Breeze, C. E. , … Horvath, S. (2021). Universal DNA methylation age across mammalian tissues. bioRxiv, 2021.01.18.426733. 10.1101/2021.01.18.426733 [DOI] [Google Scholar]
- Coyle, D. R. , Aubrey, D. P. , & Coleman, M. D. (2016). Growth responses of narrow or broad site adapted tree species to a range of resource availability treatments after a full harvest rotation. Forest Ecology and Management, 362, 107–119. 10.1016/j.foreco.2015.11.047 [DOI] [Google Scholar]
- De Paoli‐Iseppi, R. , Deagle, B. E. , Polanowski, A. M. , McMahon, C. R. , Dickinson, J. L. , Hindell, M. A. , & Jarman, S. N. (2019). Age estimation in a long‐lived seabird (Ardenna tenuirostris) using DNA methylation‐based biomarkers. Molecular Ecology Resources, 19(2), 411–425. 10.1111/1755-0998.12981 [DOI] [PubMed] [Google Scholar]
- Dubin, M. J. , Zhang, P. , Meng, D. , Remigereau, M.‐S. , Osborne, E. J. , Paolo Casale, F. , Drewe, P. , Kahles, A. , Jean, G. , Vilhjálmsson, B. , Jagoda, J. , Irez, S. , Voronin, V. , Song, Q. , Long, Q. , Rätsch, G. , Stegle, O. , Clark, R. M. , & Nordborg, M. (2015). DNA methylation in Arabidopsis has a genetic basis and shows evidence of local adaptation. eLife, 4, e05255. 10.7554/eLife.05255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubrovina, A. S. , & Kiselev, K. V. (2016). Age‐associated alterations in the somatic mutation and DNA methylation levels in plants. Plant Biology, 18(2), 185–196. 10.1111/plb.12375 [DOI] [PubMed] [Google Scholar]
- Flatt, T. , & Partridge, L. (2018). Horizons in the evolution of aging. BMC Biology, 16(1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox, T. R. , Lee Allen, H. , Albaugh, T. J. , Rubilar, R. , & Carlson, C. A. (2007). Tree nutrition and forest fertilization of pine plantations in the southern United States. Southern Journal of Applied Forestry, 31(1), 5–11. [Google Scholar]
- Friedman, J. , Hastie, T. , & Tibshirani, R. (2010). Regularization paths for generalized linear models via coordinate descent. Journal of Statistical Software, 33(1), 1–22. [PMC free article] [PubMed] [Google Scholar]
- Gehring, M. , & Henikoff, S. (2007). DNA methylation dynamics in plant genomes. Biochimica et Biophysica acta (BBA) ‐ Gene Structure and Expression, 1769(5), 276–286. 10.1016/j.bbaexp.2007.01.009 [DOI] [PubMed] [Google Scholar]
- Gendrel, A.‐V. , Lippman, Z. , Yordan, C. , Colot, V. , & Martienssen, R. (2002). Dependence of heterochromatic histone H3 methylation patterns on the Arabidopsis gene DDM1. Science, 297, 1871–1873. 10.1126/science.1074950 [DOI] [PubMed] [Google Scholar]
- Genger, R. K. , Peacock, J. W. , Dennis, E. S. , & Finnegan, J. E. (2003). Opposing effects of reduced DNA methylation on flowering time in Arabidopsis thaliana . Planta, 216(3), 461–466. 10.1007/s00425-002-0855-9 [DOI] [PubMed] [Google Scholar]
- Ghoshal, K. , Majumder, S. , Datta, J. , Motiwala, T. , Bai, S. , Sharma, S. M. , Frankel, W. , & Jacob, S. T. (2004). Role of human ribosomal RNA (rRNA) promoter methylation and of methyl‐CpG‐binding protein MBD2 in the suppression of rRNA gene expression *. Journal of Biological Chemistry, 279(8), 6783–6793. 10.1074/jbc.M309393200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez‐Benecke, C. A. , Riveros‐Walker, A. J. , Martin, T. A. , & Peter, G. F. (2015). Automated quantification of intra‐annual density fluctuations using microdensity profiles of mature Pinus taeda in a replicated irrigation experiment. Trees, 29(1), 185–197. 10.1007/s00468-014-1103-1 [DOI] [Google Scholar]
- Hamlat, E. J. , Prather, A. A. , Horvath, S. , Belsky, J. , & Epel, E. S. (2021). Early life adversity, pubertal timing, and epigenetic age acceleration in adulthood. Developmental Psychobiology, 63(5), 890–902. 10.1002/dev.22085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannum, G. , Guinney, J. , Zhao, L. , Zhang, L. , Hughes, G. , Sadda, S. , Klotzle, B. , Bibikova, M. , Fan, J. B. , Gao, Y. , Deconde, R. , Chen, M. , Rajapakse, I. , Friend, S. , Ideker, T. , & Zhang, K. (2013). Genome‐wide methylation profiles reveal quantitative views of human aging rates. Molecular Cell, 49(2), 359–367. 10.1016/j.molcel.2012.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastie, T. , Tibshirani, R. , Narasimhan, B. , & Chu, G. (2021). impute: impute: Imputation for microarray data. R package version 1.66.0.
- Horvath, D. P. , Anderson, J. V. , Chao, W. S. , & Foley, M. E. (2003). Knowing when to grow: Signals regulating bud dormancy. Trends in Plant Science, 8(11), 534–540. 10.1016/j.tplants.2003.09.013 [DOI] [PubMed] [Google Scholar]
- Horvath, S. (2013). DNA methylation age of human tissues and cell types. Genome Biology, 14(10), 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath, S. , & Raj, K. (2018). DNA methylation‐based biomarkers and the epigenetic clock theory of ageing. Nature Reviews Genetics, 19(6), 371–384. [DOI] [PubMed] [Google Scholar]
- How‐Kit, A. , Teyssier, E. , Deleuze, J.‐F. , & Gallusci, P. (2017). Locus‐specific DNA methylation analysis and applications to plants. In Plant epigenetics (pp. 303–327). Springer. [Google Scholar]
- Hsieh, W.‐Y. , Liao, J.‐C. , Chang, C.‐Y. , Harrison, T. , Boucher, C. , & Hsieh, M.‐H. (2015). The SLOW GROWTH3 pentatricopeptide repeat protein is required for the splicing of mitochondrial NADH dehydrogenase Subunit7 intron 2 in Arabidopsis . Plant Physiology, 168(2), 490–501. 10.1104/pp.15.00354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayawickrama, K. J. S. , McKeand, S. E. , Jett, J. B. , & Wheeler, E. A. (1997). Date of earlywood–latewood transition in provenances and families of loblolly pine, and its relationship to growth phenology and juvenile wood specific gravity. Canadian Journal of Forest Research, 27, 1245–1253. [Google Scholar]
- Jeddeloh, J. A. , Stokes, T. L. , & Richards, E. J. (1999). Maintenance of genomic methylation requires a SWI2/SNF2‐like protein. Nature Genetics, 22(1), 94–97. 10.1038/8803 [DOI] [PubMed] [Google Scholar]
- Jiang, C. , Mithani, A. , Belfield, E. J. , Mott, R. , Hurst, L. D. , & Harberd, N. P. (2014). Environmentally responsive genome‐wide accumulation of de novo Arabidopsis thaliana mutations and epimutations. Genome Research, 24(11), 1821–1829. 10.1101/gr.177659.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, M. , & Pfeifer, G. P. (2015). Aging and DNA methylation. BMC Biology, 13(1), 7. 10.1186/s12915-015-0118-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabacik, S. , Horvath, S. , Cohen, H. , & Raj, K. (2018). Epigenetic ageing is distinct from senescence‐mediated ageing and is not prevented by telomerase expression. Aging, 10(10), 2800–2815. 10.18632/aging.101588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellner, K. F. , & Swihart, R. K. (2016). Timber harvest and drought interact to impact oak seedling growth and survival in the central hardwood Forest. Ecosphere, 7(10), e01473. 10.1002/ecs2.1473 [DOI] [Google Scholar]
- Kilgo, J. , & Blake, J. (2005). Ecology and management of a forested landscape: Fifty years on the Savannah River site. USDA Forest Service. [Google Scholar]
- Krueger, F. , & Andrews, S. R. (2011). Bismark: A flexible aligner and methylation caller for bisulfite‐seq applications. Bioinformatics, 27(11), 1571–1572. 10.1093/bioinformatics/btr167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn, M. (2015). caret: Classification and regression training . Astrophysics Source Code Library, ascl‐1505.
- Law, J. A. , & Jacobsen, S. E. (2010). Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nature Reviews Genetics, 11(3), 204–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence, M. , Huber, W. , Pagès, H. , Aboyoun, P. , Carlson, M. , Gentleman, R. , Morgan, M. T. , & Carey, V. J. (2013). Software for computing and annotating genomic ranges. PLoS Computational Biology, 9(8), e1003118. 10.1371/journal.pcbi.1003118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaître, J.‐F. , Rey, B. , Gaillard, J.‐M. , Régis, C. , Gilot‐Fromont, E. , Débias, F. , Duhayer, J. , Pardonnet, S. , Pellerin, M. , Haghani, A. , Zoller, J. A. , Li, C. Z. , & Horvath, S. (2020). DNA methylation as a tool to explore ageing in wild roe deer populations. Molecular Ecology Resources, 22(3), 1002–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine, M. E. , Lu, A. T. , Quach, A. , Chen, B. H. , Assimes, T. L. , Bandinelli, S. , Hou, L. , Baccarelli, A. A. , Stewart, J. D. , Li, Y. , Whitsel, E. A. , Wilson, J. G. , Reiner, A. P. , Aviv, A. , Lohman, K. , Liu, Y. , Ferrucci, L. , & Horvath, S. (2018). An epigenetic biomarker of aging for lifespan and healthspan. Aging, 10(4), 573–591. 10.18632/aging.101414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. , Abecasis, G. , Durbin, R. , & 1000 Genome Project Data Processing Subgroup . (2009). The sequence alignment/map format and SAMtools. Bioinformatics, 25(16), 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo, G. P. , & Scandalios, J. G. (1969). Nuclear gene control of mitochondrial malic dehydrogenase in maize. Proceedings of the National Academy of Sciences, 62(1), 104–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeira, F. , Park, Y. M. , Lee, J. , Buso, N. , Gur, T. , Madhusoodanan, N. , Basutkar, P. , Tivey, A. R. N. , Potter, S. C. , Finn, R. D. , & Finn, R. D. (2019). The EMBL‐EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Research, 47(W1), W636–W641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney, J. , Koegler, P. , & Rood, S. (1991). The accuracy of tree Ring analysis for estimating the age of riparian poplars. In Rood S. B. & Mahoney J. M. (Eds.), The biology and management of southern Alberta's cottonwoods (pp. 25–30). University of Lethbridge, Alberta. [Google Scholar]
- Malek, O. , & Knoop, V. (1998). Trans‐splicing group II introns in plant mitochondria: The complete set of cis‐arranged homologs in ferns, fern allies, and a hornwort. RNA, 4(12), 1599–1609. 10.1017/S1355838298981262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayne, B. , Korbie, D. , Kenchington, L. , Ezzy, B. , Berry, O. , & Jarman, S. (2020). A DNA methylation age predictor for zebrafish. Aging, 12(24), 24817–24835. 10.18632/aging.202400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medlyn, B. , Barrett, D. , Landsberg, J. , Sands, P. , & Clement, R. (2003). Corrigendum to: Conversion of canopy intercepted radiation to photosynthate: A review of modelling approaches for regional scales. Functional Plant Biology, 30(7), 829. 10.1071/fp02088_co [DOI] [PubMed] [Google Scholar]
- Meek, M. H. , & Larson, W. A. (2019). The future is now: Amplicon sequencing and sequence capture usher in the conservation genomics era. Molecular Ecology Resources, 19(4), 795–803. 10.1111/1755-0998.12998 [DOI] [PubMed] [Google Scholar]
- Meer, M. V. , Podolskiy, D. I. , Tyshkovskiy, A. , & Gladyshev, V. N. (2018). A whole lifespan mouse multi‐tissue DNA methylation clock. eLife, 7, e40675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale, D. B. , Wegrzyn, J. L. , Stevens, K. A. , Zimin, A. V. , Puiu, D. , Crepeau, M. W. , Cardeno, C. , Koriabine, M. , Holtz‐Morris, A. E. , Liechty, J. D. , Martínez‐García, P. J. , Vasquez‐Gross, H. A. , Lin, B. Y. , Zieve, J. J. , Dougherty, W. M. , Fuentes‐Soriano, S. , Wu, L. S. , Gilbert, D. , Marçais, G. , … Langley, C. H. (2014). Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies. Genome Biology, 15(3), R59. 10.1186/gb-2014-15-3-r59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, H.‐H. , & Adrian, B. (1999). DNA methylation and chromatin modification. Current Opinion in Genetics & Development, 9(2), 158–163. 10.1016/S0959-437X(99)80024-0 [DOI] [PubMed] [Google Scholar]
- Ong‐Abdullah, M. , Ordway, J. M. , Jiang, N. , Ooi, S.‐E. , Kok, S.‐Y. , Sarpan, N. , Azimi, N. , Hashim, A. T. , Ishak, Z. , Rosli, S. K. , Malike, F. A. , Bakar, N. A. , Marjuni, M. , Abdullah, N. , Yaakub, Z. , Amiruddin, M. D. , Nookiah, R. , Singh, R. , Low, E. T. , … Martienssen, R. A. (2015). Loss of karma transposon methylation underlies the mantled somaclonal variant of oil palm. Nature, 525(7570), 533–537. 10.1038/nature15365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrott, B. B. , & Bertucci, E. M. (2019). Epigenetic aging clocks in ecology and evolution. Trends in Ecology & Evolution, 34(9), 767–770. [DOI] [PubMed] [Google Scholar]
- Partridge, L. , & Gems, D. (2002). Mechanisms of aging: Public or private? Nature Reviews Genetics, 3(3), 165–175. [DOI] [PubMed] [Google Scholar]
- Perls, T. , Kunkel, L. , & Puca, A. (2002). The genetics of aging. Current Opinion in Genetics & Development, 12(3), 362–369. [DOI] [PubMed] [Google Scholar]
- Perna, L. , Zhang, Y. , Mons, U. , Holleczek, B. , Saum, K.‐U. , & Brenner, H. (2016). Epigenetic age acceleration predicts cancer, cardiovascular, and all‐cause mortality in a German case cohort. Clinical Epigenetics, 8(1), 64. 10.1186/s13148-016-0228-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst, A. V. , & Mittelsten Scheid, O. (2015). Stress‐induced structural changes in plant chromatin. Current Opinion in Plant Biology, 27, 8–16. 10.1016/j.pbi.2015.05.011 [DOI] [PubMed] [Google Scholar]
- Quinlan, A. R. , & Hall, I. M. (2010). BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics, 26(6), 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raddatz, G. , Arsenault, R. J. , Aylward, B. , Whelan, R. , Böhl, F. , & Lyko, F. (2021). A chicken DNA methylation clock for the prediction of broiler health. Communications Biology, 4(1), 1–8. 10.1038/s42003-020-01608-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revelle, W. (2019). Psych: Procedures for psychological, psychometric, and personality research. Northwestern University, Evanston, Illinois. R package version 1.9.12. http://CRAN.R‐Project.Org/Package=Psych [Google Scholar]
- Richardson, B. (2003). Impact of aging on DNA methylation. Ageing Research Reviews, 2(3), 245–261. 10.1016/S1568-1637(03)00010-2 [DOI] [PubMed] [Google Scholar]
- Russell, J. , & Zomerdijk, J. C. B. M. (2005). RNA‐polymerase‐I‐directed rDNA transcription, life and works. Trends in Biochemical Sciences, 30(2), 87–96. 10.1016/j.tibs.2004.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan, C. P. (2021). “Epigenetic clocks”: Theory and applications in human biology. American Journal of Human Biology, 33(3), e23488. 10.1002/ajhb.23488 [DOI] [PubMed] [Google Scholar]
- Ryan, C. P. , Hayes, M. G. , Lee, N. R. , McDade, T. W. , Jones, M. J. , Kobor, M. S. , Kuzawa, C. W. , & Eisenberg, D. T. A. (2018). Reproduction predicts shorter telomeres and epigenetic age acceleration among young adult women. Scientific Reports, 8(1), 11100. 10.1038/s41598-018-29486-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuelson, L. , Stokes, T. , Cooksey, T. , & McLemore, P., III . (2001). Production efficiency of loblolly pine and sweetgum in response to four years of intensive management. Tree Physiology, 21(6), 369–376. [DOI] [PubMed] [Google Scholar]
- Schulze, M. D. (2003). Ecology and behavior of nine timber tree species in Pará, Brazil: Links between species life history and forest management and conservation. The Pennsylvania State University. [Google Scholar]
- Shahryary, Y. , Symeonidi, A. , Hazarika, R. R. , Denkena, J. , Mubeen, T. , Hofmeister, B. , van Gurp, T. , Colomé‐Tatché, M. , Verhoeven, K. J. F. , Tuskan, G. , Schmitz, R. J. , & Johannes, F. (2020). AlphaBeta: Computational inference of epimutation rates and spectra from high‐throughput DNA methylation data in plants. Genome Biology, 21(1), 260. 10.1186/s13059-020-02161-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, R. , & Patnaik, S. K. (1982). Differential regulation of malate dehydrogenase isoenzymes by hydrocortisone in the liver and brain of aging rats. Development, Growth & Differentiation, 24(5), 501–505. 10.1111/j.1440-169X.1982.00501.x [DOI] [PubMed] [Google Scholar]
- Simpkin, A. J. , Suderman, M. , Gaunt, T. R. , Lyttleton, O. , McArdle, W. L. , Ring, S. M. , Tilling, K. , Davey Smith, G. , & Relton, C. L. (2015). Longitudinal analysis of DNA methylation associated with birth weight and gestational age. Human Molecular Genetics, 24(13), 3752–3763. 10.1093/hmg/ddv119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin, R. K. , & Martienssen, R. (2007). Transposable elements and the epigenetic regulation of the genome. Nature Reviews Genetics, 8(4), 272–285. [DOI] [PubMed] [Google Scholar]
- Stubbs, T. M. , Bonder, M. J. , Stark, A.‐K. , Krueger, F. , BI Ageing Clock Team , von Meyenn, F. , Stegle, O. , & Reik, W. (2017). Multi‐tissue DNA methylation age predictor in mouse. Genome Biology, 18(1), 68. 10.1186/s13059-017-1203-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, M. M. , & Bird, A. (2008). DNA methylation landscapes: Provocative insights from epigenomics. Nature Reviews Genetics, 9(6), 465–476. 10.1038/nrg2341 [DOI] [PubMed] [Google Scholar]
- Sweetman, C. , Waterman, C. D. , Rainbird, B. M. , Smith, P. M. C. , Jenkins, C. D. , Day, D. A. , & Soole, K. L. (2019). AtNDB2 is the main external NADH dehydrogenase in mitochondria and is important for tolerance to environmental stress. Plant Physiology, 181(2), 774–788. 10.1104/pp.19.00877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takuno, S. , Ran, J.‐H. , & Gaut, B. S. (2016). Evolutionary patterns of genic DNA methylation vary across land plants. Nature Plants, 2(2), 1–7. 10.1038/nplants.2015.222 [DOI] [PubMed] [Google Scholar]
- Tomaz, T. , Bagard, M. , Pracharoenwattana, I. , Lindén, P. , Lee, C. P. , Carroll, A. J. , Ströher, E. , Smith, S. M. , Gardeström, P. , & Millar, A. H. (2010). Mitochondrial malate dehydrogenase lowers leaf respiration and alters photorespiration and plant growth in Arabidopsis . Plant Physiology, 154(3), 1143–1157. 10.1104/pp.110.161612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsen, E. W. J. , Sitzia, T. , & Webber, B. L. (2016). To core, or not to core: The impact of coring on tree health and a best‐practice framework for collecting dendrochronological information from living trees. Biological Reviews, 91(4), 899–924. 10.1111/brv.12200 [DOI] [PubMed] [Google Scholar]
- van der Graaf, A. , Wardenaar, R. , Neumann, D. A. , Taudt, A. , Shaw, R. G. , Jansen, R. C. , Schmitz, R. J. , Colomé‐Tatché, M. , & Johannes, F. (2015). Rate, spectrum, and evolutionary dynamics of spontaneous epimutations. Proceedings of the National Academy of Sciences of the United States of America, 112(21), 6676–6681. 10.1073/pnas.1424254112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba, R. , & Veblen, T. T. (1997). Improving estimates of total tree ages based on increment core samples. Écoscience, 4(4), 534–542. 10.1080/11956860.1997.11682433 [DOI] [Google Scholar]
- Wang, Q. , Ci, D. , Li, T. , Li, P. , Song, Y. , Chen, J. , Quan, M. , Zhou, D. , & Zhang, D. (2016). The role of DNA methylation in Xylogenesis in different tissues of poplar. Frontiers in Plant Science, 7, 1003. 10.3389/fpls.2016.01003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidner, C. I. , Lin, Q. , Koch, C. M. , Eisele, L. , Beier, F. , Ziegler, P. , Bauerschlag, D. O. , Jöckel, K. H. , Erbel, R. , Mühleisen, T. W. , Zenke, M. , Brümmendorf, T. H. , & Wagner, W. (2014). Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biology, 15(2), R24. 10.1186/gb-2014-15-2-r24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss, H. , Friedrich, T. , Hofhaus, G. , & Preis, D. (1992). The respiratory‐chain NADH dehydrogenase (complex I) of mitochondria. In Christen P. & Hofmann E. (Eds.), EJB reviews 1991 (pp. 55–68). Springer. 10.1007/978-3-642-77200-9_5 [DOI] [PubMed] [Google Scholar]
- Wong, C. M. , & Lertzman, K. P. (2001). Errors in estimating tree age: Implications for studies of stand dynamics. Canadian Journal of Forest Research, 31(7), 1262–1271. 10.1139/x01-060 [DOI] [Google Scholar]
- Xiao, F.‐H. , Wang, H.‐T. , & Kong, Q.‐P. (2019). Dynamic DNA methylation during aging: A “prophet” of age‐related outcomes. Frontiers in Genetics, 10, 107. 10.3389/fgene.2019.00107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, N. , Schmitz, R. J. , & Johannes, F. (2021). Epimutations define a fast‐ticking molecular clock in plants. Trends in Genetics, 37, 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yudina, R. S. (2012). Malate dehydrogenase in plants: Its genetics, structure, localization and use as a marker. Advances in Bioscience and Biotechnology, 3, 370–377. 10.4236/abb.2012.34053 [DOI] [Google Scholar]
- Zatsepina, O. V. , Voit, R. , Grummt, I. , Spring, H. , Semenov, M. V. , & Trendelenburg, M. F. (1993). The RNA polymerase I‐specific transcription initiation factor UBF is associated with transcriptionally active and inactive ribosomal genes. Chromosoma, 102(9), 599–611. 10.1007/BF00352307 [DOI] [PubMed] [Google Scholar]
- Zhang, H. , Lang, Z. , & Zhu, J.‐K. (2018). Dynamics and function of DNA methylation in plants. Nature Reviews Molecular Cell Biology, 19(8), 489–506. 10.1038/s41580-018-0016-z [DOI] [PubMed] [Google Scholar]
- Zheng, Y. , Joyce, B. T. , Colicino, E. , Liu, L. , Zhang, W. , Dai, Q. , Shrubsole, M. J. , Kibbe, W. A. , Gao, T. , Zhang, Z. , Jafari, N. , Vokonas, P. , Schwartz, J. , Baccarelli, A. A. , & Hou, L. (2016). Blood epigenetic age may predict cancer incidence and mortality. eBioMedicine, 5, 68–73. 10.1016/j.ebiom.2016.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberman, D. (2008). The evolving functions of DNA methylation. Current Opinion in Plant Biology, 11(5), 554–559. 10.1016/j.pbi.2008.07.004 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S2
Tables S1–S8
Appendix S1
Data Availability Statement
This article has earned an Open Data Badge for making publicly available in the NCBI Sequence Read Archive (SRA) under BioProjects PRJNA862919. Scripts used to analyse data have been made publicly available at https://github.com/stg0015/Loblolly‐scripts.