

Abstract
The role of macrophages (Mo) and their prognostic impact in diffuse large B‐cell lymphomas (DLBCL) remain controversial. By regulating the lipid metabolism, Liver‐X‐Receptors (LXRs) control Mo polarization/inflammatory response, and their pharmacological modulation is under clinical investigation to treat human cancers, including lymphomas. Herein, we surveyed the role of LXRs in DLBCL for prognostic purposes. Comparing bulk tumors with purified malignant and normal B‐cells, we found an intriguing association of NR1H3, encoding for the LXR‐α isoform, with the tumor microenvironment (TME). CIBERSORTx‐based purification on large DLBCL datasets revealed a high expression of the receptor transcript in M1‐like pro‐inflammatory Mo. By determining an expression cut‐off of NR1H3, we used digital measurement to validate its prognostic capacity on two large independent on‐trial and real‐world cohorts. Independently of classical prognosticators, NR1H3 high patients displayed longer survival compared with NR1H3 low cases and a high‐resolution Mo GEP dissection suggested a remarkable transcriptional divergence between subgroups. Overall, our findings indicate NR1H3 as a Mo‐related biomarker identifying patients at higher risk and prompt future preclinical studies investigating its mouldability for therapeutic purposes.
Keywords: deconvolution, diffuse large B‐cell lymphoma, gene expression profiling, liver X receptors, microenvironment
1. INTRODUCTION
Diffuse large B cell lymphoma (DLBCL) represents a highly heterogeneous disease calling for a more accurate risk stratification at diagnosis and improvement of first‐line immunochemotherapy. 1 , 2 , 3 Sequencing studies refined the stratification of molecular subgroups of DLBCL characterized by druggable genetic aberrations 4 , 5 , 6 and prognostic components of tumor microenvironment (TME). 7 , 8 Among these, tumor‐infiltrating macrophages (Mo) are regarded as a functionally heterogeneous immune population whose association with patient prognosis remains controversial due to the insufficient reproducibility of cell‐specific, robust functional biomarkers. 9 Moreover, owing to the lack of unique druggable targets, no data are currently available that support rationales for Mo‐directed strategies of immunomodulation in DLBCL.
Accumulating evidences indicate that critical events in TME are influenced by oxygen tensions, cytokine gradients, and nutrient alteration including lipid and cholesterol metabolism. 10 The latter is controlled by two isoforms of the Liver‐X‐Receptors (LXRs), 11 , 12 namely LXRα and LXRβ, that are mainly expressed in cells with high cholesterol turnover, such as Mo, in which they regulate viability, polarization and inflammatory response. 12 , 13 Moreover, anti‐proliferative effects have been described in solid cancers as an effect of TME changes upon direct LXR pharmacological modulation. 14 , 15 Many LXR agonists have been pre‐clinically tested with therapeutic purposes and provided controversial results due to different underlying biology in different cancers. 15 , 16 For instance, RGX‐104, a first‐in‐class oral LXR agonist, is being tested in a phase 1b/2 trial including solid tumors and aggressive lymphomas. 17 The anti‐tumor properties of RGX‐104 correlate with critical changes in TME involving myeloid‐derived suppressor cells and Mo. 18 However, no data are available on LXR biology in lymphoproliferative disorders and, particularly, in DLBCL.
Here, we explored potential association of LXRs with specialized subsets of tumor‐infiltrating immune cells in DLBCL. By systematic deconvolution of gene expression profiling (GEP) data from large patient cohorts, we revealed a striking correlation of LXRα transcript (encoded by NR1H3 gene) with a subset of pro‐inflammatory Mo in patients displaying better clinical outcome. Conversely, patients with low levels of NR1H3 expression showed inferior survival, independently of standard prognosticators. These observations were further validated by NanoString technology in two additional independent DLBCL cohorts, supporting the idea of NR1H3 as a potential biomarker of a transcriptionally‐restricted Mo subsets that might be susceptible to pharmacological agonism, paving the way for new strategies of immunomodulation in DLBCL.
2. MATERIAL AND METHODS
2.1. Patients characteristics and cohorts
Deconvolution and survival analyses were performed on a discovery set obtained by pooling 314 DLBCL with comprehensive clinical and molecular information from three different GEP datasets (GSE10846, GSE34171 and GSE98588). 2 , 6 , 19 For prognostic validation, we used data from 175 patients with advanced‐stage, nodal, de novo DLBCL, not otherwise specified, enrolled in two different clinical trials, 20 , 21 and an additional real‐world (RW, n = 146) cohort collected from four Institutions (IRCCS ‐ Istituto Tumori ‘Giovanni Paolo II’ of Bari, Italy; Istituto di Ematologia “Seràgnoli”, IRCCS Azienda Ospedaliero‐Universitaria di Bologna, Italy; IRCCS‐European Institute of Oncology of Milan, Italy; Hospital Clinic, Barcelona, Spain). Diagnostic tumor samples were obtained before any treatment.
The study was conducted according to the Declaration of Helsinki and all patients signed a dedicated informed consent. All cases were reviewed by experienced hematopathologists (CA, SF, SAP, AS, EC, VT, AFZ). An additional public DLBCL series (GSE117556) using a different microarray platform (Illumina) 22 was used to validate the prognostic value of NR1H3. Clinical characteristics of patients from different cohorts are summarized in Table 1. The whole study workflow is schematized in Figure S1.
TABLE 1.
Clinical and molecular features of diffuse large B‐cell lymphomas (DLBCL) cohorts
| Discovery dataset | DLBCL validation set (on trial) | DLBCL validation set (real‐world) | GSE117556 | |
|---|---|---|---|---|
| (N = 314) | (N = 175) | (N = 146) | (N = 893) | |
| IPI range | ||||
| High | 49 (15.6%) | 47 (26.9%) | 16 (11.0%) | 156 (17.5%) |
| High‐Int | 68 (21.7%) | 128 (73.1%) | 40 (27.4%) | 268 (30.0%) |
| Low | 121 (38.5%) | 0 (0%) | 51 (34.9%) | 244 (27.3%) |
| Low‐Int | 76 (24.2%) | 0 (0%) | 39 (26.7%) | 225 (25.2%) |
| COO | ||||
| ABC | 142 (45.2%) | 38 (21.7%) | 57 (39.0%) | 253 (28.3%) |
| GCB | 125 (39.8%) | 103 (58.9%) | 63 (43.2%) | 511 (57.2%) |
| UC | 47 (15.0%) | 34 (19.4%) | 25 (17.1%) | 129 (14.4%) |
| NR1H3 | ||||
| High | 266 (84.7%) | 154 (88.0%) | 122 (83.6%) | 837 (93.7%) |
| Low | 48 (15.3%) | 21 (12.0%) | 24 (16.4%) | 56 (6.3%) |
| Gender | ||||
| Female | 102 (32.5%) | 83 (47.4%) | 34 (23.3%) | 396 (44.3%) |
| Male | 149 (47.5%) | 92 (52.6%) | 40 (27.4%) | 497 (55.7%) |
| Age | ||||
| Median [Min, Max] | 62.0 [17.0, 92.0] | 52.0 [18.0, 65.0] | 64.2 [16.8, 87.7] | 64.7 [20.8, 86.1] |
Abbreviations: COO, cell of origin; IPI, international prognostic index.
2.2. NR1H3 and NR1H2 expression analysis
Raw data from different public datasets of DLBCL cases, cell line and normal B‐cells 2 , 6 , 19 , 23 , 24 , 25 were used to generate expression profiles by RMAExpress v1.2.0 (Robust Multi‐Array Average). Multiple probes were collapsed into unique genes by selecting those with the maximum value for each gene. Expression values were log2 transformed, and NR1H3 and NR1H2 transcripts abundance extracted for further analysis.
2.3. CIBERSORTx
CIBERSORTx (http://cibersortx.stanford.edu) was run on publicly available datasets to calculate the proportions (at 1000 permutation per run) and to obtain GEPs (by Group‐Mode imputation) of immune cytotypes in the LM22 signature. Gene expression profiling data (Affymetrix Microarray) from the discovery set were summarized and normalized using the Robust Multi‐array Averaging method by means of affy (version 1.70.0) package in R (version 4.1.0, R Core Team 2020, Vienna, Austria, https://www.R‐project.org). The GSE117556 dataset (Illumina platform) was normalized by limma (version 3.48.0) package in R before deconvolution. Bulk‐mode batch correction (B‐mode) was applied to all the datasets. GSE125966, GSE145043 26 and Schmitz et al. 5 RNA‐seq data were analyzed using the authors' normalization settings including counts per million, transcripts per million and fragments per kilobase of transcript per million (ragments per kilobase of transcript per million) space, respectively.
2.4. RNA in situ hybridization and immunohistochemistry (IHC)
RNA in situ hybridization and IHC were performed using Human NR1H3 transcript (Hs‐NR1H3; Cod.440881) and CD68‐antibody (clone PG‐M1 DAKO) respectively (Supplementary Methods).
2.5. NanoString‐based gene expression quantification
Total RNA was extracted from formalin‐fixed paraffin‐embedded (FFPE) sections of DLBCL cases as previously reported. 27 The nCounter Digital Analyzer NanoString Technology was used for Cell‐of‐origin (COO) assignment (Lymph2Cx assay) and for digital measurement of NR1H3 expression, according to manufacturer's instructions. A custom probe for NR1H3 (5′‐CCCATGGACACCTACATGCGTCGCAAGTGCCAGGAGTGTCGGCTTCGCAAATGCCGTCAGGCTGGCATGCGGGAGGAGTGTGTCCTGTCAGAAGAACAGA‐3′) and five housekeeping genes (UBXN4, TRIM56, WDR55, R3HDM1, ISY1) were used. All data were normalized using NanoStringNorm (version 1.2.1.1) package in R software, as previously described. 7
2.6. Tissue microarrays (TMA) and immunohistochemical evaluation of CD68
Tissue microarrayswere constructed by selecting three cores of 0.6‐mm diameter from representative areas of FFPE blocks relative to 64 cases selected among the on‐trial validation cohort and 2µm‐section stained with CD68 (Clone PG‐M1, dilution 1:8, courtesy of Prof. Brunangelo Falini). Further details are reported in Supplementary Methods.
2.7. Normalization and DEG
The DLBCL discovery set was generated by pooling Affymetrix‐HG133plus2 raw data from three different datasets (GSE10846, GSE34171 and GSE98588) processed as a unique expression matrix by R package affy (version 1.66) to reduce batch effect. All cases were collected in real world studies and homogeneously treated by standard immuno‐chemotherapy.
The high‐resolution mode of CIBERSORTx was applied to virtually purify macrophage GEP (M0, M1, M2 populations) and a list of differentially expressed genes (DEG) was obtained by comparing NR1H3 high and NR1H3 low cases separated using the prognostic cut‐off (Supplementary Methods). Limma R package (version 3.48.0) was used to perform DEG analysis and clusterProfiler R package (version 4.0.2) to perform Gene Ontology (GO) over‐representation tests.
2.8. In vitro macrophage polarization and treatment
THP‐1 cell line was obtained from American Type Culture Collection (Manassas, VA, USA) and grown in Roswell Park Memorial Institute 1640 (Gibco, Thermo Fisher Scientific) supplemented with 10% heat‐inactivated fetal bovine serum, 1% Penicillin‐Streptomycin (10,000 U/mL, 10mg respectively) (Sigma Aldrich), 2 mM glutamine (Gibco) and 0.05 mM of 2‐mercaptoethanol (Sigma Aldrich). M1‐ and M2‐polarized Mo were generated from THP‐1 cells as previously described. 28 The complete protocol is detailed in Supplementary Materials.
2.9. Statistical analysis
Dot plots for GO analysis representation, heatmaps, correlation matrix plots and survival analysis were produced using R statistical software. p‐values among continuous variables were calculated by Mann–Whitney test or independent Student's t‐test. Further details are provided in Supplementary Methods.
3. RESULTS
3.1. NR1H3 (LXRα) is up‐regulated in DLBCL TME and associated with M1‐like Mo
To explore the role of LXRs in DLBCL, we first analyzed the transcriptional levels of the two LXR isoforms (LXRα and LXRβ), encoded by NR1H3 and NR1H2, respectively, in different public GEP datasets of bulk DLBCL as well as in malignant cells purified from lymph node, DLBCL cell lines and normal B‐cells. We observed that NR1H3 expression was significantly higher in samples derived from whole biopsies as compared with purified tumor cells, malignant cell lines and normal B cells (p<10−3). This finding suggested that major contribution to NR1H3 expression is attributable to TME rather than tumor component. Conversely, no significant differences were observed in NR1H2 across datasets (Figure 1A).
FIGURE 1.
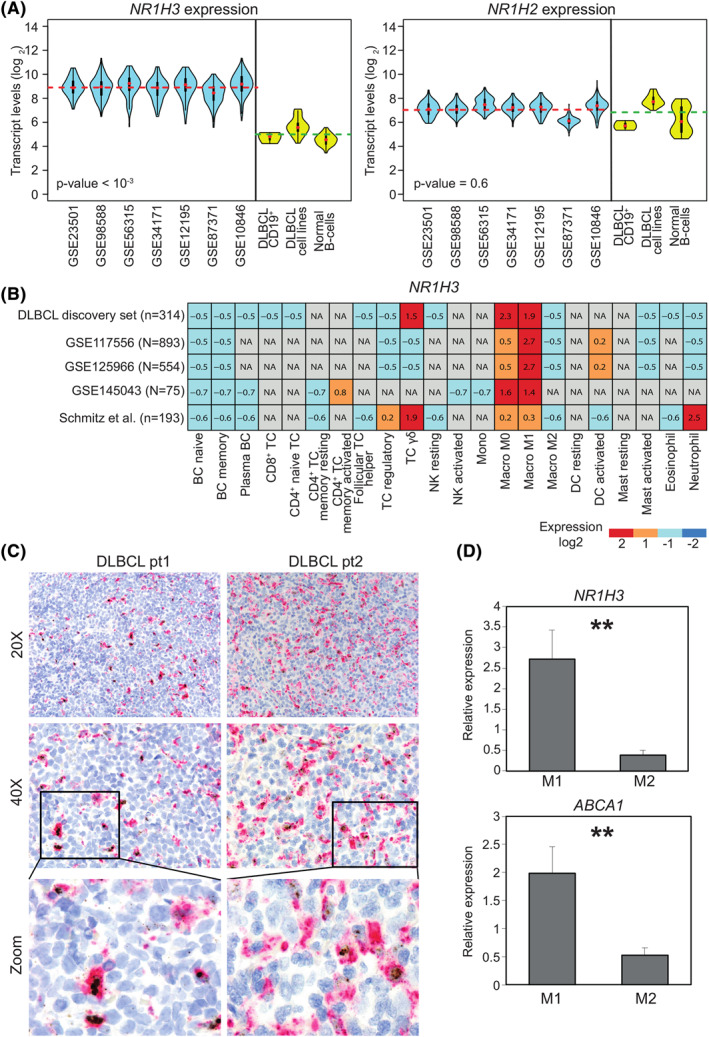
NR1H3 is expressed by macrophages in diffuse large B‐cell lymphomas (DLBCL). A, Violin‐plots representing transcript abundance of NR1H3 (left panel) and NR1H2 (right panel) in publicly‐available datasets from bulk tumor samples (light blue) or purified primary samples, DLBCL cell lines and normal B‐cells (yellow). The gene expression omnibus accession numbers of DLBCL datasets are depicted in the x‐axis. Dotted lines indicate NR1H3 averaged expression respectively from bulk DLBCL samples (red) as well as purified DLBCL cell lines and normal B‐cells (green), including germinal center centroblasts, naïve and memory B‐cells (GSE12195 and GSE56314 datasets). p‐value as derived from Mann–Whitney U test comparing bulk samples with purified cells is shown. B, Heatmap depicting NR1H3 expression from in silico purification (CIBERSORTx) of different cell types five DLBCL datasets. Signal intensities ranking from highest (red) to lowest (blue) are indicated as row‐scaled expression values. C, Representative histological sections of two DLBCL cases showing different levels of NR1H3 transcript detected by RNA in situ hybridization (brown dots) and CD68 protein (magenta) by IHC using PG‐M1 antibody (20X and 40X magnification depicted on left and right panel, respectively). The panels at the bottom are magnifications showing representative double‐positive CD68+/NR1H3 + and single‐positive CD68+/NR1H3 ‐ cells. D, Bar plot representing NR1H3 and ABCA1 expression levels determined by quantitative real time polymerase chain reaction of in vitro generated Mo. Relative quantification of gene expression was analyzed by the 2−ΔΔCt method using 18S as the endogenous control. p‐value was derived from two‐tailed t test. Data are represented as the mean of four independent experiments ± SD (standard deviation). NA, not available
To inspect which cytotype displays the highest NR1H3 expression within TME, we retrieved publicly GEP arrays from a discovery set of 314 cases and four additional independent datasets, and applied CIBERSORTx to study the gene expression patterns. Among 22 tumor‐infiltrating immune cell types resolved, Mo ‐ especially the M0/M1 fraction ‐ appeared to reproducibly express the highest levels of NR1H3. γ/δ T cells and neutrophils showed considerable NR1H3 abundance, but with no reproducibility across different cohorts. Conversely, M2 Mo, B‐ and T‐cell subsets as well as other components of innate immunity, such as natural killer, dendritic and mast cells, displayed negligible transcript levels (Figure 1B). This observation was also confirmed by a correlation analysis between NR1H3 levels and cell fractions, with the M0/M1 Mo subset displaying the highest correlation in all datasets (Supplementary Figure 2A–B). To corroborate these findings, we performed a simultaneous in situ detection of CD68 and NR1H3 in prototypical DLBCL biopsies. While no NR1H3 signal was detected in malignant B cells, we observed a variable co‐localization with CD68, as a selective histiocyte marker (Figure 1C). The amount of CD68+/NR1H3 + cells largely varied among cases, suggesting that, despite showing a similar phenotype, Mo sub‐populations with different transcriptional programs might coexist within the TME.
FIGURE 2.
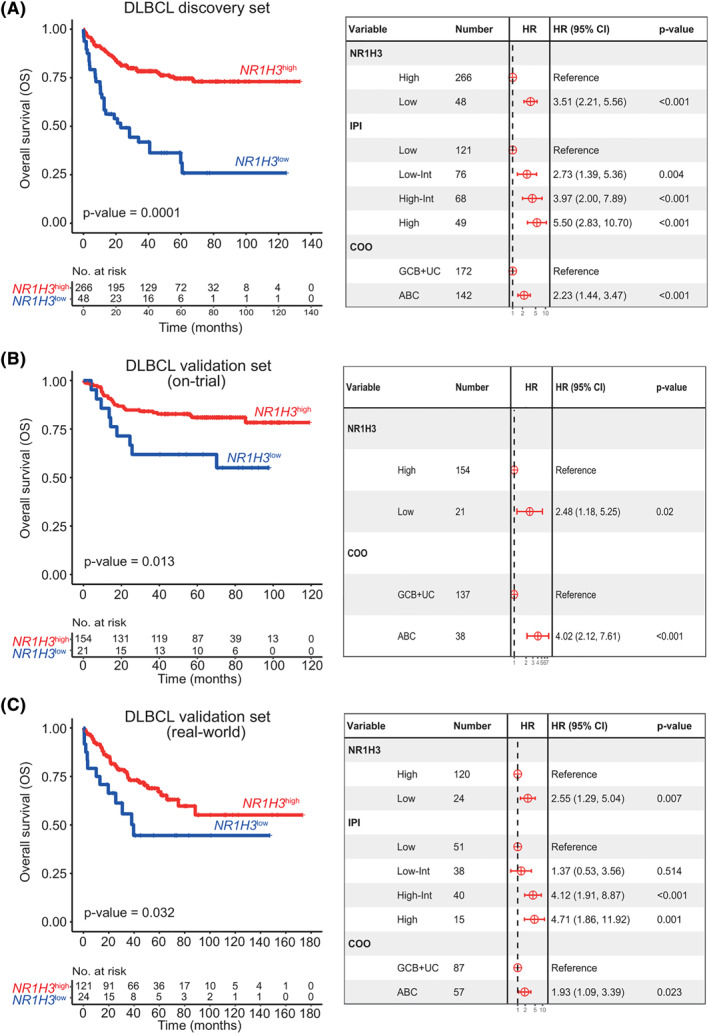
NR1H3 prognostic significance in diffuse large B‐cell lymphomas (DLBCL). A, Kaplan Meier (KM) plot (left panel) for overall survival (OS) according to NR1H3 high (red) and NR1H3 low (blue) groups derived from maximally selected rank statistic in the discovery set of 314 DLBCL. Forest plots (right panel) visualize HR and p‐value obtained from multivariate analysis of NR1H3 groups, cell of origin, COO and IPI of the discovery set. B, KM (left panel) plot of OS comparing high‐ versus low‐NR1H3 patients (NanoString Technology) in an on‐trial validation cohort (n = 175). The right panel shows a Forest plot of multivariate analysis of OS combining NR1H3 expression and COO. C, KM curves and Forest Plot for OS of the real‐world validation cohort. KM plot displays different OS according to NR1H3 expression subgroups, whereas Forest plot indicates multivariate analysis of NR1H3 stratification, COO and IPI. Abbreviations: HR, hazard ratio; COO, cell of origin; IPI, international prognostic index
We proceeded to validate the observation of NR1H3 expression being restricted to M0/M1 Mo levels in different populations of in vitro polarized Mo. Consistently, THP‐1 cells, polarized toward a M1‐like state and featuring a typical over‐expression of CXCL10 and IL‐1β, displayed significantly higher NR1H3 expression (p‐value = 0.001) as compared with the classical CD206 +/CD163 + M2 phenotype (Figure 1D, Supplementary Figure S3). This was also reflected by substantial upregulation of ABCA1, the main NR1H3 target, in M1 Mo only, thus confirming our observation of LXRα restriction to M1 Mo.
3.2. Patients expressing low NR1H3 levels display unfavorable survival
Given the potential albeit controversial prognostic impact of Mo and the TME in DLBCL, we assessed the prognostic value of NR1H3 in the discovery cohort. To do so, we applied maximally selected rank statistics to dichotomize patients on the basis of LXRα expression into NR1H3 high (n = 266) and NR1H3 low (n = 48) subgroups. Survival analysis demonstrated NR1H3 low patients associated with a significantly shorter overall survival (OS; p‐value<0.0001, median 5‐year OS, NR1H3 high 75% vs. NR1H3 low 25%) (Figure 2A). Also, a multivariate analysis indicated that the prognostic power of LXRα was independent of international prognostic index (IPI) and COO (Figure 2A). Notably, NR1H3 high and NR1H3 low subgroups (Supplementary Figure S4A) also differed in terms of Mo infiltration, as inferred by CIBERSORTx. In fact, a significant predominance of M2 Mo was determined in the NR1H3 low subgroup, whereas the M0/M1 Mo fraction prevailed in NR1H3 high cases (Supplementary Figure S4B).
The digital measurement of NR1H3 transcript in two independent DLBCL cohorts confirmed the prognostic performance of the gene according to a cut‐off built on the discovery set. Moreover, the frequency of NR1H3 low patients was consistent among both sets (12% and 16%, respectively) as well as their worse outcome compared with NR1H3 high cases (Figure 2B–C). The multivariate models validated the independence of LXRα‐based prognostication from COO and IPI risk (Figure 2B–C). Interestingly, while the transcriptional level of NR1H3 differed significantly between on‐trial cases (p‐value <10−3, Supplementary Figure S5A), the content of CD68+ cells appeared quite comparable (p‐value = 0.4176, Fisher's exact test) between NR1H3 high and NR1H3 low subgroups, supporting the idea that NR1H3 reflects a transcriptional/functional rather than phenotypical heterogeneity of Mo within TME (Supplementary Figure S5 B‐C). An additional validation attempt was carried out using a recent GEP dataset on a different platform (GSE117556, microarray technology), which highlight a dismal prognosis for NR1H3 low patients, independently of IPI and COO risk groups (Supplementary Figure S6A).
Taken together, these findings strengthened the hypothesis that NR1H3 levels characterize DLBCL with different outcomes reflecting diverse Mo subpopulations in their TME.
3.3. NR1H3 identifies DLBCL‐infiltrating Mo with peculiar transcriptomic landscapes
To explore the molecular profiles associated with NR1H3 in Mo in DLBCL, we sought to identify similarities and differences between virtually‐purified Mo fractions of the NR1H3 high and NR1H3 low subgroups (Figure 3A). Considering as DEG those displaying a log fold change (FC) > 1 and an adjusted p‐value <0.05, we obtained a total of 1040 up‐ and 169 down‐regulated genes (Supplementary Table S2). To identify putative target genes of NR1H3, we intersected the obtained DEG (both up‐ and down‐regulated) with 1079 LXRα targets from publicly available ChIP‐seq data, 29 leading to the identification of 216 downstream targets. Gene Ontology analysis revealed genes enriched not only in the cholesterol and lipid metabolism, but also in inflammatory processes such as neutrophil activation, phagocytosis, cytokine production, and stimulation of innate immunity via toll‐like receptors (Figure 3A–C). Gene set enrichment analysis also indicated that NR1H3 high patients were significantly enriched of gene sets related to M1 phenotype (normalized enrichment score, NES = 1.8, p‐value<10−3), cytokines and inflammatory response (BIOCARTA_INFLAM_PATHWAY: NES = 1.8, p‐value<103) including IL‐6 and tumor necrosis factor, and immune functions such as antigen presentation (NES = 1.7, p‐value = 0.002) (Figure 3D). A consistent enrichment of LXRα target genes (ABCA1, ABCG1, and SREBF1) was also noticeable, reflecting the transcriptional activity of the nuclear receptor (NES = 1.7, p‐value<10−3; Figure 3D).
FIGURE 3.
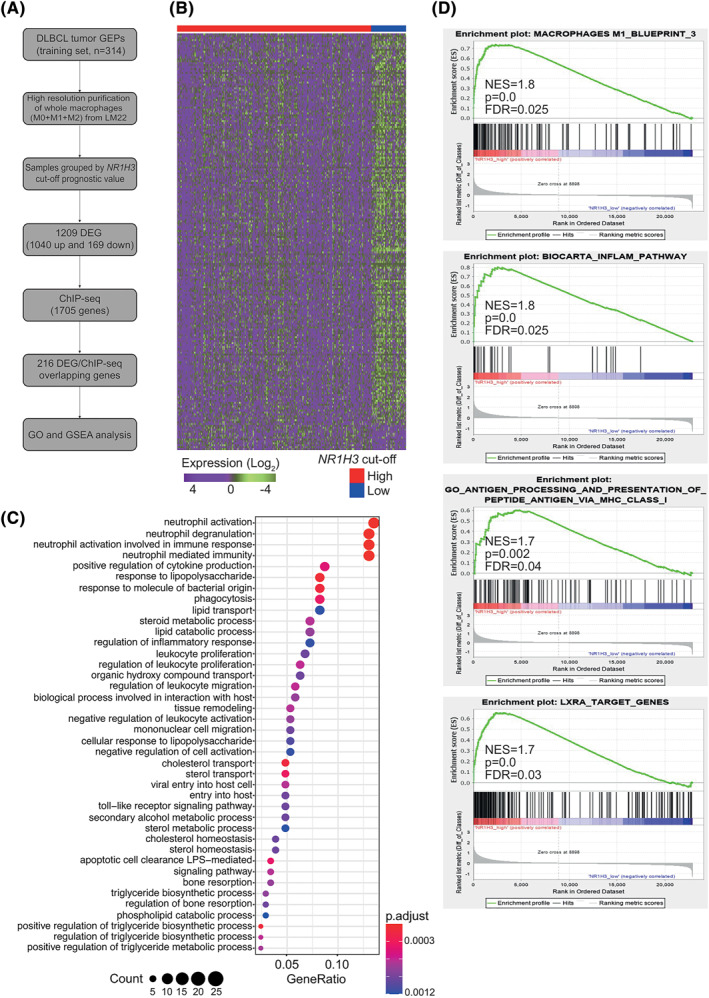
NR1H3 defines functionally‐restricted macrophages (Mo) subgroup in diffuse large B‐cell lymphomas (DLBCL). A, Left panel schematizes the methodologic worflow applied to explore Mo biology in relation to NR1H3. Differentially expressed genes (DEG) were identified comparing samples grouped by NR1H3 prognostic cut‐off and overlapped with LXRα‐target genes previously identified by ChIP‐seq. B, Heatmap indicating 216 overlapping genes obtained from the discovery set by integrating microarray and ChIP‐seq. Expression data were log2‐transformed and row‐scaled. C, Dot‐plot showing the top‐30 significantly enriched biological processes derived by Gene Ontology (GO) analysis of 216 DEG‐ChIP‐seq overlapping genes in the discovery set. p‐values adjusted using Benjamini‐Hochberg procedure and gene counts are shown in the legend at the bottom. D, gene set enrichment analysis panels showing the enrichment of gene sets related to M1‐macrophage subpopulation, inflammation and LXRα target genes in NR1H3 high cases from the discovery set. Abbreviations: DEG, differentially expressed genes; NES, normalized enrichment score; FDR, false discovery rate
Overall, these findings emphasized that LXRα‐related signaling pathways and functions are associated with M1 polarization and potentially increased immune reactivity of Mo in DLBCL.
4. DISCUSSION
Mo are essential elements of DLBCL microenvironment, but mechanisms underlying their prognostic significance remain unclear. Beyond correlating survival with the extent of Mo tumor infiltration, diverse methodologies ‐ from IHC 30 to single‐cell analyses of TME 31 , 32 ‐ unveiled a remarkable heterogeneity of DLBCL‐infiltrating Mo, at functional rather than phenotypic level. 23 , 31 , 32 However, no reproducible biomarker has been so far validated with a predictive value toward standard immunochemotherapy.
In this study, we expanded the notion of co‐existing functional subsets of Mo within TME of DLBCL, revealing striking association of the LXRα transcript (encoded by NR1H3 gene) expression with the M1 phenotype of Mo with putative anti‐tumor effect. Accumulating data highlighted the relevance of LXRs in anti‐cancer immune surveillance and described mechanisms of LXR axis activity in diverse immune cytotypes. 32 , 33 Particularly, the function of LXRα remains controversial as it can regulate a broad range of activities in specific subsets of accessory cells, such as Mo, in the context of various human cancers. 34 To dissect such biology in DLBCL, we exploited a high‐resolution deconvolution of GEP from large patient cohorts and applied a discovery‐ validation approach using additional on trial and real‐life case sets.
Compared with the reported literature, the identification of NR1H3 as a reproducible, prognostic biomarker of functional Mo features overcomes existing phenotype‐based methods to capture Mo heterogeneity. For instance, the prognostic use of CD68 immunostaining to estimate the histiocytic infiltration in DLBCL biopsies has produced inconsistent results 35 , 36 , 37 , 38 also due to the phenotypic nature of the marker, not being able to distinguish functionally‐divergent Mo subsets. Conversely, when categorized according to NR1H3 expression, DLBCL subgroups exhibit different enrichment of polarized Mo, with M1‐like cells prevailing in NR1H3 high cohort, this being reflected by a more favorable outcome. We brought additional proof to this concept by measuring CD68+ cells in our on‐trial validation set, where no significant difference emerged between prognostic subgroups despite divergent expression of LXR transcript. Such finding also stresses how the selection of functional immune biomarkers became essential to identify patient‐specific TME for therapeutic purposes.
Deeper transcriptome‐based approaches have revealed “stromal”, 19 “mesenchymal”, “inflammatory” and “lymphoma‐associated macrophage” signatures with arguable significance in terms of prognostication, underlying biological mechanisms and clinical applicability. 39 In this context, the reported “lymphoma‐associated macrophage interaction signature” (LAMIS) 9 was built in a supervised fashion on M2‐related genes. The signature characterizes poor‐outcome patients, but adds no further insights to previous evidence that greater CD163+ cell infiltration confers unfavorable prognosis. 38 A recent computational tool applied to thousands of DLBCL, named ECOTYPER, drew a high‐resolution map of functional immune cell states and remarked heterogeneity in Mo population associating M1‐like monocytes/Mo with longer survival, independently of current genomic prognosticators. 40 Such evidence is in line with ours and prompts to speculate whether the prevalence of pro‐inflammatory Mo could boost the cytotoxicity of anti‐CD20 therapy. Our results add up to this picture, providing reproducible evidence that a metabolic regulator, as LXRα, characterizes the M1‐like subset of DLBCL‐infiltrating Mo. Conversely, in low NR1H3‐expressing patients likely prevails a different Mo‐related biology which correlates with inferior outcome toward standard immunochemotherapy. We therefore envisage NR1H3 as a potential biomarker for future strategies of immunomodulation. Beyond identifying patients at higher risk, in fact, the digital measurement of the transcript, according to the validated cut‐off, may be of translational help in establishing preclinical patient‐derived screening platforms for new immunotherapies.
Novel strategies are emerging aimed at reprogramming the innate immunity in many solid tumor models. 41 In fact, the LXRs activity can be modulated by synthetic agonists, including the recent developed RGX‐104. This compound was already shown to exert remarkable anti‐cancer effect in preclinical solid tumors 17 , 18 and it is being assessed for efficacy and safety in early clinical trials including aggressive lymphomas (ClinicalTrials.Gov, NCT02922764). Beside regulation of cholesterol homeostasis in Mo, LXR modulation was shown to affect secretion of cytokines impacting immune functions of T‐regs, 42 natural killer cells, 43 myeloid‐derived suppressor cells 18 and neutrophils 44 in both inflammatory diseases and cancer. 45 We also noticed a remarkable impact of NR1H3 expression and its downstream targets on neutrophil‐related processes as degranulation, activation and inflammatory response, suggesting intriguing interplay between Mo and other inflammatory bystander cells with potential effects on tumor behavior. Such hypothesis and the notion that both physiological activity of the receptor and its pharmacological modulation are tissue‐ and disease‐specific, 46 prompt deeper mechanistic investigation in more sophisticated pre‐clinical models also resembling new genetic DLBCL subgroups. 5 , 6
Overall, our study adds new understandings on the Mo heterogeneity in DLBCL, linking their metabolic diversity to functional divergence that could be captured by NR1H3 as a reliable biomarker. The digital measurement of the receptor in diagnostic biopsy may also help in identifying NR1H3 low poor‐outcome patients deserving alternative treatments. In conclusion, we provided the first comprehensive and disease‐specific dissection of the role of LXRs in, promoting preclinical studies on the use of macrophage‐targeted therapeutic strategies in DLBCL.
AUTHOR CONTRIBUTIONS
Maria Carmela Vegliante, Antonio Moschetta, Alessandro Gulino, Sabino Ciavarella and Stefano A. Pileri conceived and planned the project; Federica Melle, Giovanna Motta, Maria Rosaria Sapienza, Giuseppina Opinto, Anna Enjuanes, Antonella Bucci and Antonio Negri performed RNA extraction, digital expression analysis and in vitro experiments; AG, GM and produced in situ data, Maria Carmela Vegliante, Saveria Mazzara, Gian Maria Zaccaria, Simona De‐Summa, Flavia Esposito, Giacomo Volpe and Grazia Gargano performed statistical analyses; Valentina Tabanelli, Stefano Fiori, Carla Minoia, Felice Clemente, Anna Scattone, Alfredo F. Zito, Stefania Tommasi, Claudio Agostinelli, Umberto Vitolo, Annalisa Chiappella, Anna Maria Barbui, Enrico Derenzini, Pier Luigi Zinzani, Beatrice Casadei, Alfredo Rivas‐Delgado, Armando López‐Guillermo, Elias Campo, Attilio Guarini and Stefano A. Pileri carried out samples collection, clinical annotation and pathology review; Maria Carmela Vegliante, Giacomo Volpe, Sabino Ciavarella and Stefano A. Pileri prepared the figures and wrote the manuscript; all authors critically reviewed the manuscript and approved the final draft for submission.
CONFLICT OF INTEREST
ED, Research funding from TG‐Therapeutics, ADC‐Therapeutics, Takeda and Advisory board for Gilead, Astra Zeneca, Takeda, BeiGene, Abbvie. The other authors declare they have no conflicts of interest.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1002/hon.3050.
Supporting information
Supplementary Information S1
ACKNOWLEDGMENTS
This work was supported by grants from Associazione Italiana per la Ricerca sul Cancro (AIRC) (Fellowship 26928‐2021 to GM, IG 23239 to AM and 5x1000 n. 21198 to SAP) and Italian Ministry of Health (Ricerca Corrente RRC‐2019‐2366660 to SC).
Open access funding provided by BIBLIOSAN.
Vegliante MC, Mazzara S, Zaccaria GM, et al. NR1H3 (LXRα) is associated with pro‐inflammatory macrophages, predicts survival and suggests potential therapeutic rationales in diffuse large b‐cell lymphoma. Hematol Oncol. 2022;40(5):864‐875. 10.1002/hon.3050
Stefano A. Pileri and Sabino Ciavarella contributed equally as last authors.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Swerdlow SH, World Health Organization, International Agency for Research on Cancer . WHO classification of tumours of haematopoietic and lymphoid tissues [Internet]. [cited 2018 Feb 15]. 585 p. http://publications.iarc.fr/Book‐And‐Report‐Series/Who‐Iarc‐Classification‐Of‐Tumours/Who‐Classification‐Of‐Tumours‐Of‐Haematopoietic‐And‐Lymphoid‐Tissues‐2017
- 2. Monti S, Chapuy B, Takeyama K, et al. Integrative analysis reveals an outcome‐associated and targetable pattern of p53 and cell cycle deregulation in diffuse large B cell lymphoma. Cancer Cell Vol 22; 2012:.3 359‐372. [Internet][cited 2018 Jan 25]http://www.ncbi.nlm.nih.gov/pubmed/22975378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Morin RD, Mungall K, Pleasance E, et al. Mutational and structural analysis of diffuse large B‐cell lymphoma using whole‐genome sequencing. Blood Vol 122; 2013.7:1256‐1265. [Internet][cited 2018 May 6]http://www.ncbi.nlm.nih.gov/pubmed/23699601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Reddy A, Zhang J, Davis NS, et al. Genetic and functional drivers of diffuse large B cell lymphoma. Cell. 2017;171(2):481‐494. e15. 10.1016/j.cell.2017.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schmitz R, Wright GW, Huang DW, et al. Genetics and pathogenesis of diffuse large B‐cell lymphoma. N Engl J Med. 2018;378(15):1396‐1407. [Internet][cited 2018 Jun 24]http://www.nejm.org/doi/10.1056/NEJMoa1801445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chapuy B, Stewart C, Dunford AJ, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med. 2018;24(5):679‐690. [Internet][cited 2018 Jun 24]Available from:. 10.1038/s41591-018-0016-8. http://www.ncbi.nlm.nih.gov/pubmed/29713087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ciavarella S, Vegliante MC, Fabbri M, et al. Corrigendum: dissection of DLBCL microenvironment provides a gene expression‐based predictor of survival applicable to formalin‐fixed paraffin‐embedded tissue. Ann Oncol. 2018;29(12):2363‐2370. 10.1093/annonc/mdy450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kotlov N, Bagaev A, Revuelta MV, et al. Clinical and biological subtypes of B‐cell lymphoma revealed by microenvironmental signatures. Cancer Discov; 2021. [Internet]4 [cited 2021 Mar 17];candisc.0839.2020. https://cancerdiscovery.aacrjournals.org/content/early/2021/02/02/2159‐8290.CD‐20‐0839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Staiger AM, Altenbuchinger M, Ziepert M, et al. A novel lymphoma‐associated macrophage interaction signature (LAMIS) provides robust risk prognostication in diffuse large B‐cell lymphoma clinical trial cohorts of the DSHNHL. Leukemia. 2019. [Internet] [cited 2019 Dec 19]; http://www.ncbi.nlm.nih.gov/pubmed/31530861 [DOI] [PubMed] [Google Scholar]
- 10. O’Neill LAJ, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med. 2016;213(1):15‐23. 10.1084/jem.20151570. [cited 2018 May 6]. http://www.ncbi.nlm.nih.gov/pubmed/26694970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Repa JJ, Mangelsdorf DJ. The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu Rev Cell Dev Biol. 2000;16(1):459‐481. [Internet][cited 2018 May 6]Available from:. 10.1146/annurev.cellbio.16.1.459. http://www.ncbi.nlm.nih.gov/pubmed/11031244 [DOI] [PubMed] [Google Scholar]
- 12. Bensinger SJ, Tontonoz P. Integration of metabolism and inflammation by lipid‐activated nuclear receptors. Nature. 2008;454(7203):470‐477. 10.1038/nature07202. [Internet][cited 2018 May 6]Available from:. http://www.ncbi.nlm.nih.gov/pubmed/18650918 [DOI] [PubMed] [Google Scholar]
- 13. Fontaine C, Rigamonti E, Nohara A, et al. Liver X receptor activation potentiates the lipopolysaccharide response in human macrophages. Circ Res Vol 101; 2007.1:40‐49. 24 [cited 2018 May 6]. http://www.ncbi.nlm.nih.gov/pubmed/17540978 [DOI] [PubMed] [Google Scholar]
- 14. Bovenga F, Sabbà C, Moschetta A. Uncoupling nuclear receptor LXR and cholesterol metabolism in cancer. Cell Metab Vol 21; 2015.4:517‐526. [Internet][cited 2019 Oct 29]https://www.sciencedirect.com/science/article/pii/S1550413115001059?via%3Dihub [DOI] [PubMed] [Google Scholar]
- 15. Pencheva N, Buss CG, Posada J, Merghoub T, Tavazoie SF. Broad‐spectrum therapeutic suppression of metastatic melanoma through nuclear hormone receptor activation. Cell Vol 156; 2014.5:986‐1001. Cell [Internet][cited 2019 Oct 29]https://www.sciencedirect.com/science/article/pii/S0092867414000907?via%3Dihub [DOI] [PubMed] [Google Scholar]
- 16. Nelson ER, Wardell SE, Jasper JS, et al. 27‐Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science. 2013;342(6162):1094‐1098. [Internet]. 2013 Nov 29 [cited 2021 Aug 26]Available from:. 10.1126/science.1241908. https://science.sciencemag.org/content/342/6162/1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mita MM, Mita AC, Chmielowski B, et al. Pharmacodynamic and clinical activity of RGX‐104, a first‐in‐class immunotherapy targeting the liver‐X nuclear hormone receptor (LXR), in patients with refractory malignancies. J Clin Oncol. 2018;36(15_Suppl l):3095. 10.1200/jco.2018.36.15_suppl.3095 [DOI] [Google Scholar]
- 18. Tavazoie MF, Pollack I, Tanqueco R, et al. LXR/ApoE activation restricts innate immune suppression in cancer. Cell. 2018;172(4):825‐840. e18. 10.1016/j.cell.2017.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lenz G, Wright G, Dave SS, et al. Stromal gene signatures in large‐B‐cell lymphomas. N Engl J Med. Vol 359; 2008.22:2313‐2323. [Internet][cited 2017 Dec 19]http://www.nejm.org/doi/abs/10.1056/NEJMoa0802885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chiappella A, Martelli M, Angelucci E, et al. Rituximab‐dose‐dense chemotherapy with or without high‐dose chemotherapy plus autologous stem‐cell transplantation in high‐risk diffuse large B‐cell lymphoma (DLCL04): final results of a multicentre, open‐label, randomised, controlled, phase 3 study. Lancet Oncol; 2017. 18(8):1076–1088. [Internet][cited 2018 Jan 8]. http://www.ncbi.nlm.nih.gov/pubmed/28668386 [DOI] [PubMed] [Google Scholar]
- 21. Cortelazzo S, Tarella C, Gianni AM, et al. Randomized trial comparing R‐CHOP versus high‐dose sequential chemotherapy in high‐risk patients with diffuse large B‐cell lymphomas. J Clin Oncol. 2016;34(33):4015–4022. [Internet][cited 2018 Jan 8];34(33):4015–22. http://www.ncbi.nlm.nih.gov/pubmed/28199143 [DOI] [PubMed] [Google Scholar]
- 22. Sha C, Barrans S, Cucco F, et al. Molecular high‐grade B‐cell lymphoma: defining a poor‐risk group that requires different approaches to therapy. J Clin Oncol. 2019;37(3):202–212. [cited 2019 Oct 29]. http://www.ncbi.nlm.nih.gov/pubmed/30523719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shaknovich R, Geng H, Johnson NA, et al. DNA methylation signatures define molecular subtypes of diffuse large B‐cell lymphoma. Blood; 2010;116(20):e81‐e89. [Internet][cited 2019 Feb 19]. http://www.ncbi.nlm.nih.gov/pubmed/20610814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dybkær K, Bøgsted M, Falgreen S, et al. Diffuse large B‐cell lymphoma classification system that associates normal B‐cell subset phenotypes with prognosis. J Clin Oncol. 2015. [cited 2022 Jan 27];33(12):1379–88. https://pubmed.ncbi.nlm.nih.gov/25800755/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dubois S, Viailly PJ, Bohers E, et al. Biological and clinical relevance of associated genomic alterations in MYD88 L265P and non‐L265P‐mutated diffuse large B‐cell lymphoma: Analysis of 361 cases. Clin Cancer Res; 2017;23(9):2232–2244. [cited 2022 Jan 27];23(9):2232–44. https://pubmed.ncbi.nlm.nih.gov/27923841/ [DOI] [PubMed] [Google Scholar]
- 26. Kotlov N, Bagaev A, Revuelta MV, et al. Clinical and biological subtypes of b‐cell lymphoma revealed by microenvironmental signatures. Cancer Discov; 2021;11(6):1468–1489. [Internet][cited 2021 Jun 8];http://cancerdiscovery.aacrjournals.org/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ciavarella S, Vegliante MC, Fabbri M, et al. Dissection of DLBCL microenvironment provides a gene expression‐based predictor of survival applicable to formalin‐fixed paraffin‐embedded tissue. Ann Oncol. 2018;29(12):2363‐2370. 10.1093/annonc/mdy450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tjiu J.‐W, Chen J.‐S, Shun C.‐T, et al. Tumor‐associated macrophage‐induced invasion and angiogenesis of human basal cell carcinoma cells by cyclooxygenase‐2 induction. J Invest Dermatol. 2009;129(4):1016–1025. [Internet][cited 2018 May 14]. http://linkinghub.elsevier.com/retrieve/pii/S0022202X15342901 [DOI] [PubMed] [Google Scholar]
- 29. Feldmann R, Fischer C, Kodelja V, et al. Genome‐wide analysis of LXRα activation reveals new transcriptional networks in human atherosclerotic foam cells. Nucleic Acids Res. 2013;41(6):3518–3531. [Internet][cited 2021 Aug 26]. https://pubmed.ncbi.nlm.nih.gov/23393188/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Croci GA, Au‐Yeung RKH, Reinke S, et al. SPARC positive macrophages are the superior prognostic factor in the microenvironment of diffuse large B‐cell lymphoma and independent of MYC rearrangement and double/triple hit status. Ann Oncol; 2021. [Internet][cited 2021 Aug 26];0(0). https://linkinghub.elsevier.com/retrieve/pii/S0923753421042757 [DOI] [PubMed] [Google Scholar]
- 31. Bruns H, Büttner M, Fabri M, et al. Vitamin D–dependent induction of cathelicidin in human macrophages results in cytotoxicity against high‐grade B cell lymphoma. Sci Transl Med. 2015;7(282). [Internet][cited 2018 May 6];7(282):282ra47‐282ra47. Available from:. 10.1126/scitranslmed.aaa3230. http://www.ncbi.nlm.nih.gov/pubmed/25855493 [DOI] [PubMed] [Google Scholar]
- 32. Bilotta MT, Petillo S, Santoni A, Cippitelli M. Liver X receptors: regulators of cholesterol metabolism, inflammation, autoimmunity, and cancer. Front Immuno. Vol 11:2020:2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brendolan A, Russo V. Targeting cholesterol homeostasis in hematopoietic malignancies. Blood; 2021;139(2). [Internet][cited 2022 Jan 14];139(2). https://pubmed.ncbi.nlm.nih.gov/34610110/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Derangère V, Chevriaux A, Courtaut F, et al. Liver X receptor β activation induces pyroptosis of human and murine colon cancer cells. Cell Death Differ. 2014;21(12):1914‐1924. [Internet][cited 2021 Dec 22];21(12):1914–24. Available from:. 10.1038/cdd.2014.117. https://pubmed.ncbi.nlm.nih.gov/25124554/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Riihijärvi S, Fiskvik I, Taskinen M, et al. Prognostic influence of macrophages in patients with diffuse large B‐cell lymphoma: a correlative study from a Nordic phase II trial. Haematologica; 2015;100(2):238–245. Haematologica [Internet][cited 2018 Jan 8];100(2):238–45. http://www.ncbi.nlm.nih.gov/pubmed/25381134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nam SJ, Go H, Paik JH, et al. An increase of M2 macrophages predicts poor prognosis in patients with diffuse large B‐cell lymphoma treated with rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone. Leuk Lymphoma. 2014;55(11):2466–2476. http://www.ncbi.nlm.nih.gov/pubmed/24397616 [DOI] [PubMed] [Google Scholar]
- 37. Wada N, Zaki MAA, Hori Y, et al. Tumour‐associated macrophages in diffuse large B‐cell lymphoma: a study of the osaka lymphoma study group. Histopathology; 2012;60(2):313–319. Histopathology [Internet][cited 2018 May 6];60(2):313–9. http://www.ncbi.nlm.nih.gov/pubmed/22211289 [DOI] [PubMed] [Google Scholar]
- 38. Marchesi F, Cirillo M, Bianchi A, et al. High density of CD68+/CD163+ tumour‐associated macrophages (M2‐TAM) at diagnosis is significantly correlated to unfavorable prognostic factors and to poor clinical outcomes in patients with diffuse large B‐cell lymphoma. Hematol Oncol; 2015;33(2):110–112. [Internet][cited 2018 May 6];33(2):110–2. http://www.ncbi.nlm.nih.gov/pubmed/24711044 [DOI] [PubMed] [Google Scholar]
- 39. Kotlov N, Bagaev A, Revuelta MV, et al. Clinical and biological subtypes of b‐cell lymphoma revealed by microenvironmental signatures. Cancer Discov; 2021;11(6):1468–1489. [Internet][cited 2021 Oct 19];11(6):1468–89. http://cancerdiscovery.aacrjournals.org/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Steen CB, Luca BA, Esfahani MS, et al. The landscape of tumor cell states and ecosystems in diffuse large B cell lymphoma. Cancer Cell; 2021:1422‐1437. [Internet][cited 2021 Oct 19];39(10)e10. https://linkinghub.elsevier.com/retrieve/pii/S1535610821004517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sun L, Kees T, Almeida AS, et al. Activating a collaborative innate‐adaptive immune response to control metastasis. Cancer Cell; 2021:1361‐1374. [Internet][cited 2021 Oct 19];39(10)e9. https://pubmed.ncbi.nlm.nih.gov/34478639/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Herold M, Breuer J, Hucke S, et al. Liver X receptor activation promotes differentiation of regulatory T cells. PLoS One Vol 12; 2017;e0184985. [Internet][cited 2021 Dec 22]https://dx.plos.org/10.1371/journal.pone.0184985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bilotta MT, Abruzzese MP, Molfetta R, et al. Activation of liver X receptor up‐regulates the expression of the NKG2D ligands MICA and MICB in multiple myeloma through different molecular mechanisms. FASEB J. 2019;33(8):9489‐9504. [Internet][cited 2021 Dec 22];33(8):9489–504. https://onlinelibrary.wiley.com/doi/10.1096/fj.201900319R [DOI] [PubMed] [Google Scholar]
- 44. Badea L, Herlea V, Dima SO, Dumitrascu T, Popescu I. Combined gene expression analysis of whole‐tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia. Hepatogastroenterology. 55(88):2016–2027. [Internet]. [cited 2018 Oct 10];55(88):2016–27. http://www.ncbi.nlm.nih.gov/pubmed/19260470 [PubMed] [Google Scholar]
- 45. González De La Aleja1 A, De La Rosa JV, Alonso B, Puig‐Kroger A, Vega MA, Corbi AL. LXR nuclear receptors prompt a pro‐innammatory gene and functional proole in human macrophages; 2021. [cited 2022 Jan 25]; https://www.researchsquare.com
- 46. Raccosta L, Fontana R, Maggioni D, et al. The oxysterol‐cxcr2 axis plays a key role in the recruitment of tumor‐promoting neutrophils. J Exp Med. 2013. 10(9):1711–1728 [Internet][cited 2021 Dec 22];210(9):1711–28. Available from: http://pmc/articles/PMC3754872/ [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information S1
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.