

Abstract
Aims
Transthyretin amyloid cardiomyopathy (ATTR‐CM) is increasingly recognized as a cause of heart failure in the elderly. Although wild‐type transthyretin amyloidosis is the most frequent form of ATTR‐CM found in the elderly, hereditary transthyretin amyloidosis (ATTRv) can also occur. We sought to determine the prevalence of ATTRv among elderly ATTR‐CM patients, identify predictors of ATTRv and evaluate the clinical consequences of positive genetic testing in this population.
Methods and results
Prevalence of ATTRv in elderly ATTR‐CM patients (≥70 years) was assessed in a cohort of 300 consecutive ATTR‐CM patients (median age 78 years at diagnosis, 82% ≥70 years, 16% female, 99% Caucasian). ATTRv was diagnosed in 35 (12%; 95% confidence interval [CI] 3.1–8.8) and 13 (5.3%; 95% CI 5.6–26.7) patients in the overall cohort and in those ≥70 years, respectively. Prevalence of ATTRv among elderly female patients with ATTR‐CM was 13% (95% CI 2.1–23.5). Univariate analysis identified female sex (odds ratio [OR] 3.66; 95% CI 1.13–11.85; p = 0.03), black ancestry (OR 46.31; 95% CI 3.52–Inf; p = 0.005), eye symptoms (OR 6.64; 95% CI 1.20–36.73; p = 0.03) and polyneuropathy (OR 10.05; 95% CI 3.09–32.64; p < 0.001) as the only factors associated with ATTRv in this population. Diagnosis of ATTRv in elderly ATTR‐CM patients allowed initiation of transthyretin‐specific drug treatment in 5 individuals, genetic screening in 33 relatives from 13 families, and identification of 9 ATTRv asymptomatic carriers.
Conclusions
Hereditary transthyretin amyloidosis is present in a substantial number of ATTR‐CM patients aged ≥70 years. Identification of ATTRv in elderly patients with ATTR‐CM has clinical meaningful therapeutic and diagnostic implications. These results support routine genetic testing in patients with ATTR‐CM regardless of age.
Keywords: Amyloidosis, Transthyretin, Genetic testing, Elderly, Age
Introduction
Transthyretin amyloid cardiomyopathy (ATTR‐CM) is a progressive heart disease caused by the extracellular deposition of transthyretin (TTR) amyloid fibrils, either in its hereditary (ATTRv) or wild‐type (ATTRwt) form. 1
Diagnosis of the hereditary nature of ATTR‐CM allows genetic counselling, permits familial screening (facilitating early diagnosis of relatives) and, if polyneuropathy is present, it enables treatment with certain TTR‐specific drugs. 1
Unfortunately, ATTR‐CM is frequently assumed to be of wild‐type origin and TTR genetic testing is not performed when ATTR‐CM is diagnosed in elderly individuals. 2
With these considerations, we sought to determine the prevalence of ATTRv among elderly ATTR‐CM patients, identify predictors of ATTRv and evaluate the clinical consequences of ATTRv diagnosis in this population.
Methods
This was a retrospective analysis of consecutive ATTR‐CM patients evaluated at Hospital Universitario Puerta de Hierro (Madrid, Spain) between December 2008 and November 2021. The study complies with the Helsinki Declaration and was approved by the institution's ethics committee.
The diagnosis of ATTR‐CM was established based on demonstration of TTR amyloid deposits on endomyocardial biopsy or by cardiac uptake grade 2 or 3 on scintigraphy associated with suggestive findings on echocardiogram or cardiac magnetic resonance and no evidence of monoclonal protein. 1 TTR genetic testing was performed by Sanger sequencing during evaluation at our centre or previously at referring centres. Other rare hereditary amyloidosis‐associated genes were not evaluated.
Patients in whom genetic testing was performed because of neurological disease or following ATTRv diagnosis in a relative were excluded.
Clinical data were extracted from medical records and age ≥70 years was used to define the elderly cohort.
Clinical implications evaluated included: (i) initiation of specific therapies as a consequence of ATTRv diagnosis, (ii) number of relatives who underwent predictive genetic testing, and (iii) relatives diagnosed as ATTRv genetic carriers.
Genetic testing policy in relatives of ATTRv patients is to offer genetic screening in adult relatives when the result of genetic screening would have a clinical impact. This includes knowing genetic status for reproductive counselling and for professional advice in certain jobs where developing polyneuropathy/cardiomyopathy could be a limiting factor. If this is not the case, in general we recommend genetic screening around the age of 40 to discharge non‐carriers and avoid unnecessary follow‐up, although this varies depending on the type of variant (in case of early‐onset Val50Met, we recommend screening around 20 years old).
Statistical analyses
Continuous variables were reported as mean ± standard deviation, or median and interquartile range (IQR). Categorical variables were reported as number and percentage. Logistic regression univariate analysis was used to identify factors associated with ATTRv in elderly patients. Data were analysed using STATA version 15.0 (StataCorp, College Station, TX, USA).
Results
During the study period, 342 patients were evaluated for ATTR‐CM. A total of 42 patients had to be excluded according to pre‐specified criteria (Figure 1 ). Therefore, the final study cohort comprised 300 subjects. Characteristics are presented in Table 1 . A total of 203 patients (67.7%) had been referred from other centres; 237 patients (79%) were referred by cardiologists while 63 (21%) were referred by other specialties (online supplementary Table S1 and Figure S1 ). Eighty‐two (40.4% of those referred) had not undergone genetic testing at the referral centre and this proportion increased with age (Figure 2 ).
Figure 1.
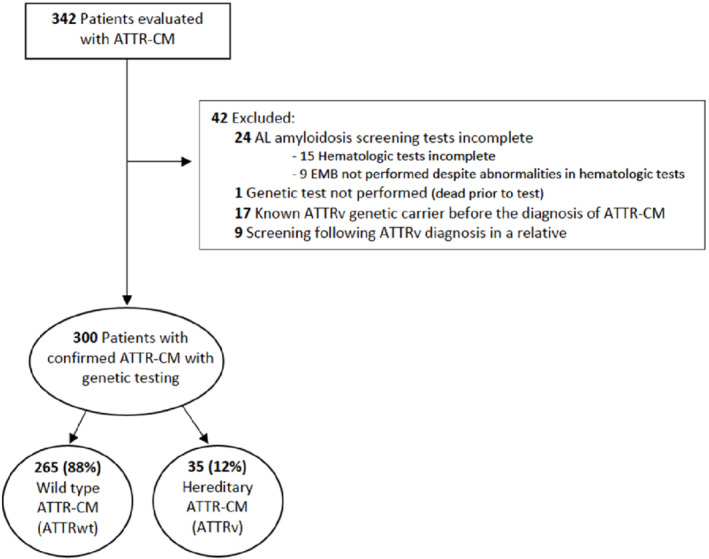
Flow‐chart of study participants. ATTR‐CM, transthyretin amyloid cardiomyopathy; EMB, endomyocardial biopsy.
Table 1.
Baseline clinical characteristics
| Total (n = 300) | Hereditary ATTR‐CM (n = 35) | Wild‐type ATTR‐CM (n = 265) | p‐value | |
|---|---|---|---|---|
| Baseline characteristics | ||||
| Female sex, n (%) | 47 (15.7) | 12 (34.3) | 35 (13.2) | 0.001 |
| Race, n (%) | 0.003 | |||
| Caucasian | 297 (99.0) | 33 (94.3) | 262 (99.6) | |
| Black | 3 (1.0) | 2 (5.7) | 1 (0.4) | |
| Age at diagnosis, years, median (IQR) | 78 (72–84) | 66 (58–75) | 79 (74–84) | <0.001 |
| ATTRv genotype (n = 35), n (%) | ||||
| Val50Met | 9 (25.7) | |||
| Val142Ile | 14 (40.0) | |||
| Glu109Gln | 1 (2.9) | |||
| Glu109Lys | 5 (14.3) | |||
| Ser43Asn | 3 (8.6) | |||
| Thr60Ala | 1 (2.9) | |||
| Val142del | 2 (5.7) | |||
| Type of diagnosis, n (%) | 0.99 | |||
| Invasive | 86 (28.7) | 10 (28.6) | 76 (28.7) | |
| Non‐invasive | 214 (71.3) | 25 (71.4) | 189 (71.3) | |
| Diagnostic era, n (%) | 0.776 | |||
| <2015 | 32 (10.7) | 4 (11.4) | 28 (10.6) | |
| 2016–2021 | 268 (89.3) | 31 (88.6) | 237 (89.4) | |
| Onset symptoms, n (%) | 0.354 | |||
| Dyspnoea | 191 (63.7) | 23 (65.7) | 168 (63.4) | |
| Peripheral oedema | 12 (4.0) | 0 (0.0) | 12 (4.5) | |
| Chest pain | 8 (2.7) | 1 (2.9) | 7 (2.6) | |
| Palpitations | 14 (4.7) | 3 (8.6) | 11 (4.2) | |
| Syncope | 19 (6.3) | 3 (8.6) | 16 (6.0) | |
| Incidental finding | 43 (14.3) | 4 (11.4) | 39 (14.7) | |
| Other | 13 (4.3) | 1 (2.9) | 12 (4.5) | |
| Polyneuropathy, n (%) | 61 (20.3) | 21 (60.0) | 40 (15.1) | <0.001 |
| Carpal tunnel syndrome, n (%) | 116 (38.7) | 16 (45.7) | 100 (37.7) | 0.362 |
| History of embolism, n (%) | 27 (9.0) | 4 (11.4) | 23 (8.7) | 0.54 |
| Atrial fibrillation, n (%) | 188 (62.7) | 12 (34.3) | 176 (66.4) | <0.001 |
| Hypertension, n (%) | 196 (65.3) | 15 (42.9) | 181 (68.3) | 0.003 |
| Coronary artery disease, n (%) | 47 (15.7) | 0 (0.0) | 47 (17.7) | 0.002 |
| Pacemaker, n (%) | 40 (13.3) | 4 (11.4) | 36 (13.6) | 1 |
| Chronic kidney disease (stage ≥3A), n (%) | 141 (47.0) | 9 (25.7) | 132 (49.8) | 0.007 |
| Baseline blood tests | ||||
| eGFR, ml/min/1.73 m2, mean ± SD | 61.3 ± 19.3 | 70.8 ± 19.4 | 60.1 ± 19.0 | 0.002 |
| NT‐proBNP, pg/ml, median (IQR) | 2400 (982–441) | 2558 (959–5236) | 2344 (991–4258) | 0.54 |
| Troponin I, μg/L, median (IQR) | 0.06 (0.03–0.11) | 0.04 (0.02–0.12) | 0.06 (0.03–0.10) | 0.45 |
| Baseline ECG, n (%) | ||||
| 1st degree AV block | 58 (19.3) | 7 (20.0) | 51 (19.3) | 0.92 |
| LBBB | 50 (19.0) | 6 (17.1) | 44 (16.6) | 0.95 |
| RBBB (n = 297) | 49 (16.3) | 4 (11.4) | 45 (17.0) | 0.69 |
| Baseline echocardiography | ||||
| Interventricular wall thickness, mm, mean ± SD | 18.0 ± 3.5 | 18.8 ± 3.7 | 17.9 ± 3.4 | 0.16 |
| End‐diastolic left ventricular diameter, mm, mean ± SD | 42.9 ± 6.5 | 40.9 ± 6.6 | 43.2 ± 6.5 | 0.06 |
| Left ventricular ejection fraction, %, mean ± SD | 53.6 ± 11.3 | 55.0 ± 9.9 | 53.7 ± 11.5 | 0.51 |
| Global longitudinal strain, −% (n = 217), mean ± SD | 12.4 ± 4.3 | 12.5 ± 3.7 | 12.4 ± 4.4 | 0.93 |
| Left atrial diameter, mm, mean ± SD | 44.6 ± 6.9 | 42.4 ± 7.0 | 44.8 ± 6.9 | 0.08 |
| Aortic stenosis (at least moderate), n (%) | 10 (3.3) | 0 (0.0) | 10 (3.8) | 0.61 |
| Pericardial effusion, n (%) | 52 (17.5) | 9 (25.7) | 43 (16.4) | 0.17 |
ATTR‐CM, transthyretin amyloid cardiomyopathy; ATTRv, hereditary transthyretin amyloidosis; AV, atrioventricular; ECG, electrocardiogram; eGFR, estimated glomerular filtration rate; IQR, interquartile range; LBBB, left bundle branch block; NT‐proBNP, N‐terminal pro‐brain natiuretic peptide; RBBB, right bundle branch block; SD, standard deviation.
Figure 2.
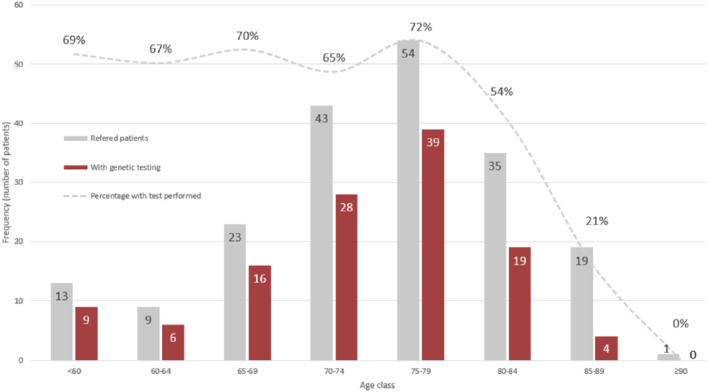
Proportion of referred patients with and without transthyretin genetic testing performed at referral centres.
Genetic testing revealed a pathogenic TTR variant in 35 patients (12%). The most common genetic variant was Va142Ile (14, 40%), followed by Val50Met (9, 26%). The proportion of ATTRv patients decreased with age, despite ATTRv cases were found in all age intervals (Figure 3 ).
Figure 3.
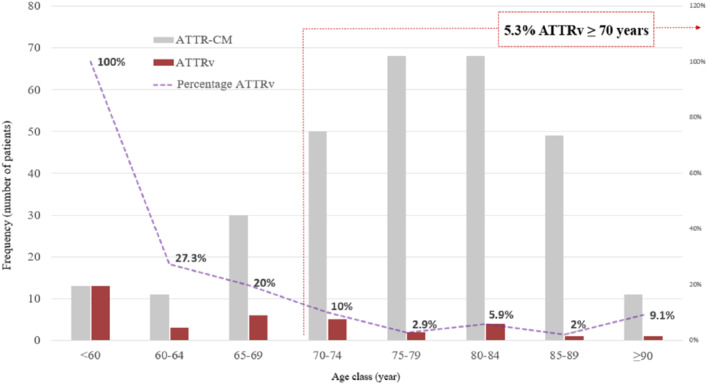
Prevalence of hereditary transthyretin amyloidosis (ATTRv) in patients with transthyretin amyloid cardiomyopathy (ATTR‐CM) according to age.
As expected, ATTRwt patients were older at diagnosis (79 [IQR 74–84] vs. 66 [IQR 58–75] years; p < 0.001) and were less frequently female (13% vs. 34%; p = 0.001). ATTRv had more neurological symptoms (60% vs. 15%; p < 0.001) and higher estimated glomerular filtration rate (70.8 ± 19.4 vs. 60.1 ± 19.0 ml/min/1.73 m2; p = 0.002). Of note, electrocardiographic and echocardiographic parameters and N‐terminal pro‐brain natriuretic peptide (NT‐proBNP) levels did not differ between groups.
Elderly patients
A total of 279 (82%) ATTR‐CM patients ≥70 years underwent evaluation at our centre during the inclusion period; 33 patients were excluded according to pre‐specified criteria. Therefore, the final elderly cohort comprised 246 patients (online supplementary Figure S2 ). Characteristics are presented in Table 2 . Among them, 13 patients (5.3%; 95% CI 3.1–8.8%) had ATTRv (Figure 3 ). The eldest ATTRv patient diagnosed was a 93‐year‐old Caucasian female with the Val142Ile variant. Prevalence of ATTRv in elderly female patients was 13% (95% CI 5.6–26.7%). Distribution of ATTRv by age and sex is shown in online supplementary Figure S3 .
Table 2.
Baseline characteristics of elderly transthyretin amyloid cardiomyopathy patients (n = 246)
| Total (n = 246) | Hereditary ATTR‐CM (n = 13) | Wild‐type ATTR‐CM (n = 233) | p‐value | |
|---|---|---|---|---|
| Baseline characteristics | ||||
| Female sex, n (%) | 39 (15.9) | 5 (38.5) | 34 (14.6) | 0.022 |
| Race, n (%) | <0.001 | |||
| Caucasian | 244 (99.2) | 11 (84.6) | 233 (100) | |
| Black | 2 (0.8) | 2 (15.4) | 0 (0.0) | |
| Age at diagnosis, years, median (IQR) | 80 (76–85) | 79 (73–82) | 80 (76–85) | 0.26 |
| ATTRv genotype (n = 35), n (%) | ||||
| Val50Met | 3 (23.1) | |||
| Val142Ile | 10 (76.9) | |||
| Type of diagnosis, n (%) | 1.00 | |||
| Invasive | 65 (26.4) | 3 (23.1) | 62 (26.6) | |
| Non‐invasive | 181 (73.6) | 10 (76.9) | 171 (73.4) | |
| Diagnosis era, n (%) | 1.00 | |||
| <2015 | 25 (10.2) | 1 (7.7) | 24 (10.3) | |
| 2016–2021 | 221 (89.8) | 12 (92.3) | 209 (89.7) | |
| Onset symptoms, n (%) | 0.802 | |||
| Dyspnoea | 191 (63.7) | 23 (65.7) | 168 (63.4) | |
| Peripheral oedema | 12 (4.0) | 0 | 12 (4.5) | |
| Chest pain | 8 (2.7) | 1 (2.9) | 7 (2.6) | |
| Palpitations | 14 (4.7) | 3 (8.6) | 11 (4.2) | |
| Syncope | 19 (6.3) | 3 (8.6) | 16 (6.0) | |
| Incidental finding | 43 (14.3) | 4 (11.4) | 39 (14.7) | |
| Other | 13 (4.3) | 1 (2.9) | 12 (4.5) | |
| Polyneuropathy, n (%) | 40 (16.3) | 8 (61.5) | 32 (13.7) | <0.001 |
| Carpal tunnel syndrome, n (%) | 87 (35.4) | 7 (53.9) | 80 (34.3) | 0.152 |
| History of embolism, n (%) | 22 (8.9) | 3 (23.1) | 19 (8.2) | 0.07 |
| Atrial fibrillation, n (%) | 166 (67.5) | 7 (53.9) | 159 (68.2) | 0.281 |
| Hypertension, n (%) | 172 (69.9) | 8 (61.5) | 164 (70.4) | 0.498 |
| Coronary artery disease, n (%) | 40 (16.3) | 0 | 40 (17.8) | 0.135 |
| Pacemaker, n (%) | 40 (22.5) | 2 (22.2) | 38 (22.5) | 1 |
| Chronic kidney disease (stage ≥3A), n (%) | 126 (51.2) | 5 (38.5) | 121 (51.9) | 0.402 |
| Baseline blood tests | ||||
| eGFR, ml/min/1.73 m2, mean ± SD | 58.6 ± 18.1 | 63.1 ± 20.3 | 58.3 ± 17.9 | 0.357 |
| NT‐proBNP, pg/ml, median (IQR) | 2592 (1020–4498) | 4794 (1650–9524) | 2544 (1020–4413) | 0.104 |
| Conventional troponin I (μg/L), median (IQR) | 0.06 (0.03–0.11) | 0.08 (0.04–0.19) | 0.06 (0.03–0.10) | 0.45 |
| Baseline ECG, n (%) | ||||
| 1st degree AV block | 42 (17.1%) | 1 (7.7%) | 41 (17.6%) | 0.70 |
| LBBB | 41 (16.7%) | 3 (23.1%) | 38 (16.3) | 0.27 |
| RBBB | 44 (17.9%) | 1 (7.7%) | 43 (18.5%) | 0.29 |
| Baseline echocardiogram | ||||
| Interventricular wall thickness, mm, mean ± SD | 17.9 ± 3.4 | 18.7 ± 3.0 | 17.9 ± 3.4 | 0.40 |
| End‐diastolic left ventricular diameter, mm, mean ± SD | 42.7 ± 6.4 | 40.5 ± 5.8 | 42.8 ± 6.5 | 0.21 |
| Left ventricular ejection fraction, %, mean ± SD | 54.2 ± 11.5 | 57.1 ± 6.8 | 54.0 ± 11.7 | 0.35 |
| Global longitudinal strain, −% (n = 173), mean ± SD | 12.3 ± 4.4 | 11.2 ± 2.5 | 12.3 ± 4.5 | 0.44 |
| Left atrial diameter, mm, mean ± SD | 44.7 ± 6.4 | 41.1 ± 6.9 | 44.9 ± 6.4 | 0.06 |
| Aortic stenosis (at least moderate), n (%) | 10 (3.3) | 0 (0.0) | 10 (3.8) | 0.61 |
| Pericardial effusion, n (%) | 52 (17.5) | 9 (25.7) | 43 (16.4) | 0.17 |
ATTR‐CM, transthyretin amyloid cardiomyopathy; ATTRv, hereditary transthyretin amyloidosis; AV, atrioventricular; ECG, electrocardiogram; eGFR, estimated glomerular filtration rate; IQR, interquartile range; LBBB, left bundle branch block; NT‐proBNP, N‐terminal pro‐brain natiuretic peptide; RBBB, right bundle branch block; SD, standard deviation.
Univariate predictors of ATTRv in ATTR‐CM elderly patients included female sex, African ancestry, polyneuropathy, and eye symptoms (Table 3 ).
Table 3.
Factors associated with hereditary transthyretin amyloidosis in elderly patients with transthyretin amyloid cardiomyopathy
| Univariate analysis | |||
|---|---|---|---|
| Odds Ratio | 95% CI | p‐value | |
| Age at diagnosis (per year) | 0.95 | 0.85–1.05 | 0.29 |
| Female sex | 3.66 | 1.13–11.85 | 0.03 |
| Black ancestry | 46.31 | 3.52–Inf | 0.005 |
| Hypertension | 0.67 | 0.21–2.13 | 0.50 |
| Diabetes | 1.37 | 0.41–4.63 | 0.61 |
| Carpal tunnel syndrome | 2.23 | 0.73–6.86 | 0.16 |
| Lumbar spinal stenosis | 0.51 | 0.06–4.01 | 0.52 |
| Coronary artery disease | 0.27 | 0.00–1.66 | 0.19 |
| Polyneuropathy | 10.05 | 3.09–32.64 | <0.001 |
| Dysautonomia | 1.14 | 0.30–4.29 | 0.85 |
| Eye symptoms | 6.64 | 1.20–36.73 | 0.03 |
| GI symptoms | 3.08 | 0.78–12.09 | 0.11 |
| NYHA class ≥ III | 0.62 | 0.13–2.87 | 0.54 |
| Atrial fibrillation | 0.54 | 0.18–1.67 | 0.29 |
| 1st degree AV block | 0.18 | 0.02–1.56 | 0.12 |
| Left bundle branch block | 1.73 | 0.87–3.42 | 0.12 |
| LVEF <50% | 0.38 | 0.08–1.79 | 0.22 |
| IVS ≥15 mm | 3.89 | 0.49–30.5 | 0.20 |
| NT‐proBNP >3000 pg/ml | 2.37 | 0.75–7.45 | 0.14 |
| Conventional troponin I >0.05 μg/L | 0.86 | 0.27–2.74 | 0.80 |
AV, atrioventricular; CI, confidence interval; GI, gastrointestinal; IVS, interventricular septum; LVEF, left ventricular ejection fraction; NT‐proBNP, N‐terminal pro‐brain natriuretic peptide; NYHA, New York Heart Association.
Implications of ATTRv diagnosis in elderly patients
All patients diagnosed with ATTRv received genetic counseling. Moreover, diagnosis of ATTRv allowed initiation of tafamidis in five subjects who had subsequent demonstration of polyneuropathy (tafamidis could not be prescribed for ATTR‐CM at that time). Furthermore, cascade genetic testing was performed in 33 relatives from 13 families leading to identification of nine asymptomatic ATTRv carriers.
Discussion
This study describes the prevalence of ATTRv among a predominantly Caucasian cohort of elderly patients with ATTR‐CM. We found that in 5.3% of elderly ATTR‐CM patients the condition had a genetic origin. Moreover, we showed that diagnosing ATTRv in elderly individuals has important clinical consequences both for patients and their families. Furthermore, we did not find any cardiac parameter to be associated with ATTRv and identified female sex, black ancestry, eye symptoms and polyneuropathy as the only factors associated with ATTRv in this population.
Transthyretin amyloid cardiomyopathy, once thought to be a rare disease, is nowadays recognized to be a frequent cause of heart failure with preserved ejection fraction and degenerative aortic stenosis. 3 , 4 Recognition of the contribution of ATTR‐CM in these clinical settings combined with advances in cardiac imaging and the appearance of specific therapies have led to a substantial increase in the number of ATTR‐CM patients currently diagnosed. 5 , 6
Unfortunately, increased awareness has also led to emerging problems in ATTR‐CM diagnosis that include lack of exclusion of light‐chain (AL) amyloidosis in patients with cardiac uptake on scintigraphy, incomplete evaluations that do not include single photon emission computed tomography and lack of genetic testing in elderly individuals. 2 , 7
The latter is a pitfall that has been favoured in non‐endemic regions by the lower frequency of ATTRv compared to ATTRwt, the wrong perception that ATTRv invariably presents with neurological manifestations and the incomplete penetrance and late disease onset of certain ATTRv variants that preclude identification of familial presentation. In fact, in our study, 40% of elderly ATTR‐CM patients referred from other centres had not undergone genetic testing at origin.
In our study, one in 20 elderly ATTR‐CM patients and one in 10 elderly females had ATTRv despite being diagnosed at ≥70 years old.
The sex imbalance found in our study is consistent with previous reports where ATTR‐CM is predominantly identified in men, particularly ATTRwt. Although still understudied, the lower prevalence of ATTR‐CM in women has been related to several factors, such as potential cardioprotective effect of female sex and underdiagnosis due to smaller heart structures and lack of awareness. 8
We compared our patients with patients included in the THAOS international registry; 9 European patients were younger than the American patients and than the patients included in our study. This could be explained by the fact that most European participants had ATTRv (91.1%), in contrast to US patients (51.5%) and ours (12%). Val30Met was the most common genetic variant in Europe in THAOS (80.7%), whereas in our study and in the US it was Val142Ile (40% and 45.3%, respectively). Cardiomyopathy was an inclusion criterion in our study, which was present in almost all the American patients included in THAOS but only in one third of the European patients in the registry. No relevant differences in echocardiographic parameters were observed between patients included in THAOS and those included in our study.
Interestingly, the Val142Ile variant was the commonest variant found in our study despite patients included in our cohort were predominantly Caucasian. Our study adds to previous reports that also showed higher than expected prevalence of the Val142Ile variant potentially suggesting that it could be an underestimated variant among Caucasian ATTR‐CM patients. 10
Transthyretin genetic testing is simple and cheap nowadays and limiting genetic evaluation in ATTR‐CM patients could have important negative effects if ATTRv is not appropriately diagnosed.
At the patient level, identification of ATTRv enables genetic counselling and better delineation of prognosis as ATTRv has worse prognosis than ATTRwt, 11 but also could lead to a more precise neurological evaluation and initiation of TTR‐specific drugs. In our study, five patients initiated tafamidis thanks to ATTRv diagnosis as tafamidis was not reimbursed for ATTR‐CM in Spain at that time. Nevertheless, even if tafamidis had been available for ATTR‐CM, other therapeutic alternatives that are only available for ATTRv patients with polyneuropathy could have been considered. 1
The positive implications of diagnosing ATTRv extend to relatives that can benefit from cascade screening and early diagnosis. In our study, nine ATTRv genetic carriers were diagnosed and will undergo periodic surveillance allowing identification of disease manifestations at early stages where specific treatments are more effective. 12
Limitations
As ATTRwt patients are old and might have comorbidities that preclude referral, a selection bias might have influenced the proportion of ATTRv found. Nevertheless, the proportion of ATTRv did not differ between elderly patients referred and those diagnosed locally (5.7% vs. 4.6%; p = 0.78). Moreover, the limited number of black patients in our cohort precludes generalization to other cohorts with higher proportion of black individuals (where 3–4% carry the Val142Ile variant). 13 However, even with a predominantly Caucasian cohort, the prevalence of ATTRv found was considerable.
Conclusions
Prevalence of ATTRv among predominantly Caucasian elderly patients with ATTR‐CM was 5.3% and its diagnosis had meaningful clinical implications. These results support routine genetic testing in ATTR‐CM regardless of age.
Funding
This study has been funded by Instituto de Salud Carlos III (ISCIII) through the projects ‘PI18/0765 & PI20/01379’ (co‐funded by European Regional Development Fund/European Social Fund ‘A way to make Europe’/‘Investing in your future’). AMB receives grant support by ISCIII (CM20/002209). The CNIC is supported by the ISCIII, MCIN, the Pro‐CNIC Foundation, and the Severo Ochoa grant (CEX2020‐001041‐S).
Conflict of interest: none declared.
Supporting information
Appendix S1. Supporting Information.
References
- 1. Garcia‐Pavia P, Rapezzi C, Adler Y, Arad M, Basso C, Brucato A, et al. Diagnosis and treatment of cardiac amyloidosis: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2021;42:1554–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Poterucha TJ, Elias P, Bokhari S, Einstein AJ, DeLuca A, Kinkhabwala M, et al. Diagnosing transthyretin cardiac amyloidosis by technetium Tc 99m pyrophosphate: a test in evolution. JACC Cardiovasc Imaging. 2021;14:1221–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. González‐López E, Gallego‐Delgado M, Guzzo‐Merello G, de Haro‐del Moral FJ, Cobo‐Marcos M, Robles C, et al. Wild‐type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36:2585–94. [DOI] [PubMed] [Google Scholar]
- 4. Castaño A, Narotsky DL, Hamid N, Khalique OK, Morgenstern R, DeLuca A, et al. Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur Heart J. 2017;38:2879–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lane T, Fontana M, Martinez‐Naharro A, Quarta CC, Whelan CJ, Petrie A, et al. Natural history, quality of life, and outcome in cardiac transthyretin amyloidosis. Circulation. 2019;140:16–26. [DOI] [PubMed] [Google Scholar]
- 6. López‐Sainz Á, Hernandez‐Hernandez A, Gonzalez‐Lopez E, Domínguez F, Restrepo‐Cordoba MA, Cobo‐Marcos M, et al. Clinical profile and outcome of cardiac amyloidosis in a Spanish referral center. Rev Esp Cardiol (Engl Ed). 2021;74:149–58. [DOI] [PubMed] [Google Scholar]
- 7. Poterucha TJ, Elias P, Ruberg FL, DeLuca A, Kinkhabwala M, Johnson LL, et al. False positive 99mTc‐pyrophosphate scanning leading to inappropriate tafamidis prescriptions. JACC Cardiovasc Imaging. 2021;14:2042–4. [DOI] [PubMed] [Google Scholar]
- 8. Bruno M, Castaño A, Burton A, Grodin JL. Transthyretin amyloid cardiomyopathy in women: frequency, characteristics, and diagnostic challenges. Heart Fail Rev. 2021;26:35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maurer MS, Hanna M, Grogan M, Dispenzieri A, Witteles R, Drachman B, et al. Genotype and phenotype of transthyretin cardiac amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J Am Coll Cardiol. 2016;68:161–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cappelli F, Frusconi S, Bergesio F, Grifoni E, Fabbri A, Giuliani C, et al. The Val142Ile transthyretin cardiac amyloidosis: not only an Afro‐American pathogenic variant? A single‐centre Italian experience. J Cardiovasc Med (Hagerstown). 2016;17:122–5. [DOI] [PubMed] [Google Scholar]
- 11. Rapezzi C, Elliott P, Damy T, Nativi‐Nicolau J, Berk JL, Velazquez EJ, et al. Efficacy of tafamidis in patients with hereditary and wild‐type transthyretin amyloid cardiomyopathy: further analyses from ATTR‐ACT. JACC Heart Fail. 2021;9:115–23. [DOI] [PubMed] [Google Scholar]
- 12. Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington‐Cruz M, et al.; ATTR‐ACT Study Investigators . Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379:1007–16. [DOI] [PubMed] [Google Scholar]
- 13. Quarta CC, Buxbaum JN, Shah AM, Falk RH, Claggett B, Kitzman DW, et al. The amyloidogenic V122I transthyretin variant in elderly black Americans. N Engl J Med. 2015;372:21–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting Information.