

Abstract
Short‐chain enoyl‐CoA hydratase 1 (ECHS1) is involved in the second step of mitochondrial fatty acid β‐oxidation (FAO), catalysing the hydration of short‐chain enoyl‐CoA esters to short‐chain 3‐hyroxyl‐CoA esters. Genetic deficiency in ECHS1 (ECHS1D) is associated with a specific subset of Leigh Syndrome, a disease typically caused by defects in oxidative phosphorylation (OXPHOS). Here, we examined the molecular pathogenesis of ECHS1D using a CRISPR/Cas9 edited human cell ‘knockout’ model and fibroblasts from ECHS1D patients. Transcriptome analysis of ECHS1 ‘knockout’ cells showed reductions in key mitochondrial pathways, including the tricarboxylic acid cycle, receptor‐mediated mitophagy and nucleotide biosynthesis. Subsequent proteomic analyses confirmed these reductions and revealed additional defects in mitochondrial oxidoreductase activity and fatty acid β‐oxidation. Functional analysis of ECHS1 ‘knockout’ cells showed reduced mitochondrial oxygen consumption rates when metabolising glucose or OXPHOS complex I‐linked substrates, as well as decreased complex I and complex IV enzyme activities. ECHS1 ‘knockout’ cells also exhibited decreased OXPHOS protein complex steady‐state levels (complex I, complex III2, complex IV, complex V and supercomplexes CIII2/CIV and CI/CIII2/CIV), which were associated with a defect in complex I assembly. Patient fibroblasts exhibit varied reduction of mature OXPHOS complex steady‐state levels, with defects detected in CIII2, CIV, CV and the CI/CIII2/CIV supercomplex. Overall, these findings highlight the contribution of defective OXPHOS function, in particular complex I deficiency, to the molecular pathogenesis of ECHS1D.
Keywords: ECHS1 deficiency, fatty acid oxidation, mitochondria, mitochondrial disease, OXPHOS, short‐chain enoyl‐CoA hydratase
Short‐Chain Enoyl‐CoA Hydratase (ECHS1) is involved in mitochondrial fatty acid β‐oxidation (left). Loss of ECHS1 causes global gene expression changes that affect the tricarboxylic acid cycle and the electron transport chain (right). This affects complex I and IV activities, resulting in reduced mitochondrial respiratory function. Furthermore, ECHS1 appears to play a role in the assembly of complex I, with loss of ECHS1 associated with reduced complex I levels.
Abbreviations
- ECHS1
short‐chain enoyl‐CoA hydratase 1
- ECHS1D
ECHS1 deficiency
- FAO
mitochondrial fatty acid β‐oxidation (FAO)
- LS
leigh syndrome
- OXPHOS
oxidative phosphorylation
Introduction
ECHS1 (also called crotonase; EC42.1.17) catalyses the second step of fatty acid β‐oxidation (FAO) in mitochondria; the hydration of C4–C6 fatty acyl‐CoA molecules. Expressed in most tissues, ECHS1 encodes eight exons, and transcribes a 1.4 kb transcript that is expressed as a 290 amino acid precursor protein. The mature 28.3 kDa ECHS1 forms a homohexamer ‘dimer of trimers’ in the mitochondria once the mitochondrial targeting sequence has been cleaved. Apart from its role in FAO, ECHS1 is also involved in the breakdown of leucine and isoleucine, and is essential for the breakdown of valine [1].
Short‐chain enoyl‐CoA hydratase 1 deficiency (ECHS1D) is a specific subset of Leigh Syndrome (subacute necrotizing encephalomyelopathy; LS) caused by genetic mutations in ECHS1 that were first identified in 2014 [2] before being characterised as a distinct disease in 2015 [3]. LS is a neurodegenerative disorder associated with over 75 different genes, including those that encode OXPHOS complex subunits and which play a role in OXPHOS complex stability and biogenesis [4]. One of the hallmarks of LS is T2 weighted bilateral hyperintensities, which have been observed in almost all reported cases of ECHS1D. Affected individuals present with a range of symptoms, including developmental delay, dystonia, cardiomyopathy, brain lesions and metabolic acidosis. Patients have a median life span of 2 years, but in severe cases, death can occur within 48 h [2, 5, 6].
Since the categorisation of ECHS1D as a distinct form of LS, more than 46 cases with confirmed ECHS1 mutations have been reported [2, 3, 5, 24]. Due to ECHS1's multi‐functional role, several techniques have been employed to identify ECHS1 deficiency. Western blotting of cultured patient cells has been used [22], as well as exome sequencing and metabolite analysis [8, 12, 14, 17]. Metabolite analysis has frequently revealed increased levels of C4 waste products, as well as the formation of amino acid by‐products such as metharcylyl‐CoA [2, 6, 10, 12].
While biochemical categorisation is still lacking, a phenotypic spectrum consisting of four specific categories, Perinatal Early Fatal (PEF), Severe Infantile (SI), Slowly Progressive Infantile (SPI) and Intermittent (I), has been developed based on current patient clinical and neurological features [25]. As almost all patients have heterozygous ECHS1 mutations, predicting disease severity based on specific genotypes remains difficult [1]. While all genotypes to date have resulted in decreased function of ECHS1, some genotypes have been found to be more pathogenic than others, with mutations such as p.Asn59Ser predicted to be ‘probably damaging’ and associated with lethality in some patients [1, 3]. However, other mutations such as p.Ala173Val are predicted to be ‘possibly damaging’ and appear to be associated with milder symptoms [18], highlighting the clinical heterogeneity of ECHS1D.
Many patients have also been found to have reduced activity of OXPHOS complex enzymes, suggesting that a primary deficiency in ECHS1 causes secondary defects in OXPHOS [3, 12, 13, 20, 22]. However, the mechanism associated with these OXPHOS defects is unknown [1]. OXPHOS complexes I, III and IV have been found to have reduced activity in patient cells, with the accumulation of fatty acid intermediates interfering with OXPHOS activity suggested as a possible mechanism [1]. In addition to reduced OXPHOS activity, western blotting has revealed decreased levels of native OXPHOS complex IV [22].
In this study, we examined nine patients diagnosed with ECHS1D, and a CRISPR/Cas9 ECHS1 ‘knockout’ cell line, to investigate ECHS1D pathogenesis. ECHS1 knockout cells exhibited global gene and protein expression changes, with several key metabolic pathways downregulated, including mitochondrial oxidoreductase activity, NAD binding, and aldehyde dehydrogenase activity. Reduction in native OXPHOS complex levels, reduction in OXPHOS subunit levels, reduced OXPHOS complex I and complex IV activities, and reduced respiratory function were also detected in ECHS1 knockout cells. Similarly, ECHS1D patient fibroblasts also exhibited reduced OXPHOS subunit and complex steady‐state levels, with common defects observed in complex III2, complex IV and the CI/CIII2/CIV supercomplex. Putative interactions between ECHS1 and the OXPHOS complex I subunits NDUFV3 and NDUFB11 were identified by co‐immunoprecipitation and mass spectrometry, suggesting that loss of ECHS1 may affect the stability of these proteins, and in turn, the stability and/or assembly of mature complex I and the CI/CIII2/CIV supercomplex.
Overall, our findings suggest that loss of ECHS1 is associated with OXPHOS complex dysfunction and instability, in particular OXPHOS complex I, which may contribute to the pathogenesis of ECHS1D.
Results
Generation of an ECHS1 ‘knockout’ cell line using CRISPR/Cas9 gene editing
To elucidate the molecular defects underlying ECHS1D, a ‘knockout’ model was generated using CRISPR/Cas9 to edit ECHS1 in 143BTK− osteosarcoma cells. Following transformation with recombinant lentiCRISPR v2‐ECHS1, single‐cell clones were generated and screened via western blot for the absence of ECHS1 protein (data not shown). Sanger sequencing confirmed a heterozygous compound mutation consisting of an 11‐bp deletion (p.Lys92GlyfsX40) and a 44‐bp deletion combined with a 21‐bp insertion (p.Iso85LeufsX34).
No ECHS1 protein was detectable by SDS/PAGE and western blotting in mitochondria isolated from the edited 143BTK− cell clone (Fig. 1A), confirming the loss of ECHS1 expression in these 143BTK− cells (now referred to as ECHS1 KO cells). BN‐PAGE also showed that no native ECHS1 was detectable via western blotting.
Fig. 1.
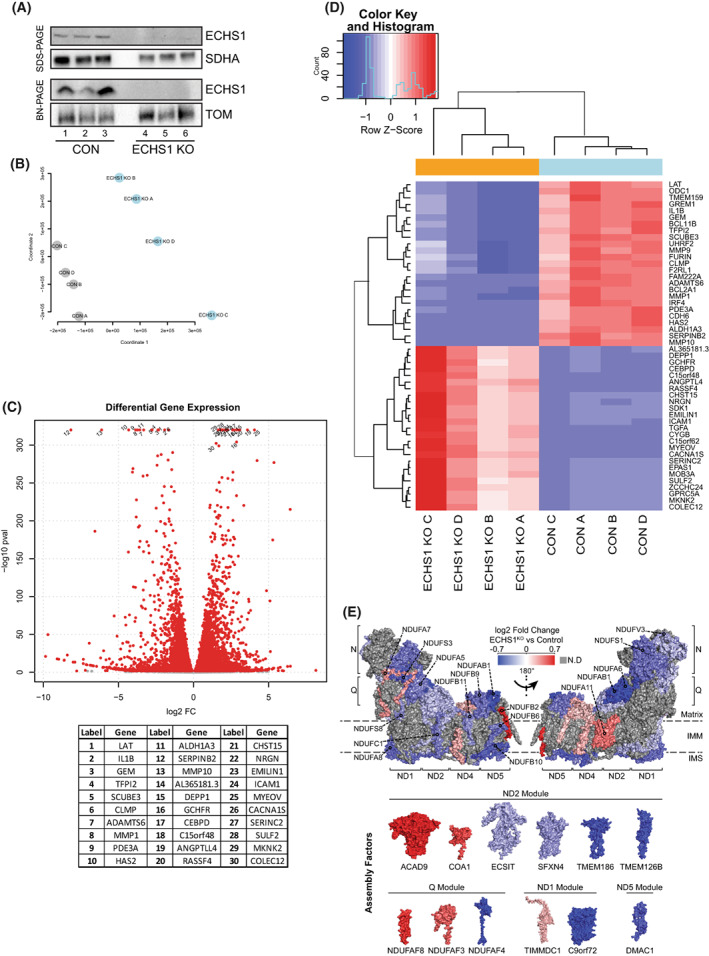
Knockout of ECHS1 impacts global gene expression. (A) Western blot analysis of ECHS1 KO cells confirming the absence of ECHS1 protein via both SDS/PAGE and BN‐PAGE. Image shown is representative of three independent experiments. (B) Multidimensional scaling analysis shows sample clustering based on sample type, either ECHS1 KO cells or control (CON) cells. (C) Volcano plot depicting gene expression changes in ECHS1 KO cells. In total, 11 278 genes were differentially expressed, with 5359 upregulated and 5919 downregulated. Red points indicate False discovery rate; false positive to total positive rate; FDR < 0.05. Top 30 genes with differential expression (as determined by P value) labelled. (D) Heatmap of the 50 most significantly differentially expressed genes in ECHS1 KO cells. (E) Topographical heatmap showing RNA‐Seq log2 fold‐changes mapped onto the structure of complex I, and its assembly factors, from ESCH1 KO cells. Complex I assembly factors are grouped according to their associated complex I assembly modules. Grey subunits had no significant differences in expression.
Complete loss of ECHS1 expression affects global gene expression
RNA‐seq analysis was performed using ECHS1 KO cells and control 143BTK− cells (CON) to determine the effects of ECHS1 loss on global gene expression. Multidimensional scaling analysis was used to cluster samples based on similarity, which showed that CON and ECHS1 KO samples cluster into separate groups (Fig. 1B). Analysis revealed 11 278 differentially expressed genes (FDR < 0.05; False Discovery Rate, false positive to total positive rate), 5359 of which are upregulated in ECHS1 KO and 5919 which are downregulated compared to CON (Fig. 1C, full list in Table S1). Shown in Fig. 1D are the top 50 differentially regulated genes, including C15orf48, a mitochondrial gene involved in replacing NDUFA4 in complex IV during inflammation to reduce ROS production [26].
Several OXPHOS transcripts were downregulated in ECHS1 KO cells, including subunits of complex II (SDHA; −0.48 log2 fold change), complex III (UQCRC2; −0.76 log2 fold change) and complex IV (COX5B; −0.22 log2 fold change, COX7B; −0.27 log2 fold change, COX8A; −0.21 log2 fold change, COX10; −0.66 log2 fold change, COX18; −0.43 log2 fold change, COX20; −0.55 log2 fold change). Notably, each of the six modules that assemble to form mature holocomplex I had reduced expression of their subunits, or their associated assembly factors, in ECHS1 KO cells (Fig. 1E). These include: NDUFB6 (−0.36 log2 fold change), NDUFB10 (−0.53 log2 fold change), NDUFB11 (−0.30 log2 fold change) and TMEM70 (−0.33 log2 fold change) in the ND4 module; NDUFC1 (−0.33 log2 fold change), ECSIT (−0.18 log2 fold change) and TMEM126B (−0.52 log2 fold change) in the ND2 module; NDUFA5 (−0.34 log2 fold change), NDUFS3 (−0.49 log2 fold change), NDUFS8 (−0.29 log2 fold change) and NDUFAF4 (−0.65 log2 fold change) in the Q/ND1 module; NDUFV2 (−0.18 log2 fold change) in the N module and NDUFAB1 (−0.68 log2 fold change)in the ND5 module. TMEM70 (−0.33 log2 fold change), a protein involved in CV biosynthesis and complex I assembly [27], as well as TMEM186 (−0.51 log2 fold change), a known chaperone involved in complex I assembly [28], were also downregulated in ECHS1 KO cells (Fig. 1E).
Interestingly, other mitochondrial genes not involved in OXPHOS or FAO were also impacted in ECHS1 KO cells (Table S1, Fig. 1C). VDAC1 (voltage dependent anion channel 1), which has a role in calcium transport and is the most abundant protein in the mitochondrial outer membrane [29], was found to be upregulated in ECHS1 KO cells (0.18 log2 fold change). The mitochondrial matrix enzyme citrate synthase (CS), which is involved in the TCA cycle [30], also had reduced transcript levels in ECHS1 KO cells (−0.23 log2 fold change). A full list of differentially regulated genes is available in Table S1.
Functional enrichment shows altered gene sets in ECHS1 KO cells
Mitch analysis [31] of Reactome gene sets was used to examine expression changes at the pathway level. After filtering, there were 1471 gene sets with 10 or more detected gene members (out of a total of 2512 gene sets), with 43 upregulated and 464 downregulated in ECHS1 KO cells (Fig. 2A). The top 20 differentially regulated gene sets as ranked by effect size included upregulated pathways such as ‘Mucopolysaccharidoses’ (NAGL4, ARSB, GALNS), ‘Hyaluronan uptake and regulation’ (HEXA), Biotin transport and metabolism (ACACA, ACACB, BTD), which is impacted in Leigh syndrome [32], and ‘unwinding of DNA’, which is comprised primarily of proteins involved in the MCM‐GINS complex (MCM2, MCM3, MCM4, GINS1, GINS2, GINS3; Fig. 2B). Downregulated pathways in the top 20 gene sets include ‘Receptor‐mediated mitophagy’ (SRC, PGAM5, ATG5) with ‘Nucleotide biosynthesis’ (ATIC, GART, PPAT, IMPDH2), ‘FOXO‐mediated transcription of cell death genes’ (NFYA, BCL2L11, CREBBP, FOXO3, PINK1) and ‘Regulation of PTEN mRNA translation’ (PTEN, PTENP1, AGO2, VAPA) in the top 50 gene sets.
Fig. 2.
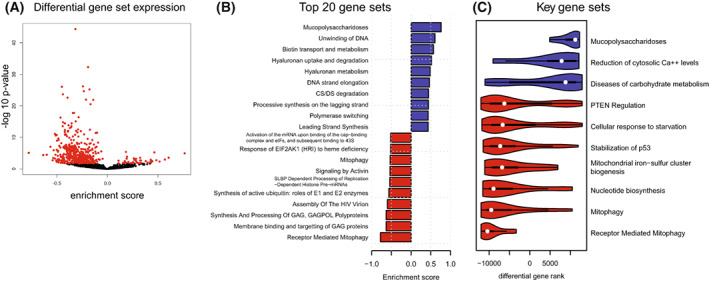
Downregulated mitochondrial metabolic pathways in ECHS1 KO cells. (A) Volcano plot of Reactome gene set enrichment. Out of 1472 gene sets analysed, 43 were upregulated and 464 were downregulated in ECHS1 KO cells. Red points indicate FDR < 0.05. (B) Mitch pathway analysis indicating top 20 differentially expressed gene sets as ranked by effect size in ECHS1 KO cells. (C) Ridge plot of 10 key gene sets that are differentially expressed in ECHS1 KO cells (FDR < 0.05).
Other significantly downregulated gene sets associated with mitochondrial processes include ‘mitochondrial biogenesis’ (−0.22 s.dist; PPARGC1B, CRTC3, SOD2, TFB1M, IDH2), ‘mitochondrial iron sulfur cluster biogenesis’ (−0.43 s.dist; HSCB, SLC25A37, GLRX5, FDX1, NFS1), ‘pyruvate metabolism and citric acid (TCA) cycle’ (−0.23 s.dist; SDHA, CS, IDH2, ME2, SLC16A1), ‘mitochondrial protein import’ (−0.20 s.dist; HSPD1, TIMM44, TOMM5, BCS1L, SAMM50), and ‘respiratory electron transport’ (−0.16 s.dist; TRAP1, UQCRC2, COQ10B, UCRFS1, NDUFA6, COX20; Fig. 2C; with significance determined by FDR < 0.05).
Mitch analysis also revealed a coordinated increased in ‘reduction of cytosolic Ca2+’ (SLC8A3, ATP2A3, CALM1), which could contribute to calcium dysregulation in ECHS1 KO cells. Additionally, ‘diseases of carbohydrate metabolism’ (GBE1, GAA, NAGLU, ARSB, GALNS) was also increased in ECHS1 KO cells (a full list of altered pathways is available in Table S2).
Knockout of ECHS1 disrupts the expression of proteins involved in key mitochondrial pathways
To determine the effects of ECHS1 expression loss at the protein level, mitochondria from ECHS1 KO cells and 143BTK− CON cells were isolated and prepared for proteomic analysis via mass spectrometry. Approximately 2500 proteins were detected (Fig. 3A, shown in grey), with 274 of these having significantly altered expression levels in ECHS1 KO mitochondria compared to control 143BTK− mitochondria (142 proteins upregulated and 132 downregulated; Table S3). Of these, 21 of the upregulated and 41 of the downregulated proteins have a known mitochondrial location [33]. Notably, 22 differentially expressed proteins had corresponding altered expression at the transcript level (Fig. 3A, shown in red, and Fig. 3C,D).
Fig. 3.
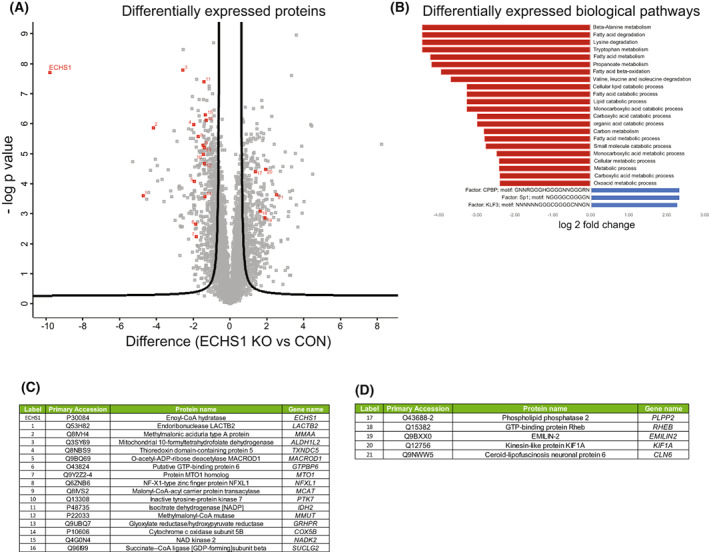
Biological pathways impacted by the loss of ECHS1 protein. (A) Volcano plot of 274 differentially expressed proteins from isolated ECHS1 KO mitochondria; 142 are upregulated and 132 are downregulated compared to control. ECHS1 is shown in red in the top left quadrant. Permutation‐based FDR set at < 1% and s0 = 1. ECHS1 was present in control (CON) cells but at negligible levels in ECHS1 KO cells. Significantly altered proteins which also had significantly altered transcript levels are numbered in red. (B) Functional enrichment of mitochondrial proteins using GO terms and KEGG pathways. Shown are top tier terms with Fisher's exact test and Bonferroni post hoc testing with P < 0.05. (C) Significantly decreased proteins in ECHS1 KO mitochondria that also had significantly reduced transcript levels detected by RNA‐seq (proteins 1–16 in Volcano plot, ranked by protein log2 fold change). (D) Significantly increased proteins in ECHS1 KO mitochondria that also had increased transcript levels detected by RNA‐seq (proteins 17–21 in Volcano plot, ranked by protein log2 fold change).
We next performed enrichment analysis of Gene Ontology proteomic data sets to detect pathway defects at a functional level (Fig. 3B). Downregulated Molecular Functions (MF) in ECHS1 KO mitochondria include ‘catalytic activity’ (GATC, COX5B, ETFA, PTK7), ‘small molecule binding’ (GATC, ALDH1B1, MMUT, PC), ‘oxidoreductase activity’ (ALDH2, COX5B, IDH2, ACAD10) and ‘aldehyde dehydrogenase (NAD+) activity’ (ALDH2, ALDH1B1, ALDH1L2). The top terms as ranked by g:Profiler of downregulated Biological Processes (BP) in ECHS1 KO mitochondria include ‘carboxylic acid metabolic process’ (GATC, ECHS1, DGLUCY, ALDH1L2) and ‘NADP Biosynthetic process’ (IDHP, NAKD2), as well as multiple terms associated with fatty acid metabolism including ‘fatty acid catabolic process’ (ACAD10, ECHS1, ETFA), ‘fatty acid beta‐oxidation’ (ETFA, ECHS1, ACAD10, MCAT) and ‘short‐chain fatty acid metabolic process’ (MMUT, MMAA, PCK2, OXSM; Fig. 3B and Table S4) as might be expected with ECHS1 expression absent.
Interestingly, COX5B (#14, Fig. 3A), a subunit of complex IV involved in the assembly of the MT‐CO2 module, was downregulated in ECHS1 KO mitochondria, corresponding with the RNA‐seq data, where COX5B transcripts were also downregulated (Fig. 3C). Other genes that had corresponding decreased expression at both the protein level and transcript level include: GTPBP6 (#6, Fig. 3A), which is required for the assembly of mitochondrial ribosomes [34], MTO1 (#7, Fig. 3A), involved in wobble base pairing of mitochondrial tRNAs [35], IDH2 (#11, Fig. 3A) and NADK2 (#15, Fig. 3A), both involved in NADPH production [36, 37], and LACTB2 (#1, Fig. 3A), which is essential for normal mitochondrial function [38] (Fig. 3C).
The only increased Gene Ontology terms for ECHS1 KO mitochondrial proteins were three transcription factors with predicted binding site motifs (Fig. 3B). Five of the upregulated proteins included in these terms (PLPP2, KIF1A, RHEB, EMILIN2 and CLN6) were also upregulated at the transcript level (Table S1). Notably, these were the only five genes to be upregulated at both the transcript and protein level in ECHS1 KO cells (Fig. 3D), with none having a known mitochondrial function. A full list of all differentially regulated proteins is available in Table S3.
Loss of ECHS1 expression results in reduced steady‐state levels of OXPHOS proteins
As the transcriptomic and proteomic data revealed the disruption of multiple mitochondrial pathways in ECHS1 KO cells, particularly those associated with OXPHOS, electron transfer and ATP generation, we next investigated whether OXPHOS protein steady‐state levels were also affected. SDS/PAGE and western blotting revealed reduced steady‐state levels of the OXPHOS complex I subunit NDUFB8 (49.8 ± 8.1%, P < 0.01) and the complex IV subunit MT‐CO2 (64.0 ± 9.8%, P < 0.05) in ECHS1 KO cells compared to control (CON) cells (Fig. 4A).
Fig. 4.
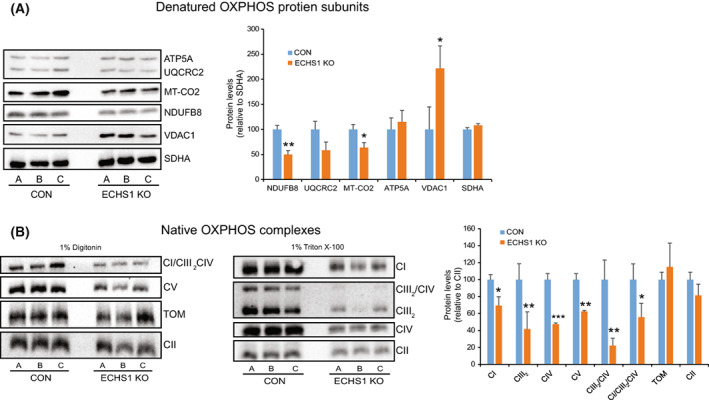
ECHS1 KO cells have decreased OXPHOS protein steady‐state levels. (A) Steady‐state levels of NDUFB8 and MT‐CO2 were decreased in ECHS1 KO mitochondria, whereas levels of the voltage‐dependent anion‐selective channel protein 1 (VDAC1) were increased. (B) Steady‐state levels of complex I (CI), the complex III dimer (CIII2), complex IV (CIV), complex V (CV), the CIII2/CIV supercomplex and the CI/CIII2/CIV supercomplex were all reduced in ECHS1 KO mitochondria compared to control (CON) mitochondria. Data shown as mean ± SD, n = 3. *P < 0.05, **P < 0.01 and ***P < 0.001 compared to control values (Student's two‐tailed t‐test).
Interestingly, increased VDAC1 steady‐state levels were observed in ECHS1 KO cells (221.7 ± 44.9%, P < 0.05), matching the increased VDAC1 transcript expression detected by RNA‐seq (0.18 log2 fold change). OXPHOS complex III subunit UQCRC2 and OXPHOS complex V subunit ATP5A levels were not altered in ECHS1 KO cells (P = 0.140 and P = 0.485 respectively).
BN‐PAGE and western blotting also revealed decreased steady‐state levels of mature OXPHOS complex I (CI, 69.4 ± 10.4%, P < 0.05), the complex III dimer (CIII2, 41.8 ± 20.1%, P < 0.05), complex IV (CIV, 47.5 ± 1.4%, P < 0.001) and complex V (CV, 62.4 ± 1.4%, P < 0.01) in ECHS1 KO cells. In addition, the steady‐state levels of the OXPHOS CI/CIII2/CIV supercomplex (55.7 ± 16.4%, P < 0.05) and the CIII2/CIV supercomplex (22.2 ± 8.7%, P < 0.01) were reduced compared to control (CON) levels (Fig. 4B). The levels of the TOM complex were unchanged between CON and ECHS1 KO cells (P = 0.420).
Interestingly, steady‐state levels of CII (P = 0.235), and its subunit SDHA (P = 0.105), were not altered in ECHS1 KO cells compared to control cells, even though transcript levels of SDHA were significantly reduced (−0.48 log2 fold change). In this case, SDHA protein levels may be stabilised to maintain complex II activity, acting as a compensatory mechanism in the face of other OXPHOS defects, as observed in some complex I deficiencies [39].
Loss of ECHS1 expression affects OXPHOS complex I and IV activity
We next investigated if the altered OXPHOS complex steady‐state levels observed in ECHS1 KO cells also impacts OXPHOS enzyme activity. When compared to mitochondrial matrix enzyme, citrate synthase (CS), complex I (42.6 ± 39.9%, P < 0.05) and complex IV (66.3 ± 9.4%, P < 0.05) had reduced activity compared to controls (Fig. 5A). Activity of complex II (P = 0.241) and complex III (P = 0.195, measured coupled to CII) were not significantly different compared to controls. Interestingly, CS also had significantly reduced activity in ECHS1 KO mitochondria (31.1 ± 4.6%, P < 0.0001), corresponding to the reduced transcript levels identified by RNA‐seq (−0.23 log2 fold change). Therefore, CS may not be an appropriate reference enzyme in this case, and as such we also report the raw enzymatic rates. Here, complex I (21.4 ± 6.6%, P < 0.01) and complex IV (28.2 ± 4.2%, P < 0.001) activity were both decreased compared to controls, whereas complex II and complex II + III activity remain unaffected (P = 0.135 and P = 0.797 respectively) (Fig. 5B).
Fig. 5.
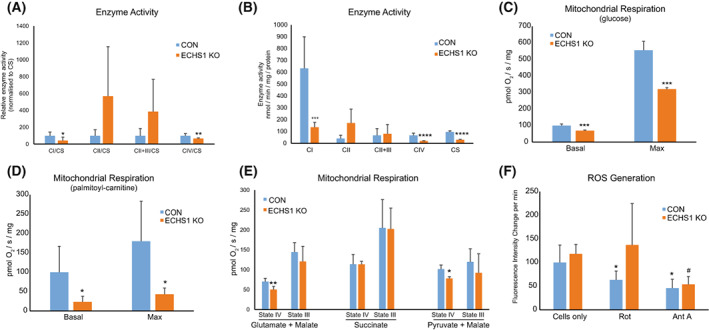
Loss of ECHS1 expression reduces mitochondrial respiratory and enzymatic function. (A) ECHS1 KO mitochondria have reduced levels of CI and CIV activity (normalised to citrate synthase, CS, activity), whereas CII and CII + CIII activity is unchanged compared to control (CON). (B) ECHS1 KO mitochondria have reduced complex I, complex IV and citrate synthase activities (raw rates) compared to control (CON) rates. (C) Basal and maximal respiration rates in ECHS1 KO cells were both significantly reduced when metabolising glucose. (D) Basal and maximal respiration rates in ECHS1 KO cells were both significantly reduced when metabolising the fatty acid ester palmitoyl‐l‐carnitine. (E) ECHS1 KO mitochondria have reduced state IV respiratory rates when metabolising glutamate and malate or pyruvate and malate, but not succinate. (F) Average change in H2DCFDA fluorescence intensity in CON and ECHS1 KO cells under basal and inhibitory conditions. Cellular H2O2 production was decreased after treatment with rotenone (Rot) or antimycin A (AntA) in CON cells. However, only antimycin A treatment reduced H2O2 production in ECHS1 KO cells. Data shown as mean ± SD, n = 3. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 relative to control (CON), # P < 0.05 compared to ECHS1 KO (Student's two‐tailed t‐test).
Loss of ECHS1 expression reduces mitochondrial respiratory capacity
As both OXPHOS complex I and complex IV activities were reduced in ECHS1 KO mitochondria, we next investigated whether these defects affect overall mitochondrial respiratory capacity.
In the presence of glucose, intact ECHS1 KO cells exhibited reduced basal respiration compared to control cells (69.2 ± 4.4%, P < 0.05; Fig. 5C). Maximal respiration in the presence of FCCP was also reduced in ECHS1 KO cells compared to control cells (57.5 ± 1.8%, P < 0.05). Leak respiration (in the presence of oligomycin) was unaffected in ECHS1 KO cells, with no difference in the cell respiratory control ratio (P = 0.702; data not shown). However, spare respiratory capacity was significantly reduced in ECHS1 KO cells compared to the control (55.7 ± 2.6%, P < 0.001).
When metabolising palmitoyl‐l‐carnitine to drive ATP production via fatty acid β‐oxidation, ECHS1 KO cells exhibited reduced basal (26.8 ± 12.3%, P < 0.05) and maximal (42.8 ± 16.5%, P < 0.05) respiration rates (Fig. 5D). This highlights the greater defect in respiratory capacity when ECHS1 KO cells are utilising fatty acids as an energy source, rather than glucose.
The effects of ECHS1 loss on respiratory capacity was also examined in isolated mitochondria using substrates that specifically drive either complex I‐ or complex II‐linked respiration. In the presence of the complex I‐linked substrates glutamate and malate, state IV respiration (ADP‐limiting) in ECHS1 KO mitochondria was 71.5 ± 21.4% (P < 0.01) of control mitochondria (Fig. 5E). State III respiration (stimulated by the addition of ADP) was not significantly different to control mitochondria (P = 0.326). Similarly, when using pyruvate and malate as complex I‐linked substrates, state IV respiration was 76.9 ± 4.4% (P < 0.05) of control mitochondria, while State III respiration was unaffected (P = 0.380; Fig. 5E). When using the complex II‐linked substrate succinate, there was no significant difference in either State IV (P = 0.985) or State III respiration (P = 0.962) between control and ECHS1 KO mitochondria (Fig. 5E).
ECHS1 knockout cells are insensitive to rotenone inhibition
As mitochondrial respiration is disrupted by the loss of ECHS1 expression, we next investigated if this defect in electron flux is associated with increased reactive oxygen species (ROS) generation (Fig. 5F). Total cellular hydrogen peroxide production rates in live cells were assessed using the fluorescent probe H2DCFDA, with no significant difference observed between ECHS1 KO cells and CON cells (Fig. 5E, ‘cells only’, P = 0.989).
Cells were next supplemented with either rotenone, an OXPHOS complex I inhibitor, or Antimycin A, an OXPHOS complex III inhibitor. After supplementation with rotenone, control cells had reduced fluorescence compared to their untreated counterparts (63.3 ± 18.9% of ‘cells only’ values, P < 0.05). In contrast, rotenone treated ECHS1 KO cells did not exhibit reduced fluorescence (P = 0.4073). After supplementation with Antimycin A, both CON cells (45.9 ± 18.9% of CON ‘cells only’ values, P < 0.01) and ECHS1 KO cells (45.4 ± 14.3% of ECHS1 ‘cells only’ values, P < 0.01) exhibited reduced fluorescence. These findings suggest that ECHS1 KO cells have reduced complex I sensitivity to rotenone, as the drug did not reduce cellular H2O2 generation as in CON cells.
Co‐immunoprecipitation of ECHS1 identifies putative interactions with OXPHOS complex I structural subunits
To identify potential interacting protein partners of ECHS1, we performed co‐immunoprecipitation (Co‐IP) analysis of digitonin‐solubilised mitochondria followed by quantitative mass spectrometry. Co‐IPs were performed in triplicate using Protein A‐Sepharose bound anti‐ECHS1 antibodies as bait (ECHS1‐PAS), or with PAS beads alone. In addition, we performed Co‐IPs using ECHS1‐PAS with ECHS1 KO mitochondria to identify proteins that bind non‐specifically to ECHS1 antibodies.
Following solubilisation of control mitochondria in 1% (w/v) digitonin, ECHS1 was captured by ECHS1‐PAS, however non‐specific, high‐molecular weight proteins (detectable by ECHS1 antibodies on western blot) were also captured (Fig. 6A, lane 1). Similar non‐specific, high‐molecular weight proteins were also captured by ECHS1‐PAS from ECHS1 KO mitochondria, although no ECHS1 was captured as would be expected (Fig. 6A, lane 2). Anti‐ECHS1 antibodies did not detect any captured proteins from either control or ECHS1 KO mitochondria following pull‐down with PAS alone (Fig. 6A, lanes 3 and 4).
Fig. 6.
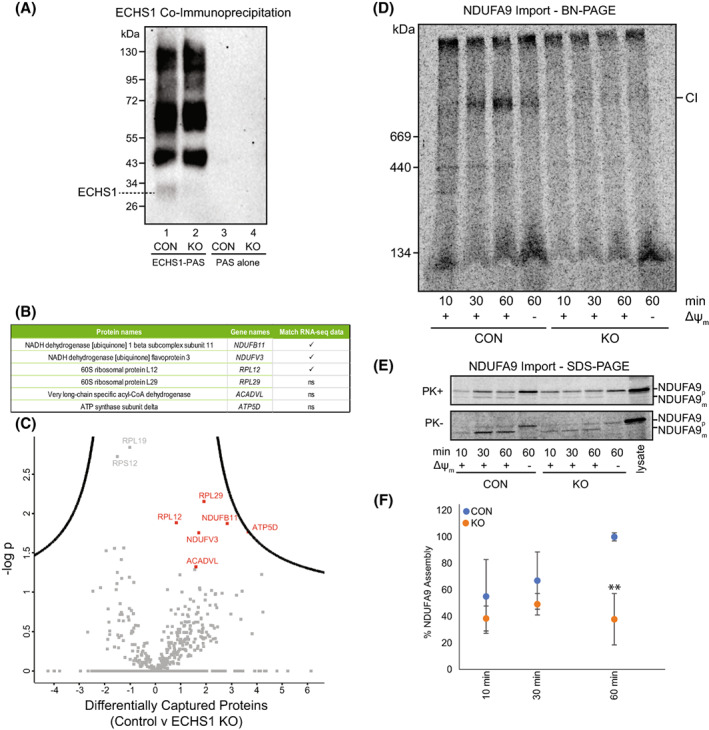
Co‐immunoprecipitation of ECHS1 identifies putative OXPHOS complex I binding partners, with loss of ECHS1 disrupting complex I assembly. Co‐immunoprecipitation with ECHS1 antibodies (ECHS1‐PAS) or PAS alone, followed by western blotting and immunodetection with anti‐ECHS1 antibodies. (A) ECHS1‐PAS pull‐down captured ECHS1 (Lane 1) from control (CON) mitochondria, with non‐specific proteins also pulled down (lane 1). ECHS1 was not captured from ECHS1 KO mitochondria as expected, however non‐specific proteins were pulled down (lane 2). PAS alone did not capture any non‐specific proteins detectable with anti‐ECHS1 antibodies (Lanes 3 and 4) from either CON or ECHS1 KO mitochondria. Image shown is representative of three independent experiments. (B) Six proteins specific for ECHS1 immunocapture identified by comparative analyses of co‐immunoprecipitated proteins captured from CON mitochondria with those captured from ECHS1 KO mitochondria. Of these, RNA‐seq identified reduced transcript levels of NDUFB11, NDUFV3 and RPL12 (ns, not significant). (C) Volcano plot of captured proteins by ECHS1 coimmunoprecipitation. Permutation‐based FDR set at < 1% and s0 = 1. RPL12, RPL29, NDUFB11, NDUFV3, ATP5D and ACADVL (shown in red) present in capture from CON mitochondria but not ECHS1 KO mitochondria (n = 3, Student's two‐tailed t‐test, P < 0.05). (D) BN‐PAGE showing the assembly of NDUFA9 into mature complex I (CI) following solubilisation of 40 μg mitochondria per lane in 1% TX‐100. (E) SDS/PAGE showing NDUFA9 following import into both CON and ECHS1 KO mitochondria. NDUFA9 is detectable in its precursor (p) form and as a proteinase K (PK+) resistant mature (m) form. (F) Quantitation of NDUFA9 assembly into mature complex I after 10, 30 and 60 min of import, normalised to maximum amount of NDUFA9 imported after 60 min in CON. The amount of NDUFA9 assembled into complex I (CI) after 60 min in ECHS1 KO mitochondria was only 37.9 ± 19.3% of CON levels. Data shown are mean ± SD with n = 3, **P < 0.01 relative to control (CON; Student's two‐tailed t‐test). Images shown are representative of three independent experiments.
Statistical analysis of proteins captured from control mitochondria by ECHS1‐PAS, compared to ECHS1 KO mitochondrial proteins captured by ECHS1, identified six putative ECHS1‐interacting proteins: the complex I subunits NDUFB11 and NDUFV3, the complex V subunit ATP5F1D, the FAO protein ACADVL and two ribosomal proteins RPL12 and RPL29 (most likely cytoplasmic contaminants; Fig. 6B,C). Notably, RNA‐seq analysis detected reduced transcript expression of NDUFB11 (−0.26 log2 fold change) and NDUFV3 (−0.30 log2 fold change) in ECHS1 KO cells, suggesting the loss of ECHS1 impacts the expression of these proteins due to the loss of important protein–protein interactions involved in their stability and/or assembly into holocomplex I.
OXPHOS complex I assembly is disrupted in ECHS1 KO cells
To confirm if the reduced complex I steady‐state levels observed in ECHS1 KO cells are due to defects in complex I biogenesis, we performed radiolabelled assembly assays using the complex I structural subunit NDUFA9 (Fig. 6D).
The amount of radiolabelled NDUFA9 assembled into mature complex I was not significantly different between CON and ECHS1 KO cells after 10 or 30 min (P = 0.38 and P = 0.25 respectively, Fig. 6D). However, after 60 min, the amount of NDUFA9 assembled into mature complex I in ECHS1 KO cells was 37.9 ± 19.4% (P < 0.01) of CON levels (Fig. 6D,F).
The mature, proteinase K (PK) resistant form of NDUFA9 (m) was detectable at similar levels in both CON and ECHS1 KO mitochondria following 60 min of import (Fig. 6E). This indicates that the decreased amount of NDUFA9 assembled into complex I is due to the disruption of complex I biogenesis, and not a processing and/or import defect of NDUFA9 due to the loss of ECHS1.
ECHS1D patients exhibit varied defects in mitochondrial OXPHOS protein steady‐state levels
As we observed a significant reduction of mitochondrial OXPHOS complex steady‐state levels in ECHS1 KO cells, we next examined if similar OXPHOS protein defects are present in fibroblasts from patients diagnosed with ECHS1D.
ECHS1 was not detected in patients P1, P2, P4, P6, P7, P8 and P9, however some protein was detectable at significantly reduced levels in patients P3 (3.9 ± 6.8%, P < 0.05) and P5 (20.8 ± 13.2%, P < 0.05; Fig. 7A).
Fig. 7.
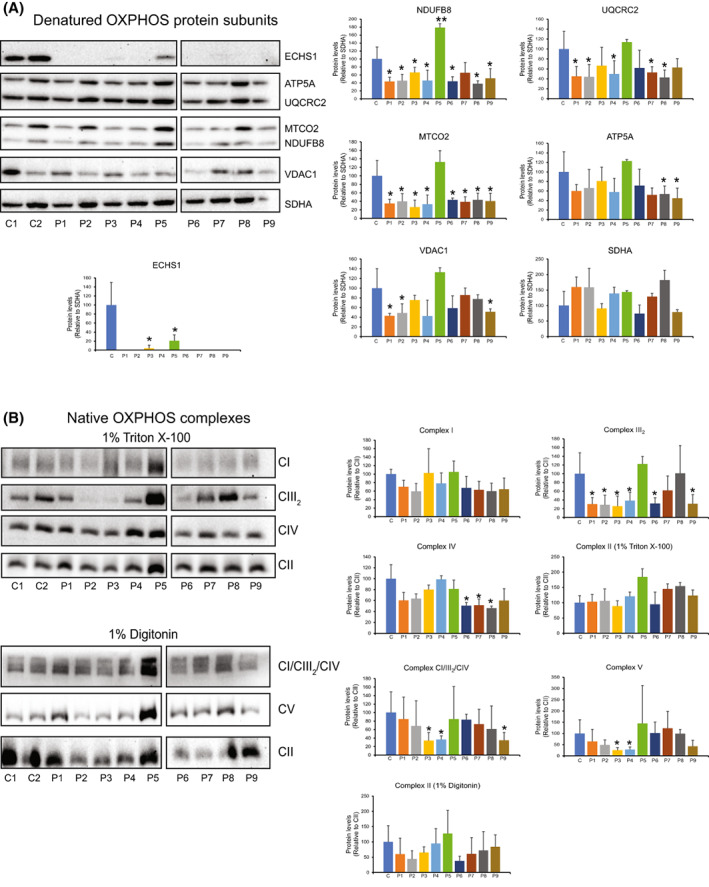
ECHS1‐deficient patient fibroblasts have reduced steady‐state levels of OXPHOS subunits and mature complexes. (A) ECHS1 was detectable in patients P3 and P5, but not in any other patients. NDUFB8 was reduced in all patients, except P5, where NDUFB8 levels were increased, and P7, where NDUFB8 levels were not reduced. UQCRC2 levels were decreased in all patients except P5 and P9. MTCO2 levels were decreased in all patients except P5. ATP5A was decreased in P8 and P9. VDAC1 levels were decreased in P1, P2 and P9. (B) Steady‐state levels of CIII2 were reduced in P2, P3, P4, P6 and P9 compared to control (CON) mitochondria. CIV levels were reduced in P6, P7 and P8. CV levels were reduced in P3 and P4. CI/CIII2/CIV supercomplex levels were reduced in P3, P4 and P9. Images shown are representative of three independent experiments. Values shown are mean ± SD, n = 3. *P < 0.05, **P < 0.01 relative to control (CON; Student's two‐tailed t‐test).
When compared to two control human skin fibroblast lines from healthy subjects, it was found that the complex I subunit, NDUFB8, was decreased in patients P1, P2, P4, P6, P8 and P9 (reduced to between 52% and 38% of CON values) Interestingly, NDUFB8 was increased in P5 to 179.0 ± 9.6% of CON levels (P < 0.01). Patients P1, P2, P4, P7 and P8 had reduced steady‐state levels of the complex III subunit UQCRC2 (reduced to between 53% and 39% of CON values). Decreased levels of the complex IV subunit, MTCO2, was additionally found in P1, P2, P3, P4 and P9 at approximately 40% of CON levels. Reduced levels of the complex V subunit, ATP5A, was detected in patients 8 and 9 only (approximately 50% of CON values).
Interestingly, patients 1, 2 and 9 also exhibited reduced levels of VDAC1 to approximately 50% of control levels. However, this is the opposite finding to ECHS1 KO cells, where the expression of VDAC1 was increased (Fig. 4A).
Native OXPHOS holocomplexes also had altered protein levels in ECHS1D patients (Fig. 7B), with the complex III dimer (CIII2) commonly reduced to approximately 30% in patient cells (P1, P2, P3, P4, P6 and P9). Reduced levels of steady‐state complex IV were detected in patients 6, 7 and 8 (approximately 50% of CON) and reduced levels of steady‐state complex V were detected in patients 3 and 4 (approximately 25% of CON). Native complex I was not found to be reduced in ECHS1D patients, however patients P3, P4 and P9 had decreased levels of the OXPHOS supercomplex CI/CIII2/CIV (approx. 35% of CON levels; Fig. 7B).
Discussion
Mitochondrial disease affects approximately one in 4800 live births and commonly impacts cardiac, skeletomuscular and central nervous systems [40]. These disorders are commonly caused by defects in either oxidative phosphorylation (OXPHOS), mitochondrial fatty acid β‐oxidation (FAO) or disorders of pyruvate metabolism, in particular pyruvate dehydrogenase complex (PDC) deficiencies [41]. However, some mitochondrial disease aetiologies exhibit a combination of defects in both OXPHOS and FAO, including LCHAD, MCAD and ACAD9 deficiencies [42, 43, 44]. LCHAD‐deficient patients exhibit defects in OXPHOS complex I activity, while MCAD‐deficient patients have presented with decreased levels of OXPHOS complexes I, III, IV and the OXPHOS ‘supercomplex’ [45]. ACAD9 has been shown to be essential for complex I biogenesis, with ACAD9 deficient patients exhibiting reduced levels of complex I [43, 46, 47]. Traditionally, combined defects in FAO and OXPHOS were thought to be due to the accumulation of intermediate FAO products that would inhibit normal OXPHOS function [1]. However, current research indicates more complex mechanisms are involved [45]. Studies have revealed that patients with some primary FAO deficiencies, including ECHS1D, also exhibit associated OXPHOS defects in enzyme activity [3, 20] and/or reduced native OXPHOS complex steady‐state levels [22]. Patients with ECHS1D have also been found to have defects in PDC [2, 3, 5, 7, 10, 12, 22]. These findings suggest that a combination of both FAO and OXPHOS defects may contribute to ECHS1D pathogenesis.
In this study, we generated an ECHS1 ‘knockout’ using CRISPR/Cas9 gene editing to examine the molecular defects associated with ECHS1D. In particular, we investigated if loss of ECHS1 not only disrupts mitochondrial FAO but whether secondary defects in OXPHOS are also induced.
Transcriptomics revealed that loss of ECHS1 causes the differential expression of multiple genes and signalling pathways. ECHS1 has been shown to be overexpressed in different human cancers, with the knock‐down of ECHS1 reducing cell proliferation via the downregulation of Akt signalling [48, 49, 50]. Here, we found that knock‐out of ECHS1 in 143B osteosarcoma cells also reduced cell proliferation by 25% (P < 0.05) (data not shown). While we only observed reduced transcripts of the Akt family members AKT1S1 and AKT2, our ontology analyses identified a significant reduction of the ‘PIP3 activates AKT signalling’ pathway, corresponding to previous findings where knock‐down of ECHS1 expression disrupts Akt signalling.
Transcriptomics also revealed the downregulation of several mitochondrial pathways, including key OXPHOS protein subunit genes. Pyruvate metabolism, fatty acid metabolism, and the tricarboxylic acid cycle were downregulated in ECHS1 KO cells, highlighting the widespread impact of the loss of ECHS1 protein. Key genes involved in OXPHOS complex I and complex IV assembly were also found to be downregulated in ECHS1 KO cells. The complex IV subunit COX5B exhibited decreased expression both at the transcript level and at the protein level (as shown by mass spectrometry). COX5B is essential for the formation of the MT‐CO2 module of complex IV [51]. Additionally, the MT‐CO2 module of complex IV requires COX18 for membrane translocation [52] and COX20 for stabilisation [53]. Both COX18 and COX20 transcripts were downregulated in ECHS1 KO cells. These findings suggest a possible defect in complex IV assembly, which is also supported by the reduced complex IV activity and steady‐state levels identified in ECHS1 KO cells.
Transcriptomics also revealed that the assembly of complex I may be compromised, with several subunits and assembly factors downregulated in ECHS1 KO cells (Fig. 1E). NDUFB10 and NDUFB11, both components of the ND4‐module of complex I [51], were downregulated. Furthermore, the assembly factor TMEM70, which is involved in the assembly of the ND4‐module with the ND2‐module, was also downregulated [27]. The ND2‐module also had decreased expression of its subunit NDUFC1 and associated assembly factors ESCIT, SFXN4, TMEM186 and TMEM126B, but interestingly had increased levels of the assembly factors COA1 and ACAD9 [28, 43]. The ND5‐module had decreased expression of NDUFAB1 and NDUFB9 and the assembly factor DMAC1 [54], but increased expression of NDUFB2 and NDUFB8. The N‐module had decreased NDUFV2, NDUFV3, NDUFS6 and NDUFA6 expression, while the Q‐module has decreased expression of its associated assembly factor NDUFAF4 [55]. The ND1‐module has decreased expression of its subunits NDUFS8 and NDUFS3, and the assembly factor C9orf72 [56]. Each of these six subassemblies of complex I had some subunits and/or assembly factors downregulated, which would contribute to the decreased functionality and steady‐state levels of complex I observed in ECHS1 KO cells.
The decreased levels of complex I transcripts, in conjunction with the reduced steady‐state levels of complexes I, IV and complex III2, may also disrupt the stability of the OXPHOS ‘supercomplex’ (CI/CIII2/CIV). Indeed, this was the case in ECHS1 KO cells, with decreased steady‐state levels of the OXPHOS ‘supercomplex’ CI/CIII2/CIV detected.
Proteomics data confirmed several of the downregulated pathways from the transcriptome analyses, including fatty acid degradation and metabolism, fatty acid beta‐oxidation, oxoacid metabolic processes and oxidoreductase activity. These pathways show defects in the FAO proteins ACAD10, ETFA, MCAT and ECHS1, as well as the OXPHOS protein COX5B. Proteomics also revealed increased biotin transport and metabolism, which is impacted in Leigh Syndrome [32].
Co‐immunoprecipitation analyses of ECHS1 revealed putative interactions with the complex I subunits NDUFB11 and NDUFV3, both of which had decreased transcript expression in ECHS1 KO cells. The loss of ECHS1 protein would result in the loss of these interactions, which may reduce the stability of NDUFB11 and NDUFV3. This in turn appears to disrupt the assembly of complex I, resulting in the reduced steady‐state complex I levels observed in ECHS1 KO cells.
Mutations in NDUFV3 have not been associated with human disease [57], with the knockout of NDUFV3 not affecting complex I activity [58]. However, knockout of NDUFV3 does alter the migration of complex I on native‐PAGE, suggesting that its loss does indeed disrupt complex I stability to some degree [58]. Conversely, NDUFB11 mutations have been associated with complex I deficiency [59], with loss of NDUFB11 severely disrupting complex I assembly [60]. Taken together, these findings highlight the requirement of these two subunits for the formation of mature complex I, and that loss of ECHS1 may disrupt the ability of NDUFV3 and NDUFB11 to assemble correctly into holocomplex I.
Previous literature has described patients with ECHS1D who also exhibit OXPHOS defects, such as reduced enzyme activity or reduced complex IV steady‐state levels [22]. Similarly, we observed reduced activity of complex I and complex IV in ECHS1 KO cells. These deficiencies contributed to mitochondrial respiratory dysfunction under standard metabolic conditions (with glucose as the substrate), with both basal and maximal respiration decreased. In addition, a much larger defect was observed when these cells switched to metabolism via FAO (with palmitoyl‐l‐carnitine as the substrate), in part likely due to the primary ECHS1 defect.
State IV respiration rates in isolated mitochondria were also reduced in the presence of complex I‐linked substrates, but not in the presence of complex II‐linked substrates. This suggests that the respiratory defect in ECHS1 KO cells is associated primarily with complex I dysfunction. This finding is consistent with the reduced complex I transcript and protein levels, as well as the complex I enzyme defect that we identified in these cells. Together, these results highlight that the loss of ECHS1 expression decreases the steady‐state levels of the mature OXPHOS complexes and their activity, in particular complexes I and IV, resulting in decreased respiratory capacity.
Nine patients diagnosed with ECHS1D were also examined to determine how pathogenic ECHS1 mutations affect OXPHOS protein levels, in comparison to the total loss of ECHS1 in KO cells. These patients were found to have varied reductions in OXPHOS complex steady‐state levels, with six of the nine patients exhibiting reduced complex III2. Notably some, but not all, of these patients had decreased levels of the complex III subunit UQCRC2, suggesting that there may be other factors, such as disruption of complex III biogenesis, playing a role in the loss of holocomplex III. Eight ECHS1D patients had reduced levels of the complex IV subunit MT‐CO2, with three of the patients having reduced levels of mature complex IV. This is possibly due to disrupted assembly of the MT‐CO2 module into complex IV, as suggested by the ECHS1 KO transcriptomic and proteomic data. Interestingly, while two patients had decreased complex V levels, they did not have decreased ATP5A subunit levels, and vice versa (two patients who had decreased ATP5A subunit levels did not have decreased complex V levels). Only one patient, patient 5, exhibited an increase in OXPHOS protein subunit levels, with elevated levels of the complex I subunit NDUFB8 compared to controls (however this did not lead to an increase in mature complex I in this patient).
Of note, ECHS1 KO cells showed increased levels of VDAC1 expression at both the transcript and protein level. While patient samples did not have increased levels of VDAC1, this may be an important protein for future investigation. VDAC1 is known to have roles in regulating Ca2+ homeostasis, oxidative stress handling and mitochondrial‐mediated apoptosis [61]. Furthermore, the Surf1 knockout mouse model of Leigh Syndrome also exhibits elevated levels of VDAC1 in both heart and skeletal muscle [62]. This suggests that the upregulation of VDAC1 plays a role in the pathogenesis of severe OXPHOS disease, or instead plays a compensatory role to counteract the OXPHOS deficiency present.
Heightened levels of VDAC1 have also been suggested to contribute to the pathology of neurodegenerative disorders, such as Alzheimer's disease [63]. It has also been found that the overexpression of VDAC1 in mice has led to increased Aβ deposits, the hallmark of Alzheimer's Disease [64]. As such, elevated expression of VDAC1 may also play a role in the neuropathological symptoms of ECHS1D, where encephalopathic white matter changes are a common feature [1].
We also examined whether the loss of ECHS1 expression increases oxidative stress under both basal and OXPHOS inhibitory conditions. ECHS1 KO cells generated the same amount of total cellular hydroxyl, peroxyl and other ROS under basal conditions as CON cells. However, once inhibited with rotenone, ROS generation was reduced in CON cells but not ECHS1 KO cells. In contrast, both ECHS1 KO and control cells had lower total ROS species when inhibited with Antimycin A. ECHS1 KO's insensitivity to rotenone may be due to defects in the assembly of the ND4 module of complex I (due to decreased NDUFB11, a possible binding partner of ECHS1), known to be a possible site for rotenone interaction [65]. This provides further evidence that loss of ECHS1 disrupts the structure and/or assembly of complex I, which in turn affects its function.
Conclusion
Diseases associated with OXPHOS complex I typically present with multisystemic symptoms such as growth defects, developmental delay, dystonia, lactic acidosis, hypotonia and neurological defects, including Leigh Syndrome. ECHS1D was first characterised in 2014, and is now considered a specific subset of Leigh Syndrome [2, 3], a disease most commonly caused by complex I disruptions [4]. Here, we confirm that defects in complex I also contribute in some part to the molecular pathogenesis of ECHS1D.
It appears that loss of ECHS1 results in decreased assembly, stability, and/or function of complex I, possibly through interactions with NDUFV3 and NDUFB11. The reduction of complex I steady‐state levels correspond with the reduction of complex I activity, with complex IV activity also reduced. These findings have been previously characterised in patients with ECHS1D. ECHS1 KO cells also have reduced basal and maximal respiratory capacity and were found to be less sensitive to complex I inhibition with rotenone. Our findings highlight a role for ECHS1 in OXPHOS complex biogenesis, and while it remains to be determined exactly how ECHS1 interacts with complex I and possibly other OXPHOS complexes (including the OXPHOS ‘supercomplex’), it is clear that loss of ECHS1 has a detrimental impact on OXPHOS function. These findings have implications for our understanding of ECHS1 deficiency and how OXPHOS dysfunction contributes to disease pathogenesis.
Materials and methods
Patient details
Patients were diagnosed with ECHS1D following clinical examination and genotyping (Table S5). All experiments using patient fibroblasts were performed in accordance with relevant guidelines and regulations, with human ethics approved by the Chiba Children's Hospital Ethics Committee (20150701), the Saitama Medical University Ethics Committee (482) and Deakin University Human Research Ethics Committee (2018‐358). Informed consent was obtained in writing from all participants and/or their legal guardians.
Cell lines and culture conditions
Cells were cultured in supplemented Dulbecco's Modified Eagle Medium (DMEM) media containing 10% (v/v) fetal bovine serum (FBS), 50 units·mL−1 penicillin and 50 μg·mL−1 streptomycin at 37 °C/5% CO2.
Mitochondrial isolation
Cell pellets were resuspended in 20 mm HEPES pH 7.6, 220 mm mannitol, 70 mm sucrose, 1 mm EDTA, 0.5 mm phenylmethylsulfonyl fluoride (PMSF), 2 mg·mL−1 fatty acid‐free BSA, and incubated on ice for 15 min. Cells were then homogenised with 30 strokes of a drill fitted Teflon pestle, then centrifuged for 10 min at 800 g at 4 °C to pellet cellular debris. The supernatant was removed and centrifuged at 10 000 g for 20 min at 4 °C. Mitochondrial pellets were when washed in 20 mm HEPES pH 7.6, 220 mm mannitol, 70 mm sucrose, 1 mm EDTA, and resuspended for downstream analysis. Mitochondrial protein was quantified by measuring the absorbance at 280 and 310 nm according to [66].
Denaturing gel electrophoresis
Proteins were separated using denaturing gel electrophoresis as previously described [67]. In brief, 40 μg of total cell protein (or isolated mitochondria) was separated on a 10–16% (w/v) Tris‐tricine continuous gradient gel at 100 V/25 mA for approximately 14 h.
Native gel electrophoresis
Blue‐native polyacrylamide gel electrophoresis (BN‐PAGE) was performed as previously described [67]. In brief, 40 μg of total cell protein or isolated mitochondria was solubilised for 30 min on ice in 50 μL of 20 mm Bis‐Tris (pH 7.4), 50 mm NaCl, and 10% (v/v) glycerol containing either 1% (v/v) Triton X‐100 (Sigma‐Aldrich, St. Louis, MO, USA) or 1% (w/v) digitonin (Merck, Branchburg, NJ, USA). Samples were spun at 18 000 g for 5 min, 4 °C to pellet insoluble material, and the supernatant was combined with 5 μL of 10× BN‐PAGE loading dye (5% (w/v) Coomassie Blue G, 500 mm Ɛ‐amino‐n‐caproic acid). Samples were resolved on a 4–13% (w/v) BN‐PAGE gel at 100 V/7 mA for 14–16 h at 4 °C.
Western blotting
Semi‐dry Western transfer and immune detection was performed as previously described [68]. Proteins were transferred to a PVDF membrane using a semi‐dry transfer method, blocked in 10% (w/v) skim milk in 1×PBS/0.05% (v/v) Tween 20, then probed overnight with primary antibodies at 4 °C. Membranes were then incubated with appropriate HRP‐coupled secondary antibody, and proteins visualised using ECL (GE Healthcare, Cincinnati, OH, USA). Proteins were visualised with a Chemidoc XRS imaging system (Bio‐rad, Hercules, CA, USA). Primary antibodies used were against mitochondrial oxidative phosphorylation (OXPHOS) proteins NDUFB8, SDHB, UQCRC2, MTCO2 and ATP5A (OXPHOS ‘cocktail’, Abcam, Cambridge, UK, ab110411), VDAC1 (Abcam, ab14734) and ECHS1 (GeneTex, GTX114375). For Native PAGE, the following antibodies were used: SDHA (Abcam, ab14715), ATP5A (Abcam, ab14748), UQCRC1 (Abcam, ab110252), MT‐COI (Abcam, ab14705), NDUFA9 (raised in rabbits, as previously described [69]), TOMM40 (Santa Cruz Biotechnology, Dallas, TX, USA, SC‐11414) and ECHS1 (GeneTex, Irvine, CA, USA, GTX114375). Protein band intensities were calculated using imagej software (National Institutes of Health, Bowie, MD, USA) from three independent, non‐saturated images. Protein complex levels were standardised to SDHA for SDS/PAGE analysis and to mature holocomplex II for BN‐PAGE analysis. Significant differences were determined using Student's two‐tailed t‐tests, with patient data being compared to the average of two controls.
Generation of ECHS1 knockout cells by CRISPR/Cas9 gene editing
ECHS1‐deficient 143BTK− osteosarcoma cells were generated by transformation with recombinant lentiCRISPR v2 vector containing the sgRNA ECHS1 target 5'‐TGC AAA GGC CTT ATC CCC GC‐3′ [70, 71]. HEK293T cells were transfected with recombinant lentiCRISPR v2‐ECHS1, pSPAX2 (Addgene, Watertown, MA, USA, 12259) and VSV‐G (Addgene 8454) to generate infectious viral particles, which were subsequently combined with 5 μL·mL−1 polybrene (Sigma‐Aldrich) to infect 143BTK− cells. Transduced cells were selected with 1 μg·mL−1 puromycin and single cell clones generated by limiting dilution. Untransformed parental 143BTK− osteosarcoma cells were used as controls.
Disruption of the ECHS1 gene was confirmed by PCR amplification of the target region (using primers forward 5′‐GGT CAC ACT GTA TCC ACC G‐3′ and reverse 5'‐CCT TGA CAC ACA CAA AGC C‐3′) and subsequent ligation of the PCR products into the pGEM‐T Easy vector. Transformed bacterial colonies were picked and plasmid clones sequenced to confirm editing of both ECHS1 alleles.
Measurement of mitochondrial oxygen consumption rates
High‐resolution respirometry was performed with an Oxygraph‐2 K oxygen electrode (Oroboros, Innsbruck, Austria). Basal respiration in intact cells was measured in supplemented DMEM (as described above), with non‐phosphorylating respiration (proton leak) measured in the presence of 5 mg·mL−1 oligomycin and maximal respiration determined by the sequential addition of 1 μm aliquots of carbonyl cyanide‐4‐(trifluoromethoxy)phenylhydrazone (FCCP). Non‐mitochondrial respiration was measured in the presence of 2 μm antimycin A. Spare respiratory capacity (maximal rate – basal rate) and cell respiratory control ratios (maximal/proton leak) were calculated according to [72] using datlab software (version 4.51, Oroboros Instruments) and expressed as pmol O2·s−1·mg−1 of whole cell protein. Significant differences were determined using Student's two‐tailed t‐tests.
High‐resolution respirometry was performed in isolated mitochondria with an Oxygraph‐2 K oxygen electrode in respiration buffer (255 mm Mannitol, 75 mm Sucrose, 10 mm KCl, 10 mm Tris HCl pH 7.2, 5 mm KH2PO4 pH 7.2) with the addition of either 5 mm glutamate plus 5 mm malate, 5 mm pyruvate plus 5 mm malate, or 5 mm succinate, and State IV respiration recorded. State III respiration was recorded after the addition of 1 mm ADP.
Assessment of reactive oxygen species (ROS) production
2 × 104 cells/well were plated into a black‐walled, clear‐bottom 96 well plate (Nunc). Cells were incubated with either 5 μm of 2,7‐dichlorodihydruofluroescein diacetate (H2DCFDA) or 5 μm MitoSOX™ Red (Thermo Fisher Scientific, Waltham, MA, USA, M36008) in Hanks Balanced Salt Solution (HBSS) for 30 min at 37 °C. Cells were then incubated with or without 2 μm Rotenone (Sigma) or 4 μm antimycin A (Sigma) and scanned using a Varioskan LUX microplate reader (Thermo Fisher Scientific VL0LATD0) at 37 °C. Fluorescence was measured every 5 min using excitation/emission wavelengths of 485/520 nm for H2DCFDA and 510/600 nm for MitoSOX™. Significant differences were determined using Student's two‐tailed t‐tests.
Assessment of OXPHOS complex activity
Spectrophotometric assessment of mitochondrial OXPHOS complex activity was performed with a Cary‐Win UV 4000 (Agilent, Santa Clara, CA, USA). Mitochondrial samples were measured with specific substrates to assess individual complex activity as previously described [73]. In brief, isolated mitochondria were incubated at 30 °C in reaction buffer [73], substrates added and change in absorbance monitored over time to calculate enzyme activity in nmol·min−1·mg−1 protein [73]. Significant differences were determined using Student's two‐tailed t‐tests.
Co‐immunoprecipitation
Fifty microgram of ECHS1 antibody (Rabbit polyclonal, GeneTex GTX114375) was bound to Protein A Sepharose (PAS, Sigma) beads in Borate Buffer (22 mm Borate, 200 mm NaCl, pH 9.0) overnight at 4 °C. ECHS1‐PAS was coupled with 19.2 mm dimethylpimelimidate for 30 min at room temp, then incubated in 0.2 m ethanolamine for 2 h at room temp. Coupled ECHS1‐PAS was washed twice in 0.1 m Glycine pH 2.5 and then twice in PBS before use.
Isolated mitochondria were prepared from mouse heart and ECHS1 KO cells as described above (with the exception of the removal of BSA), then solubilised in 1% Digitonin, 100 mm NaCl, 20 mm Tris–HCl pH 7.4 on ice for 30 min. Solubilised mitochondria were pelleted at 21 000 g for 5 min at 4 °C, then combined with ECHS1‐PAS or PAS alone. Samples were mixed overnight at 4 °C, then washed in 0.2% Digitonin, 100 mm NaCl, 20 mm Tris–HCl pH 7.4, followed by washing in 0.1% Digitonin, 10 mm NaCl, 2 mm Tris–HCl pH 7.4. Bound protein was eluted by addition of 0.1 m Glycine, pH 2.5 and precipitated in trichloroacetic acid (12% v/v final concentration) and 1.25% Deoxycholic acid for 30 min on ice. Samples were then pelleted at 18 000 g , 30 min, 4 °C. Ice cold acetone was added, and samples again pelleted.
Mass spectrometry
Samples were prepared using the S‐trap method [74]. Six hundred microgram mitochondrial protein isolates were solubilised in 5% SDS in 50 mm Triethylammonium bicarbonate (TEAB) before sonication for 5 min followed by centrifugation at 13 000 g for 8 min. Tris(2‐carboxyethyl)phosphine (TCEP) was added to a final concentration of 10 mm, incubated at 55 °C for 10 min before addition of iodoacetamide to 50 mm and 30 min incubation at room temperature. Phosphoric acid was added to reach final concentration at 2.5%. Then binding/wash buffer (50 mm TEAB in 90% methanol) was added to the sample before loading to an S‐trap column (ProtiFi, Huntington, NY, USA). S‐trap columns were washed four times with binding/wash buffer. Trypsin was added at a 1 : 10 (w/w) ratio before overnight incubation at 37 °C. Peptides were sequentially eluted by centrifugation at 4000 g in 50 mm TEAB, 0.2% formic acid and then 50% acetonitrile. Samples were freeze‐dried before being resuspended in loading buffer (2% acetonitrile (ACN), 0.05% trifluoracetic acid (TFA)).
Peptide samples were analysed by LC–MS/MS using an Orbitrap mass spectrometer coupled online to an Ultimate 3000 UHPLC (Thermo Fisher Scientific). Samples were loaded onto a PepMap C18 trap column (75 μm × 2 cm, Thermo Fisher Scientific) at 50 °C using an isocratic flow of 2% ACN/0.05% TFA at 5 μL·min−1 for 6 min, eluted and separated on a PepMap C18 analytical column (75 μm × 50 cm, Thermo Fisher Scientific) at 50 °C using a flow rate of 300 nL·min−1. The eluents used for the LC were water with 0.1% formic acid (FA) and 5% dimethyl sulfoxide (DMSO) for solvent A and ACN with 0.1% FA and 5% DMSO for solvent B. The gradient was delivered at a flow rate 300 nL·min−1.
For mitochondria‐enriched samples, solvent B was increased from 2% to 23% in 89 min, 23% to 40% in 10 min, 40% to 80% in 5 min, maintained at 80% for 5 min before dropping to 2% in 0.1 min and equilibration at 2% solvent B for 9.9 min. Q Exactive Plus Orbitrap mass spectrometer (Thermo Fisher Scientific) was used for mass spectrometry experiments. The spray voltage, temperature of ion transfer tube and S‐lens were set at 1.9 kV, 250 °C and 70% respectively. The full MS scans were acquired at m/z 375–1400, a resolving power of 70 000, an AGC target value of 3 × 106 and a maximum injection time of 50 ms. The top 15 most abundant ions in each full scan MS spectrum were subjected to higher‐energy collisional dissociation (HCD) at a resolving power of 17 500, AGC target value of 5 × 104, maximum injection time of 50 ms, isolation window of m/z 1.2 and normalised collision energy (NCE) of 30%. Dynamic exclusion of 30 s was enabled.
For co‐immunoprecipitation samples, solvent B was increased from 2% to 23% in 29 min, 23% to 40% in 5 min, 40% to 80% in 5 min, maintained at 80% for 5 min before dropping to 2% in 0.1 min and equilibration at 2% solvent B for 9.9 min. Exploris 480 Orbitrap mass spectrometer (Thermo Fisher Scientific) was used for mass spectrometry experiments. The spray voltage, temperature of ion transfer tube and RF lens were set at 1.9 kV, 275 °C and 40%, respectively. The top speed data dependent acquisition was employed with a cycle time of 3 s. The full MS scans were acquired at m/z 300–1600, a resolving power of 120 000, an AGC target value of 3.0 × 106 and a maximum injection time of 25 ms. The MS/MS experiments were performed using HCD at 30% NCE, a resolving power of 15 000, AGC target value of 7.5 × 104, maximum injection time of 23 ms and isolation window of m/z 1.2. Dynamic exclusion of 20 s was enabled.
All generated files were analysed with maxquant (version 2.0.1.0) for label free quantification (LFQ). Database searching was performed with the following parameters: cysteine carbamidomethylation as a fixed modification and methionine oxidation and protein N‐terminal acetylation as variable modifications; trypsin as enzyme and up to 2 missed cleavages allowed; 1% false discovery rate (FDR) for protein and peptide identification; human or mouse sequence database from UniProt; match‐between‐run and LFQ enabled; all other settings were default values in maxquant. The search output was further processed using perseus (Version 1.6.15.0) for data visualisation and statistical tests. Functional gene enrichment analysis of significantly changed proteins was performed against human or mouse gene database using g:Profiler [75] to study Gene Ontology (e.g. molecular function, cellular component and biological process) and biological pathways (e.g. KEGG, Reactome and WikiPathways). The significance threshold corrected for multiple testing was calculated by the built‐in g:SCS algorithm. The criteria for statistical significance are permutation‐based FDR < 1% and s0 value in volcano plot at 1 for whole cell data and Student's two‐tailed t‐test P < 0.05 and log2 transformed fold change ≥ 1 or ≤ −1 for co‐immunoprecipitation data. The list of significantly detected proteins was determined by comparison of ECHS1‐PAS captured control mitochondrial proteins with ECHS1‐PAS captured ECHS1 KO mitochondrial proteins.
RNA sequencing and differential expression analysis
Total RNA was extracted using the RNeasy Plus Kit with on‐column DNA elimination (Qiagen, Hilden, Germany) according to manufacturer's instructions. Transcriptome‐wide mRNA sequencing was performed by the Australian Genome Research Facility (Melbourne, Australia) from four independent experiments (n = 4). RNA purity and integrity were confirmed by BioAnalyser (Agilent). Libraries were prepared using the Ribo‐zero Gold protocol (Illumina) and assessed by Bioanalyser DNA 1000 chip (Agilent). Libraries were pooled and clustered through the Illumina cBot system using TruSeq PE Cluster Kit v3, followed by sequencing on the Illumina HiSeq 2500 system with TruSeq SBS Kit v3 reagents. 100 bp single‐end reads were produced with a depth of at least 30 million reads per sample. The primary sequence data was generated using the Illumina bcl2fastq 2.18.0.12 pipeline, with per base sequence quality for all samples > 94% bases above Q30. Skewer (v0.2.2 [76]) was used to trim low quality bases from the 3′ end of reads. Trimmed reads were then mapped to the Gencode (v37) human transcriptome using Kallisto [77]. Data were read into R, followed by aggregation of transcript level counts to gene level count data for downstream analysis.
Clustering between the samples was inspected using multidimensional scaling analysis in R. DESeq2 [78] version 1.32.0 was used to perform differential gene expression analysis between control 143BTK− cells and 143BTK− ECHS1 ‘knockout’ cells. DESeq2 data underwent gene set analysis with mitch v1.4.0 with the default parameters [31]. Gene sets used in the analysis were downloaded from Reactome (accessed 10 December 2021) [79]. DESeq2 and mitch results with FDR <0.05 were considered statistically significant.
Topographical mapping of RNA‐Seq log2 fold‐changes to complex I (PDB: 5LDW) and complex I assembly factors (alphafold 2.0: ACAD9 (AF‐Q9H845), COA1 (AF‐Q9GZY4), ECSIT (AF‐Q9BQ95), SFXN4 (AF‐Q6P4A7), TMEM186 (AF‐Q96B77), TMEM126B (AF‐Q8IUX1), NDUFAF8 (AF‐A1L188), NDUFAF3 (AF‐Q9BU61), NDUFAF4 (AF‐Q9P032), TIMMDC1 (AF‐Q9NPL8), C9orf72 (AF‐Q96LT7), DMAC1 (AF Q96GE9) [80, 81]) was conducted with python scripts generously provided by Dr David Stroud (Bio21) and as previously described [54].
In vitro mitochondrial import assays
CDNA encoding NDUFA9 was cloned into the pGEM4Z vector (Promega, Madison, WI, USA) and protein translated using the TnT Coupled Reticulocyte Lysate System (Promega) in the presence of [35S]‐methionine/cysteine. Translation products were incubated with freshly isolated mitochondria in 250 mm sucrose, 80 mm potassium acetate, 5 mm magnesium acetate, 10 mm sodium succinate, 1 mm dithiothreitol, 5 mm ATP and 20 mm HEPES pH 7.4 at 37 °C for the times indicated. Dissipation of the mitochondrial membrane potential (Δψm) was performed in the presence of 10 mm FCCP (with no ATP or sodium succinate). Samples subjected to protease treatment were incubated on ice for 10 min with 100 μg·mL−1 proteinase K (Sigma) before treatment with 1 mm PMSF for 10 min. Forty microgram of each sample resolved on either SDS/PAGE or BN–PAGE as described, with proteins transferred to PVDF membranes before exposure to storage phosphor screens (GE Healthcare) and detection using a Typhoon Laser Scanner (GE Healthcare). Protein band intensities were calculated using imagej (NIH) software from at least three independent experiments, normalised to the maximum amount of imported NDUFA9 after 60 min in controls, and converted to % of control levels at 60 min.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
MM involved in conceptualization; HB, AJS, SN, MZ, AO, KM and MM involved in methodology; HB, AJS, SN, MZ, JJC, DS, MZ and MM involved in formal analysis; HB, AJS, and MM involved in investigation; JP, AO, KM and MM involved in resources; HB, AJS, SN, MZ and MM involved in data curation; HB and MM involved in writing—original draft preparation; HB, AJS, SN, JJC, DS, MZ and MM involved in writing—review and editing; MM involved in supervision; MM involved in project administration; MM involved in funding acquisition. All authors have read and agreed to the published version of the manuscript.
Peer review
The peer review history for this article is available at https://publons.com/publon/10.1111/febs.16595.
Supporting information
Table S1. A full list of differentially expressed genes.
Table S2. A full list of differentially expressed pathways (Mitch).
Table S3. Differentially expressed proteins detected via MS, downregulated in ECHS1 KO.
Table S4. Differentially expressed Biological pathways detected via MS and analysed using g:Profiler, upregulated in ECHS1 KO.
Table S5. Patient genotypes.
Acknowledgements
This work was supported in part by the Practical Research Project for Rare/Intractable Diseases from the Japan Agency for Medical Research and Development, AMED (JP20ek0109468, JP20ek0109482, JP20bm0804018, JP21ek0109511), Deakin University (HJB and MM), the Australian Government, Department of Education Research Training Program Scholarship (HB) and the Australian Mito Foundation (HB). The authors wish to acknowledge the use of the services and facilities of Australian Genome Research Facility (AGRF) and the Mass Spectrometry facility at Bio21, University of Melbourne. Open access publishing facilitated by Deakin University, as part of the Wiley ‐ Deakin University agreement via the Council of Australian University Librarians. Open access publishing facilitated by Deakin University, as part of the Wiley ‐ Deakin University agreement via the Council of Australian University Librarians.
Data availability statement
The mass spectrometry proteomics data in this publication is deposited in the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD032761 and 10.6019/PXD032761.
The transcriptomic data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series accession number GSE200252 study at: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE200252.
References
- 1. Burgin HJ, McKenzie M. Understanding the role of OXPHOS dysfunction in the pathogenesis of ECHS1 deficiency. FEBS Lett. 2020;594:590–610. [DOI] [PubMed] [Google Scholar]
- 2. Peters H, Buck N, Wanders R, Ruiter J, Waterham H, Koster J, et al. ECHS1 mutations in Leigh disease: a new inborn error of metabolism affecting valine metabolism. Brain. 2014;137:2903–8. [DOI] [PubMed] [Google Scholar]
- 3. Haack TB, Jackson CB, Murayama K, Kremer LS, Schaller A, Kotzaeridou U, et al. Deficiency of ECHS1 causes mitochondrial encephalopathy with cardiac involvement. Ann Clin Transl Neurol. 2015;2:492–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lake NJ, Compton AG, Rahman S, Thorburn DR. Leigh syndrome: one disorder, more than 75 monogenic causes. Ann Neurol. 2016;79:190–203. [DOI] [PubMed] [Google Scholar]
- 5. Ferdinandusse S, Friederich MW, Burlina A, Ruiter JP, Coughlin CR 2nd, Dishop MK, et al. Clinical and biochemical characterization of four patients with mutations in ECHS1. Orphanet J Rare Dis. 2015;10:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ganetsky RD, Bloom K, Ahrens‐Nicklas R, Edmondson A, Deardorff MA, Bennett MJ, et al. ECHS1 deficiency as a cause of severe neonatal lactic acidosis. JIMD Rep. 2016;30:33–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Al Mutairi F, Shamseldin HE, Alfadhel M, Rodenburg RJ, Alkuraya FS. A lethal neonatal phenotype of mitochondrial short‐chain enoyl‐CoA hydratase‐1 deficiency. Clin Genet. 2017;91:629–33. [DOI] [PubMed] [Google Scholar]
- 8. Aretini P, Mazzanti CM, La Ferla M, Franceschi S, Lessi F, De Gregorio V, et al. Next generation sequencing technologies for a successful diagnosis in a cold case of Leigh syndrome. BMC Neurol. 2018;18:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Balasubramaniam S, Riley LG, Bratkovic D, Ketteridge D, Manton N, Cowley MJ, et al. Unique presentation of cutis laxa with Leigh‐like syndrome due to ECHS1 deficiency. J Inherit Metab Dis. 2017;40:745–7. [DOI] [PubMed] [Google Scholar]
- 10. Bedoyan JK, Yang SP, Ferdinandusse S, Jack RM, Miron A, Grahame G, et al. Lethal neonatal case and review of primary short‐chain enoyl‐CoA hydratase (SCEH) deficiency associated with secondary lymphocyte pyruvate dehydrogenase complex (PDC) deficiency. Mol Genet Metab. 2017;120:342–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carlston CM, Ferdinandusse S, Hobert JA, Mao R, Longo N. Extrapolation of variant phase in mitochondrial short‐chain enoyl‐CoA hydratase (ECHS1) deficiency. JIMD Rep. 2018;43:103–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fitzsimons PE, Alston CL, Bonnen PE, Hughes J, Crushell E, Geraghty MT, et al. Clinical, biochemical, and genetic features of four patients with short‐chain enoyl‐CoA hydratase (ECHS1) deficiency. Am J Med Genet A. 2018;176:1115–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huffnagel IC, Redeker EJW, Reneman L, Vaz FM, Ferdinandusse S, Poll‐The BT. Mitochondrial encephalopathy and transient 3‐Methylglutaconic aciduria in ECHS1 deficiency: long‐term follow‐up. JIMD Rep. 2018;39:83–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Illsinger S, Korenke GC, Boesch S, Nocker M, Karall D, Nuoffer JM, et al. Paroxysmal and non‐paroxysmal dystonia in 3 patients with biallelic ECHS1 variants: expanding the neurological spectrum and therapeutic approaches. Eur J Med Genet. 2020;63:104046. [DOI] [PubMed] [Google Scholar]
- 15. Mahajan A, Constantinou J, Sidiropoulos C. ECHS1 deficiency‐associated paroxysmal exercise‐induced dyskinesias: case presentation and initial benefit of intervention. J Neurol. 2016;264:185–7. [DOI] [PubMed] [Google Scholar]
- 16. Nair P, Hamzeh AR, Mohamed M, Malik EM, Al‐Ali MT, Bastaki F. Novel ECHS1 mutation in an Emirati neonate with severe metabolic acidosis. Metab Brain Dis. 2016;31:1189–92. [DOI] [PubMed] [Google Scholar]
- 17. Ogawa E, Shimura M, Fushimi T, Tajika M, Ichimoto K, Matsunaga A, et al. Clinical validity of biochemical and molecular analysis in diagnosing Leigh syndrome: a study of 106 Japanese patients. J Inherit Metab Dis. 2017;40:685–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Olgiati S, Skorvanek M, Quadri M, Minneboo M, Graafland J, Breedveld GJ, et al. Paroxysmal exercise‐induced dystonia within the phenotypic spectrum of ECHS1 deficiency. Mov Disord. 2016;31:1041–8. [DOI] [PubMed] [Google Scholar]
- 19. Pajares S, López RM, Gort L, Argudo‐Ramírez A, Marín JL, González de Aledo‐Castillo JM, et al. An incidental finding in newborn screening leading to the diagnosis of a patient with ECHS1 mutations. Mol Genet Metab Rep. 2020;22:100553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sakai C, Yamaguchi S, Sasaki M, Miyamoto Y, Matsushima Y, Goto Y. ECHS1 mutations cause combined respiratory chain deficiency resulting in Leigh syndrome. Hum Mutat. 2015;36:232–9. [DOI] [PubMed] [Google Scholar]
- 21. Stark Z, Tan TY, Chong B, Brett GR, Yap P, Walsh M, et al. A prospective evaluation of whole‐exome sequencing as a first‐tier molecular test in infants with suspected monogenic disorders. Genet Med. 2016;18:1090–6. [DOI] [PubMed] [Google Scholar]
- 22. Tetreault M, Fahiminiya S, Antonicka H, Mitchell GA, Geraghty MT, Lines M, et al. Whole‐exome sequencing identifies novel ECHS1 mutations in Leigh syndrome. Hum Genet. 2015;134:981–91. [DOI] [PubMed] [Google Scholar]
- 23. Uchino S, Iida A, Sato A, Ishikawa K, Mimaki M, Nishino I, et al. A novel compound heterozygous variant of ECHS1 identified in a Japanese patient with Leigh syndrome. Hum Genome Var. 2019;6:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yamada K, Aiba K, Kitaura Y, Kondo Y, Nomura N, Nakamura Y, et al. Clinical, biochemical and metabolic characterisation of a mild form of human short‐chain enoyl‐CoA hydratase deficiency: significance of increased N‐acetyl‐S‐(2‐carboxypropyl)cysteine excretion. J Med Genet. 2015;52:691–8. [DOI] [PubMed] [Google Scholar]
- 25. Masnada S, Parazzini C, Bini P, Barbarini M, Alberti L, Valente M, et al. Phenotypic spectrum of short‐chain enoyl‐Coa hydratase‐1 (ECHS1) deficiency. Eur J Paediatr Neurol. 2020;28:151–8. [DOI] [PubMed] [Google Scholar]
- 26. Clayton SA, Daley KK, MacDonald L, Fernandez‐Vizarra E, Bottegoni G, O’Neil JD et al. Inflammation causes remodeling of mitochondrial cytochrome c oxidase mediated by the bifunctional gene C15orf48. Sci Adv. 2021;7:eabl5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sánchez‐Caballero L, Elurbe DM, Baertling F, Guerrero‐Castillo S, van den Brand M, van Strien J, et al. TMEM70 functions in the assembly of complexes I and V. Biochim Biophy Acta Bioenerg. 2020;1861:148202. [DOI] [PubMed] [Google Scholar]
- 28. Formosa LE, Muellner‐Wong L, Reljic B, Sharpe AJ, Jackson TD, Beilharz TH, et al. Dissecting the roles of mitochondrial complex I intermediate assembly complex factors in the biogenesis of complex I. Cell Rep. 2020;31:107541. [DOI] [PubMed] [Google Scholar]
- 29. Camara AKS, Zhou Y, Wen P‐C, Tajkhorshid E, Kwok W‐M. Mitochondrial VDAC1: a key gatekeeper as potential therapeutic target. Front Physiol. 2017;8:460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wiegand G, Remington SJ. CITRATE SYNTHASE: structure, control, and mechanism. Annu Rev Biophys Biophys Chem. 1986;15:97–117. [DOI] [PubMed] [Google Scholar]
- 31. Kaspi A, Ziemann M. Mitch: multi‐contrast pathway enrichment for multi‐omics and single‐cell profiling data. BMC Genomics. 2020;21:447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Distelmaier F, Huppke P, Pieperhoff P, Amunts K, Schaper J, Morava E, et al. Biotin‐responsive basal ganglia disease: A treatable differential diagnosis of Leigh syndrome. In: Zschocke J, Gibson KM, Brown G, Morava E, Peters V, editors. JIMD reports – case and research reports. Volume 13. Berlin, Heidelberg: Springer Berlin Heidelberg; 2014. p. 53–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rath S, Sharma R, Gupta R, Ast T, Chan C, Durham TJ, et al. MitoCarta3.0: an updated mitochondrial proteome now with sub‐organelle localization and pathway annotations. Nucleic Acids Res. 2021;49:D1541–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lavdovskaia E, Denks K, Nadler F, Steube E, Linden A, Urlaub H, et al. Dual function of GTPBP6 in biogenesis and recycling of human mitochondrial ribosomes. Nucleic Acids Res. 2020;48:12929–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li X, Li R, Lin X, Guan MX. Isolation and characterization of the putative nuclear modifier gene MTO1 involved in the pathogenesis of deafness‐associated mitochondrial 12 S rRNA A1555G mutation. J Biol Chem. 2002;277:27256–64. [DOI] [PubMed] [Google Scholar]
- 36. Han SJ, Jang H‐S, Noh MR, Kim J, Kong MJ, Kim JI, et al. Mitochondrial NADP+‐dependent isocitrate dehydrogenase deficiency exacerbates mitochondrial and cell damage after kidney ischemia‐reperfusion injury. J Am Soc Nephrol. 2017;28:1200–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ohashi K, Kawai S, Murata K. Identification and characterization of a human mitochondrial NAD kinase. Nat Commun. 2012;3:1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Levy S, Allerston CK, Liveanu V, Habib MR, Gileadi O, Schuster G. Identification of LACTB2, a metallo‐β‐lactamase protein, as a human mitochondrial endoribonuclease. Nucleic Acids Res. 2016;44:1813–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Alston CL, Compton AG, Formosa LE, Strecker V, Oláhová M, Haack TB, et al. Biallelic mutations in TMEM126B cause severe complex I deficiency with a variable clinical phenotype. Am J Hum Genet. 2016;99:217–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Meyer JN, Hartman JH, Mello DF. Mitochondrial toxicity. Toxicol Sci. 2018;162:15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. DeBrosse SD, Kerr DS. Chapter 12 – pyruvate dehydrogenase complex deficiency. In: Saneto RP, Parikh S, Cohen BH, editors. Mitochondrial case studies. Boston, MA: Academic Press; 2016. p. 93–101. [Google Scholar]
- 42. Moczulski D, Majak I, Mamczur D. An overview of beta‐oxidation disorders. Postepy Hig Med Dosw (Online). 2009;63:266–77. [PubMed] [Google Scholar]
- 43. Nouws J, te Brinke H, Nijtmans LG, Houten SM. ACAD9, a complex I assembly factor with a moonlighting function in fatty acid oxidation deficiencies. Hum Mol Genet. 2013;23:1311–9. [DOI] [PubMed] [Google Scholar]
- 44. Salvatore D, Paola MMD. Muscle carnitine Palmityltransferase deficiency and myoglobinuria. Science. 1973;182:929. [DOI] [PubMed] [Google Scholar]
- 45. Lim SC, Tajika M, Shimura M, Carey KT, Stroud DA, Murayama K, et al. Loss of the mitochondrial fatty acid beta‐oxidation protein medium‐chain acyl‐coenzyme A dehydrogenase disrupts oxidative phosphorylation protein complex stability and function. Sci Rep. 2018;8:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nouws J, Nijtmans L, Houten SM, van den Brand M, Huynen M, Venselaar H, et al. Acyl‐CoA dehydrogenase 9 is required for the biogenesis of oxidative phosphorylation complex I. Cell Metab. 2010;12:283–94. [DOI] [PubMed] [Google Scholar]
- 47. Repp BM, Mastantuono E, Alston CL, Schiff M, Haack TB, Rötig A, et al. Clinical, biochemical and genetic spectrum of 70 patients with ACAD9 deficiency: is riboflavin supplementation effective? Orphanet J Rare Dis. 2018;13:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhao QM, Kuang F, Wu H, Zhang Y‐H. Attenuation of enoyl coenzyme A hydratase 1 expression in colorectal cancer cells using small interfering RNA inhibits cell proliferation and migration. Mol Med Rep. 2015;12:470–4. [DOI] [PubMed] [Google Scholar]
- 49. Zhu XS, Dai YC, Chen ZX, Xie JP, Zeng W, Lin YY, et al. Knockdown of ECHS1 protein expression inhibits hepatocellular carcinoma cell proliferation via suppression of Akt activity. Crit Rev Eukaryot Gene Expr. 2013;23:275–82. [DOI] [PubMed] [Google Scholar]
- 50. Xu WJ, Chen LG, Chen X, Liu YS, Zheng TH, Song JJ, et al. Silencing ECHS1 attenuates the proliferation and induces the autophagy of hepatocellular carcinoma via impairing cell metabolism and activating AMPK. Neoplasma. 2015;62:872–80. [DOI] [PubMed] [Google Scholar]
- 51. Signes A, Fernandez‐Vizarra E. Assembly of mammalian oxidative phosphorylation complexes I‐V and supercomplexes. Essays Biochem. 2018;62:255–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bourens M, Barrientos A. Human mitochondrial cytochrome c oxidase assembly factor COX18 acts transiently as a membrane insertase within the subunit 2 maturation module. J Biol Chem. 2017;292:7774–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lorenzi I, Oeljeklaus S, Aich A, Ronsör C, Callegari S, Dudek J, et al. The mitochondrial TMEM177 associates with COX20 during COX2 biogenesis. Biochim Biophys Acta Mol Cell Res. 2018;1865:323–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stroud DA, Surgenor EE, Formosa LE, Reljic B, Frazier AE, Dibley MG, et al. Accessory subunits are integral for assembly and function of human mitochondrial complex I. Nature. 2016;538:123–6. [DOI] [PubMed] [Google Scholar]
- 55. Baertling F, Sánchez‐Caballero L, van den Brand MAM, Wintjes LT, Brink M, van den Brandt FA, et al. NDUFAF4 variants are associated with Leigh syndrome and cause a specific mitochondrial complex I assembly defect. Eur J Hum Genet. 2017;25:1273–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang T, Liu H, Itoh K, Oh S, Zhao L, Murata D, et al. C9orf72 regulates energy homeostasis by stabilizing mitochondrial complex I assembly. Cell Metab. 2021;33:531–46.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rodenburg RJ. Mitochondrial complex I‐linked disease. Biochim Biophys Acta Bioenerg. 2016;1857:938–45. [DOI] [PubMed] [Google Scholar]
- 58. Dibley MG, Ryan MT, Stroud DA. A novel isoform of the human mitochondrial complex I subunit NDUFV3. FEBS Lett. 2017;591:109–17. [DOI] [PubMed] [Google Scholar]
- 59. Reinson K, Kovacs‐Nagy R, Õiglane‐Shlik E, Pajusalu S, Nõukas M, Wintjes LT, et al. Diverse phenotype in patients with complex I deficiency due to mutations in NDUFB11. Eur J Med Genet. 2019;62:103572. [DOI] [PubMed] [Google Scholar]
- 60. Kohda M, Tokuzawa Y, Kishita Y, Nyuzuki H, Moriyama Y, Mizuno Y, et al. A comprehensive genomic analysis reveals the genetic landscape of mitochondrial respiratory chain complex deficiencies. PLoS Genet. 2016;12:e1005679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shoshan‐Barmatz V, Nahon‐Crystal E, Shteinfer‐Kuzmine A, Gupta R. VDAC1, mitochondrial dysfunction, and Alzheimer's disease. Pharmacol Res. 2018;131:87–101. [DOI] [PubMed] [Google Scholar]
- 62. Pulliam DA, Deepa SS, Liu Y, Hill S, Lin A‐L, Bhattacharya A, et al. Complex IV‐deficient Surf1(−/−) mice initiate mitochondrial stress responses. Biochem J. 2014;462:359–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zaid H, Abu‐Hamad S, Israelson A, Nathan I, Shoshan‐Barmatz V. The voltage‐dependent anion channel‐1 modulates apoptotic cell death. Cell Death Differ. 2005;12:751–60. [DOI] [PubMed] [Google Scholar]
- 64. Cuadrado‐Tejedor M, Vilariño M, Cabodevilla F, Del Río J, Frechilla D, Pérez‐Mediavilla A. Enhanced expression of the voltage‐dependent anion channel 1 (VDAC1) in Alzheimer's disease transgenic mice: an insight into the pathogenic effects of amyloid‐β. J Alzheimers Dis. 2011;23:195–206. [DOI] [PubMed] [Google Scholar]
- 65. Galemou Yoga E, Schiller J, Zickermann V. Ubiquinone binding and reduction by complex I—open questions and mechanistic implications. Front Chem. 2021;9:672851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Clarke S. A major polypeptide component of rat liver mitochondria: carbamyl phosphate synthetase. J Biol Chem. 1976;251:950–61. [PubMed] [Google Scholar]
- 67. McKenzie M, Lazarou M, Ryan MT. Chapter 18 analysis of respiratory chain complex assembly with radiolabeled nuclear‐ and mitochondrial‐encoded subunits. Methods Enzymol. 2009;456:321–39. [DOI] [PubMed] [Google Scholar]
- 68. McKenzie M, Lazarou M, Thorburn DR, Ryan MT. Analysis of mitochondrial subunit assembly into respiratory chain complexes using blue native polyacrylamide gel electrophoresis. Anal Biochem. 2007;364:128–37. [DOI] [PubMed] [Google Scholar]
- 69. Johnston AJ, Hoogenraad J, Dougan DA, Truscott KN, Yano M, Mori M, et al. Insertion and assembly of human Tom7 into the preprotein translocase complex of the outer mitochondrial membrane. J Biol Chem. 2002;277:42197–204. [DOI] [PubMed] [Google Scholar]
- 70. Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, et al. Genome‐scale CRISPR‐Cas9 knockout screening in human cells. Science. 2014;343:84–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sanjana NE, Shalem O, Zhang F. Improved vectors and genome‐wide libraries for CRISPR screening. Nat Methods. 2014;11:783–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435:297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Trounce IA, Kim YL, Jun AS, Wallace DC. [42] assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Cambridge, MA: Academic Press Inc.; 1996. [DOI] [PubMed] [Google Scholar]
- 74. HaileMariam M, Eguez RV, Singh H, Bekele S, Ameni G, Pieper R, et al. S‐trap, an ultrafast sample‐preparation approach for shotgun proteomics. J Proteome Res. 2018;17:2917–24. [DOI] [PubMed] [Google Scholar]
- 75. Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, et al. G:profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019;47:W191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jiang H, Lei R, Ding SW, Zhu S. Skewer: a fast and accurate adapter trimmer for next‐generation sequencing paired‐end reads. BMC Bioinformatics. 2014;15:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bray NL, Pimentel H, Melsted P, Pachter L. Near‐optimal probabilistic RNA‐seq quantification. Nat Biotechnol. 2016;34:525–7. [DOI] [PubMed] [Google Scholar]
- 78. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Fabregat A, Sidiropoulos K, Garapati P, Gillespie M, Hausmann K, Haw R, et al. The Reactome pathway knowledgebase. Nucleic Acids Res. 2016;44:D481–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, et al. AlphaFold protein structure database: massively expanding the structural coverage of protein‐sequence space with high‐accuracy models. Nucleic Acids Res. 2022;50:D439–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. A full list of differentially expressed genes.
Table S2. A full list of differentially expressed pathways (Mitch).
Table S3. Differentially expressed proteins detected via MS, downregulated in ECHS1 KO.
Table S4. Differentially expressed Biological pathways detected via MS and analysed using g:Profiler, upregulated in ECHS1 KO.
Table S5. Patient genotypes.
Data Availability Statement
The mass spectrometry proteomics data in this publication is deposited in the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD032761 and 10.6019/PXD032761.
The transcriptomic data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series accession number GSE200252 study at: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE200252.