

Abstract
Mercury (Hg) methylation genes (hgcAB) mediate the formation of the toxic methylmercury and have been identified from diverse environments, including freshwater and marine ecosystems, Arctic permafrost, forest and paddy soils, coal‐ash amended sediments, chlor‐alkali plants discharges and geothermal springs. Here we present the first attempt at a standardized protocol for the detection, identification and quantification of hgc genes from metagenomes. Our Hg‐cycling microorganisms in aquatic and terrestrial ecosystems (Hg‐MATE) database, a catalogue of hgc genes, provides the most accurate information to date on the taxonomic identity and functional/metabolic attributes of microorganisms responsible for Hg methylation in the environment. Furthermore, we introduce “marky‐coco”, a ready‐to‐use bioinformatic pipeline based on de novo single‐metagenome assembly, for easy and accurate characterization of hgc genes from environmental samples. We compared the recovery of hgc genes from environmental metagenomes using the marky‐coco pipeline with an approach based on coassembly of multiple metagenomes. Our data show similar efficiency in both approaches for most environments except those with high diversity (i.e., paddy soils) for which a coassembly approach was preferred. Finally, we discuss the definition of true hgc genes and methods to normalize hgc gene counts from metagenomes.
Keywords: bioinformatics, hg methylation, hgcAB genes, hg‐MATE, marky‐coco, mercury, metagenomics
1. INTRODUCTION
Environmental mercury (Hg) methylation is primarily a biotic process carried out by microorganisms that transform inorganic Hg into the more toxic and bioaccumulative monomethylmercury (MeHg). The capacity to perform Hg methylation was historically associated with certain sulphate‐reducing bacteria, iron‐reducing bacteria and methanogenic archaea (Compeau & Bartha, 1985; Fleming et al., 2006; Hamelin et al., 2011; Kerin et al., 2006). Field observations revealed links between Hg methylation and sulphate‐reduction, iron‐reduction and methanogenesis in organic matter‐rich anaerobic environments (Bravo & Cosio, 2020 for review), as well as subsequent studies that tested cultured representatives of these clades for Hg methylation capability (Fleming et al., 2006; Gilmour et al., 2011, 2013, 2018). The discovery of the hgc genes (Parks et al., 2013) has facilitated the detection of novel putative Hg methylating bacteria and archaea through cultivation‐independent molecular methods (Gionfriddo et al., 2016; Podar et al., 2015). Recent studies analysing publicly available genomes and environmental metagenome‐assembled genomes (MAGs) identified hgc‐containing (hgc +) microorganisms from microbial lineages not formerly associated with Hg methylation, such as members of the PVC superphylum (Gionfriddo et al., 2019; Jones et al., 2019; Lin et al., 2021; McDaniel et al., 2020; Peterson et al., 2020). Identifying hgc genes in microbial genomes from meta‐omic data sets greatly expanded our view of the phylogenetic diversity of putative Hg methylators (Figure 1), but we still do not fully understand which microorganisms are the main drivers of Hg methylation in diverse environments, particularly outside of anoxic sediments.
FIGURE 1.
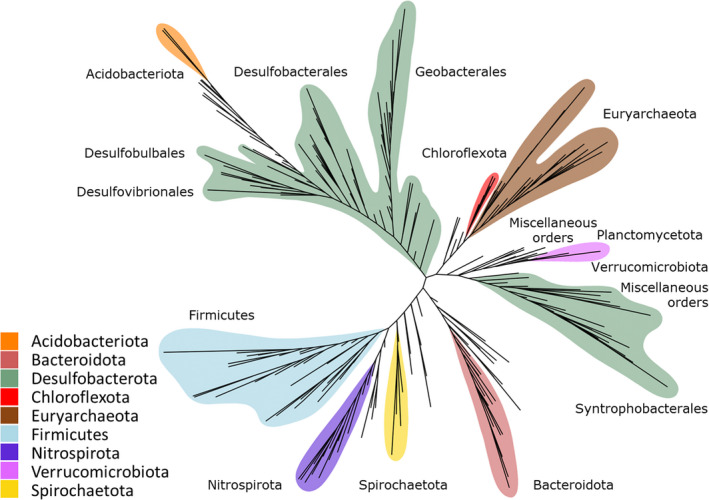
Simplified unrooted phylogenetic tree of hgcA sequences from the Hg‐MATE database. Taxonomy is based on GTDB classification Microbial groups with the highest diversity of hgc + microorganisms are denoted by colours.
Significant knowledge gaps in the identification of microorganisms capable of Hg methylation remain, largely because of the absence of hgc + cultured representatives from novel clades (i.e., outside the Desulfobacterota, Firmicutes, Euryarchaeota) with experimentally validated Hg‐methylating capability (Gilmour et al., 2018). One reason for this is the difficulty in selecting for hgc + microorganisms during cultivation, and another is the lack of a successful methodology for isolating all relevant microbes in controlled laboratory conditions. Microbes that have yet to be cultivated, and for which successful laboratory growth parameters need to be identified, are often referred to as the “unculturable” (Hug et al., 2016; Steen et al., 2019). High‐throughput meta‐omic and targeted amplicon sequencing studies have become the main methods for identifying putative Hg‐methylating microorganisms of this unculturable fraction (Bravo et al., 2018; Gionfriddo et al., 2020; Xu et al., 2021). While directly testing for Hg methylation capacity may not be a viable strategy, pairing these sequencing methods with biogeochemical measurements, Hg methylation assays, and other manipulation studies can connect a Hg‐methylating microbiome to MeHg production and metabolic activity and help to elucidate the potential contribution of these novel clades to Hg methylation (Bouchet et al., 2018; Kronberg et al., 2016; Roth et al., 2021; Schaefer et al., 2020).
The detection of hgc + MAGs provide the most precise information about the taxonomic and metabolic characteristics of putative Hg methylators (Jones et al., 2019; Lin et al., 2021; Peterson et al., 2020; Vigneron et al., 2021). However, the microbial diversity in some environments is too high and/or Hg methylators are too rare to identify them effectively (Christensen et al., 2019; Podar et al., 2015). In these cases, read‐based metagenomic analyses and hgc metabarcoding are easier and more economical. Accurately identifying Hg‐methylating clades (and metabolic guilds) from hgc sequences alone therefore requires a universally used and updated hgcAB reference database, coupled to consistent and robust bioinformatic practices, in order to identify precisely the target genes in complex meta‐omic data sets. Further, methods for quantifying hgc genes (and transcripts) from omics data are needed to predict the potential for environmental MeHg formation (Christensen et al., 2019; Capo, Feng, et al., 2022). Estimating the relative abundance of hgc sequences in a meta‐genome/transcriptome requires normalization strategies that account for differences in sequencing depth and coverage to avoid over‐ or under‐representing hgc + microorganisms and their functional importance.
In this work, we introduce the "Hg‐cycling Microorganisms in Aquatic and Terrestrial Ecosystems" (Hg‐MATE) database version 1 (https://doi.org/10.25573/serc.13105370.v1), an up‐to‐date hgcAB catalogue compiled from isolated, single‐cell and metagenome‐reconstructed genomes. Additionally, we present "marky‐coco" (https://github.com/ericcapo/marky‐coco), a ready‐to‐use bioinformatic pipeline to detect, identify and count hgc genes from metagenomes (Figure 2). We apply this pipeline to metagenomes collected from paddy soils, brackish and lake waters, as well as sediments from reservoirs and lakes, in which hgc genes have been previously detected (Capo et al., 2020; Jones et al., 2019; Liu et al., 2018; Millera Ferriz et al., 2021). Further, we specifically compared the reliability of (i) applying the marky‐coco pipeline based on de novo single assembly approach from single metagenomes with (ii) coassembling of multiple metagenomes (coassembly) prior to mapping and identification. Finally, we discuss appropriate definitions and cutoff criteria for hgc genes and also best practices to normalize data for an accurate count of hgc genes in metagenomes from environmental samples.
FIGURE 2.
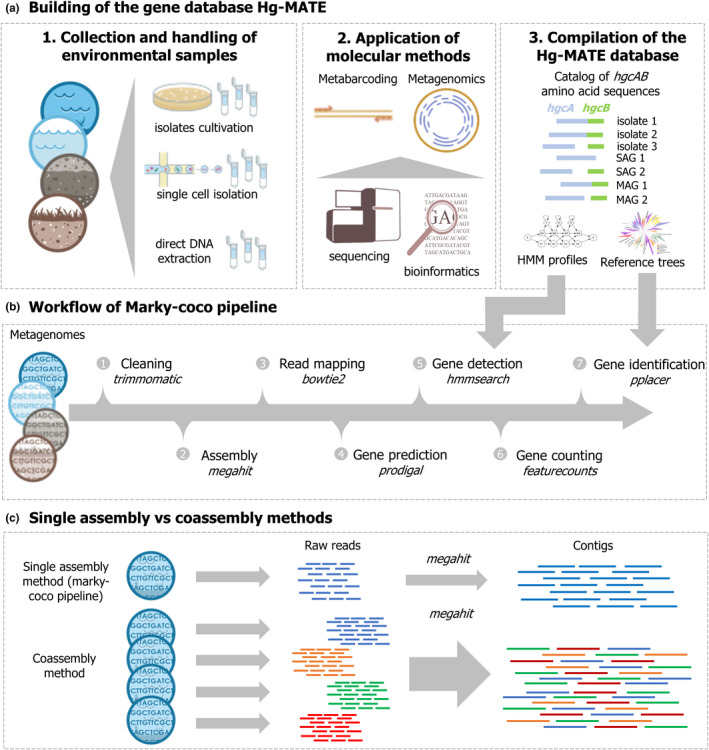
(a) Workflow illustrating how the hgcAB gene catalogue hg‐MATE database was built. (b) Simplified workflow of the marky‐coco pipeline. (c) Illustration of the two assembly approaches compared in this work: Single assembly versus coassembly.
2. MATERIALS AND METHODS
2.1. Description of the Hg‐MATE database v1
The Hg‐MATE database version 1 was released on 14 January 2021 (https://doi.org/10.25573/serc.13105370.v1), and contains an extensive hgcAB data set from a wide range of microorganisms and environments. The catalogue contains 1053 unique HgcA/B amino acid sequences (Table 1). We categorized the HgcAB amino acid sequences into four types depending on whether they were encoded in (i) pure culture/environmental microbial isolates (ISO) (ii) single‐cell genome sequences (CEL) (iii) metagenome‐assembled genomes (MAGs) (iv) or an environmental meta‐omic contig (CON). Amino acid sequences of HgcA, HgcB, and concatenated HgcA and HgcB were included in the database. If hgcB was not colocalized with hgcA in the genome and/or could not be identified, then “na” was listed in the “HgcB” sequence column. Both genes need to be present and encode functional proteins for a microbe to methylate Hg (see Parks et al., 2013; Smith et al., 2015). One reason hgcB may not be identified in some genomes carrying hgcA is because HgcB is highly homologous to other 4Fe‐4S ferredoxins. Therefore, hgcB can be difficult to differentiate from other ferredoxin‐encoding genes if not colocalized with hgcA on a contiguous sequence. In addition, hgcB may be missing from “MAGs”, “CEL” and “CON” sequences due to incomplete coverage of the genome or incomplete contig assembly, or failure to bin the contig carrying hgcB. Some hgc genes are predicted to encode a “fused HgcAB” protein which has previously been described (Podar et al., 2015), and is characterized by one gene that encodes for a 4Fe‐4S ferredoxin‐like protein with shared homology to HgcA and HgcB. This fused HgcAB protein contains the corrinoid iron–sulphur and transmembrane domains characteristic of HgcA as well as the 4Fe‐4S ferredoxin motif of HgcB (e.g., Uniprot Q8U2U9, NCBI Refseq: WP_011011854.1, Pyrococcus furiosus DSM 3638). These sequences are provided in the “HgcA” column, and labelled fused HgcAB in the HgcB column. These fused HgcAB sequences should be treated with caution because, while they share significant sequence homology to HgcA and HgcB from confirmed Hg methylators, to date all organisms with a fused HgcAB that have been tested do not seem to produce MeHg in culture (Gilmour et al., 2018; Podar et al., 2015).
TABLE 1.
Summary of HgcAB sequence types in version 1 of the hg‐MATE database
| Genome type | Total HgcA(B) sequences | Encodes both HgcA and HgcB | Encodes fused HgcAB | Only HgcA (or HgcB) present |
|---|---|---|---|---|
| ISO | 204 | 173 | 10 | 21 |
| CEL | 29 | 4 | 18 | 7 |
| MAG | 787 | 696 | 17 | 74 |
| CON | 33 | 9 | 0 | 21(3) |
The resources within the Hg‐MATE database version 1 include a catalogue with the amino acid sequences and metadata of all microorganisms. Only sequences with genomic identifying information (i.e., “ISO”, “CEL”, “MAG”) were used to compile further resources. Resources include: (i) FASTA files containing Hgc amino acid sequences; (ii) Multiple sequence alignments (MSA) in FASTA format of Hgc amino acid sequences built with MUSCLE implemented in MEGAX (Kumar et al., 2018) with the cluster method UPGMA; and (iii) Hidden Markov models (HMM) of aligned Hgc amino acid sequences built from MSAs using the hmmbuild function from the hmmer software (version 3.2.1, Finn et al., 2011). Additionally, resources include reference packages that can be used to identify and classify: (i) the corrinoid‐binding domain of HgcA which corresponds to residues ~37–156 of the HgcA sequence from Pseudodesulfovibrio mercurii ND132 and includes the characteristic cap helix domain (ii) full HgcA sequence and (iii) concatenated HgcA and HgcB. Each reference package contains sequence alignments, an HMM model, a phylogenetic tree, and NCBI taxonomy. Reference packages were constructed using the program Taxtastic (https://github.com/fhcrc/taxtastic) for HgcA(B) amino acid sequences from ISO, CEL and MAG. Phylogenetic trees were built from MSA files by RAxML using the GAMMA model of rate heterogeneity and LG amino acid substitution matrix (Le & Gascuel, 2008). Trees were rooted by HgcA paralogue sequences, carbon monoxide dehydrogenases (PF03599) from non‐HgcA coding microorganisms Candidatus Omnitrophica bacterium CG1_02_41_171 and Thermosulfurimonas dismutans. These organisms were chosen because of their distant phylogenetic relationship to hgcA+ microorganisms. Confidence values on branches were calculated from 100 bootstraps. Using the HgcA reference tree, a simplified tree of “ISO”, “CEL”, “MAG” hgcA genes was built using iTOL (Letunic & Bork, 2019) and clades were collapsed by the dominant monophyletic group, when possible, for visualization ease.
2.2. Data collection
A total of 29 metagenomes from recent studies studying hgc genes in environments with known active Hg methylation were used for the bioinformatic analyses performed in this work (Table 1, Appendix S1). Metagenomes from brackish waters (BARM8s) were collected in 2014 in the Gotland Deep basin of the Central Baltic Sea. Out of 81 available metagenomes (Alneberg et al., 2018; BioProject ID PRJEB22997), eight metagenomes where hgc genes have been detected (Capo et al., 2020) were used in the present analysis. Water depths of these metagenomes ranged from 76 to 200 m with oxygen concentrations either low (hypoxic zone) or undetectable (anoxic zone), salinity ranging between 9.2–12.1 psu and MeHg concentrations measuring up to 1640 fM (Soerensen et al., 2018). Lake sediments and water metagenomes (MANGA6s) were obtained in 2013–2014 from the sulfate‐impacted Manganika lake in Northern Minnesota (Jones et al., 2019, BioProject ID PRJNA488162). This hypereutrophic lake is characterized by dissolved oxygen approaching 16 mg/L (nearly 200% saturation) near the surface, pH exceeding 8.7 and MeHg accumulating over 3 ng/L in bottom waters. Dissolved oxygen and pH decreased with depth, and anoxic conditions were encountered below 4 m. Sulphide concentrations up to 2 mM were observed in bottom waters and sediments. Water samples were collected at these anoxic depths. Five metagenomes (RES5) were obtained from reservoir sediments from the St. Maurice River near Wemotaci, Canada in 2017 and 2018 (Millera Ferriz et al., 2021, GOLD‐JGI Ga0393614 Ga0393582, Ga0393617, Ga0393586, Ga0393589). The studied river section has been affected by the construction of two run‐of‐river power plant dams and its watershed has been disturbed by a forest fire, logging, and the construction of wetlands. MeHg concentrations in samples varied from <0.02 to 19 ng/g. Metagenomes from paddy and upland soils (PADDY10s) were collected from two historical Hg mining sites, Fenghuang (FH) and Wanshan (WS), in Southwest China in August 2016 (Liu et al., 2018, BioProject ID PRJNA450451). The pH of paddy soils ranged from 6 to 7.5. Historical discharge from Hg mining operations and ongoing atmospheric deposition contribute to high concentrations of MeHg in the soils around these areas with values up to 7.9 ng/g in the collected samples.
2.3. Bioinformatics
The detection, taxonomic identification and counting of hgc genes was done with the marky‐coco snakemake‐implemented pipeline (https://github.com/ericcapo/marky‐coco). A brief overview of this workflow is as follows: the metagenomes were trimmed and cleaned using fastp (Chen et al., 2018) with the following parameters: quality threshold of 30 (−q 30), length threshold of 25 (−l 25), and with trimming of adapters and polyG tails enabled (−‐detect_adapter_for_pe ‐‐trim_poly_g ‐‐trim_poly_x). A de novo single assembly approach, in which each metagenome was assembled individually, was applied using the assembler megahit 1.1.2 (Li et al., 2016) with default settings. The annotation of the contigs for prokaryotic protein‐coding gene prediction was done with the software Prodigal 2.6.3 (Hyatt et al., 2010). The DNA reads were mapped against the contigs with bowtie2 (Langmead & Salzberg, 2012), and the resulting .sam files were converted to .bam files using samtools 1.9 (Li et al., 2009). The .bam files and the prodigal output .gff file were used to estimate read counts by using featureCounts (Liao et al., 2014). In order to detect hgc homologues, HMM profiles derived from the Hg‐MATE database version 1 were applied to the amino acid FASTA file generated with Prodigal from each assembly with the function hmmsearch from HMMER 3.2.1 (Finn et al., 2011). The reference package “hgcA” from Hg‐MATE.db was used for phylogenetic analysis of the HgcA amino acid sequences. Briefly, the predicted amino acid sequences from gene identified as putative hgcA gene were (i) compiled in a FASTA file, (ii) aligned to the Stockholm formatted HgcA alignment from the reference package with the function hmmalign from HMMER 3.2.1 (iii) placed onto the HgcA reference tree and classified using the functions pplacer, rppr and guppy_classify from the program pplacer (Matsen et al., 2010). For more details, see the README.txt of the Hg‐MATE database version 1 (https://doi.org/10.25573/serc.13105370.v1). Additionally, to compare the efficiency of the marky‐coco pipeline to detect hgc genes from metagenomes with a coassembly approach (multiple metagenomes used for assembly), we performed coassemblies on metagenomes within each environmental system (BARM8s, MANGA6s, RES5s, PADDY10s, Table 2). Post‐assembly, all other steps of the analysis procedure were performed similarly to the marky‐coco pipeline. Detection of dsrA genes were detected in metagenomes with the function hmmsearch and HMM profile from TIGRFAM (Selengut et al., 2007). The amount of sequencing required to cover the total diversity and the estimated diversity of each metagenome were evaluated using the Nonpareil method (Rodriguez‐R & Konstantinidis, 2014).
TABLE 2.
Metagenomes collected from previously published studies investigating the presence of hg methylators in the environment
2.4. Stringency cutoffs for the definition of true hgc genes
Based on knowledge from confirmed isolated Hg methylators, we propose several stringency cutoffs that could be used to distinguish between hgcA gene which appear to be functional for Hg methylation and an hgcA‐like gene that encodes for a protein of unknown Hg methylation capability. (i) High stringency cutoff: amino acid sequence includes one of the cap‐helix motifs with the conserved cysteine (Cys93 in P. mercurii ND132), NVWCAAGK, NVWCASGK, NVWCAGGK, NIWCAAGK, NIWCAGGK or NVWCSAGK. This cutoff is based on previous findings that showed isolated microorganisms carrying HgcA proteins with the cap helix domain are capable of Hg methylation (Cooper et al., 2020; Gilmour et al., 2018; Parks et al., 2013; Smith et al., 2015). Within the high stringency cutoff, there is a possible need to distinguish between the amino acid sequences from fused HgcAB‐like proteins and those from true HgcA proteins, since isolates that encode fused HgcAB‐like genes do not have the capacity to methylate Hg in culture (Gilmour et al., 2018; Podar et al., 2015). The fused HgcAB include the cap‐helix and ferredoxin motifs of HgcA and HgcB. (ii) Moderate stringency cutoff: in addition to amino acid sequences that include the motifs described above, any sequence with a bitscore value obtained from the HMM analysis greater than or equal to 100 is included (iii) Low stringency cutoff: in addition to amino acid sequences that include the motifs described above, any sequence with a bitscore value greater than or equal to 60 is included. For hgcA‐like genes detected with medium and low‐stringency cutoffs, cap helix domains could still be identified but without the six motifs listed above and found in the amino acid sequences of HgcA proteins encoded by isolated organisms verified for their Hg methylation capacities (Gilmour et al., 2018). For hgcB gene homologues, we propose two cutoffs that could be used for their description as hgcB genes. (i) High stringency cutoff: their amino acid sequences include one of the following motifs featuring the conserved Cys (Cys73 in P. mercurii ND132, Cooper et al., 2020), C(M/I)ECGA motifs and that the genes are found on the same contig as an hgcA genes. (ii) Moderate stringency cutoff: amino acid sequences include the C(M/I)ECGA motif, but the gene are not colocated on a contig with an hgcA gene.
2.5. Estimation of hgcA abundance in metagenomes
Coverage values of hgcA genes were calculated, for each gene and each sample, as the number of reads mapping to the gene divided by the length of the gene (read/bp). We compared the reliability of four procedures for normalizing read counts of hgcA genes. Normalization metrics were (i) the total number of mapped reads (ii) the summed coverage values of rpoB genes, (iii) the median coverage values of 257 marker genes (GTDB‐Tk r89 release, Chaumeil et al., 2019), or (iv) the genome equivalents values calculated using the software MicrobeCensus (Nayfach & Pollard, 2015) which normalizes the relative abundance by the metagenomic data set size and the community average genome size of the microbial community. The coverage of each marker gene was calculated as the sum of the coverages of all the ORFs assigned to that gene (Appendix S1). The rpoB and the 256 other marker genes were detected using the function hmmsearch from hmmer software (v3.2.1, Finn et al., 2011) and applying the trusted cutoff provided in HMM files (GTDB‐Tk r89 release, Chaumeil et al., 2019).
2.6. Data analysis
A principal coordinate analysis (PCoA) was performed applying the function wcmdscale to a Bray–Curtis dissimilarity matrix built with the function vegdist from the hgcA gene coverage values table, clustered at the lowest level of NCBI taxonomic identification (txid), obtained with single assembly and coassembly approaches (Appendix S1). A Mantel test with a permutation procedure analysis (9999 permutations) and Spearman's method was performed using the function mantel from R basis to evaluate the level of concordance of the outputs between both approaches. The functions rcorr from the R package Hmisc (Harrell & Harrell, 2013), corrplot from the R package corrplot (Taiyun et al., 2017) and plot3D from the R package rgkl (Adler & Murdoch, 2003) were used to investigate correlations between normalization methods.
3. RESULTS
3.1. Data set outputs
A total of 29 single assemblies (one for each metagenome) and four coassemblies (reads from each of the BARM8s, MANGA6s, RES5S, and PADDY10s metagenome sets assembled together) were used to compare the efficiency of a single assembly using the marky‐coco pipeline and a coassembly approach to detect, identify and count hgc genes from metagenomes (Figure 2). The number of mapped reads of the analysed metagenomes ranged between 10.2–110.9 M reads (average, 29.4 ± 19.6) with single assembly and 16.6–120.7 M reads (average, 36.0 ± 19.9) with coassembly, with the percentage of mapped reads ranging between 16%–76% and 24%–89%, respectively (Appendix S1). Nonpareil diversity index values (N d ) of metagenomes were between 18.7 and 23.7 with the highest found in paddy soil metagenomes (Figure S1, Table 3). Nonpareil curves showed that paddy soil samples from this study required the highest sequencing effort for nearly complete coverage followed by reservoir sediments, and then lake sediment and lake waters and brackish waters (Figure S1). Estimated coverage of paddy soils metagenomes was relatively low (average, 0.30–0.37) compared to other metagenomes (0.49–0.83) showing that only a portion of the diversity of these environmental samples was recovered despite the relatively high sequencing depth (88.6 ± 5.6 M reads) (Table 3). Seven metagenomes (S02, S03, S19, S22, S26, S28, S29) that were used in coassemblies but with low hgcA coverage values (i.e., <0.40 obtained from coassemblies) were not used for further comparison analysis. The remaining 22 metagenomes, labelled MG01–MG22, had hgcA (unnormalized) coverage values between 0.44 and 3.06 (1.22 ± 0.79) (Appendix S1). Only hgcA genes (and not hgcB) from these metagenomes were used for comparison of the two assembly approaches as hgcAB gene pairs were not 100% similar between the two approaches (Appendix S1). Additionally, hgcAB‐like homologues that are predicted to encode for fused HgcAB proteins were excluded from further analysis.
TABLE 3.
For each metagenome, Nnonpareil diversity index values, estimated average coverage, number of mapped reads, number of hgcA genes and hgcA coverage values (reads/bp) for coassembly “c” and single assembly “s” approaches
| Environments | Metagenomes id | Nonpareil diversity index | Estimated average coverage | Number of mapped reads (millions reads) | Number of hgcA genes | hgcA coverage values | |||
|---|---|---|---|---|---|---|---|---|---|
| c | s | c | s | c | s | ||||
| Brackish water | MG01 | 19.51 | 0.83 | 120.7 | 110.9 | 38 | 14 | 2.04 | 1.85 |
| MG02 | 21.12 | 0.55 | 33.9 | 25.5 | 40 | 16 | 1.05 | 0.91 | |
| MG03 | 19.49 | 0.70 | 32.0 | 25.8 | 29 | 7 | 1.01 | 0.75 | |
| MG04 | 20.52 | 0.63 | 35.1 | 26.9 | 34 | 10 | 0.84 | 0.75 | |
| MG05 | 18.69 | 0.76 | 35.6 | 30.5 | 23 | 5 | 0.52 | 0.46 | |
| MG06 | 20.73 | 0.48 | 16.6 | 10.2 | 29 | 4 | 0.58 | 0.35 | |
| Reservoir sediment | MG07 | 22.46 | 0.59 | 28.6 | 21.8 | 147 | 69 | 3.06 | 2.36 |
| MG08 | 21.99 | 0.64 | 33.6 | 29.4 | 103 | 53 | 2.19 | 1.98 | |
| MG09 | 21.82 | 0.68 | 47.2 | 43.5 | 74 | 35 | 1.98 | 1.78 | |
| MG10 | 22.10 | 0.63 | 36.1 | 32.7 | 102 | 68 | 2.32 | 3.00 | |
| MG11 | 22.15 | 0.63 | 36.7 | 29.8 | 122 | 62 | 2.69 | 2.43 | |
| Lake sediment | MG12 | 20.55 | 0.62 | 22.7 | 26.7 | 23 | 10 | 0.83 | 0.78 |
| MG13 | 20.75 | 0.57 | 27.2 | 22.5 | 26 | 9 | 0.41 | 0.32 | |
| Lake water | MG14 | 21.57 | 0.49 | 29.6 | 24.8 | 31 | 13 | 1.19 | 1.05 |
| MG15 | 20.24 | 0.66 | 38.5 | 34.6 | 31 | 8 | 0.62 | 0.50 | |
| MG16 | 19.51 | 0.67 | 30.5 | 29.2 | 19 | 10 | 0.47 | 0.50 | |
| Paddy soils | MG17 | 23.48 | 0.34 | 31.1 | 20.4 | 77 | 21 | 0.69 | 0.45 |
| MG18 | 23.31 | 0.33 | 30.8 | 18.5 | 60 | 13 | 0.59 | 0.33 | |
| MG19 | 23.67 | 0.27 | 27.1 | 14.3 | 85 | 20 | 0.76 | 0.43 | |
| MG20 | 23.14 | 0.37 | 37.5 | 28.8 | 61 | 15 | 0.58 | 0.32 | |
| MG21 | 23.49 | 0.30 | 30.5 | 18.7 | 57 | 25 | 0.60 | 0.51 | |
| MG22 | 23.64 | 0.30 | 30.6 | 20.5 | 84 | 33 | 1.12 | 0.89 | |
Note: See Appendix S1 for extended description of the data set.
3.2. Distribution of hgcA genes with different stringency cutoffs
By definition, all hgcA genes detected with the high stringency cutoff are predicted to encode proteins that include the conserved amino acid motifs characteristic of functional HgcA proteins, while this is not the case for those additionally detected when lowering the stringency cutoffs (i.e., moderate or low). We therefore considered that gene homologues to hgcA found with bitscore values below 100 and without conserved motifs cannot with confidence be defined as true hgcA genes. Nevertheless, we wanted here to highlight how “false” hgcA genes, detected without the conserved amino acid motifs characteristic of functional HgcA proteins, were taxonomically assigned using the pplacer approach applied to the Hg‐MATE hgcA reference tree. The hgcA genes detected with a high stringency cutoff and those additionally detected with moderate stringency cutoffs were predominantly identified as Desulfobacterota, Chloroflexota and Euryarchaeota (Figure S2). In contrast, the hgcA genes additionally detected with low stringency cutoff were primarily identified as members of the PVC superphylum but were unclassified at lower taxonomic levels. For further comparison, we used information only from hgcA genes detected with the high stringency.
3.3. Comparison between coassembly versus single assembly approaches
For all metagenomes, 1.50–7.25 times more hgcA genes were detected in coassemblies (19–147 genes) compared to linked single assemblies (4–69 genes) (Table 3). We investigated the differences in hgcA gene lengths, discriminating between genes (i) found at the extremity of contigs (potentially truncated) and (ii) in contigs expected to be complete. A higher number of “complete” hgcA gene sequences were detected with the coassembly (1–17, average 6.8 ± 4.4 genes) compared to the single assembly (0–6, average 2.0 ± 2.7 genes), for example, for metagenomes from brackish and lake waters (Appendix S1). No complete genes were identified in the single assemblies that were not also identified in the coassembly. Violin plots illustrated that, overall, a higher number of “complete” hgcA sequences (>950 bp) were found with the coassembly versus the single assembly (Figure S3).
In a comparison of HgcA amino acid sequences recovered from the two assembly approaches, no HgcA sequence from the single assembly had 100% sequence identity to sequences in the coassembly (Appendix S1). The highest sequence similarity of HgcA sequences from different assemblies of the same data set was 99%. To compare, we investigated differences between assemblies for detecting dsrA gene, which encodes for dissimilatory sulphite reductase subunit A, an essential enzyme in sulphate reduction and expected to be present in these data sets. Identical amino acid sequences of DsrA‐encoding genes were found when comparing single assemblies to the related coassembly with numbers ranging from 1 to 33 depending on metagenomes (Appendix S1). Comparatively, dsrA genes were 3–34× more abundant (in coverage) than hgcA genes. This higher abundance helps explain why more identical dsrA were found between coassembly and single assembly approaches than for hgcA genes.
Distribution plots showed unnormalized coverage values of hgcA genes clustered by environment types (Figure 3a) or for each metagenome (Figure S4). Importantly, unnormalized values were used here to compare single assembly versus coassembly results for each metagenome but not to compare difference between environments for which normalization would be required (Figure S4). Overall, higher hgcA coverage values were observed with the coassembly for all types of environments (Figure 3a) and for each metagenome with the exception of reservoir sediment MG10 (Figure S4, Table 3). For each metagenome, the PCoA analysis showed a high level of similarity in taxonomy‐based hgcA inventories obtained from single assembly versus coassembly (Figure 3b). This was confirmed by Mantel tests that showed significant correlations between the hgcA inventories obtained between both assembly approaches (r = .86, p ≤ .001). Looking at each data set independently, brackish waters and paddy soils showed significant correlations (r = .85, p = .001 and r = .73, p = .01) while lake waters and reservoir sediments had nonsignificant correlations (p > .05; no statistics possible with only two metagenomes for lake sediments).
FIGURE 3.
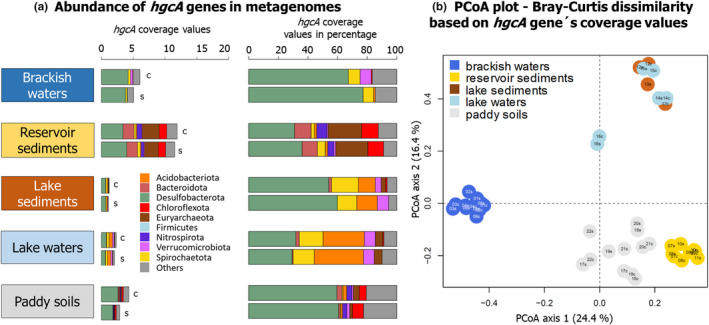
(a) Distribution of hgcA genes in the metagenomes obtained from five types of environments with the coassembly “c” and the single assembly “s” methods. For these barplots, unnormalized hgcA coverage values were used. (b) PCoA analysis showing the dissimilarities in the structure of hgcA inventories obtained with the coassembly “c” and the single assembly “s” approaches. The id of each metagenome is denoted as follows: Numbers corresponding to the metagenome id (e.g., MG01 is 01), “c” or “s” stands for analysis with the coassembly or the single assembly.
3.4. Comparison between normalization methods
In order to compare normalization methods to estimate the abundance of hgcA genes, we calculated the (i) total number mapped prokaryotic reads, (ii) rpoB genes coverage values, (iii) median coverage value of 257 marker genes and (iv) genome equivalents values (calculated as the total bp sequenced divided by average genome size in bp) (Figure 4, Appendix S1). Overall, significant correlations were observed between the total number of reads, rpoB coverage values, and the median coverage values of 257 marker genes (Figure 4a), while no significant correlations were observed between these metrics and genome equivalent values. The 3D plot shows the relationships between the total number of reads, the median coverage values of 257 marker genes and genome equivalent values (Figure 4b).
FIGURE 4.
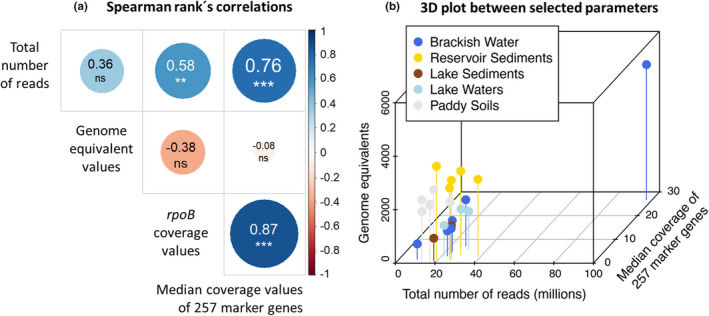
Plots showing correlations between metrics used for normalization. Outputs presented here were calculated only from data obtained with the single assembly approach.
4. DISCUSSION
4.1. Identification of true hgc genes from environmental genomic data
The absence of cultured representatives of hgc + microorganisms from novel clades (i.e., outside the Desulfobacterota, Firmicutes, Euryarchaeota) with experimentally validated Hg‐methylating capability (Gilmour et al., 2013, 2018) hampers confirmation that newly discovered hgc genes from environmental samples truly code for Hg methylating enzymes. Indeed, the recent analysis of publicly available metagenomes revealed the high diversity of microbial lineages with hgc + members, with the vast majority yet uncultured and therefore unstudied for Hg methylation activity (Gionfriddo et al., 2019; McDaniel et al., 2020). To date, all hgcA + microorganisms that have been experimentally tested have been found shown to produce MeHg (except for those with fused hgcAB‐like sequences) (Gilmour et al., 2013, 2018), and protein modelling of novel hgcA sequences suggest they have comparable active sites to HgcA sequences in experimentally verified Hg methylators. Therefore, although recent findings revealed relationships between microbial expression of hgc transcripts and MeHg formation in the environment (Capo, Feng, et al., 2022), and some putative hgcAB genes have been computationally modelled to possess functionality for methylation (Gionfriddo et al., 2016; Lin et al., 2021), we remain cautious about defining true hgc genes from environmental samples. As such, some studies have qualified hgc genes found in the environment as hgc‐like genes (e.g., Bowman et al., 2020; Capo et al., 2020; Gionfriddo et al., 2016; Villar et al., 2020).
Here, we defined three stringency cutoffs to describe hgcA genes in environmental metagenomes. By definition, the HgcA‐encoding genes detected with the high stringency cutoffs include the key amino acid residues (i.e., the cap helix motif N[V/I]WC[A/S][A/G/S]GK, Parks et al., 2013) present in HgcA from known Hg methylators. In contrast, all other hits to the HMM, from moderate and low stringency cutoffs, have a cap‐helix motif but lack these amino acid residues affecting potentially protein functionality. To date none of the isolates lacking these key amino acid residues has been found to methylate Hg, or no cultured isolate exists to test for Hg methylation capability (Gilmour et al., 2018). Substitution of some of these amino acids in the cap helix of HgcA may not result in loss of Hg methylation activity, as demonstrated by site‐directed mutagenesis experiments with P. mercurii ND132 (Smith et al., 2015). However, in addition to the cap helix domain of HgcA, the transmembrane domain of HgcA may also be required for Hg methylation activity. Unfortunately, the transmembrane region of HgcA has no detectable sequence homology (Cooper et al., 2020).
Thus, we recommend using the high stringency cutoff defined in the present study for routine identification of hgcA from environmental metagenomes. Lower stringency could reveal novel HgcA sequences that have lower similarity to HgcA from known Hg methylators, but if the lower stringency cutoff is used, we advise careful manual inspection of the sequences to ensure that they have important motifs and other HgcA features like the cap‐helix region. If the amino acid sequence in the cap helix domain is highly divergent from known sequences, we recommend protein modelling efforts to determine if the active site is similar enough to known sequences to validate classification as HgcA. Additional verification of true HgcA sequences include prediction of transmembrane domain regions (e.g., using TMHMM software, Krogh et al., 2001) and identification of other key conserved residues (Jones et al., 2019; Parks et al., 2013; Smith et al., 2015). A combination of several methods will certainly help to improve our description of hgcA genes in the coming years.
4.2. Effectiveness of the Hg‐MATE database
The Hg‐MATE database originates from the combination of two recent studies (Gionfriddo et al., 2019; McDaniel et al., 2020). The present work is a collaborative project of the Meta‐Hg working group that aimed to provide a living database that will be periodically updated. It provides several useful tools (HMM profiles and references phylogenetic trees) and a documented workflow that allows for the identification of hgc genes for easy comparison between studies. One major advantage of Hg‐MATE is the assignment of NCBI taxonomy IDs (txid) to hgcA genes allowing for easy comparison with datasets from other studies that also use the Hg‐MATE database (Appendix S1). In contrast, outputs from previous hgc‐related studies are difficult to compare with each other because hgc taxonomic identification is usually done with different in‐house databases and/or phylogenetic tools, and is based on the manual inspection of phylogenetic trees increasing the level of uncertainties and subjectivity in taxonomic identification. While the used pplacer approach here is not perfect ‐ since phylogenetic relatedness of the gene does not necessarily mean the same organismal taxonomy because of potential horizontal gene transfer (McDaniel et al., 2020) – it is a standardized approach allowing for a robust and automated identification of hgc genes from metagenomes.
A side‐by‐side comparison of previous and present taxonomic identification of putative Hg methylators is presented in this section. For water and sediment metagenomes from Lake Manganika our identification by hgcA phylogeny showed consistent results with previous identification from hgc + MAGs (Jones et al., 2019), with Desulfobacterota, Acidobacteriota, Verrucomicrobiota and Spirochaetota being the predominant putative Hg methylators. In the case of Baltic Sea water metagenomes, the comparison of our Hg‐MATE taxonomy identification with the previous identification using a set of hgc sequences from Podar et al. (2015) revealed consistency in the predominant hgc + groups detected (Desulfobacterota, Spirochaetota, Verrucomicrobiota) but noticeable differences for others, such as Planctomycetota (Appendix S1). Consistent with previous characterization, reservoir sediments were characterized by predominant hgc + Euryarchaeota, Desulfobacterota, Bacteroidota and Chloroflexota. Finally, in paddy soils, Liu et al. (2018) identified mostly hgc + Desulfobacterota, Firmicutes and Euryarchaeota while, in the present study, the two last microbial groups were found less predominant to the benefit of hgc + Nitrospirota and Chloroflexota.
In addition to using phylogenetic placements of hgc genes in reference trees from the Hg‐MATE database, a more precise approach to identification of putative Hg methylators is probably the identification of hgc + MAGs (i.e., Jones et al., 2019; Lin et al., 2021; Peterson et al., 2020). However, the recovery of MAGs from metagenomes is not always possible due to (i) the difficulty of obtaining MAGs from certain environments such as sediments and (ii) the low predominance of Hg methylators compared to other microorganisms in the environment, and therefore the lower probability of recovering hgc + MAGs. A recent study revealed the good congruence between the identification of hgc+ MAGs and a hgc phylogeny based on Hg‐MATE phylogeny (Capo, Feng, et al., 2022) highlighting that both approaches could be used to ensure the reliability in the identification of Hg methylators.
4.3. Assembly methods depend of the diversity of the metagenome
The increasing amount of publicly available environmental genomic data (Nayfach et al., 2021; Thompson et al., 2017) opens avenues to answer ecological questions related to the biogeography patterns and dispersal barriers of Hg methylators in interconnected systems (such as the global ocean and coastal systems). Coassembly of multiple metagenomes has been shown to have many important benefits compared to single assemblies including improved binning and better recovery of low abundance environmental genomes from studies that use multiple low‐coverage metagenomes. However, coassembly requires higher computational costs and potentially masks microdiversity by collapsing the genomes of multiple related strains into a single MAG (Delgado & Andersson, 2022; Narasingarao et al., 2012; Paoli et al., 2022; Ramos‐Barbero et al., 2019; Tamames et al., 2019; van der Walt et al., 2017). Here, we compared hgcA recovery from single assembled metagenomes versus coassemblies of multiple metagenomes from the same environment. Our analysis revealed that no identical amino acid sequences were obtained when comparing outputs from single assembly and coassembly methods for each metagenome for hgcA genes but not for dsrA genes for which identical amino acid sequences were found. Our results highlight the differences that can be observed between both approaches in the composition of hgcA sequences in accordance with a recent work showing the aggregation of near identical genes (i.e., 99% clustering cutoff) occurring during coassembly. In all cases except one, coassembly significantly increased the recovery of hgcA genes (Figure S4). Additionally, we showed that when the diversity and composition of the hgcA + community was compared across all the samples included in the analysis, single assemblies and coassemblies performed similarly in this regard, suggesting that also single metagenomes can provide adequate information (similar level of hgc coverage and detected diversity) on the hgc + community.
Differences in the diversity of environments can have an effect on the recovery of hgc genes from metagenomes. Nonpareil diversity index values of the metagenomes ranged between 18.7 and 23.7 with the highest being found in paddy soils metagenomes (Figure S1, Appendix S1). Here, for the paddy soils that exhibited higher Nonpareil diversity index values (Figure S1), consistently with Rodriguez‐R and Konstantinidis (2014), the coassembly approach outperforms single sample assemblies in the recovery of hgc genes (Figure 3). Noticeably, although no identical HgcA amino acid sequences were detected between single assembly and coassembly approach, identical DsrA amino acid sequences were observed. We hypothesize that the low proportion of hgcA genes in metagenomes, compared to dsrA genes, explained such discrepancies, although it did not strongly impact the overall hgcA coverage values recovery. In these situations, we recommend aiming for either higher depth of coverage or sequencing of multiple adjacent or linked metagenomes or replicates from a single sample. In contrast, we recommend avoiding the coassembly of metagenomes from different environments that could produce more misassembles and chimerism (Mikheenko et al., 2016; Sczyrba et al., 2017; Tamames et al., 2019). For other environments such as brackish and lake waters, our work highlights that using the marky‐coco pipeline based on a single assembly approach provide similar results to a coassembly approach in detecting hgc genes. Long‐read metagenomic sequencing could help reduce discrepancies between coassembly and single assembly approaches (Driscoll et al., 2017; Van Goethem et al., 2021). However, long‐read approaches require high quality intact DNA and come with a trade‐off in base‐call accuracy and assembly coverage. Also, genome assembly from long‐read HTS may be best suited for dominant members of low diversity microbiomes and therefore less applicable to hgc+ organisms due to their relative rarity in microbiomes.
4.4. Robust normalization methods are needed for quantitative inferences
The normalization of gene counts from environmental metagenomes and metatranscriptomes is a key aspect of works aiming to study the prevalence of certain microorganisms in specific environments (Pereira et al., 2018; Pierella Karlusich et al., 2022; Salazar et al., 2019). In hgcAB omics studies, the number of mapped reads and the coverage values of marker genes or housekeeping genes is usually used to normalize the coverage values of hgc genes (Capo, Broman, et al., 2022; Capo, Feng, et al., 2022; Lin et al., 2021; Tada et al., 2020; Vigneron et al., 2021). Tests here revealed that a wide range of contrasting normalization methods all provided reasonable abundance estimates that were significantly correlated with one another with the exception of genome equivalent values (Figure 4). Nonsignificant correlations found between genome equivalent values (Nayfach & Pollard, 2015) and other metrics can be explained by the weaker relationships observed for the metrics in paddy soils and reservoir sediments metagenomes, while metrics from brackish waters, lake sediment and waters appear to have linear relationships. We hypothesize that this discrepancy between normalization metrics is due to the differential contribution of DNA sequences from nonprokaryotic organisms to calculate genome equivalent values in more diverse environments (paddy soils, reservoir sediments) compared to others (brackish waters, lake waters and lake sediments). Therefore, we do not strongly recommend any single method over others. Instead, we suggest that it may be prudent to report data that employ multiple normalization methods to allow for easy comparisons to be carried out between studies. Such normalizations can without too much of an effort be included in the supporting information for later usage. Suggested normalization methods include the total number of prokaryotic reads, coverage values of rpoB genes and the median coverage values of 257 marker genes (example in Appendix S1).
5. CONCLUSION
The study of the taxonomic diversity and metabolic capacities of microorganisms involved in Hg methylation will lead to a better understanding of the environmental factors triggering microbial methylation of inorganic Hg. Although metagenomic and metatranscriptomic‐based studies have provided better insights into the environmental role of those microorganisms, there is still a need to standardize methods to detect hgc genes from environmental omic data. Furthermore, since Hg methylators often constitute such a small proportion of the microbiome, methods outlined in this study provide best practices for improving their detection and recovery from metagenomes. We provide here an up‐to‐date hgc gene catalogue, Hg‐MATE database v1, and the marky‐coco bioinformatic pipeline to detect, identify and count hgc genes from metagenomes. We recommend using our high stringency cutoff to detect hgcA genes in metagenomes and applying our protocol in future prospects of Hg methylation genes, especially for cross‐comparison between studies. Finally, although a co‐assembly approach should be chosen when analysing metagenomes from highly diverse environments (e.g., paddy soils), we recommend using marky‐coco pipeline, based on a de novo assembly for recovering hgc genes in metagenomes from aquatic environments.
CONFLICT OF INTEREST
The author declares no conflict of interest.
BENEFIT‐SHARING STATEMENT
Benefits from this research is the creation and curation of Hg‐MATE database (https://doi.org/10.25573/serc.13105370.v1) and release of the bioinformatic pipeline marky‐coco (https://github.com/ericcapo/marky‐coco).
Supporting information
Appendix S1
Figure S1‐S4
ACKNOWLEDGEMENTS
This work was funded by the Severo Ochoa Excellence Programme postdoctoral fellowship awarded in 2021 to Eric Capo (CEX2019‐000928‐S), the Swedish Research Council Formas (grant 2018‐01031), the EMFF‐Blue Economy project MER‐CLUB (grant 863584). Caitlin Gionfriddo was a Robert and Arlene Kogod Secretarial Scholar with the Smithsonian Environmental Research Center while conducting work described in this manuscript. Benjamin Peterson was funded as a postdoctoral research associate by the National Science Foundation (award 1935173) during the work in this manuscript. The computations were performed on resources provided by SNIC through Uppsala Multidisciplinary Centre for Advanced Computational Science (UPPMAX) using the compute project SNIC 2021/5‐53. Some of the computations for compiling the Hg‐MATE database were conducted on the Smithsonian High Performance Cluster (SI/HPC), Smithsonian Institution. https://doi.org/10.25572/SIHPC. Oak Ridge National Laboratory is managed by UT‐Battelle, LLC under contract no. DE‐AC05‐00OR22725 with the U.S. Department of Energy (DOE). The authors are thankful to an anonymous reviewer and the associate editor Lucie Zinger for their insightful comments, the members of the Meta‐Hg working group (https://ercapo.wixsite.com/meta‐hg) and of the Mersorcium network (https://mersorcium.github.io/).
Capo, E. , Peterson, B. D. , Kim, M. , Jones, D. S. , Acinas, S. G. , Amyot, M. , Bertilsson, S. , Björn, E. , Buck, M. , Cosio, C. , Elias, D. A. , Gilmour, C. , Goñi‐Urriza, M. , Gu, B. , Lin, H. , Liu, Y‐R , McMahon, K. , Moreau, J. W. , Pinhassi, J. … Gionfriddo, C. M. (2023). A consensus protocol for the recovery of mercury methylation genes from metagenomes. Molecular Ecology Resources, 23, 190–204. 10.1111/1755-0998.13687
Andrea G. Bravo and Caitlin Gionfriddo joint last authors.
Handling Editor: Lucie Zinger
DATA AVAILABILITY STATEMENT
All metagenomes analysed in this study are in the public domain as described in Table 2.
REFERENCES
- Adler, D. , Nenadic, O. , & Zucchini, W. (2003). Rgl: A r‐library for 3d visualization with opengl. In Proceedings of the 35th symposium of the interface: computing science and statistics, Salt Lake City. (Vol. 35, pp. 1–111). [Google Scholar]
- Alneberg, J. , Sundh, J. , Bennke, C. , Beier, S. , Lundin, D. , Hugerth, L. W. , Pinhassi, J. , Kisand, V. , Riemann, L. , Jürgens, K. , Labrenz, M. , & Andersson, A. F. (2018). Data descriptor: BARM and balticmicrobeDB, a reference metagenome and interface to meta‐omic data for the Baltic Sea. Scientific Data, 5, 1–10. 10.1038/sdata.2018.146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchet, S. , Goñi‐Urriza, M. , Monperrus, M. , Guyoneaud, R. , Fernandez, P. , Heredia, C. , Tessier, E. , Gassie, C. , Point, D. , Guédron, S. , Achá, D. , & Amouroux, D. (2018). Linking microbial activities and low‐molecular‐weight thiols to hg methylation in biofilms and periphyton from high‐altitude Tropical Lakes in the Bolivian altiplano. Environmental Science and Technology, 52(17), 9758–9767. 10.1021/acs.est.8b01885 [DOI] [PubMed] [Google Scholar]
- Bowman, K. L. , Collins, R. E. , Agather, A. M. , Lamborg, C. H. , Hammerschmidt, C. R. , Kaul, D. , Dupont, C. L. , Christensen, G. A. , & Elias, D. A. (2020). Distribution of mercury‐cycling genes in the Arctic and equatorial Pacific oceans and their relationship to mercury speciation. Limnology and Oceanography, 65(S1), S310–S320. 10.1002/lno.11310 [DOI] [Google Scholar]
- Bravo, A. , Peura, S. , Buck, M. , Ahmed, O. , Mateos‐Rivera, A. , Ortega, S. , Schaefer, J. K. , Bouchet, S. , Tolu, J. , Björn, E. , & Bertilsson, S. (2018). Methanogens and iron‐reducing bacteria: The overlooked members of mercury‐Methylating microbial communities in. Applied and Environmental Microbiology, 84(23), 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo, A. G. , & Cosio, C. (2020). Biotic formation of methylmercury: A bio–physico–chemical conundrum. Limnology and Oceanography, 65(5), 1010–1027. 10.1002/lno.11366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capo, E. , Bravo, A. G. , Soerensen, A. L. , Bertilsson, S. , Pinhassi, J. , Feng, C. , Andersson, A. F. , Buck, M. , & Björn, E. (2020). Deltaproteobacteria and Spirochaetes‐like bacteria are abundant putative mercury methylators in oxygen‐deficient water and marine particles in the Baltic Sea. Frontiers in Microbiology, 11, 574080. 10.3389/fmicb.2020.574080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capo, E. , Broman, E. , Bonaglia, S. , Bravo, A. G. , Bertilsson, S. , Soerensen, A. L. , Pinhassi, J. , Lundin, D. , Buck, M. , Hall, P. O. , Nascimento, F. J. , & Björn, E. (2022). Oxygen‐deficient water zones in the Baltic Sea promote uncharacterized hg methylating microorganisms in underlying sediments. Limnology and Oceanography, 67(1), 135–146. 10.1002/lno.11981 [DOI] [Google Scholar]
- Capo, E. , Feng, C. , Bravo, A. G. , Bertilsson, S. , Soerensen, A. L. , Pinhassi, J. , Buck, M. , Karlsson, C. , Hawkes, J. , & Björn, E. (2022). Abundance and expression of hgcAB genes and mercury availability jointly explain methylmercury formation in stratified brackish waters. BioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaumeil, P.‐A. , Mussig, A. J. , Hugenholtz, P. , & Parks, D. H. (2019). GTDB‐Tk: A toolkit to classify genomes with the genome taxonomy database. Bioinformatics, 36(6), 1925–1927. 10.1093/bioinformatics/btz848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, S. , Zhou, Y. , Chen, Y. , & Gu, J. (2018). Fastp: An ultra‐fast all‐in‐one FASTQ preprocessor. Bioinformatics, 34(17), i884–i890. 10.1093/bioinformatics/bty560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen, G. A. , Gionfriddo, C. M. , King, A. J. , Moberly, J. G. , Miller, C. L. , Somenahally, A. C. , Callister, S. J. , Brewer, H. , Podar, M. , Brown, S. D. , Palumbo, A. V. , Brandt, C. C. , Wymore, A. M. , Brooks, S. C. , Hwang, C. , Fields, M. W. , Wall, J. D. , Gilmour, C. C. , & Elias, D. A. (2019). Determining the reliability of measuring mercury cycling gene abundance with correlations with mercury and methylmercury concentrations. Environmental Science and Technology, 53(15), 8649–8663. 10.1021/acs.est.8b06389 [DOI] [PubMed] [Google Scholar]
- Compeau, G. C. , & Bartha, R. (1985). Sulfate‐reducing bacteria: Principal methylators of mercury in anoxic estuarine sediment. Applied and Environmental Microbiology, 50(2), 498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, C. J. , Zheng, K. , Rush, K. W. , Johs, A. , Sanders, B. C. , Pavlopoulos, G. A. , Kyrpides, N. C. , Podar, M. , Ovchinnikov, S. , Ragsdale, S. W. , & Parks, J. M. (2020). Structure determination of the HgcAB complex using metagenome sequence data: Insights into microbial mercury methylation. Communications Biology, 3(1), 1–9. 10.1038/s42003-020-1047-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado, L. F. , & Andersson, A. F. (2022). Evaluating metagenomic assembly approaches for biome‐specific gene catalogues. Microbiome, 10(72), 2–11. 10.1186/s40168-022-01259-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll, C. B. , Otten, T. G. , Brown, N. M. , & Dreher, T. W. (2017). Towards long‐read metagenomics: Complete assembly of three novel genomes from bacteria dependent on a diazotrophic cyanobacterium in a freshwater lake co‐culture. Standards in Genomic Sciences, 12, 9. 10.1186/s40793-017-0224-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn, R. D. , Clements, J. , & Eddy, S. R. (2011). HMMER web server: Interactive sequence similarity searching. Nucleic Acids Research, 39(suppl 2), W29–W37. 10.1093/nar/gkr367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming, E. J. , Mack, E. E. , Green, P. G. , & Nelson, D. C. (2006). Mercury methylation from unexpected sources: Molybdate‐inhibited freshwater sediments and an iron‐reducing bacterium. Applied and Environmental Microbiology, 72(1), 457–464. 10.1128/AEM.72.1.457-464.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmour, C. C. , Bullock, A. L. , McBurney, A. , Podar, M. , & Elias, D. A. (2018). Robust mercury methylation across diverse methanogenic archaea. mBio, 9(2), 1–13. 10.1128/mBio.02403-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmour, C. C. , Elias, D. A. , Kucken, A. M. , Brown, S. D. , Palumbo, A. V. , Schadt, C. W. , & Wall, J. D. (2011). Sulfate‐reducing bacterium Desulfovibrio desulfuricans ND132 as a model for understanding bacterial mercury methylation. Applied and Environmental Microbiology, 77(12), 3938–3951. 10.1128/AEM.02993-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmour, C. C. , Podar, M. , Bullock, A. L. , Graham, A. M. , Brown, S. D. , Somenahally, A. C. , Johs, A. , Hurt, R. A., Jr. , Bailey, K. L. , & Elias, D. A. (2013). Mercury methylation by novel microorganisms from new environments. Environmental Science and Technology, 47(20), 11810–11820. 10.1021/es403075t [DOI] [PubMed] [Google Scholar]
- Gionfriddo C, Podar M, Gilmour C, Pierce E, Elias D. (2019) ORNL compiled mercury methylator database https://www.osti.gov/dataexplorer/biblio/dataset/1569274
- Gionfriddo, C. M. , Tate, M. T. , Wick, R. R. , Schultz, M. B. , Zemla, A. , Thelen, M. P. , Schofield, R. , Krabbenhoft, D. P. , Holt, K. E. , & Moreau, J. W. (2016). Microbial mercury methylation in Antarctic Sea ice. Nature Microbiology, 1(10), 1–12. 10.1038/nmicrobiol.2016.127 [DOI] [PubMed] [Google Scholar]
- Gionfriddo, C. M. , Wymore, A. M. , Jones, D. S. , Wilpiszeski, R. L. , Lynes, M. M. , Christensen, G. A. , Soren, A. , Gilmour, C. C. , Podar, M. , & Elias, D. A. (2020). An improved hgcAB primer set and direct high‐throughput sequencing expand hg‐methylator diversity in nature. Frontiers in Microbiology, 11, 2275. 10.3389/fmicb.2020.541554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamelin, S. , Amyot, M. , Barkay, T. , Wang, Y. , & Planas, D. (2011). Methanogens: Principal methylators of mercury in lake periphyton. Environmental Science and Technology, 45(18), 7693–7700. 10.1021/es2010072 [DOI] [PubMed] [Google Scholar]
- Harrell, F. , & Harrell, M. (2013). Package ‘Hmisc’. CRAN, 235(6).
- Hug, L. A. , Baker, B. J. , Anantharaman, K. , Brown, C. T. , Probst, A. J. , Castelle, C. J. , Butterfield, C. N. , Hernsdorf, A. W. , Amano, Y. , Ise, K. , Suzuki, Y. , Dudek, N. , Relman, D. A. , Finstad, K. M. , Amundson, R. , Thomas, B. C. , & Ban, J. F. (2016). A new view of the tree of life. Nature Microbiology, 1, 16048. 10.1038/nmicrobiol.2016.48 [DOI] [PubMed] [Google Scholar]
- Hyatt, D. , Chen, G.‐L. , LoCascio, P. F. , Land, M. L. , Larimer, F. W. , & Hauser, L. J. (2010). Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics, 11(1), 119. 10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, D. S. , Walker, G. M. , Johnson, N. W. , Mitchell, C. P. J. , Coleman Wasik, J. K. , & Bailey, J. V. (2019). Molecular evidence for novel mercury methylating microorganisms in sulfate‐impacted lakes. ISME Journal, 13(7), 1659–1675. 10.1038/s41396-019-0376-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerin, E. J. , Gilmour, C. C. , Roden, E. , Suzuki, M. T. , Coates, J. D. , & Mason, R. P. (2006). Mercury methylation by dissimilatory iron‐reducing bacteria. Applied and Environmental Microbiology, 72(12), 7919–7921. 10.1128/AEM.01602-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh, A. , Larsson, B. , von Heijne, G. , & Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden markov model: Application to complete genomes. Journal of Molecular Biology, 305(3), 567–580. 10.1006/JMBI.2000.4315 [DOI] [PubMed] [Google Scholar]
- Kronberg, R.‐M. , Jiskra, M. , Wiederhold, J. G. , Björn, E. , & Skyllberg, U. (2016). Methyl mercury formation in hillslope soils of boreal forests: The role of Forest harvest and anaerobic microbes. Environmental Science & Technology, 50(17), 9177–9186. 10.1021/acs.est.6b00762 [DOI] [PubMed] [Google Scholar]
- Kumar, S. , Stecher, G. , Li, M. , Knyaz, C. , & Tamura, K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution, 35(6), 1547–1549. 10.1093/molbev/msy096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. , & Salzberg, S. L. (2012). Fast gapped‐read alignment with bowtie 2. Nature Methods, 9(4), 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le, S. Q. , & Gascuel, O. (2008). An improved general amino acid replacement matrix. Molecular Biology and Evolution, 25(7), 1307–1320. 10.1093/molbev/msn067 [DOI] [PubMed] [Google Scholar]
- Letunic, I. , & Bork, P. (2019). Interactive tree of life (iTOL) v4: Recent updates and new developments. Nucleic Acids Research, 47(W1), W256–W259. 10.1093/nar/gkz239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, D. , Liu, C.‐M. , Luo, R. , Sadakane, K. , & Lam, T.‐W. (2016). MEGAHIT: An ultra‐fast single‐node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics, 31(10), 1674–1676. 10.1093/bioinformatics/btv033 [DOI] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. , Abecasis, G. , & Durbin, R. (2009). The sequence alignment/map format and SAMtools. Bioinformatics, 25(16), 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, Y. , Smyth, G. K. , & Shi, W. (2014). featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics, 30(7), 923–930. 10.1093/bioinformatics/btt656 [DOI] [PubMed] [Google Scholar]
- Lin, H. , Ascher, D. B. , Myung, Y. , Lamborg, C. H. , Hallam, S. J. , Gionfriddo, C. M. , Holt, K. E. , & Moreau, J. W. (2021). Mercury methylation by metabolically versatile and cosmopolitan marine bacteria. The ISME Journal, 15, 1810–1825. 10.1038/s41396-020-00889-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. R. , Johs, A. , Bi, L. , Lu, X. , Hu, H. W. , Sun, D. , He, J. Z. , & Gu, B. (2018). Unraveling microbial communities associated with methylmercury production in Paddy soils. Environmental Science and Technology, 52(22), 13110–13118. 10.1021/acs.est.8b03052 [DOI] [PubMed] [Google Scholar]
- Matsen, F. A. , Kodner, R. B. , & Armbrust, E. V. (2010). Pplacer: Linear time maximum‐likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics, 11(1), 538. 10.1186/1471-2105-11-538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel, E. , Peterson, B. , Stevens, S. , Tran, P. , Anantharaman, K. , & McMahon, K. (2020). Expanded phylogenetic diversity and metabolic flexibility of mercury‐methylating microorganism. MSystems, 5(4), e00299‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikheenko, A. , Saveliev, V. , & Gurevich, A. (2016). MetaQUAST: Evaluation of metagenome assemblies. Bioinformatics, 32(7), 1088–1090. 10.1093/bioinformatics/btv697 [DOI] [PubMed] [Google Scholar]
- Millera Ferriz, L. , Ponton, D. E. , Storck, V. , Leclerc, M. , Bilodeau, F. , Walsh, D. A. , & Amyot, M. (2021). Role of organic matter and microbial communities in mercury retention and methylation in sediments near run‐of‐river hydroelectric dams. Science of the Total Environment, 774(4), 145686. 10.1016/j.scitotenv.2021.145686 [DOI] [PubMed] [Google Scholar]
- Narasingarao, P. , Podell, S. , Ugalde, J. A. , Brochier‐Armanet, C. , Emerson, J. B. , Brocks, J. J. , Heidelberg, K. B. , Banfield, J. F. , & Allen, E. E. (2012). De novo metagenomic assembly reveals abundant novel major lineage of archaea in hypersaline microbial communities. The ISME Journal, 6(1), 81–93. 10.1038/ismej.2011.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayfach, S. , & Pollard, K. S. (2015). Average genome size estimation improves comparative metagenomics and sheds light on the functional ecology of the human microbiome. Genome Biology, 16(1), 1–18. 10.1186/s13059-015-0611-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayfach, S. , Roux, S. , Seshadri, R. , Udwary, D. , Varghese, N. , Schulz, F. , Wu, D. , Paez‐Espino, D. , Chen, I. M. , Huntemann, M. , Palaniappan, K. , Ladau, J. , Mukherjee, S. , Reddy, T. B. K. , Nielsen, T. , Kirton, E. , Faria, J. P. , Edirisinghe, J. N. , Henry, C. S. , … Eloe‐Fadrosh, E. A. (2021). A genomic catalog of Earth's microbiomes. Nature Biotechnology, 39(4), 499–509. 10.1038/s41587-020-0718-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoli, L. , Ruscheweyh, H.‐J. , Forneris, C. C. , Hubrich, F. , Kautsar, S. , Bhushan, A. , Lotti, A. , Clayssen, Q. , Salazar, G. , Milanese, A. , Carlström, C. I. , Papadopoulou, C. , Gehrig, D. , Karasikov, M. , Mustafa, H. , Larralde, M. , Carroll, L. M. , Sánchez, P. , Zayed, A. A. , … Sunagawa, S. (2022). Biosynthetic potential of the global ocean microbiome. Nature, 607(7917), 111–118. 10.1038/s41586-022-04862-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks, J. M. , Johs, A. , Podar, M. , Bridou, R. , Hurt, R. A. , Smith, S. D. , Tomanicek, S. J. , Qian, Y. , Brown, S. D. , Brandt, C. C. , Palumbo, A. V. , Smith, J. C. , Wall, J. D. , Elias, D. A. , & Liang, L. (2013). The genetic basis for bacterial mercury methylation. Science, 339(6125), 1332–1335. 10.1126/science.1230667 [DOI] [PubMed] [Google Scholar]
- Pereira, M. B. , Wallroth, M. , Jonsson, V. , & Kristiansson, E. (2018). Comparison of normalization methods for the analysis of metagenomic gene abundance data. BMC Genomics, 19(1), 274. 10.1186/s12864-018-4637-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson, B. D. , McDaniel, E. A. , Schmidt, A. G. , Lepak, R. F. , Janssen, S. E. , Tran, P. Q. , Marick, R. A. , Ogorek, J. M. , DeWild, J. F. , Krabbenhoft, D. P. , & McMahon, K. D. (2020). Mercury methylation genes identified across diverse anaerobic microbial guilds in a eutrophic sulfate‐enriched Lake. Environmental Science & Technology, 54(24), 15840–15851. 10.1021/acs.est.0c05435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierella Karlusich, J. J. , Pelletier, E. , Zinger, L. , Lombard, F. , Zingone, A. , Colin, S. , Gasol, J. M. , Dorrell, R. G. , Henry, N. , Scalco, E. , Acinas, S. G. , Wincker, P. , de Vargas, C. , & Bowler, C. (2022). A robust approach to estimate relative phytoplankton cell abundances from metagenomes. Molecular Ecology Resources. 10.1111/1755-0998.13592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podar, M. , Gilmour, C. C. , Brandt, C. C. , Soren, A. , Brown, S. D. , Crable, B. R. , Palumbo, A. V. , Somenahally, A. C. , & Elias, D. A. (2015). Global prevalence and distribution of genes and microorganisms involved in mercury methylation. Science Advances, 1(9), e1500675. 10.1126/sciadv.1500675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos‐Barbero, M. D. , Martin‐Cuadrado, A.‐B. , Viver, T. , Santos, F. , Martinez‐Garcia, M. , & Antón, J. (2019). Recovering microbial genomes from metagenomes in hypersaline environments: The good, the bad and the ugly. Systematic and Applied Microbiology, 42(1), 30–40. 10.1016/J.SYAPM.2018.11.001 [DOI] [PubMed] [Google Scholar]
- Rodriguez‐R, L. M. , & Konstantinidis, K. T. (2014). Nonpareil: A redundancy‐based approach to assess the level of coverage in metagenomic datasets. Bioinformatics, 30(5), 629–635. 10.1093/bioinformatics/btt584 [DOI] [PubMed] [Google Scholar]
- Roth, S. , Poulin, B. A. , Baumann, Z. , Liu, X. , Zhang, L. , Krabbenhoft, D. P. , Hines, M. E. , Schaefer, J. K. , & Barkay, T. (2021). Nutrient inputs stimulate mercury methylation by Syntrophs in a subarctic peatland. Frontiers in Microbiology, 12, 741523. 10.3389/fmicb.2021.741523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar, G. , Paoli, L. , Alberti, A. , Huerta‐Cepas, J. , Ruscheweyh, H. J. , Cuenca, M. , Field, C. M. , Coelho, L. P. , Cruaud, C. , Engelen, S. , Gregory, A. C. , Labadie, K. , Marec, C. , Pelletier, E. , Royo‐Llonch, M. , Roux, S. , Sánchez, P. , Uehara, H. , Zayed, A. A. , … Wincker, P. (2019). Gene expression changes and community turnover differentially shape the Global Ocean Metatranscriptome. Cell, 179(5), 1068–1083.e21. 10.1016/j.cell.2019.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer, J. K. , Kronberg, R. M. , Björn, E. , & Skyllberg, U. (2020). Anaerobic guilds responsible for mercury methylation in boreal wetlands of varied trophic status serving as either a methylmercury source or sink. Environmental Microbiology, 22(9), 3685–3699. 10.1111/1462-2920.15134 [DOI] [PubMed] [Google Scholar]
- Sczyrba, A. , Hofmann, P. , Belmann, P. , Koslicki, D. , Janssen, S. , Dröge, J. , Gregor, I. , Majda, S. , Fiedler, J. , Dahms, E. , Bremges, A. , Fritz, A. , Garrido‐Oter, R. , Jørgensen, T. S. , Shapiro, N. , Blood, P. D. , Gurevich, A. , Bai, Y. , Turaev, D. , … McHardy, A. C. (2017). Critical assessment of metagenome interpretation—A benchmark of metagenomics software. Nature Methods, 14(11), 1063–1071. 10.1038/nmeth.4458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selengut, J. D. , Haft, D. H. , Davidsen, T. , Ganapathy, A. , Gwinn‐Giglio, M. , Nelson, W. C. , Richter, A. R. , & White, O. (2007). TIGRFAMs and genome properties: Tools for the assignment of molecular function and biological process in prokaryotic genomes. Nucleic Acids Research, 35(Database issue), D260–D264. 10.1093/nar/gkl1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, S. D. , Bridou, R. , Johs, A. , Parks, J. M. , Elias, D. A. , Hurt, R. A. , Brown, S. D. , Podar, M. , & Wall, J. D. (2015). Site‐directed mutagenesis of HgcA and HgcB reveals amino acid residues important for mercury methylation. Applied and Environmental Microbiology, 81(9), 3205–3217. 10.1128/AEM.00217-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soerensen, A. L. , Schartup, A. T. , Skrobonja, A. , Bouchet, S. , Amouroux, D. , Liem‐Nguyen, V. , & Björn, E. (2018). Deciphering the role of water column redoxclines on methylmercury cycling using speciation modeling and observations from the Baltic Sea. Global Biogeochemical Cycles, 32(10), 1498–1513. 10.1029/2018GB005942 [DOI] [Google Scholar]
- Steen, A. D. , Crits‐Christoph, A. , Carini, P. , DeAngelis, K. M. , Fierer, N. , Lloyd, K. G. , & Cameron Thrash, J. (2019). High proportions of bacteria and archaea across most biomes remain uncultured. The ISME Journal, 13(12), 3126–3130. 10.1038/s41396-019-0484-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada, Y. , Marumoto, K. , & Takeuchi, A. (2020). Nitrospina‐Like Bacteria are Potential Mercury Methylators in the Mesopelagic Zone in the East China Sea. Frontiers in Microbiology, 11, 1369. 10.3389/FMICB.2020.01369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taiyun, W. , & Viliam, S. (2017). R package “corrplot”: Visualization of a Correlation Matrix (Version 0.84). Statistician.
- Tamames, J. , Cobo‐Simón, M. , & Puente‐Sánchez, F. (2019). Assessing the performance of different approaches for functional and taxonomic annotation of metagenomes. BMC Genomics, 20(1), 960. 10.1186/s12864-019-6289-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, L. R. , Sanders, J. G. , McDonald, D. , Amir, A. , Ladau, J. , Locey, K. J. , Prill, R. J. , Tripathi, A. , Gibbons, S. M. , Ackermann, G. , Navas‐Molina, J. A. , Janssen, S. , Kopylova, E. , Vázquez‐Baeza, Y. , González, A. , Morton, J. T. , Mirarab, S. , Xu, Z. Z. , Jiang, L. , … Zhao, H. (2017). A communal catalogue reveals Earth's multiscale microbial diversity. Nature, 551(7681), 457–463. 10.1038/nature24621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Walt, A. J. , van Goethem, M. W. , Ramond, J.‐B. , Makhalanyane, T. P. , Reva, O. , & Cowan, D. A. (2017). Assembling metagenomes, one community at a time. BMC Genomics, 18(1), 521. 10.1186/s12864-017-3918-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Goethem, M. W. , Osborn, A. R. , Bowen, B. P. , Andeer, P. F. , Swenson, T. L. , Clum, A. , Riley, R. , He, G. , Koriabine, M. , Sandor, L. , Yan, M. , Daum, C. G. , Yoshinaga, Y. , Makhalanyane, T. P. , Garcia‐Pichel, F. , Visel, A. , Pennacchio, L. A. , O'Malley, R. C. , & Northen, T. R. (2021). Long‐read metagenomics of soil communities reveals phylum‐specific secondary metabolite dynamics. Communications Biology, 4(1), 1302. 10.1038/s42003-021-02809-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigneron, A. , Cruaud, P. , Aubé, J. , Guyoneaud, R. , & Goñi‐Urriza, M. (2021). Transcriptomic evidence for versatile metabolic activities of mercury cycling microorganisms in brackish microbial mats. Npj Biofilms and Microbiomes, 7(1), 1–11. 10.1038/s41522-021-00255-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villar, E. , Cabrol, L. , & Heimbürger‐Boavida, L. E. (2020). Widespread microbial mercury methylation genes in the global ocean. Environmental Microbiology Reports, 12(3), 277–287. 10.1111/1758-2229.12829 [DOI] [PubMed] [Google Scholar]
- Xu, J. , Liem‐Nguyen, V. , Buck, M. , Bertilsson, S. , Björn, E. , & Bravo, A. G. (2021). Mercury methylating microbial community structure in boreal wetlands explained by local physicochemical conditions. Frontiers in Environmental Science, 8, 518662. 10.3389/fenvs.2020.518662 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Figure S1‐S4
Data Availability Statement
All metagenomes analysed in this study are in the public domain as described in Table 2.