

Abstract
Lineage specific expansions and gene duplications are some of the most important sources of evolutionary novelty in eukaryotes. Although not as prevalent in eukaryotes compared to bacteria, horizontal gene transfer events can also result in key adaptations for insects, especially for those involved in insect‐microbe interactions. In this study we assemble the first chromosomal assembly of the psyllid Bactericera cockerelli and reveal that the B. cockerelli genome has experienced significantly more gene expansion events compared to other Hemipteran representatives with fully sequenced genomes. We also reveal that B. cockerelli's genome is the largest psyllid genome (567 Mb) sequenced to date and is ~15% larger than the other two psyllid species genomes sequenced (Pachypsylla venusta and Diaphorina citri). Structurally, B. cockerelli appears to have an additional chromosome compared to the distantly related psyllid species P. venusta due to a previous chromosomal fission or fusion event. The increase in genome size and dynamic nature of the B. cockerelli genome may largely be contributed to the widespread expansion of type I and II repeat elements that are rampant across all of B. cockerelli's. chromosomes. These repeat elements are distributed near equally in both euchromatic and heterochromatic regions. Furthermore, significant gene family expansions and gene duplications were uncovered for genes that are expected to be important in its adaptation to insect‐plant and microbe interactions, which include transcription factors, proteases, odorant receptors, and horizontally transferred genes that are involved in the nutritional symbioses with their long‐term nutritional endosymbiont Carsonella.
Keywords: Bactericera cockerelli, Candidatus Carsonella ruddii, carboxypeptidases, Psyllid genome, symbiosis‐genes, transposable elements
1. INTRODUCTION
Lineage specific gene expansions are one of the most important means of adaptation in eukaryotes. These lineage specific expansions are primarily in response to microbes, environmental stress, and species specify signalling factors, often enabling both functional and expression divergence among gene copies (Lespinet et al., 2002). Although less common in animals compared to bacteria, horizontal gene transfer events have also been recognized as important sources of new genes and evolutionary novelty in eukaryotic genomes (Keeling & Palmer, 2008; Van Etten & Bhattacharya, 2020). For example, it has been observed in some insect genomes that horizontal gene transfer has played a major role in helping orchestrate symbiotic integration between the insect host and its nutritional endosymbionts (Husnik et al., 2013; Sloan et al., 2014). Genome sequences of both insects and their obligate nutritional endosymbionts have been crucial in shedding light onto the evolution of nutritional endosymbioses (Moran et al., 2008; The International Aphid Genomics Consortium, 2010). Nevertheless, far more genome sequences have been acquired from bacterial endosymbionts compared to insect hosts (NCBI, 2022). Therefore, by sequencing of additional genomes of insects with obligate symbionts we can gain a deeper understanding of how insect host genomes evolve with their long‐term endosymbionts (Blaxter et al., 2022).
The evolution of endosymbiont genomes in sap‐feeding insects has been extensively documented revealing extreme cases of genome reduction leading to the loss of essential genes for basic cellular processes (Bennett & Moran, 2013; McCutcheon & Moran, 2012). One of the most extreme examples of genome reduction in an endosymbiont was first observed in Candidatus Carsonella ruddii (hereafter referred to as Carsonella; Nakabachi et al., 2006), a nutritional endosymbiont that is universally present in psyllids (Spaulding & von Dohlen, 2001; Thao et al., 2000). Psyllids feed on plant sap and belong to the insect superfamily Psylloidea (psyllids) within the suborder Sternorrhyncha, which includes aphids, whiteflies, and scale insects (Gullan & Martin, 2009). Carsonella supplements the psyllid's diet with essential amino acids that are deficient in the psyllid's sap diet (Baumann, 2005; McCutcheon & Moran, 2012; Moran & Wernegreen, 2000; Nakabachi et al., 2006).
At this time genome assemblies are only available for two psyllid species, Diaphorina citri (Saha et al., 2017) and Pachypsylla venusta (Li et al., 2020) which belong to two of seven psyllid families, the Psyllidae and Carsidaridae, respectively (Burckhardt et al., 2021). Therefore, in an effort to increase our understanding of psyllid genome evolution and nutritional symbioses we sequenced the potato (tomato) psyllid, Bactericera cockerelli (Šulc) genome, which belongs to a third psyllid family the Triozidae. The psyllid family Triozidae shares a more recent common ancestor to the family Psyllidae compared to the more divergent psyllid family Carsidaridae (Burckhardt et al., 2021). Like other psyllid species B. cockerelli feeds on plant sap and possesses the obligate nutritional endosymbiont Carsonella‐ BC (Riley et al., 2017). Similar to P. venusta, B. cockerelli possesses one long‐term obligate endosymbiont, Carsonella‐BC, and has not been identified with an additional cosymbiont such as Candidatus Profftella armatura in D. citri (hereafter referred to as Profftella‐DC; Nakabachi et al., 2013). The psyllid, B. cockerelli is native to North America (Crawford, 1914; Šulc, 1909) and is recognized as a major pest of solanaceous crops in its native range and as an invasive pest in New Zealand (Crosslin et al., 2010; Hansen et al., 2008; Liefting et al., 2008; Munyaneza et al., 2007). This is largely because B. cockerelli is a vector of the bacterium “Candidatus Liberibacter psyllaurous,” which is associated with psyllid yellows disease (Hansen et al., 2008). Interestingly Liberibacter species are only known to infect the insect superfamily Psylloidea (Kwak et al., 2021; Raddadi et al., 2011; Wang et al., 2017), and may influence insect‐plant interactions by playing a dual role as a plant pathogen and insect endosymbiont (Casteel et al., 2012). At this time P. venusta is not known to harbour a Liberibacter species; however, D. citri is a notorious vector of Ca. L. asiaticus, which is associated with Huanglongbing, a highly destructive disease of citrus (Bové, 2006). Sequencing additional genomes of psyllids that harbour and vector Liberibacter will increase our understanding of this unique and specialized relationship between Liberibacter taxa and Psylloidea.
In this study we sequence and annotate the first genome of B. cockerelli using short and long read sequencing technologies combined with proximity ligation to obtain a high quality chromosomal‐level genome assembly for the research community. First, to gain insight into gene expansion and contraction events and natural selection on B. cockerelli's genome we conducted evolutionary genomic analyses. Moreover, to further understand how the psyllid host and endosymbiont metabolisms integrate and evolve together we conducted comparative genomics analyses on the psyllid and endosymbiont genomes of B. cockerelli, D. citri, and P. venusta.
2. MATERIALS AND METHODS
2.1. Insect rearing
The Bactericera cockerelli culture sequenced in this study was derived ~2 years ago from a wild population in Temecula, California, USA. To prepare genomic material for genome sequencing a single mated female of B. cockerelli was used to establish an inbred line to reduce heterozygosity. The established isofemale line was maintained for over 6 generations on 4–8‐week‐old Capsicum annuum (California Wonder pepper) at 25°C under a 16L:8D light/dark. Both DNA and RNA material was obtained from this line at the same time for genome sequencing and annotation (see below). To increase coverage of the insect genome this insect line was verified to not be infected with Ca. Liberibacter psyllaurous, an insect symbiont/plant pathogen that commonly infects this psyllid species (Hansen et al., 2008), using the primers Y DRAG Q‐PCR‐IGS‐7F/R and the qPCR conditions detailed in Casteel et al. (2012).
2.2. Genome sequencing and assembly
In order to obtain a high‐quality genome of B. cockerelli, a chromosome‐level genome assembly of B. cockerelli was generated from long‐read sequencing by Pacific Biosciences (PacBio) and short‐read sequencing by Illumina with the Omni‐C proximity ligation technique developed by Dovetail Genomics. Briefly, over 200 mg of both female and male adults from the B. cockerelli inbred line were collected and were starved overnight for 8–9 h. Psyllid adults were then flash frozen in liquid nitrogen, stored at –80°C, and shipped to Dovetail Genomics for genomic DNA extraction, library preparations, sequencing, and genome assemblies.
Briefly, genomic DNA was extracted using the Qiagen DNA extraction kit following the manufacturer's instructions (Qiagen) and quantified using the Qubit 2.0 Fluorometer (Life Technologies). The PacBio SMRTbell library was constructed using the SMRTbell Express Template Prep Kit 2.0 (PacBio) following the manufacturer's protocol. The SMRTbell library was then bound to sequencing polymerase with the Sequel II Binding Kit 2.0 (PacBio). DNA sequencing was performed on PacBio Sequel II 8M SMRT cells generating 137 Gb of raw data. A de novo assembly of PacBio reads was then generated using the WTDBG2 version 2.5 pipeline (Ruan & Li, 2020). Nontarget sequences of the output assembly were filtered using blastn and blobtools version 1.1.1 (Laetsch & Blaxter, 2017), and contig overlaps and/or potential haplotypic duplications were also removed using purge_dups version 1.2.3 (Guan et al., 2020). The final PacBio de novo assembly was then used with the Omni‐C data to create the final chromosome‐level genome assembly (below).
2.3. Chromosome‐length sequencing and genome assembly
For the Omni‐C library, chromatin was fixed in place with formaldehyde in the nucleus to create a three‐dimensional scaffold. The fixed chromatin was then extracted and randomly fragmented with DNase I. The resulting chromatin fragments were end‐repaired, ligated to a biotinylated bridge adapter, and were followed by proximity ligation. After the proximity ligation, crosslinks were reversed for DNA purification, and the purified DNA was treated to remove biotin that was not internal to ligated fragments. The sequencing library was generated using NEBNext Ultra enzymes and Illumina‐compatible adapters (NEB). Biotinylated fragments were isolated using streptavidin beads before PCR enrichment of library, and the library was sequenced on an Illumina HiSeqX.
To obtain chromosome‐level scaffolds, the Omni‐C library reads were aligned to the PacBio draft assembly using bwa‐0.7.17 (Li, 2013). Read pairs mapped onto draft scaffolds were then used to generate a likelihood model using HiRise, a software pipeline designed specifically for using proximity ligation data to scaffold genome assemblies (Putnam et al., 2016). The generated likelihood model was used to identify and break putative misjoins, to score prospective joins, and make joins above a threshold. To evaluate the completeness of the HiRise assembly, Benchmarking universal single‐copy orthologues (BUSCOs) version 4.0.5 (Simão et al., 2015) was performed against the eukaryote_odb10 database (Table S1).
2.4. TAD analyses
The three‐dimensional structure of the B. cockerelli genome was analysed through the analysis of topologically associated domains (TADs) from Omni‐C data (Table S2). TADs are a fundamental unit of chromatin topology where DNA sequences within a TAD region are in proximity and physically interact more frequently with one another compared to DNA sequences that occur between TAD regions (Dixon et al., 2012). For the analysis of TADs first Omni‐C contact matrices were generated in two formats, namely cool and hic. Both contact matrices were generated from the same BAM file by using read pairs where both ends were aligned with a mapping quality of 60. TADs were identified using the Arrowhead (Rao et al., 2014) implemented with Juicer (Durand et al., 2016). TADs were called at three different resolutions: 10, 25, and 50 kbp. The parameters used were ‐k KR ‐m 2000 ‐r 10,000, ‐k KR ‐m 2000 ‐r 25,000, and ‐k KR ‐m 2000 ‐r 50,000. A/B compartments were identified at 1 Mbp using the eigenvector program implemented in Juicer (Durand et al., 2016). The parameters used were KR BP 1,000,000. Isochores were predicted using the IsoFinder (Oliver et al., 2004). The parameters used were 0.90 p2 3000. The output was post‐processed to convert into a bedpe format. CTCF sites were predicted using cread (Smith et al., 2006). The position weight matrix was downloaded from CTCFBSDB 2.0 website (Ziebarth et al., 2013). The output was then post‐processed to convert it to a bed file. Multires files were generated for visualization in HiGlass using the clodius package (Kerpedjiev et al., 2018).
2.5. RNA‐seq and annotation
To annotate the final assembly of the B. cockerelli genome, repeat sequences in the final HiRise genome assembly were identified de novo and masked using RepeatModeler version 2.01 (Flynn et al., 2020) and RepeatMasker version 4.1.0 (Smit et al., 2013). We then used coding sequences (CDS) from Acrythosiphon pisum, AL4f downloaded from NCBI, GCA_005508785.1 (Li et al., 2019), D. citri, Dcitri_OGSv2.0 downloaded from https://citrusgreening.org/organism/Diaphorina_citri/genome (Saha et al., 2017) and P. venusta, AUS‐FW‐20181119 downloaded from NCBI (GCA_012654025.1) and github (lyy005; Li et al., 2020) to train the initial ab initio models for B. cockerelli using AUGUSTUS version 2.5.5 (Stanke et al., 2006) and SNAP version 2006‐07‐28 (Korf, 2004).
We also performed RNA‐seq from the same colony DNA was collected from for genome sequencing (above) to improve the B. cockerelli genome annotation. Five pooled psyllid samples, each containing >8 mg of both female and male adults per sample, were collected and flash frozen using liquid nitrogen. Samples were then shipped to GENEWIZ for total RNA extraction and sequencing. Total RNA was extracted using Qiagen RNeasy Plus kit (Qiagen) following the manufacturer's instructions. Purified RNA integrity and quantity were measured using 4200 TapeStation (Agilent Technologies) and Qubit 2.0 Fluorometer (Life Technologies), respectively. After the rRNA depletion using Qiagen FastSelect rRNA HMR kit (Qiagen), the sequencing library was generated using NEBNext Ultra II RNA library preparation kit for Illumina by following the manufacturer's instructions (NEB). Briefly, enriched RNAs were fragmented for 15 min at 94°C. First strand and second strand cDNA were subsequently synthesized. cDNA fragments were end repaired and adenylated at 3'ends, and universal adapters were ligated to cDNA fragments, followed by index addition and library enrichment with limited cycle PCR. The sequencing libraries were then quality‐checked using 4200 TapeStation (Aglient Technologies), and quantified with Qubit 2.0 fluorometer (Invitrogen) and quantitative PCR (Applied Biosystems). The sequencing was performed on Illumina Hiseq4000 150PE platform.
RNA‐seq reads were analysed, prior to trimming, using fastqc version 0.11.7 (Andrews, 2010). Adapter sequences were then trimmed using Trimmomatic version 0.39 (Bolger et al., 2014). For the annotation of genes RNA‐seq reads were mapped onto the genome using star version 2.7 (Dobin et al., 2013), and a hint file was generated to predict intron‐exon boundaries using AUGUSTUS version 2.5.5 (Stanke et al., 2006). maker (Cantarel et al., 2008), SNAP (Korf, 2004), and the hint file generated from AUGUSTUS (Stanke et al., 2006) were then used to predict genes in the repeat‐masked reference genome. To help guide the prediction process, Swiss‐Prot peptide sequences of D. citri and P. venusta were used from UniProt to generate peptide evidence in the maker pipeline (Cantarel et al., 2008). The database was downloaded and used in conjunction with the protein sequences from A. pisum. Only genes that were predicted by both SNAP (Korf, 2004) and AUGUSTUS version 2.5.5 (Stanke et al., 2006) were retained in the final gene sets. To help assess the quality of the gene prediction, annotation edit distance (AED) scores were generated for each of the predicted genes as part of the maker pipeline (Cantarel et al., 2008). Functional annotation was further performed using blastp against the UniProt database.
To determine the relative read count for annotated genes, a custom index was created for the B. cockerelli genome using the HiSAT2 version 2.2.1 build command, and all trimmed reads were aligned to the genome using HiSAT2 version 2.2.1 (Kim et al., 2015). The outputs of the HiSAT2 alignment were compressed into bam files and then sorted using samtools version 1.14 (Li et al., 2009). Gene abundance tables were created using StringTie version 2.0.4 (Pertea et al., 2015) after inputting the sorted bam files.
2.6. Detecting contaminant proteins from the official Diaci_version 2.0 protein data set prior to orthologue analysis
To determine if the official Dcitri_OGSv2.0 (Diaci_version 2.0 protein) data set contained contaminated proteins from the common microbial contaminant Wallemia mellicola a blastp database was constructed using proteins from W. mellicola from NCBI (GCF_000263375.1_Wallemia_sebi_v1). A separate blastp database was also made with B. cockerelli proteins from our study. blastp was conducted separately on both databases using the Diaci_version 2.0 proteins as the query with an E value of 10e‐10 as a significance cutoff.
2.7. Comparative genomics and orthologue analyses
Orthologous clusters of CDSs from B. cockerelli, D. citri and P. venusta were determined using OrthoVenn2 (Xu et al., 2019) using default settings. Similar to Li et al. (2020) synteny of one‐to‐one orthologues in the chromosomes of B. cockerelli and P. venusta was determined using MCScanX_h (Wang et al., 2012), and SynVisio (Bandi & Gutwin, 2020) was used to visualize results. All B. cockerelli CDSs not assigned to clusters were screened for matches to the NCBI nr database (downloaded on 04/2021) using diamond version 2.0.13 (Buchfink et al., 2015) with an E value cutoff of 10e‐10. GO enrichment analyses were conducted within OrthoVenn2 (Xu et al., 2019). For KEGG pathway analysis of psyllid proteins, KO numbers were retrieved using KofamKoala (Aramaki et al., 2020) and analyses were conducted using KEGG‐Decoder (Graham et al., 2018) and KEGG Mapper (Kanehisa et al., 2022; Kanehisa & Sato, 2020).
To estimate rates of molecular evolution among B. cockerelli, D. citri and P. venusta we calculated nonsynonymous substitutions per nonsynonymous site (dN) and synonymous substitutions per synonymous site (dS) for one‐to‐one CDS orthologues from B. cockerelli, D. citri, and P. venusta. Similar to Degnan et al. (2010) the CDS were aligned based on the mafft (version 7.427) alignment of the amino acid sequences (Katoh & Standley, 2013), then all gaps and stop codons were removed, and pairwise estimates of (dN) and (dS) were calculated in Paml (Yang, 1997) based on the method of Goldman and Yang (1994). Gene pairs were excluded when dS was saturated (dS ≥ 3.0). In addition, this method of estimating rates of molecular evolution was used to evaluate SNP differences detected in our previously published Carsonella‐BC genome (Riley et al., 2017) and the short‐read data generated above (see below). Given the paucity of mutations in these Carsonella‐BC gene comparisons, we also excluded gene pairs when the number of synonymous or nonsynonymous changes was 0.
2.8. Gene expansion and contraction analyses
For analyses of gene contractions and expansions in psyllids, proteomes of B. cockerelli, 10 other hemipterans and 1 outgroup (Frankliniella occidentalis; Table S3) were first analysed with the busco (version 5.2.2; Manni et al., 2021) hemiptera_odb10 database to identify single‐copy orthologous genes (Table S4). Genes identified in all 12 genomes were individually aligned with mafft (version 7.427; Katoh & Standley, 2013), trimmed of gap containing positions and merged into a single alignment. This alignment was used for phylogenetic reconstruction of a species tree with RAxML‐HPC BlackBox (version 8.2.12; Stamatakis, 2014) on the CIPRES (Miller et al., 2010) webserver using the PROTCATJTT model of evolution and recommended default bootstrap parameter of “Let RAxML halt bootstrapping automatically”. Following the method outlined in the CAFÉ tutorial by Mendes et al. (2020) the species tree was converted to an ultrametric tree using r8s (version 1.7; Sanderson, 2003). This was based on an estimated divergence between the Psylloidea and Aphidoidea of 338 million years ago (Mya; Johnson et al., 2018).
The proteomes were then grouped into orthologous gene clusters using OrthoVenn2 with the default settings (Xu et al., 2019). Note that prior to clustering proteomes predicted gene isoforms were reduced to the single longest isoform. These clusters were then analysed to determine patterns of expansion and contraction over evolutionary time with café (version 5; Mendes et al., 2020). As recommended by Mendes et al. (2020), all clusters with ≥100 gene copies in a single species were initially filtered out, and a final birth‐death (λ) parameter was estimated based on an error model that accounted for the possibility of genome assembly errors. Large gene clusters were then analysed and final estimates of gene families significantly expanding or contracting across the phylogenetic tree were determined. Annotations, locations, organizations and sequences of genes belonging to expanding and contracting families within the Psylloidea were assessed. In addition to previous annotations discussed, cluster genes were also subjected to HMMER (2022) (hmmer.org) searches with the Pfam database (version 35.0, ‐‐cut_tc) (Mistry et al., 2021) and PANNZER2 (Törönen et al., 2018) rapid functional annotation tool using default parameters. Cluster functions were assigned based on a majority rule consensus of predicted functional annotations. Further, gene locations for members of rapidly expanding families predicted to be transposable elements were retrieved and identified as being euchromatic or heterochromatic based on the TAD analysis (above). The frequency of gene localizations within these distinct chromosomal regions were compared both within families and chromosomes using a two‐sample test for equality of proportions with continuity correction in r (version 4.1.2; R Core Team, 2020).
2.9. Identifying sequences from insect mitochondria and its obligate symbiont Carsonella
The mitochondrion genome sequence was acquired by mapping the short reads to the existing B. cockerelli mitochondrion reference sequence (NC_030055.1; Wu et al., 2016) from NCBI GenBank. Specifically, the OmniC Illumina paired‐end 150nt reads were trimmed using Trimmomatic 0.39 (Bolger et al., 2014) using the following settings (LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36). A total of 167,060,472 read pairs were trimmed with 92.86% surviving. Trimmed paired‐end reads were mapped to the Bactericera cockerelli mitochondrion (NC_030055.1) using bwa‐0.7.17 (Li, 2013) using default settings. A total of 132,773 reads mapped onto the reference genome and had a total of 1309× coverage. Consensus calling was performed with samtools 1.14 (Li et al., 2009) and bcftools 1.14 (Li, 2011) and the final consensus fasta file was created within bcftools 1.14 (Li, 2011) using the vcf2fq program and emboss 6.6.0 (Rice et al., 2000) using the seqret program. The Carsonella‐BC genome sequence was acquired as above for the mitochondrion by mapping the short reads to the existing Carsonella‐B. cockerelli‐reference sequence (CP019943.1; Riley et al., 2017). A total of 395,485 short reads mapped onto the reference genome and had a total of 341× coverage. Similar to Thairu et al. (2021) a custom Perl script was used to count the number of SNPs between the previously published Carsonella‐BC genome (Riley et al., 2017) and the consensus generated by short read mapping.
2.10. Identifying nutritional symbiosis‐related genes
To identify putative nutritional symbiosis‐related genes, KofamKOALA‐KEGG and NCBI‐BLAST were used to annotate amino acid metabolism pathways for all three psyllid species, B. cockerelli, D. citri and P. venusta. Specifically, we conducted both blastp and tblastn against the B. cockerelli protein data set and the initial PacBio assembly, respectively, using protein sequences from P. venusta and D. citri as the query with an E value of 10e‐10 as a significance cutoff. In addition, B. cockerelli's de novo mRNA transcripts were also investigated for putative symbiosis‐related genes in amino acid pathways based on previous symbiosis studies (Sloan et al., 2014). Specifically, the trimmed pair‐ended RNA‐seq reads from each of the five libraries (see above) were concatenated into a single de novo transcriptome assembly using Trinity version 2.13.2 (Grabherr et al., 2011). Any putative symbiosis transcripts identified using tblastn against P. venusta/D. citri query sequences were further screened against the final B. cockerelli scaffolds using nucleotide blastn to confirm that the transcripts were encoded in the psyllid's genome. If putative symbiosis genes were identified on unassembled PacBio contigs, contigs were further screened using blastx against the NCBI nr database (downloaded on 04/2021) using diamond version 2.0.13 (Buckfink et al., 2015) with an E value cutoff of 10e‐10 to determine if the contigs contained other psyllid/insect related genes, to further confirm that the transcripts were encoded in the psyllid's genome. Genes acquired through horizontal gene transfer that were initially identified in P. venusta (Sloan et al., 2014) were also searched against B. cockerelli's genome using the same blast protocol that was used for the symbiosis genes.
Amino acid biosynthetic pathways were reconstructed manually with HGT and symbiosis related genes using KEGG Mapper (Kanehisa et al., 2022; Kanehisa & Sato, 2020) for psyllids, the reference genomes for the psyllid symbionts Carsonella and Profftella (NC008512.1; CP003467.1; CP019943.1; CP003468.1), and the EcoCyc database (Keseler et al., 2013). The HGT genes were then aligned with other bacterial representative sequences using mafft (version 7.427) (Katoh & Standley, 2013). Phylogenetic analysis was performed on the alignment with RAxML‐HPC BlackBox version 8.2.12 on the CIPRES webserver (Miller et al., 2010) using the GTR model and 100 bootstrap replicates. The bipartition trees generated from RAxML were visualized and exported using figtree version 1.4.4 (Rambaut, 2018).
3. RESULTS
3.1. Whole chromosome assembly and annotation of Bactericera cockerelli
PacBio sequencing of Bactericera cockerelli DNA resulted in 5,869,245 reads with the initial filtered assembly resulting in 4411 scaffolds with 4409 scaffolds over 1 kbp resulting in a total assembly size of 567 Mb (Table 1). Using both the PacBio assembly and the Omni‐C library reads Dovetail HiRise scaffolding resulted in 51,238,984 read‐pairs with 3000 joins and 0 breaks to the initial assembly resulting in a total final assembly size of 567 Mb (Table 1). This final Dovetail HiRise Assembly was a significant improvement from the initial PacBio assembly in contiguity resulting in a 32% reduction in scaffolds (1411 total) with 1409 scaffolds >1 kbp and ~a 98X increase in N50 value compared to the initial filtered PacBio assembly (Table 1). The %GC of this final alignment was ~35% (±4.6 SD). To further evaluate the completeness of the genome Benchmarking universal single‐copy orthologues (BUSCOs; Simão et al., 2015) analysis was conducted on the final assembly and we found 93.7% of the complete BUSCOs were present based on the eukaryota database (Table S1).
TABLE 1.
Assembly statistics for the initial PacBio and the final Dovetail assembly of Bactericera cockerelli
| PacBio filtered assembly | Dovetail HiRise assembly | |
|---|---|---|
| Total length (bp) | 567,134,386 | 567,434,386 |
| Largest scaffold | 4,017,258 | 65,445,692 |
| Number of scaffolds | 4411 | 1411 |
| Number of scaffolds >1 kbp | 4409 | 1409 |
| Number of gaps | 16 | 3016 |
| Number of N's per 100 kbp | 0.06 | 52.93 |
| N50 | 423,673 | 41,634,607 |
| L50 | 331 | 6 |
| N90 | 53,963 | 21,995,751 |
| L90 | 1830 | 13 |
The TAD analysis pipeline identified 4998 CTCF Binding Sites, which are located at the boundaries of the TAD regions and are generally highly enriched in the regions of genome that are transcriptionally active (Bonev & Cavalli, 2016). TAD analysis further identified ~48% of the whole genome as active/euchromatin and ~51% as inactive/heterochromatin (Table S2).
The TAD analysis further revealed a total of 13 putative chromosomes that correspond to the 13 largest scaffolds from the final Dovetail HiRise assembly representing 92% of the final assembly length (~522 Mb; Table 2; Figure S1). We analysed the synteny of one‐to‐one orthologues from the 13 largest chromosomes/scaffolds of both B. cockerelli and P. venusta and found syntenic blocks of one‐to‐one orthologues among 11 autosomes and 1 sex chromosome (X) (Figure 1). However, all of the one‐to‐one orthologue blocks were highly rearranged within each scaffold/chromosome pair. In addition, we detected evidence of a chromosomal fission/fusion event accounting for two B. cockerelli chromosomes (bc9 and bc12) and one P. venusta chromosome (pv3; Figure 1). The 13th largest scaffold in P. venusta (pv12) was not indicated as a chromosome in Li et al. (2020); however, this scaffold shares 24 one‐to‐one orthologues with chromosome bc4 and one orthologue in both bc1 and bc3. These latter orthologues are not arranged in syntenic blocks (2 genes), however, and therefore are not indicated in Figure 1.
TABLE 2.
Chromosome scaffolds of Bactericera cockerelli identified using the TAD analysis pipeline
| Chromosomes | Scaffolds | Sizes |
|---|---|---|
| Chr. 1 | Scaffold_1 | 65,445,692 |
| Chr. 2 | Scaffold_2 | 55,905,524 |
| Chr. 3 | Scaffold_3 | 52,961,393 |
| Chr. 4 | Scaffold_4 | 50,895,300 |
| Chr. 5 | Scaffold_5 | 46,310,608 |
| Chr. 6 | Scaffold_6 | 41,634,607 |
| Chr. 7 | Scaffold_7 | 38,204,804 |
| Chr. 8 | Scaffold_8 | 33,343,378 |
| Chr. 9 | Scaffold_9 | 32,048,629 |
| Chr. 10 | Scaffold_10 | 30,660,657 |
| Chr. 11 | Scaffold_11 | 29,265,850 |
| Chr. 12 | Scaffold_12 | 23,608,477 |
| Chr. X (sex chr.) | Scaffold_13 | 21,995,751 |
FIGURE 1.
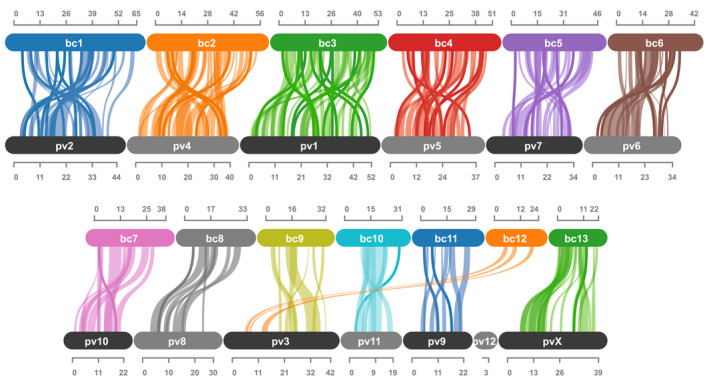
Comparison of syntenic orthologue blocks (2) between the 13 largest chromosomes/scaffolds identified in B. cockerelli (bc) and P. venusta (pv). Chromosomes/scaffold numbers correspond to the rank size of the scaffold within each species (1 is the largest). pvX represents the sex chromosome identified in Li et al. (2020). Rulers represent chromosome/scaffold length in Mb
Using our annotation pipeline (see Materials and Methods) we annotated 22,280 genes with an average length of 1726 bp within a total coding region of 38,474,975 bp. A total of 15.4% of genes were annotated as single‐exon genes. Using RepeatModeler version 2.01 (Flynn et al., 2020) and RepeatMasker version 4.1.0 (Smit et al., 2013) we found that 43.3% of the total genome was masked for repeats and 10.9%, 4.63%, 0.71%, and 2.07% of the genome is composed of Class I transposable element (TE) repeats, Class II TEs, low complexity repeats, and simple repeats, respectively. Annotation edit distance (AED) scores, were calculated for each gene (Table S5) to measure how well a predicted gene is supported by external evidence (UniProt protein and mRNA sequences). The AED score ranges from 0–1 where a lower score represents more evidence support for a gene. We found that the majority of annotated genes (80%) possess an AED score under 0.5 (Figure S2).
The 15.2 kbp mitochondrion genome sequence obtained here had 100% coverage and 100% identity to the B. cockerelli mitochondrion reference sequence (NC_030055.1) using blastn. In contrast, in the Carsonella‐BC symbiont genome we identified a total of 2143 SNPs in the 174 kbp genome. These differences were distributed among 182 of the 195 predicted CDSs. None of the differences resulted in premature stop codons, but 155 CDSs did have ≥1 nonsynonymous mutation that would alter the amino acid position. However, estimates of dN/dS indicated that all of these Carsonella‐BC CDSs are under strong purifying selection (negative selection) (dN/dS = 0.065 ± 0.0743). This is consistent with what has been previously observed among other Carsonella genomes (Sloan & Moran, 2012).
3.2. Comparative genomics of psyllid species with complete genomes
We compared B. cockerelli coding sequences annotated in this study (Table S5) to two other psyllid species with fully sequenced genomes, P. venusta (Li et al., 2020) and D. citri (Saha et al., 2017). During the preliminary analysis of proteins from the official D. citri (Diaci version 2.0) protein data set (Saha et al., 2017) that is available publicly at https://citrusgreening.org, we found hundreds of putative fungal proteins, many that are only conserved in fungi and bacteria in the psyllid protein data set. We conducted BLASTp on these suspect proteins and they had 100% amino acid identity to the fungus Wallemia mellicola (previously sebi) and did not hit any of the D. citri v. 1.0 proteins that are currently available in the NCBI database. On further inspection we also found conserved housekeeping proteins (which are present in animals as well) that had 100% amino acid identity to W. mellicola. A total of 717 Diaci_version 2.0 proteins were identified as likely fungal contaminants, with 389 having 100% identity to W. mellicola proteins. While many (n = 421) had some homology with B. cockerelli, these proteins had an average of 47% greater amino acid identity to W. mellicola. The remainder had no B. cockerelli homologue (n = 296) and ≥90% amino acid identity to W. mellicola. Wallemia mellicola is a xerophilic food‐ and airborne fungus that commonly contaminates foodstuffs, especially highly sugared materials (Zalar et al., 2005). We removed these putative contaminant W. mellicola fungal proteins (n = 717) from the official Diaci_version 2.0 protein data set for all analyses conducted in this study.
We found that all three psyllid species share 6139 orthologous clusters and genes within these clusters are significantly enriched for three GO term categories (DNA integration, GO:0015074; transposition, DNA‐mediated, GO:0006313; RNA‐directed DNA polymerase activity, GO:0003964; Table S6). Orthologous clusters that are only present in B. cockerelli (557 clusters) compared to P. venusta and D. citri are primarily related to transposition and protein processing based on GO enrichment analyses (Table S6).
Among the shared orthologous clusters found in all three psyllid species 2016 genes were identified as single‐copy 1:1:1 gene clusters (Table S7). We analysed these single‐copy genes for signatures of natural selection by calculating dN and dS values among each species pair. Overall, for all single copy orthologue pairs, where dS was not saturated the dN/dS ratio was <1, indicating that purifying selection is widespread among single‐copy 1:1:1 orthologues. Specifically, we found 1323 gene pairs between B. cockerelli and D. citri that had an average dN/dS ratio of 0.06 (± 0.04 SD). A total of 127 gene pairs between B. cockerelli and P. venusta had an average dN/dS ratio of 0.05 (± 0.05 SD), and a total of 285 gene pairs between P. venusta and D. citri had an average dN/dS ratio of 0.05 (± 0.05 SD). None of these gene pairs were significantly enriched for GO terms; however, 33, 2, and 10 gene pairs under purifying selection in B. cockerelli and D. citri, B. cockerelli and P. venusta, and P. venusta and D. citri, respectively, were related to nutrition and 19, 10, and 16 gene pairs in B. cockerelli and D. citri, B. cockerelli and P. venusta, and P. venusta and D. citri, respectively, were related to environmental response genes and immunity (Table S8).
A total of 3892 proteins in B. cockerelli were not found in any orthologous clusters and they were characterized further with DIAMOND (Buchfink et al., 2015). A total of 64.3% (1973) of these query sequences had a significant best hit to D. citri (mean amino acid identity ~69.9% [± 17.1 SD]). While the remaining 35.7% (1,094) of query sequences with significant hits primarily hit species within Insecta with a minority hitting species in Arachnida (mean amino acid identity ~58.2% [± 17.0 SD]). Most of these putative proteins were classified as “hypothetical”, “unnamed”, or “uncharacterized”. A minority of query sequences (4.7% [825]) had no significant hits to the NCBI nr database and appear to be orphan genes.
3.3. Large number of gene family expansions in Bactericera cockerelli
A fully resolved hemipteran phylogeny with strong maximum likelihood bootstrap support was estimated based on 632 complete, single‐copy genes shared by B. cockerelli and 11 additional genomes (of 2510 Hemipteran BUSCOs; Tables S3 and S4; Figure 2). After converting the tree branch lengths to ages (Mya), the expansions and contractions of 22,478 predicted gene families were estimated (Table S9; Figure 2). This analysis suggested that B. cockerelli and D. citri diverged from one another ~86 Mya and their most recent common ancestor diverged from P. venusta ~130 Mya. Note that the proteomes used were not masked for repeats prior to clustering (Table S3). Across the entire phylogeny B. cockerelli had the greatest number of significantly expanding protein families (n = 157) and zero contracting families. Nearly a third of these families (52/157) are unique to psyllids and nearly a fifth (30/157) are unique to B. cockerelli. Perhaps unsurprisingly, over three quarters of the families have annotations consistent with either type I (RNA intermediate) or type II (DNA intermediate) repetitive elements (120/157; Figure 3).
FIGURE 2.
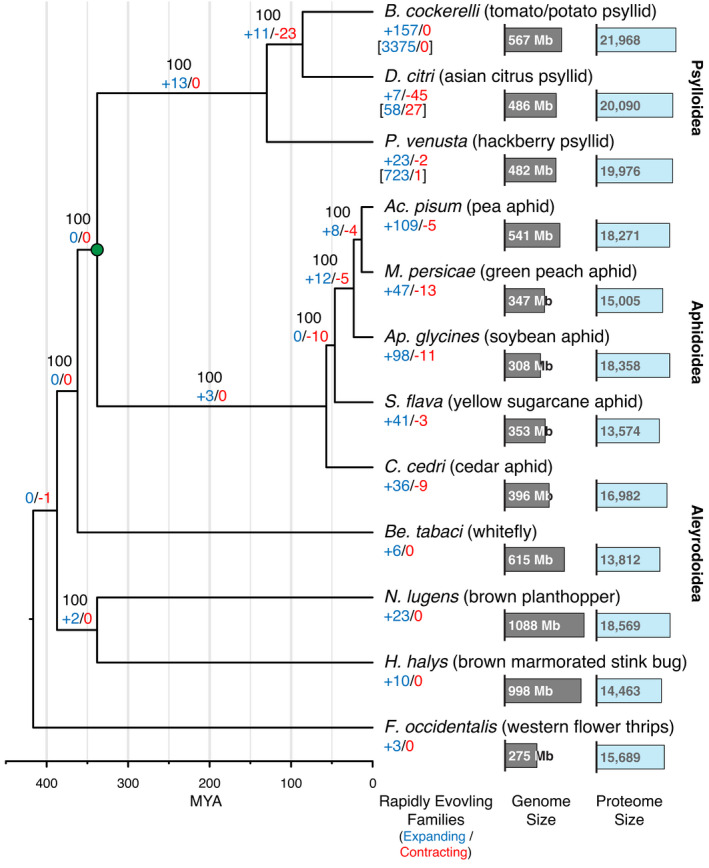
Evolutionary relationships of B. cockerelli and representative hemipterans. Maximum likelihood phylogeny of 632 conserved, single copy orthologues (a 234,854 amino acid alignment) was used as the basis to identify trends in gene family expansions and contractions (Lambda [birth‐death] = 0.0023307 – probability any gene will be gained or lost). The node marked with a green circle represents the estimated divergence between the Psylloidea and Aphidoidea of 338 Mya (Johnson et al., 2018) used to calibrate the ultrametric tree. Numbers in black at each node represent the bootstrap values, and the numbers of significantly expanding (blue) and contracting (red) gene families are also labelled. Among the Psyllodiea the number of genes represented by the expanding or contracting families are listed within brackets. Below each species name the assembled genome and proteome sizes are indicated and shown as proportionally sized horizontal bars, grey and blue, respectively
FIGURE 3.
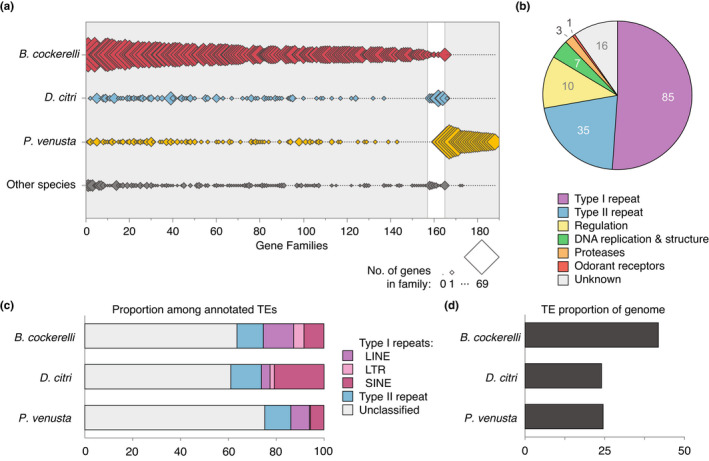
Characteristics of the significantly expanding and contracting gene families among the Psylloidea. (a) Diamond plot of the 188 total significantly expanding and/or contracting gene families in the Psylloidea. The size of the diamond represents the number of genes in each gene family and range from 0 to 69 genes. The category “other species” represent the average gene copy number among the nine other species analysed in Table S3. The horizontal axis represents rank order of homologous gene families that are present or absent in each lineage. Gene families 1–157 are expanding in B. cockerelli, families 158–164 are expanding in D. citri, and family 165 is contracting and families 166–188 are expanding in P. venusta. (b) Distribution of predicted functions of expanding gene families in B. cockerelli. (c) Proportion of different transposable element (TE) classes ≥100 nt in length and (d) total TE percent content in the genomes of B. cockerelli, D. citri, P. venusta. Data from D. citri was estimated from Gilbert et al. (2021), and data from P. venusta was retrieved from Petersen et al. (2019)
To evaluate the effect of chromosomal confirmation and activity on recent transposition, we determined the proportion of recently expanded transposon gene families within euchromatic (A) or heterochromatic (B) regions. We compared the relative proportions on each chromosome to the proportion of each chromosome comprised of euchromatin and heterochromatin. Throughout the majority of the genome, there is no significant bias in recent transposition events within euchromatic or heterochromatic regions (Figure S3). However, on chromosome/scaffold_1 we did detect a significantly greater proportion of recent transpositions within the heterochromatic regions of this chromosome/scaffold. By analysing the euchromatic or heterochromatic localization within individual gene families with more than 10 gene copies (n = 94) we identified 14 families significantly biased towards heterchromatin and 17 significantly biased towards the euchromatic regions (Proportion test, d.f. = 1, p < .05). This revealed that at least part of the trend detected on chromosome/scaffold_1 was associated with three, type II (DNA) repeat families where the vast majority of copies were in the heterochromatic regions of chromosome/scaffold_1.
While 16 families have no identifiable functions, there were several expanded families we detected in B. cockerelli with putative functional annotations that were not type I or II repeats. For instance, expansions of putative regulatory factors such as zinc finger genes, including C2H2‐ZNF domain containing homologues (PF00096) which constitutes one of the largest classes of transcription factors in eukaryotes (119 proteins in eight families) and helix‐turn‐helix motif regulators (12 proteins in 1 family) were detected. In addition, we detected expansions in several families of proteases including metalloproteases and zinc carboxypeptidases. Genes from one of these protease families consistently had a significant best blastp hit to NCBI nr for carboxypeptidase D in other insects however identity in the top 10 hits was low and ranged 36%–42% with a query coverage >90%. Finally, a family of 12 B. cockerelli specific putative seven transmembrane odorant receptors (7tm) were identified. Together these regulatory, proteolytic and odorant receptor genes may be key factors in B. cockerelli for species specific regulation and host plant interactions.
In contrast to B. cockerelli, substantially fewer gene families were identified as expanding or contracting in D. citri and P. venusta despite the general similarity in their proteome sizes (Figure 2). In fact, the entirety of contracting gene families in D. citri (n = 45) were identified as expanding families in B. cockerelli (Figure 3). Further, unlike B. cockerelli only one of the expanding families in D. citri is a type I repetitive element. The six remaining expanding families are involved in chromosome structure (n = 3; histone like proteins, kinetochore component), detoxification (n = 1; cytochrome P450), cell‐cell interactions (n = 1; fibronectin III binding domain proteins) and organic solute transport (n = 1). In P. venusta, the overwhelming majority of expanding gene families have no predicted functions (19/23). Of the remainder, three are likely type I or type II repetitive elements and one is a cysteine protease. Finally, we examined the rapidly evolving families along the internal branches of the Psylloidea clade only to find that all but one of the expanding and contracting families are represented in the terminal gene families. The only unaccounted for gene family is yet another type I repetitive element.
3.4. Identification of putative symbiosis genes in Bactericera cockerelli
We investigated the presence and absence of amino acid biosynthesis genes among the three psyllid species, B. cockerelli, D. citri and P. venusta, as well as their obligate endosymbionts (Figure 4). We identified a total of 54 B. cockerelli genes that may collaborate with Carsonella based on previous bacteriocyte gene expression studies in psyllids, aphids and other sap‐feeding insects (Hansen & Moran, 2011; Husnik et al., 2013; Sloan et al., 2014; Tables 3 and Table S10). Overall, these genes are largely conserved across all three psyllid species including those that were horizontally transferred from bacteria (Figure 4, Table 4).
FIGURE 4.
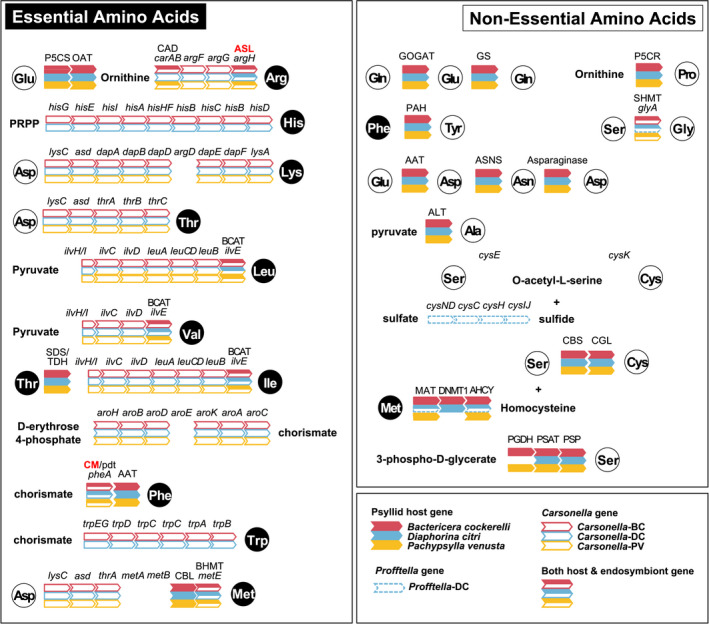
Amino acid biosynthesis pathways present in B. cockerelli, D. citri, P. venusta, and their endosymbionts. Arrows are coloured/lined accordingly for each host/endosymbiont‐encoded gene (see legend). Gene names in bold (red) indicate horizontally transferred genes of bacterial origin into the psyllids' genomes
TABLE 3.
Amino acid/symbiosis‐related genes involved in the amino acid metabolism of Bactericera cockerelli based on KEGG and blast
| Name | KO | Enzyme | B. cockerelli Protein ID a | Expression percentile b |
|---|---|---|---|---|
| P5CS | K12657 | Delta‐1‐pyrroline‐5‐carboxylate synthetase [EC:2.7.2.11/1.2.1.41] | ANN07147(8)‐RA | 98% (90%) |
| OAT | K00819 | Ornithine‐‐oxo‐acid transaminase [EC:2.6.1.13] | ANN07990(1)‐RA | 91% (84%) |
| BCAT | K00826 | Branched‐chain amino acid aminotransferase [EC:2.6.1.42] | ANN10973‐RA | 85% |
| AAT | K14455 | Aspartate aminotransferase [EC:2.6.1.1] | ANN05170‐RA | 62% |
| ANN05183‐RA | 69% | |||
| ANN05186‐RA | 65% | |||
| ANN05257‐RA | 99% | |||
| ANN17803(4)‐RA | 98% (99%) | |||
| BHMT | K00547 | Homocysteine S‐methyltransferase [EC:2.1.1.10] | ANN06058‐RA | 80% |
| ANN06059‐RA | 60% | |||
| GOGAT | K00264/K00265/K00266 | Glutamate synthase (NADH) [EC:1.4.1.13/1.4.1.14] | ANN19375‐RA | 94% |
| GS | K01915 | Glutamine synthetase [EC:6.3.1.2] | ANN10145‐RA | 52% |
| ANN10146‐RA | 97% | |||
| ANN10148‐RA | 96% | |||
| ANN10149‐RA | 42% | |||
| PAH | K00500 | Phenylalanine‐4‐hydroxylase [EC:1.14.16.1] | ANN09753‐RA | 95% |
| ASNS | K01953 | Asparagine synthase [EC:6.3.5.4] | ANN02335‐RA | 81% |
| ANN09881‐RA | 71% | |||
| ANN21723‐RA | 84% | |||
| ASPG | K01424/K13051 | L‐asparaginase [EC:3.5.1.1] | ANN07324‐RA | 68% |
| ANN21497‐RA | 17% |
Psyllid genes in tandem that were reannotated to be a single protein after manual annotation are indicated with parentheses.
The average gene expression percentile was determined based on relative gene expression (transcript per million, TPM) of the target gene relative to all genes annotated in the final assembly. RNAseq data were derived from five pooled psyllid whole body samples (n = 5; see Table S11).
TABLE 4.
Bactericera cockerelli genes of bacterial origin acquired by horizontal gene transfer
| Gene | Protein ID | Scaffold | Expression a | P. venusta unigene b | Amino acid similarity b |
|---|---|---|---|---|---|
| Argininosuccinate lyase | |||||
| ASL‐1 | ANN12874‐RA | Scaffold_7 | 56% | 116240_c1 | 68% |
| ASL‐2a | ANN10361‐RA | Scaffold_5 | 58% | 113612_c8 | 91% |
| ASL‐2b | ANN20354‐RA | Scaffold_15 | 48% | 113612_c8 | 91% |
| Chorismate mutase | |||||
| CM‐1 | ANN05927‐RA | Scaffold_2 | 35% | 113157_c6 | 30% |
| CM‐2 | ANN06705‐RA | Scaffold_2 | 93% | 113157_c6 | 33% |
| CM‐3 | ANN17115‐RA | Scaffold_9 | 81% | 113157_c6 | 62% |
| A/G‐specific adenine glycosylase | |||||
| MUTY | ANN05978‐RA | Scaffold_2 | N/A | 106533_c0 | 64% |
| AAA‐ATPase‐like | |||||
| ORF‐1a | ANN01458‐RA | Scaffold_1 | 61% | 111207_c2 | 44% |
| ORF‐1b | ANN01460‐RA | Scaffold_1 | 68% | 111207_c2 | 68% |
| ORF‐2 | ANN17656‐RA | Scaffold_10 | 30% | 115390_c0 | 25% |
| ORF‐3a | ANN18155‐RA | Scaffold_10 | 18% | 115390_c0 | 57% |
| ORF‐3b | ANN18147‐RA | Scaffold_10 | 0% | 115390_c0 | 53% |
| Riboflavin synthase | |||||
| ribC | ANN14810‐RA | Scaffold_11 | 73% | 97442_c1 | 60% |
| 16S rRNA methyltransferase | |||||
| RSMJ | ANN13802‐RA | Scaffold_8 | 10% | 107791_c0 | 41% |
| VOC family protein | |||||
| YDCJ | ANN12599‐RA | Scaffold_7 | 93% | 111380_c3 | 70% |
The average gene expression percentile was determined based on relative gene expression (transcript per million, TPM) of the target gene relative to all genes annotated in the final assembly. RNAseq data were derived from five pooled psyllid whole body samples (n = 5; see Table S11).
Amino acid similarity of B. cockerelli HGT genes using blastx against P. venusta's Trinity unigene assemblies (Sloan et al., 2014).
3.4.1. Nonessential amino acid biosynthesis pathways
Carsonella does not encode nonessential amino acid biosynthesis pathways except for glyA in the glycine pathway (Nakabachi et al., 2006). In turn, these metabolites need to be provided by either their psyllid host and/or their psyllid host's diet. All three psyllid species encode asparagine, aspartate and tyrosine biosynthesis pathways via asparagine synthase (ASNS, EC 6.3.5.4), asparaginase (ASPG, EC 3.5.1.1) and phenylalanine‐4‐hyroxylase (PAH, EC 1.14.16.1), respectively (Table 3, Figure 4). In addition, psyllid genes involved in the biosynthesis of other nonessential amino acids, such as proline, tyrosine, serine and alanine, have also been identified (Table S10, Figure 4). Moreover, all three psyllid genomes contain glutamine synthase (GS, EC 6.3.1.2) and glutamate synthase (GOGAT, EC 1.4.1.13). These latter genes are potentially crucial for recycling ammonia in bacteriocytes via the GOGAT cycle to serve as the building blocks and amino donors in EAA biosynthesis (Table 3, Figure 4; Hansen & Moran, 2011; Sloan et al., 2014). Interestingly, four intact copies of GS are encoded in B. cockerelli whereas only one copy of this ammonia recycling enzyme is present in the other two psyllid genomes (Table 3). All four gene copies are unique in identity and have an average amino acid identity of 82.8% (±7.1 SD) based on blastp. Gene copies are located in tandem in pairs (e.g., ANN10145‐RA & ANN10146‐RA; ANN10148‐RA & ANN10149‐RA) with one gene (COPA; Coatomer subunit alpha, ANN10147‐RA) located between the gene pairs on Scaffold 5. Gene expression of one gene copy in each tandem gene pair (ANN10146‐RA and ANN10148‐RA) was highly expressed in B. cockerelli (>the 96th percentile) whereas the other two gene copies were expressed moderately (42nd and 52nd percentile) relative to other genes in B. cockerelli's whole body (Table 3).
3.4.2. Psyllid genes that collaborate with Carsonella for essential amino acid biosynthesis
Similar to the nutritional endosymbionts of other plant phloem‐feeding insects, Carsonella has lost the genes argABCDE, which synthesize ornithine (Hansen & Moran, 2014). All three psyllid genomes encode the genes delta‐1‐pyrroline‐5‐carboxylate synthetase (P5CS, EC 2.7.2.11/1.2.1.41) and ornithine transaminase (OAT, EC 2.6.1.13) that synthesize ornithine from glutamate in the arginine biosynthesis pathway, and they are highly expressed (above the 80th percentile) in the whole body relative to other genes in B. cockerelli (Table 3, Figure 4). Carsonella has also lost the terminal step for phenylalanine biosynthesis (aspC), nevertheless all three psyllid species genomes encode aspartate aminotransferase (AAT, EC 2.6.1.1), which may complement Carsonella's phenylalanine biosynthesis pathway (Table 3, Figure 4). There is a total of one cytoplasmic and four mitochondrial gene copies of AAT that are expressed at a moderate to high level in B. cockerelli relative to other genes in B. cockerelli (Table 3).
In a few cases psyllid host genes are similar in enzymatic function, depending on the direction of the reaction, when compared to genes in Carsonella. For example, metE, which encodes homocysteine S‐methyltransferase is retained in all three Carsonella genomes and homocysteine S‐methyltransferase (BHMT, EC 2.1.1.10) is encoded in all three psyllid genomes as well (Figure 4). We found two BHMT gene copies in B. cockerelli and their expression levels were both above the 60th percentile relative to the expression of all other B. cockerelli genes for the whole body (Tables S10 and S11). The gene ilvE, which encodes branched‐chain aminotransferase (BCAT, EC 2.6.1.42) and catalyses the terminal step in the biosynthesis of isoleucine, leucine, and valine, is retained in all three Carsonella genomes as well (Figure 4). The BCAT gene in B. cockerelli was identified (ANN10973‐RA) and was expressed above the 85th percentile relative to the expression of all other B. cockerelli genes for the whole body (Table 3).
3.4.3. HGT in the B. cockerelli genome
We identified seven B. cockerelli genes that were horizontally transferred from bacteria into psyllids (Table 4). These results are largely consistent with the horizontally transferred genes identified initially in Sloan et al. (2014) in P. venusta, except the mutY gene (comp106533_c0_seq1) was not identified in the B. cockerelli genome; however, it is still retained in D. citri. However, we identified that multiple gene duplications occurred among four of these horizontally transferred genes in the B. cockerelli and D. citri genomes but not P. venusta (Table 4). For example, we found that both B. cockerelli and D. citri encode three copies of the arginosuccinate lyase (ASL, EC 4.3.2.1) gene while P. venusta only encodes two (Sloan et al., 2014) (Figure 5a). ASL is hypothesized to be involved in the end step of the arginine pathway with Carsonella (Figure 4; Sloan et al., 2014). Our phylogenetic reconstruction of the ASL genes reveals that all three psyllid species encode two divergent copies of ASL that share a most recent common ancestor and are most similar to ASL gene copies found within the Gammaproteobacteria (Figures 5a, S4 and S5). The most parsimonious explanation for this observation is that the ASL gene was acquired by horizontal transfer and was duplicated early in the ancestor of psyllids as suggested by Sloan et al. (2014). Curiously, B. cockerelli and D. citri both appear to have had a very recent independent duplications of one of these gene copies (Figures 5a, S4 and S5). The amino acid percent identity between these new paralogs in B. cockerelli are 100% and both paralogues have two introns. For D. citri, the two paralogs have 99% nucleotide similarity with one intron. In B. cockerelli all ASL gene copies are located on different scaffolds (Table 4) and are surrounded by different genes suggesting that the paralogs did not arise from a tandem duplication. Relative gene expression of all three gene copies are very similar to one another and range between 48th–56th percentile relative to other genes in B. cockerelli (Table 4). Sloan et al. (2014) previously suggested that the ASL gene was derived from Carsonella; however, here we find inconsistencies in our results in regard to whether this gene is derived from Carsonella or another related endosymbiont due to long branch attraction (Bergsten, 2005) of psyllid/endosymbiont sequences and lower branch support for some nodes (Figures S4 and S5).
FIGURE 5.
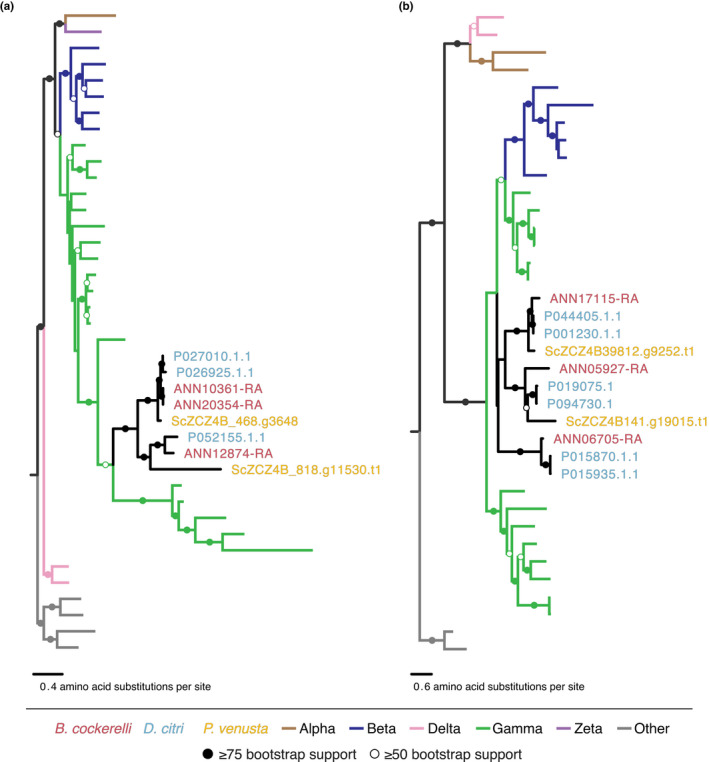
Phylogenetic analyses of gene duplications for horizontally transferred genes in the psyllid genomes of P. venusta, D. citri, and B. cockerelli for (a) argininosuccinate lysase (ASL) genes and (b) chorismite mutase (CM) genes. Branches are coloured according to their bacterial classes within the Proteobacteria and ranges of bootstrap values are indicated by filled or open circles as indicated in the key. See Figures S4, S5, and S6 for trees with detailed taxa names and accession numbers
A second horizontally transferred gene that may collaborate with Carsonella for essential amino acid biosynthesis is chorismite mutase (CM, EC 5.4.99.5), which is involved in the phenylalanine pathway (Sloan et al., 2014; Table 4, Figure 4). We identified three CM genes in the B. cockerelli genome, two genes in P. venusta (Sloan et al., 2014), and six genes in D. citri (Figure 5b). The three CM gene copies in B. cockerelli were all expressed at variable levels (35th–81st percentile) in the B. cockerelli body (Table 4). A phylogenetic analysis indicates that there were two early duplications of CM in psyllids and a likely loss of one of those copies in P. venusta (Figure 5b). Subsequently, D. citri experienced further duplications for each of these paralogues resulting in the six copies present in the proteome. These D. citri copies consist of three nearly identical sets of gene copies with 100% nucleotide sequence similarity except for the gene pair, P019075.1 and P094730.1.1 (Figure 5b). Precise origins of the transfer event are unclear; however, it is clear that a member of the Proteobacteria was the donor (Figures 5b and S6).
4. DISCUSSION
In this study we reveal that the B. cockerelli genome has experienced significantly more gene family expansion events compared to other Hemipteran representatives with fully sequenced genomes (Figure 2). Nearly 75% of the these expanding gene families in B. cockerelli are transposable elements that are rampant across all of B. cockerelli's chromosomes. These transposable elements are primarily composed of type I and II elements which occur in equal proportions throughout both the euchromatin and the heterochromatin regions of all of B. cockerelli's chromosomes except for chromosome 1, where we detected a bias of transposable elements in heterochromatic regions. Various drivers of transposable element expansions in animals including insects have been determined including the mode of reproduction, social complexity, ecology, and host mechanisms for repeat containment (Gilbert et al., 2021; Lynch, 2007). At this time, it is unclear what is driving mobile element gene family expansions in B. cockerelli. For example, B. cockerelli has a sexual mode of reproduction with a 50/50 sex ratio (Abernathy, 1991; Yang & Liu, 2009) therefore a reduced level of recombination, which can result in transposable element accumulation, is not expected for this insect species. It is also unlikely that these expansions are exclusively an artifact of laboratory rearing as 154/157 of the expanded gene families (representing ~99% of the genes) have average amino acid identities ≤90%. In insects the likeliest candidate for host mechanisms for transposon containment is the Piwi‐interacting RNA (piRNAs) pathway (Brennecke et al., 2007). Although B. cockerelli encodes the proteins Piwi and Argonaute 3 (BC, ANN11580‐RA and ANN07423‐RA, respectively) which are involved in this pathway (Dowling et al., 2016), the functional role of piRNAs in silencing transposons in B. cockerelli has not yet been described.
The expansion of mobile elements in B. cockerelli's genome has implications for impacting the evolution of insect genome architecture and adaptation to its ecological niche. For example, Petersen et al. (2019) revealed a positive correlation between transposable element content and insect genome size. We found that B. cockerelli's genome is the largest psyllid genome (567 Mb) sequenced to date and is ~15% larger than the other two psyllid species genomes (P. venusta and D. citri). The fraction of transposable element content in B. cockerelli's genome is ~2× larger than the content measured in P. venusta (Petersen et al., 2019) and D. citri (Gilbert et al., 2021; Peccoud et al., 2017), in addition to the median size of transposable element content (24.4% [±12.5% SD]) that is found in insects belonging to five orders (Petersen et al., 2019). Consequently B. cockerelli's genome size may partly be driven by the expansion of transposable elements in its genome. Transposable elements can also significantly impact genome architecture by inducing chromosomal rearrangements via nonhomologous recombination (Burns & Boeke, 2012). Structurally, B. cockerelli appears to have an additional chromosome compared to the distantly related psyllid species P. venusta due to a previous chromosomal fission or fusion event (Figure 1). When more psyllid species genome assemblies are resolved at a chromosomal‐level we will have a better understanding of the dynamics of chromosomal loss and/or gain across Psylloidea. Although we detected extensive conservation of syntenic gene blocks among the B. cockerelli and P. venusta shared chromosomes that we estimated diverged ~130 Ma, we detected a substantial amount of intrachromosomal gene block rearrangements between the homologous chromosomes (Figure 1). The dynamic nature of the B. cockerelli genome may largely be contributed to the widespread expansion of type I and II repeat elements that are omnipresent throughout all of B. cockerelli's chromosomes.
Transposable elements can also impact insect adaptation either negatively or positively depending on the environmental context (Gilbert et al., 2021). Given that transposable elements are equally expanding in the euchromatin regions of B. cockerelli's genome the disruption of coding sequences and/or the rewiring of regulatory circuits is plausible. It is hypothesized that transposable element insertions are primarily deleterious or neutral however transposable element mediated mutations can occasionally be adaptive. For example, transposable element mediated insecticide resistance, adaptation to temperate climates and oxidative stress, and faster developmental times have all been documented in insects (Gilbert et al., 2021; Petersen et al., 2019). Classic examples of natural selection in insects have also been contributed to transposable element insertions such as the industrial melanism colour polymorphism of the peppered moth Biston betularia, which helped populations evade bird predation in coal polluted environments (van't Hof et al., 2016). Interestingly, transposable elements have recently been found to be important, and sometimes essential components of antiviral immunity in insects (Gilbert et al., 2021). The psyllid B. cockerelli is known to be associated with insect viruses in field populations (Dahan et al., 2022), however it is unclear at this time what role its mobilome plays in facilitating antiviral interactions.
In addition to mobile element gene family expansions, we also found significant expansions of gene families in B. cockerelli that are associated with transcription factors, proteases, and odorant receptors. The expansion of transcription factors in B. cockerelli's genome may increase the plasticity of gene expression among environments or conversely provide more stability for the transcription of conserved genes depending on how many gene targets the transcription factors bind to (Yang & Wittkopp, 2017). The expansion of protease gene families in B. cockerelli may also be very important in the evolution of B. cockerelli by facilitating species specific responses to host plant defenses. For example, the host plants of B. cockerelli, such as tomato and potato are widely known to produce carboxypeptidase inhibitors during insect feeding and wounding (Díez Díaz et al., 2004; Graham & Ryan, 1981). These inhibitors can produce an antifeedant effect for insects (Zhu‐Salzman & Zeng, 2015), and therefore B. cockerelli may be involved in an arms race with its solanaceous host plant defences and its rapidly evolving carboxypeptidase families. Another expanding gene family in B. cockerelli that may help this psyllid species adapt to its environment especially in regard to host‐plant volatiles, mating and intraspecific communication are odor receptors. Compared to the other two psyllids species examined in this study only B. cockerelli was observed to have significant gene family expansions in odorant receptors. It will be of interest for further studies to determine how and if these expanding seven transmembrane odorant receptors influence B. cockerelli host plant recognition, as B. cockerelli is a polyphagous herbivore that can feed and reproduce on a wide variety of host plant species (Pletsch, 1947; Wallis, 1955).
The analysis of gene duplication events is largely dependent on the quality of genome assemblies. Taking advantage of our chromosomal‐level assembly we were able to detect gene duplications for several genes that are important for insect‐microbe interactions. Three of these genes that duplicated in B. cockerelli, argininosuccinate lyase (ASL), chorismate mutase (CM), and the AAA‐ATPase‐like protein, were horizontally transmitted from bacteria into psyllids before the divergence of the psyllid lineages examined (Sloan et al., 2014). The genes ASL and CM may be important in the regulation and collaboration with Carsonella for the biosynthesis of the essential amino acids arginine and phenylalanine, respectively (Figure 4; Sloan et al., 2014). Our analyses uncovered additional copies of the genes ASL and CM in B. cockerelli and D. citri compared to P. venusta (Figure 5). It will be of interest in future studies to determine if these additional copies have different functions and differential expression patterns in the symbiotic cells of psyllids compared to other body tissues. Additionally validating that these copies in D. citri are actual gene copies will be important when a chromosomal assembly for D. citri is publicly available, as several copies shared 100% amino acid identity. Another important gene duplication in B. cockerelli that is involved in the nutritional symbiosis is glutamine synthase (GS), which is hypothesized to recycle ammonia in symbiotic cells via the GOGAT cycle to provide amino donors for EAA biosynthesis (Table 3, Figure 4; Hansen & Moran, 2011; Sloan et al., 2014). Four gene copies of GS were identified in B. cockerelli and we expect future studies will be able to characterize the expression patterns and functional role of these multiple copies.
Our analysis of core single copy orthologues shared by B. cockerelli and the other psyllids identified an array of genes under strong purifying selection that are related to insect‐microbe interactions and immunity (Table S8). Genes that are maintained under purifying selection infer that these genes have an important conserved function and are candidates for future investigations. Interestingly two of these genes have homologues in pea aphids that are hypothesized to be involved in the nutritional amino acid symbiosis with its endosymbiont Buchnera (Feng et al., 2019; Pers & Hansen, 2021). This includes the amino acid transporter SLC36‐like AAAP, which is hypothesized to be important in transmembrane transport of the small nonessential amino acids such as proline, alanine, and glycine in symbiotic cells of aphids (bacteriocytes; Feng et al., 2019). This transporter is expressed the highest in first instar bacteriocytes of A. pisum and is significantly downregulated in bacteriocytes during the first to second instar transition and it is downregulated further in the second to third instar transition (Pers & Hansen, 2021). The other protein that is under purifying selection in B. cockerelli is in the kynurenine pathway (kynurenine formamidase/cyclase‐like) and is involved in the degradation of tryptophan. The homologue of this protein in A. pisum is expressed the highest in A. pisum bacteriocytes at first and second instar and is significantly downregulated from second to third instar and is further downregulated during its third to fourth instar transition (Pers & Hansen, 2021). The functional role of kynurenine intermediates in insect bacteriocytes in not well understood; however, ommochromes from this pathway are primarily found in insect ommatidia but are also known to be important in reducing oxidative stress (Figon & Casas, 2019; Lugo‐Huitrón et al., 2011), which may be crucial for allowing this symbiosis to function in metabolically active bacteriocytes.
In summary, taking advantage of our high‐quality chromosomal genome assembly we uncovered a large expansion of gene families and gene duplications events that play a large role in shaping the genome architecture and function of B. cockerelli. The significant increase of transposable elements in B. cockerelli's genome probably has significant consequences for B. cockerelli's genome architecture and adaptation through the restructuring of regulatory circuits and insertion mutations of important genes. Moreover, recent gene duplications of genes that were horizontally transferred into B. cockerelli's genome from bacteria may have species specific consequences for its nutritional symbioses with Carsonella. The genomic resources and candidate genes uncovered here will provide ample resources for future studies on this system.
AUTHOR CONTRIBUTIONS
Younghwan Kwak performed the research, analysed data, and wrote the manuscript. Jacob A. Argandona analysed data. Patrick H. Degnan analysed data, and wrote the manuscript. Allison K. Hansen designed research, analysed data, and wrote the manuscript.
CONFLICT OF INTEREST
The authors have no competing interests.
Supporting information
Figures S1–S6
Tables S1–S11
ACKNOWLEDGEMENTS
AH's research was supported by funding from the National Institute of Food and Agriculture (NIFA), United States Department of Agriculture (USDA) (Award number: 2019‐70016‐29066), the Department of Entomology at the University of California, Riverside (UCR). This research was further supported by initial complement funding to PHD from the Department of Microbiology and Plant Pathology at UCR.
Kwak, Y. , Argandona, J. A. , Degnan, P. H. , & Hansen, A. K. (2023). Chromosomal‐level assembly of Bactericera cockerelli reveals rampant gene family expansions impacting genome structure, function and insect‐microbe‐plant‐interactions. Molecular Ecology Resources, 23, 233–252. 10.1111/1755-0998.13693
Handling Editor: Kin‐Ming (Clement) Tsui
Contributor Information
Patrick H. Degnan, Email: patrick.degnan@ucr.edu.
Allison K. Hansen, Email: allison.hansen@ucr.edu.
DATA AVAILABILITY STATEMENT
Genomic data
Bioproject/biosample for the genomic data were created in NCBI under accession number PRJNA822651/SAMN27256069. Raw PacBio data have been submitted to NCBI SRA under accession number SRR18584432. Raw Illumina data have been submitted to NCBI SRA under accession number SRR18584429. Final DoveTail chromosomal assembly have been submitted to NCBI Genome under accession number JAMCCL000000000.
Annotation data
Protein sequences annotated in this study have been submitted to Githhub at https://github.com/AllisonHansenLab/BC_psyllid_Genome. The GFF file created in this study for the assembly has been submitted to Githhub at https://github.com/AllisonHansenLab/BC_psyllid_Genome.
Gene expression data
Biosamples for gene expression data were created in NCBI under accession number SAMN27256084 to SAMN27256088. Raw RNAseq Data were submitted to NCBI SRA under accession number SRR18584596 to SRR18584600. Trinity assembled unigenes NCBI TSA under accession number SRR11276723
REFERENCES
- Abernathy, R. L. (1991). Investigations into the nature of the potato psyllid toxin (M. S. Thesis). Department of Entomology, Colorado State University. [Google Scholar]
- Andrews, S. (2010). FastQC: A quality control tool for high throughput sequence data [Online] . Available from http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- Aramaki, T. , Blanc‐Mathieu, R. , Endo, H. , Ohkubo, K. , Kanehisa, M. , Goto, S. , & Ogata, H. (2020). KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics, 36(7), 2251–2252. 10.1093/bioinformatics/btz859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandi, V. , & Gutwin, C. (2020). Interactive exploration of genomic conservation . Graphics Interface.
- Baumann, P. (2005). Biology of bacteriocyte‐associated endosymbionts of plant sap‐sucking insects. Annual Review of Microbiology, 59(1), 155–189. 10.1146/annurev.micro.59.030804.121041 [DOI] [PubMed] [Google Scholar]
- Bennett, G. M. , & Moran, N. A. (2013). Small, smaller, smallest: The origins and evolution of ancient dual symbioses in a phloem‐feeding insect. Genome Biology and Evolution, 5(9), 1675–1688. 10.1093/gbe/evt118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsten, J. (2005). A review of long‐branch attraction. Cladistics, 21(2), 163–193. 10.1111/j.1096-0031.2005.00059.x [DOI] [PubMed] [Google Scholar]
- Blaxter, M. , Archibald, J. M. , Childers, A. K. , Coddington, J. A. , Crandall, K. A. , Di Palma, F. , Durbin, R. , Edwards, S. V. , Graves, J. A. M. , Hackett, K. J. , Hall, N. , Jarvis, E. D. , Johnson, R. N. , Karlsson, E. K. , Kress, W. J. , Kuraku, S. , Lawniczak, M. K. N. , Lindblad‐Toh, K. , Lopez, J. V. , … Lewin, H. A. (2022). Why sequence all eukaryotes? Proceedings of the National Academy of Sciences of the United States of America, 119(4), e2115636118. 10.1073/pnas.2115636118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A. M. , Lohse, M. , & Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics, 30(15), 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonev, B. , & Cavalli, G. (2016). Organization and function of the 3D genome. Nature Reviews Genetics, 17(11), 661–678. 10.1038/nrg.2016.112 [DOI] [PubMed] [Google Scholar]
- Bové, J. M. (2006). Huanglongbing: A destructive, newly‐emerging, century‐old disease of citrus. Journal of Plant Pathology, 88(1), 7–37. http://www.jstor.org/stable/41998278 [Google Scholar]
- Brennecke, J. , Aravin, A. A. , Stark, A. , Dus, M. , Kellis, M. , Sachidanandam, R. , & Hannon, G. J. (2007). Discrete small RNA‐generating loci as master regulators of transposon activity in Drosophila. Cell, 128(6), 1089–1103. 10.1016/j.cell.2007.01.043 [DOI] [PubMed] [Google Scholar]
- Buchfink, B. , Xie, C. , & Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nature Methods, 12(1), 59–60. 10.1038/nmeth.3176 [DOI] [PubMed] [Google Scholar]
- Burckhardt, D. , Ouvrard, D. , & Percy, D. M. (2021). An updated classification of the jumping plant‐lice (Hemiptera: Psylloidea) integrating molecular and morphological evidence. European Journal of Taxonomy, 736, 137–182. 10.5852/ejt.2021.736.1257 [DOI] [Google Scholar]
- Burns, K. H. , & Boeke, J. D. (2012). Human transposon tectonics. Cell, 149(4), 740–752. 10.1016/j.cell.2012.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarel, B. L. , Korf, I. , Robb, S. M. C. , Parra, G. , Ross, E. , Moore, B. , Holt, C. , Alvarado, A. S. , & Yandell, M. (2008). MAKER: An easy‐to‐use annotation pipeline designed for emerging model organism genomes. Genome Research, 18(1), 188–196. 10.1101/gr.6743907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casteel, C. L. , Hansen, A. K. , Walling, L. L. , & Paine, T. D. (2012). Manipulation of plant defense responses by the tomato Psyllid (Bactericerca cockerelli) and its associated endosymbiont Candidatus Liberibacter Psyllaurous. PLoS ONE, 7(4), e35191. 10.1371/journal.pone.0035191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford, D. L. (1914). A monograph of the jumping plant‐lice or Psyllidae of the New World. Bulletin of the United States National Museum, 85, 1–186 10.5479/si.03629236.85.1. [Google Scholar]
- Crosslin, J. M. , Munyaneza, J. E. , Brown, J. K. , & Liefting, L. W. (2010). Potato zebra chip disease: a phytopathological tale. Plant Health Progress, 11(1), 33. 10.1094/PHP-2010-0317-01-RV [DOI] [Google Scholar]
- Dahan, J. , Cooper, W. R. , Munyaneza, J. E. , & Karasev, A. V. (2022). A new picorna‐like virus identified in populations of the potato psyllid Bactericera cockerelli . Archives of Virology, 167(1), 177–182. 10.1007/s00705-021-05281-x [DOI] [PubMed] [Google Scholar]
- Degnan, P. H. , Leonardo, T. E. , Cass, B. N. , Hurwitz, B. , Stern, D. , Gibbs, R. A. , Richards, S. , & Moran, N. A. (2010). Dynamics of genome evolution in facultative symbionts of aphids. Environmental Microbiology, 12(8), 2060–2069. 10.1111/j.1462-2920.2009.02085.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon, J. R. , Selvaraj, S. , Yue, F. , Kim, A. , Li, Y. , Shen, Y. , Hu, M. , Liu, J. S. , & Ren, B. (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature, 485(7398), 376–380. 10.1038/nature11082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díez Díaz, M. , Conejero, V. , Rodrigo, I. , Pearce, G. , & Ryan, C. A. (2004). Isolation and characterization of wound‐inducible carboxypeptidase inhibitor from tomato leaves. Phytochemistry, 65(13), 1919–1924. 10.1016/j.phytochem.2004.06.007 [DOI] [PubMed] [Google Scholar]
- Dobin, A. , Davis, C. A. , Schlesinger, F. , Drenkow, J. , Zaleski, C. , Jha, S. , Batut, P. , Chaisson, M. , & Gingeras, T. R. (2013). STAR: Ultrafast universal RNA‐seq aligner. Bioinformatics, 29(1), 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling, D. , Pauli, T. , Donath, A. , Meusemann, K. , Podsiadlowski, L. , Petersen, M. , Peters, R. S. , Mayer, C. , Liu, S. , Zhou, X. , Misof, B. , & Niehuis, O. (2016). Phylogenetic origin and diversification of RNAi pathway genes in insects. Genome Biology and Evolution, 8(12), 3784–3793. 10.1093/gbe/evw281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand, N. C. , Shamim, M. S. , Machol, I. , Rao, S. S. P. , Huntley, M. H. , Lander, E. S. , & Aiden, E. L. (2016). Juicer provides a one‐click system for analyzing loop‐resolution Hi‐C experiments. Cell Systems, 3(1), 95–98. 10.1016/j.cels.2016.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, H. , Edwards, N. , Anderson, C. M. H. , Althaus, M. , Duncan, R. P. , Hsu, Y. C. , Luetje, C. W. , Price, D. R. G. , Wilson, A. C. C. , & Thwaites, D. T. (2019). Trading amino acids at the aphid‐Buchnera symbiotic interface. Proc Natl Acad Sci U S A, 116(32), 16003–16011. 10.1073/pnas.1906223116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figon, F. , & Casas, J. (2019). Ommochromes in invertebrates: Biochemistry and cell biology. Biological Reviews, 94(1), 156–183. 10.1111/brv.12441 [DOI] [PubMed] [Google Scholar]
- Flynn, J. M. , Hubley, R. , Goubert, C. , Rosen, J. , Clark, A. G. , Feschotte, C. , & Smit, A. F. (2020). RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences of the United States of America, 117(17), 9451–9457. 10.1073/pnas.1921046117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert, C. , Peccoud, J. , & Cordaux, R. (2021). Transposable elements and the evolution of insects. Annual Review of Entomology, 66(1), 355–372. 10.1146/annurev-ento-070720-074650 [DOI] [PubMed] [Google Scholar]
- Goldman, N. , & Yang, Z. (1994). A codon‐based model of nucleotide substitution for protein‐coding DNA sequences. Molecular Biology and Evolution, 11(5), 725–736. 10.1093/oxfordjournals.molbev.a040153 [DOI] [PubMed] [Google Scholar]
- Grabherr, M. G. , Haas, B. J. , Yassour, M. , Levin, J. Z. , Thompson, D. A. , Amit, I. , Adiconis, X. , Fan, L. , Raychowdhury, R. , Zeng, Q. , Chen, Z. , Mauceli, E. , Hacohen, N. , Gnirke, A. , Rhind, N. , di Palma, F. , Birren, B. W. , Nusbaum, C. , Lindblad‐Toh, K. , … Regev, A. (2011). Full‐length transcriptome assembly from RNA‐Seq data without a reference genome. Nature Biotechnology, 29(7), 644–652. 10.1038/nbt.1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham, E. D. , Heidelberg, J. F. , & Tully, B. J. (2018). Potential for primary productivity in a globally‐distributed bacterial phototroph. The ISME Journal, 12(7), 1861–1866. 10.1038/s41396-018-0091-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham, J. S. , & Ryan, C. A. (1981). Accumulation of a metallo‐carboxypeptidase inhibitor in leaves of wounded potato plants. Biochemical and Biophysical Research Communications, 101(4), 1164–1170. 10.1016/0006-291X(81)91570-9 [DOI] [PubMed] [Google Scholar]
- Guan, D. , McCarthy, S. A. , Wood, J. , Howe, K. , Wang, Y. , & Durbin, R. (2020). Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics, 36(9), 2896–2898. 10.1093/bioinformatics/btaa025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullan, P. J. , & Martin, J. H. (2009). Chapter 244 ‐ Sternorrhyncha: (Jumping Plant‐Lice, Whiteflies, Aphids, and Scale Insects). In Resh V. H. & Cardé R. T. (Eds.), Encyclopedia of insects (2nd ed., pp. 957–967). Academic Press. 10.1016/B978-0-12-374144-8.00253-8 [DOI] [Google Scholar]
- Hansen, A. K. , & Moran, N. A. (2011). Aphid genome expression reveals host–symbiont cooperation in the production of amino acids. Proceedings of the National Academy of Sciences of the United States of America, 108(7), 2849–2854. 10.1073/pnas.1013465108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen, A. K. , Trumble, J. T. , Stouthamer, R. , & Paine, T. D. (2008). A new Huanglongbing species, “Candidatus Liberibacter psyllaurous,” found to infect tomato and potato, is vectored by the Psyllid Bactericera cockerelli (Sulc). Applied and Environmental Microbiology, 74, 5862–5865. 10.1128/AEM.01268-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen, A. K. , & Moran, N. A. (2014). The impact of microbial symbionts on host plant utilization by herbivorous insects. Molecular Ecology., 23(6), 1473–96. [DOI] [PubMed] [Google Scholar]
- HMMER . (2022). Retrieved April 4, 2022, from http://hmmer.org/
- Husnik, F. , Nikoh, N. , Koga, R. , Ross, L. , Duncan, R. P. , Fujie, M. , Tanaka, M. , Satoh, N. , Bachtrog, D. , Wilson, A. C. C. , von Dohlen, C. D. , Fukatsu, T. , & McCutcheon, J. P. (2013). Horizontal gene transfer from diverse bacteria to an insect genome enables a tripartite nested Mealybug symbiosis. Cell, 153(7), 1567–1578. 10.1016/j.cell.2013.05.040 [DOI] [PubMed] [Google Scholar]
- Johnson, K. P. , Dietrich, C. H. , Friedrich, F. , Beutel, R. G. , Wipfler, B. , Peters, R. S. , Allen, J. M. , Petersen, M. , Donath, A. , Walden, K. K. O. , Kozlov, A. M. , Podsiadlowski, L. , Mayer, C. , Meusemann, K. , Vasilikopoulos, A. , Waterhouse, R. M. , Cameron, S. L. , Weirauch, C. , Swanson, D. R. , … Yoshizawa, K. (2018). Phylogenomics and the evolution of hemipteroid insects. Proceedings of the National Academy of Sciences of the United States of America, 115(50), 12775–12780. 10.1073/pnas.1815820115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa, M. , & Sato, Y. (2020). KEGG Mapper for inferring cellular functions from protein sequences. Protein Science, 29(1), 28–35. 10.1002/pro.3711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa, M. , Sato, Y. , & Kawashima, M. (2022). KEGG mapping tools for uncovering hidden features in biological data. Protein Science, 31(1), 47–53. 10.1002/pro.4172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K. , & Standley, D. M. (2013). MAFFT Multiple Sequence Alignment Software Version 7: Improvements in performance and usability. Molecular Biology and Evolution, 30(4), 772–780. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling, P. J. , & Palmer, J. D. (2008). Horizontal gene transfer in eukaryotic evolution. Nature Reviews Genetics, 9(8), 605–618. 10.1038/nrg2386 [DOI] [PubMed] [Google Scholar]
- Kerpedjiev, P. , Abdennur, N. , Lekschas, F. , McCallum, C. , Dinkla, K. , Strobelt, H. , Luber, J. M. , Ouellette, S. B. , Azhir, A. , Kumar, N. , Hwang, J. , Lee, S. , Alver, B. H. , Pfister, H. , Mirny, L. A. , Park, P. J. , & Gehlenborg, N. (2018). HiGlass: Web‐based visual exploration and analysis of genome interaction maps. Genome Biology, 19(1), 125. 10.1186/s13059-018-1486-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keseler, I. M. , Mackie, A. , Peralta‐Gil, M. , Santos‐Zavaleta, A. , Gama‐Castro, S. , Bonavides‐Martínez, C. , Fulcher, C. , Huerta, A. M. , Kothari, A. , Krummenacker, M. , Latendresse, M. , Muñiz‐Rascado, L. , Ong, Q. , Paley, S. , Schröder, I. , Shearer, A. G. , Subhraveti, P. , Travers, M. , Weerasinghe, D. , … Karp, P. D. (2013). EcoCyc: Fusing model organism databases with systems biology. Nucleic Acids Research, 41(D1), D605–D612. 10.1093/nar/gks1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D. , Langmead, B. , & Salzberg, S. L. (2015). HISAT: A fast spliced aligner with low memory requirements. Nature Methods, 12(4), 357–360. 10.1038/nmeth.3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf, I. (2004). Gene finding in novel genomes. BMC Bioinformatics, 5(1), 59. 10.1186/1471-2105-5-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak, Y. , Sun, P. , Meduri, V. R. , Percy, D. M. , Mauck, K. E. , & Hansen, A. K. (2021). Uncovering symbionts across the Psyllid tree of life and the discovery of a new Liberibacter species, “Candidatus” Liberibacter capsica. Frontiers in Microbiology, 12, 739763. 10.3389/fmicb.2021.739763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laetsch, D. R. , & Blaxter, M. L. (2017). BlobTools: Interrogation of genome assemblies. F1000Research, 6, 1287. 10.12688/f1000research.12232.1 [DOI] [Google Scholar]
- Lespinet, O. , Wolf, Y. I. , Koonin, E. V. , & Aravind, L. (2002). The role of lineage‐specific gene family expansion in the evolution of eukaryotes. Genome Research, 12(7), 1048–1059. 10.1101/gr.174302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics, 27(21), 2987–2993. 10.1093/bioinformatics/btr509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. ArXiv, 1303. http://arxiv.org/abs/1303.3997 [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. , Abecasis, G. , Durbin, R. , & 1000 Genome Project Data Processing Subgroup . (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25(16), 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Park, H. , Smith, T. E. , & Moran, N. A. (2019). Gene family evolution in the pea aphid based on chromosome‐level genome assembly. Molecular Biology and Evolution, 36(10), 2143–2156. 10.1093/molbev/msz138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Zhang, B. , & Moran, N. A. (2020). The Aphid X chromosome is a dangerous place for functionally important genes: Diverse evolution of Hemipteran genomes based on chromosome‐level assemblies. Molecular Biology and Evolution, 37(8), 2357–2368. 10.1093/molbev/msaa095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liefting, L. W. , Perez‐Egusquiza, Z. C. , Clover, G. R. G. , & Anderson, J. A. D. (2008). A new ‘Candidatus Liberibacter' species in Solanum tuberosum in New Zealand. Plant Disease, 92(10), 1474. 10.1094/PDIS-92-10-1474A [DOI] [PubMed] [Google Scholar]
- Lugo‐Huitrón, R. , Blanco‐Ayala, T. , Ugalde‐Muñiz, P. , Carrillo‐Mora, P. , Pedraza‐Chaverrí, J. , Silva‐Adaya, D. , Maldonado, P. D. , Torres, I. , Pinzón, E. , Ortiz‐Islas, E. , López, T. , García, E. , Pineda, B. , Torres‐Ramos, M. , Santamaría, A. , & La Cruz, V. P.‐D. (2011). On the antioxidant properties of kynurenic acid: Free radical scavenging activity and inhibition of oxidative stress. Neurotoxicology and Teratology, 33(5), 538–547. 10.1016/j.ntt.2011.07.002 [DOI] [PubMed] [Google Scholar]
- Lynch, M. (2007). The origins of genome architecture. Sinauer Associates. http://catalog.hathitrust.org/api/volumes/oclc/77574049.html [Google Scholar]
- Manni, M. , Berkeley, M. R. , Seppey, M. , & Zdobnov, E. M. (2021). BUSCO: Assessing genomic data quality and beyond. Current Protocols, 1, e323. 10.1002/cpz1.323 [DOI] [PubMed] [Google Scholar]
- McCutcheon, J. P. , & Moran, N. A. (2012). Extreme genome reduction in symbiotic bacteria. Nature Reviews Microbiology, 10(1), 13–26. 10.1038/nrmicro2670 [DOI] [PubMed] [Google Scholar]
- Mendes, F. K. , Vanderpool, D. , Fulton, B. , & Hahn, M. W. (2020). CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics, 36(22–23), 5516–5518. 10.1093/bioinformatics/btaa1022 [DOI] [PubMed] [Google Scholar]
- Miller, M. A. , Pfeiffer, W. , & Schwartz, T. (2010). Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Gateway Computing Environments Workshop (GCE), 2010, 1–8. 10.1109/GCE.2010.5676129 [DOI] [Google Scholar]
- Mistry, J. , Chuguransky, S. , Williams, L. , Qureshi, M. , Salazar, G. A. , Sonnhammer, E. L. L. , Tosatto, S. C. E. , Paladin, L. , Raj, S. , Richardson, L. J. , Finn, R. D. , & Bateman, A. (2021). Pfam: The protein families database in 2021. Nucleic Acids Research, 49(D1), D412–D419. 10.1093/nar/gkaa913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran, N. A. , McCutcheon, J. P. , & Nakabachi, A. (2008). Genomics and evolution of heritable bacterial symbionts. Annual Review of Genetics, 42(1), 165–190. 10.1146/annurev.genet.41.110306.130119 [DOI] [PubMed] [Google Scholar]
- Moran, N. A. , & Wernegreen, J. J. (2000). Lifestyle evolution in symbiotic bacteria: Insights from genomics. Trends in Ecology & Evolution, 15(8), 321–326. 10.1016/S0169-5347(00)01902-9 [DOI] [PubMed] [Google Scholar]
- Munyaneza, J. E. , Crosslin, J. M. , & Upton, J. E. (2007). Association of Bactericera cockerelli (Homoptera: Psyllidae) with “Zebra Chip,” a new potato disease in Southwestern United States and Mexico. Journal of Economic Entomology, 100(3), 656–663. 10.1093/jee/100.3.656 [DOI] [PubMed] [Google Scholar]
- Nakabachi, A. , Ueoka, R. , Oshima, K. , Teta, R. , Mangoni, A. , Gurgui, M. , Oldham, N. J. , van Echten‐Deckert, G. , Okamura, K. , Yamamoto, K. , Inoue, H. , Ohkuma, M. , Hongoh, Y. , Miyagishima, S. , Hattori, M. , Piel, J. , & Fukatsu, T. (2013). Defensive bacteriome symbiont with a drastically reduced genome. Current Biology, 23(15), 1478–1484. 10.1016/j.cub.2013.06.027 [DOI] [PubMed] [Google Scholar]
- Nakabachi, A. , Yamashita, A. , Toh, H. , Ishikawa, H. , Dunbar, H. E. , Moran, N. A. , & Hattori, M. (2006). The 160‐kilobase genome of the bacterial endosymbiont Carsonella . Science, 314(5797), 267. 10.1126/science.1134196 [DOI] [PubMed] [Google Scholar]
- NCBI . (2022). Retrieved March 8, 2022, from https://www.ncbi.nlm.nih.gov/genome/
- Oliver, J. L. , Carpena, P. , Hackenberg, M. , & Bernaola‐Galván, P. (2004). IsoFinder: Computational prediction of isochores in genome sequences. Nucleic Acids Research, 32(suppl_2), W287–W292. 10.1093/nar/gkh399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peccoud, J. , Loiseau, V. , Cordaux, R. , & Gilbert, C. (2017). Massive horizontal transfer of transposable elements in insects. Proceedings of the National Academy of Sciences of the United States of America, 114(18), 4721–4726. 10.1073/pnas.1621178114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pers, D. , & Hansen, A. K. (2021). The boom and bust of the aphid's essential amino acid metabolism across nymphal development. G3 Genes|Genomes|Genetics, 11(9), jkab115. 10.1093/g3journal/jkab115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea, M. , Pertea, G. M. , Antonescu, C. M. , Chang, T.‐C. , Mendell, J. T. , & Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA‐seq reads. Nature Biotechnology, 33(3), 290–295. 10.1038/nbt.3122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen, M. , Armisén, D. , Gibbs, R. A. , Hering, L. , Khila, A. , Mayer, G. , Richards, S. , Niehuis, O. , & Misof, B. (2019). Diversity and evolution of the transposable element repertoire in arthropods with particular reference to insects. BMC Ecology and Evolution, 19(1), 11. 10.1186/s12862-018-1324-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletsch, D. J. (1947). The potato psyllid, Paratrioza cockerelli (Sulc): Its biology and control. Montana State College, Agricultural Experiment Station. http://books.google.com/books?id=hT4nAQAAMAAJ [Google Scholar]
- Putnam, N. H. , O'Connell, B. L. , Stites, J. C. , Rice, B. J. , Blanchette, M. , Calef, R. , Troll, C. J. , Fields, A. , Hartley, P. D. , Sugnet, C. W. , Haussler, D. , Rokhsar, D. S. , & Green, R. E. (2016). Chromosome‐scale shotgun assembly using an in vitro method for long‐range linkage. Genome Research, 26(3), 342–350. 10.1101/gr.193474.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . (2020). R: A language and environment for statistical computing. R Foundation for Statistical Computing. https://www.r‐project.org/ [Google Scholar]
- Raddadi, N. , Gonella, E. , Camerota, C. , Pizzinat, A. , Tedeschi, R. , Crotti, E. , Mandrioli, M. , Attilio Bianco, P. , Daffonchio, D. , & Alma, A. (2011). ‘Candidatus Liberibacter europaeus' sp. Nov. That is associated with and transmitted by the psyllid Cacopsylla pyri apparently behaves as an endophyte rather than a pathogen. Environmental Microbiology, 13(2), 414–426. 10.1111/j.1462-2920.2010.02347.x [DOI] [PubMed] [Google Scholar]
- Rambaut A. (2018). Figtree v1.4.4 . Retrieved June 23, 2022, from http://tree.bio.ed.ac.uk/software/figtree/
- Rao, S. S. P. , Huntley, M. H. , Durand, N. C. , Stamenova, E. K. , Bochkov, I. D. , Robinson, J. T. , Sanborn, A. L. , Machol, I. , Omer, A. D. , Lander, E. S. , & Aiden, E. L. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell, 159(7), 1665–1680. 10.1016/j.cell.2014.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice, P. , Longden, I. , & Bleasby, A. (2000). EMBOSS: The European Molecular Biology Open Software Suite. Trends in Genetics, 16(6), 276–277. 10.1016/S0168-9525(00)02024-2 [DOI] [PubMed] [Google Scholar]
- Riley, A. B. , Kim, D. , & Hansen, A. K. (2017). Genome sequence of “Candidatus Carsonella ruddii” strain BC, a nutritional endosymbiont of Bactericera cockerelli . Genome Announcements, 5(17), e00236‐17. 10.1128/genomeA.00236-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan, J. , & Li, H. (2020). Fast and accurate long‐read assembly with wtdbg2. Nature Methods, 17(2), 155–158. 10.1038/s41592-019-0669-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha, S. , Hosmani, P. , Flores, M. , Hunter, W. , Brown, S. A. , & Mueller, L. (2017). Using long reads, optical maps and long‐range scaffolding to improve the Diaphorina citri genome . 10.6084/m9.figshare.5375116.v1 [DOI]
- Sanderson, M. J. (2003). r8s: Inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics, 19(2), 301–302. 10.1093/bioinformatics/19.2.301 [DOI] [PubMed] [Google Scholar]
- Simão, F. A. , Waterhouse, R. M. , Ioannidis, P. , Kriventseva, E. V. , & Zdobnov, E. M. (2015). BUSCO: Assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics, 31(19), 3210–3212. 10.1093/bioinformatics/btv351 [DOI] [PubMed] [Google Scholar]
- Sloan, D. B. , & Moran, N. A. (2012). Genome reduction and co‐evolution between the primary and secondary bacterial symbionts of Psyllids. Molecular Biology and Evolution, 29(12), 3781–3792. 10.1093/molbev/mss180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan, D. B. , Nakabachi, A. , Richards, S. , Qu, J. , Murali, S. C. , Gibbs, R. A. , & Moran, N. A. (2014). Parallel histories of horizontal gene transfer facilitated extreme reduction of endosymbiont genomes in sap‐feeding insects. Molecular Biology and Evolution, 31(4), 857–871. 10.1093/molbev/msu004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit, A. F. , Hubley, R. , & Green, P. (2013). RepeatMasker Open‐4.0 . http://www.repeatmasker.org
- Smith, A. D. , Sumazin, P. , Xuan, Z. , & Zhang, M. Q. (2006). DNA motifs in human and mouse proximal promoters predict tissue‐specific expression. Proceedings of the National Academy of Sciences of the United States of America, 103(16), 6275–6280. 10.1073/pnas.0508169103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaulding, A. W. , & von Dohlen, C. D. (2001). Psyllid endosymbionts exhibit patterns of co‐speciation with hosts and destabilizing substitutions in ribosomal RNA. Insect Molecular Biology, 10(1), 57–67. 10.1046/j.1365-2583.2001.00231.x [DOI] [PubMed] [Google Scholar]
- Stamatakis, A. (2014). RAxML version 8: A tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics, 30(9), 1312–1313. 10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke, M. , Keller, O. , Gunduz, I. , Hayes, A. , Waack, S. , & Morgenstern, B. (2006). AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Research, 34(Suppl_2), W435–W439. 10.1093/nar/gkl200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šulc, K. (1909). Trioza cockerelli n. Sp., a novelty from North America, being also of economic importance. Acta Societatis Entomologicae Bohemiaecta Societatis Entomologicae Bohemiae, 6, 102–109. [Google Scholar]
- Thairu, M. W. , Meduri, V. R. S. , Degnan, P. H. , & Hansen, A. K. (2021). Natural selection shapes maintenance of Orthologous sRNAs in divergent host‐restricted bacterial genomes. Molecular Biology and Evolution, 38(11), 4778–4791. 10.1093/molbev/msab202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thao, M. L. , Moran, N. A. , Abbot, P. , Brennan, E. B. , Burckhardt, D. H. , & Baumann, P. (2000). Cospeciation of Psyllids and their primary prokaryotic endosymbionts. Applied and Environmental Microbiology, 66(7), 2898–2905. 10.1128/AEM.66.7.2898-2905.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International Aphid Genomics Consortium . (2010). Genome sequence of the Pea Aphid Acyrthosiphon pisum . PLoS Biology, 8(2), e1000313. 10.1371/journal.pbio.1000313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Törönen, P. , Medlar, A. , & Holm, L. (2018). PANNZER2: A rapid functional annotation web server. Nucleic Acids Research, 46(W1), W84–W88. 10.1093/nar/gky350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Etten, J. , & Bhattacharya, D. (2020). Horizontal gene transfer in eukaryotes: Not if, but how much? Trends in Genetics, 36(12), 915–925. 10.1016/j.tig.2020.08.006 [DOI] [PubMed] [Google Scholar]
- van't Hof, A. E. , Campagne, P. , Rigden, D. J. , Yung, C. J. , Lingley, J. , Quail, M. A. , Hall, N. , Darby, A. C. , & Saccheri, I. J. (2016). The industrial melanism mutation in British peppered moths is a transposable element. Nature, 534(7605), 102–105. 10.1038/nature17951 [DOI] [PubMed] [Google Scholar]
- Wallis, R. L. (1955). Ecological studies on the potato psyllid as a pest of potatoes . Technical Bulletin (United States. Dept. of Agriculture); no. 1107. https://handle.nal.usda.gov/10113/CAT87201117
- Wang, N. , Pierson, E. A. , Setubal, J. C. , Xu, J. , Levy, J. G. , Zhang, Y. , Li, J. , Rangel, L. T. , & Martins, J. (2017). The Candidatus Liberibacter–host interface: Insights into pathogenesis mechanisms and disease control. Annual Review of Phytopathology, 55(1), 451–482. 10.1146/annurev-phyto-080516-035513 [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Tang, H. , DeBarry, J. D. , Tan, X. , Li, J. , Wang, X. , Lee, T. , Jin, H. , Marler, B. , Guo, H. , Kissinger, J. C. , & Paterson, A. H. (2012). MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Research, 40(7), e49. 10.1093/nar/gkr1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, F. , Cen, Y. , Wallis, C. M. , Trumble, J. T. , Prager, S. , Yokomi, R. , Zheng, Z. , Deng, X. , Chen, J. , & Liang, G. (2016). The Complete Mitochondrial Genome Sequence of Bactericera cockerelli and Comparison with Three Other Psylloidea Species. PLoS ONE, 11(5), e0155318. 10.1371/journal.pone.0155318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, L. , Dong, Z. , Fang, L. , Luo, Y. , Wei, Z. , Guo, H. , Zhang, G. , Gu, Y. Q. , Coleman‐Derr, D. , Xia, Q. , & Wang, Y. (2019). OrthoVenn2: A web server for whole‐genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Research, 47(W1), W52–W58. 10.1093/nar/gkz333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, B. , & Wittkopp, P. J. (2017). Structure of the transcriptional regulatory network correlates with regulatory divergence in Drosophila. Molecular Biology and Evolution, 34(6), 1352–1362. 10.1093/molbev/msx068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, X.‐B. , & Liu, T.‐X. (2009). Life history and life tables of Bactericera cockerelli (Homoptera: Psyllidae) on Eggplant and Bell Pepper. Environmental Entomology, 38(6), 1661–1667. 10.1603/022.038.0619 [DOI] [PubMed] [Google Scholar]
- Yang, Z. (1997). PAML: A program package for phylogenetic analysis by maximum likelihood. Computer Applications in the Biosciences: CABIOS, 13(5), 555–556. 10.1093/bioinformatics/13.5.555 [DOI] [PubMed] [Google Scholar]
- Zalar, P. , Sybren de Hoog, G. , Schroers, H.‐J. , Frank, J. M. , & Gunde‐Cimerman, N. (2005). Taxonomy and phylogeny of the xerophilic genus Wallemia (Wallemiomycetes and Wallemiales, cl. Et ord. Nov.). Antonie van Leeuwenhoek, 87(4), 311–328. 10.1007/s10482-004-6783-x [DOI] [PubMed] [Google Scholar]
- Zhu‐Salzman, K. , & Zeng, R. (2015). Insect response to plant defensive protease inhibitors. Annual Review of Entomology, 60(1), 233–252. 10.1146/annurev-ento-010814-020816 [DOI] [PubMed] [Google Scholar]
- Ziebarth, J. D. , Bhattacharya, A. , & Cui, Y. (2013). CTCFBSDB 2.0: A database for CTCF‐binding sites and genome organization. Nucleic Acids Research, 41(D1), D188–D194. 10.1093/nar/gks1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S6
Tables S1–S11
Data Availability Statement
Genomic data
Bioproject/biosample for the genomic data were created in NCBI under accession number PRJNA822651/SAMN27256069. Raw PacBio data have been submitted to NCBI SRA under accession number SRR18584432. Raw Illumina data have been submitted to NCBI SRA under accession number SRR18584429. Final DoveTail chromosomal assembly have been submitted to NCBI Genome under accession number JAMCCL000000000.
Annotation data
Protein sequences annotated in this study have been submitted to Githhub at https://github.com/AllisonHansenLab/BC_psyllid_Genome. The GFF file created in this study for the assembly has been submitted to Githhub at https://github.com/AllisonHansenLab/BC_psyllid_Genome.
Gene expression data
Biosamples for gene expression data were created in NCBI under accession number SAMN27256084 to SAMN27256088. Raw RNAseq Data were submitted to NCBI SRA under accession number SRR18584596 to SRR18584600. Trinity assembled unigenes NCBI TSA under accession number SRR11276723