

Abstract
Neutrophil granulocytes, or neutrophils, are the most abundant circulating leukocytes in humans and indispensable for antimicrobial immunity, as exemplified in patients with inborn and acquired defects of neutrophils. Neutrophils were long regarded as the foot soldiers of the immune system, solely destined to execute a set of effector functions against invading pathogens before undergoing apoptosis, the latter of which was ascribed to their short life span. This simplistic understanding of neutrophils has now been revised on the basis of insights gained from the use of mouse models and single‐cell high‐throughput techniques, revealing tissue‐ and context‐specific roles of neutrophils in guiding immune responses. These studies also demonstrated that neutrophil responses were controlled by sophisticated feedback mechanisms, including directed chemotaxis of neutrophils to tissue‐draining lymph nodes resulting in modulation of antimicrobial immunity and inflammation. Moreover, findings in mice and humans showed that neutrophil responses adapted to different deterministic cytokine signals, which controlled their migration and effector function as well as, notably, their biologic clock by affecting the kinetics of their aging. These mechanistic insights have important implications for health and disease in humans, particularly, in allergic diseases, such as atopic dermatitis and allergic asthma bronchiale, as well as in autoinflammatory and autoimmune diseases. Hence, our improved understanding of neutrophils sheds light on novel therapeutic avenues, focusing on molecularly defined biologic agents.
Keywords: autoimmunity, autoinflammation, immunodeficiency, infection, inflammation
Abbreviations
- AD
atopic dermatitis
- ANCA
anti‐neutrophil cytoplasmic antibody
- CCL
C‐C motif chemokine ligand
- CCR
C‐C motif chemokine receptor
- cDC
conventional dendritic cell
- CGD
chronic granulomatous disease
- COVID‐19
coronavirus disease 2019
- CR3
complement receptor 3
- CRAMP
cathelicidin‐related antimicrobial peptide
- CXCL
C‐X‐C motif chemokine ligand
- CXCR
C‐X‐C motif chemokine receptor
- DC
dendritic cell
- dLN
draining lymph node
- G‐CSF
granulocyte colony‐stimulating factor (also known as CSF‐3)
- GM‐CSF
granulocyte‐macrophage colony‐stimulating factor (also termed CSF‐2)
- GRK2
G‐protein‐coupled receptor kinase 2
- HEV
high endothelial venule
- ICAM
intercellular adhesion molecule
- IFN‐I
type 1 interferon
- IgE
immunoglobulin E
- IgG
immunoglobulin G
- IL
interleukin
- IL‐4R
interleukin‐4 receptor
- ILC
innate lymphoid cell
- LAD
leukocyte adhesion deficiency
- LFA‐1
lymphocyte function‐associated antigen 1
- L. major
Leishmania major
- L. monocytogenes
Listeria monocytogenes
- MPO
myeloperoxidase
- NADPH
nicotinamide adenine dinucleotide phosphate
- NE
neutrophil elastase
- NET
neutrophil extracellular trap
- PAMP
pathogen‐associated molecular pattern
- pDC
plasmacytoid DC
- PRR
pattern recognition receptor
- ROS
reactive oxygen species
- SCN
severe congenital neutropenia
- SLE
systemic lupus erythematosus
- SSc
systemic sclerosis
- TLR
Toll‐like receptor
- Th
T helper
- TNF
tumor necrosis factor (also known as TNF‐α)
1. INTRODUCTION
Neutrophils are an indispensable component of immune defense and the most abundant immune cells in human blood. 1 They crucially contribute to the first protective response against extracellular and intracellular infectious agents, including bacteria, fungi, and viruses. Immunodeficiencies resulting from quantitative or qualitative neutrophil defects, such as chronic or cyclic neutropenia and chronic granulomatous disease (CGD), provide indisputable evidence of the essential role of neutrophils in immunity (Table 1). 2 Conversely, effector functions of neutrophils are tightly controlled by several mechanisms, and dysregulation of neutrophils contributes to autoinflammatory diseases, severe allergies, and autoimmunity. 3 , 4 , 5 , 6
TABLE 1.
Neutrophils in selected primary immunodeficiencies
| Condition | Cause | Inheritance | Numbers | Functional defects | Outcome | Reference |
|---|---|---|---|---|---|---|
| Congenital neutropenias | ||||||
| Severe congenital neutropenia (SCN) | SCN1: ELANE (ELA2) deficiency | Autosomal‐dominant | Cyclic or chronic neutropenia | Early arrest of neutrophil maturation and/or increased apoptosis, possible impairment of migration | Increased susceptibility to bacterial and fungal infections, monocytosis, and possible other congenital defects | 43, 44, 45 |
| SCN2: GFI‐I deficiency | ||||||
| SCN3 (Kostmann disease): HAX1 deficiency | Autosomal‐recessive | Neutropenia | ||||
| SCN4: G6PC3 deficiency | Neutropenia and thrombocytopenia | |||||
| SCN5: VPS45 deficiency | Neutropenia | |||||
| Wiskott‐Aldrich syndrome | WAS deficiency | X‐linked | Neutropenia | Impaired maturation and increased apoptosis of myeloid cells | Increased susceptibility to bacterial and fungal infections, thrombocytopenia, eczema | 23, 43, 44 |
| G‐CSF receptor deficiency | CSF3R deficiency | Autosomal‐recessive | Neutropenia | Impaired emergency granulopoiesis | Increased susceptibility to bacterial and fungal infections, increased rate of malignancy | 45, 46 |
| Defects of chemotaxis and recruitment | ||||||
| Leukocyte adhesion deficiency (LAD) | LAD1: ITGB2 (CD18) deficiency | Autosomal‐recessive | Neutrophilia associated with poor extravasation | Impaired cell adhesion, migration, and effector function | Increased susceptibility to bacterial and fungal infections, neutrophilia | 23, 47 |
| LAD2: SLC35C1 (sialyl‐LewisX) deficiency | ||||||
| LAD3: FERMT3 (kindlin‐3) deficiency | ||||||
| WHIM syndrome | CXCR4 gain‐of‐function mutation | Autosomal‐dominant | Neutropenia | Impaired chemotaxis, impaired release to circulation | Warts, hypogammaglo‐bulinemia, bacterial and fungal infections, myelokathexis | 48 |
| CXCR2 deficiency | CXCR2 deficiency | Autosomal‐recessive | ||||
| Defects of respiratory burst | ||||||
| Chronic granulomatous disease (CGD) | CYBA deficiency | Autosomal‐recessive | Unchanged | Defective NADPH oxidase complex; cannot produce reactive oxygen species and execute phagocytosis | Increased susceptibility to bacterial and fungal infections | 2, 37, 49 |
| CYBC1 deficiency | ||||||
| NCF1 deficiency | ||||||
| NCF2 deficiency | ||||||
| NCF4 deficiency | ||||||
| CYBB deficiency | X‐linked | |||||
| G6PD deficiency | G6PD deficiency | X‐linked | Possible neutropenia | Impaired production of reactive oxygen species | Increased susceptibility to bacterial and fungal infections | 45 |
The billions of neutrophils circulating in the blood are far from homogenous. The earliest studies on neutrophil subsets date back to the 1980s and the identification of low‐density granulocytes (LDGs) in the blood of patients with systemic lupus erythematosus (SLE) and rheumatoid arthritis. 7 This particular neutrophil population exhibits distinct functional, phenotypical, and morphological features, and is associated with pathogenic effects of neutrophils in various pathologies. As the concept of neutrophil heterogeneity gained traction in the past decades (Box 1), new classifications and terminology have emerged. While there is still a lot to be learnt in this respect, these seminal studies highlighted the presence of different neutrophil subsets (reviewed by 8 , 9 , 10 ). In this article, we will outline the current state of knowledge of homeostasis, activation, and regulation of neutrophils, followed by a discussion of the role of neutrophils in allergy, autoinflammation, and autoimmunity before summarizing non‐immune functions of neutrophils.
BOX 1. Major milestone discoveries.
Neutrophils demonstrate plasticity and tissue‐specific adaptations, indicating their effector immune and non‐immune functions can be spatially distinct.
Neutrophils in various contexts contribute to collateral damage; thus, mechanisms of regulation, such as clearance in the draining lymph nodes, limit inflammation and protect tissue integrity.
Functional and numerical deficiency of neutrophils in type 2 immune diseases is driven by cell‐intrinsic IL‐4R signaling. Therapeutic approaches targeting IL‐4R can restore neutrophil‐mediated immune responses and prevent susceptibility to infections.
NET formation, ROS release, and degranulation can directly or indirectly drive autoinflammation and autoimmunity.
Neutrophils contribute to tissue‐specific non‐immune functions, such as angiogenesis in lungs and tissue repair in liver.
2. NEUTROPHILS AT HOMEOSTASIS
Neutrophils are produced in the bone marrow and, following their maturation, released to the blood stream as post‐mitotic cells. 11 Development of neutrophils in the human and mouse bone marrow has recently been elucidated using various single‐cell techniques 12 , 13 and reviewed elsewhere. 10 The human bone marrow serves as the main reservoir of neutrophils with a production rate of approximately 100 billion neutrophils each day. 10 At steady state, the bone marrow constitutes a massive reservoir for on‐demand neutrophil release and contains several fold more neutrophils than the blood circulation. 14 A tight regulation of this supply demand chain is necessary to avoid complications due to neutrophil deficiency (neutropenia) or overabundance (neutrophilia).
Release of neutrophils to the circulation is governed by a balance of C‐X‐C motif chemokine receptor (CXCR) 2 and CXCR4 signaling. 15 Osteoblasts and other bone marrow stromal cells express high levels of C‐X‐C‐motif chemokine ligand (CXCL) 12, the ligand of CXCR4, which favors neutrophil retention in the bone marrow. 14 As neutrophils mature, they lose CXCR4 and gradually increase CXCR2 expression, ligands of which are intermediary chemoattractants found in circulation and tissues. 15 Thus, the phenotypic transition from CXCR4high to CXCR2high cells allows neutrophils to leave the bone marrow, circulate via the blood stream and patrol tissues.
Disruption of tissue homeostasis by infection, inflammation, or damage induces synthesis of inflammatory signals activating leukocytes and endothelial cells. Distressed tissues release neutrophil chemoattractants, such as CXCL1, CXCL2, and leukotriene B4, promoting recruitment of neutrophils. 16 , 17 Concomitantly, systemic increase of granulocyte colony‐stimulating factor (G‐CSF, also known as colony‐stimulating factor 3 [CSF3]) concentrations accelerates granulopoiesis and egress of neutrophils from the bone marrow. 18 Free‐floating neutrophils first tether and roll on inflamed endothelium, which express a higher density of adhesion molecules due to inflammation, then firmly adhere, and finally extravasate into tissues (Figure 1). 19 Depending on the inflammatory context and the tissue involved, different selectins, such as E‐, P‐, and L‐selectin, and their ligands, sialyl‐LewisX, P‐selectin glycoprotein ligand 1, and glycosylation‐dependent cell adhesion molecule 1, respectively, play a role in neutrophil rolling. 20 Heightened chemokine sensing increases surface expression and ligand affinity of heterodimeric β2 integrins on neutrophils, such as lymphocyte function‐associated antigen 1 (LFA‐1, consisting of CD11a and CD18), and complement receptor 3 (CR3, also known as macrophage‐1 antigen or MAC‐1, and made of CD11b and CD18). Intracellular talin‐1 and kindlin‐3 molecules promote binding of β2 integrins to their interaction partners on endothelium. 21 , 22 Individuals with leukocyte adhesion deficiency (LAD) carry congenital defects in integrin subunit CD18 (causing LAD type 1 [LAD1]), sialyl‐LewisX (resulting in LAD2), or kindlin‐3 (causing LAD3) and are at greater risk of bacterial and fungal infections (Table 1). 19 , 23 Neutrophils arriving first at the affected site reinforce further neutrophil recruitment by releasing additional chemoattractants that promote neutrophil swarming. 24 This swarming behavior is a self‐amplifying process, necessary for elimination of pathogens. Once inside the tissue, neutrophils follow so‐called end‐target chemoattractants, such as complement proteins and microbial components. 17 , 25 Subsequently, neutrophils carry out their effector functions, often informed by the inflammatory microenvironment.
FIGURE 1.
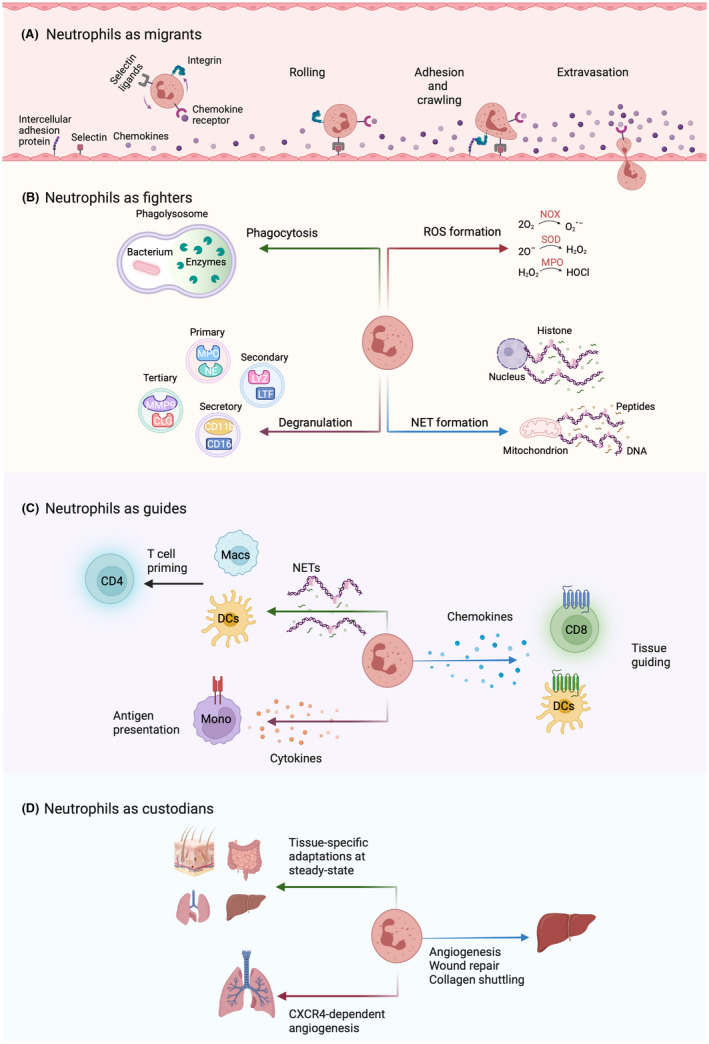
Summary of different neutrophil functions. (A) Effective migration is a prerequisite to any subsequent function executed by neutrophils. Neutrophils in circulation rely on various interactions with the endothelium. These include (1) loose binding of carbohydrate ligands on neutrophils (such as sialyl‐LewisX and P‐selectin glycoprotein ligand 1) to selectins (E‐selectin and P‐selectin, respectively) on activated endothelium, which induce rolling of neutrophils along endothelial cells; (2) sensing of chemokines (including CXCL8) by chemokine receptors on neutrophils (e.g., CXCR2), which in turn activates the integrins lymphocyte function‐associated antigen 1 (LFA‐1, consisting of CD11a and CD18) and complement receptor 3 (CR3; made of CD11b and CD18) on neutrophils; (3) the activated integrins LFA‐1 and CR3 on neutrophils strongly adhere to intercellular adhesion molecule 1 (ICAM‐1) and ICAM‐2 on activated endothelial cells, thus arresting neutrophils and allowing their transmigration through the endothelium (also called extravasation) into the tissue. Neutrophil migration, facilitated by adhesion molecules and chemotactic sensors for recruitment, is absolutely crucial, as illustrated by immunodeficiencies due to leukocyte adhesion deficiency (please see Table 1) that can lead to life‐threatening conditions. (B) Neutrophils are excellent at direct combat and feature a wide repertoire of effector functions, such as phagocytosis, degranulation of primary, secondary, tertiary, and secretory vesicles, production of reactive oxygen species (ROS; such as superoxide [O2 −], hydrogen peroxide [H2O2], and hypochloride [HOCl]), and release of neutrophil extracellular traps (NETs) consisting of either nuclear or mitochondrial DNA. (C) Their rapid mobilization to affected tissues allows neutrophils to guide the recruitment and activity of other cells involved in the inflammatory process, such as dendritic cells (DCs), monocytes (Mono), macrophages (Macs), CD4+ T helper cells, and CD8+ cytotoxic T cells by releasing or leaving “trails” of chemokines (such as CXCL12 and CCL3) and cytokines (e.g., epidermal growth factor [EGF]). (D) Whereas the importance of neutrophils for immune defense is irrefutable, recent evidence suggests they are also involved in tissue‐specific non‐immune processes. Neutrophils extravasate to various tissues at steady state and engage in non‐canonical functions, such as angiogenesis, tissue repair, and wound healing.
Upon completion of their task, neutrophils re‐upregulate CXCR4 expression, allowing them to enter tissues specialized in the clearance of neutrophils. At steady state, neutrophils are primarily eliminated in the bone marrow, spleen, and liver by macrophages and dendritic cells (DCs). 26 Upon infection or inflammation, affected peripheral tissues 27 and their draining lymph nodes 28 can also serve as clearance sites for neutrophils.
3. ACTIVATION OF NEUTROPHILS
Before neutrophils can eliminate a pathogen, they first need to sense it, either directly or indirectly, through a variety of receptors on their cell surface. Such pattern recognition receptors (PRRs) allow neutrophils to recognize pathogen‐associated molecular patterns (PAMPs), which are molecules of pathogenic origin, as well as danger‐associated molecular patterns, which comprise host‐derived factors. An important group of PRRs are Toll‐like receptors (TLRs), homologues of the Drosophila melanogaster protein Toll, 29 which bind to conserved pathogen‐derived elements. The main downstream effect of TLR engagement is activation of nuclear factor κB signaling, which promotes cell survival and release of proinflammatory cytokines. 30 Both human and mouse neutrophils are known to express various TLRs, complement receptors, and fragment‐crystallizable receptors, and thus respond to various pathogens, such as bacteria, viruses, fungi, and parasites. 1 Collectively, interaction of these receptors with their respective ligands activates neutrophils and initiates effector mechanisms.
Already in Elie Metchnikoff's description of the two morphologically distinct leukocytes, termed macrophages and microphages 31 and referring to macrophages and (mainly) neutrophil granulocytes, the ability to phagocytose infectious agents was a defining feature of neutrophils. In addition to phagocytosis, neutrophil effector functions also comprise degranulation, production of reactive oxygen species (ROS), and neutrophil extracellular trap (NET) formation (Figure 1). 32 This repertoire of effector functions makes neutrophils one of the most potent ammunitions of our immune system.
Once a pathogen is recognized and enveloped inside the phagosome, it gets exposed to the catalytic and antimicrobial content of neutrophil granules and to ROS, both of which facilitate pathogen elimination. 33 Neutrophils can also release ROS into extracellular space. 34 Fusion of granules with the cell membrane or phagosome activates the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, an enzyme complex essential for ROS production. 35 The NADPH oxidase complex is composed of two membrane‐bound subunits, called gp91phox and p22phox, three cytosolic subunits, termed p47phox, p67phox, and p40phox, and a G‐protein called Rac2. 36 Upon phosphorylation of p47phox, the cytosolic subunits translocate to the plasma membrane and activate the membrane‐bound subunits which can subsequently reduce molecular oxygen to generate different ROS. Various pathogenic variants of NADPH oxidase genes have been identified in patients with autosomal‐recessive CGD. Moreover, a defect in CYBB, which encodes the gp91phox subunit, leads to X‐linked CGD (Table 1). 2 , 37 Overall, defects in NADPH oxidase impair the machinery of phagocytosis and oxidative burst, thus, severely increasing the risk of infections.
Phagocytosis is an efficient mechanism against extracellular pathogens of relatively small size. However, against larger microbes, such as Candida albicans, neutrophils resort to an alternative means of destruction, called NET formation. 38 In order to trap and destroy the pathogen extracellularly, neutrophils eject their DNA, which can be of nuclear or mitochondrial origin and is decorated with histones, antimicrobial proteins, and other components of cytoplasmic granules. 39 , 40 Both phagocytosis and NET formation rely on mobilization of enzymes found in azurophilic granules, in particular neutrophil elastase (NE) and myeloperoxidase (MPO). During phagocytosis, azurophilic granules fuse with phagosomes, where NE and MPO participate in degradation of engulfed bacteria. 41 , 42 When pathogen size is unsuited to phagocytosis, MPO initiates translocation of NE from the azurophilic granules into the nucleus, which in turn promotes decondensation of chromatin and NET production. 38 Thus, compatibility of pathogen size with phagocytosis, at least to a certain extent, regulates release of NETs.
3.1. Neutrophils networking
As the first non‐resident cells to arrive at an infected or inflamed tissue, neutrophils have a selective advantage to orchestrate the migration or activation of other immune cells. The neutrophil proteome is rich in chemokines, cytokines, and antimicrobial peptides, which in turn enable neutrophil cell‐to‐cell communication (Figure 1). 50 Recent developments in live imaging techniques and transgenic reporter mouse models provided visual evidence for neutrophil crosstalk with various innate and adaptive immune cells in several tissues, including the skin, 27 airways, 51 and central nervous system. 52 One study combining intravital imaging with CXCL12 reporter mice demonstrated that neutrophils assisted CD8+ T cells to find their way into the tissue upon influenza A virus infection. 51 Migrating neutrophils were caught on camera leaving “trails” of CXCL12 behind. Much like the breadcrumbs in the fairytale of Hänsel and Gretel, CD8+ T cells followed the chemokine trail to enter the trachea. 51 In an independent study, apoptotic neutrophils in the trachea were shown to produce epidermal growth factor, which transformed inflammatory monocytes into efficacious antigen‐presenting cells. Thus, indirectly, neutrophils contributed to activation of influenza A virus‐specific CD8+ T cells. 27 In a model of Leishmania major (L. major) infection, neutrophils guided DCs to the infection site through a different neutrophil‐derived chemokine, C‐C motif chemokine ligand 3 (CCL3); subsequently, entry of DCs to the tissue promoted early T helper 1 (Th1) response and augmented parasite elimination. 53
While targeting pathogens, neutrophils can shape an inflammatory environment and convey activation signals to other immune cells. Of note, inflammatory outcomes of neutrophil effector functions are frequently observed and extensively documented in studies of NET formation. For instance, NET‐associated antimicrobial peptide LL‐37, or its mouse analogue cathelicidin‐related antimicrobial peptide (CRAMP), forms immune complexes with self‐nucleic acids, leading to type‐1 interferon (IFN‐I) production in target cells through activation of TLR7, TLR8, or TLR9 in endosomes. 54 , 55 Alternatively, NET‐derived CRAMP was shown to induce inflammasome assembly in alveolar macrophages and subsequent interleukin (IL)‐1β release in a model of influenza A virus infection. 56 Similar to NETs, released ROS can also influence activity of other immune cells. In a model of liver injury, deficiency of ROS production by neutrophils significantly reduced the frequency of macrophages involved in tissue repair. 57 Collectively, neutrophils activated during early infection or inflammation intensely communicate with other immune cells to guide subsequent immune responses.
3.2. Neutrophils in lymph nodes
Neutrophil migration to tissue‐draining lymph nodes (dLNs) has emerged as an eccentric mechanism by which neutrophils further partake in local immune responses. Dissemination of bacteria to the dLNs, as well as increased levels of chemoattractants and cytokines in the dLNs, promote influx of neutrophils. 58 Neutrophils can enter dLNs either via lymphatic vessels or high endothelial venules (HEVs). Thus, similar to their recruitment into tissues, neutrophils can directly enter dLNs from the circulation and execute their classical effector functions. 59 , 60 Neutrophils accessing dLNs via HEVs were also shown to contain pathogens.
Neutrophils entering dLNs via lymphatic vessels have different phenotypical and functional properties than their counterparts entering dLNs from the circulation. For instance, tissue‐experienced neutrophils in the dLNs are more prone to apoptosis and express higher levels of CD11b and lower levels of L‐selectin (CD62L), typical of activated/aged neutrophils. 28 , 61 From a functional perspective, many studies on lymphatic migration tackled the question of how tissue‐experienced neutrophils influenced adaptive immune responses in dLNs. It has been shown that neutrophils shuttle antigens between the infected tissue and its dLN via the lymphatic system and potentiate early antigen presentation to T cells. 62 While neutrophils can present antigens themselves, they can also transfer their antigen load to conventional DCs (cDCs). 63 , 64 , 65 , 66 Furthermore, neutrophils can shape adaptive immune responses independently of antigen, by conveying signals of activation or suppression (see next chapter). However, what guides neutrophils to release pro‐ or anti‐inflammatory mediators in which context is unclear. For instance, in a model of Staphylococcus aureus infection, neutrophils promoted the proliferation of T and B cells 61 yet limited antibody production by B cells. 67 How neutrophils interact with adaptive immune cells in the dLNs will need further investigation.
4. REGULATION OF NEUTROPHILS
Similar to patients with numerical or functional neutrophil deficiencies, neutropenic mice show significant defects to control infections, such as with murine cytomegalovirus 68 and Listeria monocytogenes (L. monocytogenes). 69 However, in other instances, neutrophils may do more harm than good. In L. major infection, neutrophils on the one hand augment DC migration and CD4+ T cell activation, 53 but on the other hand, shield the pathogen from other phagocytes and contribute to persistence of infection. 70 Similarly, different parasites, viruses, and bacteria can override phagocytic programming of neutrophils and evade intracellular destruction. 71 , 72 , 73 , 74 In these cases, neutrophil‐mediated damage is dictated by pathogen evolution, but more commonly, unrestrained neutrophil recruitment and target‐unspecific effector functions inflict substantial harm. The coronavirus disease 2019 (COVID‐19) pandemic, caused by severe acute respiratory syndrome coronavirus 2, revealed detrimental effects of neutrophils. Various studies identified elevated serum levels of neutrophil chemoattractants, including CXCL1, and a higher neutrophil‐to‐lymphocyte ratio in severe COVID‐19 patients. 75 , 76 , 77 Neutrophils in the airways were shown to release NETs and proinflammatory cytokines, which in turn exacerbated inflammatory tissue damage to lungs and likely contributed to an increase of systemic autoantibodies. 78 , 79 , 80
Accordingly, counts and activity of neutrophils must be kept in check to maintain a balance between essential immune responses and tissue health. Recent evidence from an intravital imaging study demonstrated that neutrophil activity and in particular the swarming can be regulated by cell‐intrinsic mechanisms. Neutrophil swarming was limited by desensitization of CXCL2 and leukotriene B4 receptors through G‐protein‐coupled receptor kinase 2 (GRK2). 81 Interestingly, deficiency of GRK2 impaired neutrophil arrest and bacterial uptake; thus, this internal control of swarming was necessary not only to prevent damage but also to achieve proper bacteria elimination.
Alternatively, an intrinsic circadian clock governing neutrophil aging and subsequent clearance may play an important role in balancing immunity versus tissue injury. At steady state, circulating neutrophils gradually upregulate CXCR4 on their surface and lose their capacity to execute important effector functions, such as degranulation and NET formation, a process termed “disarming”. 82 While CXCR4 allows clearance of aged neutrophils from circulation into healthy tissues, the functional loss prevents collateral damage. As opposed to CXCR4high “aged” neutrophils, CD62Lhigh CXCR2high “young” (or “fresh”) neutrophils recently released from the bone marrow are efficiently recruited to inflamed sites and can potently execute antimicrobial effector functions. The transition of neutrophils from CD62Lhigh CXCR2high to CD62Llow CXCR4high can be viewed as a control mechanism restricting inflammatory damage that neutrophils readily and unintentionally inflict. 11
As mentioned previously, aged neutrophils cleared out of circulation need to be safely eliminated by phagocytic cells. Interestingly, macrophages and DCs carrying out phagocytosis of apoptotic neutrophils produced low levels of proinflammatory cytokines, indicating a negative regulation between the two functions. 83 Notably, increased neutrophil elimination resulted in a significant drop of IL‐23 and IL‐17 levels, suppressing type 3 immune responses. 83 , 84 Because IL‐17 is a positive regulator of G‐CSF and granulocyte‐macrophage colony‐stimulating factor (GM‐CSF, also known as CSF2), elimination of apoptotic neutrophils created a negative feedback loop suppressing granulopoiesis. A recent study uncovered that a similar phenomenon was involved in regulation of neutrophil immune responses in cutaneous inflammation and bacterial infection. 28 Upon stimulation with PAMPs, CD11bhigh CD62Llow neutrophils in the skin upregulated C‐C motif chemokine receptor 7 (CCR7) and exited the infected tissues via the lymphatics in a CCR7‐dependent manner (Figure 2). In the dLNs, these apoptosis‐prone CD11bhigh CD62Llow CCR7+ neutrophils were readily phagocytosed by resident cDCs. 28 In line with previous publications, 83 , 84 neutrophil phagocytosis curbed IL‐23 production by cDCs and demand‐based granulopoiesis. Notably, impairment of CCR7‐mediated lymphatic migration resulted in neutrophil accumulation in the tissue, which improved anti‐bacterial immunity but exacerbated local inflammation. 28 These findings on CCR7‐ and dLN‐mediated modulation of neutrophil counts and clearance could be exploited to treat inflammatory disorders with aberrant neutrophil responses (Box 1).
FIGURE 2.
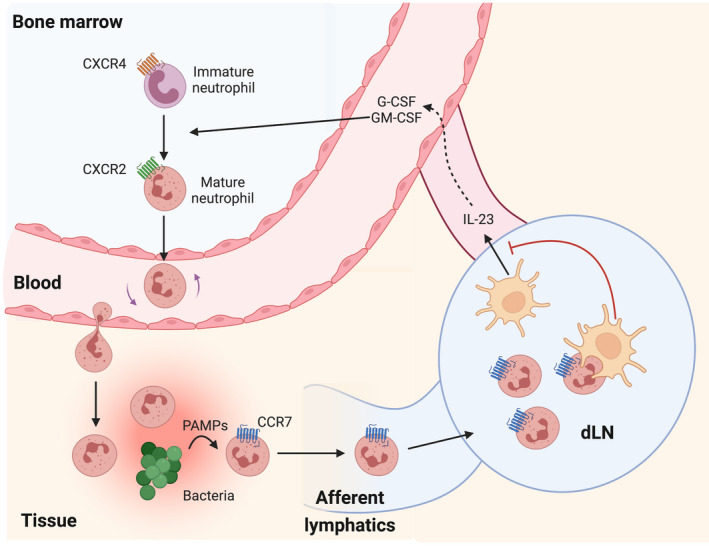
Neutrophil clearance regulates tissue inflammation. Upon infection, increased levels of granulocyte colony‐stimulating factor (G‐CSF) and granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) induce granulopoiesis and promote neutrophil release from the bone marrow. C‐X‐C motif chemokine receptor 2 (CXCR2)high neutrophils leave the bone marrow and migrate toward affected tissues, following gradients of intermediate and end‐target chemoattractants. Recognition of pathogen‐associated molecular patterns (PAMPs) in the tissue upregulates CC motif chemokine receptor 7 (CCR7) on neutrophils, allowing them to migrate via afferent lymphatics into draining lymph nodes (dLNs) where apoptosis‐prone neutrophils coming from the tissue are phagocytosed by dLN‐resident conventional DCs (cDCs). Neutrophil phagocytosis impedes interleukin (IL)‐23 production by cDCs, in turn leading to reduced synthesis of G‐CSF. Thus, CCR7‐dependent tissue neutrophil clearance provides a negative feedback loop for neutrophil immune responses and limits tissue damage.
5. NEUTROPHILS IN ALLERGY
Neutrophils prominently partake in type 1 and type 3 immune responses; however, their contribution to type 2 immunity, which is evolutionarily directed against parasites and toxins, is controversial. 85 , 86 An overshooting type 2 immune response can lead to atopic and allergic diseases, both of which have been rapidly increasing in prevalence in the past decades. 87 As first non‐resident responders to local inflammation and infection, neutrophils also participate in early phases of type 2 immunity; however, they are often absent in established or persistent settings, such as in the skin of patients afflicted with atopic dermatitis (AD). 88 , 89 Despite normal counts in circulation, neutrophils isolated from the blood of AD or allergic patients show functional deficits ex vivo, such as impaired chemotaxis or NET formation (Table 2). 90 Dysregulated neutrophil effector responses in type 2 immunity provide a likely explanation for increased susceptibility of patients to bacterial infections. 85 , 91 , 92
TABLE 2.
Neutrophils in allergic diseases
| Condition | Numbers | Involvement in pathology | Outcome | Reference |
|---|---|---|---|---|
| Atopic dermatitis | Normal in blood, low in skin | Impaired migration, phagocytosis, and NET formation, increased ROS production | Increased susceptibility to cutaneous infections | 88, 90, 108 |
| Allergic rhinitis | Elevated in sputum in severe patients | Not described in detail, however, in neutrophilic endotypes, increased cytokine production, enhanced inflammation | Not described in detail, however, possible contribution to sustained inflammation and tissue damage | 95 |
| Eosinophilic asthma | Scarcity of neutrophils in lungs and bronchoalveolar lavage | Unknown | Unknown | 90, 109, 110 |
| Neutrophilic asthma | Increased in sputum and airways | Increased propensity for NETs, ROS, and cellular survival | Neutrophilic inflammation | 93, 94, 110 |
| Food allergy | Unknown | Unclear, possible activation by IgG instead of IgE, produce platelet‐activating factor | Can induce IgG‐mediated anaphylaxis | 111 |
Interestingly, type 2 immune disorders can have neutrophilic endotypes, usually associated with a severe disease course (Table 2). 17 For instance, neutrophilic asthma, in which patients exhibit increased neutrophil frequency in the sputum and neutrophilic airway inflammation, constitutes 20%–30% of global asthma cases. 93 Increased levels of proinflammatory cues in the airways, such as CXCL8 (also known as IL‐8), IL‐6, and IL‐17, draw neutrophils into the tissue, which can be augmented by local infection. 94 Along the same lines, IL‐6 and CXCL8 are also associated with neutrophilic endotypes of chronic rhinosinusitis. 95 Furthermore, studies in mice demonstrated that neutrophils can directly or indirectly amplify type 2 immune responses. For instance, in a model of Nippostrongylus brasiliensis infection, neutrophils expressed increased levels of type 2 cytokines, including IL‐5, IL‐13, and IL‐33. Of note, neutrophil‐derived IL‐13 promoted M2 macrophage polarization, which was necessary for efficient parasite clearance. 96 Whereas neutrophil involvement in type 2 immune responses can lead to a favorable outcome in parasitic infections, it is highly detrimental in allergic diseases. In a model of allergic skin inflammation, LTB4 signaling on neutrophils significantly contributed to infiltration of CD4+ T cells, dermal and epidermal thickening, and local gene expression of il4 and il13. 97 Others discovered that suppressing neutrophil responses led to lower type 2 cytokines, immunoglobulin E (IgE) levels, and airway infiltration. 98 , 99 , 100 Nevertheless, similar approaches antagonizing neutrophil immune responses in severe asthma patients, such as by targeting CXCR2, LTB4, or IL‐17, showed none to minimal clinical benefit. 101 , 102 , 103 , 104 Recently, a study showed that neutrophils restricted allergic inflammation by reducing availability of G‐CSF for other cells, namely monocyte progenitors and type 2 innate lymphoid cells (ILC2s), both of which play prominent roles in the pathogenesis of allergy. 105 Using a mouse model of house dust mite sensitization, the authors demonstrated that G‐CSF drove antigen presentation by monocyte‐derived DCs and stimulated ILC2s to produce the type 2 cytokines IL‐5 and IL‐13. Accordingly, depletion of neutrophils in this model did not improve the pathology, but, on the contrary, exacerbated the disease by systemic elevation of G‐CSF. Nevertheless, even though the contribution of neutrophils to type 2 immune pathologies appears somewhat multifaceted and at times antagonistic (Box 2), their deficiency in atopic and allergic patients is unequivocal, as described below.
BOX 2.
The majority of novel concepts in neutrophil biology has been discovered in mouse models. It is time to fill the knowledge gap on these features in humans in both health and disease.
The previously described tissue‐specific plasticity of neutrophils can be applied to different diseases. Thus, neutrophil heterogeneity can be studied in the spectrum of different autoimmune and allergic diseases.
More effort is needed to describe functional and phenotypical aspects of neutrophils in type 2 immune diseases, which will allow better characterization of their contribution to pathology.
Mechanisms of neutrophil regulation should be explored to restore a healthy balance between immunity and inflammatory damage.
Recent studies uncovered that neutrophil defects in allergic diseases can be driven by cell‐intrinsic type 2 cytokine signaling 86 (Figure 3). A study by Woytschak et al. 106 demonstrated that IL‐4 antagonized the effects of G‐CSF, which is essential to mobilize neutrophils from the bone marrow upon infection or inflammation. More specifically, administration of exogenous IL‐4 to mice infected with Group A Streptococcus or L. monocytogenes prevented upregulation of CXCR2 on neutrophils and release of these cells into circulation. Inhibition of neutrophil recruitment by exogenous or endogenous IL‐4 signals, in turn, significantly increased bacterial burden, rendering the mice more susceptible to infection. 106 The effects of IL‐4 were shown to be mediated in a cell‐intrinsic manner via the type 2 IL‐4 receptor (IL‐4R) on neutrophils. Although both IL‐4 and IL‐13 are known to signal via this receptor, the sensitivity of mouse neutrophils to IL‐13 was substantially lower than to IL‐4 and, whereas IL‐4 significantly hampered ex vivo neutrophil migration, IL‐13 had no such effect on mouse neutrophils. 106 A subsequent study recapitulated the inhibitory effects of IL‐4R signaling on human neutrophils, which, unlike their mouse counterparts, were affected by both IL‐4 and IL‐13. 107 Incubation of human peripheral blood neutrophils with either IL‐4 or IL‐13 inhibited not only their migration but also their ability to form NETs. Thus, signaling via the IL‐4R emerges as a conserved brake for neutrophil activity in both humans and mice, albeit their sensitivity to the ligand cytokines varies (Box 1).
FIGURE 3.
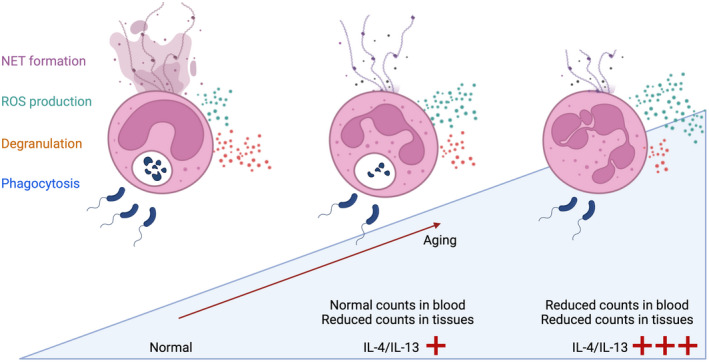
IL‐4R signaling accelerates neutrophil aging. Circulating fresh neutrophils in healthy individuals can perform a variety of effector functions, including neutrophil extracellular trap (NET) release, reactive oxygen species (ROS) production, degranulation, and phagocytosis. An increase in the type 2 immune cytokines interleukin (IL)‐4 and IL‐13 affects phenotypical and functional properties of neutrophils and accelerates their aging. Aged neutrophils lose their granularity as well as ability to perform chemotaxis, phagocytosis, and NET release, whereas they are more prone to ROS production. The functional alterations are evident in type 2 immune diseases with increased IL‐4 and IL‐13 abundance. In mild‐to‐moderate disease manifestations, neutrophil counts are reduced in the affected tissues, but are found at normal levels in the blood. In severe type 2 immune skewing (e.g., by very high systemic concentrations of IL‐4), neutrophil counts can be reduced in blood and tissues.
Very recently, using a mouse model of atopy, IL‐4R signals were demonstrated to accelerate maturation of neutrophils in the bone marrow and aging of circulating neutrophils. 69 Previous descriptions of neutrophil aging included gradual loss of granularity, phagocytic capacity and release of NETs, and increased aptitude for ROS production. 82 Interestingly, neutrophils in “atopic” mice with a predisposition to type 2 immune responses exhibited an aged phenotype, reduced phagocytic activity, and increased rate of apoptosis. 69 In line with an increased rate of apoptosis, neutrophils in atopic mice were readily phagocytosed by macrophages and DCs in bone marrow and spleen. Overall, this accelerated neutrophil aging in atopic mice was associated with increased susceptibility to bacterial infections. The findings in atopic animals were recapitulated by exogenous administration of IL‐4 to type 1 immune‐predisposed mice, exposing IL‐4 as the likely culprit behind neutrophil aging. Enhanced neutrophil aging was caused by cell‐intrinsic IL‐4R signals, which dramatically impaired in vivo anti‐bacterial responses and survival of mice. Although IL‐13 can also bind and signal via the IL‐4Rα subunit of IL‐4R, this study did not delineate the specific contribution of IL‐13 to the phenotype observed, 69 because IL‐13 has been previously shown to minimally affect mouse neutrophils ex vivo. 106 Neutrophil‐specific IL‐4Rα deficiency rescued the susceptibility of atopic animals to bacterial infections, underlining that neutrophil aging controlled by IL‐4 signaling was crucial for antimicrobial immune defense. 69
6. NEUTROPHILS IN AUTOINFLAMMATION AND AUTOIMMUNITY
Systemic autoinflammatory disorders are characterized by dysregulated innate immune responses, including neutrophilia during flares, recurrent fever episodes, and elevated proinflammatory cytokines. 112 Notably, IL‐1 family of cytokines, in particular IL‐1β, are key players of autoinflammatory pathogenesis, as they strongly enhance the activity of innate immune cells. Acting directly on neutrophils, IL‐1β promotes neutrophil effector functions, such as degranulation and NET formation, and acts as a prosurvival factor (Figure 4). 113 , 114 Moreover, locally increased IL‐1β levels induce release of neutrophil chemoattractants from stromal cells, thus reinforcing neutrophil recruitment. Neutrophils, in turn, can exacerbate autoinflammation through further release of IL‐1β, creating a positive feedback loop, and other proinflammatory cytokines, such as tumor necrosis factor (TNF) and IL‐36. 115 Thus, neutrophils are a key component in the pathogenesis of autoinflammatory diseases and are prominently found in affected tissues, such as in inflamed joints in familial Mediterranean fever and in skin lesions in cryopyrin‐associated periodic syndromes and Schnitzler's syndrome (Table 3). 116 Biologic agents targeting the IL‐1 family of cytokines have shown clinical success in autoinflammatory diseases, 117 underlining the central role of these cytokines in the pathology.
FIGURE 4.
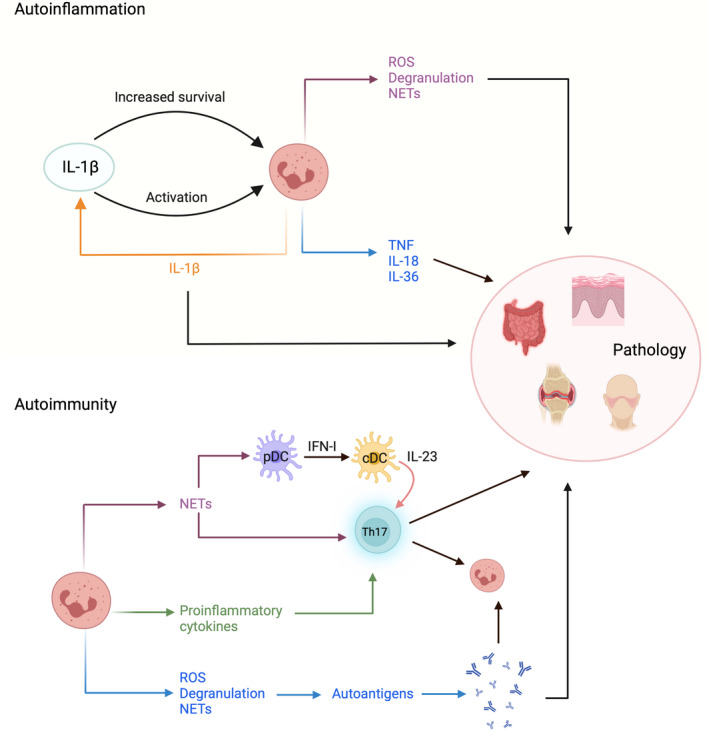
Neutrophils contribute to pathology of autoinflammatory and autoimmune diseases. A key driver of autoinflammation is IL‐1β, a cytokine that potently stimulates neutrophil survival and activity. Neutrophils can also contribute to the production of IL‐1β, thus creating an inflammatory positive feedback loop. Increased IL‐1β signaling prompts activated neutrophils to perpetuate autoinflammation through effector functions and cytokine release. In autoimmunity, neutrophils enhance activation of self‐reactive adaptive immune cells, especially T helper 17 (Th17) cells driving type 3 immune diseases. Thus, immune complexes exposed upon NET formation can stimulate plasmacytoid dendritic cells (pDCs) via endosomal Toll‐like receptors (TLRs) to produce type I interferons (IFN‐I). This, in turn, leads to IL‐23 release by conventional DCs (cDCs) and subsequent priming of Th17 cells. Neutrophils can also facilitate autoantibody production since they release various autoantigens upon activation, through degranulation, ROS production, and NET release. Both Th17 cells and autoantibodies can further activate neutrophils and cause tissue pathology.
TABLE 3.
Neutrophils in autoinflammatory and autoimmune diseases
| Condition | Numbers | Involvement in pathology | Outcome | Reference |
|---|---|---|---|---|
| Cryopyrin‐associated periodic syndromes | Neutrophilia in blood and several disease‐associated tissues during flares | Gain‐of‐function mutations leading to increased NLRP3 inflammasome activity, increased IL‐1β production and degranulation | Exacerbation of inflammation | 112, 141 |
| Familial Mediterranean fever | Neutrophilia in blood and tissues during flares | Mutations leading to increased pyrin inflammasome activity and increased IL‐1β production | Exacerbation of inflammation | 142 |
| Schnitzler syndrome | Neutrophilia in blood (60–90%) and in skin lesions | Overactivation due to increased IL‐1β signaling | Amplifying inflammation | 116, 143 |
| Psoriasis | Increased in skin lesions | Cytokine production, immune complexes activate pDC and IL‐17‐producing CD4+ and CD8+ T cells | Type 3 immune skewing, higher IL‐17, self‐amplifying inflammation | 118, 140, 144, 145 |
| Anti‐neutrophil cytoplasmic antibodies (ANCA)‐associated vasculitis | Normal counts in blood | Impaired NET clearance, increased stimulation by ANCAs | Autoantibodies against myeloperoxidase and proteinase 3 | 135, 146, 147, 148 |
| Systemic lupus erythematosus (SLE) | Higher frequency of mature and activated neutrophils in blood | Impaired phagocytosis, higher cell death and degranulation, autoantigen release, NET‐derived immune complexes activate pDCs | Higher IFN‐I, self‐amplifying inflammation, autoantibody production | 55, 64, 149, 150 |
| Rheumatoid arthritis | Most abundant cells in synovial fluid of inflamed joints | Infiltration of synovial space, delayed apoptosis, production of proinflammatory cytokines, enhanced ability to produce NETs, cartilage destruction by neutrophil elastase | Initiate inflammation in synovium, recruit other immune cells, lead to production of autoantibodies | 131, 151, 152 |
| SSc | Normal counts in blood | Reduced migration and NET formation | Increased susceptibility to infections | 130 |
Similar to autoinflammation, systemic autoimmunity is an umbrella term encompassing various diseases, commonly exhibiting self‐reactive adaptive immune cells and autoantibodies. Due to their diverse nature, the contribution of neutrophils to pathology differs across the spectrum. For instance, in SLE, neutrophils are a major source of immune complexes leading to release of IFN‐I by activated plasmacytoid DCs (pDCs) (Table 3). As mentioned previously, these immune complexes, composed of self‐nucleic acids and LL‐37, are often revealed during NET formation and signal through endosomal TLRs. Immune complexes, 55 , 118 together with other constituents of NET release, such as histones 119 , 120 and the enucleated cellular shell called the neutrophil cytoplast, 121 induce T helper 17 (Th17) cell differentiation. Th17 cells reinforce recruitment of neutrophils, creating an inflammatory positive feedback loop. 122 This mechanism involving IFN‐I, neutrophils, and Th17 cells is thought to drive the pathology of SLE, rheumatoid arthritis, and atherosclerosis (Figure 4) (Table 3). 118 , 123 , 124 , 125 , 126
Other systemic autoimmune diseases, such as systemic sclerosis, present with a predominance of type 2 cytokines, such as IL‐4 and IL‐13, either together with or instead of type 3 cytokines. 127 , 128 , 129 Recent evidence suggested that neutrophils in systemic sclerosis (SSc) patients featured functional defects, including impairment of chemotaxis, phagocytosis, and NET release, 130 as seen in allergic diseases. 90 Phenotypically, SSc neutrophils expressed lower levels of CD62L, CXCR1, and CXCR2, and lacked intracellular MPO reserves compared to neutrophils of healthy controls. 130 Interestingly, SSc neutrophils also exhibited an increase in STAT6 phosphorylation, 130 a pathway associated with IL‐4 and IL‐13 receptor signaling, indicating a possible neutrophil antagonism, similar to what has been reported in allergic diseases. 90
Another mechanism by which neutrophils perpetuate autoimmunity is the exposure of intracellular antigens to extracellular space through NETs, ROS, and degranulation (Figure 4). 1 , 131 For instance, translocation of granular proteins, such as MPO and proteinase 3, to extracellular space can cause the generation of anti‐neutrophil cytoplasmic antibodies (ANCAs) in susceptible individuals. 131 ANCAs can directly stimulate neutrophils and monocytes for the production of cytokines, but they can also induce further NET release. 6 These autoantibodies are elevated in patients with autoimmunity, especially in ANCA‐associated vasculitis (Table 3). 132 Precision therapies for ANCA‐associated vasculitis include biologic agents targeting CD20+ B cells. 133 , 134 Such therapies aim at ANCA‐producing B cells, thus indirectly interfering with pathological activation of neutrophils, whereas alternative approaches directly interfering with NET release are currently being explored. 135 Notably, ANCA levels can also increase temporarily following infections, as recently reported for COVID‐19 patients. 80
Certain diseases, such as psoriasis, share features of autoinflammation and autoimmunity (Table 3). Thus, immune complexes, IFN‐I, and Th17 cells have been implicated in the pathogenesis of psoriasis. 136 Disruption of skin homeostasis due to environmental and genetic factors results in distressed or pyroptotic keratinocytes that release self‐nucleic acids complexed with antimicrobial peptides and stimulate skin‐resident myeloid cells. 54 , 137 Psoriatic inflammation in mice can be triggered by topical application of the TLR7‐ligand imiquimod, 138 which essentially mimics innate immune activation by these immune complexes. In this experimental model, neutrophils are recruited to the skin within hours of induction of inflammation and persist until resolution. 28 Similarly, in patients, neutrophils make up a large fraction of the inflammatory infiltrate both in fresh and established psoriatic lesions. 136 In fact, an important hallmark of psoriasis is accumulation of neutrophils in the cornified layer of the epidermis, called Munro's microabscesses. Cytokines produced by neutrophils, such as TNF, IL‐1β, IL‐6, and IL‐18, foster skin‐resident IL‐17‐producing T cells, including Th17 and CD8+ cells, as well as local inflammation, culminating in tissue damage. 139 Biologics targeting Th17 cell activation or activity, including anti‐IL‐23 and anti‐IL‐17 monoclonal antibodies, have proven very efficacious in controlling disease in psoriasis patients. 136 Since IL‐17 indirectly reinforces neutrophilic responses, blocking this cytokine strongly antagonizes cutaneous neutrophilia. 140 Recently, a novel gold nanoparticle‐based methotrexate formulation has been shown to inhibit proliferation and IL‐17 production of human CD4+ T cells, thereby alleviating psoriatic inflammation in mouse models of psoriasis. 138 Similar to direct inhibition of IL‐17 signaling, methotrexate‐gold nanoparticle treatment markedly reduced neutrophil counts in the skin, highlighting the close relationship of IL‐17 production and neutrophils in the pathogenesis of disease with aberrant type 3 immune responses.
7. NEUTROPHILS WITHOUT BORDERS: NON‐IMMUNE FUNCTIONS UNVEILED
Neutrophils are short‐lived cells with limited transcriptional activity and low tissue abundance at steady state. These properties were regarded incompatible with the concept of plasticity and tissue‐specific adaptations, which, for instance, macrophages are renowned for. Development of single‐cell‐based high‐throughput technologies and transgenic reporter mouse models challenged the dogma that neutrophils are merely effector myeloid cells (Box 1). 11 , 153 , 154 Firstly, neutrophils were shown to persist in circulation and in tissues 155 for extended periods of time that would allow tissue‐specific adaptations (Figure 1). For instance, in hepatic injury, neutrophils contributed to tissue repair and their depletion significantly impaired wound healing and revascularization. 156 , 157 In another study, lung neutrophils stimulated angiogenesis upon radiation‐induced damage. 155 More recently, neutrophils were found to shuttle preexisting matrix from healthy parts of the tissue to the wounded area to promote early wound repair. 158 This fascinating study identified matrix transport to be the prevailing mode of matrix reorganization at early wound repair, preceding de novo matrix synthesis carried out by fibroblasts. Such adaptations foster non‐canonical functions, such as angiogenesis, tissue repair, and even neuronal development. These novel findings suggest there is more to discover regarding non‐canonical functions of neutrophils (Box 2).
8. CONCLUSIONS
From the description of NETs almost two decades ago, up until now, the field of immunology has become less conservative on what neutrophils are capable of. Beyond pathogen elimination, which still is their prevailing purpose, neutrophils communicate signals of activation, suppression, and migration, guide antigen presentation, and modulate local immune responses. If not regulated properly, neutrophils can inflict damage, but importantly, they can also contribute to tissue maintenance and repair. The compelling question is what guides a neutrophil to assume a specific role or function? Understanding this requires to further dissect cell‐intrinsic and extrinsic mechanisms in different immunological contexts. With each new piece of the puzzle, we get one step closer to manipulating neutrophil behavior, which opens novel therapeutic avenues for life‐threatening infections, immune‐mediated diseases, and even defects of tissue repair and regeneration.
FUNDING INFORMATION
This work was funded by the Swiss National Science Foundation (P500PB‐206852 to A.O., 310030‐200669 and CRSII5‐189950 to O.B.), the Hochspezialisierte Medizin Schwerpunkt Immunologie (HSM‐2‐Immunologie; to O.B.), and the Clinical Research Priority Program of the University of Zurich for the CRPP CYTIMM‐Z (to O.B.).
CONFLICT OF INTEREST
The authors declare no conflict of interests related to this manuscript.
ACKNOWLEDGEMENT
We thank Lukas E. M. Heeb for reading the manuscript. Open access funding provided by Universitat Zurich.
Özcan A, Boyman O. Mechanisms regulating neutrophil responses in immunity, allergy, and autoimmunity. Allergy. 2022;77:3567‐3583. doi: 10.1111/all.15505
REFERENCES
- 1. Burn GL, Foti A, Marsman G, Patel DF, Zychlinsky A. The neutrophil. Immunity. 2021;54:1377‐1391. [DOI] [PubMed] [Google Scholar]
- 2. Leiding JW, Holland SM. Chronic granulomatous disease. GeneReviews®. Published Online First: 21 April 2022. https://www.ncbi.nlm.nih.gov/books/NBK99496/
- 3. Lin B, Goldbach‐Mansky R. Pathogenic insights from genetic causes of autoinflammatory inflammasomopathies and interferonopathies. J Allergy Clin Immunol. 2022;149:819‐832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rood JE, Behrens EM. Inherited autoinflammatory syndromes. Annu Rev Pathol Mech Dis. 2021;17:227‐249. [DOI] [PubMed] [Google Scholar]
- 5. Marshall CL, Hasani K, Mookherjee N. Immunobiology of steroid‐unresponsive severe asthma. Front Allergy. 2021;2:718267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang L, Luqmani R, Udalova IA. The role of neutrophils in rheumatic disease‐associated vascular inflammation. Nat Rev Rheumatol. 2022;18:158‐170. [DOI] [PubMed] [Google Scholar]
- 7. Hacbarth E, Kajdacsy‐Balla A. Low density neutrophils in patients with systemic lupus erythematosus, rheumatoid arthritis, and acute rheumatic fever. Arthritis Rheum. 1986;29:1334‐1342. [DOI] [PubMed] [Google Scholar]
- 8. Veglia F, Sanseviero E, Gabrilovich DI. Myeloid‐derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol. 2021;21:485‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Silvestre‐Roig C, Hidalgo A, Soehnlein O. Neutrophil heterogeneity: implications for homeostasis and pathogenesis. Blood. 2016;127:2173‐2181. [DOI] [PubMed] [Google Scholar]
- 10. Ng LG, Ostuni R, Hidalgo A. Heterogeneity of neutrophils. Nat Rev Immunol. 2019;19:255‐265. [DOI] [PubMed] [Google Scholar]
- 11. Hidalgo A, Casanova‐Acebes M. Dimensions of neutrophil life and fate. Semin Immunol. 2021;57:101506. [DOI] [PubMed] [Google Scholar]
- 12. Evrard M, Kwok IWH, Chong SZ, et al. Developmental analysis of bone marrow neutrophils reveals populations specialized in expansion, trafficking, and effector functions. Immunity. 2018;48:364‐379. [DOI] [PubMed] [Google Scholar]
- 13. Kwok I, Becht E, Xia Y, et al. Combinatorial single‐cell analyses of granulocyte‐monocyte progenitor heterogeneity reveals an early uni‐potent neutrophil progenitor. Immunity. 2020;53:303‐318. [DOI] [PubMed] [Google Scholar]
- 14. De Filippo K, Rankin SM. CXCR4, the master regulator of neutrophil trafficking in homeostasis and disease. Eur J Clin Investig. 2018;48:e12949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eash KJ, Greenbaum AM, Gopalan PK, Link DC. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J Clin Invest. 2010;120:2423‐2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim ND, Luster AD. The role of tissue resident cells in neutrophil recruitment. Trends Immunol. 2015;36:547‐555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Heeb LEM, Egholm C, Impellizzieri D, Ridder F, Boyman O. Regulation of neutrophils in type 2 immune responses. Curr Opin Immunol. 2018;54:115‐122. [DOI] [PubMed] [Google Scholar]
- 18. Knudsen E, Iversen PO, Bøyum A, Seierstad T, Nicolaysen G, Benestad HB. G‐CSF enhances the proliferation and mobilization, but not the maturation rate, of murine myeloid cells. Eur J Haematol. 2011;87:302‐311. [DOI] [PubMed] [Google Scholar]
- 19. Margraf A, Lowell CA, Zarbock A. Neutrophils in acute inflammation: current concepts and translational implications. Blood. 2022;139:2130‐2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Margraf A, Ley K, Zarbock A. Neutrophil recruitment: from model systems to tissue‐specific patterns. Trends Immunol. 2019;40:613‐634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Moser M, Legate KR, Zent R, Fässler R. The tail of integrins, talin, and kindlins. Science. 2009;324:895‐899. [DOI] [PubMed] [Google Scholar]
- 22. Wen L, Moser M, Ley K. Molecular mechanisms of leukocyte β2 integrin activation. Blood. 2022;139:3480‐3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sprenkeler EGG, Webbers SDS, Kuijpers TW. When actin is not actin’ like it should: a new category of distinct primary immunodeficiency disorders. J Innate Immun. 2021;13:3‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lämmermann T, Afonso PV, Angermann BR, et al. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature. 2013;498:371‐375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159‐175. [DOI] [PubMed] [Google Scholar]
- 26. De Filippo K, Rankin SM. The secretive life of neutrophils revealed by intravital microscopy. Front Cell Dev Biol. 2020;8:603230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lim K, Kim TH, Trzeciak A, et al. In situ neutrophil efferocytosis shapes T cell immunity to influenza infection. Nat Immunol. 2020;21:1046‐1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Özcan A, Collado‐Diaz V, Egholm C, et al. CCR7‐guided neutrophil redirection to skin‐draining lymph nodes regulates cutaneous inflammation and infection. Sci Immunol. 2022;7:eabi9126. [DOI] [PubMed] [Google Scholar]
- 29. Nüsslein‐Volhard C. The Toll gene in Drosophila pattern formation. Trends Genet. 2022;38:231‐245. [DOI] [PubMed] [Google Scholar]
- 30. Kawai T, Akira S. TLR signaling. Cell Death Differ. 2006;13:816‐825. [DOI] [PubMed] [Google Scholar]
- 31. Kaufmann SHE. Elie Metchnikoff's and Paul Ehrlich's impact on infection biology. Microbes Infect. 2008;10:1417‐1419. [DOI] [PubMed] [Google Scholar]
- 32. Pérez‐Figueroa E, Álvarez‐Carrasco P, Ortega E, Maldonado‐Bernal C. Neutrophils: many ways to die. Front Immunol. 2021;12:631821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Appelberg R. Neutrophils and intracellular pathogens: beyond phagocytosis and killing. Trends Microbiol. 2007;15:87‐92. [DOI] [PubMed] [Google Scholar]
- 34. Geering B, Stoeckle C, Conus S, Simon HU. Living and dying for inflammation: neutrophils, eosinophils, basophils. Trends Immunol. 2013;34:398‐409. [DOI] [PubMed] [Google Scholar]
- 35. Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil function: from mechanisms to disease. Annu Rev Immunol. 2012;30:459‐489. [DOI] [PubMed] [Google Scholar]
- 36. Begum R, Thota S, Abdulkadir A, Kaur G, Bagam P, Batra S. NADPH oxidase family proteins: signaling dynamics to disease management. Cell Mol Immunol. 2022;19:660‐686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yu HH, Yang YH, Chiang BL. Chronic granulomatous disease: a comprehensive review. Clin Rev Allergy Immunol. 2021;61:101‐113. [DOI] [PubMed] [Google Scholar]
- 38. Branzk N, Lubojemska A, Hardison SE, et al. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol. 2014;15:1017‐1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sollberger G, Tilley DO, Zychlinsky A. Neutrophil extracellular traps: the biology of chromatin externalization. Dev Cell. 2018;44:542‐553. [DOI] [PubMed] [Google Scholar]
- 40. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. 2018;18:134‐147. [DOI] [PubMed] [Google Scholar]
- 41. Weinrauch Y, Drujan D, Shapiro SD, Weiss J, Zychlinsky A. Neutrophil elastase targets virulence factors of enterobacteria. Nature. 2002;417:91‐94. [DOI] [PubMed] [Google Scholar]
- 42. Belaaouaj AA, Kim KS, Shapiro SD. Degradation of outer membrane protein A in Escherichia coli killing by neutrophil elastase. Science. 2000;289:1185‐1187. [DOI] [PubMed] [Google Scholar]
- 43. Leiding JW. Neutrophil evolution and their diseases in humans. Front Immunol. 2017;8:1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lanini LLS, Prader S, Siler U, Reichenbach J. Modern management of phagocyte defects. Pediatr Allergy Immunol. 2017;28:124‐134. [DOI] [PubMed] [Google Scholar]
- 45. Tangye SG, Al‐Herz W, Bousfiha A, Cunningham‐Rundles C, Jose FL, et al. Human inborn errors of immunity: 2022 update on the classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2022;1:1‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Triot A, Järvinen PM, Arostegui JI, et al. Inherited biallelic CSF3R mutations in severe congenital neutropenia. Blood. 2014;123:3811‐3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Svensson L, Howarth K, McDowall A, et al. Leukocyte adhesion deficiency‐III is caused by mutations in KINDLIN3 affecting integrin activation. Nat Med. 2009;15:306‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Heusinkveld LE, Majumdar S, Gao JL, McDermott DH, Murphy PM. WHIM syndrome: from pathogenesis towards personalized medicine and cure. J Clin Immunol. 2019;39:532‐556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Arnadottir GA, Norddahl GL, Gudmundsdottir S, et al. A homozygous loss‐of‐function mutation leading to CYBC1 deficiency causes chronic granulomatous disease. Nat Commun. 2018;9:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tamassia N, Bianchetto‐Aguilera F, Arruda‐Silva F, et al. Cytokine production by human neutrophils: revisiting the “dark side of the moon”. Eur J Clin Investig. 2018;48:e12952. [DOI] [PubMed] [Google Scholar]
- 51. Lim K, Hyun YM, Lambert‐Emo K, et al. Neutrophil trails guide influenzaspecific CD8+ T cells in the airways. Science. 2015;349:aaa4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kim YR, Kim YM, Lee J, Park J, Lee JE, Hyun YM. Neutrophils return to bloodstream through the brain blood vessel after crosstalk with microglia during LPS‐induced neuroinflammation. Front Cell Dev Biol. 2020;8:613733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Charmoy M, Brunner‐Agten S, Aebischer D, et al. Neutrophil‐derived CCL3 is essential for the rapid recruitment of dendritic cells to the site of Leishmania major inoculation in resistant mice. PLoS Pathog. 2010;6:e1000755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lande R, Gregorio J, Facchinetti V, et al. Plasmacytoid dendritic cells sense self‐DNA coupled with antimicrobial peptide. Nature. 2007;449:564‐569. [DOI] [PubMed] [Google Scholar]
- 55. Lande R, Ganguly D, Facchinetti V, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self‐DNA‐peptide complexes in systemic lupus erythematosus. Sci Transl Med. 2011;3:73ra19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Peiró T, Patel DF, Akthar S, et al. Neutrophils drive alveolar macrophage IL‐1β release during respiratory viral infection. Thorax. 2018;73:546‐556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yang W, Tao Y, Wu Y, et al. Neutrophils promote the development of reparative macrophages mediated by ROS to orchestrate liver repair. Nat Commun. 2019;10:1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lok LSC, Clatworthy MR. Neutrophils in secondary lymphoid organs. Immunology. 2021;164:677‐688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bogoslowski A, Butcher EC, Kubes P. Neutrophils recruited through high endothelial venules of the lymph nodes via PNAd intercept disseminating Staphylococcus aureus. Proc Natl Acad Sci U S A. 2018;115:2449‐2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bogoslowski A, Wijeyesinghe S, Lee W‐Y, et al. Neutrophils recirculate through lymph nodes to survey tissues for pathogens. J Immunol. 2020;204:2552‐2561. [DOI] [PubMed] [Google Scholar]
- 61. Hampton HR, Bailey J, Tomura M, Brink R, Chtanova T. Microbe‐dependent lymphatic migration of neutrophils modulates lymphocyte proliferation in lymph nodes. Nat Commun. 2015;6:7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Abadie V, Badell E, Douillard P, et al. Neutrophils rapidly migrate via lymphatics after Mycobacterium bovis BCG intradermal vaccination and shuttle live bacilli to the draining lymph nodes. Blood. 2005;106:1843‐1850. [DOI] [PubMed] [Google Scholar]
- 63. Lok LSC, Dennison TW, Mahbubani KM, Saeb‐Parsy K, Chilvers ER, Clatworthy MR. Phenotypically distinct neutrophils patrol uninfected human and mouse lymph nodes. Proc Natl Acad Sci U S A. 2019;116:19083‐19089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mysore V, Cullere X, Mears J, et al. FcγR engagement reprograms neutrophils into antigen cross‐presenting cells that elicit acquired anti‐tumor immunity. Nat Commun. 2021;12:4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Alfaro C, Suarez N, Oñate C, et al. Dendritic cells take up and present antigens from viable and apoptotic polymorphonuclear leukocytes. PLoS One. 2011;6:e29300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Morel C, Badell E, Abadie V, et al. Mycobacterium bovis BCG‐infected neutrophils and dendritic cells cooperate to induced specific T cell responses in humans and mice. Eur J Immunol. 2008;38:437‐447. [DOI] [PubMed] [Google Scholar]
- 67. Kamenyeva O, Boularan C, Kabat J, et al. Neutrophil recruitment to lymph nodes limits local humoral response to Staphylococcus aureus . PLoS Pathog. 2015;11:e1004827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Stacey MA, Marsden M, Pham NTA, et al. Neutrophils recruited by IL‐22 in peripheral tissues function as TRAIL‐dependent antiviral effectors against MCMV. Cell Host Microbe. 2014;15:471‐483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Egholm C, Özcan A, Breu D, Boyman O. Type 2 immune predisposition results in accelerated neutrophil aging causing susceptibility to bacterial infection. Sci Immunol. 2022;7:eabi9733. [DOI] [PubMed] [Google Scholar]
- 70. Peters NC, Egen JG, Secundino N, et al. In vivo imaging reveals an essential role for neutrophils in leishmaniasis transmitted by sand flies. Science. 2008;321:970‐974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bai F, Kong KF, Dai J, et al. A paradoxical role for neutrophils in the pathogenesis of West Nile virus. J Infect Dis. 2010;202:1804‐1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Eruslanov EB, Lyadova IV, Kondratieva TK, et al. Neutrophil responses to Mycobacterium tuberculosis infection in genetically susceptible and resistant mice. Infect Immun. 2005;73:1744‐1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Gresham HD, Lowrance JH, Caver TE, Wilson BS, Cheung AL, Lindberg FP. Survival of Staphylococcus aureus inside neutrophils contributes to infection. J Immunol. 2000;164:3713‐3722. [DOI] [PubMed] [Google Scholar]
- 74. Zhao Y, Lu M, Lau LT, et al. Neutrophils may be a vehicle for viral replication and dissemination in human h5n1 avian influenza. Clin Infect Dis. 2008;47:1575‐1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Chevrier S, Zurbuchen Y, Cervia C, et al. A distinct innate immune signature marks progression from mild to severe COVID‐19. Cell Reports Med. 2021;2:100166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Carissimo G, Xu W, Kwok I, et al. Whole blood immunophenotyping uncovers immature neutrophil‐to‐VD2 T‐cell ratio as an early marker for severe COVID‐19. Nat Commun. 2020;11:5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Taeschler P, Adamo S, Deng Y, et al. T‐cell recovery and evidence of persistent immune activation 12 months after severe COVID ‐19. Allergy. 2022;77:2468‐2481. doi: 10.1111/all.15372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Middleton EA, He XY, Denorme F, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID‐19 acute respiratory distress syndrome. Blood. 2020;136:1169‐1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zuo Y, Yalavarthi S, Shi H, et al. Neutrophil extracellular traps in COVID‐19. JCI Insight. 2020;5:e138999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Taeschler P, Cervia C, Zurbuchen Y, et al. Autoantibodies in COVID‐19 correlate with antiviral humoral responses and distinct immune signatures. Allergy. 2022;77:2415‐2430. doi: 10.1111/all.15302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kienle K, Glaser KM, Eickhoff S, et al. Neutrophils self‐limit swarming to contain bacterial growth in vivo. Science. 2021;372:eabe7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Adrover JM, Aroca‐Crevillén A, Crainiciuc G, et al. Programmed ‘disarming’ of the neutrophil proteome reduces the magnitude of inflammation. Nat Immunol. 2020;21:135‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Stark MA, Huo Y, Burcin TL, Morris MA, Olson TS, Ley K. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL‐23 and IL‐17. Immunity. 2005;22:285‐294. [DOI] [PubMed] [Google Scholar]
- 84. Casanova‐Acebes M, Nicolás‐Ávila JA, Yao Li JL, et al. Neutrophils instruct homeostatic and pathological states in naive tissues. J Exp Med. 2018;215:2778‐2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Egholm C, Heeb LEM, Impellizzieri D, Boyman O. The regulatory effects of interleukin‐4 receptor signaling on neutrophils in type 2 immune responses. Front Immunol. 2019;10:2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Heeb LEM, Egholm C, Boyman O. Evolution and function of interleukin‐4 receptor signaling in adaptive immunity and neutrophils. Genes Immun. 2020;21:143‐149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Akdis CA. Does the epithelial barrier hypothesis explain the increase in allergy, autoimmunity and other chronic conditions? Nat Rev Immunol. 2021;21:739‐751. [DOI] [PubMed] [Google Scholar]
- 88. De Benedetto A, Agnihothri R, McGirt LY, Bankova LG, Beck LA. Atopic dermatitis: a disease caused by innate immune defects? J Invest Dermatol. 2009;129:14‐30. [DOI] [PubMed] [Google Scholar]
- 89. Choy DF, Hart KM, Borthwick LA, et al. TH2 and TH17 inflammatory pathways are reciprocally regulated in asthma. Sci Transl Med. 2015;7:301ra129. [DOI] [PubMed] [Google Scholar]
- 90. Impellizzieri D, Ridder F, Raeber ME, et al. IL‐4 receptor engagement in human neutrophils impairs their migration and extracellular trap formation. J Allergy Clin Immunol. 2019;144:267‐279. [DOI] [PubMed] [Google Scholar]
- 91. Wang V, Boguniewicz J, Boguniewicz M, Ong PY. The infectious complications of atopic dermatitis. Ann Allergy Asthma Immunol. 2021;126:3‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Wollenberg A, Wetzel S, Burgdorf WHC, Haas J. Viral infections in atopic dermatitis: pathogenic aspects and clinical management. J Allergy Clin Immunol. 2003;112:667‐674. [DOI] [PubMed] [Google Scholar]
- 93. Crisford H, Sapey E, Rogers GB, et al. Neutrophils in asthma: the good, the bad and the bacteria. Thorax. 2021;76:835‐844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Radermecker C, Louis R, Bureau F, Marichal T. Role of neutrophils in allergic asthma. Curr Opin Immunol. 2018;54:28‐34. [DOI] [PubMed] [Google Scholar]
- 95. Delemarre T, Bochner BS, Simon HU, Bachert C. Rethinking neutrophils and eosinophils in chronic rhinosinusitis. J Allergy Clin Immunol. 2021;148:327‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Chen F, Wu W, Millman A, et al. Neutrophils prime a long‐lived effector macrophage phenotype that mediates accelerated helminth expulsion. Nat Immunol. 2014;15:938‐946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Oyoshi MK, He R, Li Y, et al. Leukotriene B4‐driven neutrophil recruitment to the skin is essential for allergic skin inflammation. Immunity. 2012;37:747‐758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Cataldo DD, Tournoy KG, Vermaelen K, et al. Matrix metalloproteinase‐9 deficiency impairs cellular infiltration and bronchial hyperresponsiveness during allergen‐induced airway inflammation. Am J Pathol. 2002;161:491‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Vermaelen KY, Cataldo D, Tournoy K, et al. Matrix metalloproteinase‐9‐mediated dendritic cell recruitment into the airways is a critical step in a mouse model of asthma. J Immunol. 2003;171:1016‐1022. [DOI] [PubMed] [Google Scholar]
- 100. Hosoki K, Aguilera‐Aguirre L, Brasier AR, Kurosky A, Boldogh I, Sur S. Facilitation of allergic sensitization and allergic airway inflammation by pollen‐induced innate neutrophil recruitment. Am J Respir Cell Mol Biol. 2016;54:81‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Chaudhuri R, Norris V, Kelly K, et al. Effects of a FLAP inhibitor, GSK2190915, in asthmatics with high sputum neutrophils. Pulm Pharmacol Ther. 2014;27:62‐69. [DOI] [PubMed] [Google Scholar]
- 102. Barchuk W, Lambert J, Fuhr R, et al. Effects of JNJ‐40929837, a leukotriene A4 hydrolase inhibitor, in a bronchial allergen challenge model of asthma. Pulm Pharmacol Ther. 2014;29:15‐23. [DOI] [PubMed] [Google Scholar]
- 103. O'Byrne PM, Metev H, Puu M, et al. Efficacy and safety of a CXCR2 antagonist, AZD5069, in patients with uncontrolled persistent asthma: a randomised, double‐blind, placebo‐controlled trial. Lancet Respir Med. 2016;4:797‐806. [DOI] [PubMed] [Google Scholar]
- 104. Nair P, Gaga M, Zervas E, et al. Safety and efficacy of a CXCR2 antagonist in patients with severe asthma and sputum neutrophils: a randomized, placebo‐controlled clinical trial. Clin Exp Allergy. 2012;42:1097‐1103. [DOI] [PubMed] [Google Scholar]
- 105. Patel DF, Peiró T, Bruno N, et al. Neutrophils restrain allergic airway inflammation by limiting ILC2 function and monocyte‐dendritic cell antigen presentation. Sci Immunol. 2019;4:eaax7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Woytschak J, Keller N, Krieg C, et al. Type 2 interleukin‐4 receptor signaling in neutrophils antagonizes their expansion and migration during infection and inflammation. Immunity. 2016;45:172‐184. [DOI] [PubMed] [Google Scholar]
- 107. Grigolato F, Egholm C, Impellizzieri D, Arosio P, Boyman O. Establishment of a scalable microfluidic assay for characterization of population‐based neutrophil chemotaxis. Allergy. 2020;75:1382‐1393. [DOI] [PubMed] [Google Scholar]
- 108. Akdis CA, Arkwright PD, Brüggen MC, et al. Type 2 immunity in the skin and lungs. Allergy. 2020;75:1582‐1605. [DOI] [PubMed] [Google Scholar]
- 109. Lu Y, Huang Y, Li J, et al. Eosinophil extracellular traps drive asthma progression through neuro‐immune signals. Nat Cell Biol. 2021;23:1060‐1072. [DOI] [PubMed] [Google Scholar]
- 110. Valladao AC, Frevert CW, Koch LK, Campbell DJ, Ziegler SF. STAT6 regulates the development of eosinophilic versus neutrophilic asthma in response to alternaria alternata. J Immunol. 2016;197:4541‐4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Cianferoni A. Non–IgE‐mediated anaphylaxis. J Allergy Clin Immunol. 2021;147:1123‐1131. [DOI] [PubMed] [Google Scholar]
- 112. Krainer J, Siebenhandl S, Weinhäusel A. Systemic autoinflammatory diseases. J Autoimmun. 2020;109:102421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Pyrillou K, Burzynski LC, Clarke MCH. Alternative pathways of IL‐1 activation, and its role in health and disease. Front Immunol. 2020;11:613170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Münzer P, Negro R, Fukui S, et al. NLRP3 inflammasome assembly in neutrophils is supported by PAD4 and promotes NETosis under sterile conditions. Front Immunol. 2021;12:683803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. 2011;11:519‐531. [DOI] [PubMed] [Google Scholar]
- 116. Havnaer A, Han G. Autoinflammatory disorders: a review and update on pathogenesis and treatment. Am J Clin Dermatol. 2019;20:539‐564. [DOI] [PubMed] [Google Scholar]
- 117. Arnold DD, Yalamanoglu A, Boyman O. Systematic review of safety and efficacy of IL‐1‐targeted biologics in treating immune‐mediated disorders. Front Immunol. 2022;13:888392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Herster F, Bittner Z, Archer NK, et al. Neutrophil extracellular trap‐associated RNA and LL37 enable self‐amplifying inflammation in psoriasis. Nat Commun. 2020;11:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Tsourouktsoglou TD, Warnatsch A, Ioannou M, Hoving D, Wang Q, Papayannopoulos V. Histones, DNA, and citrullination promote neutrophil extracellular trap inflammation by regulating the localization and activation of TLR4. Cell Rep. 2020;31:107602. [DOI] [PubMed] [Google Scholar]
- 120. Wilson AS, Randall KL, Pettitt JA, et al. Neutrophil extracellular traps and their histones promote Th17 cell differentiation directly via TLR2. Nat Commun. 2022;13:528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Krishnamoorthy N, Douda DN, Brüggemann TR, et al. Neutrophil cytoplasts induce TH17 differentiation and skew inflammation toward neutrophilia in severe asthma. Sci Immunol. 2018;3:eaao4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Herrero‐Cervera A, Soehnlein O, Kenne E. Neutrophils in chronic inflammatory diseases. Cell Mol Immunol. 2022;19:177‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Yasuda K, Takeuchi Y, Hirota K. The pathogenicity of Th17 cells in autoimmune diseases. Semin Immunopathol. 2019;41:283‐297. [DOI] [PubMed] [Google Scholar]
- 124. Gupta S, Kaplan MJ. Bite of the wolf: innate immune responses propagate autoimmunity in lupus. J Clin Invest. 2021;131:e144918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Fresneda Alarcon M, McLaren Z, Wright HL. Neutrophils in the pathogenesis of rheumatoid arthritis and systemic lupus erythematosus: same foe different M.O. Front Immunol. 2021;12:649693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. 2015;349:316‐320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Parel Y, Aurrand‐Lions M, Scheja A, Dayer JM, Roosnek E, Chizzolini C. Presence of CD4+CD8+ double‐positive T cells with very high interleukin‐4 production potential in lesional skin of patients with systemic sclerosis. Arthritis Rheum. 2007;56:3459‐3467. [DOI] [PubMed] [Google Scholar]
- 128. Scaletti C, Vultaggio A, Bonifacio S, et al. Th2‐oriented profile of male offspring T cells present in women with systemic sclerosis and reactive with maternal major histocompatibility complex antigens. Arthritis Rheum. 2002;46:445‐450. [DOI] [PubMed] [Google Scholar]
- 129. Truchetet ME, Brembilla NC, Montanari E, Allanore Y, Chizzolini C. Increased frequency of circulating Th22 in addition to Th17 and Th2 lymphocytes in systemic sclerosis: association with interstitial lung disease. Arthritis Res Ther. 2011;13:R166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Impellizzieri D, Egholm C, Valaperti A, Distler O, Boyman O. Patients with systemic sclerosis show phenotypic and functional defects in neutrophils. Allergy. 2022;77:1274‐1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Liu Y, Kaplan MJ. Neutrophils in the pathogenesis of rheumatic diseases: fueling the fire. Clin Rev Allergy Immunol. 2021;60:1‐16. [DOI] [PubMed] [Google Scholar]
- 132. Chen M, Kallenberg CGM. ANCA‐associated vasculitides‐advances in pathogenesis and treatment. Nat Rev Rheumatol. 2010;6:653‐664. [DOI] [PubMed] [Google Scholar]
- 133. Kaegi C, Wuest B, Schreiner J, et al. Systematic review of safety and efficacy of rituximab in treating immune‐mediated disorders. Front Immunol. 2019;10:1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Kaegi C, Wuest B, Crowley C, Boyman O. Systematic review of safety and efficacy of second‐ and third‐generation CD20‐targeting biologics in treating immune‐mediated disorders. Front Immunol. 2022;12:788830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. O'Sullivan KM, Holdsworth SR. Neutrophil extracellular traps: a potential therapeutic target in MPO‐ANCA associated vasculitis? Front Immunol. 2021;12:635188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Griffiths CEM, Armstrong AW, Gudjonsson JE, Barker JNWN. Psoriasis. Lancet. 2021;397:1301‐1315. [DOI] [PubMed] [Google Scholar]
- 137. Nestle FO, Conrad C, Tun‐Kyi A, et al. Plasmacytoid predendritic cells initiate psoriasis through interferon‐α production. J Exp Med. 2005;202:135‐143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Özcan A, Sahin D, Impellizzieri D, et al. Nanoparticle‐coupled topical methotrexate can normalize immune responses and induce tissue remodeling in psoriasis. J Invest Dermatol. 2020;140:1003‐1014. [DOI] [PubMed] [Google Scholar]
- 139. Raeber ME, Zurbuchen Y, Impellizzieri D, Boyman O. The role of cytokines in T‐cell memory in health and disease. Immunol Rev. 2018;283:176‐193. [DOI] [PubMed] [Google Scholar]
- 140. Reich K, Papp KA, Matheson RT, et al. Evidence that a neutrophil‐keratinocyte crosstalk is an early target of IL‐17A inhibition in psoriasis. Exp Dermatol. 2015;24:529‐535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Stackowicz J, Gaudenzio N, Serhan N, et al. Neutrophil‐specific gain‐of‐function mutations in Nlrp3 promote development of cryopyrin‐associated periodic syndrome. J Exp Med. 2021;218:e20201466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Malengier‐Devlies B, Metzemaekers M, Gouwy M, et al. Phenotypical and functional characterization of neutrophils in two pyrin‐associated auto‐inflammatory diseases. J Clin Immunol. 2021;41:1072‐1084. [DOI] [PubMed] [Google Scholar]
- 143. Takimoto‐Ito R, Kambe N, Kogame T, et al. Refractory serum immunoglobulin M elevation during anti‐interleukin (IL)‐1‐ or IL‐6‐targeted treatment in four patients with Schnitzler syndrome. J Dermatol. 2021;48:1789‐1792. [DOI] [PubMed] [Google Scholar]
- 144. Sumida H, Yanagida K, Kita Y, et al. Interplay between CXCR2 and BLT1 facilitates neutrophil infiltration and resultant keratinocyte activation in a murine model of imiquimod‐induced psoriasis. J Immunol. 2014;192:4361‐4369. [DOI] [PubMed] [Google Scholar]
- 145. Di Domizio J, Gilliet M. Psoriasis caught in the NET. J Invest Dermatol. 2019;139:1426‐1429. [DOI] [PubMed] [Google Scholar]
- 146. Kitching AR, Anders HJ, Basu N, et al. ANCA‐associated vasculitis. Nat Rev Dis Primers. 2020;6:71. [DOI] [PubMed] [Google Scholar]
- 147. Kessenbrock K, Krumbholz M, Schönermarck U, et al. Netting neutrophils in autoimmune small‐vessel vasculitis. Nat Med. 2009;15:623‐625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Nozaki Y. New insights into novel therapeutic targets in ANCA‐associated vasculitis. Front Immunol. 2021;12:631055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Kahlenberg JM, Carmona‐Rivera C, Smith CK, Kaplan MJ. Neutrophil extracellular trap–associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J Immunol. 2013;190:1217‐1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Tsokos GC. Autoimmunity and organ damage in systemic lupus erythematosus. Nat Immunol. 2020;21:605‐614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Carmona‐Rivera C, Carlucci PM, Goel RR, et al. Neutrophil extracellular traps mediate articular cartilage damage and enhance cartilage component immunogenicity in rheumatoid arthritis. JCI Insight. 2020;5:e139388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Khandpur R, Carmona‐Rivera C, Vivekanandan‐Giri A, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med. 2013;5:e178ra40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Quail DF, Amulic B, Aziz M, et al. Neutrophil phenotypes and functions in cancer: a consensus statement. J Exp Med. 2022;219:e20220011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Rubio‐Ponce A, Hidalgo A, Ballesteros I. How to bridle a neutrophil. Curr Opin Immunol. 2021;68:41‐47. [DOI] [PubMed] [Google Scholar]
- 155. Ballesteros I, Rubio‐Ponce A, Genua M, et al. Co‐option of neutrophil fates by tissue environments. Cell. 2020;183:1282‐1297. [DOI] [PubMed] [Google Scholar]
- 156. Wang J, Hossain M, Thanabalasuriar A, Gunzer M, Meininger C, Kubes P. Visualizing the function and fate of neutrophils in sterile injury and repair. Science. 2017;358:111‐116. [DOI] [PubMed] [Google Scholar]
- 157. Neupane AS, Kubes P. Imaging reveals novel innate immune responses in lung, liver, and beyond. Immunol Rev. 2022;306:244‐257. [DOI] [PubMed] [Google Scholar]
- 158. Fischer A, Wannemacher J, Christ S, et al. Neutrophils direct preexisting matrix to initiate repair in damaged tissues. Nat Immunol. 2022;23:518‐531. [DOI] [PMC free article] [PubMed] [Google Scholar]