

Abstract
Cellobiohydrolases (CBHs) in the glycoside hydrolase family 7 (GH7) (EC3.2.1.176) are the major cellulose degrading enzymes both in industrial settings and in the context of carbon cycling in nature. Small carbohydrate conjugates such as p‐nitrophenyl‐β‐d‐cellobioside (pNPC), p‐nitrophenyl‐β‐d‐lactoside (pNPL) and methylumbelliferyl‐β‐d‐cellobioside have commonly been used in colorimetric and fluorometric assays for analysing activity of these enzymes. Despite the similar nature of these compounds the kinetics of their enzymatic hydrolysis vary greatly between the different compounds as well as among different enzymes within the GH7 family. Through enzyme kinetics, crystallographic structure determination, molecular dynamics simulations, and fluorometric binding studies using the closely related compound o‐nitrophenyl‐β‐d‐cellobioside (oNPC), in this work we examine the different hydrolysis characteristics of these compounds on two model enzymes of this class, TrCel7A from Trichoderma reesei and PcCel7D from Phanerochaete chrysosporium. Protein crystal structures of the E212Q mutant of TrCel7A with pNPC and pNPL, and the wildtype TrCel7A with oNPC, reveal that non‐productive binding at the product site is the dominating binding mode for these compounds. Enzyme kinetics results suggest the strength of non‐productive binding is a key determinant for the activity characteristics on these substrates, with PcCel7D consistently showing higher turnover rates (k cat) than TrCel7A, but higher Michaelis–Menten (K M) constants as well. Furthermore, oNPC turned out to be useful as an active‐site probe for fluorometric determination of the dissociation constant for cellobiose on TrCel7A but could not be utilized for the same purpose on PcCel7D, likely due to strong binding to an unknown site outside the active site.
Keywords: Cel7, cellulase, fluorescence, ligand binding, Phanerochaete chrysosporium, Trichoderma reesei
Glycoside hydrolase family 7 (GH7) cellobiohydrolases, the major workhorses in cellulose degradation in both nature and industry, are commonly studied using small soluble model substrates, yet the results are often not representative of activity on natural cellulose. Despite their similar nature, the kinetics of enzymatic hydrolysis varies greatly between the different compounds, as well as among different enzymes within the GH7 family. Through enzyme kinetics, X‐ray crystallography, molecular dynamics simulations, and fluorometric binding studies using the closely related compound o‐nitrophenyl‐β‐d‐cellobioside (oNPC), Haataja et al. examined the different hydrolysis characteristics of these compounds on two model enzymes of this class, TrCel7A and PcCel7D.
Abbreviations
- CBH
cellobiohydrolase
- GH7
glycoside hydrolase family 7
- MD
molecular dynamics
- MUC
methylumbelliferyl‐β‐d‐cellobioside
- oNPC
ortho−/2‐nitrophenyl‐β‐d‐cellobioside
- pNP
para−/4‐nitrophenol
- PcCel7D
Phanerochaete chrysosporium Cel7D
- pNPC
para−/4‐nitrophenyl‐β‐d‐cellobioside
- pNPL
para−/4‐nitrophenyl‐β‐d‐lactoside
- TrCel7A
Trichoderma reesei Cel7A
Introduction
Cellobiohydrolases catalyse the hydrolysis of polymeric cellulose and cellooligosaccharides into cellobiose. They are the major workhorses in cellulose degradation in nature, and therefore play a critical role in the carbon cycle. The most abundant cellulases produced by cellulose degrading fungi are usually cellobiohydrolases from the family 7 of glycoside hydrolases (GH7) (EC3.2.1.176). Due to their effectiveness in cellulose degradation, the GH7 enzymes have been extensively studied and engineered to better understand their action mechanism and to utilize them in various biotechnical applications [1].
The GH7 cellobiohydrolases act on cellulose chains processively from the reducing towards the non‐reducing end, with a cellulose chain threading into an active site tunnel containing 9–10 glucose unit binding subsites [1, 2, 3, 4]. The processive action is dependent on strong affinity to the product binding sites +1/+2 [5, 6, 7]. Consequently, the hydrolysis product, cellobiose, binds to this site with high affinity and is a strong inhibitor for these enzymes, with cellobiose accumulation leading to undesirable product inhibition [8, 9].
p‐ and o‐nitrophenyl glycosides are widely used artificial chromogenic substrates for kinetic studies of GH7 enzymes [10]. These substrates do in principle allow real‐time‐monitoring, but the sensitivity is greatly enhanced if a non‐continuous assay with alkalification is employed. The inhibition of nitrophenyl glycoside hydrolysis is often employed to measure equilibrium binding of nondegradable ligands. A yet more sensitive detection of glycosidase activity is achieved by the use of fluorogenic substrates such as methylumbelliferyl glycosides [8, 10, 11]. However, the use of low molecular weight model substrates for polymer degrading enzymes with long arrays of substrate binding subsites is often complicated, since the model substrates may occupy, or even prefer non‐productive positions, sometimes to such an extent that the non‐productive binding modes are dominating, meaning that these substances can be characterized as reversibly binding inhibitors [12, 13]. This leads to kinetic behaviour where both the apparent turn‐over of the hydrolysis (k cat) and binding affinity of the enzyme substrate complex (K M) are affected, with stronger non‐productive binding leading to reduction in both values. Thus the true catalytic turnover of the productive enzyme‐substrate complex cannot be determined without the dissociation constant of the non‐productively bound enzyme substrate complex [14].
Besides activity measurements, ligands with fluorogenic properties have in many cases been used as a probe for displacement titration to study biologically significant binding of non‐fluorescent molecules to proteins [15, 16]. For carbohydrate degrading enzymes, the changes in the fluorescence of methylumbelliferyl glycosides, when binding to the protein, have been used as active site probes [17, 18, 19, 20, 21]. As an alternative approach, the change of intrinsic protein fluorescence resulting from ligand binding may be monitored. Tryptophan fluorescence is sensitive to its environment and may thus be affected by ligand binding, either by contact effects or, alternatively, by means of radiationless energy transfer if the absorption spectrum of the ligand overlaps favourably with the tryptophan emission range. Indeed, substrate‐induced changes in the native fluorescence of proteins have been utilized previously for ligand binding studies [22, 23, 24, 25].
TrCel7A from the ascomycete fungus Trichoderma reesei and PcCel7D from the basidiomycete Phanerochaete chrysosporium are two model enzymes from the GH7 enzyme family. Both enzymes contain seven substrate binding subsites (−7 to −1) and three product binding subsites (+1 to +3), a conserved feature in GH7 cellobiohydrolases. Of the known GH7 cellobiohydrolases PcCel7D stands out with its open active site tunnel architecture, with significantly shorter loops in the so called A1, B2 and B3 loop regions compared to TrCel7A [26, 27, 28]. In studies conducted on the processivity and inhibition characteristics of these enzymes PcCel7D has shown weaker inhibition by cellobiose compared to TrCel7A, as well as more frequent dissociation from a cellulose chain, leading to slightly shorter processive runs but also higher overall hydrolysis rates with less enzyme bound unproductively in difficult‐to‐hydrolyse cellulose regions [13, 29]. Both attributes are likely explained by the more open active site tunnel structure.
In this work we explore the binding dynamics of common model compounds p‐nitrophenyl‐β‐d‐cellobioside (pNPC), p‐nitrophenyl‐β‐d‐lactoside (pNPL) and methylumbelliferyl‐β‐d‐cellobioside (MUC) to the active sites of TrCel7A and PcCel7D. We use enzyme kinetics measurements, X‐ray crystal structures and molecular dynamics (MD) simulations to explore the factors governing the catalytic activity of these enzymes on these model substrates by studying productive and non‐productive substrate binding, and to shed light into possible reasons for differences in the kinetics and inhibition behaviour. Furthermore, we explore the usefulness of o‐nitrophenyl‐β‐d‐cellobioside (oNPC) as an active site probe for these cellobiohydrolases.
Results
Enzyme kinetics
To compare the hydrolysis characteristics of the TrCel7A and PcCel7D cellobiohydrolases, enzyme kinetic measurements were performed with oNPC, pNPC, pNPL and MUC as substrates (Fig. 1A). The determined catalytic rate (k cat) and Michaelis–Menten (K M) constants are shown in Table 1, as well as k cat/K M, which is a measure of the catalytic efficiency of the enzyme with the substrate.
Fig. 1.

Substrates and fluorescence titrations. (A) Enzyme kinetics experiments were performed with pNPL, pNPC, oNPC and MUC as substrates. (B) Absorbance spectrum of oNPC (blue) and fluorescence spectrum of TrCel7A wildtype at an excitation wavelength of 295 nm (red). (C) Fluorescence titration of TrCel7A and PcCel7D with oNPC. The concentration of TrCel7A was 0.035 μm (□); 0.1 μm (●) or 0.35 μm (○) and the concentration of PcCel7D was 0.5 μm (■). (D) Displacement titration of TrCel7A with cellobiose at different concentrations of oNPC. The concentration of oNPC was 5 μm (○); 10 μm (●), 20 μm (□) or 40 μm (■).
Table 1.
Comparison of kinetic constants of TrCel7A and PcCel7D on different chromogenic substrates. Determined at 25 °C in 50 mm sodium acetate buffer, pH 5.0.
| Enzyme | Substrate | k cat (s−1) | K M (μm) | k cat/K M (s−1*m −1) |
|---|---|---|---|---|
| TrCel7A | oNPC | 66 × 10−6 ± 15 × 10−6 | 7.0 ± 4.5 | 9.5 |
| pNPC | 0.0026 ± 0.0001 | 26 ± 3 | 100 | |
| pNPL | 0.087 ± 0.002 | 590 ± 20 | 147 | |
| MUC | 0.013 ± 0.001 | 12 ± 1 | 1083 | |
| Ratio, TrCel7A | pNPC/oNPC | 39 | 3.7 | 11 |
| pNPL/pNPC | 33 | 23 | 1.5 | |
| MUC/pNPC | 5.0 | 0.46 | 11 | |
| PcCel7D | oNPC | 0.015 ± 0.002 | 3200 ± 100 | 4.6 |
| pNPC | 0.046 ± 0.0021 | 1300 ± 160 | 35 | |
| pNPL | 0.17 ± 0.01 | 5500 ± 400 | 31 | |
| MUC | 0.22 ± 0.01 | 210 ± 20 | 1048 | |
| Ratio, PcCel7D | pNPC/oNPC | 3.1 | 0.41 | 7.5 |
| pNPL/pNPC | 3.7 | 4.2 | 0.87 | |
| MUC/pNPC | 4.8 | 0.16 | 30 | |
| Ratio, PcCel7D/TrCel7A | oNPC | 227 | 457 | 0.50 |
| pNPC | 18 | 50 | 0.35 | |
| pNPL | 2.0 | 9.3 | 0.21 | |
| MUC | 17 | 18 | 0.97 |
The kinetic parameters differ significantly between the enzymes as well as between substrates. If we start by comparing the catalytic efficiency, MUC seems to be most efficiently hydrolysed of the substrates, with k cat/K M values about an order of magnitude higher than for pNPC and pNPL, which in turn are about an order of magnitude higher than for oNPC. Between the enzymes, k cat/K M for MUC is very similar, while for oNPC, pNPC and pNPL the values are somewhat lower with PcCel7D than with TrCel7A. Overall, PcCel7D showed much weaker apparent affinity for the substrate than TrCel7A, as reflected by higher K M, which is mostly compensated for by faster hydrolysis, i.e., higher k cat values.
It is noteworthy that the lactoside substrate (pNPL) gave much higher values for both k cat and K M than the corresponding cellobioside substrate (pNPC), although these substrates only differ by the orientation of the 4‐hydroxyl at the non‐reducing end of the molecule, being axial in pNPL and equatorial in pNPC. The k cat was about 30‐fold higher for pNPL over pNPC with TrCel7A and 4‐fold higher with PcCel7D. The activity against oNPC turned out to be significantly lower compared to the other substrates with both enzymes. However, the enzyme kinetics with oNPC were quite different for the two enzymes. With PcCel7D, k cat and K M for oNPC were about 230 and 460 times higher, respectively, than with TrCel7A. And while oNPC bound weaker than pNPC to PcCel7D (2.5 times higher K M) and k cat was about one third, oNPC bound even stronger than pNPC to TrCel7A (3.7‐fold lower K M) and k cat was 39‐fold lower.
Fluorescence titration with oNPC
The strong binding but slow hydrolysis of oNPC by TrCel7A suggests that it could in practice be utilized as a non‐reactive inhibitor for GH7 binding studies. Inspired by this, we set out to explore the possibilities of utilizing oNPC as an active site probe for GH7 CBHs. Fluorescence measurements of TrCel7A and PcCel7D solutions demonstrated that addition of oNPC quenched the intrinsic fluorescence of the enzymes because of the overlap of the absorbance spectrum of oNPC with the fluorescence spectra of the enzymes (Fig. 1B). The decrease in fluorescence was dependent on the amount of added oNPC and followed Langmuir isotherms, which enabled the derivation of dissociation constants, K d, for oNPC by regression analysis of fluorescence titration data (Fig. 1C, Table 2). With TrCel7A WT the K d value for oNPC is very close to the K M from enzyme kinetics experiments (7.4 μm vs. 7.0 μm). However, with PcCel7D we were surprised to find a much lower K d of 110 μm for oNPC, compared to the K M of 3200 μm (Table 2).
Table 2.
Dissociation constants for oNPC and cellobiose binding to TrCel7A WT and catalytic mutants and PcCel7D from fluorescence titration experiments, and inhibition constants for cellobiose a and lactose.
| Enzyme | K d for oNPC (μm) | K d for cellobiose (μm) | K i for cellobiose a (μm) | K i for lactose (μm) b |
|---|---|---|---|---|
| TrCel7A WT | 7.4 ± 0.4 | 23 ± 4 | 24 c | 180 ± 16 |
| TrCel7A D214N | 7.1 ± 0.7 | 8.9 ± 1.1 | – | – |
| TrCel7A E212Q | 4.7 ± 0.4 | 8.1 ± 0.3 | – | – |
| TrCel7A E217Q | 3.9 ± 0.4 | 14 ± 3 | – | – |
| PcCel7D | 110 ± 10 | – | 180 d | 183 ± 16 |
Previously published inhibition constants from [13]
The error margin represents the 95% confidence interval of the profile likelihood from graphpad prism 8
Competitive inhibition constant from inhibition experiments with pNPL as substrate at 30 °C, pH 5.0 [13]
Mixed‐type inhibition constant (α = 5.7) estimated from inhibition experiments with CNP‐Lac (2‐chloro‐4‐nitrophenyl‐β‐lactoside) as substrate at 33 °C, pH 5.5 [13].
Addition of cellobiose results in recovery of fluorescence of TrCel7A. Thus, competitive displacement titration could be used for indirect determination of K d for cellobiose (Fig. 1D, Table 2). The methods also allow for binding measurements with catalytically impaired mutants, and K d for oNPC and cellobiose were determined for TrCel7A WT as well as its E212Q, D214N and E217Q mutants (Table 2). The mutants gave similar K d values, albeit slightly lower with the E212Q and E217Q mutants. In the case of PcCel7D, however, fluorescence was not recovered upon addition of cellobiose up to 1 mm concentration (data not shown), which is about five times higher than the inhibition constant, K i, of cellobiose reported previously for PcCel7D [13].
Inhibition by oNPC and lactose
The binding of oNPC to TrCel7A was further analysed by inhibition assays using MUC as substrate. The activity did indeed decrease in the presence of oNPC. Regression analysis of the enzyme kinetic curves confirmed a competitive mode of inhibition with an inhibition constant K i of 5.6 ± 0.5 μm for oNPC, in good agreement with fluorescence titration and enzyme kinetics results. Furthermore, the binding of lactose was assessed by inhibition assays using pNPL as substrate. Lactose showed competitive inhibition with both TrCel7A and PcCel7D and similar inhibition constants, 180 and 183 μm, respectively (Table 2).
Structures and ligand binding
Four new X‐ray crystal structures are presented, of TrCel7A WT with oNPC, and TrCel7A E212Q mutant with pNPC, pNPL or lactose bound (PDB: 4V0Z, 4UWT, 7OC8, 7NYT, respectively). The structures were refined at 1.7, 1.2, 1.5 and 1.1 Å resolution, respectively, and are nearly identical in terms of backbone structures (pairwise RMSD α‐carbon trace values range from 0.10 to 0.28 Å). All the models contain the complete catalytic domain of Cel7A (residues 1 through 434), one N‐acetyl glucosamine residue covalently linked to Asn 270 and between two and four Co2+ ions. Interestingly, each structure contains two ligand molecules, one at the active site and one on the outside, between neighbouring protein molecules in the crystal (Fig. 2B). In the structure with lactose at the active site, which was obtained by crystal soaking with pNPL, the electron density shows that the ligand molecule on the outside is pNPL rather than lactose. Statistics from diffraction data processing and structure refinement are summarized in Table 3.
Fig. 2.
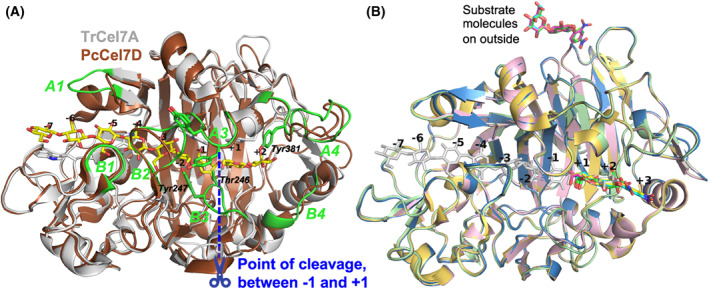
Overview of protein structures and substrate binding. (A) Crystal structure of the catalytic domain of TrCel7A (light‐grey) with cellononaose bound (yellow; PDB: 4C4C) and tunnel‐enclosing loops highlighted and labelled in green, superposed with PcCel7D (brown; PDB: 1Z3V). The point of cleavage at the catalytic center is indicated in blue, from which the glucose unit subsites are numbered, with plus‐signs towards the reducing end and minus‐signs towards the non‐reducing end of the sugar polymer. Sidechains are shown of the sugar‐binding tryptophan platforms at subsites −7, −4, −2 and +1, as well as selected residues involved in substrate binding near the catalytic center. Hydrogen bonds are indicated in cyan between Tyr247 and 6OH at subsite −2 and between Thr246 and 6OH at +1. (B) The four new crystal structures presented here, of TrCel7A showing the binding of the ligands in the product subsites +1 to +3 at the active site, and on the outside of the protein, relative to binding of cellononaose. Ligand/protein colours are as follows: pNPC, yellow/light‐yellow (PDB: 4UWT); pNPL, cyan/light‐blue (PDB: 7OC8); lactose, green/light‐green (PDB: 7NYT); oNPC, magenta/pink (PDB: 4V0Z); Cellononaose, light‐grey (PDB: 4C4C). The structure images were created with macpymol [71].
Table 3.
Statistics from X‐ray diffraction data collection and processing, structure refinement and final model.
| E212Q/pNPC | WT/oNPC | E212Q/pNPL | E212Q/lactose | |
|---|---|---|---|---|
| (A) Diffraction data | ||||
| PDB code | 4UWT | 4V0Z | 7OC8 | 7NYT |
| Beamline | MAX‐lab I911‐2 | ESRF ID14‐3 | BioMAX, MAX IV | BioMAX, MAX IV |
| Wavelength (Å) | 1.041 | 0.931 | 0.980 | 0.980 |
| Cell dimensions (Å) | 83.30, 81.78, 110.53 | 83.06, 81.38, 109.94 | 83.19, 81.51, 109.92 | 83.54, 82.21, 110.73 |
| Space group | I 2 2 2 | I 2 2 2 | I 2 2 2 | I 2 2 2 |
| Resolution range (Å) | 29.2–1.15 (1.25–1.15) | 29.1–1.70 (1.73–1.70) | 41.6–1.60 (1.63–1.60) | 41.8–1.09 (1.13–1.09) |
| No. of unique reflections a | 130 094 | 40 566 | 49 321 | 137 896 |
| Completeness (%) a | 98.1 (87.2) | 97.9 (67.6) | 99.7 (99.6) | 88.5 (41.4) |
| Multiplicity a | 7.2 (6.5) | 6.9 (5.0) | 5.4 (5.5) | 6.1 (2.7) |
| I/σ(I) a | 15.0 (3.2) | 26.6 (5.8) | 8.1 (2.3) | 13.3 (1.3) |
| R merge a , b | 0.094 (0.53) | 0.070 (0.26) | 0.15 (0.76) | 0.074 (0.68) |
| (B) Structure refinement | ||||
| Resolution used in refinement (Å) | 29.19–1.20 | 14.94–1.70 | 35.13–1.60 | 41.77–1.09 |
| No. of reflections, work set | 111 569 | 38 460 | 47 026 | 131 092 |
| No. of reflections, test set | 5905 | 2033 | 2294 | 6802 |
| R (work set) c | 0.145 | 0.129 | 0.156 | 0.123 |
| R free c | 0.159 | 0.157 | 0.184 | 0.143 |
| No. of nonhydrogen atoms | ||||
| Protein atoms | 3460 | 3400 | 3340 | 7768 |
| Solvent atoms | 681 | 525 | 405 | 588 |
| Average B factors (Å2) | ||||
| Overall | 8.0 | 8.7 | 13.0 | 12.0 |
| Protein | 5.7 | 7.91 | 11.9 | 10.2 |
| Water | 18.7 | 18.63 | 20.8 | 21.1 |
| Ligands (in active site) | 13.0 (11.6) | 20.2 (12.54) | 22.0 (29.14) | 13.1 (14.0) |
| RMSD bond lengths (Å) | 0.006 | 0.016 | 0.010 | 0.004 |
| RMSD bond angles (°) | 1.22 | 1.76 | 1.64 | 1.29 |
| Ramachandran plot outliers d | 0 | 0 | 0 | 0 |
Numbers in parentheses are for the highest resolution bin
; the final R‐factor is given
wwPDB Validation Service.
The oNPC, pNPC, pNPL and lactose bound in the product binding sites show clear and unambiguous electron density for the sugar units in subsites +1 and +2, and somewhat weaker density for the nitrophenyl group in subsite +3 (Fig. 3). The sugar units of the aryl glycoside substrates bind in the so called “unprimed” binding mode, as designated by Knott et al. [30], i.e. with the non‐reducing end sugar in subsite +1 close to the catalytic acid/base residue Glu217. This binding position of sugar units in the +1/+2 subsites is found in previous structures of both TrCel7A (PDB: 6CEL, 7CEL, 4C4C & 4C4D) [4, 30] and PcCel7D (PDB: 1Z3W) [28]. However, in the lactose complex the disaccharide is slightly tilted away from the catalytic centre towards the exit of the active site, to the “primed” binding position (Fig. 4A), similar to the sugar binding at +1/+2 in the previous TrCel7A structures PDB: 3CEL, 4PLJ and 4D5O, and PcCel7D structures PDB: 1Z3T and 1Z3V [28, 31, 32]. For the galactose residue of lactose, in subsite +1, there is clear density for the 6‐hydroxyl at two different positions and consequently two conformations were modelled of this residue. Also, two alternative conformations are seen in this structure for the B3 loop, from Asp241 to Thr255, as well as for Tyr371 at the tip of the opposing A3 loop.
Fig. 3.
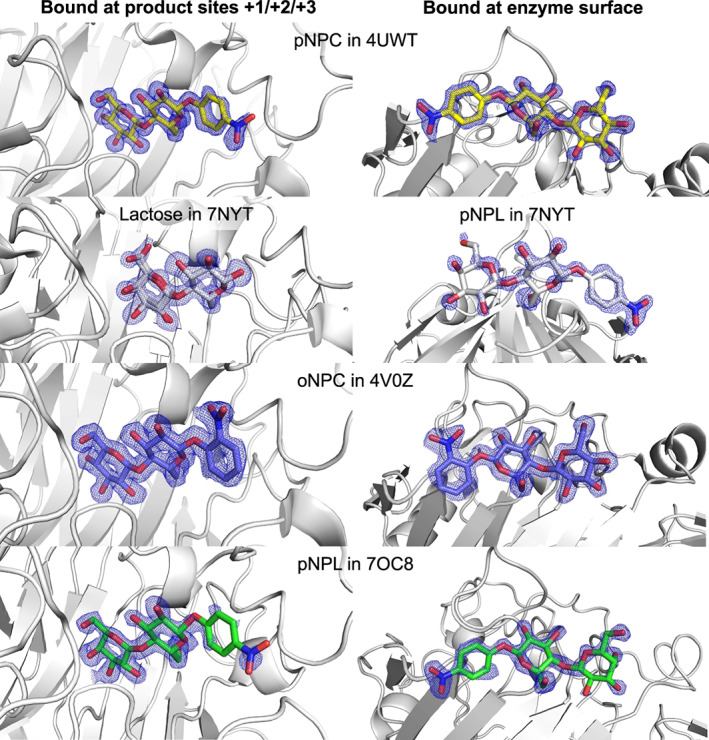
2fo‐fc electron density maps for all the sugar ligands bound in the presented TrCel7A structures, those at the product site (left) and those at the surface (right): pNPC in 4UWT (yellow), lactose and pNPL in 7NYT (white), oNPC in 4V0Z (blue), pNPL in 7OC8 (green). Electron density map contour level 1.0 σ. The structure images were created with macpymol [71].
Fig. 4.
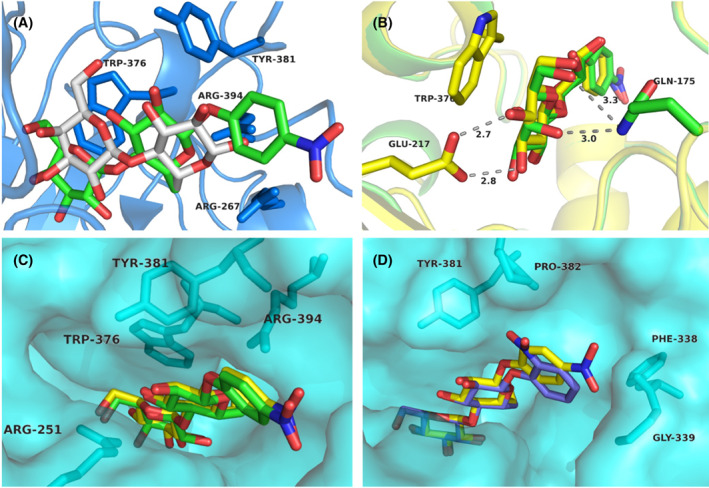
Comparison of pNPC, pNPL, oNPC and lactose binding at the product subsites of TrCel7A. (A) The pNPL molecule (green) is bound with the sugar units in the “unprimed” position at subsites +1/+2 and with the nitrophenyl group at subsite +3, whereas lactose (white) binds in the “primed” position. The protein backbone and selected residues of the lactose complex (PDB: 7NYT) are shown in blue colour. (B) An overlay of pNPL (green) and pNPC (yellow) viewed from the catalytic center shows common hydrogen bonds between sugar and protein at subsite +1 (3OH to Glu217 and 6OH to Gln175), and the difference in orientation and interactions for 4OH, with Glu217 for the glucose residue of pNPC, and with Gln175 for the galactose residue of pNPL, respectively. (C) The pNPL (green) and pNPC (yellow) ligands, viewed from the active site exit towards the catalytic center, display very similar binding positions. (D) An overlay of pNPC (yellow) and oNPC (blue) shows the difference in binding of the respective nitrophenyl moieties while the cellobiose units overlap closely. In panels (C) and (D) the protein is shown in semitransparent surface representation and selected amino acid residues as sticks. The structure images were created with macpymol [71].
The pNPC and pNPL substrates bind very similarly, as can be expected, given they only differ at the position of 4OH at the non‐reducing end of the molecule (Fig. 4B,C). However, while the 4OH of pNPC makes a favourable hydrogen bond with Glu217, the 4OH of pNPL is instead making a close contact with Gln175 (3.0 Å to NE2 atom) where neither of the atoms are well oriented for hydrogen bonding (Fig. 4B). In the pNPL structure there is a cobalt ion present at the active site with partial occupancy (0.3), appearing to interact with 3OH and 4OH of the galactose unit in pNPL and with Asp214 and His228. Given that there is electron density for only one binding position of pNPL at the product site, this Co2+ ion does not seem to significantly affect the binding position of pNPL. The sugar moieties of oNPC are virtually in the same position as in pNPC and pNPL, but the oNP unit is slightly shifted relative to pNP to avoid a clash between the 2‐nitro group and Tyr381 and Pro382 in the A4 loop region (Fig. 4D). There is no sign of electron density in any of the structures for substrate binding at the catalytic centre (i.e. with the disaccharide in subsites −2/−1 and the nitrophenyl in +1) or elsewhere along the active site. Thus, the structures clearly demonstrate that non‐productive binding at +1 to +3 is stronger and preferred over productive binding at −2 to +1 for these substrates, at least in the case of TrCel7A.
Superposition of the TrCel7A ligand structures with PcCel7D (lactose complex, PDB: 1Z3V) reveals structural differences that likely forces a different positioning of these substrates in PcCel7D. The +3 subsite is more restricted in PcCel7D due to the insertion of Asp336 in the B4 loop. The carboxylate side chain of Asp336 is pointing towards subsite +3 and overlaps partially with the nitrophenyl groups of oNPC, pNPC and pNPL in the TrCel7A structures, with close contacts of 1.0, 1.7 and 1.8 Å, respectively (Fig. 5). The same region in TrCel7A contains two glycine residues, allowing more space and possibly leading to stronger binding at the +3 site. Asp at this location is the most common motif among GH7 CBHs, but is missing in TrCel7A and a few closely related CBHs, due to a one‐residue deletion in the B4 loop.
Fig. 5.
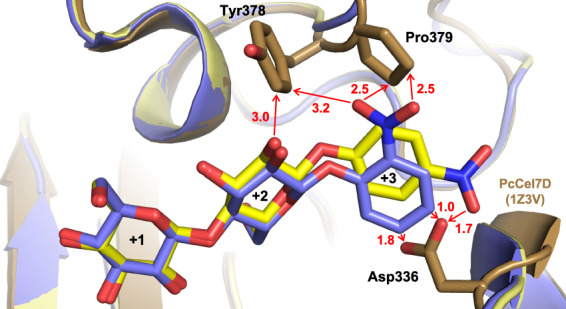
The non‐productively bound ligands pNPC (yellow; PDB: 4UWT) and oNPC (blue; PDB: 4V0Z) at subsites +1/+2/+3 in TrCel7A superposed on PcCel7D (brown; PDB: 1Z3V) showing the clash between the nitrophenyl group and Asp336 and close contacts with Tyr 378 and Pro379. The structure images were created with macpymol [71].
Molecular dynamics simulations
Since enzyme kinetics do not discriminate productive and non‐productive binding, and x‐ray crystallography only demonstrated the non‐productive binding, molecular dynamics (MD) simulations were employed to assess how the substrates would bind in the productive mode (i.e. at subsites −2 to +1). Starting models for productive binding of oNPC, pNPC and pNPL were obtained by taking the glucose residues at subsites −2 and −1 of the TrCel7A Michaelis complex with cellononaose (PDB: 4C4C) and attaching a nitrophenyl group with the glycosidic oxygen in position for protonation by the catalytic acid/base (Glu217), and with the nitrophenyl ring parallel to the Trp376 platform at subsite +1. For oNPC, two productive binding models were used, pose 1 and pose 2, with the 2‐nitro group pointing either “away” or “towards” the catalytic nucleophile (Glu212) and acid/base (Glu217) residues at the catalytic center of the enzyme. As starting models for non‐productive binding, we used the crystal structures described herein of TrCel7A in complex with oNPC, pNPC or pNPL bound at subsites +1 to +3. In this case there was only one conformation of oNPC, since only one conformation was seen in the crystal structure. Corresponding models of PcCel7D were obtained by superposition with the crystal structure of PcCel7D in complex with cellobiose (PDB: 1Z3T ).
MD simulations were run for 100 ns for TrCel7A and PcCel7D in complex with oNPC, pNPC, and pNPL in both productive and non‐productive binding modes. However, in several complexes, the substrates consistently diffused out of the active site, beginning from 500 ps. Therefore, only the first 500 ps of the production run in the simulations (after equilibration) were used for structural analyses and for computation of ligand‐binding free energies with the Molecular Mechanics Poisson‐Boltzmann Surface Area (MMPBSA) approach [33]. Fig. 6 shows cluster representations of protein backbone and ligand structures over a 500 ps trajectory for all MD simulations. Snapshots at 500 ps of all the ligand structures are shown in Figs S1 and S2, and binding free energies from the MD simulations in Table 4. Plots of distances between substrate and catalytic amino acids over the entire 100 ns MD runs are shown in Fig. S3.
Fig. 6.
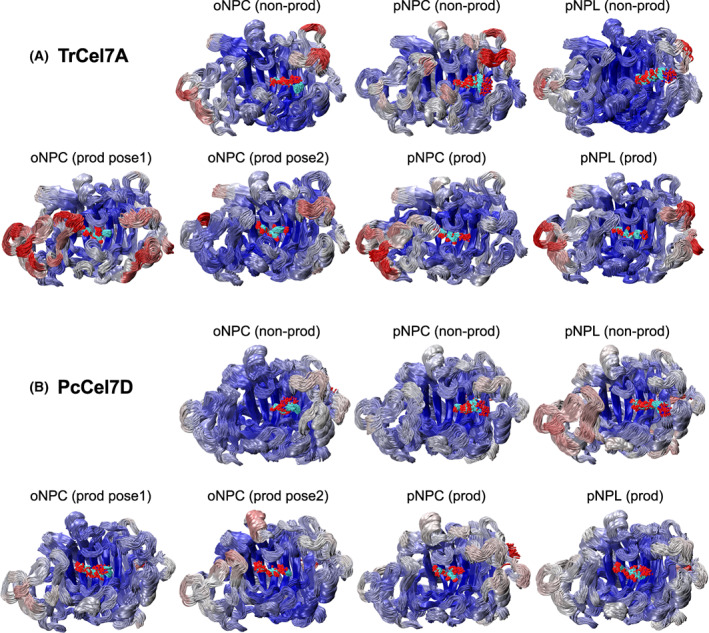
Cluster representations of (A) TrCel7A and (B) PcCel7D protein backbone and ligand structures shown over a 500 ps MD simulation trajectory. The protein backbones are coloured by RMSF (root mean square fluctuation), where red represents the largest fluctuations, and blue represents the lowest fluctuations. The structure images were created with VMD [72].
Table 4.
MMPBSA binding free energies for productive and non‐productive binding from MD simulation.
| Enzyme | Substrate | Productive binding in subsites −2 to +1 (ΔG0’, kcal·Mol−1) | Non‐productive binding in subsites +1 to +3 (ΔG0’, kcal·Mol−1) |
|---|---|---|---|
| TrCel7A | oNPC (pose1/2) | −17.6 ± 0.4/−18.1 ± 0.4 a | −14.8 ± 0.4 |
| pNPC | −18.1 ± 0.4 | −20.1 ± 0.4 | |
| pNPL | −21.2 ± 0.4 | −21.8 ± 0.3 | |
| PcCel7D | oNPC | −3.2 ± 0.4/5.2 ± 0.3 a | −1.6 ± 0.4 |
| pNPC | −4.5 ± 0.3 | −5.1 ± 0.4 | |
| pNPL | −2.0 ± 0.3 | −4.5 ± 0.4 |
Two values were calculated for oNPC, with the nitro group pointing either “away” (pose 1) or “towards” (pose 2) the catalytic center.
For the productive binding mode at subsites −2/−1/+1, Fig. 7 shows the distances between substrate and catalytic amino acids during the first 1 ns of MD simulations (glycosidic oxygen O1 to the acid/base, and anomeric carbon C1 to the nucleophile, respectively), which indicate larger ligand fluctuations with PcCel7D than with TrCel7A. This is also seen in the cluster representations of backbone and ligand structure in Fig. 6. Snapshots at 500 ps of selected substrates in the productive mode are shown in Fig. 8. For TrCel7A, the sugar units of the substrates remained close to the corresponding glucose units in the Michaelis complex (within 1 Å), and the glucose residue at subsite −1 retained the 1,4 B boat conformation. The axial 4OH of pNPL at subsite −2 seems to be readily accommodated without steric hindrance. The nitrophenyl rings lie on the Trp376 platform at subsite +1 and the glycosidic oxygen is within distance to the catalytic acid/base for protonation. However, for oNPC pose 2 the 2‐nitro group comes close to and may interfere with the catalytic acid/base Glu217 (Fig. 8A). Thus, oNPC is less likely to be hydrolyzed when bound in the pose 2 orientation.
Fig. 7.
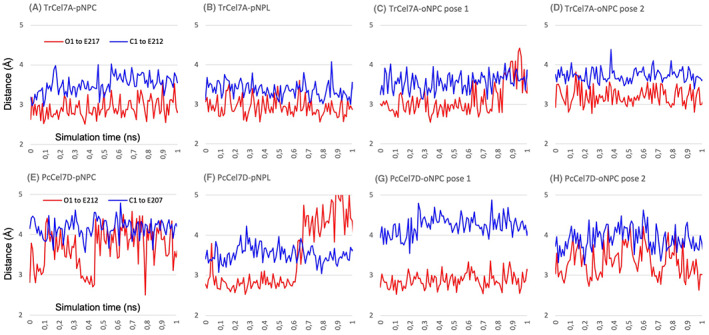
Distances between substrate and catalytic amino acids during 1 ns of MD simulations of productive binding at subsites −2/−1/+1 of pNPC, pNPL and oNPC, in TrCel7A (A–D) and in PcCel7D (E–H). The red line shows the shortest distance from the glycosidic oxygen O1 to the nearest O atom of the catalytic acid/base (Glu/E217 in TrCel7A; Glu/E212 in PcCel7D). The blue line shows the shortest distance from the anomeric carbon C1 to the nearest O atom of the catalytic nucleophile (Glu/E212 in TrCel7A; Glu/E207 in PcCel7D).
Fig. 8.
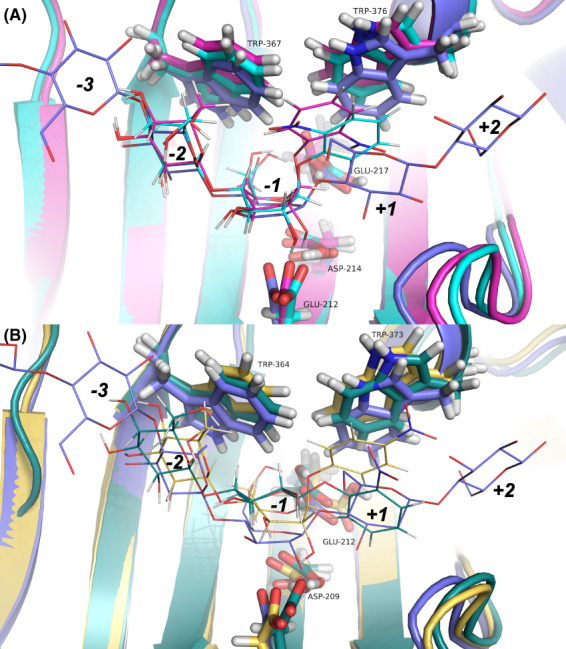
Snapshots at 500 ps from MD simulations of productive mode binding of nitrophenyl substrates at subsites −2/−1/+1 in TrCel7A and PcCel7D, superposed with the TrCel7A cellononaose complex PDB: 4C4C (blue). (A) TrCel7A with oNPC pose 1 (cyan) and oNPC pose 2 (magenta). In pose 2 the oNPC is less likely to be hydrolyzed, since the 2‐nitro group appears to obstruct protonation of the glycosidic oxygen by the catalytic acid/base Glu217. (B) PcCel7D with pNPC (yellow) and oNPC pose 2 (green). In the oNPC molecule the glucose residue at subsite −1 has flipped from boat to chair conformation. Also, the oNP ring has flipped around so that the 2‐nitro group is pointing “away,” while it was pointing towards the catalytic center in the starting model. The structure images were created with macpymol [71].
With PcCel7D productive mode, the deviation was larger from the starting models and the glucose residues were shifted further upwards from the bottom of the active site (Fig. 8B). In the 500 ps snapshots, the boat conformation of the glucose residue at subsite −1 is only retained in pNPC. In the others it is on the way to a chair in oNPC pose 1, whereas in pNPL and oNPC pose 2 it has flipped from boat and adopts a 4 C 1 chair conformation.
In the snapshots of non‐productive complexes, the sugar residues overlap at subsites +1 and +2 and the nitrophenyl groups at +3. However, the ligands deviate from each other and from the crystal structures, with up to 2–3 Å distance between corresponding sugar atoms at subsite +1 (TrCel7A/oNPC vs. PcCel7D/pNPL) and up to 5–7 Å between nitrophenyl ring atoms at subsite +3 (TrCel7A/oNPC vs. PcCel7D/pNPC). The MD simulations also show larger flexibility of the protein around the ligands than seen in the crystal structures, such as in the A4 and B4 loop regions that are flanking the +3 subsite (Fig. 6). A tyrosine residue in loop A4 (Tyr381 in TrCel7A, Fig. 4C; Tyr378 in PcCel7D, Fig. 5), which restricts nitrophenyl binding on one side, deviates up to 2 Å at the CA atom and 2.7 Å at OH. The B4 loop on the other side of the nitrophenyl moiety exhibits backbone shifts up to 3.3 Å (TrCel7A Gly339), as well as flexibility of the Asp336 side chain in PcCel7D, although it is consistently pointing towards subsite +3.
The MMPBSA binding free energy calculations do indeed indicate a large difference between the two enzymes (Table 4). As expected, TrCel7A gave more favourable free energy values (−15.0 to −22.0 kcal·mol−1) for all substrates and in both binding modes, compared to PcCel7D (−1.6 to −5.0 kcal·mol−1), consistent with stronger binding of the substrates to TrCel7A than to PcCel7D. Also, binding energies were lower for non‐productive than productive binding of pNPC and of pNPL with both enzymes. However, with TrCel7A the binding energies were lower for pNPL than for pNPC, in both binding modes, indicating stronger binding of pNPL than of pNPC, which is contradictory to the results from the enzyme kinetics experiments.
Discussion
Nitrophenyl glycosides are very popular model substrates for glycoside hydrolases, since they provide good leaving groups and also have favourable spectral properties, making the reactions easy to monitor. Consequently, p‐nitrophenyl cellobioside and p‐nitrophenyl lactoside have both found use in cellulase research. It has been found in many cases, though, that the k cat observed has been extremely low, i.e., orders of magnitude lower than that observed for the cleavage of cellulose or cellooligosaccharides. A likely explanation for this phenomenon may be based on the subsite array found in the active site of the enzymes, where model substrates may occupy several alternative positions and unproductive binding may prevent the productive one, leading to a strongly decreased apparent k cat if the non‐productive binding is stronger than that at the productive position.
Previous crystal structures have shown that the +1/+2 product binding site is the preferred binding site for cellobiose and lactose in TrCel7A and PcCel7D [28, 31, 32]. Molecular dynamics simulations of TrCel7A by Knott et al. [5] suggested that strong binding to the +1/+2 site is likely an important factor in driving the processive action in the GH7 cellobiohydrolases, making it likely that similar dynamics occur in other enzymes of this class and other processive glycosidases as well [7, 34, 35]. The crystal structures of TrCel7A with pNPL, pNPC and oNPC bound at the +1/+2/+3 sites we have presented here suggest that non‐productive binding is indeed the preferential binding mode for these small model substrates, at least in TrCel7A.
Low apparent K M‐values accompanying the low k cat observed in our experiments and in previous studies also suggest that the slow turnover of these substrates is likely caused by strong non‐productive binding, as it is expected to lower both constants by the same factor, with the efficiency constant (k cat/K M) remaining unaffected (Table 1) [13, 14]. For overlapping (competing) binding modes, the apparent kinetic parameters depend on the affinities for productive and non‐productive binding as follows (Eqns 1 and 2):
| (1) |
| (2) |
k cat app and K M app are the apparent catalytic rate and Michaelis–Menten constants, k cat prod and K M prod the intrinsic parameters for productive binding, and K d nonprod the dissociation constant for non‐productive binding. From Eqn (2) follows that K M app cannot be higher than K d nonprod (and not K M prod) if the binding modes overlap (compete). If the two modes have the same binding strength, then K M app = 0.5 * K d nonprod.
The pNPC and pNPL substrates differ only by the orientation of the 4‐hydroxyl at the non‐reducing end of the molecule, being axial in pNPL and equatorial in pNPC. As expected, they do indeed show about the same k cat/K M values indicating similar productive binding at −2/−1/+1. This is supported by MD simulations showing that the axial 4OH of pNPL is readily accommodated at subsite −2 without signs of steric hindrance. Yet, both k cat and K M were much lower for pNPC than for pNPL (about 30‐fold and 20‐fold, respectively, with TrCel7A, and about 4‐fold with PcCel7D). In the case of TrCel7A we now know that both substrates bind preferentially at the non‐productive position at subsites +1/+2/+3, which lowers the apparent catalytic constants. Hence, the difference between the substrates is mainly caused by differences in non‐productive rather than productive binding. The lower values of k cat and K M for pNPC show that it is more strongly affected by non‐productive binding and binds stronger than pNPL at subsites +1/+2/+3.
In the crystal structures the cellobiose unit of pNPC binds very similarly to cellobiose alone at subsite +1/+2, and the affinity is about the same, as reflected by similar values of K M for pNPC, and K d and K i for cellobiose (26, 23, 24 μm, respectively; Tables 1 and 2). The position of pNPL is practically identical to that of pNPC, except for the orientation of 4OH, which is thus likely the cause of the difference between the substrates. For cellobiose and pNPC, the equatorial 4OH appears to contribute favourably by hydrogen bonding to the catalytic acid/base. With pNPL this hydrogen bond is missing and instead the axial 4OH makes an unfavourably close contact with Gln175 that may rather have a negative effect on the affinity. This is further indicated by the higher apparent K M for pNPL (590 μm) compared to K i for lactose (180 μm), which suggests weaker binding of pNPL than of lactose, and by the positional deviation between the lactoside unit of pNPL and lactose alone in the complex structures. The lactose molecule prefers to bind in the “primed” position slightly tilted away from the catalytic centre (Fig. 4A) where 4OH has more space and can make hydrogen bonds with Thr246 and Arg251, whereas pNPL is not tilted to that position, presumably hindered by space limitations for the bulky nitrophenyl group at subsite +3.
A similar trend is seen with PcCel7D, i.e. similar k cat/K M values for pNPC and pNPL suggesting similar productive binding at −2/−1/+1, at the same time as the apparent catalytic constants are lower for pNPC, pointing towards stronger non‐productive binding than for pNPL. However, PcCel7D shows significantly higher k cat and K M on pNPL than TrCel7A, suggesting much weaker non‐productive binding. This could be explained by the different architectures at the +3 site, where Asp336 positioned in the B4‐loop of PcCel7D occupies a position where the nitrophenyl‐groups in oNPC, pNPC and pNPL are positioned in the TrCel7A structures, likely forcing a different positioning and weaker binding of these substrates in PcCel7D (Fig. 5). The same region in TrCel7A contains two glycine residues, allowing more space and possibly leading to stronger binding at the +3 site. Asp at this location is the most common motif among GH7 CBHs but is missing in TrCel7A and a few closely related CBHs, due to a one‐residue deletion in the B4 loop.
Interestingly, TrCel7A mutants with deletions in the B3 loop have shown pNPL hydrolysis kinetics similar to PcCel7D, with higher k cat and K M, and higher K i for cellobiose compared to the wild‐type TrCel7A, suggesting this loop plays a role in non‐productive binding and product inhibition [13, 36]. Indeed, PcCel7D is missing six residues in this loop region compared to the native TrCel7A (Fig. 2A). At the tip of the B3 loop in TrCel7A, Tyr247 contributes to productive binding through H‐bonding with 6OH of the glucose unit at subsite −2, while Thr246 promotes non‐productive binding by H‐bonding to 6OH of the sugar unit at subsite +1 (Fig. 2A). The lack of these residues in PcCel7D should result in weaker binding of the substrates, both productive and non‐productive binding.
While cellobiose inhibits TrCel7A more strongly than PcCel7D, the results from our lactose inhibition experiments suggest that with lactose the inhibitory effect is more or less equal on both enzymes (Table 2) [13]. In TrCel7A, cellobiose binds strongly at the product sites because of favourable H‐bonds with both Glu217 and Thr 246. Lactose binding is weaker due to the clash between the axial 4OH and Gln175, and the loss of the 4OH‐Glu217 H‐bond when lactose is tilted to the “primed” orientation. In PcCel7D, there is no residue corresponding to Thr246 due to the shorter B3 loop (Fig. 2A), and cellobiose binds weaker in PcCel7D than in TrCel7A. Lactose could be expected to bind even weaker due to the lack of H‐bond to the catalytic acid/base Glu217. However, when the lactose molecule is tilted to the “primed” orientation it can make compensating H‐bonds with Asp336 in the B4 loop. The affinities are actually very similar for cellobiose and lactose with PcCel7D and lactose with TrCel7A, with K i values of ~180 μm. Cellobiose binds about one order of magnitude stronger in TrCel7A, probably due to one extra hydrogen bond. The H‐bonding partners that differ between enzyme/ligand complexes may be simplified as follows: TrCel7A/cellobiose: Glu217 and Thr246; TrCel7A/lactose: Thr246; PcCel7D/cellobiose: Glu212; PcCel7D/lactose: Asp336.
While the MMPBSA calculations capture the relative differences in binding affinities between TrCel7A and PcCel7D (energy values differ by more than 10.0 kcal·mol−1), the calculations do not capture the differences between the similar substrates or between productive vs. non‐productive binding (some values are within 2.0 kcal·mol−1). Previous studies have highlighted that MMPBSA may be impractical for comparing ligands with similar affinities due to its low precision [37]. Possibly, more sensitive, computationally intensive estimation methods, such as umbrella sampling, may be needed to evaluate the relative affinities between these substrates.
The structures and MD simulations do not provide straight‐forward answers to the question why oNPC is an inferior substrate compared to pNPC (and pNPL). However, the enzyme kinetics, fluorescence titration, and oNPC inhibition studies showed low apparent K M, K d and K i values for oNPC with TrCel7A (7.0, 7.4 and 5.6 μm, respectively; Tables 1 and 2), suggesting that strong non‐productive binding might at least partially explain the slow hydrolysis in TrCel7A, whereas the higher K M and K d values for PcCel7D (3200 and 110 μm, respectively; Tables 1 and 2) imply that this effect is much less significant in PcCel7D. The fact that PcCel7D fluorescence could not be recovered after oNPC titration by the addition of cellobiose also suggests that there could in fact be another site preferential to the product site for binding oNPC on the enzyme, one which does not have as high affinity for cellobiose as for oNPC. The significantly higher apparent K M value compared to K d for oNPC with PcCel7D is in line with this hypothesis, considering that in the case of overlapping productive and non‐productive binding modes K M should not exceed the K d value. With TrCel7A the K M and K d values for oNPC are roughly equal (instead of K M app ≤0.5*K d in the case of overlapping binding sites), which implies there is some binding to a location outside the active site in TrCel7A as well, in accordance with previous titration calorimetry results from Colussi et al. [38], suggesting stoichiometry of ~ 1.5 binding sites for cellobiose and triose in the E212Q mutant of this enzyme. An additional ligand molecule is indeed seen on the outside of the protein in our crystal structures. However, that binding site is built up by crystal contacts where the ligand is bound by interactions with four neighbouring protein molecules in the crystal lattice (Fig. 9). When the enzyme is free in solution the affinity for this site would most likely be too low to be significant.
Fig. 9.
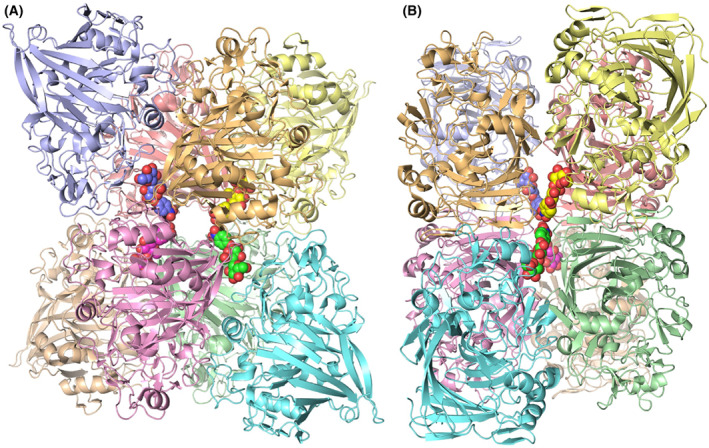
Crystal packing of protein molecules around the substrate molecules bound at the surface of the TrCel7A enzyme in the crystal structures, viewed along two of the twofold symmetry axes in the crystal. Four substrate molecules bind around the symmetry axes, each making interactions with four surrounding protein molecules. The structure shown is the TrCel7A E212Q/pNPC complex (PDB: 4UWT) with pNPC in space‐filling and protein chains in cartoon representation. Colours are arbitrarily chosen for distinction of individual molecules. The structure images were created with macpymol [71].
When considering the slower hydrolysis of oNPC compared to the other model compounds studied, it also seems clear that there is more space and conformational freedom for a nitro group at the 4‐position (as in pNPC), whereas the close proximity of the 2‐nitro group in oNPC to the glycosidic oxygen and the catalytic amino acid residues is more likely to interfere with transition‐state formation. Such interference would be more pronounced with TrCel7A since the catalytic centre is more enclosed in this enzyme compared to PcCel7D (Fig. 2A).
Another relevant question is why MUC is a much better substrate, as reflected by about an order of magnitude higher catalytic efficiency (k cat/K M) than pNPC and pNPL. Based on the dynamics of the productively bound substrates at the catalytic centre in the MD simulations, we hypothesize that this is an effect of the larger size of methylumbelliferyl compared to the nitrophenyl group. The methylumbelliferyl aglycone would fill up more of the space available in subsite +1 and will be more firmly bound. That would limit the conformational freedom for the glucose unit at subsite −1 and help to push it towards the catalytic amino acids, thereby increasing the probability to reach and pass the transition state for hydrolysis. Previous computational studies of cellulose hydrolysis and processivity by TrCel7A have shown that catalytic activation is an essential part of the catalytic mechanism. The enzyme is utilizing binding in the surrounding subsites as handles for bending the cellulose chain so that the glucose unit at subsite −1 will flip from chair to the boat/skew conformational series at the same time as it will be pushed towards the catalytic nucleophile (Glu212 in TrCel7A) [5, 30]. The tryptophan residues that serve as sugar binding platforms in the surrounding subsites −2 and +1 play an important role by acting as relatively rigid, inelastic surfaces that restrict the conformational freedom of the substrate and promote the glucopyranose ring distortion necessary for catalysis (Fig. 8A). That aromatic‐carbohydrate interactions play a role in glucopyranose distortion and TS‐stabilization has also been shown in a computational study of the other processive cellobiohydrolase of T. reesei, TrCel6A, where aromatic‐carbohydrate interactions were examined with molecular simulation [39].
Concluding remarks
In this work we have shown that, at least in the case of TrCel7A, oNPC can be utilized as an active‐site probe for fluorometric determination of the dissociation constant for cellobiose and can be used also with catalytically impaired mutants. We have also shown that the enzyme kinetics of GH7 CBHs on the convenient chromogenic substrates pNPC and pNPL is dictated by non‐productive binding in the product binding sites rather than productive binding at the catalytic centre. Structural differences distant from the catalytic centre that affect non‐productive binding may have large impact on the kinetics, as exemplified by the influence of Asp336 in subsite +3 of PcCel7D. Thus, the results of activity assays with these substrates should be interpreted with caution. One‐point measurements at a single substrate concentration are still useful for estimation of relative amount or activity of the same enzyme, e.g., to monitor protein purification or pH and temperature dependence, or in protein engineering to improve thermal stability by comparing activity before and after heat treatment (e.g., [40]). However, for comparison of homologues and/or mutants a sufficient range of substrate concentrations is needed so that enzyme kinetics parameters (k cat and K M) can be derived. Very low values of both k cat and K M are indicative of strong product binding, as with TrCel7A. The specificity constant k cat/K M is the most instrumental parameter for comparison since it is not affected by non‐productive binding but is a measure of the difference in free energy between the substrate in solution and the transition state of hydrolysis.
Materials and methods
Reagents and enzymes
Trichoderma reesei Cel7A and its catalytically inactive mutants E212Q, D214N and E217Q were purified from culture filtrate as described [31, 41]. Cel7D from Phanerochaete chrysosporium was purified as described in [42]. An additional purification step on Superose 12 gel (Pharmacia) using 0.5 m ammonium sulphate in 50 mm sodium acetate buffer, pH 5.0 was performed for all enzymes used. Catalytic domain of TrCel7A wildtype and E212Q mutant for crystallization were prepared as described [31]. The purity of the enzymes was confirmed by SDS/PAGE. Methylumbelliferyl cellobioside, o‐nitrophenyl cellobioside, p‐nitrophenyl cellobioside, p‐nitrophenyl lactoside, cellobiose and lactose were obtained from Sigma (St. Louis, MO, USA), all other chemicals were of analytical grade.
Ligand binding studies
All experiments were performed in 50 mm sodium acetate buffer, pH 5.0 at 25 °C, unless stated otherwise. Fluorescence of the protein was measured with an Aminco SPF‐500 spectrofluorometer. Fluorescence quenching experiments were performed at λex = 280 nm and λem = 340 nm with excitation band pass at 2 nm and emission band pass at 10 nm. The possible influence of cellobiose to the protein fluorescence was measured. It was found that the presence of cellobiose increases the fluorescence of TrCel7A in a hyperbolic manner with K d = 4.5 ± 1.5 mm (data not shown). Since our measurements were done at more than 10 times lower concentrations of cellobiose, we can assume that the influence from cellobiose to protein fluorescence is linear (Eqn 4).
Kinetic studies
For kinetic studies with nitrophenyl glycosides as substrates, the enzyme (0.5 or 1 μm) was incubated with substrate at various concentrations, for 2 min with pNPL, 30 min with pNPC, and 18 h (TrCel7A) or 5 h (PcCel7D) with oNPC. The reaction was stopped by the addition of equal volume of 0.1 m sodium hydroxide and the concentration of released pNP or oNP was measured spectrophotometrically at 414 nm, using ε = 16 590 m −1·cm−1 for pNP and ε = 4500 m −1·cm−1 for oNP. For kinetics with methylumbelliferyl cellobioside as a substrate, the enzyme (50 nm TrCel7A or 10 nm PcCel7D) was incubated with various concentrations of the substrate and inhibitor. The reaction was stopped by the addition of equal volume of 0.5 m sodium carbonate. The released product was quantified fluorometrically at λex = 360 nm and λem = 440 nm using methylumbelliferone as a reference. The kinetic parameters for hydrolysis of oNPC, pNPC, pNPL and MUC were calculated by nonlinear regression analysis using kyplot software package (KyensLab Inc., Tokyo, Japan).
The inhibition constant of lactose on the TrCel7A and PcCel7D was determined by kinetic measurements using pNPL as a substrate, without and with 0.2 mm lactose as inhibitor. For TrCel7A the pNPL concentrations used were 0.1, 0.2, 0.4, 0.6, 0.8, 1.5, 3, 5, 7 and 10 mm, with an enzyme concentration of 1.5 μm. For PcCel7D the pNPL concentrations used were 1, 2, 3, 4, 5, 8, 10, 14, 17 and 20 mm, with an enzyme concentration of 0.7 μm. The reactions had a volume of 150 μL and were conducted in triplicate in 10 mm sodium acetate buffer pH 5.0 at 20 °C for 30 min and stopped by adding an equal volume of 0.1 m NaOH. Substrate control samples were run with identical pNPL concentrations in triplicate, with enzyme added after the NaOH. The quantity of released pNP was determined as described above. The inhibition constants were determined by non‐linear regression using the competitive inhibitor function of graphpad prism 8 (GraphPad Software, San Diego, CA, USA).
Data treatment
Fluorescence quenching data were fitted using nonlinear regression into following equations:
Binding of the quenching ligands to the enzyme (Eqn 3):
| (3) |
Here, F 0 is the free protein fluorescence; F oNPC is the observed protein fluorescence at given concentration of the quenching ligand oNPC; F sat represents the fluorescence of the protein‐oNPC complex; [oNPC] is the oNPC concentration; K d(oNPC) is the dissociation constant for oNP binding to the protein.
Displacement binding data were fitted into the following equation (Eqn 4):
| (4) |
Here, F (oNPC,CB) is the protein fluorescence at given concentration of cellobiose and oNPC; F oNPC is the protein fluorescence at given concentration of oNPC; F 0 − F oNPC is the change in fluorescence caused by oNPC; [CB], concentration of cellobiose; K d(CB), binding constant of cellobiose to the protein; [oNPC], oNPC concentration; K d(oNPC), binding constant for the quenching ligand; B x [CB], linear component which takes into account the change of the fluorescence of the protein by adding of cellobiose.
Protein crystallization and structure determination
Crystallization experiments were carried out using the hanging‐drop vapour diffusion method [43] by mixing equal amounts of protein (6 mg·mL−1, in 10 mm sodium acetate, pH 5.0) and reservoir solution [50 or 100 mm morpholinoethane sulphonic acid (Mes), pH 6.0, 21.25% polyethylene glycol 5000 monomethyl ether (m5K), 12.5% glycerol and 5–10 mm cobalt chloride]. Crystals appeared within 1–5 days at room temperature. Ligand soaks with oNPC and pNPC were performed by transferring crystals to hanging drops containing 10 mm oNPC or pNPC, in 0.1 m NaMes, pH 6.0, 25% m5K, 12.5% glycerol and 10 mm CoCl2, with a subsequent incubation for 3 h (wildtype with oNPC) or 24 h (E212Q mutant with pNPC) before crystal picking. For pNPL soaking, a few grains of pNPL were added to a drop with TrCel7A E212Q crystals. Individual crystals were picked with 0.1–0.5 mm loops and flash‐frozen in liquid nitrogen. Synchrotron x‐ray diffraction data were recorded at 100 K. All crystals belong to space group I222 with one protein molecule per asymmetric unit.
X‐ray diffraction data for TrCel7A wildtype with oNPC were collected at beamline ID14‐3, ESRF, Grenoble, France, and for TrCel7A E212Q with pNPC at beamline I911‐2, MAX‐lab, Lund, Sweden. The data for wt/oNPC were processed with Denzo and Scalepack [44], and for E212Q/pNPC with Mosflm and Scala [45, 46]. Initial phases were obtained from the refined protein coordinates of TrCel7A E212Q in complex with cellobiose (PDB: 3CEL).
Diffraction data for the TrCel7A E212Q structures with lactose and pNPL ligands were collected at the BioMAX beamline at the MAX IV synchrotron in Lund, Sweden, using mxcube3 and ispyb software for data collection and management [47, 48, 49]. The data were indexed and integrated through automatic processing with XDS through the EDNA pipeline at MAX IV, and scaled and merged with Aimless either through the EDNA pipeline (TrCel7A E212Q with lactose) or through the CCP4i interface (TrCel7A E212Q with pNPL) [50, 51, 52, 53, 54]. This data was used as input reflections into the Dimple‐pipeline using the CCP4i2 interface [55]. The peptide chain coordinates from the TrCel7A structure PDB: 4C4C were used as search model input coordinates for Dimple, where a re‐indexing of the reflections was performed by Pointless, followed by rigid body refinement and restrained refinement by Refmac5 [56].
All the structures were further refined in several iterative cycles of model building and adjustment in Coot, and restrained refinement in Refmac5 [57]. Statistics from diffraction data processing and structure refinement are summarized in Table 3. The atomic coordinates and experimental structure factor amplitudes have been deposited in the Protein Data Bank with accession codes PDB: 7NYT, 7OC8, 4V0Z, and 4UWT for the lactose, pNPL, oNPC, and pNPC active site ligand structures respectively.
Molecular dynamics simulations and free energy calculations
MD simulations were run for 14 separate systems, which comprised TrCel7A and PcCel7D in complex with oNPC, pNPC, and pNPL, in both productive and non‐productive configurations. Two poses were simulated for the oNPC productive complex, with the nitrophenyl group pointing either away from (pose1) or towards (pose2) the catalytic center. Structure models for non‐productive binding were derived from the crystal structures presented in this study (TrCel7A models), and by superposition with PcCel7D structure PDB: 1Z3T. The models for productive binding were derived by modifying the TrCel7A/cellononaose Michaelis complex (PDB: 4C4C) [5]. The E217Q mutation in 4C4C was reverted and in all models the wildtype catalytic residues were used. Protonation states of titratable residues were determined by pKa calculations using the H++ webserver with a pH of 5.0 and internal and external dielectrics of 10 and 80 respectively [58, 59]. The systems were constructed with CHARMM, a 83 Å × 83 Å × 83 Å water box was added to solvate the system, and sodium ions were added to ensure a net neutral charge. The conformation of protein, carbohydrate, and the nitrophenyl moieties were defined with the CHARMM36 force fields, and water molecules were modelled with the TIP3P force field [60, 61, 62].
Minimization, equilibration, and production simulations were conducted with AMBER [63, 64]. The CHARMM parameter files were converted to AMBER format with the parmed package [65]. The minimization routine was performed as follows. First, the protein and ligand were restrained so that only water molecules and ions were minimized for 500 steps. Second, only the protein was fixed, and the ligand and solvent molecules were minimized for 500 steps. Finally, the entire system was minimized without any restraints for 1000 steps. Restraints were achieved by applying a 500 kcal·(mol.Å−2)−1 force constant on the desired atoms. In each case, the first 200 steps were performed with the steepest descent method (SD) and the conjugate gradient method was used for the remaining steps. After minimization, the systems were heated from 100 to 300 K over 20 ps with the NVT ensemble and a weak force restraint of 10 kcal·(mol.Å−2)−1 on protein and ligand atoms. Subsequently, the systems were equilibrated in the NPT ensemble at 300 K for 200 ps in four equal stages (50 ps each) with gradually decreasing weak restraints. Restraints of 10 and 5 kcal·(mol.Å−2)−1 were respectively used in the first two stages on both protein and ligand heavy atoms. In the third stage, restraints of 5 kcal·(mol.Å−2)−1 were applied to only ligand heavy atoms, which was followed by a final stage without restraints. The production run was performed for 100 ns with the NPT ensemble at 300 K. Long‐range electrostatics were handled with the Particle mesh Ewald algorithm (PME) [66] and hydrogen distances were fixed with the SHAKE algorithm [67]. MMPBSA calculations were performed with the ambertools package [68] using snapshots from the first 500 ps of the production simulations at an interval of 1 ps. As in previous studies, the ionic strength, external dielectric, and internal dielectric constants were set to 0.15 m, 4.0, and 80.0, respectively [69, 70], and the entropy term was excluded in the calculations [70]. Structural visualization and analyses and trajectories were done with pymol [71] and vmd [72].
Conflict of interest
The authors declare no conflict of interest.
Author contributions
TH: Solved, refined, validated and deposited protein ligand complex structures (7NYT, 7OC8). Performed lactose inhibition experiments. Analysed protein structures and made protein structure figures. Wrote final manuscript; JEG: Performed and analysed MD simulations and made corresponding figures. Contributed to final manuscript writing; AN: Performed and analysed enzyme kinetics, inhibition and fluorescence titration experiments and made corresponding figures. Wrote initial manuscript draft (as unpublished manuscript in PhD thesis). Crystallized and solved structures of protein‐ligand complexes (4UWT, 4V0Z); NTA: Set up protein structure models and systems for MD simulations; MN: Participated in initial enzyme kinetics and fluorescence titration experiments. MHM: Refined, validated and deposited protein structures (4UWT, 4V0Z); RI: Participated in project planning, experiment design and supervision; PV: Participated in enzyme kinetics and fluorescence titration methods development. Contributed to final manuscript writing; GJ: Initiated and conceptualized the investigation. PhD supervisor for Anu Nutt. Wrote final manuscript; CMP: Initiated and lead the MD simulations and subsequent analyses. JS: Co‐initiator of the investigation. Produced and purified proteins. Performed protein crystallization, x‐ray data collection and protein structure analysis. Wrote and coordinated the final manuscript.
Supporting information
Fig. S1. Snapshots at 500 ps of MD simulation of substrate binding in productive mode at subsites −2/−1/+1 in TrCel7A and PcCel7D.
Fig. S2. Snapshots at 500 ps of MD simulation of substrate binding in non‐productive mode at subsites +1/+2/+3 in TrCel7A and PcCel7D.
Fig. S3. Plots of distances between substrate and catalytic amino acids during 10 ns and 100 ns of MD simulations of productive binding mode at subsites −2/−1/+1 of pNPC, pNPL and oNPC, in TrCel7A and in PcCel7D.
Acknowledgements
We acknowledge MAX IV Laboratory for time on Beamline BioMAX under Proposal 20180025 and thank Uwe Müller and Ana Gonzalez for assistance. Research conducted at MAX IV, a Swedish national user facility, is supported by the Swedish Research council under contract 2018‐07152, the Swedish Governmental Agency for Innovation Systems under contract 2018‐04969, and Formas under contract 2019‐02496. We also thank the staff of the ESRF and EMBL Grenoble for assistance and support in using beamline ID14‐3. Funding for the research is gratefully acknowledged from the Swedish Energy Agency (Dnr 2015‐009633) and Swedish Natural Science Research Council (NFR). This work was supported in part by the National Science Foundation (NSF) award number 1552355 to CMP in support of JEG. This material is also based upon work supported by (while CMP is serving at) the NSF. Any opinion, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the NSF. This work was authored in part by Alliance for Sustainable Energy, LLC, the manager and operator of the National Renewable Energy Laboratory for the U.S. DOE under Contract No. DE‐AC36‐08GO28308. PV was supported by the Estonian Research Council (Grant PRG1540).
This is an Editor's Choice article from the 18 January 2023 issue.
Contributor Information
Gunnar Johansson, Email: gunnar.johansson@kemi.uu.se.
Christina M. Payne, Email: christy.payne@uky.edu.
Jerry Ståhlberg, Email: jerry.stahlberg@slu.se.
Data availability statement
The atomic coordinates and structure factors of the TrCel7A structures have been deposited into the Protein Data Bank with accession codes PDB: 4UWT, 4V0Z, 7NYT, and 7OC8, respectively, for the pNPC, pNPL, lactose and oNPC ligand complexes.
References
- 1. Payne CM, Knott BC, Mayes HB, Hansson H, Himmel ME, Sandgren M, et al. Fungal cellulases. Chem Rev. 2015;115:1308–448. [DOI] [PubMed] [Google Scholar]
- 2. Igarashi K, Koivula A, Wada M, Kimura S, Penttilä M, Samejima M. High speed atomic force microscopy visualizes processive movement of Trichoderma reesei cellobiohydrolase I on crystalline cellulose. J Biol Chem. 2009;284:36186–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lee I, Evans BR, Woodward J. The mechanism of cellulase action on cotton fibers: evidence from atomic force microscopy. Ultramicroscopy. 2000;82:213–21. [DOI] [PubMed] [Google Scholar]
- 4. Divne C, Ståhlberg J, Teeri TT, Jones TA. High‐resolution crystal structures reveal how a cellulose chain is bound in the 50 Å long tunnel of cellobiohydrolase I from Trichoderma reesei . J Mol Biol. 1998;275:309–25. [DOI] [PubMed] [Google Scholar]
- 5. Knott BC, Crowley MF, Himmel ME, Ståhlberg J, Beckham GT. Carbohydrate‐protein interactions that drive processive polysaccharide translocation in enzymes revealed from a computational study of cellobiohydrolase processivity. J Am Chem Soc. 2014;136:8810–9. [DOI] [PubMed] [Google Scholar]
- 6. Olsen JP, Kari J, Windahl MS, Borch K, Westh P. Molecular recognition in the product site of cellobiohydrolase Cel7A regulates processive step length. Biochem J. 2020;477:99–110. [DOI] [PubMed] [Google Scholar]
- 7. Payne CM, Jiang W, Shirts MR, Himmel ME, Crowley MF, Beckham GT. Glycoside hydrolase processivity is directly related to oligosaccharide binding free energy. J Am Chem Soc. 2013;135:18831–9. [DOI] [PubMed] [Google Scholar]
- 8. Claeyssens M, Van Tilbeurgh H, Tomme P, Wood TM, McRae SI. Fungal cellulase systems. Comparison of the specificities of the cellobiohydrolases isolated from Penicillium pinophilum and Trichoderma reesei . Biochem J. 1989;261:819–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gruno M, Väljamäe P, Pettersson G, Johansson G. Inhibition of the Trichoderma reesei cellulases by cellobiose is strongly dependent on the nature of the substrate. Biotechnol Bioeng. 2004;86:503–11. [DOI] [PubMed] [Google Scholar]
- 10. van Tilbeurgh H, Claeyssens M, de Bruyne CK. The use of 4‐methylumbelliferyl and other chromophoric glycosides in the study of cellulolytic enzymes. FEBS Lett. 1982;149:152–6. [Google Scholar]
- 11. Rabinovich ML, Melnik MS, Herner ML, Voznyi YV, Vasilchenko LG. Predominant nonproductive substrate binding by fungal Cellobiohydrolase I and implications for activity improvement. Biotechnol J. 2019;14:1–17. [DOI] [PubMed] [Google Scholar]
- 12. Becker D, Johnson KSH, Koivula A, Schülein M, Sinnott ML. Hydrolyses of α‐ and β‐cellobiosyl fluorides by Cel6A (cellobiohydrolase II) of Trichoderma reesei and Humicola insolens . Biochem J. 2000;345:315–9. [PMC free article] [PubMed] [Google Scholar]
- 13. Von Ossowski I, Ståhlberg J, Koivula A, Piens K, Becker D, Boer H, et al. Engineering the exo‐loop of Trichoderma reesei cellobiohydrolase, Cel7A. A comparison with Phanerochaete chrysosporium Cel7D. J Mol Biol. 2003;333:817–29. [DOI] [PubMed] [Google Scholar]
- 14. Kuusk S, Väljamäe P. When substrate inhibits and inhibitor activates: implications of β‐glucosidases. Biotechnol Biofuels. 2017;10:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kubala M, Plášek J, Amler E. Fluorescence competition assay for the assessment of ATP binding to an isolated domain of Na+, K(+)‐ATPase. Physiol Res. 2004;53:109–13. [PubMed] [Google Scholar]
- 16. Lin M, Nielsen K. Binding of the Brucella abortus lipopolysaccharide O‐chain fragment to a monoclonal antibody. Quantitative analysis by fluorescence quenching and polarization. J Biol Chem. 1997;272:2821–7. [DOI] [PubMed] [Google Scholar]
- 17. Zhang S, Irwin DC, Wilson DB. Site‐directed mutation of noncatalytic residues of Thermobifida fusca exocellulase Ce16B. Eur J Biochem. 2000;267:3101–15. [DOI] [PubMed] [Google Scholar]
- 18. Zhou W, Irwin DC, Escovar‐Kousen J, Wilson DB. Kinetic studies of Thermobifida fusca Cel9A active site mutant enzymes. Biochemistry. 2004;43:9655–63. [DOI] [PubMed] [Google Scholar]
- 19. van Tilbeurgh H, Loontiens FG, Engelborgs Y, Claeyssens M. Studies of the cellulolytic system of Trichoderma reesei QM 9414: binding of small ligands to the 1,4‐β‐glucan cellobiohydrolase II and influence of glucose on their affinity. Eur J Biochem. 1989;184:553–9. [DOI] [PubMed] [Google Scholar]
- 20. van Tilbeurgh H, Pettersson G, Bhikabhai R, Boeck H, Claeyssens M. Studies of the cellulolytic system of Trichoderma reesei QM 9414. Reaction specificity and thermodynamics of interactions of small substrates and ligands with the 1,4‐beta‐glucan cellobiohydrolase II. Eur J Biochem. 1985;148:329–34. [DOI] [PubMed] [Google Scholar]
- 21. Barr BK, Wolfgang DE, Piens K, Claeyssens M, Wilson DB. Active‐site binding of glycosides by Thermomonospora fusca endocellulase E2. Biochemistry. 1998;37:9220–9. [DOI] [PubMed] [Google Scholar]
- 22. Cogswell LP, Raines DE, Parekh S, Jonas O, Maggio JE, Strichartz GR. Development of a novel probe for measuring drug binding to the F1*S variant of human alpha 1‐acid glycoprotein. J Pharm Sci. 2001;90:1407–23. [DOI] [PubMed] [Google Scholar]
- 23. Nordenman B, Danielsson Å, Björk I. The binding of low‐affinity and high‐affinity heparin to antithrombin: fluorescence studies. Eur J Biochem. 1978;90:1–6. [DOI] [PubMed] [Google Scholar]
- 24. Westerlund B, Saarinen M, Person B, Ramaswamy S, Eaker D, Eklund H. Crystallographic investigation of the dependence of calcium and phosphate ions for notexin. FEBS Lett. 1997;403:51–6. [DOI] [PubMed] [Google Scholar]
- 25. Røjel N, Kari J, Sørensen TH, Badino SF, Morth JP, Schaller K, et al. Substrate binding in the processive cellulase Cel7A: transition state of complexation and roles of conserved tryptophan residues. J Biol Chem. 2020;295:1454–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Haddad Momeni M, Payne CM, Hansson H, Mikkelsen NE, Svedberg J, Engström A, et al. Structural, biochemical, and computational characterization of the glycoside hydrolase family 7 cellobiohydrolase of the tree‐killing fungus Heterobasidion irregulare . J Biol Chem. 2013;288:5861–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Muñoz IG, Ubhayasekera W, Henriksson H, Szabó I, Pettersson G, Johansson G, et al. Family 7 cellobiohydrolases from Phanerochaete chrysosporium: crystal structure of the catalytic module of Cel7D (CBH58) at 1.32 Å resolution and homology models of the isozymes. J Mol Biol. 2001;314:1097–111. [DOI] [PubMed] [Google Scholar]
- 28. Ubhayasekera W, Muñoz IG, Vasella A, Ståhlberg J, Mowbray SL. Structures of Phanerochaete chrysosporium Cel7D in complex with product and inhibitors. FEBS J. 2005;272:1952–64. [DOI] [PubMed] [Google Scholar]
- 29. Kurasin M, Valjamae P. Processivity of cellobiohydrolases is limited by the substrate. J Biol Chem. 2011;286:169–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Knott BC, Haddad Momeni M, Crowley MF, MacKenzie LF, Götz AW, Sandgren M, et al. The mechanism of cellulose hydrolysis by a two‐step, retaining cellobiohydrolase elucidated by structural and transition path sampling studies. J Am Chem Soc. 2014;136:321–9. [DOI] [PubMed] [Google Scholar]
- 31. Ståhlberg J, Divne C, Koivula A, Piens K, Claeyssens M, Teeri TT, et al. Activity studies and crystal structures of catalytically deficient mutants of cellobiohydrolase I from Trichoderma reesei . J Mol Biol. 1996;264:337–49. [DOI] [PubMed] [Google Scholar]
- 32. Haddad Momeni M, Ubhayasekera W, Sandgren M, Ståhlberg J, Hansson H. Structural insights into the inhibition of cellobiohydrolase Cel7A by xylo‐oligosaccharides. FEBS J. 2015;282:2167–77. [DOI] [PubMed] [Google Scholar]
- 33. Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, et al. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res. 2000;33:889–97. [DOI] [PubMed] [Google Scholar]
- 34. Kurasin M, Kuusk S, Kuusk P, Sørlie M, Väljamäe P. Slow off‐rates and strong product binding are required for processivity and efficient degradation of recalcitrant chitin by family 18 chitinases. J Biol Chem. 2015;290:29074–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nakamura A, Okazaki KI, Furuta T, Sakurai M, Iino R. Processive chitinase is Brownian monorail operated by fast catalysis after peeling rail from crystalline chitin. Nat Commun. 2018;9:3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schiano‐di‐Cola C, Røjel N, Jensen K, Kari J, Sørensen TH, Borch K, et al. Systematic deletions in the cellobiohydrolase (CBH) Cel7A from the fungus Trichoderma reesei reveal flexible loops critical for CBH activity. J Biol Chem. 2019;294:1807–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand‐binding affinities. Expert Opin Drug Discov. 2015;10:449–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Colussi F, Sorensen TH, Alasepp K, Kari J, Cruys‐Bagger N, Windahl MS, et al. Probing substrate interactions in the active tunnel of a catalytically deficient cellobiohydrolase (Cel7). J Biol Chem. 2015;290:2444–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Payne CM, Bomble YJ, Taylor CB, McCabe C, Himmel ME, Crowley MF, et al. Multiple functions of aromatic‐carbohydrate interactions in a processive cellulase examined with molecular simulation. J Biol Chem. 2011;286:41028–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Goedegebuur F, Dankmeyer L, Gualfetti P, Karkehabadi S, Hansson H, Jana S, et al. Improving the thermal stability of cellobiohydrolase Cel7A from Hypocrea jecorina by directed evolution. J Biol Chem. 2017;292:17418–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bhikhabhai R, Johansson G, Pettersson G. Isolation of cellulolytic enzymes from Trichoderma reesei QM 9414. J Appl Biochem. 1984;6:336–45. [PubMed] [Google Scholar]
- 42. Uzcategui E, Raices M, Montesino R, Johansson G, Pettersson G, Eriksson KE. Pilot‐scale production and purification of the cellulolytic enzyme system from the white‐rot fungus Phanerochaete chrysosporium . Biotechnol Appl Biochem. 1991;13:323–34. [Google Scholar]
- 43. McPherson A. Preparation and analysis of protein crystals. New York, NY: Wiley; 1982. [Google Scholar]
- 44. Otwinowski Z, Minor W. Processing of X‐ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–26. [DOI] [PubMed] [Google Scholar]
- 45. Battye TGG, Kontogiannis L, Johnson O, Powell HR, Leslie AGW. iMOSFLM: a new graphical interface for diffraction‐image processing with MOSFLM. Acta Crystallogr Sect D Biol Crystallogr. 2011;67:271–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Evans P. Scaling and assessment of data quality. Acta Crystallogr Sect D Biol Crystallogr. 2006;62:72–82. [DOI] [PubMed] [Google Scholar]
- 47. Ursby T, Hnberg KA, Appio R, Aurelius O, Barczyk A, Bartalesi A, et al. BioMAX the first macromolecular crystallography beamline at MAX IV laboratory. J Synchrotron Radiat. 2020;27:1415–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mueller U, Thunnissen M, Nan J, Eguiraun M, Bolmsten F, Milàn‐Otero A, et al. MXCuBE3: a new era of MX‐beamline control begins. Synchrotron Radiat News. 2017;30:22–7. [Google Scholar]
- 49. Delagenière S, Brenchereau P, Launer L, Ashton AW, Leal R, Veyrier S, et al. ISPyB: an information management system for synchrotron macromolecular crystallography. Bioinformatics. 2011;27:3186–92. [DOI] [PubMed] [Google Scholar]
- 50. Kabsch W. XDS. Acta Crystallogr Sect D Biol Crystallogr. 2010;66:125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr Sect D Biol Crystallogr. 2011;67:235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Evans PR, Murshudov GN. How good are my data and what is the resolution? Acta Crystallogr Sect D Biol Crystallogr. 2013;69:1204–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Incardona MF, Bourenkov GP, Levik K, Pieritz RA, Popov AN, Svensson O. EDNA: a framework for plugin‐based applications applied to X‐ray experiment online data analysis. J Synchrotron Radiat. 2009;16:872–9. [DOI] [PubMed] [Google Scholar]
- 54. Potterton E, Briggs P, Turkenburg M, Dodson E. A graphical user interface to the CCP4 program suite. Acta Crystallogr Sect D Biol Crystallogr. 2003;59:1131–7. [DOI] [PubMed] [Google Scholar]
- 55. Potterton L, Agirre J, Ballard C, Cowtan K, Dodson E, Evans PR, et al. CCP 4 i 2: the new graphical user interface to the CCP 4 program suite. Acta Crystallogr Sect D Struct Biol. 2018;74:68–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kovalevskiy O, Nicholls RA, Long F, Carlon A, Murshudov GN. Overview of refinement procedures within REFMAC 5: utilizing data from different sources. Acta Crystallogr Sect D Struct Biol. 2018;74:215–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of coot. Acta Crystallogr Sect D Biol Crystallogr. 2010;66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gordon JC, Myers JB, Folta T, Shoja V, Heath LS, Onufriev A. H++: a server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005;33:368–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Anandakrishnan R, Aguilar B, Onufriev AV. H++ 3.0: automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012;40:537–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Best RB, Zhu X, Shim J, Lopes PEM, Mittal J, Feig M, et al. Optimization of the additive CHARMM all‐atom protein force field targeting improved sampling of the backbone φ, ψ and side‐chain χ1 and χ2 dihedral angles. J Chem Theory Comput. 2012;8:3257–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Guvench O, Mallajosyula SS, Raman EP, Hatcher E, Vanommeslaeghe K, Foster TJ, et al. CHARMM additive all‐atom force field for carbohydrate derivatives and its utility in polysaccharide and carbohydrate‐protein modeling. J Chem Theory Comput. 2011;7:3162–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–35. [Google Scholar]
- 63. Pearlman DA, Case DA, Caldwell JW, Ross WS, Cheatham TE, DeBolt S, et al. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput Phys Commun. 1995;91:1–41. [Google Scholar]
- 64. Case DA, Aktulga HM, Belfon K, Ben‐Shalom IY, Brozell SR, Cerutti DS, et al. Amber 2021. San Francisco, CA: University of California; 2021. p. 957. [Google Scholar]
- 65. Shirts MR, Klein C, Swails JM, Yin J, Gilson MK, Mobley DL, et al. Lessons learned from comparing molecular dynamics engines on the SAMPL5 dataset. J Comput Aided Mol Des. 2017;31:147–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Darden T, York D, Pedersen L. Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–92. [Google Scholar]
- 67. Ryckaert J‐P, Ciccotti G, Berendsen HJ. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n‐alkanes. J Comput Phys. 1977;23:327–41. [Google Scholar]
- 68. Miller BR, McGee TD, Swails JM, Homeyer N, Gohlke H, Roitberg AE. MMPBSA.Py: an efficient program for end‐state free energy calculations. J. Chem. Theory Comput. 2012;8:3314–21. [DOI] [PubMed] [Google Scholar]
- 69. Wang Y, Song X, Zhang S, Li J, Shu Z, He C, et al. Improving the activity of Trichoderma reesei cel7B through stabilizing the transition state. Biotechnol Bioeng. 2016;113:1171–7. [DOI] [PubMed] [Google Scholar]
- 70. Hou T, Wang J, Li Y, Wang W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model. 2011;51:69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Schrödinger L. The PyMOL molecular graphics system 1.5.0.4. 2010.
- 72. Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Snapshots at 500 ps of MD simulation of substrate binding in productive mode at subsites −2/−1/+1 in TrCel7A and PcCel7D.
Fig. S2. Snapshots at 500 ps of MD simulation of substrate binding in non‐productive mode at subsites +1/+2/+3 in TrCel7A and PcCel7D.
Fig. S3. Plots of distances between substrate and catalytic amino acids during 10 ns and 100 ns of MD simulations of productive binding mode at subsites −2/−1/+1 of pNPC, pNPL and oNPC, in TrCel7A and in PcCel7D.
Data Availability Statement
The atomic coordinates and structure factors of the TrCel7A structures have been deposited into the Protein Data Bank with accession codes PDB: 4UWT, 4V0Z, 7NYT, and 7OC8, respectively, for the pNPC, pNPL, lactose and oNPC ligand complexes.