

Abstract
TATA‐binding protein associated factor 4 (TAF4) is a subunit of the Transcription Factor IID (TFIID) complex, a central player in transcription initiation. Other members of this multimeric complex have been implicated previously as monogenic disease genes in human developmental disorders. TAF4 has not been described to date as a monogenic disease gene. We here present a cohort of eight individuals, each carrying de novo putative loss‐of‐function (pLoF) variants in TAF4 and expressing phenotypes consistent with a neuro‐developmental disorder (NDD). Common features include intellectual disability, abnormal behavior, and facial dysmorphisms. We propose TAF4 as a novel dominant disease gene for NDD, and coin this novel disorder “TAF4‐related NDD” (T4NDD). We place T4NDD in the context of other disorders related to TFIID subunits, revealing shared features of T4NDD with other TAF‐opathies.
Keywords: human genetics, mendelian disorders, neurodevelopmental disorder, TAF4, TFIID
1.
TATA‐binding protein‐associated factor 4 (TAF4; MIM# 601796) encodes for a subunit of Transcription Factor IID (TFIID). This multimeric protein complex acts as a central player in transcriptional regulation by directing the assembly of the pre‐initiation complex (PIC) to specific promoters. Besides TATA‐binding Protein (TBP) and TAF4, the complex is made up of more than 10 other TBP‐associated factors (TAFs) (Patel et al., 2018).
The TAF4 gene comprises 15 exons, encoding for a 1085 amino acid protein which contains two conserved domains. The first is the TAF‐homology domain (TAFH), which binds several different transcriptional regulators and thereby plays an essential role in the co‐activating function of TAF4 (Brunkhorst et al., 2005; Kazantseva et al., 2013, 2015; Wang et al., 2007). The TAF4 domain is located at the C‐terminus of the protein and is encoded by exons 10–15. This domain facilitates the binding of TAF4 to TAF12, which creates a heterodimer that in turn forms part of a subcomplex required for the assembly of the TFIID complex (Patel et al., 2018; Thuault et al., 2002; Werten et al., 2002; Wright et al., 2006). At the distal C‐terminal end of TAF4, within the TAF4‐domain, resides a short‐conserved C‐terminal domain (CCTD), a region described to be important for optimal TAF4‐TAF12 dimerization (Thuault et al., 2002). The TAF4‐TAF12 heterodimer forms a subcomplex completed by TAF5, −6, and −9. The TFIID complex consists of three main lobes (A B and C), two of which (A and B) contain the TAF4/5/6/9/12 complex, and thus TFIID contains two copies of TAF4 (Bieniossek et al., 2013; Patel et al., 2018; Sanders et al., 2002).
TFIID and TAF4 have been widely implicated in organismal development, consistent with their central role in transcriptional regulation (Langer et al., 2016; Pijnappel et al., 2013). TAF4 expression fluctuates during neural development and cellular differentiation is regulated by different isoforms (Brunkhorst et al., 2005; Kazantseva et al., 2013, 2015, 2016). Moreover, mouse embryonic stem cells deficient for Taf4a fail to differentiate (Bahat et al., 2013) and Taf4a knock‐out results in embryonic lethality in mice (Langer et al.,2016).
Several TAFs have been identified as monogenic disease genes, including TAF1, TAF2, TAF6, and TAF13 (Alazami et al., 2015; Gudmundsson et al., 2019; Halevy et al., 2012; Hellman‐Aharony et al., 2013; Hurst et al., 2018; O'Rawe et al., 2015; Tawamie et al., 2017; Yuan et al., 2015). In general, pathogenic variants in these genes cause neurodevelopmental disorders (NDDs), typically associated with intellectual disability. To date, TAF4 has not yet been causally implicated as a monogenic disease gene. We present a cohort of eight individuals carrying heterozygous, de novo putative loss‐of‐function (pLoF) TAF4 variants.
All variants were identified through clinical trio‐exome or ‐genome sequencing efforts. This study is performed within the ethical framework of the University Medical Centre Utrecht (details in Online Supporting Information).
The present study was initiated after the identification of a de novo c.2185C>T [p.(Gln729*)] variant in TAF4 (NM_003185.3) by clinical trio‐exome sequencing (trio‐ES) testing in proband 3. TAF4 presented as an outstanding candidate disease gene based on three main arguments. Firstly, TAF4 is well conserved against pLoF variants in the general population as no pLoF variants are reported in the gnomAD database and the LoF observed/expected upper bound fraction (LOEUF) is 0.08, which suggests heterozygous pLoF variants in TAF4 might cause disease (Karczewski et al., 2020). Utilizing the LOEUF metric in gnomAD, we have previously identified TAF4 as a putative novel dominant disease gene in an independent analysis (Seaby et al., 2022). Secondly, several other TFIID complex members are known monogenic disease genes in NDDs (see below), consistent with TFIID malfunction resulting in NDDs. Finally, TAF4 function has been implicated in cellular differentiation and organismal development. Most notably, Taf4a knock‐out is embryonically lethal in mice and respective embryonic stem cells fail to differentiate (Langer et al., 2016).
We thus suspected TAF4 to be a monogenic disease gene and initiated the formation of the present cohort by patient matchmaking through the Genematcher platform (Sobreira et al., 2015). Consultation of other commonly used patient databases (e.g., Decipher and ClinVar) did not result in additional cases.
The present cohort comprises eight unrelated probands (henceforth referred to as P1–8), who all had clinical genetic testing that led to the identification of a heterozygous, de novo pLoF variant in TAF4 as the prime candidate to explain the phenotypes (Figure 1). No relevant family history or other candidate gene variants were reported in any of the probands. In four probands (P1–3 and 5), nonsense variants were identified, including three single base substitutions (P1: c.1348C>T [p.(Gln450*)]; P2: c.2029C>T [p.(Gln677*)]; P3: c.2185C>T [p.(Gln729*)]) and one single base deletion (P5: c.2570_2571del [p.(Ser857*)]). The remaining four variants were indel variants leading to a frameshift (P4: c.2453dup [p.(Asn818Lysfs*2)]; P6: c.2664delA [p.(Lys888Asnfs*4)]; P7: c.3096_3097insCGAC [p.(Gly1033Argfs*39)]; P8: c.3101_3102del [p.(Pro1034Argfs*36)]) (Figure 1a). All variants were expected to result in nonsense‐mediated decay (NMD), except for the two frameshifts in exon 15, and were thus expected to produce no functional gene product. Whilst the two frameshifts located in the last exon may escape NMD, the resulting products are expected to disrupt the architecture of the C‐terminus of TAF4 and thereby the CCTD motif. This motif is known to facilitate the binding of TAF4 to TAF12 (Thuault et al., 2002), which in turn is essential for the function of the TFIID complex (Patel et al., 2018; Thuault et al., 2002). We thus expect these variants to have a profound impact on gene function even in the absence of NMD, either by interfering with the normal function of the TAF4/CCTD domain, or by impacting protein stability. In summary, eight de novo pLoF variants in TAF4 were identified in eight unrelated individuals (Figure 1a).
Figure 1.
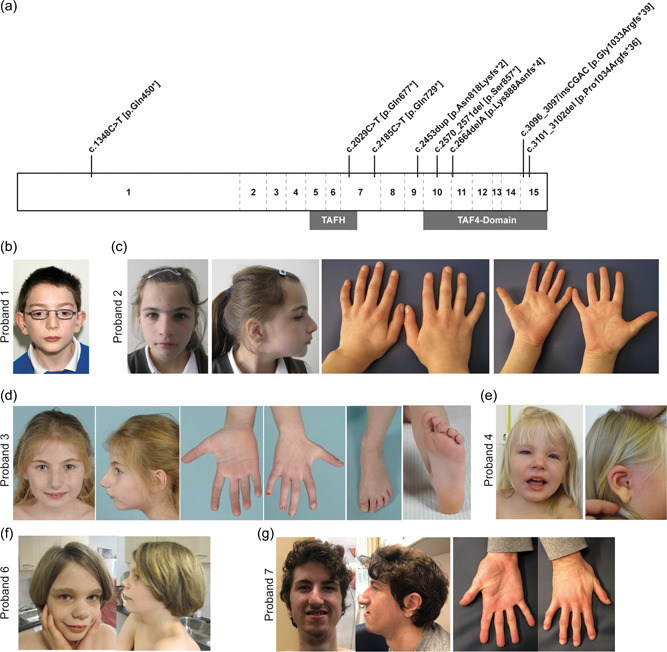
De novo pLoF variants in TAF4 and associated dysmorphologies. (a) Schematic representation of TAF4. The TAF4 gene comprises 15 exons (separated by dashed lines). The protein contains two domains, TAFH and TAF4‐domain, depicted by gray boxes below the gene. All variants are annotated at the respective location with the predicted amino acid changes. All annotations are based on transcript NM_003185.3. b–g) Photographs of probands 1 (b), 2 (c), 3 (d), 4 (e), 6 (f), and 7 (g). TAF4, TATA‐binding protein‐associated factor 4.
The cohort comprises six females and two males, ranging in age from two to 18 years old. Individual phenotypes are presented in Table S1 and clinical histories for five probands is available as supplementary data as well. Summarized findings are presented in Table 1. Seven probands were diagnosed with intellectual disability (7/8), and seven probands have delayed speech development (7/8), although P2's speech showed improvement over time. Interestingly, P2 is also the only proband in the cohort that has no ID. Other commonly reported phenotypes include behavioral abnormalities (6/8) (e.g., shy behavior in 3/8), joint laxity (4/8), and spine anomalies (3/8). An MRI of the brain was performed for five (5/8) probands and detected anomalies in 3/5 probands (Table S1).
Table 1.
Clinical features of patients with variants in TAF4 in comparison with phenotypes associated with other TFIID NDDs
| Features (HPO code) | TAF1 | TAF2 | TAF6 | TAF13 | TAF4 |
|---|---|---|---|---|---|
| Development | |||||
| Postnatal growth retardation (HP:0008897) | + (11/15) | + (1/5) | + (2/4) | ||
| Delayed gross motor development (HP:0002194) | + (15/15) | + (1/5) | + (4/4) | ||
| Delayed speech and language development (HP:0000750) | + (14/15) | + (1/5) | + (4/4) | + (7/8) | |
| Delayed puberty (HP:0000823) | + (3/4) | ||||
| Delayed bone age (HP:0002750) | + (3/4) | + (1/8) | |||
| Intellectual disability (HP:0001249) | + (14/15) | + (6/6) | + (5/5) | + (4/4) | + (7/8) |
| None of the above | 0/15 | 0/6 | 0/5 | 0/4 | 0/8 |
| Craniofacial | |||||
| Long face (HP:0000276) | + (10/15) | ||||
| Prominent forehead (HP:0011220) | + (9/15) | ||||
| Low frontal hairline (HP:0000294) | + (4/5) | ||||
| Arched eyebrows (HP:0002553) | + (5/5) | + (5/8) | |||
| Prominent supraorbital ridges (HP:0000336) | + (11/15) | ||||
| Downslanted palpebral fissures (HP:0000494) | + (10/15) | + (1/8) | |||
| Deeply set eyes (HP:0000490) | + (8/15) | + (1/8) | |||
| Long (curly) eyelashes (HP:0000527) | + (1/5) | ||||
| Sunken cheeks (HP:0009938) | + (9/15) | ||||
| Prominent nose/nasal bridge (HP:0000448 & HP:0000426) | + (5/5) | + (6/8) | |||
| Bulbous nasal tip (HP:0000414) | + (7/15) | ||||
| Anteverted nares (HP:0000463) | + (11/15) | + (2/8) | |||
| Long philtrum (HP:0000343) | + (12/15) | + (1/5) | + (1/8) | ||
| Prominent philtrum (HP:0002002) | + (2/8) | ||||
| Thin upper lip (HP:0000219) | + (12/15) | + (1/5) | + (1/8) | ||
| Microstomia (HP:0000160) | + (1/5) | ||||
| High palate (HP:0000218) | + (10/15) | + (1/5) | + (1/8) | ||
| Pointed chin (HP:0000307) | + (10/15) | ||||
| Low‐set ears (HP:0000369) | + (12/15) | + (1/8) | |||
| Thickened/Overfolded Helices (HP:0000391 & HP:0000396) | + (9/15) | + (1/8) | |||
| Protruding ears (HP:0000411) | + (11/15) | ||||
| None of the above | 0/15 | 6/6 | 0/5 | 4/4 | 0/8 |
| Eyes | |||||
| Myopia (HP:0000545) | + (5/15) | ||||
| Hypermetropia (HP:0000540) | + (2/8) | ||||
| Strabismus (HP:0000486) | + (9/15) | + (1/5) | |||
| Nystagmus (HP:0000639) | + (2/15) | + (1/6) | |||
| None of the above | 5/15 | 5/6 | 4/5 | 4/4 | 7/8 |
| Ears | |||||
| Hearing impairment (HP:0000365) | + (8/15) | + (1/8) | |||
| Chronic otitis media (HP:0000389) | + (9/15) | ||||
| None of the above | (5/15) | 6/6 | 5/5 | 4/4 | 7/8 |
| Skin | |||||
| Hirsutism (HP:0001007) | + (5/15) | + (4/5) | |||
| None of the above | 10/15 | 6/6 | 1/5 | 4/4 | 8/8 |
| Neurological | |||||
| Microcephaly (HP:0000252) | + (10/15) | + (6/6) | + (1/5) | + (4/4) | + (1/8) |
| Hypoplasia of the corpus callosum (HP:0002079) | + (11/15) | + (4/6) | |||
| Hypoplasia of the cerebellar vermis (HP:0001320) | + (5/15) | + (1/8) | |||
| Gait imbalance (HP:0002141) | + (6/15) | + (1/8) | |||
| Gait disturbance (HP:0001288) | + (6/15) | ||||
| Generalized hypotonia (HP:0001290) | + (13/15) | + (1/5) | |||
| Pyramidal signs (HP:0007256) | + (4/6) | ||||
| Seizures (HP:0001250) | + single seizure (2/4) | + (1/8) | |||
| None of the above | 0/15 | 0/6 | 3/5 | 0/4 | 4/8 |
| Musculoskeletal | |||||
| Unusual gluteal crease (No HP code available) | + (12/15) | ||||
| Narrow shoulders (HP:0000774) | + (1/5) | ||||
| Joint hypermobility (HP:0001382) | + (8/15) | + (4/8) | |||
| Digital anomalies (HP:0011297) | + (4/15) | + (4/8) | |||
| Single transverse palmar crease (HP:0000954) | + (1/5) | + (1/8) | |||
| Foot deformity (HP:0001760) | + (2/6) | + (3/5) | |||
| None of the above | 2/15 | 4/6 | 1/5 | 4/4 | 3/8 |
| Other | |||||
| Cardiac abnormalities (HP:0001627) | + (5/15) | + (1/6) | + (1/8) | ||
| Cryptorchidism (HP:0000028) | + (3/15) | + (2/4) | + (1/8) | ||
| Wide intermamillary distance (HP:0006610) | + (1/5) | + (1/8) | |||
| Autistic behaviors (HP:0000729) | + (9/15) | + (2/8) |
Note: Phenotypic comparison of known TAF‐opthies. The table summarizes the published characteristics of patients with known TAF‐opathies (Alazami et al., 2015; Halevy et al., 2012; Hurst et al., 2018; Najmabadi et al., 2011; O'Rawe et al., 2015; Tawamie et al., 2017). The characteristics identified in the current cohort are presented for reference and contain the same data as presented throughout this report.
Abbreviations: + = present; HPO, Human Phenotype Ontology; TAF4, TATA‐binding protein‐associated factor 4; TFIID, transcription Factor IID.
Mild facial dysmorphisms were reported for all probands (8/8), however, no recognizable facial gestalt could be identified. Photographs of selected probands are provided (P1‐4, 6 & 7; Figure 1b–g). Recurrent dysmorphic features, including a prominent nose or nasal bridge (6/8), arched eyebrows (5/8), and prominent midface (2/8) were present.
The phenotypes associated with pLoF variants in TAF4 thus appear nonspecific, consisting of ID, behavioral abnormalities, skeletal anomalies, and variable facial dysmorphologies. We propose to refer to this newly identified disorder as “TAF4‐related NDD” (T4NDD).
Given the central role of TAF4 in the TFIID complex, we investigated if the reported disorders associated with other complex members showed similar phenotypes. Therefore, we searched the current literature for TFIID‐associated disorders (TAF‐opathies) and compared the phenotypes with T4NDD (Table 1). We found congenital disorders associated with four other TFIID complex members: TAF1 (Intellectual developmental disorder, X‐linked syndromic 33; MIM# 300966) (Cheng et al., 2019; Gudmundsson et al., 2019; Hurst et al., 2018; Morton et al., 2020; O'Rawe et al., 2015), TAF2 (Intellectual developmental disorder, autosomal recessive 40; MIM# 615599) (Halevy et al., 2012; Hellman‐Aharony et al., 2013), TAF6 (Alazami‐Yuan syndrome; MIM# 617126) (Alazami et al., 2015; Yuan et al., 2015) and TAF13 (Intellectual developmental disorder, autosomal recessive 60; MIM# 617432) (Tawamie et al., 2017). When comparing the phenotypes of all five reported TAF‐opathies, some commonalities were noted, but many differences as well. First, according to the central role of TFIID in neuronal development, all TAF‐opathies are associated with ID. Three out of five (i.e., TAF1, 4, and 6) TAF‐opathies are reported with some form of craniofacial dysmorphology. We conclude that each TAF‐opathy is associated with ID/NDD but additional shared features could not be identified.
We present a cohort of eight probands each harboring heterozygous, de novo pLoF variants in TAF4. Disruptive variants in TAF4 are expected to be pathogenic since the gene is highly constrained for pLoFs in the general population (Karczewski et al., 2020; Seaby et al., 2022). Furthermore, other TAF‐opathies have been associated with NDDs before. Common phenotypes observed in this cohort are ID, skeletal anomalies, facial dysmorphologies and behavioral abnormalities. The TAF4 related phenotypes however appear nonspecific, exemplified by P2, who is the only individual with normal intellectual ability. We propose to refer to this novel disorder as “TAF4 related NDD” (T4NDD).
Given the limited number of probands included in the present cohort, it is possible that more common characteristics will present when more individuals with pLoF TAF4 variant are identified. As our cohort consist exclusively out of pLoF variants, copy number variants (CNV) leading to disruption or deletion of TAF4 might be relevant for future collection of patient cohorts as well. To provide a more comprehensive description of T4NDD, we encourage anyone with relevant cases to contact us.
Through its role in regulating TFIID function, TAF4 is a central player in cellular differentiation and organismal development. Accordingly, knock‐out of Taf4a in mice is embryonically lethal at E9.5 and the embryos show severe developmental retardation (Langer et al., 2016). This study furthermore showed that Taf4a ‐/‐ embryonic stem cells are unable to differentiate properly, most notably into glutamatergic neurons. These phenotypes are most likely to result from the improper formation of the PIC, which is compensated by the inclusion of the TAF4 paralogue TAF4B. Whereas TAF4 has been reported to promote differentiation, TAF4B acts to retain stemness of pluripotent stem cells (Brunkhorst et al., 2004; Brunkhorst et al., 2005; Gazit et al., 2009; Kazantseva & Palm, 2014; Kazantseva et al., 2013, 2015; Langer et al., 2016; Pijnappel et al., 2013). Within this model, one could hypothesize that imbalances in TAF4/TAF4B gene dosage, such as resulting from disruptive TAF4 variants, will impact on the regulation of cellular differentiation by relative increase of TAF4B incorporation in the TFIID complex.
As an alternative hypothesis, NMD‐related mechanisms resulting from premature stopcodons in TAF4 might shift the expression profile of different TAF4 splicing products. Since different alternative splicing variants of TAF4 have been reported to direct cellular proliferation (Kazantseva et al., 2013, 2015, 2016), such a mechanism is also expected to impact on development. Future studies will have to unravel the mechanism by which TAF4 pLoF variants cause NDD.
Variants in other members of the TFIID complex have been reported previously as monogenic causes for congenital NDDs. We compared reported phenotypes for TAF1, TAF2, TAF6, and TAF13‐related disorders with T4NDD. This revealed no commonalities other than ID, and the TAF4‐related disorder is so far the only disorder that presents with an autosomal dominant inheritance; this is in keeping with its much lower LOEUF score when compared to other known TAF‐opathy genes. Future insights into the patho‐molecular mechanisms underlying the different TAF‐opathies will be needed to explain the differences in inheritance and clinical presentation. In general, the phenotypes observed across and within TAF‐opathies are diffuse and nonspecific. This is likely to be the result of the broad functions of the TFIID complex and expression of specific phenotypes is likely to be influenced by secondary, modifying factors such as genetic variants affecting other TFIID members, other interacting genes or the DNA‐binding sites for the TFIID complex. However, the current limited number of patients with TAF‐opathies precludes determining contributing factors. Therefore, to provide patients with a clearer prognosis, future studies involving larger patient cohorts and in‐depth analyses of geno‐phenotype relationships will be needed to determine the range and severity of phenotypes encompassing T4NDD and other TAF‐opathies.
WEB RESOURCES
Online Mendelian Inheritance in Man: https://omim.org/
Genome Aggregation Database: https://gnomad.broadinstitute.org/
Genematcher: https://genematcher.org/
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Supporting information.
Supporting information.
ACKNOWLEDGEMENTS
We wish to thank the patients and families described in this manuscript. We thank the genetic diagnostic labs for providing technical support and data presentation. We thank the van Haaften Lab for fruitful discussions and manuscript review. This study was supported by funding from the Genetics Department at UMC Utrecht, the Netherlands; the Australian NHMRC Centre for Research Excellence in Neurocognition (Grant Number 1117394); LR, SP, and KÕ are supported by the Estonian Research Council grants PRG471 and MOBTP175. This study was furthermore made possible through access to the data and findings generated by the 100,000 Genomes Project. The 100,000 Genomes Project is managed by Genomics England Limited (a wholly owned company of the Department of Health and Social Care). The 100,000 Genomes Project is funded by the National Institute for Health Research and NHS England. The Wellcome Trust, Cancer Research UK, and the Medical Research Council have also funded research infrastructure. The 100,000 Genomes Project uses data provided by patients and collected by the National Health Service as part of their care and support.
Janssen, B. D. E. , van den Boogaard, M.‐J. H. , Lichtenbelt, K. , Seaby, E. G. , Stals, K. , Ellard, S. , Newbury‐Ecob, R. , Dixit, A. , Roht, L. , Pajusalu, S. , Õunap, K. , Firth, H. V. , Buckley, M. , Wilson, M. , Roscioli, T. , Tidwell, T. , Mao, R. , Ennis, S. , Holwerda, S. J. , van Gassen, k. , van Jaarsveld, R. H. (2022). De novo putative loss‐of‐function variants in TAF4 are associated with a neuro‐developmental disorder. Human Mutation, 43, 1844–1851. 10.1002/humu.24444
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions. Variant data has been submitted to the Global Variome shared LOVD: https://databases.lovd.nl/shared/references/DOI:10.1002/humu.24444
REFERENCES
- Alazami, A. M. , Patel, N. , Shamseldin, H. E. , Anazi, S. , Al‐Dosari, M. S. , Alzahrani, F. , Hijazi, H. , Alshammari, M. , Aldahmesh, M. A. , Salih, M. A. , Faqeih, E. , Alhashem, A. , Bashiri, F. A. , Al‐Owain, M. , Kentab, A. Y. , Sogaty, S. , Al Tala, S. , Temsah, M. H. , Tulbah, M. , … Alkuraya, F. S. (2015). Accelerating novel candidate gene discovery in neurogenetic disorders via whole‐exome sequencing of prescreened multiplex consanguineous families. Cell Reports, 10(2), 148–161. 10.1016/j.celrep.2014.12.015 [DOI] [PubMed] [Google Scholar]
- Bahat, A. , Kedmi, R. , Gazit, K. , Richardo‐Lax, I. , Ainbinder, E. , & Dikstein, R. (2013). TAF4b and TAF4 differentially regulate mouse embryonic stem cells maintenance and proliferation. Genes to Cells, 18(3), 225–237. 10.1111/gtc.12030 [DOI] [PubMed] [Google Scholar]
- Bieniossek, C. , Papai, G. , Schaffitzel, C. , Garzoni, F. , Chaillet, M. , Scheer, E. , Papadopoulos, P. , Tora, L. , Schultz, P. , & Berger, I. (2013). The architecture of human general transcription factor TFIID core complex. Nature, 493(7434), 699–702. 10.1038/nature11791 [DOI] [PubMed] [Google Scholar]
- Brunkhorst, A. , Karlen, M. , Shi, J. , Mikolajczyk, M. , Nelson, M. A. , Metsis, M. , & Hermanson, O. (2005). A specific role for the TFIID subunit TAF4 and RanBPM in neural progenitor differentiation. Molecular and Cellular Neuroscience, 29(2), 250–258. 10.1016/j.mcn.2005.02.015 [DOI] [PubMed] [Google Scholar]
- Brunkhorst, A. , Neuman, T. , Hall, A. , Arenas, E. , Bartfai, T. , Hermanson, O. , & Metsis, M. (2004). Novel isoforms of the TFIID subunit TAF4 modulate nuclear receptor‐mediated transcriptional activity. Biochemical and Biophysical Research Communications, 325(2), 574–579. 10.1016/j.bbrc.2004.10.078 [DOI] [PubMed] [Google Scholar]
- Cheng, H. , Capponi, S. , Wakeling, E. , Marchi, E. , Li, Q. , Zhao, M. , Weng, C. , Piatek, S. G. , Ahlfors, H. , Kleyner, R. , Rope, A. , Lumaka, A. , Lukusa, P. , Devriendt, K. , Vermeesch, J. , Posey, J. E. , Palmer, E. E. , Murray, L. , Leon, E. ,, … Lyon, G. J. (2019). Missense variants in TAF1 and developmental phenotypes: Challenges of determining pathogenicity. Human Mutation, 41, 449–464. 10.1002/humu.23936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazit, K. , Moshonov, S. , Elfakess, R. , Sharon, M. , Mengus, G. , Davidson, I. , & Dikstein, R. (2009). TAF4/4b x TAF12 displays a unique mode of DNA binding and is required for core promoter function of a subset of genes. Journal of Biological Chemistry, 284(39), 26286–26296. 10.1074/jbc.M109.011486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudmundsson, S. , Wilbe, M. , Filipek‐Górniok, B. , Molin, A. M. , Ekvall, S. , Johansson, J. , Allalou, A. , Gylje, H. , Kalscheuer, V. M. , Ledin, J. , Annerén, G. , & Bondeson, M. L. (2019). TAF1, associated with intellectual disability in humans, is essential for embryogenesis and regulates neurodevelopmental processes in zebrafish. Scientific Reports, 9(1), 10730. 10.1038/s41598-019-46632-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halevy, A. , Basel‐Vanagaite, L. , Shuper, A. , Helman, S. , Har‐Zahav, A. , Birk, E. , & Straussberg, R. (2012). Microcephaly‐thin corpus callosum syndrome maps to 8q23.2‐q24.12. Pediatric Neurology, 46(6), 363–368. 10.1016/j.pediatrneurol.2012.03.0 [DOI] [PubMed] [Google Scholar]
- Hellman‐Aharony, S. , Smirin‐Yosef, P. , Halevy, A. , Pasmanik‐Chor, M. , Yeheskel, A. , Har‐Zahav, A. , Maya, I. , Straussberg, R. , Dahary, D. , Haviv, A. , Shohat, M. , & Basel‐Vanagaite, L. (2013). Microcephaly thin corpus callosum intellectual disability syndrome caused by mutated TAF2. Pediatric Neurology, 49(6), 411–416 e411. 10.1016/j.pediatrneurol.2013.07.017 [DOI] [PubMed] [Google Scholar]
- Hurst, S. E. , Liktor‐Busa, E. , Moutal, A. , Parker, S. , Rice, S. , Szelinger, S. , Senner, G. , Hammer, M. F. , Johnstone, L. , Ramsey, K. , Narayanan, V. , Perez‐Miller, S. , Khanna, M. , Dahlin, H. , Lewis, K. , Craig, D. , Wang, E. H. , Khanna, R. , & Nelson, M. A. (2018). A novel variant in TAF1 affects gene expression and is associated with X‐linked TAF1 intellectual disability syndrome. Neuronal Signal, 2(3), NS20180141. 10.1042/NS20180141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski, K. J. , Francioli, L. C. , Tiao, G. , Cummings, B. B. , Alföldi, J. , Wang, Q. , Collins, R. L. , Laricchia, K. M. , Ganna, A. , Birnbaum, D. P. , Gauthier, L. D. , Brand, H. , Solomonson, M. , Watts, N. A. , Rhodes, D. , Singer‐Berk, M. , England, E. M. , Seaby, E. G. , Kosmicki, J. A. , … Salomaa, V. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581(7809), 434–443. 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazantseva, J. , Kivil, A. , Tints, K. , Kazantseva, A. , Neuman, T. , & Palm, K. (2013). Alternative splicing targeting the hTAF4‐TAFH domain of TAF4 represses proliferation and accelerates chondrogenic differentiation of human mesenchymal stem cells. PLoS One, 8(10), e74799. 10.1371/journal.pone.0074799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazantseva, J. , & Palm, K. (2014). Diversity in TAF proteomics: Consequences for cellular differentiation and migration. International Journal of Molecular Sciences, 15(9), 16680–16697. 10.3390/ijms150916680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazantseva, J. , Sadam, H. , Neuman, T. , & Palm, K. (2016). Targeted alternative splicing of TAF4: A new strategy for cell reprogramming. Scientific Reports, 6, 30852. 10.1038/srep30852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazantseva, J. , Tints, K. , Neuman, T. , & Palm, K. (2015). TAF4 controls differentiation of human neural progenitor cells through hTAF4‐TAFH activity. Journal of Molecular Neuroscience, 55(1), 160–166. 10.1007/s12031-014-0295-6 [DOI] [PubMed] [Google Scholar]
- Langer, D. , Martianov, I. , Alpern, D. , Rhinn, M. , Keime, C. , Dollé, P. , Mengus, G. , & Davidson, I. (2016). Essential role of the TFIID subunit TAF4 in murine embryogenesis and embryonic stem cell differentiation. Nature Communications, 7, 11063. 10.1038/ncomms11063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton, S. U. , Agarwal, R. , Madden, J. A. , Genetti, C. A. , Brownstein, C. A. , López‐Giráldez, F. , Choi, J. , Seidman, C. E. , Seidman, J. G. , Lyon, G. J. , & Agrawal, P. B. (2020). Congenital heart defects due to TAF1 missense variants. Circulation Genomic & Precision Medicine, 13(3), e002843. 10.1161/CIRCGEN.119.002843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najmabadi, H. , Hu, H. , Garshasbi, M. , Zemojtel, T. , Abedini, S. S. , Chen, W. , Hosseini, M. , Behjati, F. , Haas, S. , Jamali, P. , Zecha, A. , Mohseni, M. , Püttmann, L. , Vahid, L. N. , Jensen, C. , Moheb, L. A. , Bienek, M. , Larti, F. , Mueller, I. , … Ropers, H. H. (2011). Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature, 478(7367), 57–63. 10.1038/nature10423 [DOI] [PubMed] [Google Scholar]
- O′Rawe, J. A. , Wu, Y. , Dörfel, M. J. , Rope, A. F. , Au, P. Y. B. , Parboosingh, J. S. , Moon, S. , Kousi, M. , Kosma, K. , Smith, C. S. , Tzetis, M. , Schuette, J. L. , Hufnagel, R. B. , Prada, C. E. , Martinez, F. , Orellana, C. , Crain, J. , Caro‐Llopis, A. , Oltra, S. ,, … Lyon, G. J. (2015). TAF1 variants are associated with dysmorphic features, intellectual disability, and neurological manifestations. American Journal of Human Genetics, 97(6), 922–932. 10.1016/j.ajhg.2015.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, A. B. , Louder, R. K. , Greber, B. J. , Grünberg, S. , Luo, J. , Fang, J. , Liu, Y. , Ranish, J. , Hahn, S. , & Nogales, E. (2018). Structure of human TFIID and mechanism of TBP loading onto promoter DNA. Science , 362(6421). 10.1126/science.aau8872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pijnappel, W. W. , Esch, D. , Baltissen, M. P. , Wu, G. , Mischerikow, N. , Bergsma, A. J. , van der Wal, E. , Han, D. W. , Bruch, H. v , Moritz, S. , Lijnzaad, P. , Altelaar, A. F. , Sameith, K. , Zaehres, H. , Heck, A. J. , Holstege, F. C. , Schöler, H. R. , & Timmers, H. T. (2013). A central role for TFIID in the pluripotent transcription circuitry. Nature, 495(7442), 516–519. 10.1038/nature11970 [DOI] [PubMed] [Google Scholar]
- Sanders, S. L. , Garbett, K. A. , & Weil, P. A. (2002). Molecular characterization of Saccharomyces cerevisiae TFIID. Molecular and Cellular Biology, 22(16), 6000–6013. 10.1128/MCB.22.16.6000-6013.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaby, E. G. , Smedley, D. , Taylor Tavares, A. L. , Brittain, H. , van Jaarsveld, R. H. , Baralle, D. , Rehm, H. L. , O′Donnell‐Luria, A. , & Ennis, S. (2022). A gene‐to‐patient approach uplifts novel disease gene discovery and identifies 18 putative novel disease genes. Genetics in Medicine . 10.1016/j.gim.2022.04.019 [DOI] [PubMed]
- Sobreira, N. , Schiettecatte, F. , Valle, D. , & Hamosh, A. (2015). GeneMatcher: A matching tool for connecting investigators with an interest in the same gene. Human Mutation, 36(10), 928–930. 10.1002/humu.22844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawamie, H. , Martianov, I. , Wohlfahrt, N. , Buchert, R. , Mengus, G. , Uebe, S. , Janiri, L. , Hirsch, F. W. , Schumacher, J. , Ferrazzi, F. , Sticht, H. , Reis, A. , Davidson, I. , Colombo, R. , & Abou Jamra, R. (2017). Hypomorphic pathogenic variants in TAF13 are associated with autosomal‐recessive intellectual disability and microcephaly. The American Journal of Human Genetics, 100(3), 555–561. 10.1016/j.ajhg.2017.01.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuault, S. , Gangloff, Y. G. , Kirchner, J. , Sanders, S. , Werten, S. , Romier, C. , Weil, P. A. , & Davidson, I. (2002). Functional analysis of the TFIID‐specific yeast TAF4 (yTAF(II)48) reveals an unexpected organization of its histone‐fold domain. Journal of Biological Chemistry, 277(47), 45510–45517. 10.1074/jbc.M206556200 [DOI] [PubMed] [Google Scholar]
- Wang, X. , Truckses, D. M. , Takada, S. , Matsumura, T. , Tanese, N. , & Jacobson, R. H. (2007). Conserved region I of human coactivator TAF4 binds to a short hydrophobic motif present in transcriptional regulators. Proceedings of the National Academy of Sciences of the United States of America, 104(19), 7839–7844. 10.1073/pnas.0608570104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werten, S. , Mitschler, A. , Romier, C. , Gangloff, Y. G. , Thuault, S. , Davidson, I. , & Moras, D. (2002). Crystal structure of a subcomplex of human transcription factor TFIID formed by TATA binding protein‐associated factors hTAF4 (hTAF(II)135) and hTAF12 (hTAF(II)20. Journal of Biological Chemistry, 277(47), 45502–45509. 10.1074/jbc.M206587200 [DOI] [PubMed] [Google Scholar]
- Wright, K. J. , Marr, M. T., 2nd , & Tjian, R. (2006). TAF4 nucleates a core subcomplex of TFIID and mediates activated transcription from a TATA‐less promoter. Proceedings of the National Academy of Sciences of the United States of America, 103(33), 12347–12352. 10.1073/pnas.0605499103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, B. , Pehlivan, D. , Karaca, E. , Patel, N. , Charng, W. L. , Gambin, T. , Gonzaga‐Jauregui, C. , Sutton, V. R. , Yesil, G. , Bozdogan, S. T. , Tos, T. , Koparir, A. , Koparir, E. , Beck, C. R. , Gu, S. , Aslan, H. , Yuregir, O. O. , Al Rubeaan, K. , Alnaqeb, D. , … Lupski, J. R. (2015). Global transcriptional disturbances underlie cornelia de Lange syndrome and related phenotypes. Journal of Clinical Investigation, 125(2), 636–651. 10.1172/JCI77435 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions. Variant data has been submitted to the Global Variome shared LOVD: https://databases.lovd.nl/shared/references/DOI:10.1002/humu.24444