

Abstract
Multidrug resistance is a major public health problem that requires the urgent development of new antibiotics and therefore the identification of novel bacterial targets. The activity of nicotinamide adenine dinucleotide kinase, NADK, is essential in all bacteria tested so far, including many human pathogens that display antibiotic resistance leading to the failure of current treatments. Inhibiting NADK is therefore a promising and innovative antibacterial strategy since there is currently no drug on the market targeting this enzyme. Through a fragment‐based drug design approach, we have recently developed a NAD+‐competitive inhibitor of NADKs, which displayed in vivo activity against Staphylococcus aureus. Here, we show that this compound, a di‐adenosine derivative, is inactive against the NADK enzyme from the Gram‐negative bacteria Pseudomonas aeruginosa (PaNADK). This lack of activity can be explained by the crystal structure of PaNADK, which was determined in complex with NADP+ in this study. Structural analysis led us to design and synthesize a benzamide adenine dinucleoside analogue, active against PaNADK. This novel compound efficiently inhibited PaNADK enzymatic activity in vitro with a K i of 4.6 μm. Moreover, this compound reduced P. aeruginosa infection in vivo in a zebrafish model.
Keywords: antibiotics, benzamide adenine dinucleoside analogue, ESKAPE, new target enzyme, zebrafish
The previously synthesized di‐adenosine derivative NKI1 inhibits S. aureus NADK but not P. aeruginosa NADK. P. aeruginosa NADK crystal structure explains the lack of effect of NKI1. A new rationally designed NAD+ analogue containing a benzamide inhibits P. aeruginosa NADK. This compound displays activity against P. aeruginosa in a zebrafish infection model.

Abbreviations
- APTS
p‐toluene sulfonic acid
- CFU
colony‐forming unit
- DCM
dichloromethane
- DMF
N,N‐dimethylformamide
- DMSO
dimethylsulfoxide
- G6PDH
glucose‐6‐phosphate dehydrogenase
- HPLC
high‐ performance liquid chromatography
- HRMS
high‐resolution mass spectra
- IPTG
isopropyl β‐D‐1‐thiogalactopyranoside
- k ss
steady‐ state rate constant
- LB
Luria broth
- LmNADK
NADK 1 from Listeria monocytogenes
- NAD+
nicotinamide adenine dinucleotide
- NADK
NAD kinase
- NADP+
nicotinamide adenine dinucleotide phosphate
- OD
optical density
- PaNADK
NADK from Pseudomonas aeruginosa
- PBS
Phosphate Bbuffered Ssaline
- RMSD
root‐mean‐square deviation
- ROS
reactive oxygen species
- R t
retention time
- SaNADK
NADK from Staphylococcus aureus
- SEC
size exclusion chromatography
- TEAA
triethylammonium acetate
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- TLC
thin‐layer chromatography
- TMSOK
potassium trimethylsilanolate
- WHO
World Health Organization
Introduction
Pseudomonas aeruginosa is an environmental Gram‐negative bacterium and an opportunistic human pathogen that can cause serious acute infections. It is also responsible for chronic infections in patients with cystic fibrosis, being a major cause of mortality. Indeed, the infection maintains the inflammation, worsening the state of health of infected patients. Due to the increased resistance of bacteria to antibiotics, the World Health Organization (WHO) has listed pathogenic bacteria for which new antibiotics or alternative therapeutic strategies are urgently needed and P. aeruginosa is in the priority 1 list defined as critical [1].
Bacterial nicotinamide adenine dinucleotide kinase (NADK, EC2.7.1.23) has been identified as a promising target for the development of new antibiotics [2], NADK being the sole enzyme that phosphorylates nicotinamide adenine dinucleotide (NAD+) to NADP+. NADP+ is an essential coenzyme converted by different redundant enzymes into NADPH, to provide reducing power in many processes such as oxidative stress response, fatty acid and nucleotide biosynthesis [3, 4]. NADK is therefore an essential enzyme for rapidly dividing cells, which need both high macromolecular turnover and an efficient response to oxidative stress, and therefore require high level of NADPH [5]. This is particularly important for bacteria in the context of antibiotic treatment. In fact, three main classes of antibiotics (quinolones, beta‐lactams and aminoglycosides) stimulate the production of reactive oxygen species (ROS), which can participate in bacterial cell death [6]. In addition, inactivation of the gene encoding NADK is fatal for all bacteria tested so far [7, 8, 9, 10, 11, 12].
By a structural approach, we have shown the originality of the enzymatic mechanism of NADK and of its NAD+ binding mode [13]. Based on the analysis of the structure of the NADK 1 from Listeria monocytogenes (LmNADK), we developed a new class of antibacterial compounds that specifically mimics the particular conformation that NAD+ adopts in the NADK binding site [14]. This led us to synthesize original compounds targeting bacterial NADKs [2, 15]. Optimization of a propargyl‐linked di‐adenosine led to NKI1 (Fig. 1), a new lead compound active against the Gram‐positive bacterium Staphylococcus aureus both in vitro and in vivo in a mouse animal model of infection [16].
Fig. 1.
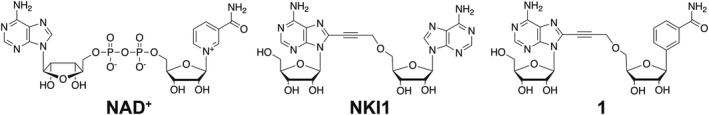
Chemical structures of compounds used in this study. Structures of NAD+ (NADK substrate), NKI1 (inhibitor of NADKs from S. aureus and L. monocytogenes, [16]) and compound 1 (PaNADK inhibitor, this study).
Here, we demonstrate the lack of effect of NKI1 in vitro on the enzymatic activity of P. aeruginosa NADK (PaNADK). We provide an explanation for the absence of PaNADK inhibition by NKI1 based on the crystallographic structure of the enzyme complexed with NADP+ and propose to replace one adenine of NKI1 with a benzamide. This leads to a new molecule, compound 1 (Fig. 1), active in vitro on PaNADK enzyme. This new inhibitor is then tested against P. aeruginosa in vitro and in vivo in zebrafish embryos, which is now an established vertebrate model of infection to validate novel anti‐Pseudomonas compounds [17, 18].
Results
Enzymatic activity of PaNADK is not inhibited by NKI1
The overexpression and purification of the NADK from P. aeruginosa was readily obtained using standard protocols (Fig. 2). The enzymatic activity of PaNADK was determined using a coupled system using G6PDH as previously performed for LmNADK [16]. First, we looked for optimal experimental conditions for the in vitro activity of PaNADK. The activity of the enzyme was constant over a wide range of pH, from 7 to 10, but was sensitive to the presence of NaCl (Fig. 3). Based on these results, the conditions retained for further in vitro measurements of enzyme activity and inhibition were 50 mm Tris pH 7.5, 1 mm MgCl2 and 5 mm DTT.
Fig. 2.
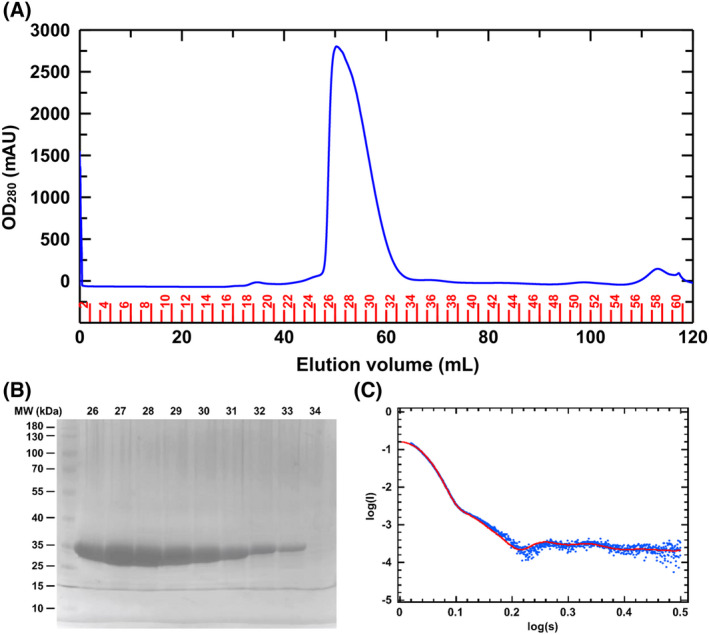
Purification and biophysical characterization of PaNADK. (A) Elugram from the gel filtration column. OD at 280 nm is shown as blue line. Fraction numbers are shown in red. (B) Verification of the purity of the recombinant PaNADK by SDS/PAGE gel. The numbers of the fractions obtained from the GF are indicated at the top of the gel. (C) Scattering profiles obtained by SAXS on PaNADK and fitting with a tetrameric model. The fitting of homo‐tetrameric protein solution scattering obtained from X‐ray crystallography (red line) to the experimental scattering obtained from SAXS experiments (blue dots).
Fig. 3.
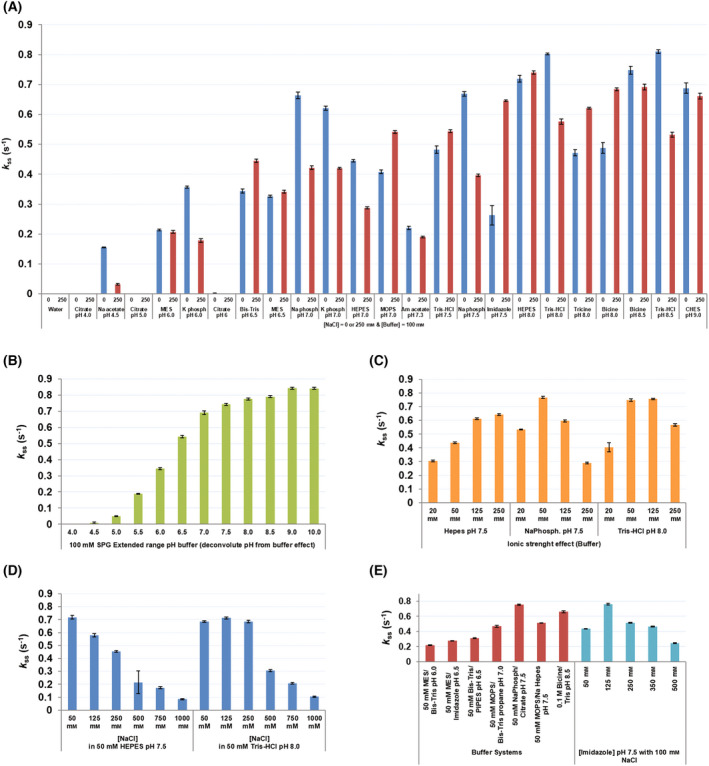
Screening of optimal in vitro experimental conditions for the measurement of PaNADK enzymatic activity. PaNADK activity was measured by an enzyme‐coupled system in different conditions using the RUBIC buffer kit. Final concentrations were 1 μm PaNADK, 5 mm MgATP, 5 mm NAD+, 5 mm glucose‐6‐phosphate and 1 U·mL−1 G6PDH. Conditions tested were as follows: (A) buffers (100 mm) of different pHs at low (0 mm NaCl) or high (250 mm NaCl) ionic strength, (B) composite buffer with extended range of pH deconvoluting pH from buffer effect, (C) buffers at different concentrations (20–250 mm), (D) different ionic strengths by variation of NaCl concentration (50–1000 mm) and (E) combination of buffers and effect of imidazole (50–500 mm) in the presence of 100 mm NaCl. Error bars represent calculated standard deviation.
We determined the values for k cat at 30 °C of 1 s−1 and K m for MgATP of 2 mm. These two parameters are comparable to those determined with other bacterial or eukaryotic cytosolic NADKs [19]. However, in contrast to what was described for NADKs from Bacillus subtilis [20], Salmonella typhimurium [21] or L. monocytogenes [13], PaNADK does not display any cooperative activity. Strikingly, the value of K m determined for NAD+ with PaNADK (0.15 mm) is clearly lower than the K m (or K m app in the case of cooperative activity) measured with other bacterial NADKs (1–4 mm) from Escherichia coli [22], Salmonella enterica [5], Listeria monocytogenes [13], Corynebacterium glutamicum [23] or Bacillus subtilis [20]. The highest sensitivity of PaNADK to NAD+ compared to that of NADKs from other micro‐organisms may be related to the relatively low concentration of NAD+ in Pseudomonas [24].
NKI1, whose chemical structure is shown in Fig. 1, was designed based on the crystal structure of the LmNADK [16]. This lead compound was shown to act as a NAD+ competitive inhibitor of the enzymatic activity of LmNADK with a K i of 25 μm. NKI1 was also active on the NADK from Staphylococcus aureus (SaNADK) with a K i of 5 μm. Surprisingly, when tested on PaNADK, no significant inhibition was observed up to 50 μm (Fig. 4), suggesting that NKI1 is unable to bind into the active site of PaNADK despite a rather good sequence conservation within the NADP+ binding site and to a lower extent on the whole sequence (Fig. 5).
Fig. 4.
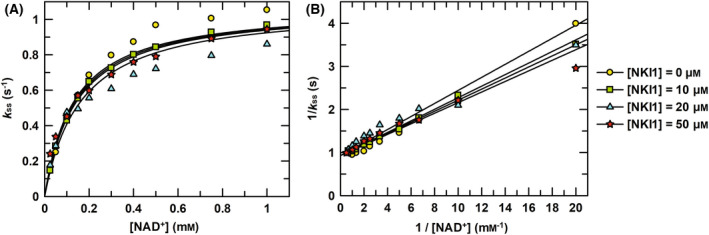
No significant effect of NKI1 on the enzymatic activity of PaNADK. (A) The steady‐state rate constants of PaNADK activity were determined at concentrations of 4 mm of MgATP, 0.05 to 1 mm of NAD+ and at different concentrations of NKI1, as indicated in the figure. The global fitting of competitive inhibition is shown as lines. (B) Lineweaver–Burk representation of the data showing no significant inhibition of PaNADK by NKI1.
Fig. 5.
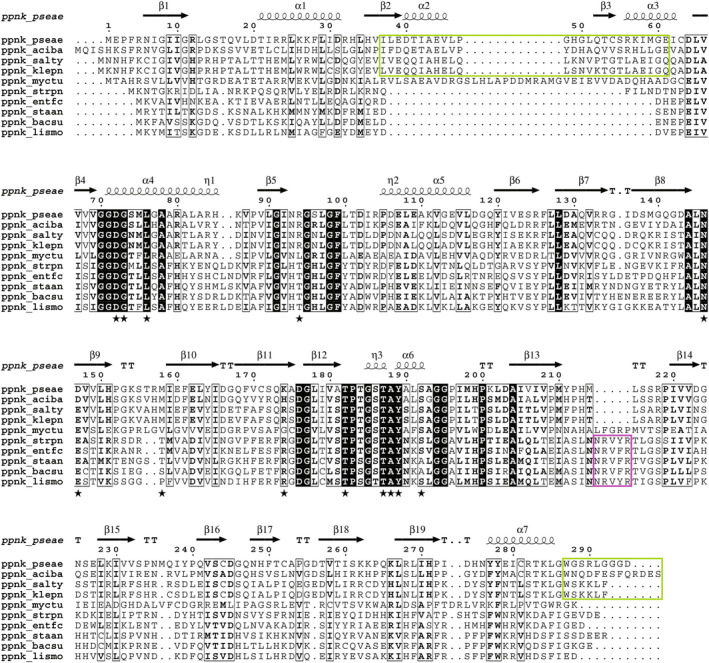
Sequence alignment of bacterial NADKs. Sequences of bacterial NADKs were aligned and classified from top to bottom as follows: NADKs of Gram‐negative bacteria (Pseudomonas aeruginosa, Acinetobacter baumannii, Salmonella typhimurium and Klebsiella pneumoniae), of Mycobacterium tuberculosis and of Gram‐positive bacteria (Streptococcus pneumoniae, Enterococcus faecium, Staphylococcus aureus, Bacillus subtilis and Listeria monocytogenes). Conserved residues are highlighted by black boxes, and conservative substitutions are shown in bold. Extensions specific to Gram‐negative and Gram‐positive bacteria are highlighted by green and pink boxes, respectively. Residues important for NADP+ binding are indicated by a black star symbol. Secondary structures in PaNADK are shown above the alignment. The figure was drawn using ESPript [44].
The crystal structure of PaNADK explains why NKI1 does not inhibit its activity
As for the other bacterial NADKs described so far, PaNADK appeared to be tetrameric in solution from size exclusion chromatography (SEC) analysis (Fig. 2A) and SAXS measurement (Fig. 2C). The SAXS curve showed little trace of structure unfolding and can be nicely fitted with a tetrameric model deduced from homologous structure (e.g. PDB 2AN1) or subsequently from the refined PaNADK structure (see below).
To better understand the lack of effect of NKI1 on PaNADK activity, we determined the atomic structure of the protein bound to NADP+ by X‐ray crystallography (Table 1). Crystals were grown in 0.1 m Tris/HCl pH 8.5 and 0.8 m ammonium sulphate and diffracted to 2.3 Å. The structure was solved using molecular replacement and a comparative model extracted from the homologous structure from Salmonella typhimurium (PDB 2AN1; 48% of sequence identity). During the course of our study, another structure of PaNADK complexed with NADP+ at a resolution of 2.6 Å was deposited on the PDB (ID 7MH7) without associated publication. It shows no major difference compared with our crystal structure despite a distinct space group. This suggests no major impact from the crystal packing on the two crystal structures.
Table 1.
Data collection, phasing and refinement statistics for NADP+‐bound PaNADK (PDB ID 7QVS).
| Ligand | Nicotinamide adenine dinucleotide phosphate (NADP+) |
|---|---|
| Data collection statistics | |
| Space group | P 41 2 2 |
| Unit cell dimensions | |
| a, b, c (Å) | 88.8, 88.8, 174.2 |
| α, β, γ (°) | 90.0, 90.0, 90.0 |
| Resolution range (Å) | 79.10–2.30 (2.34–2.30) a |
| R merge | 0.069 (0.656) a |
| R meas | 0.071 (0.669) a |
| R pim | 0.014 (0.131) a |
| I/σI | 39.2 (5.4) a |
| Completeness (%) | 100.0 (99.7) a |
| Avg multiplicity | 26.2 (25.3) a |
| CC1/2 | 1.00 (0.96) a |
| Total no. of reflections | 832 502 (39 222) a |
| No. of unique reflections | 31 752 (1551) a |
| Refinement statistics | |
| Resolution (Å) | 29.59–2.30 |
| No. reflections | 31 706 |
| R work/R free (%) | 19.7/23.6 |
| No. atoms | |
| Protein | 4284 (Chain A, Chain B) |
| Ligands | 144 |
| Water | 231 |
| B‐wilson/B‐average | 31.5/41 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.01 |
| Bond angles (°) | 1.52 |
| Ramachandran statistics | |
| Preferred region (%) | 97.00 |
| Allowed region (%) | 2.83 |
| Outliers (%) | 0.18 |
| Experimental conditions | |
| Wavelength (Å) | 1.00 |
| Temperature (K) | 100 |
| Beamline | SLS, X06DA, PXIII |
| Date and time of collection | 18 November 2019, 03:25:15 |
These statistics are for all measurements used during scale determination—this corresponds to the traditional isotropic analysis.
Values in parentheses are for the highest‐resolution shell. Note that data collection and refinement statistics have different highest‐resolution shells.
Two monomers were present in the asymmetric unit and highlighted the same fold as the other NADKs. Each monomer is made up of two domains, while the active site is formed at the interface between these two domains where the NADP+ molecule seats. Crystal symmetry allowed to build a tetramer predicted to be stable in solution according to the size of the buried interface according to PISA [25] and in agreement with our SEC and SAXS data (Fig. 2). The crystal structure of PaNADK is a tetramer consisting of a dimer of dimers (Fig. 6A,B), a conserved organization among all NADKs known so far (except the dimeric human mitochondrial NADK2 described recently [26]). Compared with the structure of the closely related NADK from S. typhimurium (which was solved in its apo state and not complexed to NADP+), the overall root‐mean‐square deviation (RMSD) per monomer and per tetramer is of 1.1 Å (over a common core of 270 residues) and 2.4 Å, respectively. When comparing the structure of PaNADK to the one of LmNADK (PDB 2I2A), which shares only 22% sequence identity, in the same NADP+‐liganded state, the RMSD rises to 1.9 Å (243 residues) and 2.5 Å (914 residues) for the monomer and the tetramer, respectively. The PaNADK harbours some specific extensions (Fig. 6C), especially an insertion in the N‐terminal domain (residues 37–61) and a distorted C‐terminal extension that is partially visible in the electron density (residues 287–294). On the contrary, it lacks the small insertion specific to the Gram‐positive bacteria (residues 188–193 in LmNADK). These features of PaNADK sequence are shared by NADKs from other Gram‐negative ESKAPE nosocomial pathogens (Fig. 5).
Fig. 6.
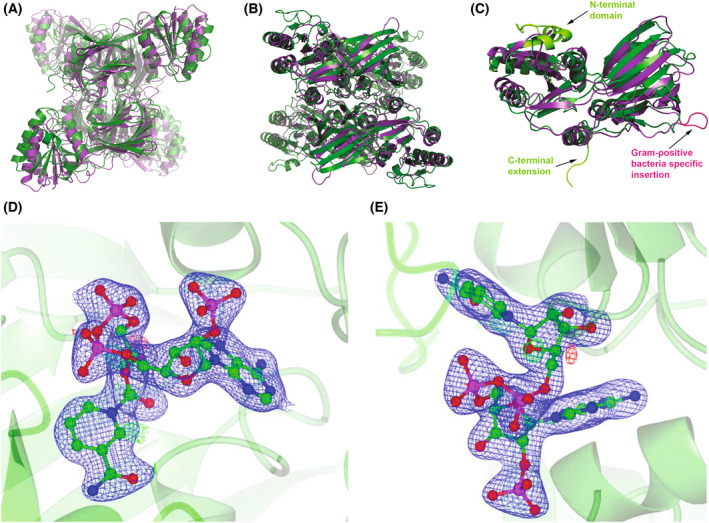
Comparison of the structure of PaNADK and LmNADK. In all panels, PaNADK and LmNADK structures are shown in green and purple, respectively. (A, B) Superimposed tetrameric form of PaNADK (PDB ID 7QVS) and LmNADK (PDB ID 2I2A) in two different orientations. (C) Superimposed monomeric form of PaNADK and LmNADK highlighting the main structural differences between the two structures, corresponding to extensions or insertions indicated on the sequence alignment (Fig. 5). (D, E) Electron density corresponding to the NADP+ bound to PaNADK. The 2F o − F c omit map (blue) at a sigma level of 3 is shown in two different orientations. Images of the structures were generated using pymol version 2.5.2, Molecular Graphics System, Schrödinger, LLC. The electron density was generated using ccp4mg‐2.10.11 software.
The NADP+ electron density is well defined in our crystal structure (Fig. 6D,E) and provides accurate structural information on the NADP+ binding site of PaNADK. Comparison with the LmNADK‐NADP+ complex (PDB ID 2I2A) showed that the sub‐site A, which accommodates the adenosine moiety of NADP+, is well conserved while the sub‐site N accommodating the nicotinamide ribose moiety showed more variations. In the sub‐site A, the main interactions correspond to hydrogen bonds between the adenine moiety and residues N146, T184 and T187 (Fig. 7A) to form the same network as in other known NADKs. As with LmNADK, in the PaNADK sub‐site N, NADP+ interacts with the side chains of the conserved D176’ (from a second monomer), S192 and D147 residues through hydrogen bonds and with the side chain of Y189 via aromatic stacking (Fig. 7B). The main differences between LmNADK and PaNADK lie in the region of the diphosphate group and the sub‐site N. In PaNADK, a two‐residue long insertion brings new residues from a second monomer into the active site such as R157′ and M158′. The side chain of R157′ interacts through a salt bridge with one phosphate group of the diphosphate moiety of NADP+ (Fig. 7C). In addition, the following residue, M158′ also interacts with the same phosphate moiety of NADP+ group through a hydrogen bond with its backbone amine while its hydrophobic side chain stacks onto the nicotinamide. Meanwhile, Q247 interacts with the other phosphate of the diphosphate moiety of NADP+. In addition, R94 forms a stabilizing salt bridge with the incoming 2′‐phosphate group and K174′ (from a second monomer) forms a hydrogen bond with the 3′ hydroxyl of the phosphorylated ribose, two features not seen before. Of note, R94 appears conserved in NADKs from other Gram‐negative bacteria (see alignment). These additional interactions likely explain the higher sensitivity to NAD+ of PaNADK (K m = 0.15 mm) than of LmNADK (cooperative kinetics with S 0.5 = 1.11 mm and Hill coefficient of 1.27 [13]).
Fig. 7.
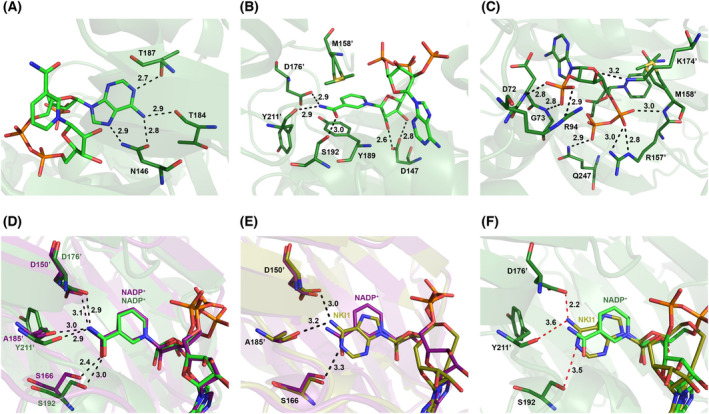
Detailed analysis of ligand binding sites of PaNADK and LmNADK. In all panels, PaNADK‐NADP+ and LmNADK‐NADP+ structures are shown in green and purple, respectively. PaNADK interactions with the different groups of NADP+: (A) adenine, (B) nicotinamide ribose and (C) diphosphate and phosphorylated ribose. (D) Comparison of the NADP+ binding sites of PaNADK and LmNADK showing a similar position of the ligand. (E) Superimposition of the structure of the LmNADK‐NADP+ (PDB ID 2I2A, in purple) and LmNADK‐NKI1 (PDB ID 6RG9, in gold) structure showing favourable interactions with both ligands. (F) Superimposition of the structure of the PaNADK‐NADP+ structure (PDB ID 7QVS, green) and PaNADK‐NKI1 model (in gold) showing unfavourable interactions with the adenine of NKI1. Images of the structures were generated using pymol version 2.5.2, Molecular Graphics System, Schrödinger, LLC.
The PaNADK structure allowed further comparisons with LmNADK to address NKI1 binding. Although the conformation of NADP+ is identical in both proteins (Fig. 7D), the compound NKI1 does not fit into the PaNADK active site as perfectly as it does in LmNADK. While the adenine moiety replacing the nicotinamide perfectly reproduced the network of hydrogen bonds involving the carbonyl of A185′ and the side chains of S166 and D150′ in LmNADK (Fig. 7E), a 1.1 Å shift in PaNADK leads to two contact losses with the equivalent residues Y211′ and S192 (3.6 and 3.5 Å, respectively), and one important clash with residue D176′ (2.2 Å) (Fig. 7F). These unfavourable interactions may explain the absence of inhibition of PaNADK by NKI1 and led us to propose a new derivative of NKI1 to fit better the active site of PaNADK.
Structure‐based design and synthesis of a new potential inhibitor of PaNADK
Considering the differences in the structure of the PaNADK binding site compared to that of LmNADK, an analogue of NKI1 with a benzamide in place of the adenine in the N sub‐site (hereinafter called 1) was synthesized. Because benzamide is an isosteric analogue of nicotinamide, it was already used for the design of inhibitors targeting NAD+‐utilizing enzymes, in particular IMP dehydrogenase and NADK [27, 28, 29, 30, 31]. It should adapt better than adenosine to the nicotinamide binding sub‐site of PaNADK.
The synthesis of compound 1 was achieved via a Sonogashira cross‐coupling reaction between the 8‐bromoadenosine derivative 2 and the benzamide riboside derivative 3 bearing a terminal alkyne functional group at the 5′‐position (Scheme 1).
Scheme 1.
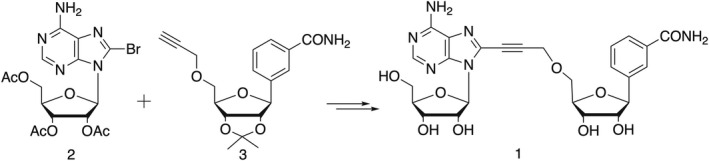
Synthesis of NAD+ analogue 1 via Sonogashira reaction between bromide 2 and alkyne 3.
The synthesis of the required alkyne 3 is depicted in Scheme 2. The intermediate benzonitrile 6 was obtained using previously reported procedures with slight modifications [27, 30]. Thus, Grignard reaction of commercially available 3‐bromobenzonitrile with the ribonolactone 4, followed by triethylsilane reduction of the anomeric hydroxyl group provided the C‐riboside 6 in 73% overall yield.
Scheme 2.
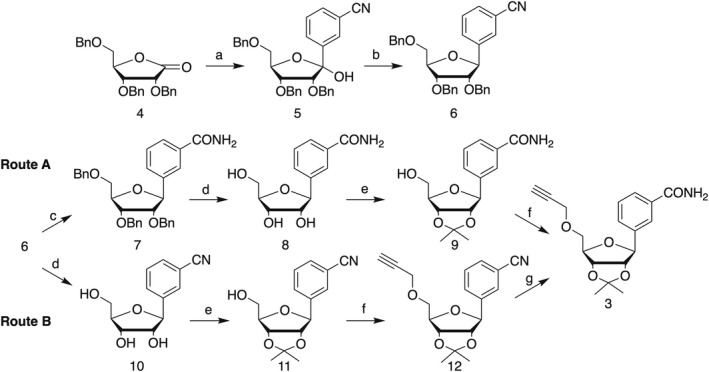
Synthesis of required alkyne 3. Reagents and conditions: (a) 5‐bromobenzonitrile, n‐BuLi, THF, −78 °C; MeOH – 78 °C to rt; (b) BF3–OEt2, Et3SiH, −0 °C to rt; (c) TMSOK, THF, reflux; (d) 1 m BBr3 in DCM, −78 °C to rt; (e) APTS, acetone, 2,2‐dimethoxypropane; (f) propargyl bromide, NaH, DMF, −0 °C; (g) H2O2, K2CO3, DMSO.
Next, following route A, conversion of the cyano group into carboxamide 7 by potassium trimethylsilanolate reaction, followed by benzyl ether cleavage using boron tribromide, afforded the benzamide riboside 8 in high overall yield [30]. After protection of the secondary hydroxyl groups as isopropylidene ether, the alkyne was introduced by reaction of 9 with propargyl bromide in the presence of sodium hydride in DMF affording 3 in moderate yield, along with propargylation of carboxamide group. Alternative route B involved debenzylation of 6 into benzonitrile riboside 10, followed by 2′,3′‐hydroxyl protection and 5′‐O‐propargylation into 12 in 87% over two steps. Next, conversion of the cyano group into the carboxyamide derivative 3 was achieved by treatment of 12 with hydrogen peroxide under basic conditions.
Finally, Sonogashira cross‐coupling reaction between bromide 2 and alkyne 3, followed by two‐step deprotection of the resulting coupling product 13 afforded the target dinucleotide 1 in 20% overall yield.
Compound 1 inhibits the enzymatic activity of PaNADK and is not cytotoxic
The effect of compound 1 on the enzymatic activity of PaNADK was tested in the same experimental conditions as the ones used for NKI1. Compound 1 was found to be a NAD+‐competitive inhibitor of PaNADK with a K i value of 4.6 ± 0.3 μm, as shown in Fig. 8.
Fig. 8.
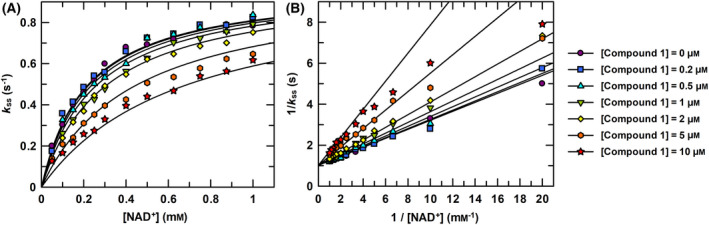
Inhibition of the enzymatic activity of PaNADK by compound 1. (A) The steady‐state rate constants of PaNADK activity were determined at 4 mm MgATP, 0.05–1 mm NAD+ and at different concentrations of 1, as indicated in the figure. Global competitive inhibition fittings are shown as lines. (B) Lineweaver–Burk representation of the data showing a significant competitive inhibition of PaNADK by compound 1.
Given its inhibitory effect on PaNADK enzymatic activity, compound 1 appears as a suitable molecule that may be active against the bacterium P. aeruginosa. Before testing the anti‐infectious properties of compound 1, we have first evaluated its cytotoxicity on human cells using a standard protocol. No cytotoxicity was observed up to concentrations of 50 μm of compound 1 (data not shown), supporting the relevance of further in vivo experimentations.
Compound 1 has no effect on P. aeruginosa growth in vitro but displays an antivirulence effect in vivo
The effect of compound 1 was first tested on P. aeruginosa growth in liquid cultures. We used PAO1 strain, which is the most commonly P. aeruginosa used strain for research. Various concentrations of compound 1 (ranging from 2 to 200 μm, dissolved in 0.01–1% DMSO) were tested, and the corresponding concentrations of DMSO were used as control. No difference in growth curves was found between compound 1‐treated bacteria and DMSO‐treated bacteria (Fig. 9). Hence, compound 1 does not affect P. aeruginosa growth in these experimental conditions. The lack of antibacterial action in liquid cultures may be linked to conditions (LB medium) that allow growth on a sub‐optimal pool of NADP(H) and/or to a poor penetration of compound 1 through the bacterial envelope.
Fig. 9.
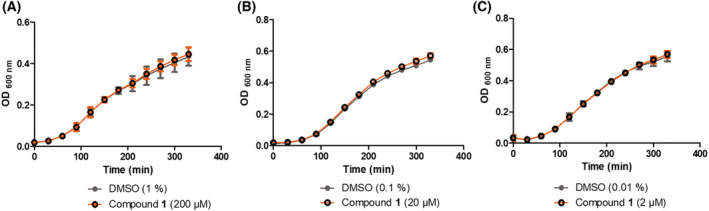
Effect of compound 1 on P. aeruginosa growth in liquid medium. P. aeruginosa growth curves in LB medium in the presence of compound 1 at (A) 200, (B) 20 and (C) 2 μm (in 1%, 0.1% or 0.01% DMSO, respectively) or in the presence of the indicated percentage of DMSO as control. Graphs represent the mean of three independent experiments. Error bars represent standard errors of the mean.
We then evaluated the effect of compound 1 in vivo in the zebrafish embryo infection model. Zebrafish (Danio rerio) is a vertebrate that offers several advantages, in terms of methodological, financial and ethical issues, over mammalians models. This model is suitable for in vivo chemical screening [32, 33] and drug toxicity assays [34], with the advantage that permeability of the larvae allows the entry of small compounds by balneation exposure. Zebrafish embryo has been used with various bacterial pathogens, including P. aeruginosa [18, 35], and we have recently developed a novel mode of infection, based on bath immersion of injured embryos, to screen anti‐infectious compounds efficient against P. aeruginosa [17].
We have thus used this bath immersion infection mode with tail‐injured zebrafish embryos to address the effect of compound 1 on P. aeruginosa virulence. The effect of compound 1, dissolved in DMSO, was tested at various concentrations on injured embryos immersed with bacterial inoculum at approximately 8.107 CFU·mL−1. Compound 1 was added simultaneously with the bacterial cells following a protocol previously described [17]. In non‐treated embryos immersed with PAO1, DMSO was added to reflect the amount of DMSO in compound 1‐treated embryos. Compound 1 at 20 μm exhibited a protective effect since a significant reduction in embryo mortality was observed comparatively to DMSO‐treated embryos (Fig. 10A). Importantly, the addition of 20 μm compound 1 was not associated with toxicity for embryos, as shown by the lack of significant mortality upon addition of the sole molecule (Fig. 10B).
Fig. 10.
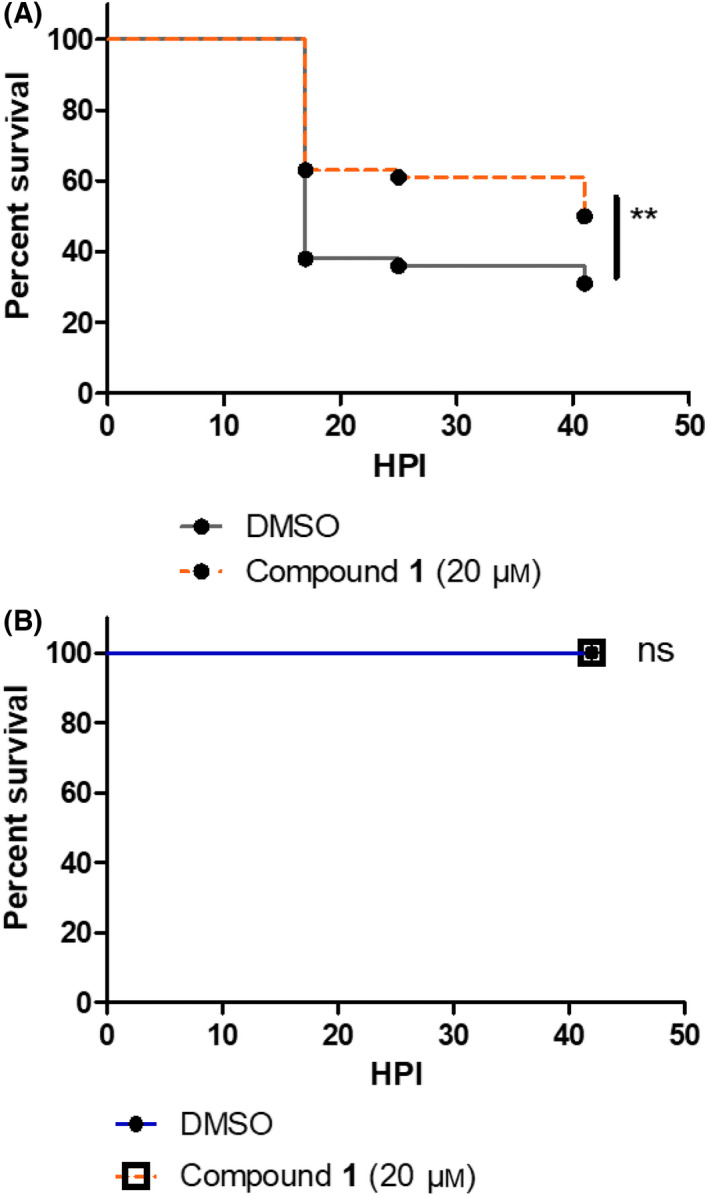
Evaluation of compound 1 on Pseudomonas virulence in vivo. (A) The efficacy of compound 1 against P. aeruginosa was tested with embryos injured in the tail fin and bath infected with PAO1 suspension at approximately 8.107 CFU·mL−1 in the presence of compound 1 at 20 μm (in 0.1% DMSO). Injured embryos in the control group were treated with PAO1 suspension in the presence of DMSO at 0.1%. A significant difference (P < 0.01) is found in the survival curve of treated versus non‐treated embryos. (B) The toxicity of compound 1 at 20 μm was monitored after immersion of embryos injured in the tail fin. For all experiments, the embryo survival was monitored for 45 h and survival curves were represented with a Kaplan–Meier representation. Graphs represent the pool of three minimum independent experiments (n = 120 larvae for compound 1 test or 60 larvae for toxicity test, in total per condition). ** means P value <0.01, ns means no significant difference.
Thus, our in vivo results indicate an anti‐infective potential of this novel compound 1, without exhibiting any toxicity. Because compound 1 does not display antibacterial activity in vitro in the conditions tested, the in vivo effect can be qualified as antivirulence effect.
Discussion
Inhibiting bacterial NADK is a promising and innovative antimicrobial strategy. A NAD+‐competitive inhibitor, NKI1, which is active against NADK from Gram‐positive bacteria, appeared inactive against PaNADK. The structure of PaNADK suggested that, unlike LmNADK, the sub‐site N of PaNADK cannot accommodate a ligand such as NKI1, which has an adenine in place of the nicotinamide of NAD+. In agreement, a novel analogue of NKI1 harbouring a benzamide instead of the clashing adenine (compound 1) had an inhibition constant value on PaNADK that is comparable to that of NKI1 on LmNADK and SaNADK [16]. NAD+ analogues with a benzamide moiety in place of the nicotinamide have been previously reported [30], but the most potent competitive inhibitor exhibited only a moderate activity on NADK from Mycobacterium tuberculosis (37% inhibition at 2 mm). In the case of compound 1, the introduction of a propargyl‐link contributes to its strong inhibitory activity on purified enzyme, probably due to an increased rigidity of the molecule and a better mimic of the NAD+ conformation in its binding site.
Compound 1 displays an antivirulence effect in vivo in P. aeruginosa‐infected zebrafish model, without displaying antibacterial activity in vitro under the conditions used in this study. Further studies will be required to better understand the mechanism by which compound 1 increases the defensive ability of zebrafish embryos against P. aeruginosa. Compound 1 may have an effect on P. aeruginosa specifically during infection due to the stressing conditions encountered within the host, such as reactive oxygen species. In addition, or alternatively, we cannot exclude that compound 1 may have other targets than PaNADK in vivo.
To a better understanding of its mechanism of action in vivo, optimization of compound 1 should be done in the future and its synergy with other antibacterial compounds should be investigated. Compound 1 could also serve as a basis to develop fluorescent or clickable probes that could be used as molecular tools to detect or modulate the activity of NAD+‐dependent enzymes or identify putative alternative targets [36, 37, 38]. Importantly, NADKs from Gram‐negative bacteria from the ESKAPE group are closely related to PaNADK, suggesting that compound 1 might have an inhibitory effect on the enzymes from other bacterial pathogens as well. According to the World Health Organization, the most critical group of bacteria for which new antibiotics are urgently needed includes multi‐resistant Acinetobacter, Pseudomonas and various Enterobacteriaceae such as Klebsiella, E. coli, Serratia and Proteus that can cause severe and often deadly infections. In addition, we do not exclude an efficacy of compound 1 against Gram‐positive bacteria since it is active on LmNADK with a K i of 3.2 ± 0.4 μm (data not shown).
In conclusion, we have designed a new inhibitor of bacterial NADK, which is a still orphan therapeutic target. Efficient NADK inhibitors could have a broad activity spectrum and therefore an important impact on the treatment of multiple infectious diseases, including those triggered by these multidrug‐resistant and critical bacteria.
Materials and methods
Molecular cloning of the PaNADK gene
A synthetic gene encoding PaNADK (corresponding to ppnK gene in PAO1) was optimized for expression in E. coli and ordered from Integrated DNA Technologies, BVBA (Leuven, Belgium). It was amplified with forward primer 5′‐CCCTTTCATATGATGGAGCCCTTCCGCAAT‐3′ and reverse primer 5′‐CCCAAACTCGAGGTCACCCCCTCCTAAACG‐3′ including cleavage sites of restriction enzymes for cloning into pET‐22b(+) vector (Q5 Hot Start High‐Fidelity 2× Master Mix). The PCR product was then digested using the NdeI and Xho1 restriction enzymes, purified (NucleoSpin Gel and PCR clean‐up kit, Macherey‐Nagel, Hoerdt, France), and ligated (T4 ligase) into linearized pET‐22b(+) vector at the same restriction sites. At the C terminus, the construct codes for two additional amino acids (LE) followed by a 6His‐tag. The gene sequence of PaNADK was checked by sequencing (Genewiz, Paris, France).
PaNADK expression and purification
PaNADK protein was overexpressed in E. coli BL21 (DE3) cells in 2‐YT broth with 100 μg·mL−1 ampicillin. The protein expression was induced at OD600 of 0.7–0.8 with 1 mm isopropyl β‐D‐1‐thiogalactopyranoside (IPTG) overnight at 25 °C. The harvested cells were re‐suspended in buffer A (50 mm sodium phosphate pH 8.3, 150 mm NaCl, 10 mm imidazole, 2 mm DTT) supplemented with 1 tablet of cOmplete™ EDTA‐free protease inhibitor cocktail (Roche), 5 μg·mL−1 of lysozyme (Euromedex), 5 μg·mL−1 of DNase (Sigma Aldrich). Bacterial cell disruption was achieved by sonication at 40% amplitude during 5 min (5 s on, 5 s off) with a 6 mm probe on ice. Cellular debris were removed by centrifugation at 20 000 g for 30 min and followed by filtration through 0.45‐μm syringe filter. The bacteria lysate was loaded onto a HisTrap FF column equilibrated with buffer A using Äkta Pure system (GE Healthcare) at 4 °C. The column was washed with buffer A during 15 column volumes to remove unspecific binding proteins, followed by elution using buffer B (50 mm sodium phosphate pH 8.3, 150 mm NaCl, 500 mm imidazole, 2 mm DTT) with a linear gradient of imidazole from 10 to 500 mm in 20 column volumes. The fractions containing the protein were pooled and concentrated using Amicon Ultra 15 centrifugal filters (3 kDa, Merck Millipore) at 4 °C. The concentrated protein was loaded onto a 16/60 Superdex 200 pre‐equilibrated with buffer C (50 mm NaH2PO4/Na2HPO4 pH 8.3, 100 mm NaCl, 5 mm DTT). Finally, the protein was concentrated till 30 mg·mL−1, aliquoted, flash‐frozen in liquid nitrogen and stored at −80 °C.
The sequence of the recombinant protein is shown below and corresponds to the sequence of P. aeruginosa PAO1 NADK (UniProtKB Q9HZC0) with a LEHHHHHH tag at the C terminus:
MEPFRNIGIIGRLGSTQVLDTIRRLKKFLIDRHLHVILEDTIAEVLPGHGLQTCSRKIMGEICDLVVVVGGDGSMLGAARALARHKVPVLGINRGSLGFLTDIRPDELEAKVGEVLDGQYIVESRFLLDAQVRRGIDSMGQGDALNDVVLHPGKSTRMIEFELYIDGQFVCSQKADGLIVATPTGSTAYALSAGGPIMHPKLDAIVIVPMYPHMLSSRPIVVDGNSELKIVVSPNMQIYPQVSCDGQNHFTCAPGDTVTISKKPQKLRLIHPIDHNYYEICRTKLGWGSRLGGGDLEHHHHHH.
The expression, solubility and purity of PaNADK were tested on SDS/PAGE gel (Invitrogen™ NuPAGE™ 4–12% gel). The molar absorption coefficient and the absorbance value for 0.1% of the 6His‐tagged protein, hereafter named PaNADK, were estimated with the abim software (http://bioserv.cbs.cnrs.fr/ABIM/w3bb/d_abim/) at 14 650 m −1 cm−1 and 0.441 (g·L−1)−1·cm−1, respectively.
SAXS
Scattering curves were recorded at the SWING beamline of SOLEIL (Paris‐Saclay, France) at X‐ray wavelength of 1 Å with the EIGER‐4M detector positioned 2 m from the sample using 12 keV X‐ray flux. The resulting range of the scattering vector was 0.038 < q < 6.3 nm−1. The protein samples dialysed against 50 mm Tris/HCl pH 8, 100 mm NaCl, 5 mm DTT buffer during 3 h at 4 °C and centrifuged at 12 000 g before measurements. Batch measurements were recorded at room temperature over 38 exposures and carefully averaged. To eliminate the contribution of the mobile phase, buffer scattering was collected before and after each sample measurement and subtracted from sample scattering. A dilution series consisting of three protein concentrations (9, 3 and 1 mg·mL−1) was recorded. Additionally, the scattering curves were recorded in the presence of 2 mm NAD+ or 2 mm NADP+. Guinier analysis, distance distribution functions P(r) and Kratky analysis of the scattering curves were performed using the ATSAS suite [39]. Crysol was used to fit experimental SAXS data to the protein structure obtained from x‐ray crystallography. The goodness of fit obtained from the dimer and its assembly as a tetramer (calculated via PISA) were compared [40].
In vitro enzymatic activity measurements and inhibition assays
The optimum pH, buffer and salt concentration for the in vitro enzymatic activity of the recombinant PaNADK were screened using the RUBIC Buffer kit (Molecular Dimensions). The enzymatic reaction was followed at 30 °C in a CLARIOstar plate reader (BMG LABTECH) by measuring the absorbance at 340 nm (OD340) using an enzymatic coupled system involving glucose‐6‐phosphate dehydrogenase (G6PDH). The reaction mixture was made in a half‐area 96‐well microplate (Greiner Bio‐One Clear UV‐Star). It consisted of 42 μL of the RUBIC kit buffers, 4 μL of 12.5 μm PaNADK in Buffer C and 4 μL of Mix (12.5 U·mL−1 G6PDH, 62.5 mm NAD+, 62.5 mm MgATP, 62.5 mm glucose‐6‐phosphate) added at time 0. The final protein and ligand concentrations were 1 μm PaNADK, 1 U·mL−1 G6PDH, 5 mm NAD+, 5 mm MgATP and 5 mm glucose‐6‐phosphate. OD340 values were converted to concentrations of produced NADH (i.e. NAD+ consumed) using a molar absorption coefficient for NADH of 6220 m −1·cm−1. The value of the steady‐state rate constant (k ss) corresponds to the initial slope of NAD+ consumed per second divided by the concentration of PaNADK. The values of k cat and K m for NAD+ were determined from the dependence of k ss on the initial concentration of NAD+ using the Michaelis–Menten equation.
Inhibition assays were performed as described above at 30 °C in a buffer consisting of 50 mm Tris pH 7.5, 1 mm MgCl2 and 5 mm DTT and a final volume of 100 μL. The final concentrations in the reaction mixtures were 25 nm PaNADK, 1 U·mL−1 G6PDH, 4 mm MgATP, 5 mm glucose‐6‐phosphate, 0.05–1 mm NAD+ and various concentrations of inhibitor (see the legends of the figures). Buffer, NAD+ and inhibitor solutions (total volume 25 μL, concentration 4×) were dispensed into the microplate using an OT‐2 lab robot (Opentrons). The 25 μL of mixture containing G6PDH, MgATP and glucose‐6‐phosphate at a 4× concentration was dispensed using a manual repeating pipette. Finally, the reaction was started at time 0 by adding and mixing 50 μL of PaNADK at a 2× concentration using a multichannel pipette.
The values of the inhibition constants were determined from the raw values of k ss at different concentrations of NAD+ and inhibitor using a competitive inhibition equation (Eqn 1). A Lineweaver–Burk transformation of the raw values was used to produce an illustrative double‐reciprocal plot of the data.
| (1) |
Fits and graphical representations were made using GraFit software (version 7.0.3, Erithacus Software Ltd).
Crystallization and structure refinement
The protein solution at 10 mg·mL−1 in 50 mm NaH2PO4/Na2HPO4 pH 8.3, 100 mm NaCl and 5 mm DTT was pre‐incubated 2 h with 10 mm NADP+ (100 mm NADP+ stock solution in 50 mm Tris, pH 6.5) and cleared using 0.22 μm centrifugal filter by centrifugation at 12 000 g. Subsequently, a crystallization screen was applied using six commercially available kits from Qiagen (PEG 1 & 2, pH‐Clear 1 & 2, AmSO4 Suite and Classics) handled by crystallization robot (Mosquito, SPT Labtech). 0.1 μL of PaNADK‐NADP+ solution and 0.1 μL of reservoir were mixed to equilibrate against 45 μL reservoir solution using the sitting‐drop vapour‐diffusion method at 18 °C. Crystals appeared within a week in condition N° 77 of AmSO4 Suite (0.1 mm Tris pH 8.5, 1 m ammonium sulphate). Crystal growth was further manually reproduced and optimized in an EasyXtal 15‐well plate (NeXtal) by mixing 1 μL of PaNADK‐NADP+ solution with 1 μL of reservoir and equilibrating against 500 μL reservoir solution using the hanging‐drop vapour‐diffusion method at 18 °C. Crystals formed in the optimal condition (0.1 mm Tris pH 8.5, 0.8 mm ammonium sulphate) were flash‐frozen in liquid nitrogen. X‐ray diffraction data were collected in the X06DA (PXIII) macromolecular crystallography beamline at the Swiss Light Source. The structure was solved by molecular replacement (CCP4, PHENIX) using as a search model, a comparative model derived from PDB 2AN1, the NADK from Salmonella typhimurium LT2. Global Phasing Limited software packages (Buster and Rhofit) were used for a final step of structure refinement [41]. The statistics of X‐ray data processing and structure refinement are summarized in Table 1.
Sequence alignment of bacterial NADKs
The primary sequences of bacterial NADKs were extracted from UniProtKB. UniProt entry IDs were Q9HZC0 for P. aeruginosa (ppnk_pseae), A0A059ZQ75 for Acinetobacter baumannii (ppnk_aciba), P65774 for Salmonella typhimurium (ppnk_salty), A6TCM2 for Klebsiella pneumoniae (ppnk_klepn), P9WHV7 for Mycobacterium tuberculosis (ppnk_myctu), P9WHV7 for Streptococcus pneumoniae (ppnk_strpn), Q3XWF9 for Enterococcus faecium (ppnk_entfc), H6UH60 for Staphylococcus aureus (ppnk_staau), O31612 for Bacillus subtilis (ppnk_bacsu) and Q8Y8D7 for Listeria monocytogenes (ppnk_lismo). Sequences were aligned using ViTO software [42].
Chemical synthesis
General
The general methods have already been described in [15]. All commercially available reagents and solvents, unless otherwise stated, were used without purification. Anhydrous reactions were carried out under an argon atmosphere. Analytical thin‐layer chromatography (TLC) was performed on TLC plates pre‐coated with silica gel 60 F254. Compounds were visualized with UV light (254 nm) and by spraying with a mixture of ethanol/anisaldehyde/sulfuric acid/acetic acid (90/5/4/1), followed by heating. Reactions were also monitored using an HPLC system (Agilent 1100 equipped with a C18 reverse phase column) coupled to a mass spectrometer (ESI source). Flash chromatography was performed with silica gel 60 (230–400 mesh). HPLC purification was carried out on an Agilent system (1100 Series) equipped with a diode array detector using a C18 reverse phase column (Kromasil, 5 μm, 100 Å, 150 × 4.6 mm) and a linear gradient of acetonitrile in 10 mm triethylammonium acetate (TEAA) buffer over 15 or 20 min at a flow rate of 4 mL·min−1. Retention time (R t) and elution conditions are specified. The purity of tested compounds was at least 96% pure as determined by HPLC. NMR spectra (Appendix S1) were recorded on Bruker Avance 400 (1H at 400.13 MHz and13C at 100.62 MHz). Chemical shifts (δ) are reported in ppm relative to the solvent signals. Coupling constants (J values) are reported in Hz. The complete assignment of 1H and 13C signals was performed by analysis of the correlated homonuclear {H,H}‐COSY and heteronuclear {H,C}‐HMBC, {H,C}‐HSQC spectra. High‐resolution mass spectra (HRMS) were recorded with a Q‐Tof Micro mass spectrometer under electrospray ionization in positive ionization mode using a mobile phase of acetonitrile/water with 0.1% formic acid.
NKI1 was prepared as described previously [16].
3‐(2,3,5‐tri‐O‐benzyl‐β‐D‐ribofuranosyl)benzonitrile (6)
The title compound was prepared following reported procedures with slight modifications [27, 30]. To a solution of 3‐bromobenzonitrile (3.81 g, 21 mmol) in dry tetrahydrofuran (THF) (72 mL) containing activated 4 Å molecular sieves (5 g) was slowly added n‐BuLi (2.5 m in hexane, 9.2 mL) at –78 °C under an argon atmosphere. Stirring was maintained for 20 min after addition was completed, and then a solution of ribonolactone 4 (2.51 g, 6 mmol) in THF (24 mL) was slowly added to the reaction mixture at –78 °C. After 30 min, anhydrous MeOH (7.3 mL) was added at the temperature allowed to warm to room temperature. The reaction mixture was neutralized by adding 2 N HCl (12 mL) and extracted with sat. NaHCO3 (150 mL). The organic phase was dried and concentrated under reduced pressure, and the residue was purified by flash column chromatography (180 g silica gel, 5–15% ethyl acetate in cyclohexane) affording hemiacetal 5 (2.77 g, 88.5%). To a solution of 5 (2.75 g, 5.3 mmol) in dry dichloromethane (DCM) (26.5 mL) at 0 °C was added Et3SiH (3.37 mL, 21.1 mmol). After stirring for 5 min, BF3·Et2O (1.31 mL, 10.6 mmol) was slowly added and the resulting mixture was warmed to room temperature over 10 min. Then, E3N (7.39 mL, 53 mmol) was added, and the reaction mixture was concentrated under reduced pressure. The residue was purified by flash column chromatography (5–10% ethyl acetate in cyclohexane) to give 6 (2.23 g, 83.2%). 1H NMR (400 MHz, DMSO‐d 6 ): δ 3.64 (dd, J = 3.9 Hz, J = 10.6 Hz, 1H, H‐5′), 3.71 (dd, J = 3.7 Hz, J = 10.6 Hz, 1H, H‐5″), 3.93 (dd, J = 5.2 Hz, J = 6.4 Hz, 1H, H‐2′), 4.11 (t, J = 4.4 Hz, 1H, H‐3′), 4.29 (q, J = 3.6 Hz, 1H, H‐4′), 4.44–4.61 (m, 6 H, CH2 Bn), 4.95 (d, J = 6.6 Hz, 1H, H‐1′), 7.18–7.40 (m, 15 H, H Bn), 7.50 (t, 1H, H Ph), 7.71 (d, 1H, H Ph), 7.75 (d, 1H, H Ph), 7.78 (s, 1H, H Ph); 13C NMR (100 MHz, DMSO‐d 6 ): δ 70.64 (C‐5′), 71.55, 71.71 and 72.95 (CH2 Bn), 77.72 (C‐3′), 81.28 (C‐1′), 82.03 (C‐4′), 83.85 (C‐2′), 111.74 and 119.18 (Cq), 127.87, 127.93, 128.02, 128.09, 128.29, 128.59, 128.67, 128.75, 129.86, 130.10, 131.63 and 131.89 (CH Ph), 138.37, 138.66 and 142.78 (Cq Ph); HRMS (ESI+‐TOF): m/z calcd for [C33H31NO4 + H]+ 506.2326, found 506.2320.
3‐(2,3,5‐tri‐O‐benzyl‐β‐D‐ribofuranosyl)benzamide (7)
To compound 6 (2.22 g, 4.4 mmol) in anhydrous THF (11 mL) was added TMSOK (0.845 g, 6.6 mmol) in a sealed tube. After stirring at 70 °C for 24 h, ethanol was added to the reaction mixture, then volatiles were removed under reduced pressure and the residue was purified by flash column chromatography (100 g silica gel, 0–2% MeOH in DCM) affording 7 (1.92 g, 83.5%). 1H NMR (400 MHz, DMSO‐d 6 ): δ 3.60–3.72 (m, 2H, H‐5′ and H‐5″), 3.94 (dd, J = 5.4 Hz, J = 7.1 Hz, 1H, H‐2′), 4.06–4.11 (m, 1H, H‐4′), 4.24–4.30 (m, 1H, H‐3′), 4.45–4.61 (m, 6H, CH2 Bn), 4.93 (d, J = 6.8 Hz, 1H, H‐1′), 7.19–7.40 (m, 18H, 15H Bn, H Ph and CONH2), 7.55 (d, 1H, H Ph), 7.79 (d, 1H, H Ph), 7.92 (br, 1H, H Ph); 13C NMR (100 MHz, DMSO‐d 6 ): δ 70.82 (C‐5′), 71.51, 71.69 and 72.93 (CH2 Bn), 77.89 (C‐3′), 81.79 (C‐1′), 82.13 (C‐4′), 83.91 (C‐2′), 111.74 (Cq), 126.10, 127.17, 127.87, 127.91, 127.93, 128.01, 128.27, 128.51, 128.59, 128.67, 128.73 and 129.44 (CH), 134.83 (Cq Ph), 138.48, 138.71 and 138.96 (Cq Bn), 141.11 (Cq Ph), 168.33 (CO); HRMS (ESI+‐TOF): m/z calcd for [C33H33NO5 + H]+ 524.2431, found 524.2429.
3‐(β‐D‐ribofuranosyl)benzamide (8)
A 1 M solution of BBr3 in DCM (14.6 mL) was added dropwise to a stirred and cooled to −78 °C solution of 7 (1.91 g, 3.6 mmol) in anhydrous DCM (72 mL) containing molecular sieves. After a 1 h at −78 °C, the mixture was allowed to warm to room temperature and stirred overnight. The reaction was quenched by adding diethyl ether/water (4/1, 180 mL) at 4 °C for 20 min, and then the volatiles were removed under reduced pressure. Purification by flash column chromatography (115 g silica gel, 10 to 12% MeOH in DCM) afforded 8 (0.76 g, 83%). 1H NMR (400 MHz, DMSO‐d 6 ): δ 3.54 (dd, J = 4.8 Hz, J = 11.6 Hz, 1H, H‐5′), 3.59 (dd, J = 4.4 Hz, J = 11.6 Hz, 1H, H‐5″), 3.71 (dd, J = 5.4 Hz, J = 7.1 Hz, 1H, H‐2′), 3.83 (t, J = 4.6 Hz, J = 8.3 Hz, 1H, H‐4′), 3.90 (dd, J = 3.6 Hz, J = 5.4 Hz, 1H, H‐3′), 4.60 (d, J = 7.1 Hz, 1H, H‐1′), 7.32 (br, 1H, CONH2), 7.40 (t, 1H, H Ph), 7.55 (d, 1H, H Ph), 7.77 (d, 1H, H Ph), 7.86 (s, 1H, H Ph), 7.94 (br, 1H, CONH2); 13C NMR (100 MHz, DMSO‐d 6 ): δ 62.51 (C‐5′), 71.85 (C‐3′), 77.94 (C‐2′), 83.33 (C‐1′), 85.71 (C‐4′), 125.91, 126.84, 128.36 and 129.56 (CH Ph), 134.59 and 141.93 (Cq Ph), 168.46 (CO); HRMS (ESI+‐TOF): m/z calculated for [C12H15NO5 + H]+ 254.1023, found 254.1025.
3‐(2,3‐O‐isopropylidene‐β‐D‐ribofuranosyl)benzamide (9)
To a suspension of 8 (0.76 g, 3.0 mmol) in acetone (30 mL) were added 2,2‐dimethoxypropane (1.11 mL, 9.0 mmol) and APTS (0.57 g, 3.0 mmol). After stirring overnight at room temperature, the reaction was quenched by the addition of 1 M Na2CO3 (15 mL), concentrated under reduced pressure and then extracted twice with ethyl acetate. The organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure. Purification by flash column chromatography (40 g silica gel, 5% MeOH in DCM) afforded 9 (0.58 g, 66%). 1H NMR (400 MHz, DMSO‐d 6 ): δ 1.30 (s, 3H, CH3 isop), 1.55 (s, 3H, CH3 isop), 3.56 (dd, 1H, H‐5′), 3.61 (dd, 1H, H‐5″), 4.01 (q, J = 4.7 Hz, J = 8.8 Hz, 1H, H‐4′), 4.53 (dd, J = 5.4 Hz, J = 6.7 Hz, 1H, H‐2′), 4.69 (dd, J = 3.8 Hz, J = 6.7 Hz, 1H, H‐3′), 4.79 (d, J = 5.4 Hz, 1H, H‐1′), 4.92 (t, 1H, OH), 7.36 (br, 1H, CONH2), 7.44 (t, 1H, H Ph), 7.53 (d, 1H, H Ph), 7.80 (d, 1H, H Ph), 7.85 (s, 1H, H Ph), 7.96 (br, 1H, CONH2); 13C NMR (100 MHz, DMSO‐d 6 ): δ 25.86 (CH3 isop), 27.86 (CH3 isop), 61.99 (C‐5′), 82.22 (C‐3′), 84.99 (C‐1′), 85.23 (C‐4′), 86.54 (C‐2′), 114.34 (Cq isop), 125.70, 127.24, 128.69 and 129.34 (CH Ph), 134.85 and 140.55 (Cq Ph), 168.23 (CO); HRMS (ESI+‐TOF): m/z calcd for [C15H19NO5 + Na]+ 316.1155, found 316.1151.
3‐(β‐D‐ribofuranosyl)benzonitrile (10)
A 1 M solution of BBr3 in DCM (0.8 mL) was added dropwise to a stirred and cooled to −78 °C solution of 6 (100 mg, 0.2 mmol) in anhydrous DCM (4 mL) containing molecular sieves. After a 1 h at −78 °C, the mixture was allowed to warm to room temperature and stirred overnight. The reaction was quenched by adding diethyl ether/water (4/1, 10 mL) at 4 °C for 20 min, and then the volatiles were removed under reduced pressure. Purification by flash column chromatography (82 g silica gel, 5–8% MeOH in DCM) afforded 10 (37 mg, 79%). 1H NMR (400 MHz, DMSO‐d 6 ): δ 3.54 (dd, J = 4.3 Hz, J = 11.7 Hz, 1H, H‐5′), 3.60 (dd, J = 4.0 Hz, J = 11.7 Hz, 1H, H‐5″), 3.68 (dd, J = 5.3 Hz, J = 7.4 Hz, 1H, H‐2′), 3.84–3.88 (m, 1H, H‐4′), 3.93 (dd, J = 3.2 Hz, J = 5.2 Hz, 1H, H‐3′), 4.63 (d, J = 7.4 Hz, 1H, H‐1′), 7.55 (t, 1H, H Ph), 7.72 (d, 1H, H Ph), 7.74 (d, 1H, H Ph), 7.85 (br, 1H, H Ph); 13C NMR (100 MHz, DMSO‐d 6 ): δ 62.30 (C‐5′), 71.88 (C‐3′), 78.27 (C‐2′), 82.23 (C‐1′), 86.06 (C‐4′), 111.56 (CN), 119.40 (Cq), 129.73, 129.96, 131.51 and 131.53 (CH Ph), 143.71 (Cq Ph); HRMS (ESI+‐TOF): m/z calcd for [C12H13NO4‐H]+ 234.0772, found 234.0767.
3‐(2,3‐O‐isopropylidene‐β‐D‐ribofuranosyl)benzonitrile (11)
To a suspension of 10 (0.30 g, 1.28 mmol) in acetone (13 mL) were added 2,2‐dimethoxypropane (0.47 mL, 3.84 mmol) and APTS (0.24 g, 1.28 mmol). After stirring overnight at room temperature, the reaction was quenched by the addition of 1 m Na2CO3 (6.5 mL) and volatiles were removed under reduced pressure and then extracted twice with DCM (3 × 50 mL). The organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure. Purification by flash column chromatography (25 g silica gel, 0–1.5% MeOH in DCM) afforded 11 (0.30 g, 86%). 1H NMR (400 MHz, DMSO‐d 6 ): δ 1.30 (s, 3H, CH3 isop), 1.55 (s, 3H, CH3 isop), 3.56 (dd, J = 4.7 Hz, J = 5.4 Hz, 2H, H‐5′ and H‐5″), 4.04 (q, J = 4.4 Hz, 1H, H‐4′), 4.50 (dd, J = 5.7 Hz, J = 6.4 Hz, 1H, H‐2′), 4.70 (dd, J = 3.6 Hz, J = 6.6 Hz, 1H, H‐3′), 4.83 (d, J = 5.4 Hz, 1H, H‐1′), 4.96 (t, 1H, OH), 7.58 (t, 1H, H Ph), 7.71 (d, 1H, H Ph), 7.78 (dt, 1H, H Ph), 7.85 (s, 1H, H Ph); 13C NMR (100 MHz, DMSO‐d 6 ): δ 25.87 (CH3 isop), 27.85 (CH3 isop), 61.87 (C‐5′), 82.12 (C‐3′), 84.51 (C‐1′), 85.08 (C‐4′), 86.51 (C‐2′), 111.89 (CN), 114.25 (Cq isop), 119.18 (Cq), 129.97, 130.05, 131.45 and 131.99 (CH Ph), 142.26 (Cq Ph); HRMS (ESI+‐TOF): m/z calcd for [C15H17NO4 + H]+ 276.1230, found 276.1231.
3‐(2,3‐O‐isopropylidene‐5‐O‐propargyl‐β‐D‐ribofuranosyl)benzonitrile (12)
Sodium hydride (60% in oil, 32 mg, 0.8 mmol) was added to an ice‐cooled solution of 11 (55 mg, 0.2 mmol) in DMF (2 mL) containing molecular sieves. After stirring at 0 °C for 2 h, a solution of propargyl bromide (80% in toluene, 45 μL, 0.4 mmol) was added dropwise. After stirring at 0 °C for 1 h, acetic acid (60 μL) was added and the reaction solution was stirred for an additional 1 h. Water (3 mL) was added, and the mixture was extracted with DCM (3 × 30 mL). The organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure. Purification by flash column chromatography (10 g silica gel, 0–1% MeOH in DCM) afforded 12 (54 mg, 86%). 1H NMR (400 MHz, DMSO‐d 6 ): δ 1.30 (s, 3H, CH3 isop), 1.55 (s, 3H, CH3 isop), 3.45 (t, J = 2.4 Hz, 1H, C ≡ CH), 3.62–3.70 (m, 2H, H‐5′ and H‐5″), 4.17 (q, J = 4.5 Hz, 1H, H‐4′), 4.21 (d, J = 2.4 Hz, 2H, CH2‐C≡), 4.56 (dd, J = 5.2 Hz, J = 6.5 Hz, 1H, H‐2′), 4.68 (dd, J = 3.8 Hz, J = 6.6 Hz, 1H, H‐3′), 4.87 (d, J = 5.0 Hz, 1H, H‐1′), 7.60 (t, 1H, H Ph), 7.51 (d, 1H, H Ph), 7.70 (d, 1H, H Ph), 7.78 (s, 1H, H Ph), 7.79 (d, 1H, H Ph); 13C NMR (100 MHz, DMSO‐d 6 ): δ 25.85 (CH3 isop), 27.78 (CH3 isop), 58.41 ( C H2‐C≡), 69.90 (C‐5′), 77.91 ( C ≡ CH), 80.42 (C ≡ C H), 82.23 (C‐3′), 83.07 (C‐4′), 84.68 (C‐1′), 86.37 (C‐2′), 111.93 (CN), 114.49 (Cq isop), 119.10 (Cq Ph), 129.92, 130.15, 131.39 and 132.08 (CH Ph), 140. 28 (Cq Ph), 141.99 (Cq Ph); HRMS (ESI+‐TOF): m/z calcd for [C18H19NO4 + H]+ 314.1387, found 314.1385.
3‐(2,3‐O‐isopropylidene‐5‐O‐propargyl‐β‐D‐ribofuranosyl)benzamide (3)
From 9 (route A): Sodium hydride (60% in oil, 84 mg, 2.1 mmol) was added at −10 °C to a solution of 8 (0.21 g, 0.7 mmol) in DMF (21 mL) containing molecular sieves. After stirring at −10 °C for 2 h, a solution of propargyl bromide (80% in toluene, 78 μL, 0.7 mmol) in DMF (1 mL) was added dropwise. After 2 h at −10 °C, acetic acid (210 μL) was added and the reaction solution was stirred for an additional 1 h. Water was added, and the mixture was extracted with DCM (3 times). The organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure. Purification by flash column chromatography (25 g silica gel, 0 to 3% MeOH in DCM) afforded 3 (94 mg, 41%).
From 12 (route B): To an ice‐cooled solution of 11 (94 mg, 0.3 mmol) in DMSO (5 mL) in the presence of K2CO3 (180 mg, 1.3 mmol) was added H2O2 (35% wt in H2O, 0.166 mL, 1.9 mmol). After stirring for 2 h at room temperature, volatiles were removed by lyophilization. Purification by flash column chromatography (12 g silica gel, 3% MeOH in DCM) afforded 3 (69 mg, 70%).
1H NMR (400 MHz, DMSO‐d 6 ): δ 1.31 (s, 3H, CH3 isop), 1.55 (s, 3H, CH3 isop), 3.45 (t, J = 2.4 Hz, 1H, C ≡ CH), 3.66 (d, J = 5.0 Hz, 2H, H‐5′ and H‐5″), 4.14 (dd, J = 5.0 Hz, J = 9.2 Hz, 1H, H‐4′), 4.22 (d, J = 2.4 Hz, 2H, CH2‐C≡), 4.57 (dd, J = 5.2 Hz, J = 6.7 Hz, 1H, H‐2′), 4.67 (dd, J = 4.0 Hz, J = 6.8 Hz, 1H, H‐3′), 4.82 (d, J = 5.2 Hz, 1H, H‐1′), 7.36 (br, 1H, CONH2), 7.45 (t, 1H, H Ph), 7.51 (d, 1H, H Ph), 7.83 (d, 1H, H Ph), 7.85 (s, 1H, H Ph), 7.99 (br, 1H, CONH2); 13C NMR (100 MHz, DMSO‐d 6 ): δ 25.85 (CH3 isop), 27.80 (CH3 isop), 58.42 ( C H2‐C≡), 69.93 (C‐5′), 77.90 ( C ≡ CH), 80.50 (C ≡ C H), 82.25 (C‐3′), 85.01 (C‐4′), 85.40 (C‐1′), 86.46 (C‐2′), 114.64 (Cq isop), 125.74, 127.28, 128.76 and 129.24 (CH Ph), 134.90 and 140.28 (Cq Ph), 168.24 (CO); HRMS (ESI+‐TOF): m/z calcd for [C18H21NO5 + Na]+ 354.1312, found 354.1299.
5‐O ‐[3‐(2′,3′,5′‐tri‐O ‐acetyl‐adenosin‐8‐yl)prop‐2‐yn‐1‐yl]‐1‐deoxy‐1‐(3‐carbamoylphenyl)‐2,3‐O‐isopropylidene‐β‐D‐ribofuranose (13)
To an argon‐purged flask containing bromide 2 (284 mg, 0.6 mmol) and alkyne 3 (133 mg, 0.4 mmol) in THF (4 mL) were added under argon atmosphere triethylamine (167 μL, 1.2 mmol), CuI (8 mg, 0.04 mmol) and tetrakis(triphenylphosphine)palladium (23 mg, 0.02 mmol). The reaction mixture was heated at 60 °C for 1 h under argon atmosphere and concentrated under reduced pressure, and the residue was purified by flash column chromatography (40 g SiO2, 0–5% MeOH in DCM) affording 13 (145 mg, 50%). 1H NMR (400 MHz, DMSO‐d 6 ): δ 1.30 (s, 3H, CH3 isop), 1.55 (s, 3H, CH3 isop), 1.94 (s, 3H, CH3 OAc), 2.05 (s, 3H, CH3 OAc), 2.10 (s, 3H, CH3 OAc), 3.80 (d, J = 5.3 Hz, 2H, H‐5′b and H‐5″b), 4.13–4.22 (m, 2H, H‐4′b and H‐5′a), 4.32–4.38 (m, 1H, H‐4′a), 4.42 (dd, J = 3.5 Hz, J = 12.0 Hz, 1H, H‐5″a), 4.59 (dd, J = 5.2 Hz, J = 6.6 Hz, 1H, H‐2′b), 4.65 (br, 2H, CH2‐C≡), 4.72 (dd, J = 4.1 Hz, J = 6.8 Hz, 1H, H‐3′b), 4.82 (d, J = 5.2 Hz, 1H, H‐1′b), 5.73–5.78 (m, 1H, H‐3′a), 6.16 (d, J = 4.3 Hz, 1H, H‐2′a), 6.21 (dd, J = 4.3 Hz, J = 6.0 Hz, 1H, H‐1′a), 7.40 (br, 1H, CONH2), 7.44 (t, J = 7.5 Hz, 1H, H Ph), 7.53 (d, 1H, H Ph), 7.66 (br, 2H, NH2), 7.82 (dd, J = 6.3 Hz, J = 6.3 Hz, 1H, H Ph), 7.86 (s, 1H, H Ph), 8.02 (br, 1H, CONH2), 8.22 (s, 1H, H‐2a); 13C NMR (100 MHz, DMSO‐d 6 ): δ 20.65 (CH3 OAc), 20.76 (CH3 OAc), 20.82 (CH3 OAc), 25.84 (CH3 isop), 27.79 (CH3 isop), 58.83 ( C H2‐C≡), 62.87 (C‐5′a), 70.17 (C‐3′a), 70.43 (C‐5′b), 71.88 (C‐2′a), 75.30 (CH2‐ C ≡ C), 79.64 (C‐4′a), 82.19 (C‐3′b), 82.89 (C‐4′b), 85.42 (C‐1′b), 86.45 (C‐2′b), 87.29 (C‐1′a), 93.12 (CH2‐C ≡ C ), 114.75 (Cq isop), 119.36 (C‐5), 125.80, 127.29, 128.78 and 129.22 (CH Ph), 132.73 (C‐8), 134.92 and 140.19 (Cq Ph), 149.14 (C‐4), 154.56 (C‐2), 156.53 (C‐6), 168.17 (CONH2), 169.86 (2C, CO Ac), 170.42 (CO Ac); HRMS (ESI+‐TOF): m/z calcd for [C34H38N6O12 + H]+ 723.2620, found 723.2618.
5‐O ‐[3‐(adenosin‐8‐yl)prop‐2‐yn‐1‐yl]‐1‐deoxy‐1‐(3‐carbamoylphenyl)‐β‐D‐ribofuranose (1)
Compound 13 (133 mg, 0.18 mmol) was treated with 28% aq. ammonia (1 mL) in MeOH (2 mL) at room temperature for 5 h. After removal of the volatiles, the residue was then treated with an ice‐cooled solution of 70% aqueous TFA (5 mL) for 4 h, then the volatiles were removed by lyophilization, and the residue was purified by reverse phase HPLC (linear gradient of 15–25% acetonitrile in 10 mm TEAA over 15 min) affording 1 as a white foam (40 mg, 40%). 1H NMR (400 MHz, DMSO‐d 6 ): δ 3.49–3.60 (m, 1H, H‐5′a), 3.66–3.84 (m, 4H, H‐5″a, H‐2′b, H‐5′b and H‐5″b), 3.87–3.94 (m, 1H, H‐3′b), 3.97–4.04 (m, J = 6.3 Hz, 2H, H‐4′a and H‐4′b), 4.18–4.24 (m, 1H, H‐3′a), 4.64 (s, 2H, CH2‐C≡), 4.65 (d, 1H, H‐1′b), 4.95–5.15 (m, 3H, H‐2′a, OH‐2′b and OH‐3′b), 5.20 (br, 1H, OH‐3′a), 5.45 (br, 1H, OH‐2′a), 5.52 (dd, 1H, OH‐5′), 5.97 (d, J = 6.8 Hz, 1H, H‐1′a), 7.32 (br, 1H, CONH2), 7.42 (t, J = 7.8 Hz, 1H, H Ph), 7.55 (d, 1H, H Ph), 7.64 (br, 2H, NH2), 7.77 (d, 1H, H Ph), 7.86 (s, 1H, H Ph), 7.95 (br, 1H, CONH2), 8.17 (s, 1H, H‐2a); 13C NMR (100 MHz, DMSO‐d 6 ): δ 58.79 ( C H2‐C≡), 62.64 (C‐5′a), 71.29 (C‐5′b), 71.45 (C‐3′a), 72.07 (C‐2′a and C‐3′b), 75.58 (CH2‐ C ≡ C), 77.61 (C‐2′b), 83.27 (C‐4′b), 83.91 (C‐1′b), 87.17 (C‐4′a), 89.90 (C‐1′a), 92.68 (CH2‐C ≡ C ), 119.83 (C‐5), 126.03, 126.88, 128.53 and 129.37 (CH Ph), 133.48 (C‐8), 134.67 and 141.58 (Cq Ph), 148.89 (C‐4), 153.89 (C‐2), 156.64 (C‐6), 168.45 (CONH2); HRMS (ESI+‐TOF): m/z calcd for [C25H28N6O9 + H]+ 557.1991, found 557.1987.
Compound 1 was dissolved at 20 mm in dimethylsulfoxide (DMSO) 100%. For activity measurements, the stock solution was first diluted at 200 μm in DMSO and then further diluted in the experimental buffer at the indicated concentrations (final concentration of DMSO was kept constant at 5%). For bath infection, the stock solution was first diluted at 2 mm by adding phosphate buffered saline (PBS) drop by drop and then further diluted at the indicated concentration.
Cytotoxicity
The synthesized compounds were assayed for cytotoxicity on MRC‐5 (human foetal lung fibroblasts) cells. The cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 25 mm glucose, 10% (v/v) serum, 1% penicillin/streptomycin and kept under 5% CO2 at 37 °C. The cell viability was assessed using the CellTiter Glo kit from Promega (G7572) after 72‐h incubation time using four concentrations of compounds (1, 10, 25 and 50 μm) in triplicate.
Bacterial growth curves
Overnight culture of PAO1 strain was diluted 1 : 10 in LB and grown until OD600 = 0.7 and further diluted to OD600 = 0.1. For bacterial growth curve, compound 1 solution was first diluted at 2 mm by adding LB:phosphate buffered saline (PBS) (75 : 25) and then diluted at the indicated concentration in 96‐well plate by adding drop by drop 100 μL of bacteria suspension by well. The same protocol was done for DMSO with a first dilution to 10% in LB:PBS. Bacterial growth was monitored in 96‐well plates by measuring the OD600 with a spectrophotometer (TECAN Spark®, Männedorf, Switzerland) during 6 h at 37 °C.
Experiments on zebrafish embryos
Bath infection of zebrafish embryos with P. aeruginosa
Experiments were performed using the GAB zebrafish line, which is maintained at 28 °C under standard conditions in fish water [43]. Preparation of bacteria and immersion infection of injured zebrafish embryos was realized as described [17] with the following modifications. All experiments were conducted on groups of 20 injured embryos into 96‐well plates (two embryos per well) containing 200 μL of fish water supplemented with bacterial suspension and compound 1 or DMSO as control. The compound 1 stock solution was first diluted at 2 mm by adding PBS drop by drop. Bacterial suspension was then added drop by drop to compound 1 to reach a final concentration of 20 μm before immersion of injured embryos. Toxicity was addressed with non‐infected injured embryos treated with compound 1. For survival kinetics, the number of dead embryos was determined visually based on the absence of heartbeat.
Statistical analysis
Statistical analyses were performed using graphpad prism 5 (GraphPad Software, San Diego, CA, USA). Comparisons between survival curves were performed using the log‐rank test.
Conflict of interest
A patent on the molecule series described here and their applications have been deposited by INSERM Transfert on the 15 July 2022 under the reference EP22185280.9.
Author contributions
RR performed experiments, analysed data, prepared figures and wrote the paper. PN and DAC performed experiments, analysed data and prepared figs. VH and MG performed experiments and analysed data. GL and SP planned experiments, analysed data, prepared figures, wrote the paper and raised funds. ABBP planned experiments, analysed data, wrote the paper and raised funds. CL planned experiments, performed experiments, analysed data, prepared figures, wrote the paper and raised funds.
Ethics statement
All animal experiments described in the present study were conducted at the University of Montpellier according to European Union guidelines for handling of laboratory animals and were approved by the Direction Sanitaire et Vétérinaire de l'Hérault and Comité d'Ethique pour l'Expérimentation Animale under reference CEEA‐LR‐ B4‐172‐37 and APAFIS#5737‐2 016 061 511 212 601 v3.
Peer review
The peer review history for this article is available at https://publons.com/publon/10.1111/febs.16604.
Supporting information
Appendix S1. NMR spectra of the original compounds synthesized here.
Acknowledgements
This work was funded by the Agence Nationale de la Recherche (ANR‐17‐CE18‐0011) and supported by Institut Pasteur, the Centre National de la Recherche Scientifique (CNRS), Institut National de la Santé et de la Recherche Médicale (INSERM) and University of Montpellier. R.R., P.N. and D.A.C. acknowledge ANR for financial support. The authors would like to thank the Chemogenomic and Biological Screening Platform (Institut Pasteur, France) for carrying out the cytotoxicity experiments and Frédéric Bonhomme (CNRS, UMR3523, Paris) for assisting with HRMS analysis. We thank Dr. Catherine Dunyach‐Remy and Andréa Clary (INSERM U1047) for helpful discussion. We also acknowledge support from the French Infrastructure for Integrated Structural Biology (FRISBI) ANR‐10‐INSB‐0005.
Contributor Information
Sylvie Pochet, Email: sylvie.pochet@pasteur.fr.
Anne‐Béatrice Blanc‐Potard, Email: anne.blanc-potard@umontpellier.fr.
Corinne Lionne, Email: lionne@cbs.cnrs.fr.
References
- 1. Tacconelli E, Carrara E, Savoldi A, Harbarth S, Mendelson M, Monnet DL, et al. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic‐resistant bacteria and tuberculosis. Lancet Infect Dis. 2018;18:318–27. [DOI] [PubMed] [Google Scholar]
- 2. Gelin M, Poncet‐Montange G, Assairi L, Morellato L, Huteau V, Dugué L, et al. Screening and in situ synthesis using crystals of a NAD kinase lead to a potent antistaphylococcal compound. Structure. 2012;20:1107–17. [DOI] [PubMed] [Google Scholar]
- 3. Agledal L, Niere M, Ziegler M. The phosphate makes a difference: cellular functions of NADP. Redox Rep. 2010;15:2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pollak N, Dölle C, Ziegler M. The power to reduce: pyridine nucleotides – small molecules with a multitude of functions. Biochem J. 2007;402:205–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grose JH, Joss L, Velick SF, Roth JR. Evidence that feedback inhibition of NAD kinase controls responses to oxidative stress. Proc Natl Acad Sci USA. 2006;103:7601–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Van Acker H, Coenye T. The role of reactive oxygen species in antibiotic‐mediated killing of bacteria. Trends Microbiol. 2017;25:456–66. [DOI] [PubMed] [Google Scholar]
- 7. Gerdes SY, Scholle MD, D'Souza M, Bernal A, Baev MV, Farrell M, et al. From genetic footprinting to antimicrobial drug targets: examples in cofactor biosynthetic pathways. J Bacteriol. 2002;184:4555–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thanassi JA, Hartman‐Neumann SL, Dougherty TJ, Dougherty BA, Pucci MJ. Identification of 113 conserved essential genes using a high‐throughput gene disruption system in Streptococcus pneumoniae . Nucleic Acids Res. 2002;30:3152–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zalacain M, Biswas S, Ingraham KA, Ambrad J, Bryant A, Chalker AF, et al. A global approach to identify novel broad‐spectrum antibacterial targets among proteins of unknown function. J Mol Microbiol Biotechnol. 2003;6:109–26. [DOI] [PubMed] [Google Scholar]
- 10. Kobayashi K, Ehrlich SD, Albertini A, Amati G, Andersen KK, Arnaud M, et al. Essential Bacillus subtilis genes. Proc Natl Acad Sci USA. 2003;100:4678–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003;48:77–84. [DOI] [PubMed] [Google Scholar]
- 12. Spaans SK, Weusthuis RA, van der Oost J, Kengen SWM. NADPH‐generating systems in bacteria and archaea. Front Microbiol. 2015;6:742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Poncet‐Montange G, Assairi L, Arold S, Pochet S, Labesse G. NAD kinases use substrate‐assisted catalysis for specific recognition of NAD. J Biol Chem. 2007;282:33925–34. [DOI] [PubMed] [Google Scholar]
- 14. Labesse G, Douguet D, Assairi L, Gilles AM. Diacylglyceride kinases, sphingosine kinases and NAD kinases: distant relatives of 6‐phosphofructokinases. Trends Biochem Sci. 2002;27:273–5. [DOI] [PubMed] [Google Scholar]
- 15. Paoletti J, Assairi L, Gelin M, Huteau V, Nahori M‐A, Dussurget O, et al. 8‐thioalkyl‐adenosine derivatives inhibit Listeria monocytogenes NAD kinase through a novel binding mode. Eur J Med Chem. 2016;124:1041–56. [DOI] [PubMed] [Google Scholar]
- 16. Gelin M, Paoletti J, Nahori M‐A, Huteau V, Leseigneur C, Jouvion G, et al. From substrate to fragments to inhibitor active in vivo against Staphylococcus aureus . ACS Infect Dis. 2020;6:422–35. [DOI] [PubMed] [Google Scholar]
- 17. Nogaret P, El Garah F, Blanc‐Potard A‐B. A novel infection protocol in zebrafish embryo to assess Pseudomonas aeruginosa virulence and validate efficacy of a quorum sensing inhibitor in vivo. Pathogens. 2021;10:401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pont S, Blanc‐Potard A‐B. Zebrafish embryo infection model to investigate Pseudomonas aeruginosa interaction with innate immunity and validate new therapeutics. Front Cell Infect Microbiol. 2021;11:745851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li B‐B, Wang X, Tai L, Ma T‐T, Shalmani A, Liu W‐T, et al. NAD kinases: metabolic targets controlling redox co‐enzymes and reducing power partitioning in plant stress and development. Front Plant Sci. 2018;9:379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Garavaglia S, Galizzi A, Rizzi M. Allosteric regulation of Bacillus subtilis NAD kinase by quinolinic acid. J Bacteriol. 2003;185:4844–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Orsak T, Smith TL, Eckert D, Lindsley JE, Borges CR, Rutter J. Revealing the allosterome: systematic identification of metabolite‐protein interactions. Biochemistry. 2012;51:225–32. [DOI] [PubMed] [Google Scholar]
- 22. Kawai S, Mori S, Mukai T, Hashimoto W, Murata K. Molecular characterization of Escherichia coli NAD kinase: E. coli ATP‐NAD kinase. Eur J Biochem. 2001;268:4359–65. [DOI] [PubMed] [Google Scholar]
- 23. Shi F, Huan X, Wang X, Ning J. Overexpression of NAD kinases improves the l‐isoleucine biosynthesis in Corynebacterium glutamicum ssp. lactofermentum . Enzyme Microb Technol. 2012;51:73–80. [DOI] [PubMed] [Google Scholar]
- 24. London J, Knight MY. Concentrations of nicotinamide nucleotide coenzymes in micro‐organisms. Microbiology. 1966;44:241–54. [DOI] [PubMed] [Google Scholar]
- 25. Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–97. [DOI] [PubMed] [Google Scholar]
- 26. Mary C, Soflaee MH, Kesavan R, Gelin M, Brown H, Zacharias G, et al. Crystal structure of human NADK2 reveals a dimeric organization and active site occlusion by lysine acetylation. Mol Cell. 2022;82:3299–3311.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krohn K, Heins H, Wielckens K. Synthesis and cytotoxic activity of C‐glycosidic nicotinamide riboside analogues. J Med Chem. 1992;35:511–7. [DOI] [PubMed] [Google Scholar]
- 28. Pankiewicz KW, Zeidler J, Ciszewski LA, Bell JE, Goldstein BM, Jayaram HN, et al. Synthesis of isosteric analogues of nicotinamide adenine dinucleotide containing C‐nucleotide of nicotinamide or picolinamide. J Med Chem. 1993;36:1855–9. [DOI] [PubMed] [Google Scholar]
- 29. Franchetti P, Cappellacci L, Perlini P, Jayaram HN, Butler A, Schneider BP, et al. Isosteric analogues of nicotinamide adenine dinucleotide derived from furanfurin, thiophenfurin, and selenophenfurin as mammalian inosine monophosphate dehydrogenase (type I and II) inhibitors. J Med Chem. 1998;41:1702–7. [DOI] [PubMed] [Google Scholar]
- 30. Bonnac L, Chen L, Pathak R, Gao G, Ming Q, Bennett E, et al. Probing binding requirements of NAD kinase with modified substrate (NAD) analogues. Bioorg Med Chem Lett. 2007;17:1512–5. [DOI] [PubMed] [Google Scholar]
- 31. Pankiewicz KW, Petrelli R, Singh R, Felczak K. Nicotinamide adenine dinucleotide based therapeutics, update. Curr Med Chem. 2015;22:3991–4028. [DOI] [PubMed] [Google Scholar]
- 32. Rennekamp AJ, Peterson RT. 15 years of zebrafish chemical screening. Curr Opin Chem Biol. 2015;24:58–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zon LI, Peterson RT. In vivo drug discovery in the zebrafish. Nat Rev Drug Discov. 2005;4:35–44. [DOI] [PubMed] [Google Scholar]
- 34. Eimon PM, Rubinstein AL. The use of in vivo zebrafish assays in drug toxicity screening. Expert Opin Drug Metab Toxicol. 2009;5:393–401. [DOI] [PubMed] [Google Scholar]
- 35. Gomes MC, Mostowy S. The case for modeling human infection in zebrafish. Trends Microbiol. 2020;28:10–8. [DOI] [PubMed] [Google Scholar]
- 36. Clément DA, Leseigneur C, Gelin M, Coelho D, Huteau V, Lionne C, et al. New chemical probe targeting bacterial NAD kinase. Molecules. 2020;25:E4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pergolizzi G, Butt JN, Bowater RP, Wagner GK. A novel fluorescent probe for NAD‐consuming enzymes. Chem Commun (Camb). 2011;47:12655–7. [DOI] [PubMed] [Google Scholar]
- 38. Depaix A, Kowalska J. NAD analogs in aid of chemical biology and medicinal chemistry. Molecules. 2019;24:E4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Manalastas‐Cantos K, Konarev PV, Hajizadeh NR, Kikhney AG, Petoukhov MV, Molodenskiy DS, et al. ATSAS 3.0: expanded functionality and new tools for small‐angle scattering data analysis. J Appl Cryst. 2021;54:343–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Franke D, Petoukhov MV, Konarev PV, Panjkovich A, Tuukkanen A, Mertens HDT, et al. ATSAS 2.8: a comprehensive data analysis suite for small‐angle scattering from macromolecular solutions. J Appl Cryst. 2017;50:1212–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vonrhein C, Keller P, Smart O, Womack T, Bricogne G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr D Biol Crystallogr. 2011;67:293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Catherinot V, Labesse G. ViTO: tool for refinement of protein sequence‐structure alignments. Bioinformatics. 2004;20:3694–6. [DOI] [PubMed] [Google Scholar]
- 43. Belon C, Soscia C, Bernut A, Laubier A, Bleves S, Blanc‐Potard A‐B. A macrophage subversion factor is shared by intracellular and extracellular pathogens. PLoS Pathog. 2015;11:e1004969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42:W320–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. NMR spectra of the original compounds synthesized here.