

Abstract
The capsid assembly modulator JNJ‐56136379 (bersacapavir) disrupts hepatitis B virus replication. It is metabolized via cytochrome P450 (CYP) 3A, but little is known about the drug‐drug interactions of JNJ‐56136379 when combined with drugs that inhibit or are metabolized by CYP3A. In a phase 1, open‐label trial (NCT03945539), healthy adults received 1 dose of JNJ‐56136379 with and without 21 days of prior exposure to itraconazole 200 mg (CYP3A inhibitor). In a second phase 1, open‐label trial (NCT03111511), healthy women received 1 dose of drospirenone/ethinyl estradiol and midazolam before and after 15 days of JNJ‐56136379. Itraconazole increased the area under the plasma concentration–time curve (AUC) of JNJ‐56136379 by 38%. JNJ‐56136379 reduced the maximum observed concentration and AUC of midazolam (CYP3A substrate) by 42%–54%, increased AUC of ethinyl estradiol by 1.6‐fold, but had no effect on drospirenone pharmacokinetics. Overall, these results demonstrated that a strong CYP3A inhibitor (itraconazole) modestly increased JNJ‐56136379 exposure. Furthermore, JNJ‐56136379 was a weak inducer of CYP3A (midazolam) and increased ethinyl estradiol exposure; coadministration of high‐dose estrogen‐based contraceptives and JNJ‐56136379 is not recommended.
Keywords: capsid assembly modulator, CYP3A, drug‐drug interactions, hepatitis B, JNJ‐56136379
JNJ‐56136379 (bersacapavir) is a hepatitis B virus (HBV) capsid assembly modulator (CAM) that induces the formation of structurally normal, but empty, nonfunctional HBV capsids. 1 , 2 , 3 Through binding of the HBV core protein, JNJ‐56136379 disrupts early‐ and late‐stage processes in the HBV life cycle, thus disrupting viral replication via 2 mechanisms. 2 , 4 , 5 The primary mechanism is to accelerate capsid assembly kinetics, thereby preventing encapsidation of polymerase‐bound pregenomic RNA, which leads to the formation of empty capsids, a late step in the viral life cycle. 2 , 4 , 5 The secondary mechanism of disrupting viral replication is the inhibition of de novo formation of covalently closed circular DNA, an early‐stage process in the HBV life cycle. 2 , 4 , 5
The pharmacokinetics of orally administered JNJ‐56136379 have been evaluated in both healthy participants and patients with chronic hepatitis B (CHB), with dose‐proportional pharmacokinetics observed at doses up to 300 mg. 4 , 5 In patients with CHB, the terminal half‐life of JNJ‐56136379 is long, ranging from 103 to 148 hours, with low to moderate fluctuations in pharmacokinetic parameters. 5 Potent antiviral activity of JNJ‐56136379 has been observed in patients with CHB, with reductions in HBV DNA and RNA but only marginal declines in hepatitis B surface antigen. 5 , 6
Approximately 50% of JNJ‐56136379 undergoes metabolic elimination, with the remainder eliminated unchanged in urine (20%) or via intestinal secretion (30%; data on file). In vitro data have shown that metabolism of JNJ‐56136379 is almost exclusively mediated via cytochrome P450 (CYP) 3A. 5 Metabolism of JNJ‐56136379 was assessed with supersomes (Corning Life Sciences, Tewksbury, Massachusetts) expressing the single enzymes CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, or CYP3A4. Metabolism could be picked up in supersomes overexpressing CYP2C19 or CYP3A4. The CYP3A4 intrinsic metabolic clearance was found to be 10‐fold higher than the CYP2C19 intrinsic clearance. The major contribution of CYP3A4 in hepatic metabolism was confirmed by the addition of troleandomycin to primary human hepatocyte incubations with JNJ‐56136379 resulting in 80% inhibition of metabolic turnover.
Investigations using human liver microsomes have shown that the half maximal inhibitory concentration (IC50) values for all major human CYP isoforms (CYP 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4) were >15 μM. No mechanism‐based inactivation toward CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, or 3A4 was observed up to 75 μM, the highest concentration tested. JNJ‐56136379 was found to inhibit uridine diphosphate glucuronosyltransferase (UGT) 1A1 activity with an unbound IC50 value of 24.4 μM and UGT2B7 with an unbound IC50 of 47.3 μM (see Table S1 for a comparison of the IC50 values for UGT1A1 and UGT2B7 to total and unbound maximum observed concentration [Cmax] of JNJ‐56136379). The unbound IC50 of JNJ‐56136379 toward sulfotransferase 1E1 and 2A1 in liver and intestinal cytosol was above 63.8 μM. The effect of JNJ‐56136379 on the expression of CYP1A2, CYP2B6, and CYP3A4 was evaluated in vitro in cultures of cryopreserved primary human hepatocytes from 3 different donors. JNJ‐56136379 was found to be an inducer of CYP3A4 at concentrations above 2 μM. At 10 μM, induction of CYP3A4 amounted to 10%–31% of the induction observed with the positive control inducer rifampin (data on file). These findings suggest that JNJ‐56136379 has the potential to induce the CYP3A4 pathway, thus affecting the pharmacokinetics of CYP3A4‐metabolized comedications.
Itraconazole is a strong CYP3A4, breast cancer resistance protein (BCRP), and P‐glycoprotein inhibitor with an acceptable safety profile. It is considered a suitable replacement for ketoconazole (no longer manufactured) due to its extensive pharmacologic characterization and lack of induction properties (compared to ritonavir). 7 Midazolam is a sensitive CYP3A4 substrate and is commonly used to probe CYP3A induction/inhibition effects, and drospirenone, the progesterone component of oral contraceptives (OCs), is sensitive to potential drug‐drug interactions (DDIs) involving CYP3A4 induction and inhibition. 8 , 9 Ethinyl estradiol, a synthetic estrogen also used in OCs, is primarily metabolized by CYP3A in the liver by 2‐hydroxylation. Considering the CYP3A induction potential of JNJ‐56136379 and as a substrate of CYP3A, interactions may occur if these drugs are given in combination.
The current studies sought to assess the effect of steady‐state itraconazole on the single‐dose pharmacokinetics of JNJ‐56136379 in healthy participants, to evaluate the effect of steady‐state concentrations of JNJ‐56136379 on the single‐dose pharmacokinetics of midazolam and drospirenone/ethinyl estradiol in healthy female participants, and to assess the safety and tolerability of JNJ‐56136379 alone or in combination with itraconazole, midazolam, or drospirenone/ethinyl estradiol.
Methods
Study Designs
Two phase 1, open‐label trials in healthy adults were conducted at the Clinical Pharmacology Unit of Janssen Research & Development in Merksem, Belgium, in accordance with the ethical principles of the Declaration of Helsinki and were consistent with Good Clinical Practice. The protocols were reviewed by an Independent Ethics Committee, Ethisch Comité UZA (Drie Eikenstraat 655, B‐2650 Edegem, Antwerp, Belgium), and participants provided informed written consent.
In the itraconazole study (NCT03945539), participants received either a single oral dose of JNJ‐56136379 250 mg on day 1 followed by a washout of ≥33 days or once‐daily oral itraconazole 200 mg on days 1 to 21 with a single oral dose of JNJ‐56136379 250 mg on day 5 (immediately following the itraconazole dose). Study drugs were administered under fed conditions.
In the midazolam/OC study (NCT03111511), all participants received a single oral dose of drospirenone/ethinyl estradiol 3 mg/0.02 mg with a single oral dose of midazolam 2 mg on day 1 under fasted conditions, twice‐daily oral doses of JNJ‐56136379 250 mg on days 6 and 7 under fed conditions, once‐daily oral JNJ‐56136379 170 mg on days 8–25 under fed conditions (except for day 21), and finally a single oral dose of drospirenone/ethinyl estradiol 3 mg/0.02 mg with a single oral dose of midazolam 2 mg on day 21 under fasted conditions. The dosing regimen of JNJ‐56136379 was implemented to reach steady state early in the dosing interval and to maximize induction potential at this dose.
Participants
The itraconazole study included healthy men and women aged 18–55 years with a body mass index of 18–30 kg/m2, a body weight ≥50 kg, and a normal 12‐lead electrocardiogram (ECG) and who were deemed healthy on the basis of medical history, physical examination, vital signs, and clinical laboratory tests at screening.
The midazolam/OC study included healthy women aged 18–50 years with a body mass index of 18–30 kg/m2, a body weight ≥50 kg, and who were deemed healthy on the basis of medical history, physical examination, normal 12‐lead ECG, vital signs, and clinical laboratory tests conducted at screening. Participants were either of non‐childbearing potential or using acceptable contraception, with a negative highly sensitive serum β‐human chorionic gonadotropin pregnancy test at screening (all participants except for postmenopausal participants) and a negative highly sensitive urine pregnancy test on day –1 (all participants).
Pharmacokinetic Evaluations
Venous blood samples were collected for determination of JNJ‐56136379, itraconazole, midazolam, 1‐OH‐midazolam, 4‐β‐hydroxycholesterol (4‐β‐OH‐cholesterol; a potential biomarker of CYP3A4 induction), ethinyl estradiol, and drospirenone plasma concentrations using validated, specific, and sensitive achiral liquid chromatography–tandem mass spectrometry methods. Separation between potential metabolites and interfering endogenous compounds was achieved by high‐performance liquid chromatography, detection was done on a triple quadrupole mass spectrometer, and quantification was based on multiple reaction monitoring. Sample processing was done by protein precipitation for JNJ‐56136379 with a range of 10.0–1000 ng/mL. Sample processing for midazolam, 4‐β‐OH‐cholesterol, ethinyl estradiol, and drospirenone were done by liquid/liquid extraction with ranges of 0.100–100 ng/mL, 5.00–500 ng/mL, 2.00–800 pg/mL, and 0.500–200 ng/mL, respectively. For itraconazole, sample processing was done by solid‐phase extraction with a range of 20.0–5000 ng/mL. Urine samples were collected for determination of JNJ‐56136379 concentrations in the itraconazole study. Sample processing was done by stabilization (2% of solution 10% Tween 80 in water) and dilution, with concentrations determined using a validated, specific, and sensitive achiral liquid chromatography–tandem mass spectrometry method with a range of 20.0–20,000 ng/mL in stabilized urine. Further details on the methodology used to analyze each analyte can be found in Table S2.
For the itraconazole study, standard pharmacokinetic parameters were derived, including Cmax, time to maximum concentration (tmax), area under the plasma concentration–time curve (AUC) from time 0 to 72 hours, AUC from time 0 to 408 hours, AUC from time 0 to infinity (AUC∞), terminal elimination half‐life (t1/2), total apparent oral clearance (CL/F), renal clearance (urine), and amount excreted in urine from time 0 to 72 hours.
For the midazolam/OC study, standard pharmacokinetic parameters were derived for JNJ‐56136379, including Cmax and tmax measured on days 6, 20, and 21, minimum observed concentration, observed concentration 24 hours after dosing, AUC from time 0 to 24 hours after dosing, average concentration at steady state over the treatment interval, percentage fluctuation, Cmax, tmax, and CL/F measured on days 20 and 21. The standard pharmacokinetic parameters derived for drospirenone, ethinyl estradiol, midazolam, and 1‐OH‐midazolam include Cmax, tmax, AUC from time 0 to the last measurable concentration (AUClast), AUC∞, t1/2, and CL/F (not for 1‐OH‐midazolam) measured on days 1 and 21 and ratio Cmax, metabolite/parent, ratio AUClast, metabolite/parent, ratio AUC∞, metabolite/parent (metabolite/parent = 1‐OH‐midazolam/midazolam).
For both studies, pharmacokinetic analysis was performed using WinNonlin version 6.2.1 (Certara, Princeton, New Jersey).
Safety Evaluations
Adverse events (AEs) were assessed by the investigator for severity and relationship to study medication. Follow‐up was conducted for specific toxicities, such as rash, acute systemic allergic reactions, aspartate aminotransferase/alanine transaminase, clinical hepatitis, renal complications, nausea, and diarrhea using clinical laboratory tests, ECGs, vital signs, and physical examination.
Statistical Methods
All participants who received ≥1 dose of study measurement and had ≥1 pharmacokinetic parameter were included in pharmacokinetic analysis. All participants who received ≥1 dose of study drug and had ≥1 plasma concentration were included in descriptive statistics for plasma concentrations.
For the itraconazole study, assuming a within‐participant coefficient of variation (%CV) of ≈15% for Cmax and a sample size of 14 participants, the point estimate of the geometric mean ratio (GMR) of Cmax of JNJ‐56136379 was calculated to fall within 91%–110% of the true value with 90% confidence. Assuming a %CV of ≈13%, the point estimate of the GMR of AUClast was calculated to fall within 92%–109% of the true value with 90% confidence. To account for potential dropout, 16 participants were enrolled.
For the midazolam/OC study, using an estimated intraparticipant %CV of 22% for all analytes and a sample size of 16 participants, the point estimate of the GMR, with and without coadministration of JNJ‐56136379, was calculated to fall within 88% and 114% of the true value with 90% confidence. To account for potential dropout, 18 participants were enrolled.
Descriptive statistics were used to calculate plasma concentrations and pharmacokinetic parameters. Comparisons of interest were performed for differences in geometric mean values and 90%CI of Cmax and AUC.
Results
Participants
Sixteen participants were enrolled in the itraconazole study, and 13 (81%) completed the study. Of the 3 participants who discontinued, 1 withdrew for personal reasons, 1 due to increased amylase levels (grade 1), and 1 due to a serious AE (SAE) of pneumothorax that was not related to the study drugs. Participants were primarily women (75%) and White (94%), with a mean age of 38 years and a mean weight of 70 kg (Table 1).
Table 1.
Summary of Participant Demographics and Baseline Characteristics
| Itraconazole Study | Midazolam/OC Study | |
|---|---|---|
| Parameter | (n = 16) | (n = 18) |
| Age, y, mean (SD) | 37.7 (10.4) | 38.9 (8.8) |
| Women, n (%) | 12 (75.0) | 18 (100) |
| Race, n (%) | ||
| White | 15 (93.8) | 16 (88.9) |
| Black or African American | 0 | 2 (11.1) |
| Multiple | 1 (6.3) | 0 |
| Ethnicity, n (%) | ||
| Hispanic/Latino | 0 | 1 (5.6) |
| Not Hispanic/Latino | 16 (100) | 17 (94.4) |
| Body weight, kg, mean (SD) | 70.4 (8.8) | 66.0 (9.0) |
| BMI, kg/m2, mean (SD) | 24.5 (2.6) | 23.6 (3.3) |
BMI, body mass index; OC, oral contraceptive; SD, standard deviation.
Eighteen participants were enrolled in the midazolam/OC study, and 16 (89%) completed the study. Of the 2 participants who discontinued, 1 withdrew consent and 1 was lost to follow‐up. All the participants were women and most were White (89%), with a mean age of 39 years and a mean weight of 66 kg (Table 1).
Pharmacokinetic Results
Itraconazole Study
The plasma concentration–time curves for JNJ‐56136379 with and without itraconazole are illustrated in Figure 1, and the pharmacokinetic parameters are summarized in Table 2. After a single dose of JNJ‐56136379 with or without itraconazole, JNJ‐56136379 Cmax was reached 4 hours after dosing followed by a multiphase decrease with an initial rapid decline and a subsequent slower decline. Mean JNJ‐56136379 plasma concentrations were slightly higher in the presence of itraconazole. AUC from time 0 to 408 hours was 38% higher when a single dose of JNJ‐56136379 was coadministered with itraconazole versus alone (GMR, 138.2; 90%CI, 129.6–147.3); however, there was no significant effect on Cmax (GMR, 111.1; 90%CI, 100.2–123.2) or AUC from time 0 to 72 hours (GMR, 109.5; 90%CI, 103.8–115.6). Mean t1/2 was longer when JNJ‐56136379 was administered in the presence of itraconazole versus alone (180 vs 163 hours; Table 2). The apparent clearance and volume of distribution of JNJ‐56136379 were 45% and 28% lower, respectively, after coadministration with itraconazole, whereas the renal clearance was slightly higher (1.15‐fold) in the presence of itraconazole compared to JNJ‐56136379 alone (Table 2). Mean itraconazole predose plasma concentrations increased before coadministration with JNJ‐56136379 and continued to increase after coadministration of JNJ‐56136379 on day 5 (Figure S1).
Figure 1.
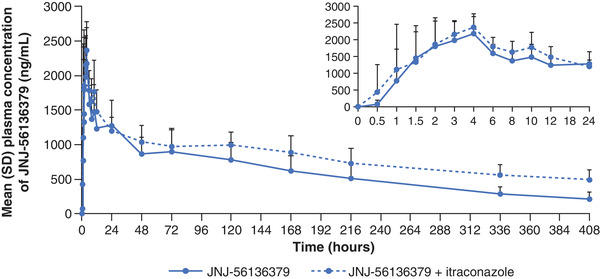
Mean (SD) plasma concentration–time profile of JNJ‐56136379 after a single dose administered alone or in combination with multiple‐dose itraconazole. SD, standard deviation.
Table 2.
Pharmacokinetic Parameters of JNJ‐56136379 After a Single Dose Administered Alone or in Combination With Multiple‐Dose Itraconazole
| JNJ‐56136379 Alone | JNJ‐56136379 + Itraconazole | |
|---|---|---|
| Pharmacokinetic Parametera | (n = 16) | (n = 14) |
| Cmax, ng/mL | 2357 (647) | 2620 (752) |
| tmax, h | 4.00 (1.00–4.02) | 3.50 (1.00–6.00) |
| AUC0–72h, ng · h/mL | 79,926 (20,538) | 87,448 (16,391) |
| AUC0–408h, ng · h/mL | 247,730 (74,999) | 335,245 (65,421) |
| AUC∞, ng · h/mL | 281,768 (103,768) b | 488,626 (112,732) b |
| t1/2, h | 162.7 (58.9) | 180.2 (33.6) |
| CL/F, L/h | 0.983 (0.304) b | 0.541 (0.144) b |
| CLR, L/h | 0.155 (0.0485) c | 0.179 (0.0520) d |
| Ae0–72h, % dose | 4.74 (1.94) c | 6.19 (2.09) d |
Ae0–72h, amount excreted in urine from time 0 to 72 hours; AUC0–72h, area under the plasma concentration–time curve from time 0 to 72 hours; AUC0–408h, area under the plasma concentration–time curve from time 0 to 408 hours; AUC∞, area under the plasma concentration–time curve from time 0 to infinity; CL/F, total apparent oral clearance; CLR, renal clearance; Cmax, maximum observed concentration; SD, standard deviation; t1/2, terminal elimination half‐life; tmax, time to maximum concentration.
Data are mean (SD) except for tmax, which is median (range).
n = 12.
n = 15.
n = 13.
Midazolam/OC Study
The plasma concentration–time curves for midazolam, drospirenone, and ethinyl estradiol are illustrated in Figure 2, and the pharmacokinetic parameters are summarized in Table 3. The mean plasma concentration profile of midazolam was lower when midazolam was coadministered with JNJ‐56136379 (day 21) versus alone (day 1; Figure 2A). When given in combination with JNJ‐56136379, Cmax, AUClast, and AUC∞ for midazolam were 42% (GMR, 58.4; 90%CI, 52.2–65.2), 53% (GMR, 47.4; 90%CI, 43.1–52.2), and 54% (GMR, 45.9; 90%CI, 41.2–51.2) lower, respectively. Mean apparent clearance of midazolam was higher in the presence of JNJ‐56136379 versus alone (239 vs 103 L/h), and mean t1/2 of midazolam was 4.3 hours when given alone and 3.2 hours when coadministered with JNJ‐56136379 (Table 3).
Figure 2.
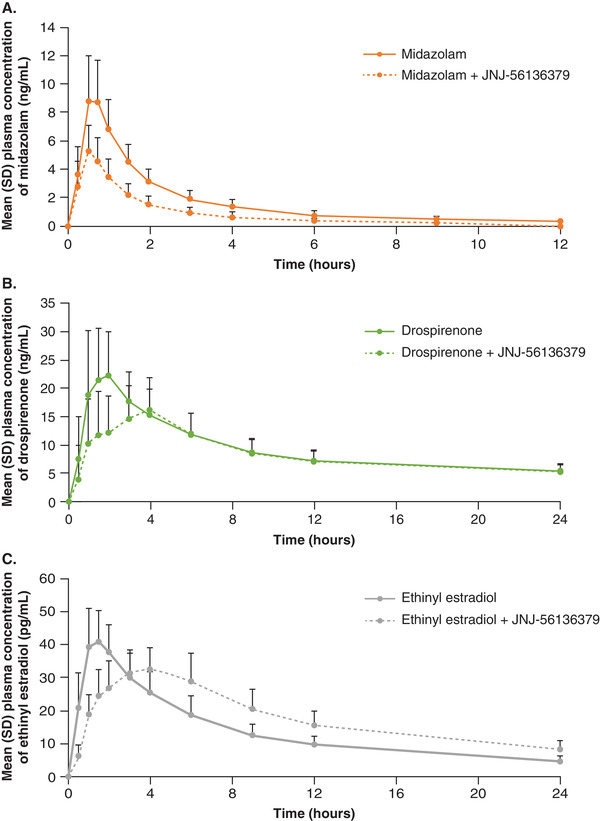
Mean (SD) plasma concentration–time profiles of (A) midazolam, (B) drospirenone, and (C) ethinyl estradiol after a single dose of midazolam/OC administered alone (day 1) or in combination with multiple‐dose JNJ‐56136379 (day 21). OC, oral contraceptive; SD, standard deviation.
Table 3.
Pharmacokinetic Parameters of Midazolam, Drospirenone, and Ethinyl Estradiol After a Single Dose Administered Alone or in Combination With Multiple‐Dose JNJ‐56136379
| Midazolam | 1‐OH‐Midazolam | Drospirenone | Ethinyl Estradiol | |||||
|---|---|---|---|---|---|---|---|---|
| Midazolam Alone Day 1 | Midazolam + JNJ‐56136379 Day 21 | Midazolam Alone Day 1 | Midazolam + JNJ‐56136379 Day 21 | OC Alone Day 1 | OC + JNJ‐56136379 Day 21 | OC Alone Day 1 | OC + JNJ‐56136379 Day 21 | |
| (n = 18) | (n = 17) | (n = 18) | (n = 17) | (n = 18) | (n = 16) | (n = 18) | (n = 16) | |
| Pharmacokinetic Parameter a | ||||||||
| Cmax, ng/mL (pg/mL for ethinyl estradiol) | 9.46 (3.08) | 5.55 (1.85) | 3.56 (1.01) | 3.51 (1.41) | 27.9 (6.99) | 20.2 (6.52) | 43.7 (9.94) | 35.6 (7.61) |
| tmax, h | 0.50 (0.50–1.00) | 0.50 (0.25–0.75) | 0.75 (0.50–1.00) | 0.50 (0.25–0.75) | 1.53 (0.50–6.00) | 3.03 (1.02–4.00) | 1.50 (0.50–2.02) | 3.50 (1.50–6.02) |
| AUC∞, ng · h/mL (pg · h/mL for ethinyl estradiol) | 20.5 (4.91) b | 9.37 (3.01) c | 6.53 (1.65) d | 6.37 (3.33) | 464 (85.7) c | 447 (83.7) | 407 (100) e | 647 (165) b |
| t1/2, h | 4.3 (1.4) f | 3.2 (2.3) | 1.7 (0.4) d | 1.9 (1.7) | 28.6 (7.4) g | 30.7 (10.1) | 11.7 (2.3) c | 15.8 (3.9) |
| CL/F, L/h | 103 (26.4) b | 239 (89.1) c | … | … | 6.70 (1.36) c | 6.97 (1.54) | 51.7 (11.4) e | 32.5 (6.93) b |
AUC∞, area under the plasma concentration–time curve from time 0 to infinity; CL/F, total apparent oral clearance; Cmax, maximum observed concentration; OC, oral contraceptive; SD, standard deviation; t1/2, terminal elimination half‐life; tmax, time to maximum concentration.
Data are mean (SD) except for tmax, which is median (range).
n = 14.
n = 16.
n = 13.
n = 12.
n = 15.
n = 17.
Cmax (GMR, 99.2; 90%CI, 83.9–117.3), AUClast (GMR, 89.7; 90%CI, 79.3–101.5), and AUC∞ (GMR, 92.8; 90%CI, 79.5–108.3) for 1‐OH‐midazolam were not significantly changed when it was given in combination with JNJ‐56136379. Metabolite to parent ratios, 1‐OH‐midazolam/midazolam, of Cmax, AUClast, and AUC∞ were 1.7‐fold (GMR, 170.8; 90%CI, 153.3–190.4), 1.9‐fold (GMR, 188.7; 90%CI, 166.9–213.3), and 1.9‐fold (GMR, 187.2; 90%CI, 160.7–218.0) higher in the presence of JNJ‐56136379.
Mean plasma concentration profile of drospirenone was lower, and the peak occurred later after coadministration with multiple‐dose JNJ‐56136379 (day 21) versus alone (day 1; Figure 2B). Cmax was 29% (GMR, 71.2; 90%CI, 59.5–85.3) lower, but AUClast (GMR, 93.7; 90%CI, 87.5–100.4) and AUC∞ (GMR, 95.5; 90%CI, 88.7–102.8) were unchanged when given in combination with JNJ‐56136379. Mean apparent clearance of drospirenone was similar after a single dose alone (6.70 L/h) or in the presence of JNJ‐56136379 (6.97 L/h; Table 3). Mean t1/2 of drospirenone was also similar when given alone (28.6 hours) and in the presence of JNJ‐56136379 (30.7 hours; Table 3).
Mean plasma concentrations of ethinyl estradiol were lower and peak concentrations occurred later when it was coadministered with JNJ‐56136379 (day 21) versus alone (day 1; Figure 2C). When given in combination with JNJ‐56136379, Cmax for ethinyl estradiol was 16% (GMR, 83.9; 90%CI, 78.2–89.9) lower while AUClast (GMR, 162.5; 90%CI, 146.3–180.4) and AUC∞ (GMR, 158.9; 90%CI, 136.2–185.5) were ≈60% higher than when ethinyl estradiol was given alone. Mean apparent clearance of ethinyl estradiol was lower in the presence of JNJ‐56136379 versus alone (32.5 vs 51.7 L/h), and mean t1/2 was 11.7 hours when ethinyl estradiol was given alone and 15.8 hours when coadministered with JNJ‐56136379 (Table 3).
The pharmacokinetic parameters of JNJ‐56136379 are summarized in Table 4. The mean Cmax for JNJ‐56136379 increased between day 6 (2155 ng/mL) and day 20 (8032 ng/mL). The mean AUC from time 0 to 24 hours after dosing for JNJ‐56136379 also increased between day 6 (19,371 ng · h/mL) and day 20 (160,904 ng · h/mL) and was similar on days 20 and 21 (161,176 ng · h/mL), indicating that steady state was reached before day 20.
Table 4.
Pharmacokinetic Parameters of JNJ‐56136379 in the Midazolam/OC Study
| JNJ‐56136379 250 mg Day 6 | JNJ‐56136379 170 mg Day 20 | JNJ‐56136379 170 mg + OC and Midazolam Day 21 | |
|---|---|---|---|
| (n = 17) | (n = 17) | (n = 17) | |
| Pharmacokinetic Parameter a | |||
| Cmin, ng/mL | … | 5715 (1497) | 5451 (1480) |
| Cmax, ng/mL | 2155 (649) | 8032 (2168) | 8798 (2753) |
| tmax, h | 4.00 (2.00–11.92) | 4.00 (4.00–23.77) | 4.00 (1.00–4.05) |
| C24h, ng/mL | … | 6256 (1808) | 6879 (1700) |
| AUC24h, ng · h/mL | 19,371 (5897) | 160,904 (41,024) | 161,176 (42,732) |
| Cavg, ng/mL | … | 6762 (1728) | 6733 (1790) |
| FI, % | … | 34.1 (15.9) | 48.7 (17.3) |
| CL/F, L/h | … | 1.12 (0.283) | 1.13 (0.308) |
AUC24h, area under the plasma concentration–time curve from time 0 to 24 hours after dosing; C24h, observed concentration 24 hours after dosing; Cavg, average concentration at steady state over the treatment interval; CL/F, total apparent clearance; Cmax, maximum observed concentration; Cmin, minimum observed concentration; FI, percentage fluctuation; OC, oral contraceptive; SD, standard deviation; tmax, time to reach maximum concentration.
Data are mean (SD) except for tmax, which is median (range).
Increases in 4‐β‐OH‐cholesterol levels were observed following repeated administration of JNJ‐56136379 170 mg, with mean (standard deviation) of 40.0 (13.4) ng/mL at day 20 compared with 29.4 (11.3) ng/mL at baseline. The baseline‐corrected increase in 4‐β‐OH‐cholesterol level on day 20 was similar over the measured JNJ‐56136379 plasma concentration range, with no apparent relation between JNJ‐56136379 concentration just before the start of a dosing interval and the increase in 4‐β‐OH‐cholesterol.
Safety Results
Itraconazole Study
All AEs that were observed in >1 participant in the itraconazole study are shown in Table 5. There were no deaths or treatment‐emergent AEs with grade 3 or 4 severity. One participant terminated the study prematurely due to a grade 2 SAE of pneumothorax not considered to be related to either itraconazole or JNJ‐56136379, and 1 participant prematurely discontinued after increased amylase levels were observed (grade 1). The decision to discontinue this participant from study participation was made as a precautionary measure. They had received 1 dose of JNJ‐56136379 250 mg followed by 4 doses of itraconazole 200 mg. More AEs were experienced by participants during the first part of the study when JNJ‐56136379 was given alone. The most frequently reported AEs after treatment with JNJ‐56136379 alone were headaches (31%), vertigo (13%), abdominal discomfort (13%), and back pain (13%). One participant had a treatment‐emergent AE of hypercholesterolemia that was considered possibly related to itraconazole.
Table 5.
Summary of AEs for JNJ‐56136379 and Itraconazole Alone or Combined
| JNJ‐56136379 | Itraconazole | Itraconazole + JNJ‐56136379 | |
|---|---|---|---|
| Participants, n (%) | (n = 16) | (n = 15) | (n = 14) |
| With ≥1 AE | 10 (62.5) | 4 (26.7) | 8 (57.1) |
| With ≥1 SAE | 0 (0) | 0 (0) | 1 (7.1)a |
| TEAEs seen in >1 participant | |||
| Headache | 5 (31.3) | 2 (13.3) | 3 (21.4) |
| Abdominal discomfort | 2 (12.5) | 1 (6.7) | 0 (0) |
| Abdominal distension | 1 (6.3) | 1 (6.7) | 0 (0) |
| Back pain | 2 (12.5) | 0 (0) | 0 (0) |
| Diarrhea | 1 (6.3) | 0 (0) | 1 (7.1) |
| Hypercholesterolemia | 1 (6.3) | 0 (0) | 1 (7.1) |
| Myalgia | 1 (6.3) | 0 (0) | 1 (7.1) |
| Nasopharyngitis | 1 (6.3) | 0 (0) | 1 (7.1) |
| Vertigo | 2 (12.5) | 0 (0) | 0 (0) |
AE, adverse event; SAE, serious adverse event; TEAE, treatment‐emergent adverse event.
aSAE of pneumothorax that led to premature study discontinuation.
Midazolam/OC Study
Table 6 summarizes all AEs that were observed in >1 participant in the midazolam/OC study. There were no deaths; SAEs; AEs leading to early discontinuation of the study; or AEs of moderate, severe, or life‐threatening severity. AEs occurred most frequently during treatment with JNJ‐56136379 alone. The most commonly reported AEs after any treatment in the study were fatigue and headache, which were reported by 47.1% and 29.4% of participants, respectively, during treatment with JNJ‐56136379 alone. Five participants reported ≥1 rash occurring during the study, all of which were of mild severity. The investigator considered 1 participant's event of maculopapular rash to be possibly related to JNJ‐56136379 but not likely related to other study treatments.
Table 6.
Summary of AEs for JNJ‐56136379 and Midazolam + OC Alone or Combined
| Midazolam + OC | JNJ‐56136379 | Midazolam + OC + JNJ‐56136379 | |
|---|---|---|---|
| Participants, n (%) | (n = 18) | (n = 17) | (n = 17) |
| With ≥1 AE | 12 (66.7) | 15 (88.2) | 9 (52.9) |
| With ≥1 SAE | 0 (0) | 0 (0) | 0 (0) |
| TEAEs seen in >1 participant | |||
| Fatigue | 6 (33.3) | 8 (47.1) | 0 (0) |
| Headache | 4 (22.2) | 5 (29.4) | 0 (0) |
| Nausea | 1 (5.6) | 3 (17.6) | 1 (5.9) |
| Abdominal pain | 1 (5.6) | 3 (17.6) | 0 (0) |
| Abdominal distension | 1 (5.6) | 2 (11.8) | 0 (0) |
| Acne | 1 (5.6) | 2 (11.8) | 0 (0) |
| Neck pain | 0 (0) | 3 (17.6) | 0 (0) |
| Palpitations | 0 (0) | 3 (17.6) | 0 (0) |
| Rash, maculopapular | 0 (0) | 2 (11.8) | 1 (5.9) |
| Abdominal pain, upper | 0 (0) | 1 (5.9) | 1 (5.9) |
| Application‐site erythema | 1 (5.6) | 1 (5.9) | 0 (0) |
| Constipation | 1 (5.6) | 1 (5.9) | 0 (0) |
| Decreased appetite | 1 (5.6) | 1 (5.9) | 0 (0) |
| Pain in extremity | 1 (5.6) | 1 (5.9) | 0 (0) |
| Pruritus | 0 (0) | 2 (11.8) | 0 (0) |
| Rash, erythematous | 0 (0) | 1 (5.9) | 1 (5.9) |
| Somnolence | 0 (0) | 2 (11.8) | 0 (0) |
| Vaginal discharge | 0 (0) | 2 (11.8) | 0 (0) |
| Vaginal hemorrhage | 0 (0) | 2 (11.8) | 0 (0) |
| Vomiting | 0 (0) | 1 (5.9) | 1 (5.9) |
AE, adverse event; OC, oral contraceptive; SAE, serious adverse event; TEAE, treatment‐emergent adverse event.
Discussion
The current studies were designed to evaluate the DDI potential of itraconazole, a BCRP, CYP3A, and P‐glycoprotein inhibitor, on the pharmacokinetics of JNJ‐56136379 and the effect of JNJ‐56136379 on agents that are metabolized by CYP3A, including midazolam, drospirenone, and ethinyl estradiol. Itraconazole and midazolam were selected in the DDI analyses because these are drugs that are mentioned in the US Food and Drug Administration guidance as examples of perpetrator and substrate drugs, respectively, for DDI studies, there is a large body of information regarding their effects on metabolic pathways, and they have acceptable safety profiles.
When given in combination, itraconazole had a modest effect on JNJ‐56136379 pharmacokinetics. The extent of JNJ‐56136379 exposure (AUC) was 38% higher when coadministered with itraconazole, but Cmax was not significantly affected. Renal clearance of JNJ‐56136379 and the amount of JNJ‐56136379 excreted in urine over 72 hours after dosing were both higher (1.15‐ and 1.30‐fold, respectively) when JNJ‐56136379 was administered in the presence of itraconazole. This observation suggests that itraconazole promotes a shift in the clearance of JNJ‐56136379 from metabolic pathways to renal elimination. Importantly, this study could provide guidance for participants who receive coadministration of JNJ‐56136379 and drugs that inhibit JNJ‐56136379 disposition pathways, such as CYP3A4, P‐glycoprotein, and BCRP.
When JNJ‐56136379 was coadministered with midazolam, the rate (as assessed by Cmax) and extent (as assessed by AUC) of exposure to midazolam were decreased by 42% and 54%, respectively. The increase in mean 4‐β‐OH‐cholesterol level was observed after repeated dosing of JNJ‐56136379 and confirmed the potential for CYP3A4 induction, as has been observed in nonclinical experiments. 10 , 11 The changes in exposure to midazolam appeared to be more consistent, confirming its value as an exogenous marker of CYP3A4 induction.
The effects of JNJ‐56136379 on drospirenone were less pronounced. The Cmax of drospirenone was reduced by 29%, but AUC parameters were not significantly affected, suggesting no clinically significant effect of JNJ‐56136379 on drospirenone. As progestin mainly contributes to the efficacy of human chorionic gonadotropin, and because drospirenone is more sensitive to CYP3A induction, it was concluded that there was no interaction in terms of efficacy when JNJ‐56136379 is coadministered with a combined OC.
Exposure of ethinyl estradiol increased by ≈1.6‐fold as a result of coadministration with JNJ‐56136379, but there was little effect on Cmax. As the risk of events of venous thromboembolism or cardiovascular disease is the highest in the first 3 months of combined OC use, 12 participants should be stable for 3 months on combination OCs with ethinyl estradiol 20 μg before the study or start with a progestin‐containing OC, if OCs become necessary during the study. Additionally, as increased exposure to ethinyl estradiol has been associated with increased cardiovascular complications, 13 coadministration of high‐dose (>20 μg) ethinyl estradiol and JNJ‐56136379 should be avoided. Regarding mechanisms that may have contributed to this increase in ethinyl estradiol when it was combined with JNJ‐56136379, inhibition of glucuronidation or sulfation pathways by JNJ‐56136379 was explored but could not be confirmed. Similarly, the role of a main excipient was explored for potentially increasing absorption of ethinyl estradiol; however, comparing across other drug products did not reveal a consistent pattern in relation to the increase in ethinyl estradiol when coadministered with JNJ‐56136379.
Based on the results from the midazolam/OC study, JNJ‐56136379 can be classified as a mild to moderate inducer of CYP3A4 and, as such, JNJ‐56136379 may enhance the metabolism of drugs that are metabolized via CYP3A, leading to a potential for decreased therapeutic effect of the victim drugs.
Overall, treatment regimens in both studies were generally well tolerated with no unexpected safety concerns given the relatively short duration of exposure. Thus, these studies suggest that JNJ‐56136379 can be safely used in combination with drugs that may interact with JNJ‐56136379 disposition pathways, at least over a short time frame, though some interaction effects may occur due to the mild to moderate CYP3A4 induction by JNJ‐56136379. Importantly, coadministration of high‐dose (>20 μg) ethinyl estradiol and JNJ‐56136379 should be avoided.
Financial Interest Disclosures
Joris Vandenbossche, Vera Hillewaert, Freya Rasschaert, Willem Talloen, Jeike Biewenga, Jan Snoeys, Thomas N. Kakuda, Martyn Palmer, and Michael Biermer are employees of Janssen Pharmaceuticals and may be Johnson & Johnson stockholders. Jeysen Yogaratnam is a former employee of Janssen Pharmaceuticals. Julius Nangosyah is a consultant at Janssen Pharmaceuticals.
Supporting information
Supporting Information
Acknowledgments
This study was sponsored by Janssen Research & Development, with medical writing support provided by Kim Caldwell, PhD, of Cello Health Communications/MedErgy, and funded by Janssen.
References
- 1. Verbinnen T, Hodari M, Talloen W, et al. Virology analysis of chronic hepatitis B virus–infected patients treated for 28 days with JNJ‐56136379 monotherapy. J Viral Hepat. 2020;27(11):1127‐1137. [DOI] [PubMed] [Google Scholar]
- 2. Verbinnen T, Tan Y, Wang G, et al. Anti‐HBV activity of the HBV capsid assembly modulator JNJ‐56136379 across full‐length genotype A‐H clinical isolates and core site‐directed mutants in vitro. J Antimicrob Chemother. 2020;75(9):2526‐2534. [DOI] [PubMed] [Google Scholar]
- 3. Berke JM, Dehertogh P, Vergauwen K, et al. Antiviral properties and mechanism of action studies of the hepatitis B virus capsid assembly modulator JNJ‐56136379. Antimicrob Agents Chemother. 2020;64(5):e02439‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vandenbossche J, Jessner W, van den Boer M, et al. Pharmacokinetics, safety and tolerability of JNJ‐56136379, a novel hepatitis B virus capsid assembly modulator, in healthy subjects. Adv Ther. 2019;36(9):2450‐2462. [DOI] [PubMed] [Google Scholar]
- 5. Zoulim F, Lenz O, Vandenbossche JJ, et al. JNJ‐56136379, an HBV capsid assembly modulator, is well‐tolerated and has antiviral activity in a phase 1 study of patients with chronic infection. Gastroenterology. 2020;159(2):521‐533.e9. [DOI] [PubMed] [Google Scholar]
- 6. Janssen HLA, Hou J, Asselah T, et al. Efficacy and safety results of the phase 2 JNJ‐56136379 JADE study in patients with chronic hepatitis B: interim week 24 data. Presented at: EASL, The Digital International Liver Congress; 27–29 August 2020. Poster LBP‐012.
- 7. Liu L, Bello A, Dresser MJ, et al. Best practices for the use of itraconazole as a replacement for ketoconazole in drug‐drug interaction studies. J Clin Pharmacol. 2016;56(2):143‐151. [DOI] [PubMed] [Google Scholar]
- 8. Ayala RC, Arya V, Younis IR. Design features of drug‐drug interaction trials between antivirals and oral contraceptives. J Clin Pharmacol. 2016;56(5):541‐547. [DOI] [PubMed] [Google Scholar]
- 9. Zhang N, Shon J, Kim MJ, et al. Role of CYP3A in oral contraceptives clearance. Clin Transl Sci. 2018;11(3):251‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mangold JB, Wu F, Rebello S. Compelling relationship of CYP3A induction to levels of the putative biomarker 4β‐hydroxycholesterol and changes in midazolam exposure. Clin Pharmacol Drug Dev. 2016;5(4):245‐249. [DOI] [PubMed] [Google Scholar]
- 11. Penzak SR, Rojas‐Fernandez C. 4β‐hydroxycholesterol as an endogenous biomarker for CYP3A activity: literature review and critical evaluation. J Clin Pharmacol. 2019;59(5):611‐624. [DOI] [PubMed] [Google Scholar]
- 12. van Hylckama Vlieg A, Helmerhorst FM, Vandenbroucke JP, Doggen CJ, Rosendaal FR. The venous thrombotic risk of oral contraceptives, effects of oestrogen dose and progestogen type: results of the MEGA case‐control study. BMJ. 2009;339:b2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stanczyk FZ, Archer DF, Bhavnani BR. Ethinyl estradiol and 17β‐estradiol in combined oral contraceptives: pharmacokinetics, pharmacodynamics and risk assessment. Contraception. 2013;87(6):706‐727. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information