

Abstract
Monkeypox virus (MPXV) has generally circulated in West and Central Africa since its emergence. Recently, sporadic MPXV infections in several nonendemic countries have attracted widespread attention. Here, we conducted a systematic analysis of the recent outbreak of MPXV‐2022, including its genomic annotation and molecular evolution. The phylogenetic analysis indicated that the MPXV‐2022 strains belong to the same lineage of the MPXV strain isolated in 2018. However, compared with the MPXV strain in 2018, in total 46 new consensus mutations were observed in the MPXV‐2022 strains, including 24 nonsynonymous mutations. By assigning mutations to 187 proteins encoded by the MPXV genome, we found that 10 proteins in the MPXV are more prone to mutation, including D2L‐like, OPG023, OPG047, OPG071, OPG105, OPG109, A27L‐like, OPG153, OPG188, and OPG210 proteins. In the MPXV‐2022 strains, four and three nucleotide substitutions are observed in OPG105 and OPG210, respectively. Overall, our studies illustrated the genome evolution of the ongoing MPXV outbreak and pointed out novel mutations as a reference for further studies.
Keywords: molecular evolution, monkeypox, MPXV endemic, orthopoxvirus
1. INTRODUCTION
The unexpected monkeypox virus (MPXV‐2022) emergence in several nonendemic countries has raised global concerns recently. Since the smallpox virus was eliminated in 1980, MPXV was largely considered the most severe orthopoxvirus circulating in humans as an incidental host. Epidemiological records showed that MPXV was initially identified around the countries within the Central and West Africa. 1 The first MPXV virus‐associated monkeys were firstly found in Copenhagen. 2 In contrast, the first human infection case was found in the Democratic Republic of Congo in 1970 and then spread to neighboring countries. 3 The first human MPXV infection reported outside Africa was in the United States. 4 In 2017, an MPXV outbreak was reported in Nigeria, followed by a case in the United Kingdom with a Nigeria sojourn. 5 Then, another three cases appeared in the United Kingdom, with a history of visits to Nigeria between 2019 and 2021. However, unlike previous cases with clear epidemiological relationships, the MPXV‐2022 cases occurred sporadically across multiple nonendemic countries. Most patients had no travel history to MPXV‐endemic areas. 6 As of July 15, 2022, there were 11 128 confirmed cases in 75 countries, 7 including 1051 confirmed cases in the United States. The WHO has recently highlighted MPXV as a virus that requires close attention.
MPXV belongs to the genus Orthopoxvirus with a double‐stranded DNA genome, whose clinical manifestations are similar to the smallpox virus. 1 The orthopoxvirus encodes ~200 genes, whereas MPXV encodes about 190 genes which could be assigned to the genome as three parts, a core region, a left arm, and a right arm. Viral replication and assembly genes are encoded by the core region, which is relatively conserved in the genome. The MPXV's left and right variable regions were known to be more involved in the host range and the pathogenicity of the orthopoxvirus. 8 These regions contain an identical but opposite sequence called inverted terminal repeats (ITR), which is prone to forming hair‐pin loop‐outs. 9 Although a similar genome composition was observed in other viruses, the variation across different poxviruses is apparent. 8
Poxvirus was considered to undergo high‐frequency recombination and a gradual process that starts with nonsense mutations and small indels, which leads to gene gain or loss. Inter‐species recombination between cowpox virus and ectromelia virus 10 and intra‐species recombination of vaccinia viruses 11 were documented. Recombination within the variola viruses might cause gene loss resulting in a lower virulence. 12 The gain or loss of genetic material had also been reported in the MPXV. The West Africa (WA) lineage and the Congo Basin (CB) lineage differed by about 900 bp in genome length. 5 The WA lineage has a case fatality rate of 3.6%, whereas the CB lineage has a case fatality rate of 10.6%. 6 The MPXV should, in principle, have a low mutation rate based on the stability of the double‐strand DNA genome. However, 46 single nucleotide polymorphisms (SNPs) have been observed in the MPXV‐2022 strains compared to the NCBI Monkeypox reference sequence NC_063383. 13 Therefore, an in‐depth genomic annotation and evolutionary analysis of the MPXV are urgent to understand better the molecular mechanism of its sudden outbreak.
2. MATERIAL AND METHODS
2.1. Data sources
The genome and CDS sequence of the MPXV was obtained from the National Center for Biotechnology Information (NCBI) on June 22, 2022. A complete list of the MPXV is shown in Supporting Information: Table S1. The specific strains used for pairwise comparisons were MVA‐BN (DQ983238.1), MPXV (MT903344.1), Zaire‐96‐I‐16 (AF380138.1), and VAR‐IND (X69198.1), VAC‐COP (M35027.1), CPV‐GRI (X94355.2). The aligned file of MPXV genomes and their spatiotemporal metadata were acquired from the mpox‐spectrum (https://mpox.gen-spectrum.org/explore) database on June 22, 2022.
2.2. Phylogenetic analysis
The full‐length genome of the MPXV and the MPXV‐2022 phylogenetic tree was constructed using FastTree 2.1.11 (www.microbesonline.org/fasttree/) on June 22, 2022. The phylogenetic analysis infers approximately maximum likelihood with the GTR model of nucleotide evolution. The trees were visualized in R by ggplot2 v3.3.6, ggtree v3.2.1, and ggtreeExtra v1.4.2.
2.3. Nucleotide mutations and sectionalization
The nucleotide mutations, including nucleotide substitutes, deletions, and insertions of each MPXV strain, were called by Nextclade (https://master.clades.nextstrain.org) on June 22, 2022. The sequence quality was evaluated by four metrics (Missing data, Mixed sites, Private mutations, and SNP clusters), which were used in subsequent analysis. NCBI MPXV reference sequence NC_063383 (MPXV‐M5312_HM12_Rivers) was used as the coordinate sequence. The nucleotide mutations occurred more than 20 times and were clustered by hclust from R packages stats v4.1.2 with cluster method “single” and showed on the tree by ggtree v3.2.1. These nucleotide mutations were sectioned referring to sequence lineages from NextStrain on June 27, 2022, and the clustering result above. The de novo mutations in B.1 were listed when they appeared more than three times in all MPXV sequences. The complete list of mutations in various lineages and clades is shown in Supporting Information: Table S3.
2.4. Amino acid mutations and APOBEC3‐like mutations
The genome annotation files of NC_063383 and AF380138.1 were acquired from NCBI on June 22, 2022. The missing protein in the NC_063383 was fixed by AF380138.1 annotation by aligning these two sequences. Each open reading frame's initiation and termination codons were checked while naming the missing proteins in NC_063383. These proteins were called AF380139.1 proteins‐like, such as D2L‐like. The 46 B.1‐specific mutations (2022 outbreak) (excluding two contained in NC_063383) were mapped on the reference NC_063383 with founder A.1‐specific mutations. The influence of single nucleotide mutations on amino acids was calculated by package findout_NTtoAA (github.com/wuaipinglab/genome_treatment). APOBEC3‐like mutations (GA > AA or TC > TT) were found on the same reference.
2.5. MPXV‐2022 consensus genome and protein identity scoring
We extracted the highest proportion of bases at each site to construct consensus sequences from the multiple sequence alignment file of the 2022 outbreak sequence. The open reading frame (ORF) of the consensus sequences was identified by ORFfinder (https://www.ncbi.nlm.nih.gov/orffinder/). The genetic code was set as the standard, and ATG was selected as the starting sequence. The minimum predicted length was 120 bp. The credible ORFs were identified by BLAST searching. NC_063383 and AF380138.1 were used to build the blast database. The across‐species protein identity score was also calculated by BLAST. The BLAST results were filtered by an e value less than or equal to 0.01. All the results were manually checked.
3. RESULTS
To understand the genomic evolution of the MPXV‐2022, here we performed a full‐genome phylogenetic analysis using the sequenced MPXV strains from 1958 to 2022 from NCBI (Figure 1A and Supporting Information: Table S1). The MPXV is currently divided into three large clades, and the strains in 2018 and the current outbreak strains (n = 143) belong to Clade 3. Variation analysis showed that each clade has its unique variations. According to different lineages, we divided these consensus variations into six groups: CB‐specific, Clade 2‐specific, WA‐common, A‐specific, A.1‐specific, and B.1‐specific (Supporting Information: Tables S2 and S3). Notably, the number of SNPs in the current outbreak exceeded previous estimates of the substitution rate for Orthopoxviruses as of 1−2 substitutions per site per year. 14 Phylogenetic analysis of the 2022 outbreak strain showed that the current infections exhibit distinct regional characteristics (Figure 1B). Since most cases were reported from May to June 2022, multiple introductions of the MPXV may have been likely. This may point to the endemic regions where the MPXV may have already spread without surveillance.
Figure 1.

Phylogenetic characterization of the MPXV lineage. (A) Phylogenetic analysis using FastTree of 224 isolated MPXV sequences between October 1958 to June 2022. Branches showing different lineages/clades were colored using orange (2022‐outbreak—West Africa—Clade 3—B.1), pink and black (West Africa Lineage—Clades 2 and 3), and Blue (Congo Basin lineage—Clade 1). In‐depth SNP analysis was performed to determine evolutionary trends at a nucleotide resolution. The term WA‐common refers to shared mutations within the clade; **‐specific refers to group‐specific mutations compared to the coordinate genome (NCBI: NC_063383). (B) Sequence divergence and geographical comparison map of the MPXV‐2022 outbreak sequences (updated as of June 22, 2022).
Mapping B.1‐specific mutations to the reference genome (NC_063383 strain), 13 mutations were densely contained in the right variable region and 8 mutations in the left, particularly in the region of 189−200 Kb (Figure 2A and Supporting Information: Table S4). O'Toole and Rambaut 15 first hypothesized a deaminase editing in driving MPXV with GA > AA or TC > TT mutations with APOBEC3‐like protein. Here, we showed that APOBEC3‐like mutation could account for 16 of 18 synonymous mutations and 23 of 24 nonsynonymous mutations. Further, we identified de novo mutations in the MPXV‐2022 strains, including 23 deletions, 13 insertions, and 22 nucleotide substitutions (Figure 2B and Supporting Information: Table S5). Interestingly, the 17 of 22 de novo nucleotide substitution has a conserved feature associated with APOBEC3 enzymes (Figure 2C and Supporting Information: Table S6). Of these, 19 de novo nucleotide substitutions were in protein‐coding regions, resulting in nonsynonymous mutations in 13 proteins, in which 5 proteins are involved in immunity, ankyrin‐like, and membrane glycoprotein (Figure 2D).
Figure 2.
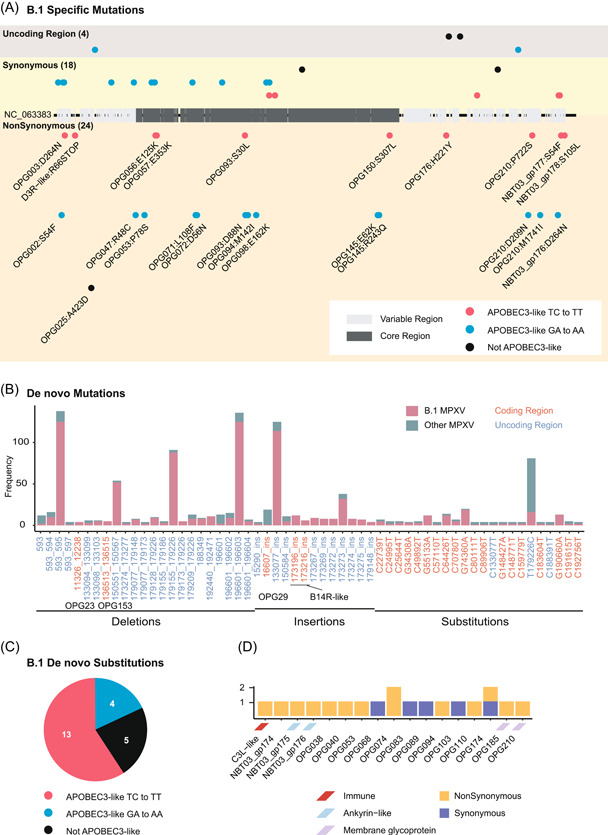
Characterization of the mutation of MPXV‐2022 strains. (A) Mutational analysis map of the MPXV‐2022 outbreak strains. B.1‐specific mutations were characterized and mapped based on their genomic regions (left and right variable region or core region). Nonsynonymous mutations were labeled with the protein and the mutation. APOBEC3‐like mutations were also colored red (for TC > TT mutations) and blue (for GA > AA mutations). (B) de novo mutations in the MPXV‐2022 strains. (C) Association analysis of de novo nucleotide substitutions with APOBEC3‐like in the MPXV‐2022 strains. (D) Protein annotation of de novo nucleotide substitutions in the MPXV‐2022 strains.
Then, the MPXV‐2022 genome was annotated based on its consensus sequence. Its genome, with 187 ORFs could be divided into three regions, including a left variable region (segment 1−31 205 bp, from MPXV‐2022 protein 001 to 030), a conserved core region (segment 31 206−132 419 bp, from MPXV‐2022 protein 031 to 132), and a right variable region (segment 132 420−197 148 bp, from MPXV‐2022 protein 133 to 187) (Figure 3A). The functional annotation of encoded ORFs showed that most of the previously identified immune‐related and membrane‐related ORFs were densely distributed in the left or right variable regions. 9 The predicted host‐range‐related ORFs were mainly in the left variable region.
Figure 3.
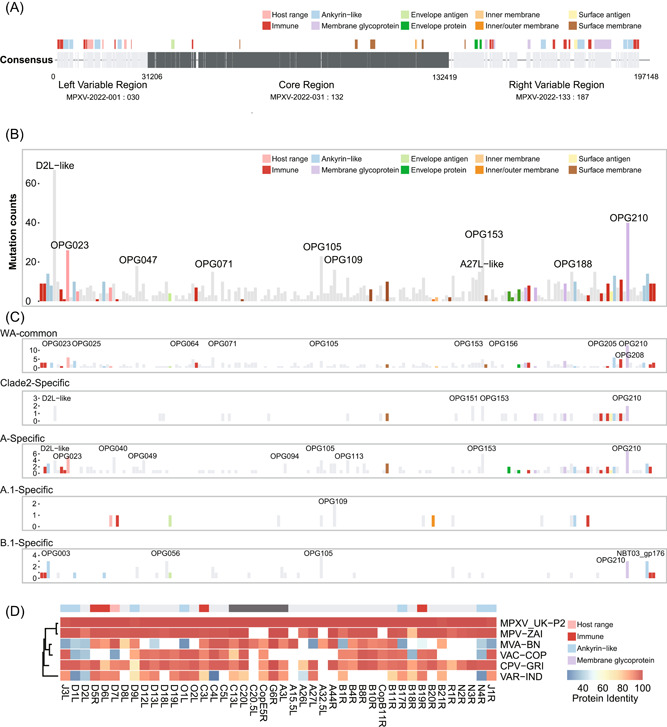
Genomic annotation of the MPXV‐2022 outbreak. (A) Illustration of previously identified MPXV protein annotations. Functions of homologous proteins are color‐coded. (B) A cumulative number of variants type greater than three times on the protein. All the protein annotations were colored accordingly, and the most variable proteins (top 10 percentile) were labeled. (C) A cumulative number of variants type 20 times more in the WA lineage at different sublineages. Genome composition changes were colored accordingly. (D) The identified protein sequence similarity between consensus outbreak sequences and other strains (MPXV‐UK‐P2, MPV‐ZAI, MVA‐BN, VAC‐COP, CPV‐GRI, and VAR‐IND).
Next, we assigned 1121 identified SNP through all monkeypox strains, which occur more than three times, to all 187 ORFs. Although most proteins in the core region have less than 20 mutations, we found that the top 10 proteins (D2L‐like, OPG023, OPG047, OPG071, OPG105, OPG109, A27L‐like, OPG153, OPG188, and OPG210) with the highest number of mutations are distributed throughout the whole genome (Figure 3B). The homologous protein D7L of OPG023 is an ankyrin‐like protein, which the Cowpox virus requires multiplication in Chinese hamster ovary (CHO) cells. 16 The homologous protein B21R of OPG210 is a surface membrane glycoprotein with MPXV‐specific antibody epitopes against Vaccinia. 17 Furthermore, we counted the cumulative number of group‐specific variations on the encoded protein (Figure 3C and Supporting Information: Figure S1 and Table S3). We found four proteins possessed variations among different groups, including OPG023, OPG105, OPG153, and OPG210 proteins (Figure 3C and Supporting Information: Figure S2).
We also compared the protein identity score between the MPXV‐2022 consensus sequence against Zaire‐96‐I‐16, Modified Vaccinia Ankara (MVA‐BN), Vaccinia (VAC‐COP), Cowpox (CPV‐GRI), and Variola (VAR‐IND). We found that 44 proteins, mainly in the left or right variable regions, showed ~80% identified sequence similarities against at least one strain among MVA‐BN, VAC‐COP, CPV‐GRI, and VAR‐IND (Figure 3D and Supporting Information: Table S7). Ten proteins were missing in the MVA‐BN vaccine strain, including D12L, D18L, D19L, O2L, A26L, B18R, B20R, R1R, N2R, and N3R. Two (D9L and B21R) of the 20 mutated proteins in B.1 specific showed similarities between the MPXV‐2022 consensus sequence and MVA‐BN of 92% and 85%, respectively. Further study will be required to examine if these genetic differences will have an impact on the virus.
4. DISCUSSION
There are increasing concerns about monkeypox's global distribution among human populations. Although the human‐to‐human transmissions of the WA lineage have been observed previously, the recent outbreak of novel MPXV sporadically across multiple nonendemic countries has raised significant concerns. Our data suggest that 2022 outbreak strains have most likely been derived from WA‐clade 3. However, it is still unclear if the MPXV‐2022 outbreak strain originated from humans or other hosts.
MPXV is a double‐stranded DNA virus with a low mutation frequency. 18 However, 46 common mutations among the MPXV‐2022 outbreak strains were identified. Although the mechanisms responsible for generating these mutations are unclear, recent studies pointed out the possible contribution of the host APOBEC3‐like deaminase to many of these SNPs. 15 Although we showed that 41/46 mutations could be classified as APOBEC3‐like driving mutations, how a host antiviral gene could lead to mutations accumulated in the outbreak of MPXV remains an open question. Therefore, the continued evolution of the transmission ability of the MPXV requires further attention.
The present mutations in the MPXV‐2022 strains affected at least 20 proteins. It is crucial to determine whether these mutations contribute to the transmission or pathogenesis of the virus or help the virus evade host immunity. Our results showed that OPG105 and OPG210 have nucleotide substitutions in the 2022 outbreak strain. Both proteins have been mutated in multiple lineages of the MPXV. Previous experimental studies have shown that the homologous protein L6R of OPG105 has the epitopes conserved among vaccinia and variola viruses. 19 And the homologous protein of OPG210 was also shown to be associated with monkeypox‐specific antibody epitopes. 17 More investigation is needed to further evaluate the impact of these two protein mutations on viral function.
The smallpox vaccine was considered the most effective way to fight against MPXV. The US CDC has suggested the FDA‐approved MVA‐BN as a potential vaccine strain for MPXV. 20 We found that the vaccine strain differed by 10 proteins by comparing the protein composition and identifying sequence identities between the vaccine strain (MVA‐BN) and the MPXV‐2022 consensus sequence. Together, our study provided an initial assessment of the genetic composition of the 2022 outbreak and showed comprehensive mutation profiling that may serve as a reference for future studies on the transmission and pathogenesis of the current strains.
AUTHOR CONTRIBUTIONS
All authors contributed to the study designing, data analysis, and manuscript writing. Lulan Wang, Jingzhe Shang, and Shenghui Weng performed the research and wrote the manuscript. All authors have revised and approved the final draft of the paper.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
ACKNOWLEDGMENTS
This study was supported by the National Key Research and Development Program (2021YFC2301300, A. W.); the CAMS Innovation Fund for Medical Sciences (2021‐I2M‐1‐061, A. W.); the National Natural Science Foundation of China (92169106, A. W., 31900472, J. S.); the special research fund for central universities, Peking Union Medical College (2021‐PT180‐001, A. W.); Suzhou Science and Technology Development Plan (szs2020311, A. W.), as well as the Research Funds from US National Institute of Health funds (AI069120, AI158154, AI149718, and AI155232), the UCLA AIDS Institute, UCLA David Geffen School of Medicine—Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research Award Program, and Microbial Pathogenesis Training Grant (AI7323‐31).
Wang L, Shang J, Weng S, et al. Genomic annotation and molecular evolution of monkeypox virus outbreak in 2022. J Med Virol. 2022;95:e28036. 10.1002/jmv.28036
Lulan Wang, Jingzhe Shang, and Shenghui Weng contributed equally to this study.
Contributor Information
Genhong Cheng, Email: gcheng@mednet.ucla.edu.
Aiping Wu, Email: wap@ism.cams.cn.
DATA AVAILABILITY STATEMENT
The original contributions presented in the study are included in the article/Supporting Information; further inquiries can be directed to the corresponding author.
REFERENCES
- 1. McCollum AM, Damon IK. Human monkeypox. Clin Infect Dis. 2014;58(2):260‐267. [DOI] [PubMed] [Google Scholar]
- 2. Marennikova S, Gurvich E, Shelukhina E. Comparison of the properties of five pox virus strains isolated from monkeys. Arch Gesamte Virusforsch. 1971;33(3):201‐210. [DOI] [PubMed] [Google Scholar]
- 3. Ladnyj I, Ziegler P, Kima E. A human infection caused by monkeypox virus in Basankusu Territory, Democratic Republic of the Congo. Bull World Health Organ. 1972;46(5):593‐597. [PMC free article] [PubMed] [Google Scholar]
- 4. Control CfD, Prevention . Multistate outbreak of monkeypox—Illinois, Indiana, and Wisconsin, 2003. MMWR Morb Mort Wkly Rep. 2003;52(23):537‐540. [PubMed] [Google Scholar]
- 5. Alakunle E, Moens U, Nchinda G, Okeke MI. Monkeypox virus in Nigeria: infection biology, epidemiology, and evolution. Viruses. 2020;12(11):1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. WHO . Disease outbreak news; multi‐country monkeypox outbreak in non‐endemic countries. 2022. Accessed May 29, 2022. https://wwwwhoint/emergencies/disease-outbreak-news/item/2022-DON388
- 7. team Gh . Monkeypox 2022 global epidemiology. 2022. Accessed July 15, 2022. https://wwwmonkeypoxglobalhealth/
- 8. Lefkowitz E, Wang C, Upton C. Poxviruses: past, present and future. Virus Res. 2006;117(1):105‐118. [DOI] [PubMed] [Google Scholar]
- 9. Shchelkunov SN, Totmenin AV, Safronov PF, et al. Analysis of the monkeypox virus genome. Virology. 2002;297(2):172‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gubser C, Hué S, Kellam P, Smith GL. Poxvirus genomes: a phylogenetic analysis. J Gen Virol. 2004;85(1):105‐117. [DOI] [PubMed] [Google Scholar]
- 11. Coulson D, Upton C. Characterization of indels in poxvirus genomes. Virus Genes. 2011;42(2):171‐177. [DOI] [PubMed] [Google Scholar]
- 12. Esposito JJ, Sammons SA, Frace AM, et al. Genome sequence diversity and clues to the evolution of variola (smallpox) virus. Science. 2006;313(5788):807‐812. [DOI] [PubMed] [Google Scholar]
- 13. nextstrain . monkeypox‐nextstrain. 2022. Accessed May 28, 2022. https://nextstrainorg/monkeypox?l=scatter&scatterY=displayOrder
- 14. Firth C, Kitchen A, Shapiro B, Suchard MA, Holmes EC, Rambaut A. Using time‐structured data to estimate evolutionary rates of double‐stranded DNA viruses. Mol Biol Evol. 2010;27(9):2038‐2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. O'Toole Á, Rambaut A. Initial Observations about Putative APOBEC3 Deaminase Editing Driving Short‐Term Evolution of MPXV Since 2017. ARTIC Network; 2022. [Google Scholar]
- 16. Spehner D, Gillard S, Drillien R, Kirn A. A cowpox virus gene required for multiplication in Chinese hamster ovary cells. J Virol. 1988;62(4):1297‐1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hammarlund E, Lewis MW, Carter SV, et al. Multiple diagnostic techniques identify previously vaccinated individuals with protective immunity against monkeypox. Nat Med. 2005;11(9):1005‐1011. [DOI] [PubMed] [Google Scholar]
- 18. Elde NC, Child SJ, Eickbush MT, et al. Poxviruses deploy genomic accordions to adapt rapidly against host antiviral defenses. Cell. 2012;150(4):831‐841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Terajima M, Cruz J, Leporati AM, Demkowicz Jr., WE , Kennedy JS, Ennis FA. Identification of vaccinia CD8+ T‐cell epitopes conserved among vaccinia and variola viruses restricted by common MHC class I molecules, HLA‐A2 or HLA‐B7. Hum Immunol. 2006;67(7):512‐520. [DOI] [PubMed] [Google Scholar]
- 20. Rao AK, Petersen BW, Whitehill F, et al. Use of JYNNEOS (smallpox and monkeypox vaccine, live, nonreplicating) for preexposure vaccination of persons at risk for occupational exposure to orthopoxviruses: recommendations of the Advisory Committee on Immunization Practices—United States, 2022. MMWR Morb Mort Wkly Rep. 2022;71(22):734‐742. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Supplementary information.
Data Availability Statement
The original contributions presented in the study are included in the article/Supporting Information; further inquiries can be directed to the corresponding author.