

Abstract
Ocean ecosystems are inhabited by a diverse set of viruses that impact microbial mortality and evolution. However, the distribution and abundances of specific viral lineages, particularly those from the large bank of rare viruses, remains largely unknown. Here, we assessed the diversity and abundance of the TIM5‐like cyanophages. The sequencing of three new TIM5‐like cyanophage genomes and environmental amplicons of a signature gene from the Red Sea revealed highly conserved gene content and sequence similarity. We adapted the polony method, a solid‐phase polymerase chain reaction assay, to quantify TIM5‐like cyanophages during three 2000 km expeditions in the Pacific Ocean and four annual cycles in the Red Sea. TIM5‐like cyanophages were widespread, detected at all latitudes and seasons surveyed throughout the photic zone. Yet they were generally rare, ranging between <100 and 4000 viruses·ml−1. Occasional peaks in abundance of 10‐ to 100‐fold were observed, reaching 71,000 viruses·ml−1. These peaks were ephemeral and seasonally variable in the Red Sea. Infection levels, quantified during one such peak, were very low. These characteristics of low diversity and abundance, as well as variable outbreaks, distinguishes the TIM5‐like lineage from other major cyanophage lineages and illuminates that rare virus lineages can be persistent and widespread in the oceans.
INTRODUCTION
Global surveys of ocean ecosystems have revealed that viruses influence the mortality and population dynamics of microbial communities and harbour vast genomic diversity (Breitbart et al., 2018; Rohwer & Thurber, 2009; Suttle, 2005; Zimmerman et al., 2020). Viral assemblages have repeatedly been found to be characterized by long‐term stability and distinctive community composition in which many viral taxa are relatively rare with only a few dominant taxa (Angly et al., 2006; Aylward et al., 2017; Breitbart et al., 2002; Brum et al., 2015; Culley et al., 2006; Goldsmith et al., 2015; Ignacio‐Espinoza et al., 2020). As such, the persistence of rare taxa appears paradoxical considering that viruses usually have narrow host ranges, are host specific (Flores et al., 2011; Koskella & Meaden, 2013; Wang & Chen, 2008), decay rapidly, particularly in sunlit waters (Bongiorni et al., 2005; Heldal & Bratbak, 1991; Hewson et al., 2012; Noble & Fuhrman, 1997; Suttle & Chen, 1992), and are faced with diverse host populations wielding numerous resistance mechanisms (Avrani et al., 2011; Kashtan et al., 2014; Zborowsky & Lindell, 2019). However, the abundances and distribution patterns of most rare viral taxa are unknown but essential to understanding their ecological dynamics.
Cyanophages, viruses that infect cyanobacteria, particularly the globally important unicellular marine phototrophs, Prochlorococcus and Synechococcus (Biller et al., 2015; Mann, 2003) are important members of marine virioplankton communities (Baran et al., 2018; Gainer et al., 2017; Parsons et al., 2012). Through infecting and lysing their hosts, cyanophages can substantially impact the abundances, biogeography, and diversification of picocyanobacteria (Ahlgren et al., 2019; Carlson et al., 2022; Garza & Suttle, 1998; Marston et al., 2012; Proctor & Fuhrman, 1990). They are a diverse group of tailed dsDNA viruses that belong to the order Caudovirales and are divided into three families based on tail morphology: the Myoviridae with long contractile tails, the Podoviridae with short tails and the Siphoviridae with long non‐contractile tails (Mann, 2003; Sullivan et al., 2003; Suttle & Chan, 1993; Wang & Chen, 2008; Waterbury & Valois, 1993; Wilson et al., 1993). A cyanophage genus within each of the Myoviridae and Podoviridae families, the T4‐like cyanophages and T7‐like cyanophages, respectively (Lavigne et al., 2008, 2009) are recognized as important components of virioplankton communities and are abundant throughout the euphotic zone (Baran et al., 2018; Carlson et al., 2022; Goldin et al., 2020; Maidanik et al., 2022; Marston & Sallee, 2003; Matteson et al., 2013; Mruwat et al., 2021; Suttle & Chan, 1993; Waterbury & Valois, 1993). However, several cyanophage lineages, groups of genetically related viruses derived from a common ancestor, including lambdoid‐like siphoviruses, non‐T4‐like myoviruses, and non‐T7‐like podoviruses have been discovered through isolation and metagenomic surveys (Chenard et al., 2015; Flores‐Uribe et al., 2019; Nishimura et al., 2017; Sabehi et al., 2012; Sullivan et al., 2009), but their distribution and contributions to cyanobacterial ecology have yet to be evaluated.
More than a decade ago, viruses belonging to a new lineage within the Myoviridae family, the TIM5‐like cyanophages, were isolated from the Gulf of Aqaba, Red Sea (Sabehi et al., 2012). These TIM5‐like cyanophages are lytic and have a narrow host range being found to infect only two marine Synechococcus hosts out of 17 cyanobacterial strains tested (Dekel‐Bird et al., 2015; Sabehi et al., 2012). The genome of the type strain, cyanophage S‐TIM5, carries a distinctive set of structural proteins not found in other phage types and encodes a unique assortment of replication and DNA metabolism genes, including a mitochondrial‐like DNA polymerase gene (Sabehi et al., 2012), and is thus a genus distinct from other cyanophages, and was recently named Aurunviruses (Schoch et al., 2020; https://www.genome.jp/virushostdb/1137745). However, similar to other cyanophages, the S‐TIM5 phage has a number of bacterial‐like metabolism genes (Sabehi et al., 2012) including phosphate acquisition (pstS), photosynthesis (psbA, psbD, PTOX), and carbon metabolism genes (talC, gnd, zwf). Initial reports suggested that TIM5‐like cyanophages may be abundant as over 100 infective phages belonging to this genus were enriched from Red Sea samples collected over the years 2008–2010 (Dekel‐Bird et al., 2015; Sabehi et al., 2012). Additionally, during the Global Ocean Sampling Expedition (Yooseph et al., 2007), sequences similar to TIM5‐like phages were found in metagenomes collected in other ocean basins suggesting this cyanophage type may be widespread (Sabehi et al., 2012). However, TIM5‐like cyanophage genome sequences are limited, as are data on their abundance and distribution, precluding an understanding of their dynamics and impact on the picocyanobacteria.
Here, we analyse of the genomic diversity of three new TIM5‐like cyanophages from the Red Sea. We also adapt a single‐molecule solid‐phase PCR‐based polony method for the quantification of diverse environmental virus populations, the collective of viruses belonging to a specific lineage in a given area, and the extent of infection to investigate the potential impact of TIM5‐like cyanophages on the picocyanobacteria. We then used this method to assess the diversity, abundance, and dynamics of TIM5‐like cyanophage populations across diverse conditions in two marine environments: seasonally over 4 years in the Gulf of Aqaba, Red Sea and spatially across transects in the North Pacific Ocean from cruises in spring or early summer in three subsequent years. We found that this virus genus is widespread in the oceans, yet has limited genomic and allelic diversity, at least in the Red Sea. Furthermore, TIM5‐like cyanophages are typically rare but exhibit peaks that are locally or seasonally ephemeral. This illuminates the dynamics of a virus genus representative of the large bank of rare viruses that exist in the oceans.
EXPERIMENTAL PROCEDURES
Phage isolation and sequencing
Three new TIM5‐like cyanophages, S‐TIM54, S‐TIM61 and S‐TIM66, were isolated from the Gulf of Aqaba, Red Sea in March or August 2009 (Table 1). The 0.22 μm filtrate of seawater samples was serially diluted and plated on Synechococcus sp. strains WH8102 and CC9605 to obtain spatially separated plaques as described previously (Dekel‐Bird et al., 2015). After identification as TIM5‐like phages by PCR (Dekel‐Bird et al., 2015), the phages were plaque purified twice and then propagated on Synechococcus sp. strain WH8102. The host range of the S‐TIM61 and S‐TIM66 phages was determined on 6 Prochlorococcus and 11 Synechococcus strains (Sabehi et al., 2012). Host range experiments were conducted in liquid culture in 96‐well plates with three wells per phage‐host pair and repeated twice. Infection was considered successful only if lysis was observed in both experiments.
TABLE 1.
Isolation and genomic characterization of TIM5‐like cyanophages
| Isolate | Collection location | Collection date | Synechococcus host strain of isolation | Genome length (bp) | % GC | # ORFs | # tRNAs |
|---|---|---|---|---|---|---|---|
| S‐TIM5 | Red Sea, IUI pier, 0 m | October 2008 | WH8102 | 172,474 | 41 | 213 | 10 |
| S‐TIM54 | Red Sea, St. A 20 m | March 2009 | WH8102 | 179,860 | 42 | 212 | 10 |
| S‐TIM61 | Red Sea, St. A 20 m | August 2009 | WH8102 | 171,618 | 41 | 210 | 10 |
| S‐TIM66 | Red Sea, IUI pier, 0 m | August 2009 | CC9605 | 172,357 | 41 | 213 | 10 |
To generate DNA for genome sequencing, cell debris from lysates were removed by centrifugation at 13,131 × g at 21°C for 15 min, then filtered through a 0.2 μm filter (Nalgene). The filtered lysate was concentrated using Centricon Plus 70 centrifugal filters (100 kDa, Millipore). Phage DNA was extracted from 0.9 ml of concentrated lysate at a concentration of approximately 109 viruses·ml−1. The lysate was treated with RNase A and DNase I by adding 2.7 μl of the following solution: 300 mM NaCl, 100 mM Tris‐Cl, pH 7.5, 10 mM EDTA, 0.2 mg·ml−1 BSA, 20 mg·ml−1 RNase A, 6 mg·ml−1 DNase I, and incubating at 37°C for 30 min. The lysate was then treated with Proteinase K by adding 23 μl of 20% SDS and 9 μl of 10 mg/ml Proteinase K and incubating for 30 min at 37°C. DNA was then extracted twice with equal volumes of a 1:1 phenol (Tris saturated, pH 8.0): chloroform mix, and twice with chloroform. DNA was precipitated by adding 45 μl of 3 M Na‐acetate (pH 5.2) and 0.5 ml isopropanol to 0.5 ml of the aqueous phase and allowed to precipitate at room temperature for 20 min. The mixture was then spun at ≥15,000 × g for 30 min at 4°C. The pellet was washed twice with 70% ethanol and resuspended in 30 μl of Tris‐Cl pH 8.5. DNA libraries were prepared using the Illumina TruSeq LT kit for S‐TIM54, S‐TIM61, S‐TIM66 and Illumina TruSeq Nano kit for the resequencing of S‐TIM5 (see subsection ‘Genome analysis’). Genomes were sequenced on the Illumina MiSeq platform (Technion Genome Centre, Israel) as 250 bp paired‐end (PE) reads for three new cyanophages and as 150 bp PE reads for S‐TIM5. Fastp v0.23.2 (Chen et al., 2018) was used to remove adapters and indexes and perform sequence quality control.
The S‐TIM5 genome was additionally sequenced using Nanopore technology (Oxford Nanopore Technologies, ONT, Oxford, UK) following the SQK‐NBD112.24 v. NBE_9134_v112_revF_01Dec2021 protocol to verify its genome structure. The sequencing library was prepared using the NEB Blunt/TA Ligase Master Mix, NEBNext FFPE Repair Mix, NEBNext Ultra II End repair/dA‐tailing Module, NEBNext Quick Ligation Module (New England Biolabs, USA), and Native Barcoding Kit 24 (SQK‐NBD112.24, ONT, Oxford, UK) and loaded onto the MinION flow cell FLO‐MIN112 (R10.4 SpotON). Adapter sequences were trimmed using Porechop (v0.2.3) and low‐quality reads were removed using NanoFilt (v.2.8.0).
Genome analysis
Quality controlled S‐TIM54, S‐TIM61 and S‐TIM66 short‐read sequences were first digitally normalized (Brown et al., 2012) then assembled de novo using velvet‐1.2.10 and velvetoptimiser‐1.2.10. Resulting assemblies had 60–70× coverage. A 10 kb region was identified in these genomes that were not found in the original assembly of the S‐TIM5 phage genome (JQ245707.1). After PCR verification that this region was present in S‐TIM5, the S‐TIM5 genome was resequenced (see subsection ‘Phage isolation and sequencing’) and then assembled using Unicycler v0.4.7 (Wick et al., 2017) with an average genome coverage by Illumina reads of ×130 and by Nanopore reads of ×397. For the genome comparison, query coverage was calculated with blastn (e‐value ≤ 1e‐5) and average nucleotide identity (ANI) was obtained using fastANI (‐‐fragLen 1000; https://github.com/ParBLiSS/FastANI).
Annotation of the re‐sequenced S‐TIM5 genome was based on the previously submitted and manually curated gene repertoire of the S‐TIM5 phage (Sabehi et al., 2012). Gene calling for the three new cyanophage genomes and the additional 10 kb region of S‐TIM5 was performed by Prodigal 2.6.3 with flags ‘‐c ‐m ‐g 1’ (Hyatt et al., 2010). The predicted open reading frames (ORFs) were annotated using blastp (e‐value ≤1e‐5, query coverage >50%) against the non‐redundant protein NCBI database (13 March 2022) and HMMER 3.3.2 hmmscan (options: ‐‐notextw, e‐value ≤ 1e‐5) against the Pfam database (November 2021) and Prokaryotic Virus Remote Homologous Groups database v3 (July 2021) (Terzian et al., 2021). tRNAs were identified by Aragorn v1.2.38 (Laslett & Canback, 2004) with flags ‘‐l ‐gc1’. The predicted ORFs of the four TIM5‐like cyanophages were compared all‐to‐all using blastp and divided into ORF families based on the pairwise homology found (e‐value ≤ 1e‐5, >50% of the amino acid sequence coverage). Genomic comparison and gene calling results were visualized with an R package ‘gggenomes’ (https://github.com/thackl/gggenomes).
For phylogenic analyses, DNA polymerase nucleotide sequences were aligned using muscle v3.8.1551 (Edgar, 2004). Sequences were between 319 and 339 nt in length with the exception of 5 polony amplicons that were 211 nt after PCR primer removal. A maximum likelihood tree was inferred using IQ‐TREE v2.0.3 (Minh et al., 2020) using the empirically determined best‐fit model K2P + G4 with 1000 iterations of ultrafast bootstrapping. The tree was visualized using the interactive Tree of Life (Letunic & Bork, 2021).
Viral metagenomes that were collected in parallel with polony samples in the Red Sea in 2015 and 2016, in the North Pacific Ocean in the 2016 and 2017 cruises, and in the North Pacific Subtropical Gyre in 2015 (Aylward et al., 2017) were used to assess the diversity of TIM5‐like cyanophages in the Pacific Ocean. Sequence reads were mapped to the S‐TIM5 phage DNA polymerase gene using bbmap v38 (Bushnell et al., 2017). Percent identity was calculated for each read based on the number of shared nucleotide positions relative to the reference S‐TIM5 phage sequence (Table S1).
Preparation of S‐TIM5 cyanophage lysate for use in the polony method
Adaptation of the polony method for quantification of TIM5‐like cyanophages was carried out with the S‐TIM5 cyanophage. S‐TIM5 lysates were prepared by growing the phage on Synechococcus sp. strain WH8102. Cell debris was removed by filtration through 0.22 μm filters. Filtrates were collected and stored at 4°C.
S‐TIM5 lysates at 2 × 107 phage·ml−1 were treated with DNase I to degrade free S‐TIM5 phage DNA present in lysates. DNase digestion was carried out by incubation with 5 Kunitz units·ml−1 enzyme at 37°C for 1 h. Under these conditions, naked DNA was completely digested while encapsidated viral DNA remained intact (Baran et al., 2018). The enzyme was inactivated with 50 mM EDTA (pH 8.0). DNase‐treated lysates were stored at −80°C for polony assays (see subsection ‘The polony method’). Quantification of S‐TIM5 phages in the lysates was carried out using the SYBR® Green I virus‐like particle (VLP) enumeration method (Patel et al., 2007).
Sample collection and environmental conditions
Red Sea samples were collected at approximately monthly intervals in the Gulf of Aqaba, at Station A (29°28′ N, 34°55′ E) aboard the RV Sam Rothberg from February 2013 to December 2016. North Pacific Ocean samples were collected along a transect from the coastal Eastern Pacific Ocean off Oregon (44°2′ N, 127°00′ W) to the central subtropical gyre near Hawai'i (24°32′ N, −153°7′ W) from 21 March to 29 March 2015 aboard the RV Thomas G. Thompson and from Hawai'i north along the 158° W longitudinal line between 23–43° N in 20 April– and 4 May 2016 aboard the RV Ka'imikai O Kanaloa and from 26 Mayto 13 June 2017 aboard the RV Marcus G. Langseth (Carlson et al., 2022). Water‐column salinity, temperature, and in situ chlorophyll fluorescence levels were determined from a Sea‐Bird Scientific CTD.
Seawater samples were collected from Niskin® bottles attached to a CTD‐rosette. For virus abundance analyses, seawater samples were filtered through a 0.22 μm filter (Millipore, Durapore) and stored at −80°C. To assess percent infection, seawater samples were filtered through a 20 μm mesh, amended with glutaraldehyde at a final concentration of 0.1%, incubated for 30 min in the dark, frozen in liquid nitrogen and stored at −80°C.
Enumeration of Synechococcus was performed using a flow cytometer as previously described (Carlson et al., 2022; Maidanik et al., 2022). Briefly, seawater samples were fixed with 0.125% glutaraldehyde, frozen in liquid nitrogen, and stored at −80°C until analysis. The samples were analysed with an LSR‐II flow cytometer (BD Biosciences) for Red Sea samples or a continuous underway system, Seaflow, for Pacific Ocean samples. For discrete samples, 1 μm diameter yellow‐green microspheres (Fluoresbrite) were added as an internal reference for size and fluorescence intensity. Abundances of Synechococcus were determined based on their orange fluorescence of phycoerythrin and size based on forward scatter. Chlorophyll‐a concentrations were determined after extraction with cold acetone (90%) using a Turner TD700 fluorometer (Maidanik et al., 2022).
Ancillary data for the North Pacific are available through the Simons Collaborative Marine Atlas Project (Ashkezari et al., 2021) at https://simonscmap.com/catalog/cruises/ in the directories KM1502, KOK1606, MGL1704 and for the Gulf of Aqaba, Red Sea through the National Monitoring Program in Eilat at http://www.meteo-tech.co.il/EilatYam_data/ey_data.asp.
The polony method
Cyanophage abundances were quantified using the polony method as previously described for T7‐like cyanophages (Baran et al., 2018). This protocol was adapted for use with TIM5‐like cyanophages. Briefly, S‐TIM5 phage lysate or 0.2 μm filtrate of seawater samples (up to 2.3 μl per polony gel) were mixed with 12% acrylamide/bisacrylamide solution and 20 μM of the 5′‐acrydite‐modified forward primer (for sequence, see subsection ‘Primers and probes for polony analyses'). The acrydite modification of the primer covalently binds to polyacrylamide and serves to anchor the primers and consequently the resulting viral amplicons to the gel. Gels (11.6 μl each) were poured into 40 μm deep oval wells printed on glass microscope slides (Thermo Fisher Scientific) treated with Bind Silane. Following polymerization under argon gas, gels were washed to remove residual polymerizing reagents. Next, PCR reagents including Taq Polymerase reaction buffer, 10 μM reverse unmodified primer (for sequence, see subsection ‘Primers and probes for polony analyses'), 0.25 mM dNTPs, and 6.8 units of Jumpstart Taq Polymerase (Sigma‐Aldrich) were diffused into the gels. Slide PCR was then performed in a twin‐tower slide thermocycler (BioRad). Thermal cycling started with a heat step that facilitated viral DNA accessibility by disrupting capsids (94°C for 4 min) followed by 50 amplification cycles of denaturation at 94°C for 45 s, annealing at 50°C for 45 s, and extension 72°C for 2 m, and finally at 72°C for 6 m extension step.
For detection of polonies, hybridization was carried out on slides by denaturing amplicons in 70% formamide with SSC buffer (150 mM NaCl, 15 mM sodium citrate) for 15 min at 70°C. Unbound template was removed by washing with buffer E (10 mM Tris (pH 7.5), 50 mM KCl, 2 mM EDTA (pH 8.0), 0.01% Triton X‐100). The gel‐anchored DNA strand was then hybridized with a combination of two Cy5 fluorescently labelled probes (for sequence, see subsection ‘Primers and probes for polony analyses’) located internal to the primer sequences, at 0.6 μM each for 30 min at 42°C. Slides were washed with buffer E and then scanned on a GenePix 4000B microarray scanner using the 635 nm laser. Polonies were enumerated using the ImageJ v1.0 software. All polony experiments were carried out at least in technical duplicates and included a positive control and a ‘no template’ control of 0.02 μm‐filtered seawater. Analyses were repeated when >20% variation existed between technical replicates.
In some cases, phages needed to be concentrated prior to polony assays to increase accuracy of quantification. To do this, seawater samples were 40‐ or 200‐fold concentrated using the iron chloride (FeCl3) flocculation method (John et al., 2011) that we slightly modified to accommodate precipitation in small volumes and to optimize the iron resuspension buffer as described previously (Baran et al., 2018). Iron chloride was added to 0.2 μm filtrates of the samples at a final concentration of 0.18 mM. Samples were then incubated for at least 1 h at room temperature or for 2 h at 4°C. Flocculated phages were precipitated by centrifugation at 17,000 × g at 10°C for 10 min. The flocculants were dissolved in a solution containing 0.125 M Tris, 0.1 M Na2EDTA and 0.125 M oxalic acid, pH 6.0 and stored at −80°C. Flocculation was performed on environmental samples that had been thawed only once and were analysed immediately after concentration. The efficiency of concentration was determined using the polony method, by enumeration of TIM5‐like phages in a seawater sample collected on 6 June 2016 at 20 m depth, before and after 40‐fold concentration. Efficiency of concentration of TIM5‐like phages in seawater samples was 0.82 ± 0.066 (n = 12). This allowed accurate detection of as few as 40 phages·ml−1 seawater (with 10 polonies per gel).
For quantification of infection levels, Prochlorococcus and Synechococcus cells were sorted into separate tubes based on their autofluorescence and forward angle light scatter using a BD Influx cytometer equipped with a small particle detector and 488 and 457 nm lasers. The concentration of cells was volumetrically quantified on the cytometer after sorting. Thousands of sorted cells were screened for intracellular viral DNA by applying the iPolony procedures (Mruwat et al., 2021) with the TIM5‐like primers and probe. In this procedure, known numbers of sorted cells are added to gels and serve as the template rather than free virus samples. Thermal cycling was initiated with 15 min at 94°C to lyse cells followed by 25 min at 25°C to allow entry of PCR reagents into the gel and followed by standard polony protocols described above. Percent infection was calculated by dividing the number of polonies observed by the number of input cells.
Primers and probes for polony analyses
Degenerate primers specific for the DNA polymerase (DNApol) gene of the TIM5‐like cyanophage lineage were previously designed using the S‐TIM5 cyanophage sequence and homologous sequences from environmental databases (Sabehi et al., 2012). To account for limited knowledge of TIM5‐like diversity and in an attempt to detect more diverse members of this genus if they exist, primer and probe sequences were designed using both cultured and environmental sequences and then back translated to amino acid sequences. Degeneracies in the nucleotide sequences of the primers were added based on all possible nucleotide combinations corresponding to each codon. In this study, the same 18 nt reverse primer as used in the study by Sabehi et al. (2012) was used while the forward primer was shifted 4 nucleotides downstream and shortened to 18 nucleotides to reduce degeneracy. Degeneracies of the forward primer, DPOL2_2F (YWYGCNTAYAAYGARATG), and the reverse primer, DPOL2_R (CCANGCRTTNGCNWSNGG), were 256 and 2048, respectively. The amplicon size was 372 bp. For detection of TIM5‐like polonies, we designed a combination of two degenerate probes that target a conserved locus that encompasses all previously reported TIM5‐like sequences and TIM5‐like phage homologues in metagenomes but excludes siphoviruses similar to those that infect Acaryochloris (Dekel‐Bird et al., 2015; Sabehi et al., 2012): 5′‐Cy5‐TIM5‐C(d + 2i) (GAYTGiATRCACCAiTTRTT) and 5′‐Cy5‐TIM5‐V(d + 2i) (GAYTGRATiACCCAiTTRTT). The inclusion of inosines reduced the degeneracy in the position of the probe from the observed 96 to 16 for the two probes combined (8 for each probe). The effective degeneracy that is covered by these combined probes is 248. The probes were complementary to an internal region within the 5′‐acrydite‐anchored strand of the amplicon to prevent detection of nonspecific polonies and primer dimers. Primers and probes were purified by high‐performance liquid chromatography.
Polony picking and sequencing
We picked polonies from samples collected in the Red Sea on 17 June 2013 at three depths of 20, 60, and 80 m. The picking procedure for TIM5‐like polonies was similar to that described previously for T7‐like polonies (Baran et al., 2018) but used primers and PCR conditions for TIM5‐like cyanophages. A piece of gel containing a single, well‐separated polony was excised and placed into 30 μl of PCR reaction mixture containing DPOL2_2F as a forward primer and DPOL2_R as a reverse primer at 2 μM each and 2 units of BIO‐X‐ACT Short DNA polymerase (Bioline). The PCR programme started with a denaturation step of 5 min at 95°C followed by 40 cycles of denaturation at 95°C for 45 s, annealing at 50°C or 54°C for 45 s and elongation at 70°C for 1 min and finished with an elongation step at 70C for 5 min. In some cases, semi‐nested PCR was carried out by replacing the DPOL2_2F primer with the primer sn_F (AAYAAYTGGKKNATYCARWS), an additional forward primer that was complementary to combined probe sequences, resulting in a 249 bp amplicon. The annealing temperature in the PCR programme was 45°C. The resulting 372 or 249 bp amplicons were extracted from agarose gels, cloned using the PCRII TOPO TA cloning kit (Invitrogen) and sequenced.
RESULTS
Diversity of TIM5‐like cyanophages
To begin assessing the genomic diversity within the TIM5‐like cyanophage genus, we isolated three new TIM5‐like cyanophages from different seasons in the Gulf of Aqaba, Red Sea (Table 1). The cyanophage S‐TIM54 was isolated from a sample collected during the onset of stratification after the annual mixing event in March 2009 while the cyanophages S‐TIM61 and S‐TIM66 were isolated from two different sites in August 2009 near the peak of water column stratification (Carlson et al., 2014). Both S‐TIM54 and S‐TIM61 were isolated on Synechococcus sp. strain WH8102, whereas S‐TIM66 was isolated on Synechococcus sp. strain CC9605. S‐TIM61 and S‐TIM66 were able to infect both Synechococcus sp. strains CC9605 and WH8102 but none of the other 17 picocyanobacterial strains tested (see Experimental Procedures).
The genome sequences of all three TIM5‐like cyanophage isolates revealed that they were highly similar (Figure 1). All genomes had ~41% GC content and were between 171 and 180 kbp in length (Table 1). All genomes had >87% average nucleotide identity to the S‐TIM5 genome (isolated in October 2008 in the Red Sea; Sabehi et al., 2012) with S‐TIM61 and S‐TIM66 having >99% identity to S‐TIM5, and to each other, across 98% of their genomes. The TIM5‐like phages encode between 210 and 213 predicted ORFs (Table 1, Figure 1A). The vast majority of the cumulative ORFs (801/848) and ORF families (181/205) were shared by all TIM5‐like cyanophages (Figure 1B). Of the small number of genes that were unique to the S‐TIM54 phage (11 genes) or the S‐TIM66 phage (1 gene), most had no functional annotation, with the exception of a DNA endonuclease V protein and a Fe2OG dioxygenase domain‐containing protein found in the S‐TIM54 genome. The S‐TIM54 genome also lacks 10 genes shared by the other 3 genomes, including the phosphate binding protein, pstS. Thus, TIM5‐like cyanophages isolated months apart in different environmental conditions on different Synechococcus hosts reveal limited diversity in the Red Sea.
FIGURE 1.
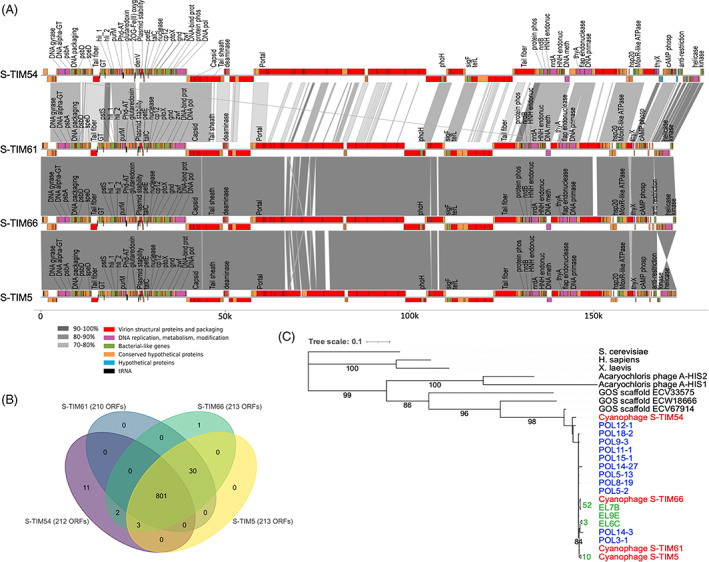
Genomic diversity of TIM5‐like cyanophages. (A) Genome alignment of four TIM5‐like cyanophages. Genome maps show predicted open reading frames (ORFs) and their functional category by colour. ORFs that are homologous to known proteins are labelled. Alignment blocks are shaded by percent nucleotide identity. The hypothetical genes (in blue) are those unique to a single phage genome. DNA pol, DNA polymerase; DNA meth, DNA methylase; DNA‐bind prot, DNA‐binding protein; cAMP phosph, cAMP phosphodiesterase; GT, glucosyl transferase; DNA alpha‐GT, DNA alpha glucosyl transferase; Phd‐AT, Phd‐like antitoxin; protein phos, protein phosphatase; HNH endonuc, HNH endonuclease; flat endonuc, flat endonuclease; 2OG‐Fe(II) oxyg, 2OG‐Fe(II) dioxygenase domain‐containing protein. (B) Venn diagram of shared ORFs by TIM5‐like cyanophages. Numbers are the cumulative shared ORFs out of a total of 848 ORFs in the 4 phage genomes. (C) Maximum likelihood phylogenetic tree inferred from 345 positions of an alignment of DNA polymerase nucleotide sequences. TIM5‐like cyanophage sequences from phage isolates (red), polony amplicons from natural Red Sea communities (blue), and enriched TIM5‐like phage lysates from Dekel‐Bird et al. (2015) (green). Note that numbers in green indicate the number of identical sequences from enriched lysates within each collapsed branch. Nodes with support of >75% are shown with bootstrap values.
To assess additional diversity beyond that found in the four genomes, we analysed sequences of TIM5‐like cyanophage DNA polymerase gene homologues in viromes collected from the North Pacific Subtropical Gyre in 2015 (Aylward et al., 2017), along latitudinal transects between the subtropical gyre and the inter‐gyre transition zone in the North Pacific Ocean in 2016 and 2017 and from the Gulf of Aqaba in the Red Sea in 2015 and 2016. In the Pacific Ocean, only 10 sequence reads (44–56 nt long) were found that mapped to the DNA polymerase gene and were between 34% and 71% identical to the S‐TIM5 phage sequence (Table S1). It is unclear whether sequence identities at such levels belong to cyanophages or other bacterial viruses (see Discussion) (Sabehi et al., 2012). In the Red Sea, all 657 sequence reads that mapped to the DNA polymerase gene were >80% identical to S‐TIM5, 94% had three or fewer mismatches, and only 1% were <90% identical to the S‐TIM5 sequence. This suggests relatively low allelic diversity in the DNA polymerase gene in TIM5‐like cyanophages in the Red Sea while the lack of sequence data for TIM5‐like cyanophages in the Pacific Ocean precludes the ability to address TIM5‐like diversity in that region.
Adaptation of the polony method for TIM5‐like cyanophages
Next, we wanted to assess the distribution and seasonal patterns of TIM5‐like cyanophages and the extent to which they infected picocyanobacteria in the environment. We used the polony method that we previously developed for T7‐like (Baran et al., 2018) and T4‐like (Goldin et al., 2020) cyanophages that allows quantification of viral populations using degenerate primers and probes to capture the diversity within a target viral lineage. We adapted this method for TIM5‐like cyanophages by targeting the mitochondrial‐like DNA polymerase gene, a signature gene for this group (Dekel‐Bird et al., 2015; Sabehi et al., 2012).
In the polony method, viruses or sorted picocyanobacterial cells from natural communities are dispersed in thin polyacrylamide gels cast on microscope slides. The viruses or cells are screened for the presence of the viral target DNA in a solid‐phase PCR. Amplification results in a PCR product similar to a bacterial colony and is thus called a polony for PCR colony. Polonies are detected by hybridization with a fluorescently labelled probe located within the amplicon and enumerated. Polony numbers are then converted to absolute abundances based on an empirically determined virus‐to‐polony conversion efficiency.
Using the S‐TIM5 cyanophage for system optimization and calibration, the efficiency of polony formation (defined as the fraction of input phage particles that formed polonies) was 0.41 ± 0.064 (n = 23). Virus‐to‐polony conversion was reproducible and linear for 2 orders of magnitude (Figure 2). All abundances of TIM5‐like cyanophages in seawater samples were within the linear range of the virus‐to‐polony conversion curve, up to 1000 polonies per gel, which corresponds to 106 phages·ml−1. This limit was lower than that for T7‐like and T4‐like phages because of S‐TIM5's considerably larger polony size likely due to more efficient amplification of S‐TIM5's smaller amplicon (372 bp). To verify that the polonies detected in field samples were likely derived from TIM5‐like cyanophages, we picked single, well‐separated polonies from gels of Red Sea seawater samples collected in July 2013. The 372 bp or 249 bp DNApol amplicons (see Experimental Procedures) of 11 polonies were sequenced following PCR and cloning. These nucleotide sequences were >97% identical to that of polonies originating from the S‐TIM5 phage.
FIGURE 2.
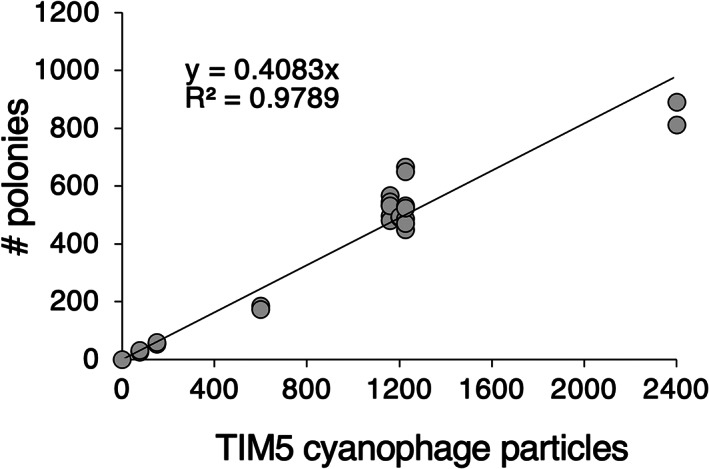
Virus to polony conversion curve. Standard curve of polony formation using degenerate primers and probes determined for the S‐TIM5 cyanophage (n = 23). The number of input viruses was determined by epifluorescence microscopy counts of lysates.
Seasonal distribution patterns of TIM5‐like cyanophages in the Red Sea
Using the polony method, we quantified TIM5‐like cyanophages over the annual cycle in the Red Sea at 20 m depth in 4 years between 2013 and 2016 (Figure 3). During this period, the water column exhibited typical seasonal dynamics for the Red Sea (Carlson et al., 2014; Lindell & Post, 1995; Maidanik et al., 2022). In the winter, the upper layers of the water column were colder (Figure 3A), which drove deep convective mixing and the influx of nutrients into the photic zone that stimulated phytoplankton productivity observed in increased chlorophyll concentrations (Figure 3B). As the water column warmed from spring to early autumn (Figure 3A), the water column stratified. This is generally associated with succession of the phototrophic community from picoeukaryotes in winter, Synechococcus in early spring and Prochlorococcus in summer (Lindell & Post, 1995; Maidanik et al., 2022) and is reflected in the decreasing chlorophyll‐a concentrations (Figure 3B). Maximum Synechococcus abundances, observed with monthly resolution, occurred in March following the onset of stratification after convective mixing. The spring peak in abundances was twice as high in 2013 and 2016 compared to 2014 and 2015. After this period, Synechococcus persisted at lower abundances as stratification intensified from spring to summer (Figure 3C). During this seasonal transition, the deep chlorophyll maximum intensified and deepened reaching 100 m in the late summer (Figure 4).
FIGURE 3.
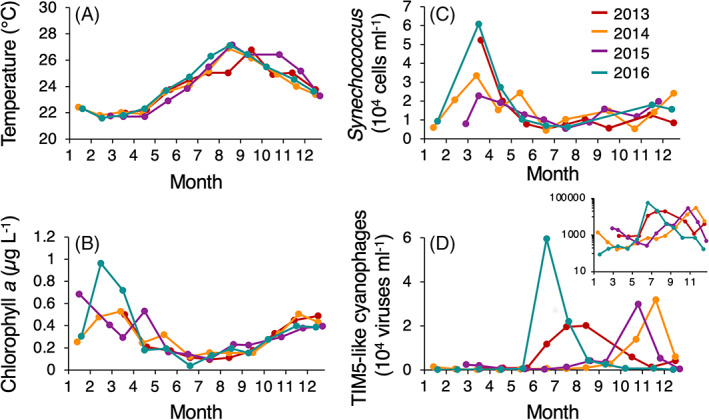
Seasonal abundances of TIM5‐like cyanophages at 20 m depth in the Red Sea during 2013–2016. (A) Temperature, (B) chlorophyll‐a concentration, (C) Synechococcus abundances, and (D) TIM5‐like cyanophages sampled monthly at 20 m depth. The inset shows TIM5‐like cyanophage abundances plotted on a log scale.
FIGURE 4.
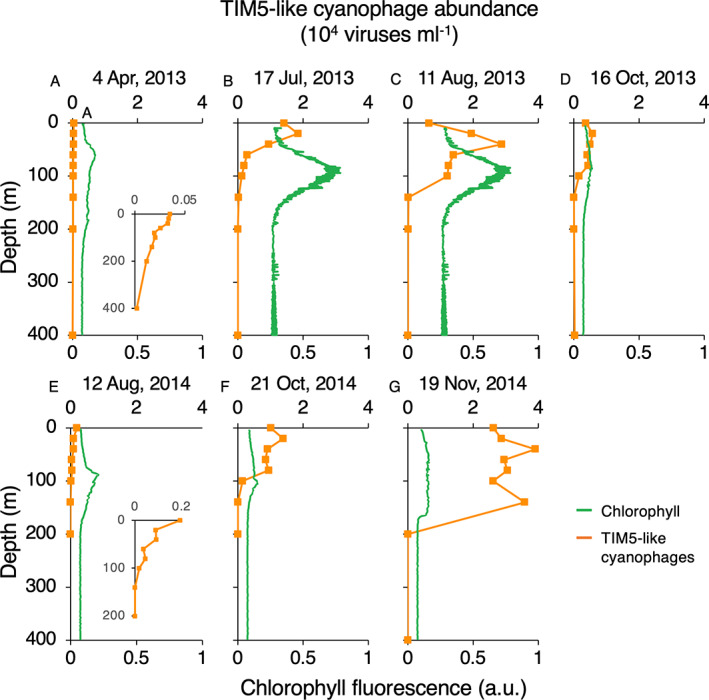
Depth distributions of TIM5‐like cyanophages in the Red Sea. TIM5‐like cyanophage abundances (orange) and chlorophyll fluorescence (green) with depth on different dates during 2013 (A–D) and 2014 (E–G). The dates of sample collection are shown above each panel. Insets show virus distributions on an enlarged scale. Chlorophyll fluorescence is shown in arbitrary units (a.u.).
We measured TIM5‐like cyanophage abundances at 20 m depth over these 4 years at monthly intervals (Figure 3D). During winter and spring, TIM5‐like cyanophages ranged between 59 and 2400 viruses·ml−1 with minimal fluctuation in abundances (Figure 3D). During summer and autumn, TIM5‐like cyanophage populations exhibited dramatic peaks in abundances, reaching a maximum of 59,000 ± 4800 viruses·ml−1 in June 2016. During these summer/autumn peaks, TIM5‐like cyanophages were 10‐to 100‐fold more abundant than in the spring and winter months. The timing of these peaks was seasonally variable with peaks in 2013 and 2016 occurring in the early summer whereas peaks in 2014 and 2015 occurred in the late autumn. These increases in abundances were generally short‐lived as abundances typically dropped back to baseline levels by the next month's sampling.
To assess patterns throughout the water column, we quantified the abundances of TIM5‐like cyanophages along depth profiles that were representative of seasonal patterns and that captured the cyanophage peaks occurring at different times in 2013 (summer peak) and 2014 (autumn peak) (Figure 4). TIM5‐like phages were present throughout the top 100 m of the water column in all profiles (Figure 4). Spring and summer depth profiles all had cyanophage maxima in the upper 40 m of the water column, which was always shallower than the deep chlorophyll maximum (Figure 4A–C,E). Autumn profiles in October and November (Figure 4D,F,G) had much broader subsurface maxima indicative of the deeper mixed layer depths during these periods based on temperature profiles. Consistent with the monthly observations at 20 m, TIM5‐like cyanophages were most abundant in summer 2013 and autumn 2014 both with maxima at 40 m depth of 29,000 viruses·ml−1 and 39,000 viruses·ml−1, respectively, whereas they were much less abundant in spring (≤250 viruses·ml−1 in April 2013). Indeed, integrated water column abundances of TIM5‐like cyanophages confirmed that TIM5‐like cyanophages were least abundant in the spring (April 2013) at 5.7 × 1010 viruses·m−3 compared to summer of 2013 and autumn of 2014, when abundances peaked at 1.94 × 1012 viruses·m−3 (June 2013) and 5.4 × 1012 viruses·m−3 (November 2014).
We then evaluated whether TIM5‐like cyanophages contributed substantially to picocyanobacteria mortality at times when TIM5‐like cyanophages were most abundant in the water column. Infection levels were determined for Prochlorococcus and Synechococcus along the November 2014 depth profile. Infection was detectable at two depths (40 and 140 m) for Synechococcus although infection levels were below the limit of accurate quantification (<0.05%). (Note that in two additional samples, Synechococcus at 60 m and Prochlorococcus at 40 m, a single polony was found, but we do not consider these to be robust observations.) Even if TIM5‐like cyanophages could complete four infection cycles in a day with an estimated 6‐h latent period (Sabehi et al., 2012), TIM5‐like cyanophages likely contributed to less than 0.2% of daily picocyanobacterial mortality at this time.
Spatial distribution of TIM5‐like cyanophages in the North Pacific Ocean
We compared the temporal variability of TIM5‐like cyanophages in the Red Sea to their spatial distribution along three ~2000 km latitudinal transects in the spring and early summer of 2015, 2016, and 2017 in the North Pacific Ocean (Figure 5) (Carlson et al., 2022). These transects transited from the oligotrophic subtropical gyre across the North Pacific Transition Zone Front to more productive waters at higher latitudes influenced by the subpolar gyre (Figure 5A,B). TIM5‐like cyanophages were found to be lowest in abundance in surface samples from subtropical waters at densities generally ranging between 75 and 3750 viruses·ml−1 (Figure 5D). Abundances increased towards more productive waters. TIM5‐like cyanophages increased 2‐ to 10‐fold relative to abundances in the subtropics between 34 and 38° N, where sharp increases in Synechococcus and other cyanophage lineage populations were also observed (Figure 5C,D) (Carlson et al., 2022). Northward of 38° N, TIM5‐like cyanophage abundances in 2015 and 2016 increased further, reaching their transect‐wide maxima with highest TIM5‐like abundances observed across all samples tested at 71,000 viruses·ml−1 at the highest latitude location in March 2015 (Figure 5D).
FIGURE 5.
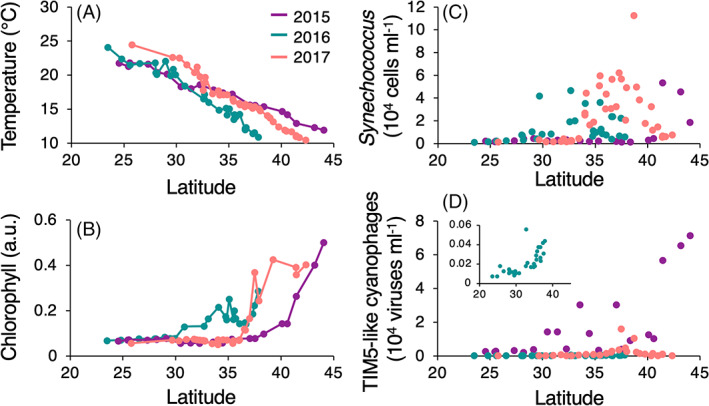
Latitudinal distributions of TIM5‐like cyanophages at 5 m depth in the North Pacific Ocean. (A) Temperature, (B) chlorophyll fluorescence, (C) Synechococcus, and (D) TIM5‐like cyanophages abundances sampled at 5 m depth along three transects in the North Pacific Ocean in March 2015, April 2016, and June 2017. The inset shows TIM5‐like cyanophage abundances from the April 2016 transect on an enlarged vertical axis scale. Chlorophyll fluorescence is shown in arbitrary units (a.u.). Source: Data shown here are reproduced from Carlson et al. (2022).
We also assessed the abundances of TIM5‐like cyanophages from depth profiles across the North Pacific Ocean from the 2016 and 2017 transects. Trophic changes along these transects were apparent from depth profiles of chlorophyll fluorescence in which the deep chlorophyll maximum (DCM) shoaled and increased in magnitude moving northward (Figure 6). TIM5‐like cyanophages were generally <200 viruses·ml−1 along depth profiles in oligotrophic waters south of 30° N and increased in the northernmost stations reaching up to 19,300 viruses·ml−1 at 25 m depth (Figure 6). Abundance profiles had little structure with depth except for the three northernmost profiles in 2017. In most profiles, the TIM5‐like cyanophage maximum was located in the upper 30 m of the water column, which was generally shallower than the DCM.
FIGURE 6.
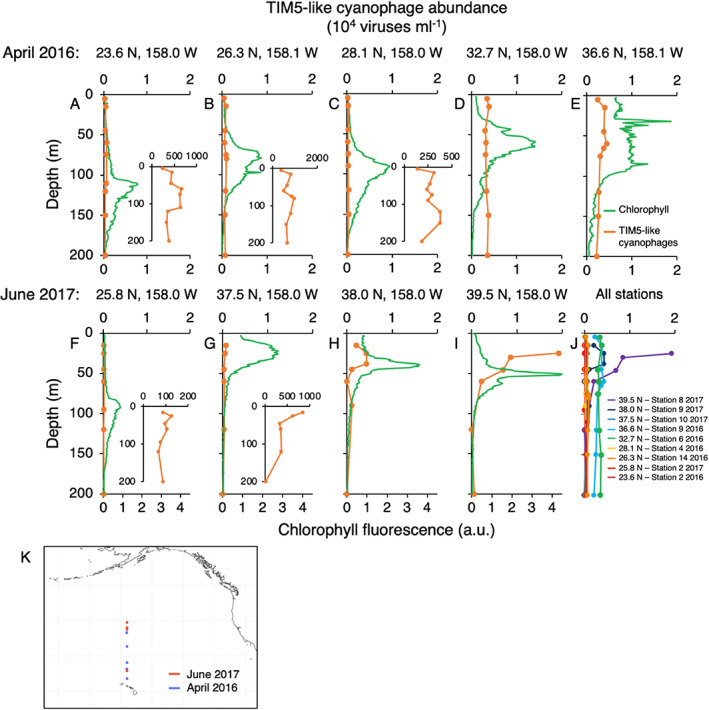
Depth distributions of TIM5‐like cyanophages in the North Pacific Ocean. TIM5‐like cyanophage abundances (orange) and chlorophyll fluorescence (green) with depth in April 2016 (A–E) and June 2017 (F–I). Sample locations are marked above the individual panels. Insets show virus distributions on an expanded scale. (J) Combined depth profiles of TIM5‐like cyanophages from 2016 and 2017. Latitude of sampling is indicated by colour with southern latitudes represented by warm colours and northern latitude by cool colours. (K) Geographic locations of depth profiles in the North Pacific coloured by year. Chlorophyll fluorescence is shown in arbitrary units (a.u.).
DISCUSSION
Diversity of TIM5‐like cyanophages
The isolation and genomic characterization of new TIM5‐like cyanophages revealed that this genus has traits and ecological dynamics that are distinctive from other cyanophage genera. Despite our attempts to isolate TIM5‐like cyanophages from the Red Sea on 17 different Synechococcus and Prochlorococcus hosts and under different environmental conditions to maximize putative diversity, only those capable of infecting two Synechococcus strains were found (this study and Dekel‐Bird et al., 2015). Thus, TIM5‐like cyanophages appear to have a limited set of hosts, at least out of those currently in our culture collection. The three isolated phages reported here have a high degree of genetic similarity and gene conservation relative to S‐TIM5 despite being isolated over the period of a year in different seasons from each other and from the original S‐TIM5 phage. Only 14 genes were identified on these TIM5‐like genomes that were not encoded on the S‐TIM5 genome (Figure 1B). In comparison, the T4‐like and T7‐like cyanophage genera are highly diverse and collectively encode a large repertoire of genes (Crummett et al., 2016; Dekel‐Bird et al., 2013; Fuchsman et al., 2020; Gregory et al., 2016; Huang et al., 2015; Ignacio‐Espinoza & Sullivan, 2012; Labrie et al., 2013; Marston & Martiny, 2016; Sullivan et al., 2008). Isolation attempts for the T4‐like and T7‐like cyanophages typically reflect this diversity, with isolated phages within a genus varying from being near identical to belonging to numerous, genomically diverse clades even when isolations are limited to a single host and single water sample (Gregory et al., 2016; Labrie et al., 2013; Marston & Martiny, 2016). The genomic diversity among the TIM5‐like cyanophage isolates observed here is less than that of individual clades within the T4‐like and T7‐like cyanophages and is more similar to discrete lineages within clades where there is high intra‐clade nucleotide identity and gene conservation (Gregory et al., 2016; Labrie et al., 2013; Marston & Martiny, 2016).
Sequences of the TIM5‐like cyanophage DNA polymerase gene from the Red Sea also showed a high degree of sequence similarity despite sample collection occurring over a ~5‐year period (Figure 1C; Dekel‐Bird et al., 2015; Sabehi et al., 2012). The high nucleotide identity of TIM5‐like DNA polymerase sequences observed in viral metagenomes from the Red provides independent support for the low genomic and allelic diversity from the phage isolations and amplicon‐based assays in this same region. In comparison, polony sequences from T7‐like and T4‐like cyanophages originating from a single sampling date fell into multiple clades across the diversity of the respective genera (Baran et al., 2018; Goldin et al., 2020). These T4‐like and T7‐like cyanophage polonies each had a median nucleotide sequence identity of 58%, much lower than the >97% identity for TIM5‐like cyanophage polonies. Moreover, polony sequences of cyanophages belonging to the T4‐like clade II and T7‐like clade A had median sequence identities of 71% and 67% and were thus more diverse than the polony sequences from the TIM5‐like genus. Thus, the TIM5‐like cyanophages in the Red Sea likely have limited diversity compared to the other cyanophage genera that also inhabit these waters.
Nucleotide similarity and gene sharing are strongly correlated in other cyanophage lineages (Gregory et al., 2016; Marston & Martiny, 2016). The low allelic diversity observed in the sequences of the TIM5‐like cyanophage DNA polymerase in this study, Dekel‐Bird et al. (2015) and Sabehi et al. (2012) suggest that the TIM5‐like phage populations in the Red Sea have equivalently low genomic diversity. The combination of this apparently limited intra‐group diversity, narrow host range, and specificity for Synechococcus would be expected to pose a challenge for TIM5‐like cyanophages against a background of immense host diversity (Ahlgren et al., 2019; Kashtan et al., 2014) as well as during times of host scarcity.
The limited diversity we observed in the Red Sea from the phage genomes and polony amplicons was surprising relative to the diversity observed in environmental gene homologues of the S‐TIM5 phage in the Global Ocean Sampling (GOS) Expedition (Sabehi et al., 2012). However, since we were unable to isolate viruses with more dissimilar genomic sequences, it is unclear whether such sequences come from cyanophages or other viruses that infect other host types. Indeed, the DNA polymerase sequences that had <80% amino acid identity in the GOS dataset originated from hypersaline lagoons or lakes (table S4 of Sabehi et al., 2012) suggesting there may be TIM5‐like phages that infect other bacteria (see supplementary text in Sabehi et al., 2012). In addition, DNA polymerase sequences with up to 66% amino acid identity and ~34%–49% nucleotide identity with TIM5‐like cyanophages cluster with the DNA polymerase gene from siphoviruses that infect a cyanobacterial symbiont of ascidians, Acaryochloris (Chan et al., 2011; Sabehi et al., 2012). We note that the PCR primers and probes used here for TIM5‐like cyanophages were degenerate and designed to detect all codon options of the target region in the DNA polymerase gene for the S‐TIM5 cluster while discriminating against the cyanosiphovirus lineage (see Experimental procedures). In silico, the primers and probes amplify environmental sequences that have >90% amino acid identity or >60% nucleotide identity to TIM5‐like cyanophages. At this stage, it is not possible to rule out that more diverse TIM5‐like cyanophages exist in nature that have yet to be isolated and/or cannot be captured by our polony primers and probes.
Ecological dynamics of TIM5‐like cyanophages
TIM5‐like cyanophages were widespread in the oceans as they were present in every sample we analysed in the photic zone across multiple years, in different ocean basins, and in distinct environmental conditions. However, TIM5‐like cyanophage abundances, which ranged between 101 and 104 viruses·ml−1, were typically 2–3 orders of magnitude lower than cyanophages of T4‐like and T7‐like genera in the same samples (Baran et al., 2018; Carlson et al., 2022; Maidanik et al., 2022). TIM5‐like abundances were more comparable to those of the T7‐like clade A cyanophages (Baran et al., 2018; Carlson et al., 2022; Maidanik et al., 2022), which have similar host range and host specificity as TIM5‐like cyanophages. The observation of infection levels that were barely detectable (<0.05%) even in samples with the highest TIM5‐like cyanophage abundances may explain such low abundances in general and indicates that this cyanophage genus plays a minor role in virus‐mediated mortality of cyanobacteria compared to the T7‐like and T4‐like cyanophage genera, which have been observed to infect between 0.1% and 2% of picocyanobacteria in oligotrophic conditions of the Red Sea and North Pacific Subtropical gyre and up to ~10% of picocyanobacteria in mesotrophic conditions of the North Pacific Transition Zone (Carlson et al., 2022; Maidanik et al., 2022; Mruwat et al., 2021). It is possible that we missed the peak in infection that produced such high virus abundances. Nevertheless, encounters between hosts and viruses provide opportunity for the generation of diversity such as through gene transfer or point mutations during replication (Avrani et al., 2011; Marston et al., 2012; Marston & Martiny, 2016). Therefore, low levels of infection stemming from putatively low host‐virus encounter rates could be a key factor contributing to the relatively low allelic diversity and apparently low genomic diversity within the TIM5‐like lineage, at least in the Red Sea.
The combination of low levels of infection, generally low abundances, and putative specificity for Synechococcus, which is often at low abundances during summer and fall in the Red Sea and the North Pacific Subtropical Gyre, raises the question of how TIM5‐like cyanophages persist in such environments. Decay of viruses in sunlit waters is often considered to occur on the order of days (Bongiorni et al., 2005; Heldal & Bratbak, 1991; Pasulka et al., 2018; Suttle & Chen, 1992). Specifically, turnover times of the cyanophage community have been estimated to occur in approximately 1 week based on infectivity measurements (Garza & Suttle, 1998), though cyanophage particles may turnover on the order of a month in subtropical conditions (Mruwat et al., 2021). Based on theoretical rates of contact based on particle diffusion (Mruwat et al., 2021; Murray & Jackson, 1992), at the average Synechococcus and TIM5‐like cyanophage abundances in the Red Sea (20,000 Synechococcus cells·ml−1 and 6000 TIM5‐like viruses·ml−1), a TIM5‐like cyanophage would encounter one Synechococcus cell (susceptible or not) approximately every 3 months. This interval between contacts is far longer than previously reported and suggests TIM5‐like cyanophages would be more likely to decay before they could find a host and propagate. This raises the possibility that diffusion‐based contact rates may not accurately capture the movement of TIM5‐like cyanophages or Synechococcus through water or the physical mechanisms that putatively mediate their interactions (Aguilo‐Ferretjans et al., 2021). Alternatively, TIM5‐like cyanophages may not decay as quickly as other previously reported cyanophages, and, rather, maintain infectivity for long periods of time. In fact, infectious plaques were found in March of 2009 and 2010 (Dekel‐Bird et al., 2015) when TIM5‐like cyanophages are expected to be low in abundance based on our current findings, as well as in August 2009 and 2010 (Dekel‐Bird et al., 2015) when Synechococcus abundances are expected to be low. These findings support such hypotheses of potentially long‐lived infectious particles.
Another notable feature of TIM5‐like cyanophage distributions was the observation of sharp peaks in abundances at monthly resolution in the Red Sea and over narrow spatial scales in the North Pacific Ocean. However, this phenomenon appeared to be related to different environmental parameters in the Red Sea and Pacific Ocean. In the Red Sea, peaks were seasonally variable occurring over a 6‐month period from early summer to late autumn and were not associated with overall abundances of Synechococcus at the time of sampling. In fact, these increases in TIM5‐like cyanophage abundances occurred during times of typically low‐to‐medium productivity in summer at the height of stratification, when Synechococcus was at or near their annual low in abundance or soon after the onset of mixing in the late autumn. In the North Pacific Transition Zone, TIM5‐like cyanophage peaks occurred in similar geographic regions to where Synechococcus peaked in abundance in regions of higher productivity.
We hypothesize that the potential for unusually long‐term persistence may enable TIM5‐like cyanophages to exploit limited or ephemeral increases in host abundances leading to the observed peaks in their abundance. Given that highly similar TIM5‐like cyanophage genotypes were found in Red Sea samples collected in different seasons, we hypothesize that peaks in abundance may happen due to a seasonal increase in specific Synechococcus subpopulations that are susceptible to TIM5‐like cyanophages, potentially belonging to clades II and III since these are hosts for the isolated TIM5‐like cyanophages, CC9605 and WH8102, respectively. Synechococcus clade III has been observed to increase in relative abundance in July and August in the Red Sea (Post et al., 2011). Synechococcus clade II is dominant throughout the year (Post et al., 2011). We wonder whether a currently unknown clade II subpopulation may also increase in summer/fall. We speculate that the timing of a putative increase in susceptible Synechococcus hosts is related to the extent of winter mixing that initiates the seasonal succession of phytoplankton (Lindell & Post, 1995) as TIM5‐like cyanophage abundances peaks occurred earlier in years when Synechococcus populations were substantially higher in March (2013 and 2016 compared to 2014 and 2015). Analogous successional processes in Synechococcus subpopulations may be occurring over spatial scales in the North Pacific Ocean. However, TIM5‐like cyanophage abundances were highest in regions where Synechococcus clades II and III have not been detected (Sohm et al., 2016) suggesting that a potentially different Synechococcus population drives TIM5‐like cyanophage distribution patterns in that region.
TIM5‐like cyanophage depth distributions in both the Red Sea and the Pacific Ocean were coherent despite the apparent differences in abundance patterns in surface waters. The distribution of TIM5‐like cyanophages along depth profiles was largely influenced by physical processes that structure the water column. TIM5‐like cyanophage abundance patterns mirrored that of well‐mixed water columns in the northern regions of the North Pacific Ocean transects and during convective mixing events in the Red Sea. In more stratified water columns such as in the North Pacific subtropics or after the onset of stratification in the Red Sea, maximum TIM5‐like abundances were typically observed in the upper 50 m. This pattern follows abundances of Synechococcus, which typically have maximum abundances at shallower depths relative to Prochlorococcus (Kent et al., 2019; Lindell & Post, 1995; Maidanik et al., 2022; Zwirglmaier et al., 2008). This is shallower than the maximum abundances of T4‐like and T7‐like clade B cyanophages, which typically have maxima at or below 50 m depth in stratified water columns and more similar to the depth distributions of T7‐like clade A cyanophages (Baran et al., 2018; Goldin et al., 2020; Maidanik et al., 2022). Thus, different cyanophage lineages appear to differentially partition the water column.
Conclusions
Our quantitative exploration of the distributions of the TIM5‐like genus have illuminated that these cyanophages appear to employ an ecological strategy of long‐term persistence that sustains low abundances yet widespread distributions in the oceans. In the Red Sea and perhaps elsewhere, these phages appear to endure with minimal levels of infection due to putatively few encounters between host and phage, which may slow the evolutionary arms race and result in limited diversification and gene exchange. This is vastly different to findings for the T7‐like and T4‐like cyanophage genera, which are both very abundant and highly diverse. The ephemeral outbreaks of TIM5‐like cyanophages may be an important process that seeds the water column during times of relatively high host abundance. Thus, the lifestyle of TIM5‐like cyanophages may shed light on how rare viral taxa survive in the oceans.
CONFLICT OF INTEREST
The author declares that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Supporting information
Table S1: TIM5‐like homologous sequences from Red Sea and Pacific Ocean viral metagenomes
ACKNOWLEDGEMENTS
The authors thank Ilia Maidanik, Nitzan Shamir, Yotam Hulata, and the Armbrust and White labs for assistance in sample collection, Svetlana Goldin for assistance in S‐TIM5 phage quantification, and Lindell lab members for discussions. The authors thank the Interuniversity Institute for Marine Sciences in Eilat and the Israel National Monitoring Program of the Gulf of Eilat (Aqaba) for access to sampling facilities and provision of environmental data. Funding was provided by the European Research Council (grant no. ERC‐CoG 646868), the Israel Science Foundation (grant no. 749/11 and 2679/20) and the Simons Foundation (Gradients grant No. 426570 and SCOPE grant no. 329108 and LIFE grant nos. 529554 and 721254) to Debbie Lindell. Michael C. G. Carlson was supported by a Fulbright Postdoctoral Fellowship. This manuscript is a contribution of the Simons Collaboration on Ocean Processes and Ecology (SCOPE).
Baran, N. , Carlson, M.C.G. , Sabehi, G. , Peleg, M. , Kondratyeva, K. , Pekarski, I. et al. (2022) Widespread yet persistent low abundance of TIM5‐like cyanophages in the oceans. Environmental Microbiology, 24(12), 6476–6492. Available from: 10.1111/1462-2920.16210
Nava Baran and Michael C. G. Carlson contributed equally to this work.
Funding information H2020 European Research Council, Grant/Award Number: ERC‐CoG 646868; Israel Science Foundation, Grant/Award Numbers: 2679/20, 749/11; Simons Foundation, Grant/Award Numbers: 329108, 426570, 529554, 721254
DATA AVAILABILITY STATEMENT
The revised S‐TIM5 genome is available under the accession number JQ245707.2. The other TIM5‐like cyanophage genomes were deposited under accession numbers OP053360–OP053362, and polony amplicon sequences are available under accession numbers ON014506–ON014516 at the NCBI GenBank.
REFERENCES
- Aguilo‐Ferretjans, M.D.M. , Bosch, R. , Puxty, R.J. , Latva, M. , Zadjelovic, V. , Chhun, A. et al. (2021) Pili allow dominant marine cyanobacteria to avoid sinking and evade predation. Nature Communications, 12, 1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahlgren, N.A. , Perelman, J.N. , Yeh, Y.C. & Fuhrman, J.A. (2019) Multi‐year dynamics of fine‐scale marine cyanobacterial populations are more strongly explained by phage interactions than abiotic, bottom‐up factors. Environmental Microbiology, 21, 2948–2963. [DOI] [PubMed] [Google Scholar]
- Angly, F.E. , Felts, B. , Breitbart, M. , Salamon, P. , Edwards, R.A. , Carlson, C. et al. (2006) The marine viromes of four oceanic regions. PLoS Biology, 4, 2121–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkezari, M.D. , Hagen, N.R. , Denholtz, M. , Neang, A. , Burns, T.C. , Morales, R.L. et al. (2021) Simons collaborative marine atlas project (Simons CMAP): an open‐source portal to share, visualize, and analyze ocean data. Limnology and Oceanography: Methods, 19, 488–496. [Google Scholar]
- Avrani, S. , Wurtzel, O. , Sharon, I. , Sorek, R. & Lindell, D. (2011) Genomic Island variability facilitates Prochlorococcus‐virus coexistence. Nature, 474, 604–608. [DOI] [PubMed] [Google Scholar]
- Aylward, F.O. , Boeuf, D. , Mende, D.R. , Wood‐Charlson, E.M. , Vislova, A. , Eppley, J.M. et al. (2017) Diel cycling and long‐term persistence of viruses in the ocean's euphotic zone. Proceedings of the National Academy of Sciences of the United States of America, 114, 11446–11451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baran, N. , Goldin, S. , Maidanik, I. & Lindell, D. (2018) Quantification of diverse virus populations in the environment using the polony method. Nature Microbiology, 3, 62–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biller, S.J. , Berube, P.M. , Lindell, D. & Chisholm, S.W. (2015) Prochlorococcus: the structure and function of collective diversity. Nature Reviews. Microbiology, 13, 13–27. [DOI] [PubMed] [Google Scholar]
- Bongiorni, L. , Magagnini, M. , Armeni, M. , Noble, R. & Danovaro, R. (2005) Viral production, decay rates, and life strategies along a trophic gradient in the North Adriatic Sea. Applied and Environmental Microbiology, 71, 6644–6650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbart, M. , Bonnain, C. , Malki, K. & Sawaya, N.A. (2018) Phage puppet masters of the marine microbial realm. Nature Microbiology, 3, 754–766. [DOI] [PubMed] [Google Scholar]
- Breitbart, M. , Salamon, P. , Andresen, B. , Mahaffy, J.M. , Segall, A.M. , Mead, D. et al. (2002) Genomic analysis of uncultured marine viral communities. Proceedings of the National Academy of Sciences of the United States of America, 99, 14250–14255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, C.T. , Howe, A. , Zhang, Q. , Pyrkosz, A.B. & Brom, T.H. (2012) A reference‐free algorithm for computational normalization of shotgun sequencing data. arXiv prepint, arXiv:1203.4802, 1–18. [Google Scholar]
- Brum, J.R. , Ignacio‐Espinoza, J.C. , Roux, S. , Doulcier, G. , Acinas, S.G. , Alberti, A. et al. (2015) Patterns and ecological drivers of ocean viral communities. Science, 348, 1261498. [DOI] [PubMed] [Google Scholar]
- Bushnell, B. , Rood, J. & Singer, E. (2017) BBMerge – accurate paired shotgun read merging via overlap. PLoS One, 12, e0185056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson, D.F. , Fredj, E. & Gildor, H. (2014) The annual cycle of vertical mixing and restratification in the northern Gulf of Eilat/Aqaba (Red Sea) based on high temporal and vertical resolution observations. Deep‐Sea Research Part I: Oceanographic Research Papers, 84, 1–17. [Google Scholar]
- Carlson, M.C.G. , Ribalet, F. , Maidanik, I. , Durham, B.P. , Hulata, Y. , Ferron, S. et al. (2022) Viruses affect picocyanobacterial abundance and biogeography in the North Pacific Ocean. Nature Microbiology, 7, 570–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, Y.‐W. , Mohr, R. , Millard, A.D. , Holmes, A.B. , Larkum, A.W. , Whitworth, A.L. et al. (2011) Discovery of cyanophage genomes which contain mitochondrial DNA polymerase. Molecular Biology and Evolution, 28, 2269–2274. [DOI] [PubMed] [Google Scholar]
- Chen, S. , Zhou, Y. , Chen, Y. & Gu, J. (2018) Fastp: an ultra‐fast all‐in‐one FASTQ preprocessor. Bioinformatics, 34, i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenard, C. , Chan, A.M. , Vincent, W.F. & Suttle, C.A. (2015) Polar freshwater cyanophage S‐EIV1 represents a new widespread evolutionary lineage of phages. The ISME Journal, 9, 2046–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crummett, L.T. , Puxty, R.J. , Weihe, C. , Marston, M.F. & Martiny, J.B.H. (2016) The genomic content and context of auxiliary metabolic genes in marine cyanomyoviruses. Virology, 499, 219–229. [DOI] [PubMed] [Google Scholar]
- Culley, A.I. , Lang, A.S. & Suttle, C.A. (2006) Metagenomic analysis of coastal RNA virus communities. Science, 312, 1795–1798. [DOI] [PubMed] [Google Scholar]
- Dekel‐Bird, N.P. , Avrani, S. , Sabehi, G. , Pekarsky, I. , Marston, M.F. , Kirzner, S. et al. (2013) Diversity and evolutionary relationships of T7‐like podoviruses infecting marine cyanobacteria. Environmental Microbiology, 15, 1476–1491. [DOI] [PubMed] [Google Scholar]
- Dekel‐Bird, N.P. , Sabehi, G. , Mosevitzky, B. & Lindell, D. (2015) Host‐dependent differences in abundance, composition and host range of cyanophages from the Red Sea. Environmental Microbiology, 17, 1286–1299. [DOI] [PubMed] [Google Scholar]
- Edgar, R.C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores, C.O. , Meyer, J.R. , Valverde, S. , Farr, L. & Weitz, J.S. (2011) Statistical structure of host–phage interactions. Proceedings of the National Academy of Sciences of the United States of America, 108, E288–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores‐Uribe, J. , Philosof, A. , Sharon, I. , Fridman, S. , Larom, S. & Béjà, O. (2019) A novel uncultured marine cyanophage lineage with lysogenic potential linked to a putative marine Synechococcus ‘relic’ prophage. Environmental Microbiology Reports, 11, 598–604. [DOI] [PubMed] [Google Scholar]
- Fuchsman, C.A. , Carlson, M.C.G. , Garcia Prieto, D. , Hays, M.D. & Rocap, G. (2020) Cyanophage host‐derived genes reflect contrasting selective pressures with depth in the oxic and anoxic water column of the eastern tropical North Pacific. Environmental Microbiology, 23, 2782–2800. [DOI] [PubMed] [Google Scholar]
- Gainer, P.J. , Pound, H.L. , Larkin, A.A. , LeCleir, G.R. , DeBruyn, J.M. , Zinser, E.R. et al. (2017) Contrasting seasonal drivers of virus abundance and production in the North Pacific Ocean. PLoS One, 12, e0184371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garza, D.R. & Suttle, C.A. (1998) The effect of cyanophages on the mortality of Synechococcus spp. and selection for UV resistant viral communities. Microbial Ecology, 36, 281–292. [DOI] [PubMed] [Google Scholar]
- Goldin, S. , Hulata, Y. , Baran, N. & Lindell, D. (2020) Quantification of T4‐like and T7‐like cyanophages using the polony method show they are significant members of the virioplankton of the North Pacific subtropical gyre. Frontiers in Microbiology, 11, 1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith, D.B. , Parsons, R.J. , Beyene, D. , Salamon, P. & Breitbart, M. (2015) Deep sequencing of the viral phoH gene reveals temporal variation, depth‐specific composition, and persistent dominance of the same viral phoH genes in the Sargasso Sea. PeerJ, 3, e997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory, A.C. , Solonenko, S.A. , Ignacio‐Espinoza, J.C. , LaButti, K. , Copeland, A. , Sudek, S. et al. (2016) Genomic differentiation among wild cyanophages despite widespread horizontal gene transfer. BMC Genomics, 17, 930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldal, M. & Bratbak, G. (1991) Production and decay of viruses in aquatic environments. Marine Ecology Progress Series, 72, 205–212. [Google Scholar]
- Hewson, I. , Barbosa, J.G. , Brown, J.M. , Donelan, R.P. , Eaglesham, J.B. , Eggleston, E.M. et al. (2012) Temporal dynamics and decay of putatively Allochthonous and autochthonous viral genotypes in contrasting Freshwater Lakes. Applied and Environmental Microbiology, 78, 6583–6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, S. , Zhang, S. , Jiao, N. & Chen, F. (2015) Comparative genomic and phylogenomic analyses reveal a conserved core genome shared by estuarine and oceanic cyanopodoviruses. PLoS One, 10, e0142962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt, D. , Chen, G.‐L. , LoCascio, P.F. , Land, M.L. , Larimer, F.W. & Hauser, L.J. (2010) Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics, 11, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignacio‐Espinoza, J.C. , Ahlgren, N.A. & Fuhrman, J.A. (2020) Long‐term stability and red queen‐like strain dynamics in marine viruses. Nature Microbiology, 5, 265–271. [DOI] [PubMed] [Google Scholar]
- Ignacio‐Espinoza, J.C. & Sullivan, M.B. (2012) Phylogenomics of T4 cyanophages: lateral gene transfer in the 'core' and origins of host genes. Environmental Microbiology, 14, 2113–2126. [DOI] [PubMed] [Google Scholar]
- John, S.G. , Mendez, C.B. , Deng, L. , Poulos, B. , Kauffman, A.K.M. , Kern, S. et al. (2011) A simple and efficient method for concentration of ocean viruses by chemical flocculation. Environmental Microbiology Reports, 3, 809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashtan, N. , Roggensack, S.E. , Rodrigue, S. , Thompson, J.W. , Stocker, R. , Follows, M.J. et al. (2014) Single‐cell genomics reveals hundreds of coexisting subpopulations in wild Prochlorococcus . Science, 416, 416–420. [DOI] [PubMed] [Google Scholar]
- Kent, A.G. , Baer, S.E. , Mouginot, C. , Huang, J.S. , Larkin, A.A. , Lomas, M.W. et al. (2019) Parallel phylogeography of Prochlorococcus and Synechococcus . The ISME Journal, 13, 430–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koskella, B. & Meaden, S. (2013) Understanding bacteriophage specificity in natural microbial communities. Viruses, 5, 806–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrie, S.J. , Frois‐Moniz, K. , Osburne, M.S. , Kelly, L. , Roggensack, S.E. , Sullivan, M.B. et al. (2013) Genomes of marine cyanopodoviruses reveal multiple origins of diversity. Environmental Microbiology, 15, 1356–1376. [DOI] [PubMed] [Google Scholar]
- Laslett, D. & Canback, B. (2004) ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Research, 32, 11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavigne, R. , Darius, P. , Summer, E.J. , Seto, D. , Mahadevan, P. , Nilsson, A.S. et al. (2009) Classification of Myoviridae bacteriophages using protein sequence similarity. BMC Microbiology, 9, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavigne, R. , Seto, D. , Mahadevan, P. , Ackermann, H.W. & Kropinski, A.M. (2008) Unifying classical and molecular taxonomic classification: analysis of the Podoviridae using BLASTP‐based tools. Research in Microbiology, 159, 406–414. [DOI] [PubMed] [Google Scholar]
- Letunic, I. & Bork, P. (2021) Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Research, 49, W293–W296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindell, D. & Post, A.F. (1995) Ultraphytoplankton succession is triggered by deep winter mixing in the Gulf of Aqaba (Eilat), Red Sea. Limnology and Oceanography, 40, 1130–1141. [Google Scholar]
- Maidanik, I. , Kirzner, S. , Pekarski, I. , Arsenieff, L. , Tahan, R. , Carlson, M.C.G. et al. (2022) The less virulent cyanophage clade dominates over its sister clade in the oceans. The ISME Journal, 16, 2169–2180. 10.1038/s41396-41022-01259-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann, N.H. (2003) Phages of the marine cyanobacterial picophytoplankton. FEMS Microbiology Reviews, 27, 17–34. [DOI] [PubMed] [Google Scholar]
- Marston, M.F. & Martiny, J.B. (2016) Genomic diversification of marine cyanophages into stable ecotypes. Environmental Microbiology, 18, 4240–4253. [DOI] [PubMed] [Google Scholar]
- Marston, M.F. , Pierciey, F.J. , Shepard, A. , Gearin, G. , Qi, J. , Yandava, C. et al. (2012) Rapid diversification of coevolving marine Synechococcus and a virus. Proceedings of the National Academy of Sciences of the United States of America, 109, 4544–4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marston, M.F. & Sallee, J.L. (2003) Genetic diversity and temporal variation in the cyanophage community infecting marine Synechococcus species in Rhode Island's coastal waters. Applied and Environmental Microbiology, 69, 4639–4647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matteson, A.R. , Rowe, J.M. , Ponsero, A.J. , Pimentel, T.M. , Boyd, P.W. & Wilhelm, S.W. (2013) High abundances of cyanomyoviruses in marine ecosystems demonstrate ecological relevance. FEMS Microbiology Ecology, 84, 223–234. [DOI] [PubMed] [Google Scholar]
- Minh, B.Q. , Schmidt, H.A. , Chernomor, O. , Schrempf, D. , Woodhams, M.D. , von Haeseler, A. et al. (2020) IQ‐TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Molecular Biology and Evolution, 37, 1530–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mruwat, N. , Carlson, M.C.G. , Goldin, S. , Ribalet, F. , Kirzner, S. , Hulata, Y. et al. (2021) A single‐cell polony method reveals low levels of infected Prochlorococcus in oligotrophic waters despite high cyanophage abundances. The ISME Journal, 15, 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray, A.G. & Jackson, G.A. (1992) Viral dynamics ‐ a model of size, shape, motion and abundance of single‐celled planktonic organisms and other particles. Marine Ecology Progress Series, 89, 103–116. [Google Scholar]
- Nishimura, Y. , Watai, H. , Honda, T. , Mihara, T. , Omae, K. , Roux, S. et al. (2017) Environmental viral genomes shed new light on virus‐host interactions in the ocean. mSphere, 2, e00359–e00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble, R.T. & Fuhrman, J.A. (1997) Virus decay and its causes in coastal waters. Applied and Environmental Microbiology, 63, 77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons, R.J. , Breitbart, M. , Lomas, M.W. & Carlson, C.A. (2012) Ocean time‐series reveals recurring seasonal patterns of virioplankton dynamics in the northwestern Sargasso Sea. The ISME Journal, 6, 273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasulka, A.L. , Thamatrakoln, K. , Kopf, S.H. , Guan, Y. , Poulos, B. , Moradian, A. et al. (2018) Interrogating marine virus‐host interactions and elemental transfer with BONCAT and nanoSIMS‐based methods. Environmental Microbiology, 20, 671–692. [DOI] [PubMed] [Google Scholar]
- Patel, A. , Noble, R.T. , Steele, J.A. , Schwalbach, M.S. , Hewson, I. & Fuhrman, J.A. (2007) Virus and prokaryote enumeration from planktonic aquatic environments by epifluorescence microscopy with SYBR green I. Nature Protocols, 2, 269–276. [DOI] [PubMed] [Google Scholar]
- Post, A. , Penno, S. , Zandbank, K. , Paytan, A. , Huse, S. & Mark Welch, D. (2011) Long term seasonal dynamics of Synechococcus population structure in the Gulf of Aqaba, northern Red Sea. Frontiers in Microbiology, 2, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor, L.M. & Fuhrman, J.A. (1990) Viral mortality of marine bacteria and cyanobacteria. Nature, 343, 60–62. [Google Scholar]
- Rohwer, F. & Thurber, R.V. (2009) Viruses manipulate the marine environment. Nature, 459, 207–212. [DOI] [PubMed] [Google Scholar]
- Sabehi, G. , Shaulov, L. , Silver, D.H. , Yanai, I. , Harel, A. & Lindell, D. (2012) A novel lineage of myoviruses infecting cyanobacteria is widespread in the oceans. Proceedings of the National Academy of Sciences of the United States of America, 109, 2037–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch, C.L. , Ciufo, S. , Domrachev, M. , Hotton, C.L. , Kannan, S. , Khovanskaya, R. et al. (2020) NCBI taxonomy: a comprehensive update on curation, resources and tools. Database: The Journal of Biological Databases and Curation, 2020, baaa062, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohm, J.A. , Ahlgren, N.A. , Thomson, Z.J. , Williams, C. , Moffett, J.W. , Saito, M.A. et al. (2016) Co‐occurring Synechococcus ecotypes occupy four major oceanic regimes defined by temperature, macronutrients and iron. The ISME Journal, 10, 333–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan, M.B. , Coleman, M.L. , Quinlivan, V. , Rosenkrantz, J.E. , Defrancesco, A.S. , Tan, G. et al. (2008) Portal protein diversity and phage ecology. Environmental Microbiology, 10, 2810–2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan, M.B. , Krastins, B. , Hughes, J.L. , Kelly, L. , Chase, M. , Sarracino, D. et al. (2009) The genome and structural proteome of an ocean siphovirus: a new window into the cyanobacterial 'mobilome'. Environmental Microbiology, 11, 2935–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan, M.B. , Waterbury, J.B. & Chisholm, S.W. (2003) Cyanophages infecting the oceanic cyanobacterium Prochlorococcus . Nature, 424, 1047–1051. [DOI] [PubMed] [Google Scholar]
- Suttle, C.A. (2005) Viruses in the sea. Nature, 437, 356–361. [DOI] [PubMed] [Google Scholar]
- Suttle, C.A. & Chan, A.M. (1993) Marine cyanophages infecting oceanic and coastal strains of Synechococcus ‐ abundance, morphology, cross‐infectivity and growth‐characteristics. Marine Ecology Progress Series, 92, 99–109. [Google Scholar]
- Suttle, C.A. & Chen, F. (1992) Mechanisms and rates of decay of marine viruses in seawater. Applied and Environmental Microbiology, 58, 3721–3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzian, P. , Olo Ndela, E. , Galiez, C. , Lossouarn, J. , Bucio, P. , Rubén, E. et al. (2021) PHROG: families of prokaryotic virus proteins clustered using remote homology. NAR Genomics and Bioinformatics, 3, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, K. & Chen, F. (2008) Prevalence of highly host‐specific cyanophages in the estuarine environment. Environmental Microbiology, 10, 300–312. [DOI] [PubMed] [Google Scholar]
- Waterbury, J.B. & Valois, F.W. (1993) Resistance to co‐occurring phages enables marine Synechococcus communities to coexist with cyanophages abundant in seawater. Applied and Environmental Microbiology, 59, 3393–3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick, R.R. , Judd, L.M. , Gorrie, C.L. & Holt, K.E. (2017) Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLOS Computational Biology, 13, e1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, W.H. , Joint, I.R. , Carr, N.G. & Mann, N.H. (1993) Isolation and molecular characterization of 5 marine cyanophages propagated on Synechococcus sp. strain WH7803. Applied and Environmental Microbiology, 59, 3736–3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yooseph, S. , Sutton, G. , Rusch, D.B. , Halpern, A.L. , Williamson, S.J. , Remington, K. et al. (2007) The sorcerer II Global Ocean sampling expedition: expanding the universe of protein families. PLoS Biology, 5, e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zborowsky, S. & Lindell, D. (2019) Resistance in marine cyanobacteria differs against specialist and generalist cyanophages. Proceedings of the National Academy of Sciences of the United States of America, 116, 16899–16908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman, A.E. , Howard‐Varona, C. , Needham, D.M. , John, S.G. , Worden, A.Z. , Sullivan, M.B. et al. (2020) Metabolic and biogeochemical consequences of viral infection in aquatic ecosystems. Nature Reviews. Microbiology, 18, 21–34. [DOI] [PubMed] [Google Scholar]
- Zwirglmaier, K. , Jardillier, L. , Ostrowski, M. , Mazard, S. , Garczarek, L. , Vaulot, D. et al. (2008) Global phylogeography of marine Synechococcus and Prochlorococcus reveals a distinct partitioning of lineages among oceanic biomes. Environmental Microbiology, 10, 147–161. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: TIM5‐like homologous sequences from Red Sea and Pacific Ocean viral metagenomes
Data Availability Statement
The revised S‐TIM5 genome is available under the accession number JQ245707.2. The other TIM5‐like cyanophage genomes were deposited under accession numbers OP053360–OP053362, and polony amplicon sequences are available under accession numbers ON014506–ON014516 at the NCBI GenBank.