

Abstract
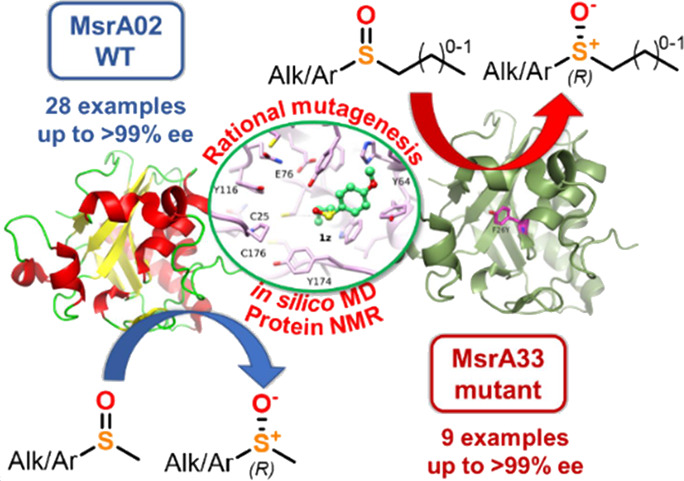
Methionine sulfoxide reductase A (MsrA) enzymes have recently found applications as nonoxidative biocatalysts in the enantioselective kinetic resolution of racemic sulfoxides. This work describes the identification of selective and robust MsrA biocatalysts able to catalyze the enantioselective reduction of a variety of aromatic and aliphatic chiral sulfoxides at 8–64 mM concentration with high yields and excellent ees (up to 99%). Moreover, with the aim to expand the substrate scope of MsrA biocatalysts, a library of mutant enzymes has been designed via rational mutagenesis utilizing in silico docking, molecular dynamics, and structural nuclear magnetic resonance (NMR) studies. The mutant enzyme MsrA33 was found to catalyze the kinetic resolution of bulky sulfoxide substrates bearing non-methyl substituents on the sulfur atom with ees up to 99%, overcoming a significant limitation of the currently available MsrA biocatalysts.
Keywords: sulfoxide, methionine reductase, MsrA, biocatalysis, mutagenesis
Introduction
Chiral sulfoxides are a ubiquitous class of organic compounds which find broad applications in organic and medicinal chemistry.1 Indeed, chiral sulfoxides can be used in organic chemistry as chiral ligands and auxiliaries for asymmetric reactions.2 They are also important as pharmaceutical ingredients, for example, in the blockbuster antacid esomeprazole,3 namely, the (S)-enantiomer of omeprazole, or the sleep disorder drug armodafinil.4 The chirality of sulfoxides has a key impact on their chemical and pharmacological properties, which has driven interest in the scientific community toward developing asymmetric strategies5 to access these molecules in enantiomerically pure form. The two most common methods to synthesize enantiomerically pure chiral sulfoxides involve (1) the formation of a new C–S bond by nucleophilic substitution of nonracemic sulfinyl substrates using Grignard reagents2b,6 and (2) the formation of the S–O bond through asymmetric oxidation of prochiral sulfides with chiral oxidants or auxiliaries.2,7 While catalytic C–S bond formation reactions are rare, several efficient oxidation systems have been developed for the preparation of enantiomerically pure sulfoxides. In the last decades, enzymes such as monooxygenases1,8 and peroxygenases9 have shown their potential as biocatalysts in the stereoselective oxidation of sulfides into sulfoxides, offering advantages over classic chemical oxidation methods due to the mild reaction conditions and sustainability of the processes used. However, although biocatalytic oxidative methodologies are highly efficient and stereoselective, their industrial applicability may be limited by some factors. Monooxygenases require expensive NAD(P)H to activate the FAD or FMN prosthetic groups and nonatom-economical systems to recycle NAD(P)H itself, while peroxygenases require the use of stoichiometric amounts of H2O2. Monooxygenases can also generate peroxide side products which, if not removed from the reaction mixtures, may cause overoxidation of the sulfoxide products and degradation of the enzyme (Figure 1). Recently, methionine sulfoxide reductase (Msr) enzymes have emerged as alternative nonoxidative biocatalysts able to catalyze the stereoselective reduction of racemic sulfoxides.10 Msrs are a large class of reductive enzymes found in many organisms,11 which selectively reduce the methionine sulfoxide (MetSO) residues found in proteins as a consequence of oxidative stress in cells, back to methionine (Met). Three subclasses of Msr enzymes have been identified to date, namely, MsrA and MsrB, which reduce, respectively, (S)-MetSO and (R)-MetSO residues in proteins, and the free (R)-Msr (frMsr), which reduces free (R)-MetSO amino acid.12,13 To date, out of the three subclasses, only a few MsrA enzymes from Pseudomonas sp. (pmMsrA, paMsr, and pmMsr),14−16E. coli,17a and a mammalian MsrA,18 as well as a MsrB from Acidovorax sp.,17 have shown potential as biocatalysts in the kinetic resolution (KR) of racemic aryl methyl sulfoxides.
Figure 1.

Biocatalytic approaches for the synthesis of enantiomerically pure sulfoxides.
The number of Msr biocatalysts currently available in the biocatalysis toolbox is still limited and additional investigations are required to expand their synthetic scope and fully disclose their industrial potential. Compared to oxidative biocatalysts, Msr enzymes have the advantage of not requiring any additional expensive cofactors and being regenerated with the cheap sacrificial co-substrate dithiothreitol (DTT).18 On the other hand, a current major limitation of available MsrA biocatalysts is their limited substrate scope as they are only able to reduce sulfoxides bearing a methyl, and in a few cases an ethyl,16a substituent on the sulfur atom, while they are generally inactive on bulkier substrates. To date, only one example of a mutant MsrA enzyme with expanded substrate scope, identified via a high-throughput assay for directed evolution, has been reported.16e
Following up on our studies on the synthesis of chiral sulfur compounds via biocatalysis,19 herein, we report the preparation, screening, and development of novel MsrA biocatalysts from different microorganisms. Moreover, through a combination of in silico docking and molecular dynamics, protein NMR, and rational mutagenesis studies, a library of MsrA mutant enzymes has been rationally designed and prepared. This process resulted in a new mutant enzyme able to reduce stereoselectively sulfoxide substrates bearing non-methyl S-substituents, in turn allowing the expansion of the substrate scope of this class of biocatalysts. To the best of our knowledge, this is the first example of a rational mutagenesis study of Msr enzymes resulting in the successful development of a mutant biocatalyst with enhanced substrate scope.
Results and Discussion
First, a library of 15 Msr enzymes was cloned and overexpressed. Msr enzymes were selected from literature and homology searching in public databases to build a panel of diverse biocatalysts from different organisms as well as different Msr subclasses (Table 1). Msr encoding genes were cloned into pET28a and expressed in the E. coli BL21(DE3) expression strain. The gene expression was induced by the addition of 1 mM IPTG at 25 °C. After expression, the 15 Msr enzymes were prepared as lyophilized cell free extracts (CFE) and screened for the KR of the racemic sulfoxide 1a to identify the best biocatalysts to afford the enantiomer (R)-1a in the highest yield and enantiomeric excess (ee). The initial biotransformation conditions for the screening were adopted from the literature.16 Each enzyme was resuspended in a 100 mM KPi buffer solution at pH = 8.0, together with 4.0 equiv of dithiothreitol (DTT), to regenerate the Msr enzymes (details on the role of DTT are reported below and in Figure 2a). An excess of DTT was initially used to promote full regeneration of the biocatalyst. Then, the substrate 1a was dissolved in iPrOH (IPA), added to the enzyme mixture to initiate the reaction, and was left stirring at 30 °C for 24 h, after which the yields and ees of the enantiomer (R)-1a(20) were calculated using chiral HPLC (Table 1).
Table 1. Screening of the Msr Panel Using Substrate 1a.

| entry | Msr | Msr subclass | organism | 1a yielda % | (R)/(S)1a eeb % | 1a enantb |
|---|---|---|---|---|---|---|
| 1 | 01 | A | Escherichia coli | 51 | >99 | (R) |
| 2 | 02 | A | Saccharomyces cerevisiae | 50 | >99 | (R) |
| 3 | 03 | B | Escherichia coli | 90 | 4 | (S) |
| 4 | 05 | Free R | Saccharomyces cerevisiae | >99 | <1 | n.d.c |
| 5 | 07 | A | Mycobacterium tuberculosis | 44 | >99 | (R) |
| 6 | 08 | AB hybrid | Neisseria meningitidis | 44 | >99 | (R) |
| 7 | 09 | A | Staphylococcus aureus | 55 | >99 | (R) |
| 8 | 10 | AB hybrid | Thermococcus kodakarensis | 31 | >99 | (R) |
| 9 | 11 | A | Streptomyces griseochromogenes | 54 | >99 | (R) |
| 10 | 13 | AB hybrid | Streptococcus pneumoniae | 48 | >99 | (R) |
| 11 | 15 | AB hybrid | Treponema denticola | 54 | >99 | (R) |
| 12 | 16 | A | Klebsiella pneumoniae | 50 | >99 | (R) |
| 13 | 17 | A | Salmonella schwarzengrund | 53 | >99 | (R) |
| 14 | 18 | A | Serratia symbiotica | >99 | 4 | (R) |
| 15 | 21 | B | Klebsiella oxytoca | >99 | 1 | n.d.c |
HPLC yields are reported. Yields were calculated using an Agilent Eclipse Plus C18 column and methyl phenyl sulfoxide as the internal standard.
Determined by chiral HPLC using a Chiralpak IG column.
Not determined.
Figure 2.

(a) Mechanism for the reduction of sulfoxides by MsrA02 biocatalysts; (b) comparison between the active sites of the wild-type (WT) MsrA02 enzyme (blue) and the C23S variant (pink). The serine hydroxide sidechain makes an H bond with threonine 29, significantly changing the loop containing the nucleophilic cysteine (C25).
Most MsrA enzymes showed excellent ees and conversions. Only MsrA18 (Table 1, entry 14), MsrB03, and MsrB21 (Table 1, entries 3 and 15) showed no activity. Interestingly, while frMsr05 from Saccharomyces cerevisiae showed no activity in the KR of 1a (Table 1, entry 4), its MsrA variant, namely, MsrA02, from the same microorganism,21 furnished (R)-1a in high ee (>99%) and excellent HPLC yield (50%) (Table 1, entry 2). Since no MsrA biocatalyst from yeasts has been described to date, and due to the crystallographic data available in the literature,21,22 MsrA02 was selected for further optimization studies.
First, the optimal concentration of the biocatalyst, used as CFE, in the kinetic resolution of 1a was investigated (Table 2). The concentration of 1a was initially fixed at 8.0 mM and 4.0 equiv of DTT was used. The reactions (Table 2, entries 1–5) were carried out at 30 °C and stopped after 4 h when a 50% 1H-NMR yield of the enantiomer (R)-1a was observed, suggesting the completion of the KR reaction. The best concentration for MsrA02 was found at 1.0 gL–1, while lower concentrations led to poorer ees and yields. When the reaction was carried out in the absence of an enzyme, the sulfoxide 1a was fully recovered as a racemate (Table 2, entry 5). Remarkably, biocatalytic transformation proved to tolerate concentrations of the substrate 1a in the range of 32–64 mM (Table 2, entries 6–9), while at higher concentrations, 1a was recovered as a racemic mixture (entry 9). Higher temperatures (37 °C) led to 1a after 1 h with yield and ee (Table 2, entry 10) similar to those observed at 30 °C. Changing the cosolvent IPA to MeOH, EtOH or CH3CN did not affect the biotransformation outcome (Table 2, entries 11–14). Interestingly, when the reaction was carried out without any cosolvent or in the presence of DMSO, the sulfoxide 1a was recovered as a racemate, indicating that no reduction took place (Table 2, entries 15–16). It is plausible that DMSO could act as a competitive substrate for MsrA02 or as an oxidant. Instead, in the absence of an organic cosolvent, 1a showed poor solubility in aqueous buffer. The optimal amount of DTT used for the regeneration of MsrA02 was set at 1.1 equiv (compared to 1a, Table 2, entry 17). Significantly, no biocatalytic transformation occurs in the absence of DTT, confirming its crucial role in the regeneration of MsrA02 (Table 2, entry 19 and Figure 2a).
Table 2. Optimization of the KR of 1a with MsrA02.

| entry | MsrA02 (gL–1)a | 1a (mM) | DTT (equiv) | cosolvent | temp °C | (R)-1a yieldb % | eec % |
|---|---|---|---|---|---|---|---|
| 1 | 1.6 | 8.0 | 4.0 | IPA | 30 | 51 (51)d | 98 |
| 2 | 1.0 | 8.0 | 4.0 | IPA | 30 | 54 (51)d | 99 |
| 3 | 0.4 | 8.0 | 4.0 | IPA | 30 | 53 (52)d | 98 |
| 4 | 0.1 | 8.0 | 4.0 | IPA | 30 | 71 (86)d | 40 |
| 5 | 8.0 | 4.0 | IPA | 30 | >99 | <1 | |
| 6 | 1.6 | 16 | 4.0 | IPA | 30 | 54 | 99 |
| 7 | 1.6 | 32 | 4.0 | IPA | 30 | 52 | 99 |
| 8 | 1.6 | 64 | 4.0 | IPA | 30 | (48)d | 99 |
| 9 | 1.6 | 128 | 4.0 | IPA | 30 | 54 | 5 |
| 10e | 1.6 | 8.0 | 4.0 | IPA | 37 | 47 | 99 |
| 11 | 1.6 | 8.0 | 4.0 | MeOH | 30 | 46 | >99 |
| 12 | 1.6 | 8.0 | 4.0 | EtOH | 30 | 50 | >99 |
| 13 | 1.6 | 8.0 | 4.0 | IPA | 30 | 54 | >99 |
| 14 | 1.6 | 8.0 | 4.0 | CH3CN | 30 | 49 | >99 |
| 15 | 1.6 | 8.0 | 4.0 | DMSO | 30 | 90 | 5 |
| 16 | 1.6 | 8.0 | 4.0 | Neat | 30 | >99 | 5 |
| 17 | 1.6 | 8.0 | 1.0 | IPA | 30 | 45 | 99 |
| 18 | 1.6 | 8.0 | 0.5 | IPA | 30 | 69 (62)d | 64 |
| 19 | 1.6 | 8.0 | IPA | 30 | >99 | <1 | |
| 20 | 1.0 | 32 | 1.1 | IPA | 30 | 53 (48)f | 99 |
| 21 | 1.0 | 64 | 1.1 | IPA | 30 | 46 | 99 |
| 22 | 2.3g | 32 | 1.1 | IPA | 30 | (52)d | >99 |
| 23 | 10h | 32 | 1.1 | IPA | 30 | (52)d | >99 |
| 24i | 1.6 | 8.0 | 4.0 | IPA | 30 | 52 | >99 |
MsrA02 used as CFE.
1H-NMR yields are reported.
Determined by chiral HPLC using the Chiralpak IG column.
HPLC yield is reported.
1 h reaction time.
Isolated yield.
Pure MsrA02 enzyme was used.
Whole cell MsrA02 was used.
Reaction time was 18 h.
The optimal reaction conditions were finally combined and set at 1 gL–1 of the biocatalyst, 32 mM of 1a, 1.1 equiv of DTT, and 30 °C, leading to (R)-1a with 48% isolated yield and 99% ee (Table 2, entry 20). Remarkably, similar results were obtained when a higher concentration of the substrate (64 mM) was used (Table 2, entry 21), proving the robustness of this biotransformation. The use of MsrA02 as a whole cell biocatalyst (Table 2, entry 23) or as pure enzyme (Table 2, entry 22, see the Supporting Information for the purification procedure) led to enantiomerically pure (R)-1a in identical conversion and ee to the CFE, confirming that the observed reactivity was solely a result of MsrA02 and not from any other proteins present in the cell lysate. Finally, longer reaction times up to 18 h (Table 2, entry 24) proved to not affect the biotransformation outcome. With the best conditions for the MsrA02 biocatalyzed KR of 1a in hand (Table 2, entry 20), the substrate scope of the reaction was then investigated. For some substrates where MsrA02 performed poorly, the biotransformation was also attempted with other biocatalysts selected from the initial screening (Table S3). All results are reported in Table 3. Isolated yields are reported for the reaction products.
Table 3. Substrate Scope of the KR of Sulfoxides 1b-ab.

| entry | cmpd. | R1 | R2 | Msr | time (h) | isolated yield %c | ee %b |
|---|---|---|---|---|---|---|---|
| 1 | 1b | Ph | Me | 02 | 4 | 35 | >99 |
| 2 | 1c | 4-F-Ph | Me | 02 | 6 | 26 | >99 |
| 3 | 1d | 4-Br-Ph | Me | 02 | 6 | 41 | >99 |
| 4 | 1e | 4-Cl-Ph | Me | 02 | 4 | 38 | >99 |
| 5 | 1f | 3-Cl-Ph | Me | 02 | 6 | 40 | >99 |
| 6 | 1g | 2-Cl-Ph | Me | 02 | 28 | 30 | >99 |
| 7 | 1h | 4-Ac-Ph | Me | 02 | 4 | 50 | >99 |
| 8 | 1i | 4-MeO-Ph | Me | 02 | 4 | 43 | >99 |
| 9 | 1j | 3-MeO-Ph | Me | 02 | 6 | 35 | >99 |
| 10 | 1k | 3-Me-Ph | Me | 02 | 6 | 48 | >99 |
| 11 | 1l | 4,2-Me-Ph | Me | 02 | 81 | 41 | >99 |
| 12 | 1m | 2-Naph | Me | 02c | 28 | n.d.d | 74 |
| 10 | 24 | 69e | 30 | ||||
| 10f | 24 | 45e | >99 | ||||
| 13 | 1n | 1-Naph | Me | 02 | 24 | n.d.d | 16 |
| 14 | 1o | Bn | Me | 02 | 24 | 29 | 94 |
| 15 | 1p | Dodecyl | Me | 02 | 24 | 46 | >99 |
| 16 | 1q | CH3CO(CH2)2 | Me | 02 | 48 | 40 | >99 |
| 17 | 1r | PhSO(CH2)2 | Me | 02 | 8 | 39 | >99j |
| 18 | 1s | Ph | Et | 02 | 48 | 47 | 90 |
| 19 | 1tc | 4-Me-Ph | Et | 02 | 48 | 36 | 98 |
| 20 | 1uc | 4-Br-Ph | Et | 02 | 48 | 55 | 80 |
| 21 | 1vg | Bn | Et | 02 | 24 | 36 | 96 |
| 22 | 1w | Ph(CH2)2 | Et | 02 | 8 | 40 | 99 |
| 23 | 1xg | 2-PyCH2 | Et | 02 | 24 | n.d.d | 54 |
| 01 | 24 | n.d.d | 20 | ||||
| 24 | 1yh | Ph | nPr | 02 | 24 | n.d.d | <1 |
| 25 | 1zh | 4-MeO-Ph | nPr | 02 | 24 | n.d.d | <1 |
| 01 | 24 | n.d.d | 10 | ||||
| 26 | 1aai | Ph | Vinyl | 02 | 48 | <5e | >99 |
| 27 | 1ab | Ph | Allyl | 02 | 7d | n.d.d | 16 |
| 01 | 24 | n.d.d | 24 |
Isolated yields after chromatographic purification of the (R)-sulfoxide.
Determined by chiral HPLC using Chiralpak column IG or IC or Chiracel OD-H.
2.0 gL–1 CFE MsrA02.
Not determined.
Conversion determined by HPLC.
8 mM substrate, 1.0 gL–1.
10 gL–1 CFE MsrA02.
8 mM substrate, 10 gL–1 CFE MsrA02.
10 μM purified MsrA02.
Two diastereoisomers, each one with >99% ee.
Sulfoxides 1b–l were all resolved by MsrA02 with excellent ee (99%) and high isolated yields (Table 3, entries 1–11). Even if the maximum yield expected for these KR reactions was 50%, the isolated yield detected after chromatographic purification was, in a few cases, slightly lower due to the difficulties associated with the extraction of the sulfoxide compounds from the buffer media with organic solvents like DCM or AcOEt. Nevertheless, in most cases, the isolated yields obtained were well above 40%. The (R)-enantiomer of the 2-naphtyl derivative 1m was obtained with high ee (74%) using MsrA02 (Table 3, entry 12). Interestingly, another AB hybrid Msr enzyme, namely, MsrA10 from Thermococcus kodakarensis, which was more active but less stereoselective on substrate 1a (Table 1), proved to be highly effective in the KR of 1m when used at 8 mM concentration, affording the (R)-enantiomer with excellent >99% ee and 45% conversion. On the other hand, the regioisomer 1-naphtyl derivative 1n, proved to be a poor substrate for both MsrA02 and MsrA10 (Table 3, entry 13). The sulfoxides 1o–q bearing benzyl or alkyl group on the sulfur atom were also reduced by MsrA02 (Table 3, entries 14–16), affording the corresponding (R)-enantiomers with high isolated yields (up to 46%) and excellent ee (up to 99%). Substrate 1r bears two sulfoxide groups, and thus, it exists as two pairs of enantiomers, (R,S)-1r and (S,R)-1r, and (R,R)-1r and (S,S)-1r (Scheme S1). MsrA02 was able to reduce only the (S)-sulfoxide moiety adjacent to the methyl group, leaving the others unreacted. Thus, the diastereoisomers (R,S)-1r and (S,S)-1r were reduced by MsrA02 into the enantiomers (R)-2r and (S)-2r, while the diastereoisomers (S,R)-1r and (R,R)-1r were recovered in a 1:1 ratio, each one with excellent >99% ee (Table 3, entry 17).
Remarkably, MsrA02 showed excellent activity also on the substrates 1s–w bearing an Et substituent on the sulfur atom with high yields and excellent ees (Table 3, entries 18–22). The pyridyl derivative (R)-1x was obtained with only 54% ee when MsrA02 was used. Attempts to find another suitable MsrA biocatalyst to resolve 1x failed and only MsrA01 showed little enantioselectivity, affording the (R)-enantiomer with 20% ee (Table 3, entry 23). Similarly, the bulkier propyl derivatives 1y and 1z as well as allyl substrates 1ab were not or poorly resolved by MsrA02 (Table 3, entries 24–25 and entry 27), while high ee (>99%) was detected for the smaller vinyl derivative 1aa (Table 3, entry 26). However, 1aa proved to be unstable and it was obtained in low HPLC yields.
Mechanism of MsrA Biocatalytic Reduction
The substrate scope study of this biocatalytic transformation clearly showed the ability of MsrA02 to catalyze the KR of sulfoxides bearing a methyl and, remarkably, an ethyl substituent on the sulfur atom. However, bulkier substrates bearing a propyl or an allyl substituent were not reduced by MsrA02 or other MsrA wild-type (WT) enzymes. Thus, the possibility to mutate MsrA enzymes enabling them to catalyze the reduction of bulkier substrates was investigated. To design improved MsrA mutants via rational mutagenesis, the mechanism of the MsrA02 biocatalyzed reaction was first investigated in depth. The generally accepted mechanism for the reduction of MetSO and other sulfoxides by MsrA enzymes involves at least two catalytic cysteine (Cys) residues (Figure 2a).23 The first cysteine attacks and reduces the sulfoxide group forming a sulfenic acid intermediate (Cys-SOH), which then reacts with the second cysteine residue forming an intramolecular disulfide bond via dehydration. In vivo, the oxidoreductase thioredoxin (Trx) breaks the disulfide bond, leading to the regeneration of the MsrA enzymes, while in vitro Trx is replaced with the inexpensive sacrificial co-substrate DTT.
The biocatalyst MsrA02 contains five cysteine residues (Cys23, Cys25, Cys44, Cys68, and Cys176) which could be involved in the reduction of sulfoxides. According to the crystal structure analysis,21 the two residues involved in the catalytic cycle in MsrA02 are likely to be Cys25 and Cys176. To confirm the role of the five cysteines in the reduction of 1a, single point mutations were carried out on MsrA02. Each cysteine was mutated to serine and all five MsrA02 mutants (MsrA02 C25S, C176S, C23S, C44S, and C68S) were then overexpressed and purified as N-terminal His-tagged fusion proteins. The mutants were assayed with sulfoxide 1a as either CFE or pure enzymes (Table 4). Experiments were carried out at different concentrations of the biocatalysts. Mutants C25S and C176S dramatically affected the outcome of the sulfoxide reduction, and in both cases, 1a was recovered as a racemate (Table 4, entries 2 and 5). This clearly confirms that these two Cys residues are involved in the catalytic cycle and in the sulfoxide reduction. Mutation C44S did not affect the biotransformation and (R)-1a was obtained with 99% ee, confirming that Cys44 has no role in the catalytic cycle (Table 4, entry 3). The mutation C68S also had no effect on the catalytic activity of the enzyme, as even at low concentrations (0.23 gL–1 of pure enzyme), >99% ee of (R)-1a was obtained (Table 4, entry 4). Interestingly, the mutation C23S, led to (R)-1a with poor ee, especially at low concentration of the enzyme (Table 4, entry 1).
Table 4. Single Point Mutation of MsrA02.

| entry | single point mutation | enzyme conc. (gL–1)a,b | (R)-1a ee% |
|---|---|---|---|
| 1 | C23S | 1.0 | 26 |
| 10 | 67 | ||
| 40 | 80 | ||
| 2 | C25Sc | 0.23 | <1 |
| 2.3 | 8 | ||
| 9.2 | 21 | ||
| 3 | C44S | 1.0 | 9 |
| 10 | 99 | ||
| 40 | >99 | ||
| 4 | C68Sc | 0.23 | >99 |
| 2.3 | >99 | ||
| 9.2 | >99 | ||
| 5 | C176S | 1.0 | 4 |
| 10 | 26 | ||
| 40 | 36 | ||
| 6 | Wild type | 1.0 | >99 |
Enzyme used as CFE.
gL–1 of protein concentration calculated from 10, 100, and 400 μM.
Purified enzyme was used.
Computational studies were carried out to show that even if Cys23 is not directly involved in the catalytic cycle or in the attack and reduction of the sulfoxide 1a, the replacement of this cysteine with a serine alters the binding pocket. In the C23S mutant, the smaller serine hydroxide makes a new hydrogen bond with Thr29 (Figure 2b), leading to a significant change in the size and position of the loop containing the nucleophilic Cys25, which explains the poor ee observed.
To further investigate and elucidate the dynamics of the catalytic cycle, an NMR structural study of the biocatalyst MsrA02 was carried out. 15N- and 13C-labeled MsrA02 enzymes were expressed and purified and the biocatalytic reduction of the sulfoxide 1a was studied by 15N-1H NMR, allowing residue-specific information to be collected. Using a standard suit of triple resonance NMR experiments, around 70% of the backbone resonances were identified, allowing us to identify residues involved in the catalytic activity (Figure 3a).
Figure 3.

15N HSQC spectrum of MsrA02. (a) Assigned 15N HSQC of MsrA02 in its free, reduced form. (b) 15N SOFAST HMQC of MsrA02 collected at increasing 0 (black), 0.5 (blue), and 1 mM (red) of 1a.
SOFAST HMQC experiments were subsequently collected at increasing substrate 1a concentrations in the presence of DTT, resulting in the modulation of both intensities and chemical shift positions of a subset of NMR resonances following a slow exchange regime (Figure 3b). A comparison of the decay of intensities upon substrate addition at 0.5 and 1 mM show that at a lower concentration of 1a, the most affected region of the protein is around Cys25, including the beta strand at positions 74–76 (Figure 4, blue), suggesting that the catalytic activity starts with the engagement of Cys25 with the substrate. At 1 mM of 1a, large intensity changes extend from the Cys25 to the whole beta strand between residues 74–80 and the area around it, reaching Cys176 and its neighboring residues (Figure 4, red).
Figure 4.
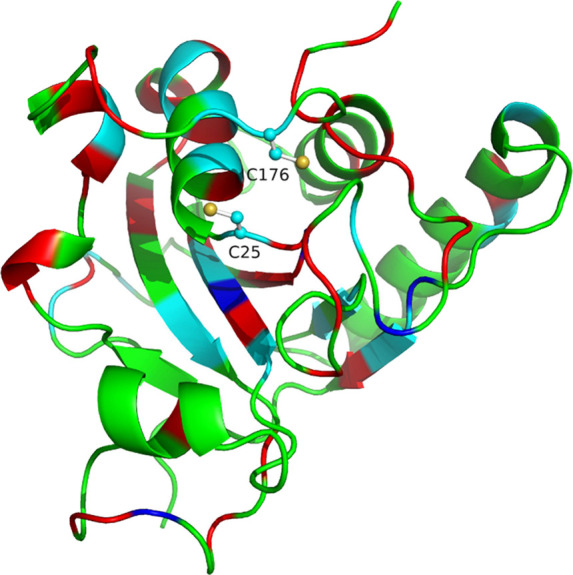
Cartoon representation of the MsrA02 structure (PDB: 3PIL) with the residues involved in the catalytic cycle of 1a highlighted. Residues with resonances undergoing significant intensity losses upon the addition of 0.5 mM or 1 mM of 1a are labeled in blue or red, respectively. Residues with resonances undergoing significant chemical shift differences between the free and bound forms are labeled in cyan. C25 and C176 sidechains are shown for clarity.
Such changes suggest that once the substrate is bound to Cys25, the area around Cys176 is involved in the catalytic cycle, supported by the changes occurring in the helical region between residues 161–172. The largest chemical shift changes between the free and bound state are also found in the patch surrounding Cys25, Cys176, and the central beta strand (Figure 4, cyan). The spectrum for the bound state of the protein shows broader peaks, indicating that substrate binding induces significant dynamics on the structure of MsrA02. These titration experiments present a slow exchange binding regime, characteristic of high affinity binding. The specificity of these spectral changes due to binding to substrate 1a was confirmed by the addition of DTT, which reverts the spectrum of MsrA02 back to the apo form (Figure S2). This indicates that the chemical shift changes observed are specific to substrate 1a and not caused by alterations of pH, salt concentrations, or any other effect.
Rational Mutagenesis
The generation of mutant enzymes able to accept sulfoxide substrates bearing substituents on the sulfur atom other than a methyl group was then investigated. Molecular dynamics simulations were performed on the WT enzyme MsrA02.
The substrates 1i and 1z bearing, respectively, a methyl and a n-propyl substituent on the sulfur atom, were chosen for docking into the active site of WT MsrA02 (Figure 5). Initial MD studies show that, when bonded with substrate 1i, Cys25 is in proximity (3.55 Å) to the sulfoxide group for nucleophilic attack (Figure 5a). Conversely, when complexed with the bulkier 1z, the n-propyl group hinders the binding in sufficient proximity to Cys25 (6.6 Å), disfavoring the nucleophilic attack on the sulfoxide group (Figure 5b). This explains the lack of activity of MsrA02 on 1z. Previous crystallographic studies on MsrA from S. cerevisiae and MetSO have shown that, in addition to Cys25 and Cys176, other essential residues for the reaction mechanism are Tyr64, Glu76, and Tyr116, as they bind and stabilize the oxygen of the sulfoxide, and Phe26, Trp27, which stabilize the methyl group of MetSO instead (Figure S1).21,24−27 Additionally, as shown in Figure 5, Tyr174 seems crucial to keep the Cys176 close to the adduct between the enzyme and the substrate to facilitate sulfide release and enzyme regeneration. Since the amino acids Phe26 and Trp27 are responsible for the interaction of the methyl group of MetSO with the MsrA02 binding pocket, we hypothesized that mutations of these residues would help accommodate larger alkyl substituents. Moreover, the mutation of other amino acid residues in the C-terminal and the α2-helix regions of MsrA02 were also explored (Table S1). Therefore, a series of nine mutant enzymes was computationally designed by modifying the WT MsrA02 amino acid residues to allow a shorter distance between the sulfoxide group of 1z and the Cys25 residue. The design of appropriate mutants was complicated by the fact that in silico mutations in the C-terminal region of MsrA02 seemed deleterious for the stability of the enzyme. Hence, enzyme MsrA10, which showed good activity on the substrate 1m, was also investigated for rational mutagenesis. In fact, since the second cysteine residue involved in the catalytic cycle of MsrA10 (corresponding to Cys176 in MsrA02 from a catalytic point of view) is not in the C-terminal region of the enzyme (Figure 5c), we hypothesized that mutations in this area would not affect the stability of the mutants.
Figure 5.
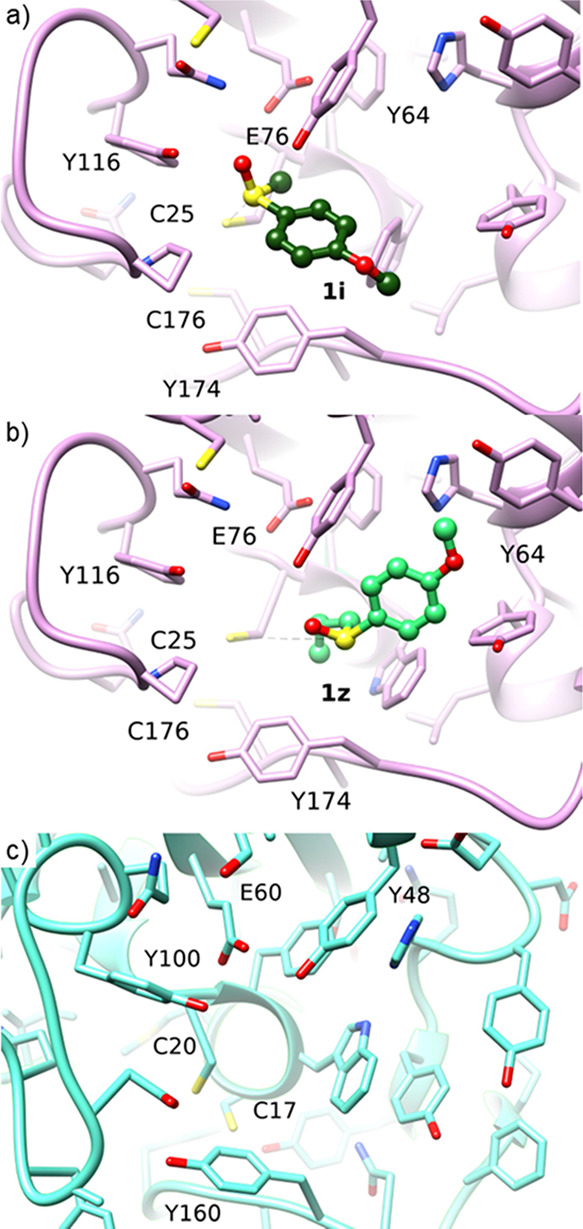
(a) docking of sulfoxide 1i in MsrA02; the Tyr174 residue seems important to keep the Cys25 close to the sulfoxide group. (b) docking of the sulfoxide 1z in MsrA02. (c) MsrA10 enzyme. The second cysteine residue of the enzymes is not in the C-terminal region.
Thus, three additional mutants of MsrA10 were designed and produced. Finally, seven new wild-type MsrA enzymes showing different C-terminal regions from MsrA02 were selected from the literature and homology searching in public databases and prepared for screening on substrate 1z. The new WT enzymes and MsrA02 and MsrA10 mutants were cloned and expressed in E. coli under the same conditions as the original Msr enzyme panel and screened, as CFE, with the n-propyl substrate 1z (Table S1). Among all the mutants, only mutant F26Y (MsrA33) was able to reduce 1z, providing the (R)-enantiomer with 43% ee. Figure 6 shows that the F26Y mutation creates additional space for the n-propyl substituent of substrate 1z, bringing Cys25 and the sulfoxide at 5.0 Å distance.
Figure 6.
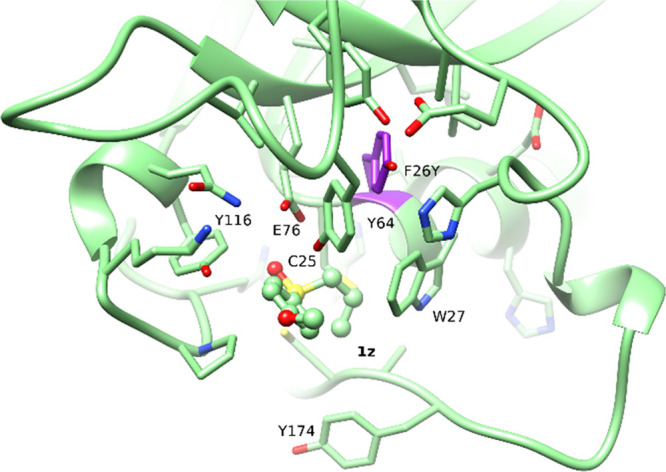
1z binding pose for the mutant MsrA33 (F26Y). In the WT enzyme, the substrate is far from Cys25, caused by steric hindrance between the propyl group of 1z and the enzyme (Figure 5b). In the mutant, the substrate is in proximity with Cys25 favoring the nucleophilic attack.
The biocatalyzed reduction of 1z with the MsrA02-mutant MsrA33, as CFE biocatalyst, was then optimized (Table S2). Enantiomer (R)-1z was obtained in 42% HPLC yield and 99% ee when the biotransformation was carried out for 48 h using 8 mM of racemic 1z, 40 gL–1 of MsrA33, and 4 equiv DTT28 (Table S2, entry 16). The substrate scope of the biocatalytic reduction of various sulfoxides with the mutant MsrA33 was finally investigated. The results are reported in Table 5. All the sulfoxides 1z, 1y, and 1ac–af bearing an n-propyl substituent on the sulfur atom were obtained with good-high conversion (Table 5, entries 1–6). Remarkably, the (R)-enantiomers of the derivatives 1z, 1ac, and 1ad were obtained with excellent ees (up to 99%). The bulkier sulfoxide 1ad bearing an n-butyl substituent was also reacted with MsrA33, but lower ee was detected for (R)-1ad. On the other hand, the ethyl-substituted sulfoxides 1u and 1x (Table 5, entries 8–9), which were poorly reduced by MsrA02, were formed with good conversion and excellent ee (>99 and 96%).
Table 5. Scope of the MsrA33 Biocatalyzed Deracemization of Sulfoxides.

| entry | substrate | R1 | R2 | (R)-sulfoxide conv. %a | ee %b |
|---|---|---|---|---|---|
| 1 | 1z | 4-MeOPh | nPr | 42 | >99 |
| 2 | 1y | Ph | nPr | 37 | 80 |
| 3 | 1ac | 4-MePh | nPr | 40 | 99 |
| 4 | 1ad | 4-ClPh | nPr | 46 | 92 |
| 5 | 1ae | 4-BrPh | nPr | 56 | 76 |
| 6 | 1af | 3-MeOPh | nPr | 51 | 64 |
| 7 | 1ad | 4-MeOPh | nBu | 87 | 12 |
| 8 | 1u | 4-BrPh | Et | 39 | >99 |
| 9 | 1x | 2-PyCH2 | Et | 44 | 96 |
Conversions determined by reverse-phase HPLC.
Determined by chiral HPLC using Chiralpak column IG, IC, ID, or Chiracel OD-H.
Conclusions
MsrAs are a promising class of enzymes able to catalyze the stereoselective reduction of chiral sulfoxides into enantiomerically pure products. The use of reductive instead of oxidative enzymes in the synthesis of enantiomerically pure sulfoxides offers substantial advantages from a synthetic point of view, such as the use of cheap cosubstrate DTT in place of the expensive cofactor NAD(P)H and related recycling systems or avoiding peroxide reagents which can lead to overoxidation byproducts. Thus, the discovery and preparation of new MsrA enzymes is an important development as it expands the biocatalysis toolbox and it provides an alternative biocatalytic strategy to access enantiomerically pure sulfoxides especially from substrates bearing multiple oxidation sites, which are incompatible with existing oxidizing biocatalysts.
To date, only few MsrA biocatalysts have been described in the literature and little is still known on their potential and industrial application. This work has expanded the range of MsrA enzymes suitable for biocatalytic applications, leading to the identification of MsrA02 from Saccharomyces cerevisiae. MsrA02 proved to be a robust and efficient biocatalyst in the KR of a large variety of aromatic and aliphatic racemic sulfoxides and to work well at high substrate concentrations as CFE, pure enzyme or whole cell biocatalyst. Moreover, in this work, a study of the catalytic mechanism of the biotransformation has been carried out, through mutagenesis and structural biology NMR studies, highlighting the amino acid residues and the dynamics involved in the reduction of sulfoxides. Such studies have led to the in silico design and preparation of a novel MsrA mutant enzyme, which is remarkable in its ability to catalyze the reduction of sulfoxides bearing a diverse range of non-methyl substituents on the sulfur atom. This development significantly expands the substrate scope of MsrAs and thus overcomes a major limitation of currently available MsrA biocatalysts. To the best of our knowledge, this is the first example of rational mutagenesis of MsrA enzymes, and it can pave the way to exploit and expand the scope and application of such biocatalysts both in academia and industry.
Acknowledgments
We gratefully acknowledge BBSRC LIDo (BB/M009513/1) for PhD Studentships to SA and IO. SMB acknowledges BBSRC (BB/P019811/1) for financial support. NMR structural study was supported by the Francis Crick Institute through provision of access to the MRC Biomedical NMR Centre. The Francis Crick Institute receives its core funding from Cancer Research UK (FC001029), the UK Medical Research Council (FC001029), and the Wellcome Trust (FC001029).
Glossary
Abbreviations
- Msr
methionine sulfoxide reductase
- MetSO
methionine sulfoxide
- KR
kinetic resolution
- FAD
flavin adenine dinucleotide
- FMN
flavin mononucleotide
- WT
wild type
- CFE
cell free extract
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.3c00372.
Detailed experimental procedure; compound characterization data; copies of NMR spectra of sulfoxides; and copies of HPLC traces of biocatalytic reaction products (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
BBSRC LIDo (BB/M009513/1), BBSRC (BB/P019811/1).
The authors declare no competing financial interest.
Supplementary Material
References
- Han J.; Soloshonok V. A.; Klika K. D.; Drabowicz J.; Wzorek A. Chiral sulfoxides: advances in asymmetric synthesis and problems with the accurate determination of the stereochemical outcome. Chem. Soc. Rev. 2018, 47, 1307–1350. 10.1039/C6CS00703A. [DOI] [PubMed] [Google Scholar]
- a Trost B. M.; Rao M. Development of chiral sulfoxide ligands for asymmetric catalysis. Angew. Chem., Int. Ed. 2015, 54, 5026–5043. 10.1002/anie.201411073. [DOI] [PubMed] [Google Scholar]; b Salom-Roig X.; Bauder C. Recent applications in the use of sulfoxides as chiral auxiliaries for the asymmetric synthesis of natural and biologically active products. Synthesis 2020, 52, 964–978. 10.1055/s-0039-1690803. [DOI] [Google Scholar]; c Jia T.; Wang M.; Liao J. Chiral Sulfoxide Ligands in Asymmetric Catalysis. Top. Curr. Chem. 2019, 377, 1. 10.1007/s41061-019-0232-9. [DOI] [PubMed] [Google Scholar]; d Sipos G.; Drinkel E. E.; Dorta R. The emergence of sulfoxides as efficient ligands in transition metal catalysis. Chem. Soc. Rev. 2015, 44, 3834–3860. 10.1039/C4CS00524D. [DOI] [PubMed] [Google Scholar]
- Kendall M. J. Esomeprazole–the first proton pump inhibitor to be developed as an isomer. Aliment. Pharmacol. Ther. 2003, 17, 1–4. 10.1046/j.1365-2036.17.s1.1.x. [DOI] [PubMed] [Google Scholar]
- Loland C. J.; Mereu M.; Okunola O. M.; Cao J.; Prisinzano T. E.; Mazier S.; Kopajtic T.; Shi L.; Katz J. L.; Tanda G.; Hauck Newman A. R-modafinil (armodafinil): a unique dopamine uptake inhibitor and potential medication for psychostimulant abuse. Biol. Psychiatr. 2012, 72, 405–413. 10.1016/j.biopsych.2012.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Fernández I.; Khiar N. Recent Developments in the synthesis and utilization of chiral sulfoxides. Chem. Rev. 2003, 103, 3651–3706. 10.1021/cr990372u. [DOI] [PubMed] [Google Scholar]; b Wojaczyńska E.; Wojaczyński J. Enantioselective synthesis of sulfoxides: 2000– 2009. Chem. Rev. 2010, 110, 4303–4356. 10.1021/cr900147h. [DOI] [PubMed] [Google Scholar]
- Cogan D. A.; Liu G.; Kim K.; Backes B. J.; Ellman J. A. Catalytic asymmetric oxidation of tert-butyl disulfide. synthesis of tert-butanesulfinamides, tert-butyl sulfoxides, and tert-butanesulfinimines. J. Am. Chem. Soc. 1998, 120, 8011. 10.1021/ja9809206. [DOI] [Google Scholar]
- Huang Q. Y.; Zhu J.; Deng J. G. Recent development in asymmetric catalytic oxidation of prochiral sulfides by metal-based catalysts. Chin. J. Org. Chem. 2005, 25, 496–506. [Google Scholar]
- a Zhang Y.; Wu Y. Q.; Xu N.; Zhao Q.; Yu H. L.; Xu J. H. Engineering of cyclohexanone monooxygenase for the enantioselective synthesis of (S)-omeprazole. ACS Sustainable Chem. Eng. 2019, 7, 7218–7226. 10.1021/acssuschemeng.9b00224. [DOI] [Google Scholar]; b de Gonzalo G.; Franconetti A. Enantioselective sulfoxidations employing the thermostable cyclohexanone monooxygenase from Thermocrispum municipal. Enzyme Microb. Technol. 2018, 113, 24–28. 10.1016/j.enzmictec.2018.02.006. [DOI] [PubMed] [Google Scholar]; c de Gonzalo G.; Fürst M. J. L. J.; Fraaije M. W. Polycyclic ketone monooxygenase (PockeMO): a robust biocatalyst for the synthesis of optically active sulfoxides. Catalysts 2017, 7, 288. 10.3390/catal7100288. [DOI] [Google Scholar]; d Gul T.; Krzek M.; Permentier H. P.; Fraaije M. W.; Bischoff R. Microbial flavoprotein monooxygenases as mimics of mammalian flavin-containing monooxygenases for the enantioselective preparation of drug metabolites. Drug Metab. Dispos. 2016, 44, 1270–1276. 10.1124/dmd.115.069104. [DOI] [PubMed] [Google Scholar]; e Rioz-Martínez A.; de Gonzalo G.; Pazmiño D. E. T.; Fraaije M. W.; Gotor V. Enzymatic synthesis of novel chiral sulfoxides employing Baeyer–Villiger monooxygenases. Eur. J. Org. Chem. 2010, 2010, 6409–6416. 10.1002/ejoc.201000890. [DOI] [Google Scholar]; f Fürst M. J. L. J.; Gran-Scheuch A.; Aalbers F. S.; Fraaije M. W. Baeyer-Villiger monooxygenases: tunable oxidative biocatalysts. ACS Catal. 2019, 9, 11207–11241. 10.1021/acscatal.9b03396. [DOI] [Google Scholar]; g Kamerbeek N. M.; Janssen D. B.; van Berkel W. J. H.; Fraaije M. W. Baeyer–Villiger monooxygenases, an emerging family of flavin-dependent biocatalysts. Adv. Synth. Catal. 2003, 345, 667–678. 10.1002/adsc.200303014. [DOI] [Google Scholar]; h Heine T.; Scholtissek A.; Hofmann A.; Koch R.; Tischler D. Accessing enantiopure epoxides and sulfoxides: related flavin-dependent monooxygenases provide reversed enantioselectivity. ChemCatChem 2020, 12, 199–209. 10.1002/cctc.201901353. [DOI] [Google Scholar]
- a Bassanini I.; Ferrandi E. E.; Vanoni M.; Ottolina G.; Riva S.; Crotti M.; Brenna E.; Monti D. Peroxygenase-catalyzed enantioselective sulfoxidations. Eur. J. Org. Chem. 2017, 2017, 7186–7189. 10.1002/ejoc.201701390. [DOI] [Google Scholar]; b Li Y.; Ma Y.; Li P.; Zhang X.; Ribitsch D.; Alcalde M.; Hollmann F.; Wang Y. Enantioselective sulfoxidation of thioanisole by cascading a choline oxidase and a peroxygenase in the presence of natural deep eutectic solvents. ChemPlusChem 2020, 85, 254–257. 10.1002/cplu.201900751. [DOI] [PubMed] [Google Scholar]; c Wei X.; Zhang C.; Gao X.; Gao Y.; Yang Y.; Guo K.; Du X.; Pu L.; Wang Q. Enhanced activity and substrate specificity by site-directed mutagenesis for the P450 119 peroxygenase catalyzed sulfoxidation of thioanisole. ChemistryOpen 2019, 8, 1076–1083. 10.1002/open.201900157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Anselmi S.; Aggarwal N.; Moody T. S.; Castagnolo D. Unconventional biocatalytic approaches to the synthesis of chiral sulfoxides. ChemBioChem 2021, 22, 298–307. 10.1002/cbic.202000430. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Peng T.; Cheng X.; Chen Y.; Yang J. Sulfoxide reductases and applications in biocatalytic preparation of chiral sulfoxides: a mini-review. Front. Chem. 2021, 9, 714899 10.3389/fchem.2021.714899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boschi-Muller S.; Olry A.; Antoine M.; Branlant G. The enzymology and biochemistry of methionine sulfoxide reductases. Biochim. Biophys. Acta Proteins Proteom. 2005, 1703, 231–238. 10.1016/j.bbapap.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Tarrago L.; Kaya A.; Weerapana E.; Marino S. M.; Gladyshev V. N. Methionine sulfoxide reductases preferentially reduce unfolded oxidized proteins and protect cells from oxidative protein unfolding. J. Biol. Chem. 2012, 287, 24448–24459. 10.1074/jbc.M112.374520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le D. T.; Lee B. C.; Marino S. M.; Zhang Y.; Fomenko D. E.; Kaya A.; Hacioglu E.; Kwak G. H.; Koc A.; Kim H. Y.; Gladyshev V. N. Functional analysis of free methionine-R-sulfoxide reductase from Saccharomyces cerevisiae. J. Biol. Chem. 2009, 284, 4354–4364. 10.1074/jbc.M805891200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Yuan Z.; Zhou Y.; Zhao J.; Yang M.; Cheng X.; Ou G.; Chen Y. Asymmetric reductive resolution of racemic sulfoxides by recombinant methionine sulfoxide reductase from a pseudomonas monteilii strain. J. Mol. Catal. B: Enzym. 2016, 133, S588–S592. 10.1016/j.molcatb.2017.02.005. [DOI] [Google Scholar]
- Peng L.; Wen Y.; Chen Y.; Yuan Z.; Zhou Y.; Cheng X.; Chen Y.; Yang J. Asymmetric reductive resolution of racemic sulfoxides by recombinant methionine sulfoxide reductase from a pseudomonas monteilii strain. ChemCatChem 2018, 10, 3284–3290. 10.1002/cctc.201800279. [DOI] [Google Scholar]
- a Yang J.; Wen Y.; Peng L.; Chan Y.; Cheng X.; Chen Y. Identification of MsrA homologues for the preparation of (R)-sulfoxides at high substrate concentrations. Org. Biomol. Chem. 2019, 17, 3381–3388. 10.1039/C9OB00384C. [DOI] [PubMed] [Google Scholar]; b Peng T.; Tian J.; Zhao Y.; Jiang X.; Cheng X.; Deng G.; Zhang Q.; Wang Z.; Yang Y.; Chen Y. Multienzyme redox system with cofactor regeneration for cyclic deracemization of sulfoxides. Angew. Chem., Int. Ed. 2022, 61, e202209272 10.1002/anie.202209272. [DOI] [PubMed] [Google Scholar]; c Nosek V.; Míšek J. Chemoenzymatic Deracemization of Chiral Sulfoxides. Angew. Chem., Int. Ed. 2018, 57, 9849–9852. 10.1002/anie.201805858. [DOI] [PubMed] [Google Scholar]; d Bierbaumer S.; Schmermund L.; List A.; Winkler C. K.; Glueck S. M.; Kroutil W. Synthesis of enantiopure sulfoxides by concurrent photocatalytic oxidation and biocatalytic reduction. Angew. Chem. Int. Ed. 2022, 61, e2021171 10.1002/anie.202117103. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Tarallo V.; Sudarshan K.; Nosek V.; Misek J. Development of a Simple High-Throughput Assay for Directed Evolution of Enantioselective Sulfoxide Reductases. Chem. Commun. 2020, 56, 5386–5388. 10.1039/D0CC01660H. [DOI] [PubMed] [Google Scholar]
- a Wang P.; Han X.; Liu X.; Lin R.; Chen Y.; Sun Z.; Zhang W. Synthesis of enantioenriched sulfoxides by an oxidation-reduction enzymatic cascade. Chem. – Eur. J. 2022, 28, e202201997 10.1002/chem.202201997. [DOI] [PubMed] [Google Scholar]; b Wen Y.; Peng L.; Zhou Y.; Peng T.; Chen Y.; Cheng X.; Chen Y.; Yang J. Discovery and application of methionine sulfoxide reductase B for preparation of (S)-sulfoxides through kinetic resolution. Catal. Commun. 2020, 136, 105908 10.1016/j.catcom.2019.105908. [DOI] [Google Scholar]
- Achilli C.; Ciana A.; Minetti G. Kinetic resolution of phenyl methyl sulfoxides by mammalian methionine sulfoxide reductase A. Tetrahedron Lett. 2017, 58, 4781–4782. 10.1016/j.tetlet.2017.11.022. [DOI] [Google Scholar]
- a Zhao F.; Lauder K.; Liu S.; Finnigan J. D.; Charnock S. B. R.; Charnock S. J.; Castagnolo D. Chemoenzymatic cascades for the enantioselective synthesis of β-hydroxysulfides bearing a stereocentre at C–O or C–S bonds by ketoreductases (KREDs). Angew. Chem., Int. Ed. 2022, 61, e202202363 10.1002/anie.202202363. [DOI] [PubMed] [Google Scholar]; b Lauder K.; Toscani A.; Qi Y.; Lim J.; Charnock S. J.; Korah K.; Castagnolo D. Photo-biocatalytic one-pot cascades for the enantioselective synthesis of 1,3-mercaptoalkanol volatile sulfur compounds. Angew. Chem., Int. Ed. 2018, 57, 5803–5807. 10.1002/anie.201802135. [DOI] [PubMed] [Google Scholar]; c Anselmi S.; Liu S.; Kim S.-H.; Barry S. M.; Moody T. S.; Castagnolo D. A mild and chemoselective CALB biocatalysed synthesis of sulfoxides exploiting the dual role of AcOEt as solvent and reagent. Org. Biomol. Chem. 2020, 19, 156–161. 10.1039/D0OB01966F. [DOI] [PubMed] [Google Scholar]; d Lauder K.; Anselmi S.; Finnigan J. D.; Qi Y.; Charnock S. J.; Castagnolo D. Enantioselective synthesis of α-thiocarboxylic acids by nitrilase biocatalysed dynamic kinetic resolution of α-thionitriles. Chem. – Eur. J. 2020, 26, 10422–10426. 10.1002/chem.202001108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The absolute configuration of (R)-1a was determined by comparison of HPLC data with the commercially available enantiomer. The configuration of all other sulfoxides was established by comparison of the [α]D values with those reported in the literature (Table S4).
- The crystallographic data for S. cerevisiae MsrA is reported (PDB 3PIL).; Ma X.-X.; Guo P.-C.; Shi W. W.; Luo M.; Tan X.-F.; Chen Y.; Zhou C.-Z. Structural plasticity of the thioredoxin recognition site of yeast methionine S-sulfoxide reductase Mxr1. J. Biol. Chem. 2011, 286, 13430–13437. 10.1074/jbc.M110.205161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Even if other MsrA enzymes proved to be efficient in the reduction of 1a, they were not selected for further investigation due to the lack of crystallographic information which might have complicated the mutagenesis studies. However, some of the MsrA enzymes active on 1a, have been also tested on other sulfoxide substrates (see Table 3).
- a Lim J. C.; You Z.; Kim G.; Levine R. L. Methionine sulfoxide reductase A is a stereospecific methionine oxidase. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 10472–10477. 10.1073/pnas.1101275108. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Weissbach H.; Etienne F.; Hoshi T.; Heinemann S. H.; Lowther W. T.; Matthews B.; St John G.; Nathan C.; Brot N. Peptide methionine sulfoxide reductase: structure, mechanism of action, and biological function. Arch. Biochem. Biophys. 2002, 397, 172–178. 10.1006/abbi.2001.2664. [DOI] [PubMed] [Google Scholar]; c Lowther W. T.; Brot N.; Weissbach H.; Matthews B. W. Structure and mechanism of peptide methionine sulfoxide reductase, an “anti-oxidation” enzyme. Biochemistry 2000, 39, 13307–13312. 10.1021/bi0020269. [DOI] [PubMed] [Google Scholar]
- Dokainish H. M.; Gauld J. W. A molecular dynamics and quantum mechanics/molecular mechanics study of the catalytic reductase mechanism of methionine sulfoxide reductase a: formation and reduction of a sulfenic acid. Biochemistry 2013, 52, 1814–1827. 10.1021/bi301168p. [DOI] [PubMed] [Google Scholar]
- Quinternet M.; Tsan P.; Neiers F.; Beaufils C.; Boschi-Muller S.; Averlant-Petit M. C.; Branlant G.; Cung M. T. Solution, structure and dynamics of the reduced and oxidized forms of the N-terminal domain of PilB from Neisseria meningitidis. Biochemistry 2008, 47, 8577–8589. 10.1021/bi800884w. [DOI] [PubMed] [Google Scholar]
- Moskovitz J.; Poston J. M.; Berlett B. S.; Nosworthy N. J.; Szczepanowski R.; Stadtman E. R. Identification and characterization of a putative active site for peptide methionine sulfoxide reductase (MsrA) and its substrate stereospecificity. J. Biol. Chem. 2000, 275, 14167–14172. 10.1074/jbc.275.19.14167. [DOI] [PubMed] [Google Scholar]
- The primary sequences of the MsrA enzymes from E. coli, M. tuberculosis, N. meningitidis and S. cerevisiae shows a great degree of similarity, with the greatest differences in the C-terminal loop, while the amino acids in the binding pocket are conserved. Thus, previous studies on the mechanism of action of MsrA from these species were taken into account in the design of MsrA02 mutants.
- An excess of DTT was required due to the higher biocatalyst loading used in the optimization reaction.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.