

Abstract
Tuning solubility and mechanical activation alters the stereoselectivity of the [2+2] photochemical cycloaddition of acenaphthylene. Photomechanochemical conditions produce the syn cyclobutane, whereas the solid-state reaction in the absence of mechanical activation provides the anti. When the photochemical dimerization occurs in a solubilizing organic solvent, there is no selectivity. Dimerization in H2O, where acenaphthylene is insoluble, provides the anti product. DFT calculations reveal that insoluble and solid-state reactions proceed via a covalently bonded excimer, which drives anti selectivity. Alternatively, the noncovalently bound syn conformer is more mechanosusceptible than the anti, meaning it experiences greater destabilization, thereby producing the syn product under photomechanochemical conditions. Cyclobutanes are important components of biologically active natural products and organic materials, and we demonstrate stereoselective methods for obtaining syn or anti cyclobutanes under mild conditions and without organic solvents. With this work, we validate photomechanochemistry as a viable new direction for the preparation of complex organic scaffolds.
Keywords: Mechanochemistry, Photochemistry, Cycloaddition, Cyclobutanes, Density Functional Theory
Graphical Abstract
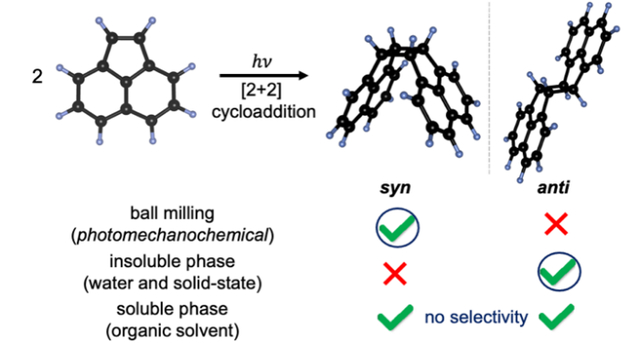
Unique and mutually orthogonal photomechanochemical and photosolvochemical stereoselective routes for cycloaddition of acenaphthylene with high yields have been reported. These methods provide scalable and environmentally benign approaches for stereoselective photocycloaddition reactions.
Introduction
Mechanochemistry involves1–3 the use of force/strain/curvature to make or break chemical bonds, and such methods are attractive because they satisfy all twelve principles of green chemistry.4 In addition to the minimal environmental impact, the benefits of mechanochemical reactions can include faster reaction kinetics,5 improved stereoselectivities and yields, or access to stereo- and regioisomers, or other chemical products,6 that are not accessible by solvothermal methods. Recognizing the advantages of mechanochemistry and the growing suite of tools for performing and studying reactions under force, the diversity of reactions that have been performed mechanochemically is rapidly expanding, and includes the formation of metal-organic frameworks,7 synthetic polymers,8 and small molecules.1, 9 Amongst the reactions that have been most thoroughly studied under mechanochemical activation is the thermally allowed [4+2] Diels-Alder3, 10, 11 reaction, and it is now known that this reaction can be driven forward with force because of its negative volume of activation.12–14 The Diels-Alder reaction is an example of a broader class of reactions, the pericyclic cycloadditions,15 that also have negative activation volumes, and many of these have been carried out mechanochemically as well.10, 16, 17 A class of pericyclic reactions which should also possess negative activation volumes, and thus should be accelerated by the application of force, are symmetry-allowed [4n] photochemical cycloadditions, which are widely exploited in organic synthesis to prepare complex molecular scaffolds.14, 18, 19 The potential benefits of performing photochemical cycloadditions mechanochemically include improved yields and stereochemical control, the latter of which is particularly important as a mixture of stereoisomers are often obtained.20 Despite these potential advantages, [4n] cycloadditions have not yet been investigated under photomechanochemical conditions – where both illumination and force are applied to the reactants simultaneously – because of challenges associated with developing instrumentation that is capable of illuminating the reactants during compression. To this end, here we implement a new reactor to drive a [2+2] cycloaddition reaction under compression, and, in doing-so, show that such conditions are highly stereoselective as a result of a unique photomechanochemical reaction pathway.
The symmetry-allowed [2+2] photochemical cycloadditions of alkenes to form cyclobutanes,20 are frequently used to form primary and secondary metabolites21, 22 with attractive antimicrobial,23, 24 antibacterial,24 and analgesic properties,24 and are increasingly explored as a responsive component of functional materials.25–27 As a consequence, developing methods for their construction remains a focus of the synthetic community.23 One challenge that still remains, however, is controlling the stereoselectivity of these photoreactions because an undesired mixture of cyclobutane stereoisomers is often produced.20, 28–30 For example, the dimerization of acenaphthylene is considered a model reaction for understanding how experimental conditions affect the yields and stereochemical outcomes of [2+2] photocycloadditions.31–36 In organic solvents, a mixture of syn and anti isomers of the acenaphthylene dimer is obtained,37 and, generally, the former is the major product.34 Strategies to alter the reaction outcome so that the primary product is anti include the use of heavy atom solvents,34 photosensitizers,34 or a combination of a photosensitizer and a molecular cage,38 but such conditions often require undesirable solvents, use expensive catalysts, or continue to produce a mixture of isomers. As such, further efforts are needed to understand the drivers of stereoselectivity, which could result in design rules that could be deployed to develop simple, scalable, and widely accessible methods for the stereochemical control of [2+2] photochemical cycloadditions that minimize the use of organic solvents and complex and expensive reagents.
Results and discussion
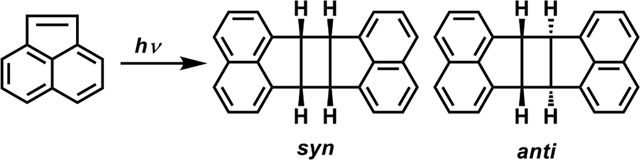
Here we study the [2+2] photodimerization of acenaphthylene under four different conditions, (i) illumination of the solid-state acenaphthylene under ball milling, (ii) illumination of the solid-state crystal in the absence of ball milling, (iii) illumination in organic solvents (soluble phase), and (iv) illumination in H2O (insoluble phase). For (i), we find that primarily the syn dimer is selectively formed, whereas (ii) and (iv) selectively form the anti dimer. Interestingly, we do not observe any noticeable selectivity for (iii). First-principles Density Functional Theory (DFT) calculations reveal that the extent of solubility changes the cycloaddition pathway: for insoluble and solid-state conditions (b and d), the cycloaddition proceeds via the formation of a covalently bonded excimer, which is geometrically and energetically closer to the anti transition state, thereby explaining the anti selectivity observed in (ii) and (iv). Additionally, we reveal that the noncovalently bound syn dimer is more mechanosusceptible, as it experiences more destabilization upon mechanical compression, which underpins the formation of syn product under ball milling (a). Therefore, in addition to providing mild conditions for the selective production of both syn and anti cyclobutanes that avoid the use of organic solvents, this work lays the foundation for understanding photomechanochemical stereoselectivity, which in turn, could lead to environmentally benign methods for the production of important chemical reagents.
Solid-state [2+2] photocycloaddition of acenaphthylene
We first investigated whether the [2+2] dimerization of acenaphthylene can be driven photomechanochemically and how these conditions affect reaction yield and stereoselectivity. To do so, a ball-mill reactor (SPEXSamplePrep, 8000M) was modified (Figure S1) with a blue LED (HepatoChem, DX Series light 30 W, λmax = 450 nm), and the reaction was run in a glass vial (20 mL) with two methacrylate balls (SPEXSamplePrep, 8006A, 9.5 mm diameter, 350 mg) so that light could reach the reagents during milling. In addition, we found it necessary to fluorinate the vial39 and add silica gel (SILICYCLE, SilicaFlash® GE60, 70–230 mesh) to the reaction to prevent the reagents from adhering to the side walls. Acenaphthylene (1.2 g) and silica gel (1.2 g) were added to the vial under inert Ar atmosphere, unless otherwise noted, and milled for 20 h at a frequency of 17.7 Hz. Upon completion of the milling, yields and anti:syn ratios were determined by 1H NMR spectroscopy by dissolving a portion of the crude mixture in CDCl3 followed by filtration to remove silica from the CDCl3 solution. We found (Table 1, entries 1 – 2) that, photomechanochemically, the dimerization reaction proceeded with yields up to 96 % with 6:94 anti:syn selectivity. Stereochemical assignments were confirmed by 1H NMR spectroscopy and microcrystal electron diffraction40, 41 on the crystallites isolated from the reactions (Figure S26 and Table S2). This syn selectivity and yield were retained in ambient atmosphere. In the absence of the illumination, no product was observed. To put this result in the context, in organic solvents, this reaction favors the syn product but with poor selectivity, and high syn selectivity has only been obtained using expensive and complex reagents or undesirable organic solvents.42–45 For example, Kaanumalle et al.46 reported 99 % syn product (99 % yield) in borate buffer using an organometallic molecular cage, while Cowan et al.34 obtained 98 % syn (39 % yield) selectivity in O2-saturated benzene. Thus, we find that photomechanochemical conditions provide the mildest route towards the syn product of the acenapthylene dimer yet reported.
Table 1.
Solid-state [2+2] photocycloaddition of acenaphthylene.
| |||
|---|---|---|---|
|
| |||
| Entry | Atm. | Total Product Yield (%) |
Anti : Syn |
|
| |||
| 1 a | Ar | 95 | 6:94 |
| 2 a | air | 87 | 8:92 |
| 3 b | Ar | 65 | 70:30 |
| 4 b | air | 99 | 46:54 |
All reactions were performed at 20 °C. Acenaphthylene samples were irradiated with a blue LED (HepatoChem, DX Series light 30 W, λmax = 450 nm) for 20 h. Yields and selectivities were obtained from 1H NMR in CDCl3.
Ball-mill reactions performed in the presence of silica as an additive to prevent the reagents from adhering to the side walls of the reaction vessel.
Reactions were performed in a petri dish with ground acenaphthylene crystals and no milling during illumination.
All reactions were performed at 20 °C. Acenaphthylene samples were irradiated with a blue LED (HepatoChem, DX Series light 30 W, λmax = 450 nm) for 20 h. Yields and selectivities were obtained from 1H NMR in CDCl3. [a] Ball-mill reactions performed in the presence of silica as an additive to prevent the reagents from adhering to the side walls of the reaction vessel. [b] Reactions were performed in a petri dish with ground acenaphthylene crystals and no milling during illumination.
From these experiments it was unclear whether the syn selectivity was the result of carrying out the reaction in the solid state or whether force played an active role in dictating stereoselectivity. To determine whether force has a role in stereoselectivity, the reaction was carried out on acenaphthylene crystals by illuminating them in the absence of force. The acenaphthylene crystals (100 mg) were ground with a mortar and pestle, sealed in a petri dish under either inert or ambient atmosphere, and the crystals were then irradiated with the same blue LED for 20 h (Table 1, entries 3 – 4). Under Ar, we found that the reaction favored the anti isomer (70:30 anti:syn), while ambient atmosphere provided a mixture with a 46:54 anti:syn ratio. So, in the absence of O2, more anti product forms, but as O2 is introduced, such selectivity is diminished. This was also observed by Haga et al.,36 who noted that the presence of a triplet quencher (here O2) reduces the anti-selectivity. These results show that the absence of force provides substantially different products than when the products were milled during illumination. Therefore, we find that running the [2+2] photodimerization of acenaphthylene under Ar while milling results in the opposite stereoselectivity than when the reaction is carried out without milling. These observations confirm that a unique photomechanochemical reaction pathway exists for the dimerization of acenaphthylene that is distinct from the pathway followed during irradiation in the absence of force.
[2+2] photocycloaddition of acenaphthylene in solution-state
We then studied the photodimerization of acenaphthylene in different organic solvents with variable H2O:organic solvent ratios at the room temperature. Here we have examined the photodimerization with 5 different organic solvents that are miscible with H2O – DMSO, EtOH, MeOH, DMF, and MeCN – along with H2O itself, under blue light illumination. As acenaphthylene is insoluble in H2O, these experiments provide insight into the role of solubility on the stereoselectivity. To maximize solvent/acenaphthylene interaction (especially for the solvent mixture where acenaphthylene is poorly soluble), 5 M solution of acenaphthylene was prepared in DMSO, and 4 μL of this stock solution was then added to 1 mL of the desired solvent system to prepare 20 mM solutions of acenaphthylene. The solution was then sonicated for 1 min. The summary of the stereoselectivity observed in these experiments is given in Figure 1C and Table S1. Although, we find that the absolute value of the anti:syn ratio varies depending on the nature of the organic solvent, we observe the conspicuous trend that the anti selectivity increases with the amount of H2O, where using just H2O selectively provides the anti product. Because of the insolubility of the acenaphthylene in H2O, the solution was sonicated to disperse the reactant, which led to a cloudy, opaque solution. Following irradiation, the anti isomer was recovered as the major product (84:16 anti:syn) in 84 % yield. In other words, the insolubility of acenaphthylene favors the anti product. This observation is in good agreement with the solid-phase reactions on the crystal in the absence of milling, which also resulted in anti stereoselectivity (anti:syn 70:30). Thus, we conclude that solubility also plays an important role in determining the stereoselectivity. For photocycloadditions, it is known that lowering the reaction temperature can improve regio- and stereoselectivity.47 As such, the reaction temperature was then lowered to 10 °C, and running the reaction for 16 h in H2O resulted in quantitative conversion, with anti:syn reaching 91:9. The anti selectivity observed here is similar to those reported by Guo et al.38 who used a molecular cage composed of a Ru2+ photocatalyst to achieve 89:11 anti:syn. So a major challenge in stereocontrolled photochemical cycloadditions – obtaining the anti isomer of acenaphthylene as the major product – was resolved by simply running the reaction in H2O, which, in addition to providing an otherwise elusive product, is both atom-efficient and environmentally benign.
Figure 1.
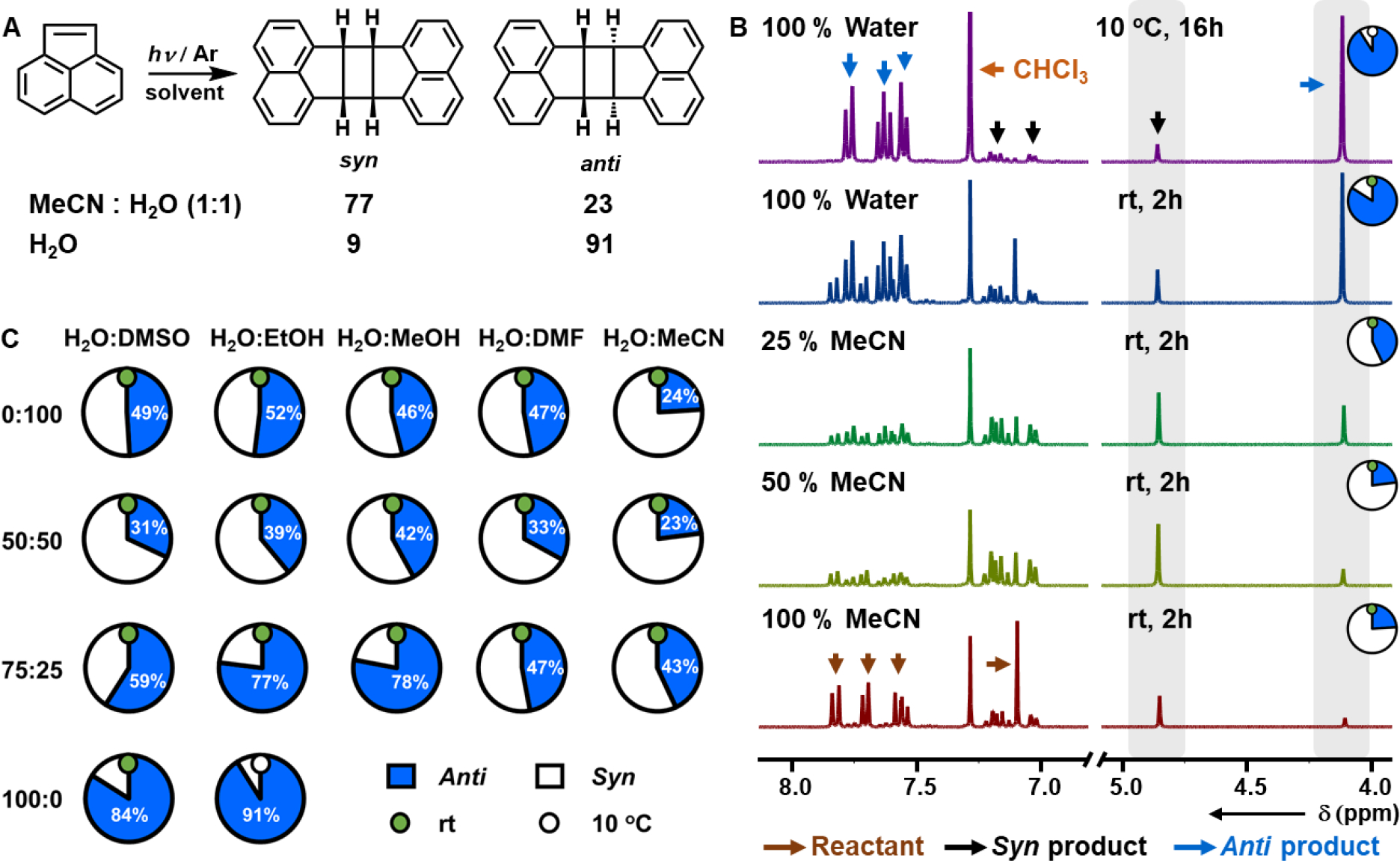
(A) [2+2] Photocycloaddition of acenaphthylene, with the most stereoselective conditions for syn and anti dimers noted. (B) 1H NMR (300 MHz, CDCl3) of the products of the [2+2] photocycloaddition of acenaphthylene in a binary solvent mixture composed of different ratios of MeCN and H2O. (C) Change in selectivity with changing solvent composition for the [2+2] photocycloaddition of acenaphthylene in binary solvent mixtures.
DFT calculations for the [2+2] photocycloaddition of acenaphthylene in solution-state
We performed DFT calculations48 to understand why liquid and solid phase reactions result in different stereoisomers. In doing so, we explored the potential energy surfaces by considering the photodimerization pathways from three different initial states– syn and anti conformers, which are noncovalently bound supramolecular dimers, and a state that consists of two separated monomers (Figure 2). On the other hand, the noncovalently bound syn and anti conformers correspond to the insoluble solid-state acenaphthylene (and state present in H2O), when the monomers are close to each other and experience a net stabilizing interaction as compared to separated monomers. We find that the thermal activation barrier for the anti product formation (179 kJ/mol) is lower than the syn product formation (269 kJ/mol) (Figure 2). Such high barriers suggest that the reactions cannot be performed at room temperature without photoexcitation, which we also observe experimentally. In the soluble-phase scenario, we find that the excited triplet (T) and singlet (S) states of the separated monomers (yellow dotted lines in Figure 2) are above the syn and anti transition states (TS). Thus, when completely solvated, photoexcitation injects sufficient energy to access both the anti and syn TS; this in turn, leads to both syn and anti product formations, explaining the loss of selectivity for this reaction in organic solvents. In the solid state or in H2O, where the poor solubility keeps the acenaphthylene aggregated, the molecules exist as noncovalently bound syn and anti supramolecular conformers. We find that the spin-flipped T states for both the syn and anti supramolecular conformer lead to the formation (shaded cyan region in Figure 2) of a covalently bonded excimer (excimer in Figure 2). Formation of excimers in photochemical reactions have been reported previously49–51 to have a role in determining the selectivity of the products. Therefore, as the photoexcitation occurs in the insoluble phase, the supramolecular dimers form a covalent bond in the excimer, gaining a stabilization of nearly 117 kcal‧mol−1 per dimer compared to the vertically excited T state. We find that the geometry of this bonded excimer resembles more closely to the anti transition state than the syn. Additionally, as the anti TS has lower energy than the syn TS, energetically it is also easier to access the anti TS from the bonded excimer. Thus, the formation of the bonded excimer preferentially leads to the anti product. This explains the selective formation of the anti product in the insoluble phase, i.e. in H2O and in crystals in the absence of ball milling.
Figure 2.
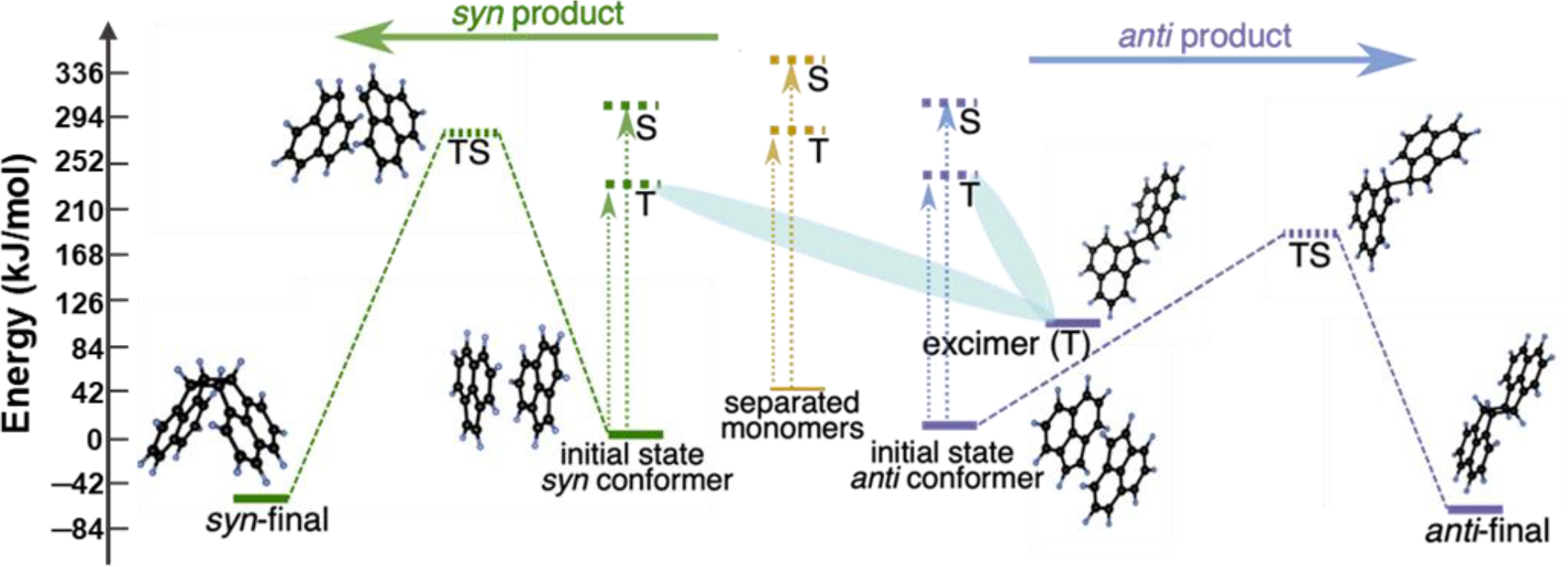
Potential energy surface obtained using the geometries at the B3LYP-D3/6-31G(d) level of theory showing the pathways for syn and anti product formation. The abbreviations are as follows: T: Triplet; S: Singlet; TS: Transition state. Dotted arrows represent vertical photoexcitation, and cyan shading illustrates relaxation of the triplet conformer states to the excimer.
DFT calculations for the [2+2] photocycloaddition of acenaphthylene in solid-state
To understand the origin of syn selectivity that occurs under the photomechanochemical conditions, we have applied force (F) along the C–C bond forming vector of the syn and anti conformations using the external force explicitly induced (EFEI) method.52, 53 In this simulation, the intertwined effect of F and photoexcitation is not incorporated, rather only the effect of compressive F on stability is studied. We envisage that ball milling exerts compression by bringing the monomers in the syn and the anti supramolecular conformers closer to each other. Thus, applying forces along the C–C bonds in the simulation (as shown in Figure 3A) can have an effect similar to what occurs in a ball mill. We find that the syn conformer undergoes bond formation at lower F (between 8 nN and 9 nN), compared to the anti dimer (between 12 nN and 13 nN), which is shown by the grey shaded regions in Figure 3B. To understand why the syn supramolecular conformer is more susceptible to F, we examined the F range of 5–8 nN with more datapoints. The reaction coordinate (RC) in Figure 3C is defined based on the C–C distance, wherein the bond distances at 5 nN and 8.55 nN correspond to RC of 0 and 1, respectively. We find that the slope of RC vs. F (black dashed lines in Figure 3C) is higher for the syn supramolecular conformer. This shows that a more significant change in RC is attained for syn than anti at the same applied F. In other words, the results directly confirm that syn supramolecular conformer is more mechanosusceptible, meaning that its RC has a stronger dependence on F. In addition, the syn supramolecular conformer experiences more destabilization for the same applied F than the anti (filled markers in Figure 3C). To analyze these effects, we implement the energy decomposition analysis (EDA)54 to understand the origin of the mechanosusceptibility by quantifying the interaction energy between the acenaphthylene monomers (Figure 3D). EDA consists of three terms: frozen density (EFZ), charge transfer (ECT), and polarization (EPol).54 We find that the EFZ dominates in the higher F regime, and it increases drastically near 7–8.55 nN for the syn, but anti does not exhibit such behavior (Figure 3D). As EFZ captures the interaction between the unrelaxed electron density between the monomers, we conclude that the face-to-face orientation of the π-clouds of the syn supramolecular conformer, and the resulting repulsion at smaller distances, make it more mechanosusceptible, whereas for the anti supramolecular dimer, π-clouds interact to a lesser extent. Additionally, it is worth noting that the TS for the syn product formation corresponds solely to compression along the C–C bonds participating in the [2+2] cycloaddition (syn TS in Figure 2). On the other hand, for the anti product formation, shear force also plays an important role along with compression as the monomers need to slide to form the anti product (anti TS in Figure 2). We conjecture that as attaining shear and compression simultaneously is statistically less probable under ball milling than just compression, syn selectivity is boosted. Thus, these results explain qualitatively the stereoselectivity of solid-state photomechanochemical conditions.
Figure 3.
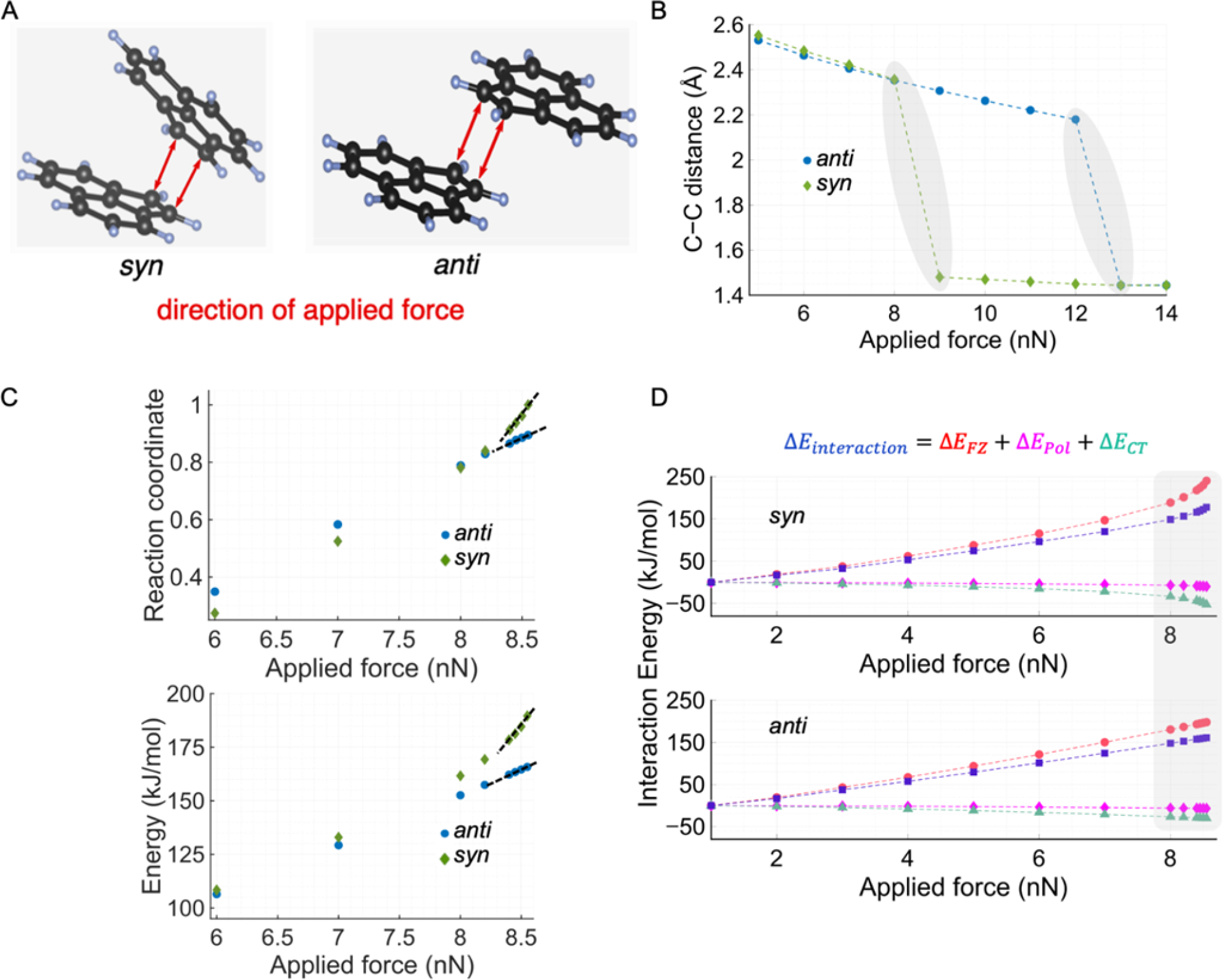
Effect of force on the dimerization of syn and anti supramolecular conformers. (A) The directions of the applied forces in simulations using the external force explicitly included (EFEI) method are shown. (B) The effect of applied force on C–C bond formation for syn and anti conformers. The shaded regions indicate where the syn and/or anti dimers undergo covalent C–C bond formation from a noncovalent conformer. (C) The change in reaction coordinate (RC) and energy (E) as a function of the applied force. The RC is defined based on the C–C distance, wherein distances at 5 nN and 8.55 nN correspond to RC 0 and 1, respectively. The filled circles/diamonds correspond to the E values, whereas unfilled markers indicate the change in the RC. (D) Energy decomposition analysis showing the effect of force on different inter-action terms between the monomers. The abbreviations are: FZ: frozen density, CT: charge transfer, Pol: polarization.
Conclusion
In conclusion, we have studied the [2+2] dimerization of acenaphthylene and explored how light, force, and solvent can be combined to achieve both syn and anti stereoselective photodimerization. We find that syn stereoselectivity is observed under photomechanochemical conditions because of the greater mechanosusceptibility of the noncovalently bound syn supramolecular conformer compared to the anti conformer. In studying how solvents and solubility affect yields and stereoselectivity, we have shown that simply running the photodimerization in H2O provides the anti dimer in high yields, and these conditions provide among the highest anti stereoselectivity yet observed because of the formation of a bonded excimer in insoluble phases. Thus, we report scalable and mild conditions for obtaining both syn and anti [2+2] cycloaddition products without necessitating organic solvent or complex additional reagents. More importantly, we demonstrate that photomechanochemical pathways exist that are distinct from solvothermal or solid-state reaction pathways, and we explain stereochemistry based upon mechanosusceptibility, a concept that may be used to predict stereoselectivity for other mechanochemical reactions as well. In doing so, have shown that photomechanochemistry and photosolvochemistry can provide mutually orthogonal stereoselective routes for making complex organic scaffolds. Therefore, photomechanochemistry merits further investigation as a scalable and environmentally benign approach to stereoselective photochemistry.
Experimental Section
Experimental procedures, synthesis, and characterization of all new compounds, NMRs, Microcrystalline electron diffraction are provided in the supporting information file.
Supplementary Material
Acknowledgement
A.B.B. and A.M. R. acknowledge support from the National Science Foundation Center for Mechanical Control of Chemistry (CHE-2023644). The NMR data (Bruker Avance 300 MHz) presented herein were collected in part at the CUNY ASRC Biomolecular NMR facility and the NMR facility of the City College of the CUNY. Mass spectra were collected at the CUNY ASRC Biomolecular Mass Spectrometry Facility. S.B.2. acknowledges a graduate fellowship from the Vagelos Institute of Energy Science and Technology. B.L.N. acknowledges support from the National Institutes of Health grant number R21GM135784, and the use of the Titan Krios within the Eyring Materials Center at Arizona State University (NSF DBI 1531991).
Footnotes
Conflicts of interest
The authors declare no competing financial interests.
References
- 1.O’Neill RT and Boulatov R, Nat. Rev. Chem, 2021, 5, 148–167. [DOI] [PubMed] [Google Scholar]
- 2.Do J-L and Friščić T, ACS Cent. Sci, 2017, 3, 13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hernández JG and Bolm C, J. Org. Chem, 2017, 82, 4007–4019. [DOI] [PubMed] [Google Scholar]
- 4.Mateti S, Mathesh M, Liu Z, Tao T, Ramireddy T, Glushenkov AM, Yang W and Chen YI, Chem. Commun, 2021, 57, 1080–1092. [DOI] [PubMed] [Google Scholar]
- 5.Qi Y, Yang J and Rappe AM, ACS Appl. Mater. Interfaces, 2016, 8, 7529–7535. [DOI] [PubMed] [Google Scholar]
- 6.Yang F, Yang J, Qi Y, de Boer MP, Carpick RW, Rappe AM and Srolovitz DJ, ACS Appl. Mater. Interfaces, 2019, 11, 39238–39247. [DOI] [PubMed] [Google Scholar]
- 7.Chen D, Zhao J, Zhang P and Dai S, Polyhedron, 2019, 162, 59–64. [Google Scholar]
- 8.Chen Y, Mellot G, van Luijk D, Creton C and Sijbesma RP, Chem. Soc. Rev, 2021, 50, 4100–4140. [DOI] [PubMed] [Google Scholar]
- 9.Achar TK, Bose A and Mal P, Beilstein J. Org. Chem, 2017, 13, 1907–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han X, Bian S, Liang Y, Houk KN and Braunschweig AB, J. Am. Chem. Soc, 2014, 136, 10553–10556. [DOI] [PubMed] [Google Scholar]
- 11.Bian S, Scott AM, Cao Y, Liang Y, Osuna S, Houk KN and Braunschweig AB, Journal of the American Chemical Society, 2013, 135, 9240–9243. [DOI] [PubMed] [Google Scholar]
- 12.Breslow R, Acc. Chem. Res, 1991, 24, 159–164. [Google Scholar]
- 13.Meijer A, Otto S and Engberts JBFN, J. Org. Chem, 1998, 63, 8989–8994. [Google Scholar]
- 14.Chen B, Hoffmann R and Cammi R, Angewandte Chemie International Edition, 2017, 56, 11126–11142. [DOI] [PubMed] [Google Scholar]
- 15.Herndon WC, Chem. Rev, 1972, 72, 157–179. [Google Scholar]
- 16.Mehlich J and Ravoo BJ, Org. Biomol. Chem, 2011, 9, 4108–4115. [DOI] [PubMed] [Google Scholar]
- 17.Rozkiewicz DI, Jańczewski D, Verboom W, Ravoo BJ and Reinhoudt DN, Angew. Chem. Int. Ed, 2006, 45, 5292–5296. [DOI] [PubMed] [Google Scholar]
- 18.CRC Handbook of Organic Photochemistry and Photobiology, CRC Press, Boca Raton, 2012. [Google Scholar]
- 19.Klärner F-G and Diedrich MK, in The Chemistry of Dienes and Polyenes, 1997, pp. 547–617. [Google Scholar]
- 20.Poplata S, Tröster A, Zou Y-Q and Bach T, Chem. Rev, 2016, 116, 9748–9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sergeiko A, Poroikov VV, Hanus LO and Dembitsky VM, Open Med Chem J, 2008, 2, 26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dembitsky VM, Journal of Natural Medicines, 2008, 62, 1–33. [DOI] [PubMed] [Google Scholar]
- 23.Li J, Gao K, Bian M and Ding H, Organic Chemistry Frontiers, 2020, 7, 136–154. [Google Scholar]
- 24.Sergeiko A, Poroikov VV, Hanus LO and Dembitsky VM, Open Med Chem J, 2008, 2, 26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang J, Horst M, Romaniuk JAH, Jin Z, Cegelski L and Xia Y, J. Am. Chem. Soc, 2019, 141, 6479–6483. [DOI] [PubMed] [Google Scholar]
- 26.Chen Z, Mercer JAM, Zhu X, Romaniuk JAH, Pfattner R, Cegelski L, Martinez TJ, Burns NZ and Xia Y, Science, 2017, 357, 475–479. [DOI] [PubMed] [Google Scholar]
- 27.Chen Z, Zhu X, Yang J, Mercer JAM, Burns NZ, Martinez TJ and Xia Y, Nat. Chem, 2020, 12, 302–309. [DOI] [PubMed] [Google Scholar]
- 28.Ramamurthy V and Mondal B, J. Photochem. Photobiol. C: Photochem. Rev, 2015, 23, 68–102. [Google Scholar]
- 29.Yoon TP, Acc. Chem. Res, 2016, 49, 2307–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang J-S, Wu K, Yin C, Li K, Huang Y, Ruan J, Feng X, Hu P and Su C-Y, Nat. Commun, 2020, 11, 4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Samanta A, Devadoss C and Fessenden RW, J Phys Chem, 1990, 94, 7106–7110. [Google Scholar]
- 32.Bowen EJ and Marsh JDF, J. Am. Chem. Soc, 1947, DOI: 10.1039/JR9470000109, 109–110. [DOI] [Google Scholar]
- 33.Livingston R and Wei KS, J Phys Chem, 1967, 71, 541–547. [Google Scholar]
- 34.Cowan DO and Drisko RLE, J. Am. Chem. Soc, 1970, 92, 6286–6291. [Google Scholar]
- 35.White EH, Wildes PD, Wiecko J, Doshan H and Wei CC, J. Am. Chem. Soc, 1973, 95, 7050–7058. [Google Scholar]
- 36.Haga N, Takayanagi H and Tokumaru K, J. Org. Chem, 1997, 62, 3734–3743. [Google Scholar]
- 37.Dziewoński K and Rapalski G, Ber. Dtsch. Chem. Ges, 1912, 45, 2491–2495. [Google Scholar]
- 38.Guo J, Fan Y-Z, Lu Y-L, Zheng S-P and Su C-Y, Angew. Chem. Int. Ed, 2020, 59, 8661–8669. [DOI] [PubMed] [Google Scholar]
- 39.Eichelsdoerfer DJ, Liao X, Cabezas MD, Morris W, Radha B, Brown KA, Giam LR, Braunschweig AB and Mirkin CA, Nat. Protoc, 2013, 8, 2548–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levine AM, Bu G, Biswas S, Tsai EHR, Braunschweig AB and Nannenga BL, Chem. Commun, 2020, 56, 4204–4207. [DOI] [PubMed] [Google Scholar]
- 41.Nannenga BL and Gonen T, Nat Methods, 2019, 16, 369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karthikeyan S and Ramamurthy V, Tetrahedron Lett, 2005, 46, 4495–4498. [Google Scholar]
- 43.Yoshizawa M, Takeyama Y, Okano T and Fujita M, J. Am. Chem. Soc, 2003, 125, 3243–3247. [DOI] [PubMed] [Google Scholar]
- 44.Takaoka K, Kawano M, Ozeki T and Fujita M, Chem. Commun, 2006, DOI: 10.1039/B600812G, 1625–1627. [DOI] [PubMed] [Google Scholar]
- 45.Yoshizawa M, Takeyama Y, Kusukawa T and Fujita M, Angew. Chem. Int. Ed, 2002, 41, 1347–1349. [DOI] [PubMed] [Google Scholar]
- 46.Kaanumalle LS and Ramamurthy V, Chem. Commun, 2007, DOI: 10.1039/B615937K, 1062–1064. [DOI] [PubMed] [Google Scholar]
- 47.Yabuno Y, Hiraga Y, Takagi R and Abe M, J. Am. Chem. Soc, 2011, 133, 2592–2604. [DOI] [PubMed] [Google Scholar]
- 48.Shao Y, Molnar LF, Jung Y, Kussmann J, Ochsenfeld C, Brown ST, Gilbert ATB, Slipchenko LV, Levchenko SV, O’Neill DP, DiStasio RA Jr, Lochan RC, Wang T, Beran GJO, Besley NA, Herbert JM, Yeh Lin C, Van Voorhis T, Hung Chien S, Sodt A, Steele RP, Rassolov VA, Maslen PE, Korambath PP, Adamson RD, Austin B, Baker J, Byrd EFC, Dachsel H, Doerksen RJ, Dreuw A, Dunietz BD, Dutoi AD, Furlani TR, Gwaltney SR, Heyden A, Hirata S, Hsu C-P, Kedziora G, Khalliulin RZ, Klunzinger P, Lee AM, Lee MS, Liang W, Lotan I, Nair N, Peters B, Proynov EI, Pieniazek PA, Min Rhee Y, Ritchie J, Rosta E, David Sherrill C, Simmonett AC, Subotnik JE, Lee Woodcock Iii H, Zhang W, Bell AT, Chakraborty AK, Chipman DM, Keil FJ, Warshel A, Hehre WJ, Schaefer Iii HF, Kong J, Krylov AI, Gill PMW and Head-Gordon M, Phys. Chem. Chem. Phys, 2006, 8, 3172–3191. [DOI] [PubMed] [Google Scholar]
- 49.Wang Y, Haze O, Dinnocenzo JP, Farid S, Farid RS and Gould IR, J. Org. Chem, 2007, 72, 6970–6981. [DOI] [PubMed] [Google Scholar]
- 50.Mattay J, Angew. Chem. Int. Ed, 2007, 46, 663–665. [DOI] [PubMed] [Google Scholar]
- 51.Wilzbach KE and Kaplan L, J. Am. Chem. Soc, 1971, 93, 2073–2074. [Google Scholar]
- 52.Ribas-Arino J and Marx D, Chem. Rev, 2012, 112, 5412–5487. [DOI] [PubMed] [Google Scholar]
- 53.Ribas-Arino J, Shiga M and Marx D, Angew. Chem. Int. Ed, 2009, 48, 4190–4193. [DOI] [PubMed] [Google Scholar]
- 54.Khaliullin RZ, Cobar EA, Lochan RC, Bell AT and Head-Gordon M, J. Phys. Chem. A, 2007, 111, 8753–8765. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.