

Abstract
Background:
The phenotype of peripheral neuropathy (PN) associated with glial fibrillary acidic protein-immunoglobulin G (GFAP-IgG) has not been well described.
Objectives:
The aim of this study was to report the frequency, clinical, and electrophysiological characteristics of PN in GFAP-IgG-positive patients.
Design:
This study is a single-center retrospective observational study.
Data Sources and methods:
GFAP-IgG-positive patients with PN were retrospectively identified from the Huashan Hospital Autoimmune Encephalitis Cohort between 2017 and 2021. Eight patients who presented with PN from other published studies were also included in the analysis. The clinical and electrophysiological characteristics of GFAP-IgG-related PN were described.
Results:
A total of 21 (31%) patients (7 females, 14 males; Mage: 42 ± 16 years) from a cohort of 68 GFAP-IgG-positive patients presented with PN. Twenty of 21 patients had symmetrical weakness. Sensory and autonomic symptoms were present in 16 and 15 patients, respectively. Lower extremities were the most frequently involved regions for both motor (20/21) and sensory (15/21) symptoms. Moreover, 13 patients (4 females, 9 males; Mage: 43 ± 13 years) had electrodiagnostic study data, and 12 of 13 patients had abnormal findings. Regarding clinical features, motor nerve fibers were predominantly involved (12/13), and symmetric lower extremities (12/13) were the most commonly affected regions. Axonal neuropathy is the typical underlying pathophysiologic process of PN. All 21 patients responded to immunotherapy. However, four patients with tetraplegia had poor outcomes, and PN was the major determinant of their long-term disability. Most cases (6/8) from the literature presented with similar clinical and electrophysiological features to those from our cohort.
Conclusion:
Peripheral nerves could be involved in autoimmune GFAP astrocytopathy. Predominant motor axonal neuropathy mainly involving the lower extremities is the most common PN phenotype in this disorder. GFAP-IgG-related PN is responsive to immunotherapy.
Registration:
Chinese Clinical Trial Registry: ChiCTR2000029115 (http://www.chictr.org).
Keywords: astrocytopathy, autoantibody, glial fibrillary acidic protein, peripheral neuropathy
Introduction
Autoimmune glial fibrillary acidic protein (GFAP) astrocytopathy is a recently identified autoimmune neurological disease associated with immunoglobulin G to GFAP (GFAP-IgG).1,2 Central nervous system (CNS) syndromes, including meningitis, encephalitis, myelitis, and their combinations, are widely recognized as the common phenotypes of this disorder. 2 Whether GFAP-IgG-related astrocytopathy can also affect the peripheral nervous system (PNS) is unclear. More recently, several studies have reported GFAP-IgG-positive patients who presented with peripheral neuropathy (PN).3–5 However, the frequency and phenotypes of PN associated with GFAP-IgG have not been well described.
Here, we report a case series of GFAP-IgG-positive patients with PN. Clinical, electrophysiological, and imaging characteristics were comprehensively described to raise awareness that autoimmune GFAP astrocytopathy is a disorder involving both the CNS and PNS.
Methods
Participants
We screened for GFAP-IgG in patients from the Huashan Hospital Autoimmune Encephalitis cohort between 2017 and 2021. All patients had a clinical syndrome of meningoencephalomyelitis or limited forms (i.e. at least one of the following presentations: meningitis, encephalitis, and myelitis). A total of 68 cases were GFAP-IgG-positive and their clinical information was retrospectively reviewed. Patients included in this study fulfilled the following three criteria: (1) GFAP-IgG was positive in the patient’s cerebrospinal fluid (CSF); (2) patient presented with symptom(s) and sign(s) of PN; and (3) PN could not be attributed to alternative etiologies (e.g. diabetes mellitus, drugs, alcohol, and critical illness neuropathy).
Clinical features of PN, nerve conduction study (NCS) and electromyography (EMG) data, and neuroimaging of these patients were comprehensively reviewed. Specifically, the Medical Research Council (MRC) scale was used to assess muscle strength. Sensory dysfunction was classified into small-fiber dysfunction (symptoms of pain, paresthesia, hyperesthesia, and signs of reduction in light touch or pinprick) and large-fiber dysfunction (reduction or loss of vibratory sensation or proprioceptive sensation). Otherwise unexplained gastrointestinal tract dysfunction (abdominal distension, diarrhea, and constipation), cardiovascular symptoms (orthostatic hypotension, bradycardia, and tachycardia), and skin symptoms (hyperhidrosis, hypohidrosis, and dry skin) were regarded as autonomic symptoms. Bladder and bowel incontinence were not included as autonomic symptoms of PN in this study because myelopathy of autoimmune GFAP astrocytopathy could also cause bladder and bowel dysfunction.
Evidence of demyelinating neuropathy includes prolonged distal motor latency, conduction slowing, conduction block, markedly dispersed compound muscle action potential (CMAP), or very prolonged F-wave latency. 6 Evidence of axonal neuropathy included reduced or absent CMAP or sensory nerve action potential (SNAP) amplitude with normal or slightly slow conduction velocity, and EMG findings of denervation potential, high amplitude, and long duration motor unit potential. Evidence of nerve root involvement included symptoms or signs indicating radiculopathy, prolonged or absent F/H waves, abnormal EMG findings of proximal and paraspinal muscles, and nerve root enhancement or hypertrophy on magnetic resonance imaging (MRI). We used the modified Rankin Scale (mRS) to assess patients’ disease severity and outcomes.
Detection of autoantibodies
GFAPα-IgG was tested by tissue-based assays (TBA) and cell-based assays (CBA) as previously reported.2,7 Briefly, for CBA, the GFAPα isoform was expressed in HEK293-T cells. Cells were fixed, permeabilized, blocked, and then incubated with patient serum (1:100) or CSF (1:2). For TBA, commercially available monkey cerebellum or hippocampus slides (Biosystems S.A., Barcelona, Spain) were used to detect autoantibodies in CSF. After incubation with the patient’s sample overnight at 4°C, Alexa Fluor 488 goat anti-human IgG (Invitrogen, Eugene, Oregon, USA) was used to label autoantibodies. Positive staining of GFAP-expressing cells on CBA and an astrocytic pattern of IgG staining on TBA were interpreted as positive according to the literature.1,2 Since coexisting autoimmunity is not rare in autoimmune GFAP astrocytopathy,2,8 we also evaluated autoantibodies to aquaporin-4, myelin oligodendrocyte glycoprotein, and N-methyl-D-aspartate receptor in patients’ serum or CSF by live CBA.
Literature review
We searched PubMed [search terms: (‘Peripheral Nervous System’[Mesh] OR ‘Peripheral Nervous System Diseases’[Mesh] OR *Neuropath* OR Radiculopath* OR Neuritis) AND (‘Glial Fibrillary Acidic Protein’[Mesh] OR ‘ Glial Fibrillary Acidic Protein’ OR GFAP) AND (Astrocytopathy OR Autoimmu* OR Autoantibod*); time limitation: between January, 2016 and April, 2022] to screen GFAP-IgG-positive cases with PN in the literature. Eight patients with sufficient demographic and clinical data from three studies were identified. We summarized the pattern of PN in autoimmune GFAP astrocytopathy by analyzing all cases from our center and the literature.
Statistical analysis
We used SPSS, version 20.0 (SPSS, Chicago, Illinois, USA) to perform statistical analysis. Normally distributed data, non-normally distributed data, and categorical data are expressed as the mean ± standard deviation (SD), median (range), and frequencies (percentages), respectively. We used the paired data Wilcoxon rank-sum test to compare the muscle strength between the lower and upper extremities. A two-tailed p-value less than 0.05 was considered statistically significant.
Results
Frequency and clinical features
Of 68 GFAP-IgG-positive patients, 21 (7 females, 14 males; Mage: 42 ± 16 years) presented with PN during their clinical course (Figure 1). All 21 patients had GFAP-IgG in CSF, 17 of whom also had seropositive GFAP-IgG. All 21 patients presented with PN in addition to CNS syndromes (meningoencephalomyelitis, 13; encephalomyelitis, 2; meningomyelitis, 3; meningoencephalitis, 2; and encephalitis, 1). Symmetric extremity weakness (paraplegia, 13; tetraplegia, 7) was the most common PN symptom. Sensory symptoms were present in 16 patients. Lower extremities were the most frequently involved regions for both motor (20/21) and sensory (15/21) symptoms. Autonomic nerve involvement occurred in 15 patients, 10 of whom had gastrointestinal motility disorder. Seven patients had clinical evidence of nerve root involvement. Two patients had concurrent cranial neuropathies. Physical examination revealed that the median muscle strength of the lower extremities was weaker than that of the upper extremities [3 (0, 5) versus 5 (2, 5); p < 0.001]. A reduced or absent tendon reflex was present in 15 patients (ankle, 15; knee, 12; biceps, 6). Interestingly, more than half of them (8/15) had concurrent Babinski signs. Examination of sensory deficits included impaired light touch and pinprick sensation in 16 patients, and impaired vibration sensation in 2 patients. Table 1 summarizes the demographic information and clinical and electrophysiological characteristics of each patient. The distribution of motor and sensory involvement is shown in Figure 2.
Figure 1.
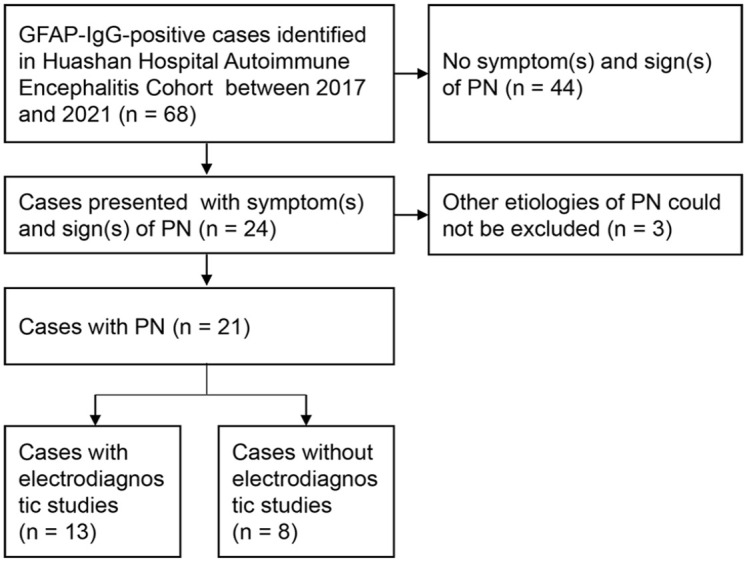
Flowchart of patient inclusion.
Table 1.
Demographic, PN phenotype, and clinical characteristics of 13 GFAP-IgG-positive patients.
| Pt. no. | Age (y) / sex | Pattern of PN | Symptoms of autonomic and cranial neuropathy | CNS syndromes | Electrodiagnostic studies | Brain or spinal lesions | Nerve root enhancement | Treatment | Max. mRS/last follow-up mRS | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Axonal neuropathy | Demyelinating neuropathy | Abnormal F or H reflexes | |||||||||
| 1 | 29/M | Sensorimotor axonal neuropathy | Tachycardia, abdominal distension | Meningoencephalomyelitis | Yes | No | Yes | Yes | No | Steroids, IVIG | 5/3 |
| 2 | 55/M | Sensorimotor axonal and demyelinating neuropathy | Abdominal distension | Encephalomyelitis | Yes | Yes | No | No | Yes | Steroids, IVIG | 4/2 |
| 3 | 54/M | Predominant motor axonal neuropathy | Bradycardia | Meningoencephalomyelitis | Yes | No | Yes | Yes | NA | Steroids, IVIG, PE | 5/4 |
| 4 | 59/M | Predominant motor axonal neuropathy | None | Meningoencephalomyelitis | Yes | No | Yes | Yes | NA | Steroids, IVIG | 4/4 |
| 5 | 43/M | Predominant motor axonal neuropathy | Orthostatic hypotension, abdominal distension | Meningoencephalomyelitis | Yes | No | Yes | Yes | Yes | Steroids, IVIG | 5/4 |
| 6 | 29/F | Predominant motor axonal neuropathy | Hyperhidrosis, diplopia | Meningoencephalitis | Yes | No | Yes | No | NA | Steroids | 3/1 |
| 7 | 47/F | Predominant motor axonal neuropathy | None | Encephalitis | Yes | No | No | Yes | NA | Steroids | 2/1 |
| 8 | 68/M | Motor axonal neuropathy | Abdominal distension | Meningoencephalomyelitis | Yes | No | No | Yes | NA | Steroids | 5/2 |
| 9 | 48/M | Motor axonal neuropathy | Constipation, diarrhea, abdominal distension, hyperhidrosis | Meningomyelitis | Yes | No | Yes | No | Yes | Steroids | 4/1 |
| 10 | 35/M | Motor axonal neuropathy | None | Meningoencephalomyelitis | Yes | No | No | No | Yes | Steroids, IVIG | 3/1 |
| 11 | 36/F | Sensorimotor neuropathy | Abdominal distension | Encephalomyelitis | No | No | Yes | No | Yes | Steroids | 4/3 |
| 12 | 30/F | Sensorimotor neuropathy | Abdominal distension | Meningoencephalomyelitis | No | No | Yes | Yes | No | Steroids, IVIG | 3/0 |
| 13 | 30/M | Sensorimotor neuropathy | Diplopia | Meningomyelitis | No | No | No | Yes | NA | Steroids | 2/0 |
CNS, central nervous system; F, female; IVIG, intravenous immunoglobulins; M, male; Max., maximum; mRS, modified Rankin scale; NA, not available; PE, plasma exchange; PN, peripheral neuropathy; Pt., patient; y, years.
Figure 2.
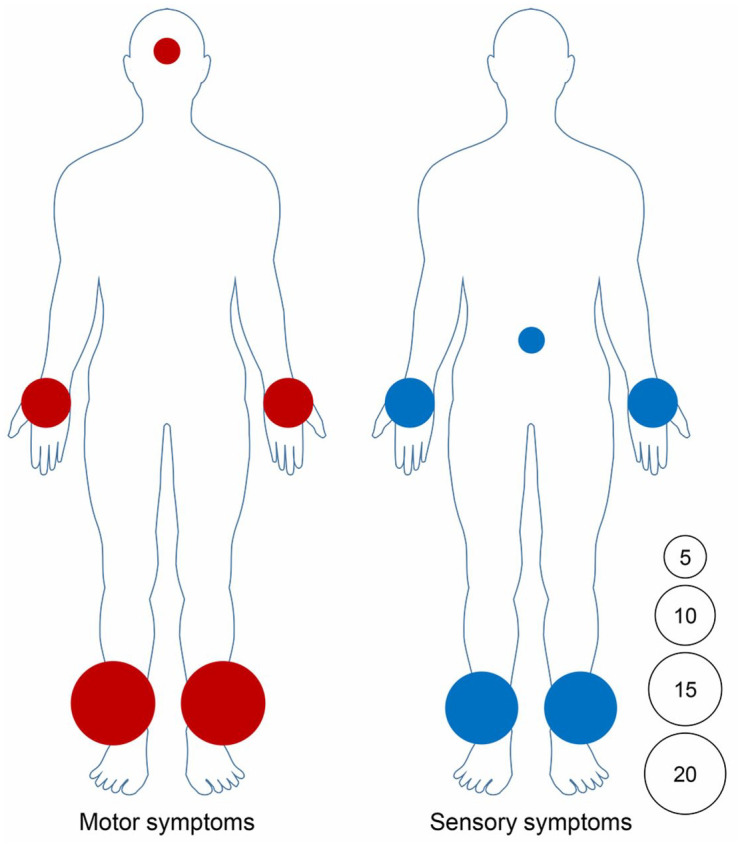
Motor and sensory symptoms distribution in 21 patients.
The area of circle represents the relative frequency of motor and sensory symptoms involvement in different body areas. The number in the circle represents the number of affected patients.
Electrophysiological characteristics
Overall, 13 of 21 patients underwent electrodiagnostic studies. The mean age of these 13 patients (4 females, 9 males) was 43 ± 13 years. Twelve patients had abnormal findings. Nine patients had axonal neuropathy (reduced/absent CMAP, 7; reduced/absent CMAP and SNAP, 2). One patient presented with evidence of both axonal and demyelinating neuropathies (reduced CMAP and slowing of motor conduction velocities). Denervation activity was found in three patients. Prolonged or absent F or H reflexes were found in eight patients. Abnormal NCS findings involving the lower extremities (12/13) were much more common than those involving the upper extremities (2/13). Overall, most patients (10/13) had electrophysiological features consistent with predominant motor axonal neuropathy. The only patient with normal electrodiagnostic studies was presented with typical glove-and-stocking reduction of sensation to pinprick and mild weakness in the lower extremities. Detailed neurophysiological results are summarized in Tables 1 and 2.
Table 2.
Summary of demographic, clinical, and electrophysiological characteristics of patients from this and previous studies.
| Items | Cases in this cohort (n = 21 a ) | Cases in literatures (n = 8) | Total cases (n = 29) |
|---|---|---|---|
| Female, n | 7/21 | 0/8 | 7/29 |
| Age (y), M± SD | 42 ± 16 | 57 ± 24 | 46 ± 19 |
| Symptoms during clinical course | |||
| Muscle weakness | 20/21 | 8/8 | 28/29 |
| Lower extremity | 13/21 | 5/8 | 18/29 |
| Both lower and upper extremities | 7/21 | 3/8 | 10/29 |
| Sensory symptoms | 16/21 | 7/8 | 23/29 |
| Lower extremity | 9/21 | 6/8 | 15/29 |
| Upper extremity | 1/21 | 0/8 | 1/29 |
| Both lower and upper extremities | 4/21 | 1/8 | 5/29 |
| Limbs and trunk | 2/21 | 0/8 | 2/29 |
| Cranial neuropathy | 2/21 | 1/8 | 3/29 |
| Electrodiagnostic studies | |||
| Nerve fiber involvement | |||
| Motor nerve fiber | 10/13 | 0/8 | 10/21 |
| Motor and sensory nerve fiber | 2/13 | 8/8 | 10/21 |
| Distribution of nerve involvement | |||
| Lower extremity | 10/13 | 7/8 | 17/21 |
| Lower and upper extremities | 2/13 | 1/8 | 3/21 |
| Pathophysiologic process of PN | |||
| Axonal neuropathies | 9/13 | 4/8 | 13/21 |
| Demyelinating neuropathies | 0/13 | 2/8 | 2/21 |
| Axonal and demyelinating neuropathies | 1/13 | 2/8 | 3/21 |
| Abnormal F or H reflexes | 8/13 | 2/8 | 10/21 |
| Denervation activity | 3/13 | 5/8 | 8/21 |
PN, peripheral neuropathy; PNS, peripheral nervous system; SD, standard deviation; y, years.
Overall, 13 of 21 patients from our center had electrodiagnostic study data.
MRI and CSF
MRI scans revealed that all 21 patients had brain or spinal abnormalities (leptomeningeal enhancement, 21; brain or spinal T2-Flair hyperintense lesions, 14). Thirteen patients had MRI data available to review nerve roots, 8 of whom presented with lumbosacral root enhancement. Representative nerve root enhancement is shown in Figure 3. All 21 patients had lymphocytic-predominant pleocytosis and elevated protein in CSF.
Figure 3.
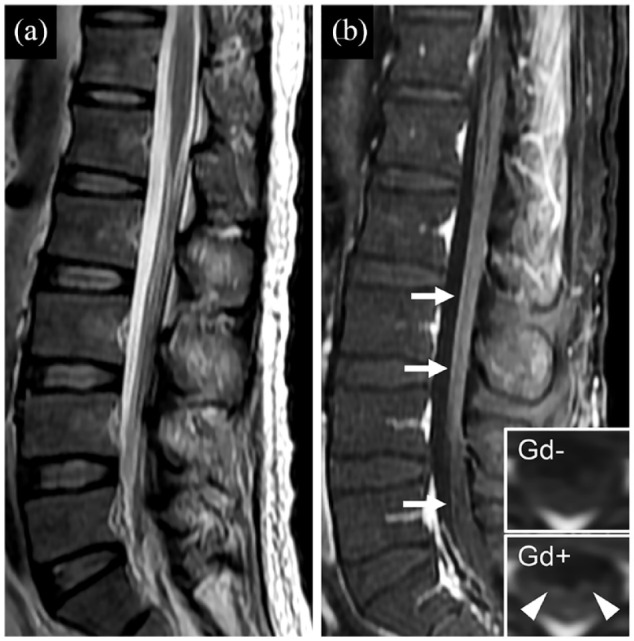
Representative image of nerve root gadolinium enhancement in patients with autoimmune GFAP astrocytopathy. (a) Sagittal T2-weighted scan shows nerve root disorganization. (b) Sagittal T1-weighted scan with gadolinium shows enhancement alone nerve roots (b; white arrows). Inset in (b) is the axial image before (Gd−) and after (Gd+) gadolinium enhancement, which also shows nerve root enhancement (b; white arrowheads). Gd, gadolinium.
Treatments and outcomes
All 21 patients received at least one first-line immunotherapy (steroids, 10; steroids and intravenous immunoglobulins, 10; steroids, intravenous immunoglobulins, and plasma exchange, 1). Patients’ median mRS at disease nadir was 4 (2–5), which was reduced to 2 (0–4) after a median follow-up of 4 (1–25) months. At the time of the last follow-up, 17 patients were independently ambulatory, 14 of whom achieved good outcomes (mRS ⩽ 2). However, four patients were wheelchair-bound or bedridden due to poor recovery of muscle strength in the lower extremities. These patients were presented with tetraplegia and had severe weakness in the lower extremities at disease nadir (MRC grade 0). Detailed treatment and outcome data are shown in Table 1 and Figure 4.
Figure 4.
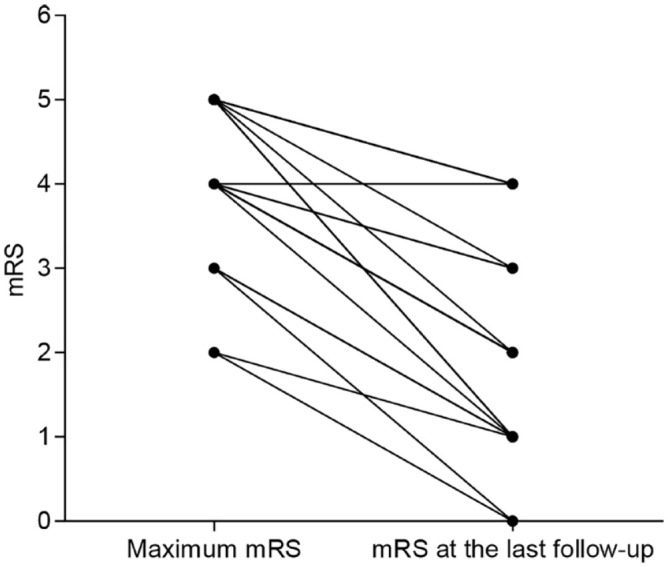
Changes of patients’ mRS at disease nadir and the last follow-up time.
All 21 patients improved with immunotherapies. Median mRS at disease nadir was 4 (2–5), which reduced to 2 (0–4) at the last follow-up time. mRS, modified Rankin scale.
Literature review and pooled analysis of PN phenotype in GFAP-IgG-positive patients
A total of 37 GFAP-IgG-positive patients from six studies were reported to have PN, 8 of whom had sufficient demographic and clinical data (Supplementary Figure S1). The majority of these cases (6/8) had similar clinical and electrophysiological features to patients from our cohort, that is, prominent motor symptoms mainly involving the lower extremities and axonal neuropathy mainly affecting motor nerve fibers. A summary of these cases is presented in Table 2.
Discussion
This study demonstrated that PN involvement was a notable clinical manifestation of autoimmune GFAP astrocytopathy. The major PN phenotype in these patients was axonal neuropathy with predominant lower extremity motor fiber involvement. In addition, we found that PN in autoimmune GFAP astrocytopathy often responds to immunotherapy.
The occurrence of PN in our center for patients with autoimmune GFAP astrocytopathy was 31% (21/68). The frequency in our cohort is similar to that in a French cohort, which reported a frequency of 24% (11/46). 8 The frequency reported in a Mayo cohort was 8% (8/102) to 11% (10/90).2,7 Notably, the data from the Mayo cohort were published in 2017 and 2018, while the French cohort was published in 2021, indicating that PN is increasingly recognized in this disorder. In summary, these results have clearly shown that PN is not rare in autoimmune GFAP astrocytopathy. Future studies should pay attention to the involvement of the PNS in this disorder, which could advance the timely diagnosis and treatment of patients.
Our cases shared several common clinical characteristics of PN. (1) Weakness was the most common symptom of PN, which occurred in nearly all patients. Bilateral lower extremities were the most frequently and severely involved regions. Previous studies also showed that motor symptoms involving the lower extremities were the typical PN phenotype in patients with autoimmune GFAP astrocytopathy.3–5 (2) Symptoms of pain, paresthesia, and signs of reduction in light touch and pinprick sensation were common, supporting small-fiber dysfunction in GFAP-IgG-related astrocytopathy. (3) Autonomic nerve involvement was often combined. Gastrointestinal motility disorder represented the most common autonomic symptom. The abovementioned clinical features could be useful diagnostic clues for PN in this disorder.
Electrophysiological abnormalities were observed in most (12/13) GFAP-IgG-positive patients with PN in this study, supporting the involvement of large-fiber neuropathy. Several electrophysiological features can also be summarized: (1) motor nerve fibers were predominantly involved; (2) symmetric lower extremities were the most commonly affected regions, which was in line with clinical findings; and (3) axonal neuropathy represented the typical underlying pathophysiologic process of our patients. Most patients identified in the literature also had similar electrophysiological features to those from our cohort.3–5 In summary, these results, along with clinical findings, demonstrated that GFAP-IgG was associated with predominant motor axonal neuropathy mainly involving the lower extremities.
GFAP-IgG-associated PN was presented with the electrophysiological characteristics of predominant axonal neuropathy. Nevertheless, a substantial portion of our patients and previously reported cases could benefit from immunotherapies. It should still be noted that four patients in our study and two patients in previous studies responded poorly to treatment.4,5 These patients had similar features, that is, severe flaccid paralysis (lower extremities MRC grade 0–1), and reduction of CMAP, and some patients had delayed diagnosis and treatment.4,5 Importantly, PN was the major determinant of long-term disability in these patients. These results indicated that recognizing peripheral nerve involvement in autoimmune GFAP astrocytopathy was important, as early immunotherapy might modify the clinical course of the disease. Research efforts are needed to identify markers that could predict poor PN outcome in further studies.
Previous studies have demonstrated that PN can be an isolated symptom or accompanied by CNS symptoms in autoimmune GFAP astrocytopathy.2,3,8 In our cohort, we found that mixed clinical symptoms and signs of CNS and PNS involvement were commonly occurred (e.g. combination of encephalitis and flaccid paralysis; persistent cooccurrence of reduced/absent tend on reflexes, and Babinski sign). The abovementioned paradoxical clinical findings of autoimmune GFAP astrocytopathy were also reported in a recent study. 9 Notably, overlap of PN and myelitis was very common in our patients. Although motor and sensory symptoms caused by myelitis could not be excluded, the typical clinical picture of PN (i.e. length-dependent, but not segmental sensorimotor polyneuropathy; motor and sensory fiber involvement revealed by electrodiagnostic studies) in most of our patients suggested that the PNS was affected. In addition, no significant difference in PN prevalence was found between patients with and without myelitis in our cohort of autoimmune GFAP astrocytopathy patients (18/47 versus 3/21, p > 0.05). These findings supported that PN in autoimmune GFAP astrocytopathy might not be due to myelitis or selective impairment of lower motor neurons in the anterior gray column. Since PN and myelitis could lead to similar motor and sensory symptoms, our study indicated that it was necessary to conduct thorough physical examination and electrodiagnostic studies to verify the involvement of the PNS when some symptoms appeared in patients with autoimmune GFAP astrocytopathy that could not be reasonably attributed to CNS involvement.
GFAP is mainly detected in the CNS as a marker of astrocytes. 10 However, GFAP could also be expressed in the PNS, including nonmyelinating Schwann cells and enteric glial cells,10,11 which might partly explain why the PNS could be involved in autoimmune GFAP astrocytopathy. Immunohistochemical studies of brain, meninges, and peripheral nerve biopsies demonstrated similar T cell-predominant perivascular inflammatory infiltrates, supporting the common pathophysiology of central and peripheral involvement in this disorder.3,12,13 However, GFAP is an intracellular protein and is not expressed in axons. The role of GFAP-IgG in the pathogenesis of PN and the relationship between glial dysfunction and axonal neuropathy in autoimmune GFAP astrocytopathy warrant further exploration.
Our study has several limitations. (1) Due to the retrospective nature of the study, some clinical manifestations of PN may have been overlooked. Some common CNS symptoms in autoimmune GFAP astrocytopathy, that is, reduced consciousness, psychosis, cognitive deficit, cerebellar ataxia, and myelitis, could also disturb accurate evaluation of PN. Therefore, the frequency of PN in autoimmune GFAP astrocytopathy might be underestimated. Our data still await further independent confirmation. (2) Since myelitis was common in our cohort, we could not exclude PN secondary to lower motor neuron involvement. (3) Although all patients in this study presented with symptoms or signs of PN before ICU treatment or long immobilization, critical illness neuropathy could also contribute to some patients’ long-term outcomes. (4) We did not conduct follow-up electrodiagnostic studies to validate whether PN could be reversed by immunotherapies. (5) Peripheral nerve biopsy and pathologic studies were lacking in our study.
Supplemental Material
Supplemental material, sj-pdf-1-tan-10.1177_17562864231164806 for Clinical and electrophysiological characteristics of peripheral neuropathy in autoimmune glial fibrillary acidic protein astrocytopathy: an observational study and literature review by Bo Deng, Jingguo Wang, Yue Qiu, Xiaoni Liu, Lei Jin, Dongqing Zhu and Xiangjun Chen in Therapeutic Advances in Neurological Disorders
Acknowledgments
Not applicable.
Footnotes
ORCID iD: Xiangjun Chen  https://orcid.org/0000-0002-8108-9013
https://orcid.org/0000-0002-8108-9013
Supplemental material: Supplemental material for this article is available online.
Contributor Information
Bo Deng, Department of Neurology, Huashan Hospital and Institute of Neurology, Fudan University, Shanghai, China; National Center for Neurological Disorders, Shanghai, China.
Jingguo Wang, Department of Neurology, Huashan Hospital and Institute of Neurology, Fudan University, Shanghai, China; National Center for Neurological Disorders, Shanghai, China.
Yue Qiu, Department of Neurology, Huashan Hospital and Institute of Neurology, Fudan University, Shanghai, China; National Center for Neurological Disorders, Shanghai, China.
Xiaoni Liu, Department of Neurology, Huashan Hospital and Institute of Neurology, Fudan University, Shanghai, China; National Center for Neurological Disorders, Shanghai, China.
Lei Jin, Department of Neurology, Huashan Hospital and Institute of Neurology, Fudan University, Shanghai, China; National Center for Neurological Disorders, Shanghai, China.
Dongqing Zhu, Department of Neurology, Huashan Hospital, Fudan University, 12 Wulumuqi Zhong Road, Shanghai 200040, China; National Center for Neurological Disorders, Shanghai, China.
Xiangjun Chen, Department of Neurology, Huashan Hospital, Fudan University, 12 Wulumuqi Zhong Road, Shanghai 200040, China; National Center for Neurological Disorders, Shanghai, China; Human Phenome Institute, Fudan University, Shanghai, China.
Declarations
Ethics approval and consent to participate: The study was approved by the Institutional Review Board of Huashan Hospital, Fudan University (KY2019-428) and was registered on a WHO-approved Chinese Clinical Trial Registry site (http://www.chictr.org, number ChiCTR2000029115). Written informed consent was obtained from study participants.
Consent for publication: Not applicable.
Author contributions: Bo Deng: Data curation; Formal analysis; Investigation; Methodology; Visualization; Writing – original draft.
Jingguo Wang: Data curation; Methodology; Writing – review & editing.
Yue Qiu: Data curation; Methodology; Writing – review & editing.
Xiaoni Liu: Data curation; Methodology; Writing – review & editing.
Lei Jin: Data curation; Writing – review & editing.
Dongqing Zhu: Conceptualization; Formal analysis; Supervision; Writing – review & editing.
Xiangjun Chen: Conceptualization; Formal analysis; Funding acquisition; Supervision; Writing – review & editing.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the Clinical Research Plan of SHDC (Grant No. SHDC2020CR2027B), 2022 Medical Service and Support Capacity Improvement Project: Construction of the Cohort-Based Multidisciplinary Accurate Diagnosis and Treatment Platform for Neurological Autoimmune and Infectious Diseases, and Shanghai Municipal Science and Technology Major Project (Grant No. 2017SHZDZX01), and State Key Laboratory of Genetic Engineering, Human Phenome Institute, Zhangjiang Fudan International Innovation Center, Fudan University.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Availability of data and materials: Anonymized data of this study will be shared on reasonable request from a qualified investigator.
References
- 1.Fang B, McKeon A, Hinson SR, et al. Autoimmune glial fibrillary acidic protein astrocytopathy a novel meningoencephalomyelitis. JAMA Neurol 2016; 73: 1297–1307. [DOI] [PubMed] [Google Scholar]
- 2.Flanagan EP, Hinson SR, Lennon VA, et al. Glial fibrillary acidic protein immunoglobulin G as biomarker of autoimmune astrocytopathy: analysis of 102 patients. Ann Neurol 2017; 81: 298–309. [DOI] [PubMed] [Google Scholar]
- 3.Paul P, McKeon A, Pittock SJ, et al. GFAP IgG associated inflammatory polyneuropathy. J Neuroimmunol 2020; 343: 577233. [DOI] [PubMed] [Google Scholar]
- 4.Allen A, Gulhar S, Haidari R, et al. Autoimmune glial fibrillary acidic protein astrocytopathy resulting in treatment-refractory flaccid paralysis. Mult Scler Relat Disord 2020; 39: 101924. [DOI] [PubMed] [Google Scholar]
- 5.Li J, Wang C, Cao Y, et al. Autoimmune glial fibrillary acidic protein astrocytopathy mimicking acute disseminated encephalomyelitis: a case report. Medicine 2021; 100: e26648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bowley MP, Chad DA. Clinical neurophysiology of demyelinating polyneuropathy. Handb Clin Neurol 2019; 161: 241–268. [DOI] [PubMed] [Google Scholar]
- 7.Dubey D, Hinso SR, Jolliffe EA, et al. Autoimmune GFAP astrocytopathy: prospective evaluation of 90 patients in 1 year. J Neuroimmunol 2018; 321: 157–163. [DOI] [PubMed] [Google Scholar]
- 8.Dumonceau AG, Ameli R, Rogemond V, et al. Glial fibrillary acidic protein autoimmunity: a French cohort study. Neurology 2021; 98: e653–e668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patel NM, Bronder J, Motta M, et al. Mystery case: a 23-year-old man with headaches, confusion, and lower extremity weakness. Neurology 2019; 92: 863–866. [DOI] [PubMed] [Google Scholar]
- 10.Yang Z, Wang KK. Glial fibrillary acidic protein: from intermediate filament assembly and gliosis to neurobiomarker. Trends Neurosci 2015; 38: 364–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jessen KR, Morgan L, Stewart HJ, et al. Three markers of adult non-myelin-forming Schwann cells, 217c(Ran-1), A5E3 and GFAP: development and regulation by neuron-Schwann cell interactions. Development 1990; 109: 91–103. [DOI] [PubMed] [Google Scholar]
- 12.Yuan Z, Li H, Huang L, et al. CD8(+) T-cell predominance in autoimmune glial fibrillary acidic protein astrocytopathy. Eur J Neurol 2021; 28: 2121–2125. [DOI] [PubMed] [Google Scholar]
- 13.Smith KM, Amin S, Jones LK, et al. Glial fibrillary acidic protein (GFAP) autoimmunity in the setting of seropositive rheumatoid arthritis treated with Etanercept. Neurologist 2019; 24: 152–154. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-pdf-1-tan-10.1177_17562864231164806 for Clinical and electrophysiological characteristics of peripheral neuropathy in autoimmune glial fibrillary acidic protein astrocytopathy: an observational study and literature review by Bo Deng, Jingguo Wang, Yue Qiu, Xiaoni Liu, Lei Jin, Dongqing Zhu and Xiangjun Chen in Therapeutic Advances in Neurological Disorders