

Abstract
Recent studies have reported early cerebellar and subcortical impact in the disease progression of genetic frontotemporal dementia (FTD) due to microtubule‐associated protein tau (MAPT), progranulin (GRN) and chromosome 9 open reading frame 72 (C9orf72). However, the cerebello‐subcortical circuitry in FTD has been understudied despite its essential role in cognition and behaviors related to FTD symptomatology. The present study aims to investigate the association between cerebellar and subcortical atrophy, and neuropsychiatric symptoms across genetic mutations. Our study included 983 participants from the Genetic Frontotemporal dementia Initiative including mutation carriers and noncarrier first‐degree relatives of known symptomatic carriers. Voxel‐wise analysis of the thalamus, striatum, globus pallidus, amygdala, and the cerebellum was performed, and partial least squares analyses (PLS) were used to link morphometry and behavior. In presymptomatic C9orf72 expansion carriers, thalamic atrophy was found compared to noncarriers, suggesting the importance of this structure in FTD prodromes. PLS analyses demonstrated that the cerebello‐subcortical circuitry is related to neuropsychiatric symptoms, with significant overlap in brain/behavior patterns, but also specificity for each genetic mutation group. The largest differences were in the cerebellar atrophy (larger extent in C9orf72 expansion group) and more prominent amygdalar volume reduction in the MAPT group. Brain scores in the C9orf72 expansion carriers and MAPT carriers demonstrated covariation patterns concordant with atrophy patterns detectable up to 20 years before expected symptom onset. Overall, these results demonstrated the important role of the subcortical structures in genetic FTD symptom expression, particularly the cerebellum in C9orf72 and the amygdala in MAPT carriers.
Keywords: frontotemporal dementia, genetics, magnetic resonance imaging, neuropsychiatry
The present study aims to investigate the association between cerebellar and subcortical atrophy, and neuropsychiatric symptoms across genetic mutations. Voxel‐wise analysis of the thalamus, striatum, globus pallidus, amygdala, and the cerebellum was performed, and partial least squares analyses were used to link morphometry and behavior. Our results demonstrated the important role of the subcortical structures in genetic FTD symptom expression, particularly the cerebellum in C9orf72 and the amygdala in MAPT carriers.

1. INTRODUCTION
Frontotemporal dementia (FTD) is the second most common form of neurodegenerative dementia in people under 65 years of age (Coyle‐Gilchrist et al., 2016). Behavioral and personality alterations encompassing “negative” symptoms (apathy, loss of empathy) and “positive” symptoms (disinhibition, inappropriate behavior) are symptoms of behavioral variant FTD (Mendez et al., 2008; Santamaría‐García et al., 2017; Sturm et al., 2017) and also commonly encountered in primary progressive aphasia. While most FTD cases are sporadic, 10%–30% of cases are caused by three well known autosomal dominant full penetrance mutations: microtubule‐associated protein tau (MAPT) and progranulin (GRN) and the hexanucleotide expansion of chromosome 9 open reading frame 72 (C9orf72) (Greaves & Rohrer, 2019a). Studying presymptomatic mutation carriers can provide valuable insight into the neuroanatomical changes that occur during the preclinical phase of FTD. Mounting evidence has demonstrated that FTD‐related pathophysiology starts several years before the clear onset of the disease (Bertrand et al., 2018; Rohrer et al., 2015). Furthermore, psychosis‐related symptoms can constitute the prodrome of genetic FTD (Ducharme et al., 2017), as evidenced by findings demonstrating that presymptomatic carriers have subtle increases in neuropsychiatric symptoms (Tavares et al., 2020).
To date, FTD studies have related cognitive and behavioral impairment with cortical atrophy (Du et al., 2007; J. D. Rohrer et al., 2009; Hartikainen et al., 2012; Blanc, Gabriella, et al., 2020; Borrego‐Écija et al., 2021; Ratti et al., 2021), white matter abnormalities (Dadar et al., 2022; Zhang et al., 2009), subcortical structures and with the cerebellum (Bocchetta et al., 2021). Further, atrophy of the cerebellum and subcortical structures in genetic FTD appears earlier than the cortical atrophy in the disease progression and therefore may provide important biomarkers at the earliest stages of disease onset (Cash, 2018). Moreover, numerous studies in healthy controls indicate that these structures have a central role in cognitive and behavioral processes (attention, mood, language and memory) (Balleine et al., 2007; Klostermann et al., 2013; Packard & Knowlton, 2002; Sherman, 2016). Thus, this study aims to examine association between the atrophy of the cerebello‐subcortical structures and diverse FTD‐specific cognitive and behavioral metrics across genetic mutations and whether this association starts during the preclinical phase of the disease.
We included 983 participants from the Genetic Frontotemporal dementia Initiative (GENFI [Jonathan D. Rohrer et al., 2015]). Voxel‐wise analysis in a region of interest encompassing the thalamus, striatum, globus pallidus, amygdala, and the cerebellum was performed using deformation‐based morphometry (DBM). These regions were selected because of their implication in cerebello‐subcortical circuitry (Palesi et al., 2017) and for their potential implication in psychiatric symptoms (Blithikioti et al., 2022; Phillips et al., 2015). We first examined the relationship between voxel‐wise measures and FTD‐relevant demographic and clinical data, namely: genetic mutation group, age, estimated years before the age of symptom onset (EYO; calculated as the difference between the parental age of symptom onset and the individual's current age) and symptomatic status (symptomatic or presymptomatic). Next, a multivariate technique was used to derive linked dimensions of covariation between voxel‐wise morphometry with cognitive and behavioral symptoms for each mutation group. We finally investigated if these brain/behavioral patterns could be explained by different demographic and clinical information.
The issue of heterogeneity in clinical presentations of FTD mutation is complex and we believe that our approach can help us understand this heterogeneity by approaching brain‐behavior relationships in a data driven way. Instead of selecting only subjects with FTD symptoms or neuropsychiatric symptoms for example, we attempt to find those relationships across all disease stages and clinical manifestation to identify why some patients develop these symptoms while others do not. We believe that the selected structures of interest could play an important role in neuropsychiatric phenotype, explaining some of the different phenotypic expressions.
2. METHODS
2.1. Participants
The 983 participants were included from the GENFI2 dataset data release five which includes data from participants across multiple research sites. The participants were either known carriers of a pathogenic mutation in MAPT (n = 104), GRN (n = 243), C9orf72 (n = 256), TANK‐binding kinase 1 (TBK1; n = 5) or first‐degree relatives of known symptomatic carriers (“noncarriers” group; n = 375). TBK1 carriers were excluded because of the low number of carriers of this mutation. Inclusion and exclusion criteria are detailed in the Supplementary material and information on the participants selected for further analyses can be found in Table 1. The protocol was approved by each participating centers' research ethics board and informed written consent was obtained from each participant or an approved surrogate decision maker.
TABLE 1.
Complete demographic and clinical information of the 724 individuals who passed QC
| Noncarriers (n = 281) | C9orf72 presympt (n = 122) | GRN presympt (n = 135) | MAPT presympt (n = 56) | C9orf72 sympt (n = 62) | GRN sympt (n = 44) | MAPT sympt (n = 24) | |
|---|---|---|---|---|---|---|---|
| Age at visit | |||||||
| Mean (SD) | 46.1 (13.5) | 44.0 (11.9) | 46.1 (11.9) | 38.9 (11.4) | 63.7 (7.23) | 63.2 (7.07) | 58.5 (8.23) |
| Median [Min, Max] | 44.6 [19.4, 85.0] | 43.7 [20.1, 69.3] | 46.0 [20.2, 75.5] | 37.0 [20.5, 74.0] | 64.7 [46.5,77.9] | 63.4 [50.1,77.0] | 59.2 [31.9,69.2] |
| EYO | |||||||
| Mean (SD) | −13.1 (14.1) | −14.90 (12.2) | −14.1 (12.4) | −13.6 (12.4) | 4.72 (5.96) | 1.96 (6.20) | 5.25 (4.48) |
| Median [Min, Max] | −15.3 [−50.0,27.6] | −15.8 [−47.4, 18.3] | −15.6 [−39.0, 14.0] | −14.4 [−39.4, 15.7] | 5.33 [−15.3,15.6] | 1.59 [−11.9,19.1] | 5.46 [−5.44,14.3] |
| Gender | |||||||
| Female | 164 (58.4%) | 71 (58.2%) | 89 (65.9%) | 32 (57.1%) | 25 (40.3%) | 25 (56.8%) | 8 (33.3%) |
| Male | 117 (41.6%) | 51 (41.8%) | 46 (34.1%) | 24 (42.9%) | 37 (59.7%) | 19 (43.2%) | 16 (66.7%) |
| Education | |||||||
| Mean (SD) | 14.3 (3.62) | 14.3 (3.13) | 14.7 (3.57) | 14.3 (3.18) | 12.5 (3.77) | 13.6 (3.77) | 14.0 (3.63) |
| Median [Min, Max] | 14.0 [5.0, 24.0] | 15.0 [5.0,20.0] | 15.0 [8.0, 24.0] | 15.0 [5.0,20.0] | 12.0 [4.0,22.0] | 13.5 [5.0,20.0] | 14.0 [4.0,24,0] |
| Memory | |||||||
| Mean (SD) | 0.8 (1.9) | 1.06 (2.34) | 0.5 (1.27) | 0.8 (1.6) | 11.6 (8.0) | 9.8 (7.1) | 12.8 (9.2) |
| Median [Min, Max] | 0 [0,13.0] | 0 [0,14.0] | 0 [0,10.0] | 0[0,7.0] | 12.5 [0,28.0] | 8.0 [0,32.0] | 13.0 [0,30.0] |
| Everyday skills | |||||||
| Mean (SD) | 0.1 (0.6) | 0.1 (0.5) | 0.1 (0.6) | 0.1 (0.6) | 6.5 (6.2) | 5.6 (5.9) | 4.13 (5.17) |
| Median [Min, Max] | 0 [0,9.0] | 0 [0,4.0] | 0 [0,6.0] | 0 [0,4.0] | 5.5 [0,20.0] | 4.0 [0,20.0] | 2.0 [0,14.0] |
| Self‐care | |||||||
| Mean (SD) | 0.2 (0.8) | 0.4 (0.9) | 0.19 (0.7) | 0.3 (0.9) | 5.6 (4.7) | 4.3 (5.6) | 3.4 (5.3) |
| Median [Min, Max] | 0 [0,7.0] | 0 [0,4.0] | 0 [0,6.0] | 0 [0,5.0] | 5.0 [0,19.0] | 1.5 [0,18.0] | 1.0 [0,20.0] |
| Abnormal behaviour | |||||||
| Mean (SD) | 0.6 (1.2) | 1.2 (2.0) | 0.6 (1.5) | 1.5 (2.6) | 6.3 (5.5) | 4.1 (4.7) | 6.6 (5.3) |
| Median [Min, Max] | 0 [0,6.0] | 0 [0,9.0] | 0 [0,10.0] | 0 [0,13.0] | 5.5 [0,18.0] | 3.0 [0,23.0] | 6.0 [0,18.0] |
| Sleep | |||||||
| Mean (SD) | 0.4 (1.0) | 0.5 (1.2) | 0.4 (1.2) | 0.7 (1.8) | 3.6 (3.3) | 2.9 (2.9) | 3.3 (4.2) |
| Median [Min, Max] | 0 [0,8.0] | 0 [0,8.0] | 0 [0,10.0] | 0 [0,10.0] | 3.0 [0,14.0] | 3.0 [0,10.0] | 2.0 [0,16.0] |
| Eating | |||||||
| Mean (SD) | 0.3 (1.0) | 0.4 (1.0) | 0.2 (0.7) | 0.6 (1.6) | 5.3 (5.2) | 3.6 (3.7) | 5.4 (5.1) |
| Median [Min, Max] | 0 [0,12.0] | 0 [0,6.0] | 0 [0,4.0] | 0 [0,8.0] | 3.0 [0,16.0] | 2.0 [0,14.0] | 3.5 [0,15.0] |
| Mood | |||||||
| Mean (SD) | 0.6 (1.4) | 0.8 (1.6) | 0.5 (1.1) | 1.0 (1.8) | 3.1 (3.4) | 2.4 (3.0) | 3.0 (3.0) |
| Median [Min, Max] | 0 [0,9.0] | 0 [0,7.0] | 0 [0,5.0] | 0 [0,7.0] | 2.0 [0,13.0] | 2.0 [0,12.0] | 2.0 [0,10.0] |
| Beliefs | |||||||
| Mean (SD) | 0.1 (0.3) | 0.1 (0.7) | 0.1 (0.4) | 0.2 (1.3) | 2.5 (3.5) | 2.6 (3.8) | 1.0 (2.7) |
| Median [Min, Max] | 0 [0,3.0] | 0 [0,6.0] | 0 [0,4.0] | 0 [0,9.0] | 1.0 [0,16.0] | 0 [0,13.0] | 0 [0,12.0] |
| Stereotypic behavior | |||||||
| Mean (SD) | 0.5 (1.2) | 0.9 (1.7) | 0.4 (1.1) | 1.1 (2.4) | 7.4 (5.4) | 5.0 (4.6) | 7.1 (5.7) |
| Median [Min, Max] | 0 [0,8.0] | 0 [0,8.0] | 0 [0,10.0] | 0 [0,13.0] | 7.0 [0,20.0] | 4.0 [0,20.0] | 6.5 [0,16.0] |
| Motivation | |||||||
| Mean (SD) | 1.6 (3.9) | 2.9 (5.1) | 1.7 (4.0) | 3.43 (10.1) | 26.1 (26.9) | 26.3 (29.9) | 15.5 (20.7) |
| Median [Min, Max] | 0 [0,38.0] | 0 [0,28.0] | 0 [0,30.0] | 0 [0,72.0] | 14.0 [0,108.0] | 14.0 [0,106.0] | 9.5 [0,80.0] |
2.2. Cognitive, behavioral and symptom assessments
All participants underwent clinical, cognitive and behavioral assessments. A “symptomatic status” binary variable was defined by clinicians (based on the clinician assessment at the time of the participant's first GENFI visit), where the individuals were either assigned as “presymptomatic” if they did not demonstrate overall symptoms, or “symptomatic” if they expressed FTD symptoms. The Cambridge Behavioral Inventory Revised version (CBI‐R; a validated scale for FTD behavioral assessment), was completed by an informant to evaluate memory (memory, attention and orientation), everyday skills, self‐care, abnormal behavior (challenging behavior and disinhibition), mood (depression and agitation), beliefs (auditory and visual hallucinations; considered to be psychotic symptoms), eating, sleep, stereotypic behavior (repetitive behavior and motor movement) and motivation. The CBI‐R questionnaire also rates the frequency of any particular behavior on a scale of 0–4. A score of 0 denotes no impairment, a score of 1 an occasional occurrence defined as a few times per month, 2 a repeated occurrence defined as a few times per week, 3 a daily occurrence, and 4 a constant occurrence. Scores of three or four are indicative of a severe behavioral deficit. The rating of each question is summed to give a final score per CBI‐R category and these itemized scores were used in subsequent analyses (see supplementary methods for more details).
2.3. Image analysis
2.3.1. Image acquisition
All participants were recruited and scanned at one of the 34 GENFI2 sites. T1‐weighted (T1w) images were acquired using an MPRAGE sequence (for parameters https://www.genfi.org/study). The 724 participants were included in the statistical analyses (see 2.3.2 and 3.1 sections). Acquisition parameters (median and ranges) included slice thickness 1.1 mm (1–1.2 mm), repetition time 2.0 ms (6.6–2.4), echo time 2.9 ms (2.2–9 ms), flip angle 8 (8–11), and number of slices 208 (140–208).
2.3.2. Preprocessing
Preprocessing steps were performed on the raw T1 images to standardize images being input into the deformation‐based analysis (see 2.3.3 section; Figure 1). Motion quality control and the minc‐bpipe‐library pipeline (https://github.com/CobraLab/minc-bpipe-library) were used to perform standard preprocessing steps (described in the Supplementary methods).
FIGURE 1.
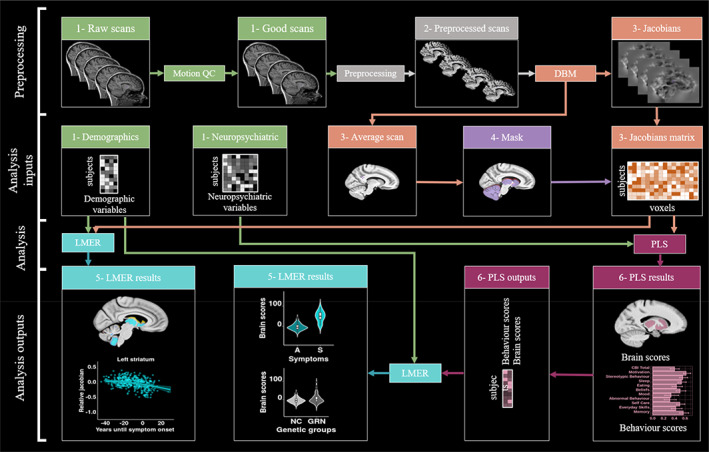
Chart flow of the step by step methods and analyses used in this paper. 1‐Green: Raw inputs (2.3.1. Image acquisition); 2‐Gray: preprocessing (2.3.2 Preprocessing); 3‐Orange: Deformation based morphometry (2.3.3. Deformation based morphometry); 4‐Purple: Mask creation (2.3.4. Mask creation); 5‐Turquoise: linear mixed effect models (2.4.1. Linear mixed effect models); 6‐Pink: Partial least squares (2.4.2. Partial least squares analysis). DBM, deformation based morphometry; LMER, linear mixed effect models; PLS, partial least squares; Preprocessing, minc‐bpipe‐library; QC, quality control; SVD, singular value decomposition.
2.3.3. Deformation based morphometry
We used the two‐level deformation‐based morphometry (DBM) python pipeline developed in the CoBrA Lab (https://github.com/CoBrALab/twolevel_ants_dbm and described in more details in the Supplementary methods) to investigate voxel‐wise morphometry (Figure 1). Each individual image was warped using affine and nonlinear registration to create an unbiased average using ANTs tools and a group‐wise registration strategy (Avants et al., 2011). Relative voxel volume increases and decreases were determined from the deformation fields by estimating the Jacobian determinant at each voxel (Chung et al., 2001). This mathematical transformation allows easier statistical analyses and interpretation: positive values indicate that the voxel in template space must be expanded to get to the subject space, and negative values indicate that the voxel in template space must be reduced. Importantly, Relative Jacobian determinants explicitly model only the nonlinear part of the deformations and remove residual global linear transformations (attributable to differences in total brain size).
2.3.4. Mask creation
In order to focus our voxel‐wise analysis within our regions of interest (ROI), a mask of the subcortical structures (striatum [caudate and putamen], globus pallidus, thalamus), the amygdala and the cerebellum was manually created on the average brain obtained from DBM analyses. This mask will have the goal to restrict our analyses to the 3,609,356 voxels of interest within those structures (note that all our analyses throughout the paper are voxel‐wise and never based on a unitary measure of those ROIs).
2.4. Statistics
2.4.1. Linear mixed effect models
Vertex‐wise linear mixed‐effects models (vertexLmer from RMINC_1.5.2.2 package in R 3.6.3) were used to test the significance of our relative Jacobians within our mask (Figure 1). This model included one group variable assigning the FTD genetic mutation carrier and their clinical status (referred to hereafter as genetic mutation group) of each individual (seven groups; noncarriers, presymptomatic C9orf72, symptomatic C9orf72, symptomatic GRN, presymptomatic GRN, symptomatic MAPT or presymptomatic MAPT carriers), sex (females as reference), education, age at visit and EYO as fixed effects and, scanner and family ID as random effects (to control for potential relatives). A 5% false‐discovery rate (FDR) correction was applied to correct for the potential proportion of false positives at a given FDR threshold (Benjamini & Hochberg, 1995) (FDR correction applied to all predictors). In this analysis, we were specifically interested by the genetic mutation group predictor (different mutation carriers vs. noncarriers), age at visit, and sex (females vs. males). The model equations can be found in the Supplementary methods. Similar linear models were tested including voxels of the whole brain to test whether our results in the subcortical and cerebellar regions were still significant when including the entire brain (see supplementary Figure 1).
2.4.2. Partial least squares
Partial least squares (PLS) correlation, a multivariate technique, was used to detect covariance patterns across two matrices (Figure 1). The goal of PLS is to identify a set of orthogonal latent variables (LVs) that explain patterns of covariance between brain (relative Jacobians) and behavior data (CBI‐R scores). Theoretically, each LV depicts a linear combination of the brain and behavior matrices. Here, one PLS analysis was run for each group (C9orf72, GRN, MAPT and noncarriers separately; four total), to examine if distinct brain/behavior patterns are present in each mutation group. Our brain data included the relative Jacobian of each voxel for each subject (matrix size 3609356 × 184 for C9orf72; 3609356 × 185 for GRN, 3609356 × 80 for MAPT and 3609356 × 281 for noncarriers). Our behavior data contained 10 CBI‐R scores for each subject. The values in the behavior matrices were z‐scored along each column prior to performing PLS (i.e., z‐score the values of each behavior score across all subjects). Each LV was tested statistically using permutation testing and bootstrap resampling following a similar protocol (detailed in the Supplementary methods) as in previous studies (Anthony Randal McIntosh & Lobaugh, 2004; Zeighami et al., 2017; Patel et al., 2020; Bussy et al., 2021).
2.4.3. Post‐hoc analyses of PLS outputs
Once the PLS results were obtained and tested for significance, the brain and behavior scores were further analyzed to determine if they were associated with key demographic and clinical variables. Linear mixed effect models including age, EYO, sex, education and symptomatic status as fixed effects and scanner and family ID as random effects to examine the brain or the behavior scores (see equations in Supplementary methods). FDR correction was applied to correct for multiple comparisons. The goal of these analyses is to see if the LVs found from PLS capture patterns of brain and behavior which are specific to disease‐related demographics and symptomatic status. Indeed, PLS was blind to the EYO, sex, education and to the symptomatic status. These models were also tested on the presymptomatic carriers only (Supplementary Figure 2).
2.5. Flat map visualization
Matlab (version R 2014b) and SUIT toolbox were used to display lmer and PLS results into a surface‐based flatmap representation of the cerebellum (Jörn Diedrichsen, 2006; Jörn Diedrichsen et al., 2009; J. Diedrichsen et al., 2011; Jörn Diedrichsen & Zotow, 2015). Details on the commands used are described in the supplementary methods.
3. RESULTS
3.1. Demographic and clinical information
After motion quality control (criteria described in the supplementary methods), from 983 initial scans, 130 scans were excluded (supplementary Table 1). From 853 scans, 8 failed our preprocessing and 121 individuals were excluded due to missing CBI‐R information. Table 1 describes the demographic and clinical information of the 724 individuals used in subsequent analyses (184 C9orf72 [122 presymptomatic and 62 symptomatic], 179 GRN [135 presymptomatic and 44 symptomatic], 80 MAPT [56 presymptomatic and 24 symptomatic] carriers and 281 controls).
3.2. Linear mixed effect model
3.2.1. Genetic mutation group
Figure 2 a2 illustrates the brain regions showing significant volume differences between genetic mutation groups versus noncarrier participants. When controlling for variables including sex, age and EYO, presymptomatic C9orf72 expansion carriers demonstrated significant volume decrease in the bilateral thalamus. No significant differences were found between presymptomatic GRN carriers and presymptomatic MAPT carriers versus noncarriers. All symptomatic groups demonstrated significant volume decrease compared to noncarriers. Symptomatic C9orf72 expansion carriers demonstrated the largest volume decrease, in the thalamus, striatum, globus pallidus, amygdala and a large extent of the cerebellum including lobules Crus I, Crus II, IX and X. Symptomatic GRN carriers mostly expressed decreased volume in the striatum and thalamus and symptomatic MAPT carriers demonstrated decreased amygdalar volumes compared to controls. Figure 2b illustrates four peak voxels for each group in the left thalamus, striatum, amygdala and cerebellum.
FIGURE 2.
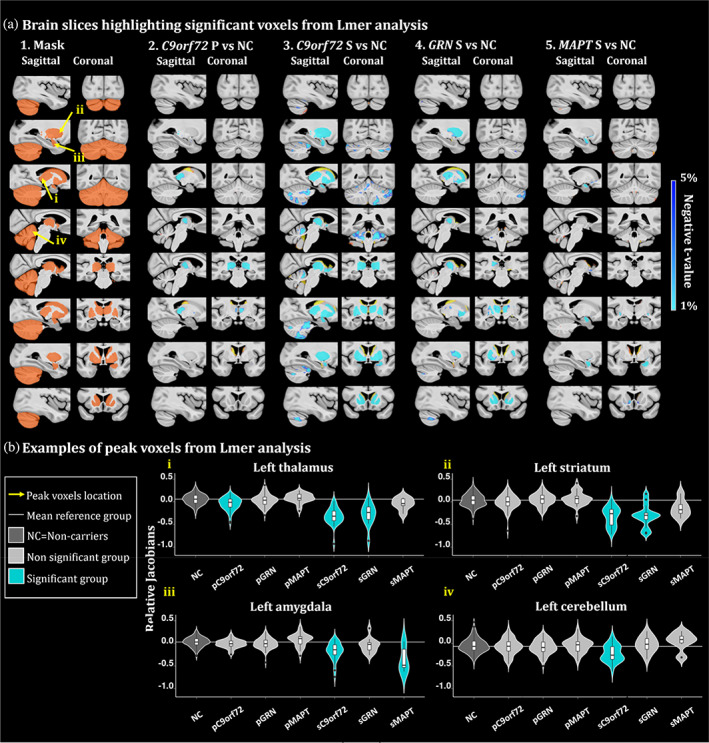
(a) Brain slices of the brain highlighting significant group differences from the lmer analyses. t‐value maps correspond to significant p‐values between 5% and 1% after FDR correction. Axial slices represented from left to right and coronal slices represented from posterior to anterior. The t‐statistics color maps for the significant expansion are in yellow to red and for the significant contraction are in turquoise to blue. Yellow arrows were used to highlight the peak voxels selected for the plots in Figure 2b. A1. Sagittal and coronal slices of the mask used to focus the analyses in the regions of interest. Slices of the brain showing significant differences between the relative Jacobians of the A2. presymptomatic (P) C9orf72 carriers; A3. symptomatic (S) C9orf72 carriers; A4. symptomatic GRN carriers and A5. MAPT carriers versus the relative Jacobians of the noncarrier participants. (b) Examples of peak voxels from lmer analyses. White horizontal line highlights the mean relative Jacobian of the reference group (i.e., noncarrier participants). Violin plots illustrate the group difference of a peak voxel in the i. left thalamus, ii. left striatum, iii. left amygdala and iv. left cerebellum.
Our whole brain analyses (Supplementary Figure 1) demonstrated that presymptomatic C9orf72 carriers have a specific significant volume reduction in the thalamus, putamen and globus pallidus, demonstrating that those structures are the first (before the cortex or other regions) to be impacted in C9orf72 mutation carriers. Further, symptomatic carriers revealed frontal and temporal atrophy in C9orf72 and GRN carriers and mostly temporal atrophy in MAPT carriers. In addition to this global atrophy, significant volume reductions were also found in the subcortical and cerebellar regions, showing that those structures are impacted even when whole brain atrophy is accounted for in our statistical analyses.
3.2.2. Age and sex
In Figure 3 a2 and a3, the brain maps exhibit the voxels demonstrating significant second order volume decrease with age, independently of mutation status. These voxels are mostly situated in the thalamus and in the cerebellum, particularly in lobule IX. Figure 3 a4 highlights the differences of volume between men and women, demonstrating lower cerebellar WM and thalamic volumes in men. Figure 3b exhibits three peak voxels of the right thalamus, left cerebellum lobule IX and left cerebellum white matter, highlighting a second order volume decrease with advanced age and a sex effect. Interestingly, the pattern of decrease with age is present for the entire age range and not only for the symptomatic participants.
FIGURE 3.
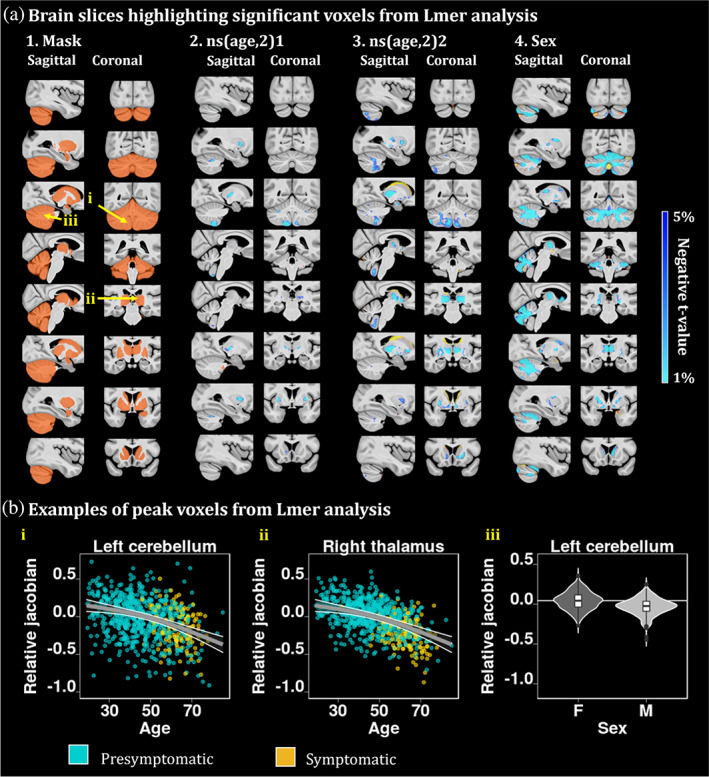
(a) Brain slices of the brain highlighting significant age effects from the lmer analyses. t‐value maps of the age variable corresponding to significant p‐values between 5% and 1% after FDR correction. Axial slices represented from left to right and coronal slices represented from posterior to anterior. The t‐statistics color maps for the significant expansion are in yellow to red and for the significant contraction are in turquoise to blue. Yellow arrows were used to highlight the peak voxels selected for the plots in Figure 3b. A.4. Map of the relative Jacobians difference between males versus females. (b) (i) and (ii) second order relationships between the relative Jacobians and age, using the predicted Jacobians between age 19 and 85 for a subject of mean EYO and unweighted averages over the levels of sex and group. These plots highlight a second order volume decrease with advanced age in two peak voxels of the left cerebellum lobule IX and right thalamus. (iii) peak voxel from the white matter cerebellum showing sex difference atrophy between males and females.
3.3. PLS results
3.3.1. Latent variables
PLS results demonstrate one significant LV per genetic group, except for the noncarriers PLS which had no significant LVs (Figure 4a). For all mutation groups, we found consistent brain regions associated with behavior scores, such as the thalamus, striatum and globus pallidus. However, C9orf72 and GRN groups demonstrated large cerebellar reduction while MAPT individuals expressed very local cerebellar atrophy but larger amygdalar volume reduction. Interestingly, all the PLS runs exhibited almost similar worsen behavior scores in all CBI‐R categories (Figure 4b).
FIGURE 4.
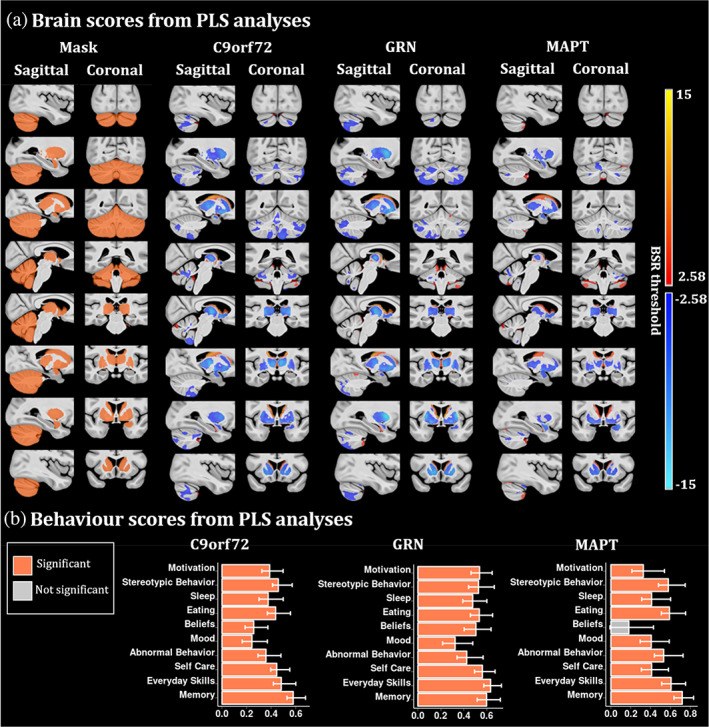
PLS analyses between the voxel‐wise relative Jacobians and the CBI‐R variables for each mutation group separately. (a) Brain scores of each latent variable (LV) were plotted using the vertex wise BSR thresholded at 2.58 (p < .01). The range of BSR values was (−12.4, 11.7) for C9orf72, (−14.8, 14.5) for GRN and (−8.4, 9.1) for MAPT LV. A common minimum/maximum BSR threshold was selected (−15, 15) to have a similar color scale between each brain map. Each group demonstrated one significant LV except the noncarriers group (not shown). The LV explained 91.8% of the variance for C9orf72, 93.2% of the variance for GRN and 84.4% of the variance for MAPT. (b) Bar plots describe the correlation of each CBI‐R variable with each LV, with error bars denoting the 95% confidence interval. Orange color represents CBI‐R variables that significantly participate in the LV while grey color represents nonsignificant CBI‐R variables.
Chromosome 9 open reading frame 72
C9orf72 LV explained 91.8% of the variance. C9orf72 brain map demonstrates a pattern of volume reduction in the thalamus, globus pallidus, striatum and in a large area in the cerebellum, including superior posterior lobules, and lobules IX associated with higher behavior scores (worse cognition and behavior symptoms).
Progranulin
GRN LV explained 93.2% of the variance and the GRN map illustrates a volume reduction of the thalamus, globus pallidus, striatum and very subtle parts of the cerebellum, mostly in lobules Crus I‐II. The GRN behavior scores plot demonstrates significantly worse behavior scores being linked to a volume reduction in the structures described above.
Microtubule‐associated protein tau
Finally, the MAPT LV explained 84.4% of the variance and its map demonstrates a volume reduction in the thalamus, globus pallidus, striatum, larger reduction in the amygdala and very subtle volume decrease in the cerebellum. The MAPT behavior scores plot demonstrated significantly worse behavior scores in all CBI‐R categories, except for the abnormal beliefs.
3.3.2. Post‐hoc analyses of PLS results
Brain and behavior scores from PLS analyses were analysed to examine if they could be explained by the demographic and clinical information of the participants.
Chromosome 9 open reading frame 72
Figure 5a demonstrates that C9orf72 brain scores were significantly reduced with increased age and EYO (p = 8.7 × 10−8 and p = 9.2 × 10−5 respectively); however, it was not significantly different between presymptomatic and symptomatic carriers. Age is not related to behavior scores while increasing EYO and being symptomatic are linked to higher behavior scores in all subcategories (p = 3.9 × 10−2 and p = 4.7 × 10−22 respectively).
FIGURE 5.
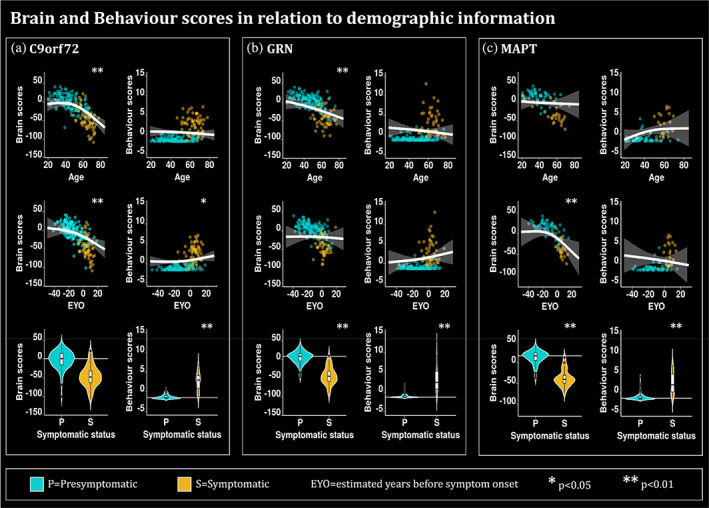
Plots describing the relationship of the brain and behavior scores for (a) C9orf72, (b) GRN and (c) MAPT mutation carriers with demographic and clinical information such as age, EYO, and symptomatic status. The plots for age and EYO either demonstrate the second order relationships between the relative Jacobians and age using the predicted Jacobians between age 19 and 85 for a subject of mean EYO or using the predicted Jacobians between EYO‐50 and 30 for a subject of mean age, respectively. These models were computed using the unweighted averages over the levels of sex, education and symptomatic status. Turquoise is used to highlight the presymptomatic (P) individuals while gold is used to highlight the symptomatic (S) individuals. * is used to show significant variables (p < .05 after FDR correction) and ** to show significant variables (p < .01 after FDR correction. The age and EYO relationships were plotted based on the lmer model. White horizontal lines highlight the mean relative Jacobian of the presymptomatic individuals (reference group).
Progranulin
In Figure 5b, GRN brain scores are reduced with increasing age and being symptomatic (p = 6.8 × 10−3 and 5.0 × 10−16 respectively), while EYO is not related to the brain scores. GRN behavior scores are only related to being presymptomatic or symptomatic (p = 2.6 × 10−19).
Microtubule‐associated protein tau
Finally, the brain scores of MAPT are significantly reduced with increased EYO and for symptomatic individuals (p = 3.6 × 10−3 and p = 6.0 × 10−7 respectively), while the behavior scores are only increased for the symptomatic individuals compared to presymptomatic individuals (p = 4.3 × 10−5). Interestingly, for both C9orf72 and MAPT, the brain scores are starting to decrease up to 20 years before the expected symptom onset, when most of the individuals are still presymptomatic (see supplementary Figure 3).
3.4. Cerebellar contribution
In order to discuss our cerebellar results, we used well‐defined and previously published anatomical and functional maps of the cerebellum. Anatomically, we can see that our C9orf72 map is principally located on lobules Crus I‐II, VIIIB and IX (Figure 6a). The GRN map is mainly localized in lobules Crus I‐II while the MAPT map only includes a very small part of lobule VI (Jörn Diedrichsen, 2006; Jörn Diedrichsen et al., 2009). Interestingly, when looking at a simplified resting‐state network atlas (Buckner et al., 2011) and a simplified task processing atlas (Guell et al., 2023), we observe that the C9orf72 map overlaps mostly with the default mode network (Figure 6b), and corresponds with the working memory, language, and social tasks (Figure 6c). On the other hand, the GRN map overlaps mostly with the frontoparietal network and the working memory task.
FIGURE 6.
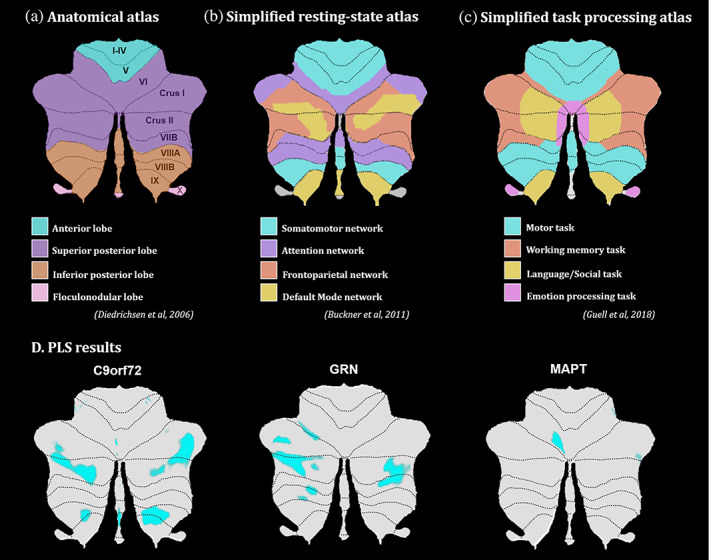
Flatmaps of the (a) cerebellar anatomical atlas Diedrichsen et al., 2009), (b) simplified resting‐state network atlas Buckner et al., 2011), (c) simplified task processing atlas (Guell et al., 2023) and (d) LV1 brain map results from PLS analyses for each mutation group.
4. DISCUSSION
This paper confirms the role of subcortical brain atrophy in behavioral symptoms in each of the main genetic forms of FTD. We used DBM, univariate voxel‐wise analyses and multivariate techniques to identify these relationships. First, we demonstrated that in presymptomatic participants, only C9orf72 expansion carriers demonstrated significant atrophy in the thalamus compared to noncarriers. However, all the genetic mutation groups demonstrated brain atrophy compared to noncarriers in the symptomatic group. Differences in atrophy patterns were found; C9orf72 expansion carriers have largest volume decrease in the thalamus, striatum, globus pallidus, amygdala and a large extent of the cerebellum including lobules Crus I, Crus II, IX and X. Symptomatic GRN carriers mostly expressed decreased volume in the striatum and thalamus while symptomatic MAPT carriers mainly demonstrated decreased amygdalar volumes compared to controls. Further, second order age atrophy was found in the cerebellum in the thalamus and cerebellum (IX). We also found significantly lower volumes in the thalamus and white matter of the cerebellum in the men compared to the women. Overall, these univariate analyses demonstrate direct effects of age, gene expansion and symptomatic status on the subcortical volumes.
The PLS analyses demonstrated overlapping gene‐specific associations between brain and behavior. While all groups' brain maps included the thalamus, globus pallidus and striatum, the C9orf72 expansion carriers had specific atrophy of a large area of the cerebellum, which is in agreement with a recent study showing preferential cerebellar involvement in C9orf72 expansion carriers (Bocchetta et al., 2021). GRN cerebellum map demonstrated more restricted atrophy of the cerebellar lobules Crus I‐II. MAPT LV included a very limited atrophy of the cerebellum but a larger atrophy of the amygdala compared to the other two genetic groups. These findings are in line with previous studies showing thalamic and cerebellar symmetrical and widespread patterns of atrophy in C9orf72 expansion carriers (Bocchetta et al., 2016; Sha et al., 2012), while amygdalar atrophy seemed to be more related to MAPT carriers (Bocchetta et al., 2021; Cash et al., 2018).
While we saw brain map differences between mutation groups, the PLS behavioral results did not demonstrate gene‐specific effects. Therefore, the variations in atrophy observed across genetic groups were not associated with different symptom profiles. This is consistent with clinical observations which suggest that different mutations have major overlap in their clinical presentations, despite regional specificity in atrophy profiles (Jonathan D. Rohrer et al., 2015). Additionally, our PLS analysis on the noncarrier group did not lead to a significant LV demonstrating that these patterns of brain/behavior are specific to mutation carriers, as opposed to normative anatomical correlates of behavioral traits in healthy adults. Given the significant prevalence of psychotic symptoms in C9orf72 and to a lesser extent in GRN, we would have expected to find a specific latent variable for psychosis‐related categories. However, the absence of such results could be explained by the relatively small number of symptomatic carriers with psychotic symptoms. Of note, this lack of brain/behavior differences between genetic groups, contrasts with some previous reports that link specific symptoms to specific anatomical correlates (Benussi et al., 2021; Sellami et al., 2018). However, while these previous studies have found significant association between behavior symptoms and genetic groups, they ran a series of ANOVA or linear models for each symptom. On the contrary, here we used PLS analyses which is a recommended technique when the number of explanatory variables is high, and likely correlated (Anthony R. McIntosh & Mišić, 2013), which is the case in our study.
As expected, the presence of symptoms was consistently a strong predictor of the PLS‐derived brain and behavior scores. However, while C9orf72 and MAPT brain scores were related to EYO and expressed a second order relationship highlighting a more rapid brain score decline in the 20 years before symptom onset, GRN brain scores were related to age and not EYO. These results are consistent with previous reports that GRN mutation carrier symptom onset is only weakly predictable using parental age of onset (Moore et al., 2020). Also, while variant‐specific grey matter atrophy (mostly in the thalamus) has been shown to be detectable up to 20 years before EYO in C9orf72 expansion carriers, atrophy is detectable 15 years before EYO for MAPT carriers (mostly fronto‐parietal) and only 10 years before EYO for GRN carriers (mostly medial‐temporal) (Greaves & Rohrer, 2019b). Further, EYO was a better predictor at explaining the covariance between morphometry and CBI‐R variables (at least for C9orf72 expansion carriers and MAPT carriers), but it was not directly associated with morphometry. These results suggest that future clinical and research studies might benefit from using brain atrophy and behavior variables simultaneously to identify subjects at higher risk of short‐term clinical conversion to the dementia stage. In addition, our results suggest that the brain regions that we found to be significantly associated with the CBI‐R scores could be particularly sensitive to changes related to EYO.
While we identified a similar behavioral cluster in all the genetic mutation groups, we hypothesize that different patterns of cerebellar and amygdalar atrophy could be responsible for symptomatic differences in each mutation group later in the disease progression. For example, pronounced psychotic symptoms in C9orf72 expansion carriers (potentially linked to the preferential cerebellar lobule Crus I‐II and lobule IX atrophy) while MAPT carriers demonstrate more depression and anxiety (potentially linked to the preferential amygdalar atrophy).
Of interest, the cerebellar structures found to be associated with the CBI‐R variables (Crus I‐II and IX) correspond to the ones associated with frontoparietal and default mode networks (DMN; (Guell et al., 2018)). In our results, cerebellar atrophy in C9orf72 expansion carriers was predominantly related to the DMN, which relates to previous findings (Lee et al., 2014), while in GRN carriers, cerebellar regions were primarily functionally correlated to the fronto‐parietal network, which ties in to the well documented component of parietal involvement in this disease (Cash et al., 2018). Interestingly, reduction of Crus I‐II volumes has previously been associated with behavioral and cognitive deficits in FTD (Chen et al., 2018; Chen et al., 2019). These results suggest that while our study focused on the anatomical changes in FTD in relation with behavior, we demonstrated that the structures covarying the most with behavioral changes colocalize specifically with the DMN and fronto‐parietal networks, motivating the relevance of using fMRI techniques to study brain/behavior changes in familial FTD.
Some limitations in our study need to be highlighted and discussed. Biases in our results may arise from the limited number of MAPT mutation carriers in the GENFI dataset, explaining the apparent reduced brain involvement in this group. Only about 18% of our mutation carriers were MAPT carriers. However worldwide, MAPT mutation is the least frequent (23.2%), followed by GRN mutation (34.6%) and C9orf72 expansion (42.1%) (Moore et al., 2020). Therefore, our dataset was representative of the general mutation distribution in FTD. Further, while GENFI is the largest dataset of carriers for FTD‐linked mutations to date and offers a sufficient number of participants to conduct complex statistical analyses, only 18% of those participants were symptomatic mutation carriers.
Secondly, this study aims to understand the brain and CBI‐R variables impairment during the presymptomatic phase of the disease, and uses EYO as an estimator of age of symptom onset. However, as discussed above, EYO accuracy is limited and varies between genetic mutation groups (Moore et al., 2020). Therefore, to better characterize the association between brain and behavior during this presymptomatic phase, longitudinal data with documentation of the actual age at onset is necessary. This will eventually be available from GENFI from ongoing follow‐up brain imaging and CBI‐R assessments. Finally, it is important to note that while our results allowed straightforward qualitative comparisons between each PLS findings, no test has been performed to quantitatively compare the different brain maps. To verify that our differences in brain maps were not driven by a threshold effect, we also plotted the different brain maps using a less stringent significant threshold (Supplementary Figure 1), demonstrating that even with a more lenient threshold, different parts of the cerebellum were associated with the behavior scores for each genetic mutation group.
To conclude, our study shows that the subcortical and cerebellar circuitry is involved in the relationship between FTD pathology and the emergence of neuropsychiatric symptoms, even in the presymptomatic stage. Furthermore, while all brain maps included the thalamus, globus pallidus and striatum, C9orf72 expansion demonstrated a specific involvement of a large area of the cerebellum. Variations in atrophy across genetic groups did not explain symptomatic profile differences, suggesting that these differences in cerebellar and amygdalar atrophy pattern only lead to behavioral differences later in the disease progression.
Our study suggests the importance of cerebellar and subcortical involvement during the earliest stage of FTD progression and a relationship to the appearance of neuropsychiatric symptoms. Clinically, this can help clinicians provide an explanatory model to patients and their family for their behavioral symptoms. It further highlights the importance of these structures in early disease stages. The longitudinal follow‐up of these volumes factoring differences across mutation subtypes could provide useful biomarker information on disease progression in future clinical trials.
FUNDING INFORMATION
A. Bussy receives support from the Alzheimer Society of Canada. Dr Chakravarty is funded by the Weston Brain Institute, the Canadian Institutes of Health Research, the Natural Sciences and Engineering Research Council of Canada, Healthy Brains Healthy Lives and Fondation de Recherches Santé Québec. Dr Ducharme received salary funding from the Fonds de Recherche du Québec ‐ Santé.
This work was also supported by the MRC UK GENFI grant (MR/M023664/1), the Italian Ministry of Health (CoEN015 and Ricerca Corrente), the Canadian Institutes of Health Research as part of a Centres of Excellence in Neurodegeneration grant, a Canadian Institutes of Health Research operating grant, the Alzheimer's Society grant (AS‐PG‐16‐007), the Bluefield Project and the JPND GENFI‐PROX grant (2019–02248). MB is supported by a Fellowship award from the Alzheimer's Society, UK (AS‐JF‐19a‐004‐517). MB's work was also supported by the UK Dementia Research Institute which receives its funding from DRI Ltd., funded by the UK Medical Research Council, Alzheimer's Society and Alzheimer's Research UK. JDR is an MRC Clinician Scientist (MR/M008525/1) and has received funding from the NIHR Rare Diseases Translational Research Collaboration (BRC149/NS/MH), the Bluefield Project and the Association for Frontotemporal Degeneration. This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy within the framework of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy–ID 390857198). Several authors of this publication (JCvS, MS, RSV, AD, MO, JDR) are members of the European Reference Network for Rare Neurological Diseases (ERN‐RND) ‐ Project ID No 739510. This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy within the framework of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy–ID 390857198).
This research was supported by the NIHR Cambridge Biomedical Research Centre (BRC‐1215‐20014). The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care.
CONFLICT OF INTEREST STATEMENT
The authors report no competing interests related to this paper.
Supporting information
Data S1: Supporting information
Appendix
Bussy, A. , Levy, J. P. , Best, T. , Patel, R. , Cupo, L. , Van Langenhove, T. , Nielsen, J. E. , Pijnenburg, Y. , Waldö, M. L. , Remes, A. M. , Schroeter, M. L. , Santana, I. , Pasquier, F. , Otto, M. , Danek, A. , Levin, J. , Le Ber, I. , Vandenberghe, R. , Synofzik, M. , Moreno, F. , de Mendonça, A. , Sanchez‐Valle, R. , Laforce, R. , Langheinrich, T. , Gerhard, A. , Graff, C. , Butler, C. R. , Sorbi, S. , Jiskoot, L. , Seelaar, H. , van Swieten, J. C. , Finger, E. , Tartaglia, M. C. , Masellis, M. , Tiraboschi, P. , Galimberti, D. , Borroni, B. , Rowe, J. B. , Bocchetta, M. , Rohrer, J. D. , Devenyi, G. A. , Chakravarty, M. M. , Ducharme, S. , & GENetic Frontotemporal dementia Initiative (GENFI) (2023). Cerebellar and subcortical atrophy contribute to psychiatric symptoms in frontotemporal dementia. Human Brain Mapping, 44(7), 2684–2700. 10.1002/hbm.26220
M. Mallar Chakravarty and Simon Ducharme equally contributed.
List of consortium authors in appendix.
Contributor Information
Aurélie Bussy, Email: aureliee.bussy@gmail.com.
Jørgen E. Nielsen, Email: joergen.erik.nielsen.01@regionh.dk.
Robert Laforce, Email: robert.laforce@fmed.ulaval.ca.
Simon Ducharme, Email: simon.ducharme@mcgill.ca.
GENetic Frontotemporal dementia Initiative (GENFI):
Aitana Sogorb Esteve, Annabel Nelson, Arabella Bouzigues, Carolin Heller, Caroline V Greaves, David Cash, David L Thomas, Emily Todd, Hanya Benotmane, Henrik Zetterberg, Imogen J Swift, Jennifer Nicholas, Kiran Samra, Lucy L Russell, Martina Bocchetta, Rachelle Shafei, Rhian S Convery, Carolyn Timberlake, Thomas Cope, Timothy Rittman, Alberto Benussi, Enrico Premi, Roberto Gasparotti, Silvana Archetti, Stefano Gazzina, Valentina Cantoni, Andrea Arighi, Chiara Fenoglio, Elio Scarpini, Giorgio Fumagalli, Vittoria Borracci, Giacomina Rossi, Giorgio Giaccone, Giuseppe Di Fede, Paola Caroppo, Pietro Tiraboschi, Sara Prioni, Veronica Redaelli, David Tang‐Wai, Ekaterina Rogaeva, Miguel Castelo‐Branco, Morris Freedman, Ron Keren, Sandra Black, Sara Mitchell, Christen Shoesmith, Robart Bartha, Rosa Rademakers, Jackie Poos, Janne M. Papma, Lucia Giannini, Rick van Minkelen, Yolande Pijnenburg, Benedetta Nacmias, Camilla Ferrari, Cristina Polito, Gemma Lombardi, Valentina Bessi, Michele Veldsman, Christin Andersson, Hakan Thonberg, Linn Öijerstedt, Vesna Jelic, Paul Thompson, Tobias Langheinrich, Albert Lladó, Anna Antonell, Jaume Olives, Mircea Balasa, Nuria Bargalló, Sergi Borrego‐Ecija, Ana Verdelho, Carolina Maruta, Catarina B. Ferreira, Gabriel Miltenberger, Frederico Simões do Couto, Alazne Gabilondo, Ana Gorostidi, Jorge Villanua, Marta Cañada, Mikel Tainta, Miren Zulaica, Myriam Barandiaran, Patricia Alves, Benjamin Bender, Carlo Wilke, Lisa Graf, Annick Vogels, Mathieu Vandenbulcke, Philip Van Damme, Rose Bruffaerts, Koen Poesen, Pedro Rosa‐Neto, Serge Gauthier, Agnès Camuzat, Alexis Brice, Anne Bertrand, Aurélie Funkiewiez, Daisy Rinaldi, Dario Saracino, Olivier Colliot, Sabrina Sayah, Catharina Prix, Elisabeth Wlasich, Olivia Wagemann, Sandra Loosli, Sonja Schönecker, Tobias Hoegen, Jolina Lombardi, Sarah Anderl‐Straub, Adeline Rollin, Gregory Kuchcinski, Maxime Bertoux, Thibaud Lebouvier, Vincent Deramecourt, Beatriz Santiago, Diana Duro, Maria João Leitão, Maria Rosario Almeida, Miguel Tábuas‐Pereira, Sónia Afonso, Annerose Engel, and Maryna Polyakova
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request via https://www.genfi.org/study/ or by emailing genfi@ucl.ac.uk. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- Avants, B. B. , Tustison, N. J. , Song, G. , Cook, P. A. , Klein, A. , & Gee, J. C. (2011). A reproducible evaluation of ANTs similarity metric performance in brain image registration. NeuroImage, 54(3), 2033–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balleine, B. W. , Delgado, M. R. , & Hikosaka, O. (2007). The role of the dorsal striatum in reward and decision‐making. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 27(31), 8161–8165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini, Y. , & Hochberg, Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. Series B, Statistical Methodology, 57(1), 289–300. [Google Scholar]
- Benussi, A. , Premi, E. , Gazzina, S. , Brattini, C. , Bonomi, E. , Alberici, A. , Jiskoot, L. , van Swieten, J. C. , Sanchez‐Valle, R. , Moreno, F. , Laforce, R. , Graff, C. , Synofzik, M. , Galimberti, D. , Masellis, M. , Tartaglia, C. , Rowe, J. B. , Finger, E. , Vandenberghe, R. , … Zulaica, M. (2021). Progression of behavioral disturbances and neuropsychiatric symptoms in patients with genetic frontotemporal dementia. JAMA Network Open, 4(1), e2030194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand, A. , Wen, J. , Rinaldi, D. , Houot, M. , Sayah, S. , Camuzat, A. , Fournier, C. , Fontanella, S. , Routier, A. , Couratier, P. , Pasquier, F. , Habert, M.‐O. , Hannequin, D. , Martinaud, O. , Caroppo, P. , Levy, R. , Dubois, B. , Brice, A. , Durrleman, S. , … for the Predict to Prevent Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis (PREV‐DEMALS) Study Group . (2018). Early cognitive, structural, and microstructural changes in Presymptomatic C9orf72 carriers younger than 40 years. JAMA Neurology, 75(2), 236–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc, Gabriella, L. , Pomerleau, V. J. , McCarthy, J. , Borroni, B. , van Swieten, J. , Galimberti, D. , Sanchez‐Valle, R. , LaForce, R., Moreno, F., Synofzik, M., Graff, C., Masellis, M., Tartaglia, M., Rowe, J., Vandenberghe, R., Finger, E., Tagliavini, F., de Mendonca, A, Santana, I., … Genetic Frontotemporal Dementia Initiative (GENFI). (2020). Faster cortical thinning and surface area loss in Presymptomatic and symptomatic C9orf72 repeat expansion adult carriers. Annals of Neurology, 88(1), 113–122. [DOI] [PubMed] [Google Scholar]
- Blithikioti, C. , Nuño, L. , Guell, X. , Pascual‐Diaz, S. , Gual, A. , Balcells‐Olivero, Μ. , & Miquel, L. (2022). The cerebellum and psychological trauma: A systematic review of neuroimaging studies. Neurobiology of Stress, 17, 100429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocchetta, M. , Jorge Cardoso, M. , Cash, D. M. , Ourselin, S. , Warren, J. D. , & Rohrer, J. D. (2016). Patterns of regional cerebellar atrophy in genetic frontotemporal dementia. NeuroImage. Clinical, 11, 287–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocchetta, M. , Todd, E. G. , Peakman, G. , Cash, D. M. , Convery, R. S. , Russell, L. L. , Thomas, D. L. , Eugenio Iglesias, J. , van Swieten, J. C. , Jiskoot, L. C. , Seelaar, H. , Borroni, B. , Galimberti, D. , Sanchez‐Valle, R. , Laforce, R. , Moreno, F. , Synofzik, M. , Graff, C. , Masellis, M. , … Zulaica, M. (2021). Differential early subcortical involvement in genetic FTD within the GENFI cohort. NeuroImage, 30, 102646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrego‐Écija, S. , Sala‐Llonch, R. , van Swieten, J. , Borroni, B. , Moreno, F. , Masellis, M. , Tartaglia, C. , Graff, C. , Galimberti, D. , Laforce, R. , Rowe, J. B. , Finger, E. , Vandenberghe, R. , Tagliavini, F. , de Mendonça, A. , Santana, I. , Synofzik, M. , Ducharme, S. , Levin, J. , … Anderl‐Straub, S. (2021). Disease‐related cortical thinning in Presymptomatic Granulin mutation carriers. NeuroImage. Clinical, 29, 102540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner, R. L. , Krienen, F. M. , Castellanos, A. , Diaz, J. C. , & Thomas Yeo, B. T. (2011). The Organization of the Human Cerebellum Estimated by intrinsic functional connectivity. Journal of Neurophysiology, 106, 2322–2345. 10.1152/jn.00339.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussy, A. , Patel, R. , Plitman, E. , Tullo, S. , Salaciak, A. , Bedford, S. A. , Farzin, S. , et al. (2021). Hippocampus shape across the healthy lifespan and its relationship with cognition. Neurobiology of Aging, 106, 153–168. 10.1101/2020.10.30.362921 [DOI] [PubMed] [Google Scholar]
- Cash, D. M. (2018). Patterns of gray matter atrophy in genetic frontotemporal dementia: Results from the GENFI study. Neurobiology of Aging, 62, 191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y. , Kumfor, F. , Landin‐Romero, R. , Irish, M. , Hodges, J. R. , & Piguet, O. (2018). Cerebellar atrophy and its contribution to cognition in frontotemporal dementias. Annals of Neurology, 84(1), 98–109. [DOI] [PubMed] [Google Scholar]
- Chen, Y. , Kumfor, F. , Landin‐Romero, R. , Irish, M. , & Piguet, O. (2019). The cerebellum in frontotemporal dementia: A meta‐analysis of neuroimaging studies. Neuropsychology Review, 29(4), 450–464. [DOI] [PubMed] [Google Scholar]
- Chung, M. K. , Worsley, K. J. , Paus, T. , Cherif, C. , Collins, D. L. , Giedd, J. N. , Rapoport, J. L. , & Evans, A. C. (2001). A unified statistical approach to deformation‐based morphometry. NeuroImage, 14(3), 595–606. [DOI] [PubMed] [Google Scholar]
- Coyle‐Gilchrist, I. T. S. , Dick, K. M. , Patterson, K. , Rodríquez, P. V. , Wehmann, E. , Wilcox, A. , Lansdall, C. J. , et al. (2016). Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology, 86(18), 1736–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadar, M. , Manera, A. L. , Ducharme, S. , & Louis Collins, D. (2022). White matter Hyperintensities are associated with Grey matter atrophy and cognitive decline in Alzheimer's disease and frontotemporal dementia. Neurobiology of Aging, 111, 54–63. [DOI] [PubMed] [Google Scholar]
- Diedrichsen, J. , Maderwald, S. , Küper, M. , Thürling, M. , Rabe, K. , Gizewski, E. R. , Ladd, M. E. , & Timmann, D. (2011). Imaging the deep cerebellar nuclei: A probabilistic atlas and normalization procedure. NeuroImage, 54(3), 1786–1794. [DOI] [PubMed] [Google Scholar]
- Diedrichsen, J. (2006). A spatially unbiased atlas template of the human cerebellum. NeuroImage, 33(1), 127–138. [DOI] [PubMed] [Google Scholar]
- Diedrichsen, J. , Balsters, J. H. , Flavell, J. , Cussans, E. , & Ramnani, N. (2009). A probabilistic MR atlas of the human cerebellum. NeuroImage, 46(1), 39–46. [DOI] [PubMed] [Google Scholar]
- Diedrichsen, J. , & Zotow, E. (2015). Surface‐based display of volume‐averaged cerebellar imaging data. PloS One, 10(7), e0133402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, A.‐T. , Schuff, N. , Kramer, J. H. , Rosen, H. J. , Gorno‐Tempini, M. L. , Rankin, K. , Miller, B. L. , & Weiner, M. W. (2007). Different regional patterns of cortical thinning in Alzheimer's disease and frontotemporal dementia. Brain: A Journal of Neurology, 130(Pt 4), 1159–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducharme, S. , Bajestan, S. , Dickerson, B. C. , & Voon, V. (2017). Psychiatric presentations of C9orf72 mutation: What are the diagnostic implications for clinicians? The Journal of Neuropsychiatry and Clinical Neurosciences, 29(3), 195–205. [DOI] [PubMed] [Google Scholar]
- Greaves, C. V. , & Rohrer, J. D. (2019a). An update on genetic frontotemporal dementia. Journal of Neurology, 266(8), 2075–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guell, X. , Gabrieli, J. D. E. , & Jeremy, D. S. (2023). Triple Representation of Language, Working Memory, Social and Emotion Processing in the Cerebellum: Convergent Evidence from Task and Seed‐Based Resting‐State Fmri Analyses in a Single Large Cohort. 10.1101/254110 [DOI] [PMC free article] [PubMed]
- Guell, X. , Schmahmann, J. D. , Gabrieli, J. D. E. , & Ghosh, S. S. (2018). Functional gradients of the cerebellum. eLife, 7, e36652. 10.7554/eLife.36652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartikainen, P. , Räsänen, J. , Julkunen, V. , Niskanen, E. , Hallikainen, M. , Kivipelto, M. , Vanninen, R. , Remes, A. M. , & Soininen, H. (2012). Cortical thickness in frontotemporal dementia, mild cognitive impairment, and Alzheimer's disease. Journal of Alzheimer's Disease: JAD, 30(4), 857–874. [DOI] [PubMed] [Google Scholar]
- Klostermann, F. , Krugel, L. K. , & Ehlen, F. (2013). Functional roles of the thalamus for language capacities. Frontiers in Systems Neuroscience, 7. 10.3389/fnsys.2013.00032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S. E. , Khazenzon, A. M. , Trujillo, A. J. , Guo, C. C. , Yokoyama, J. S. , Sha, S. J. , Leonel, T. , et al. (2014). Altered network connectivity in frontotemporal dementia with C9orf72 Hexanucleotide repeat expansion. Brain: A Journal of Neurology, 137(Pt 11), 3047–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh, A. R. , & Mišić, B. (2013). Multivariate statistical analyses for neuroimaging data. Annual Review of Psychology, 64, 499–525. [DOI] [PubMed] [Google Scholar]
- McIntosh, A. R. , & Lobaugh, N. J. (2004). Partial least squares analysis of neuroimaging data: Applications and advances. NeuroImage, 23(Suppl 1), S250–S263. [DOI] [PubMed] [Google Scholar]
- Mendez, M. F. , Shapira, J. S. , Woods, R. J. , Licht, E. A. , & Saul, R. E. (2008). Psychotic symptoms in frontotemporal dementia: Prevalence and review. Dementia and Geriatric Cognitive Disorders, 25(3), 206–211. [DOI] [PubMed] [Google Scholar]
- Moore, K. M. , Nicholas, J. , Grossman, M. , McMillan, C. T. , Irwin, D. J. , Massimo, L. , van Deerin, V. M. , et al. (2020). Age at symptom onset and death and disease duration in genetic frontotemporal dementia: An international retrospective cohort study. Lancet Neurology, 19(2), 145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packard, M. G. , & Knowlton, B. J. (2002). Learning and memory functions of the basal ganglia. Annual Review of Neuroscience, 25, 563–593. [DOI] [PubMed] [Google Scholar]
- Palesi, F. , De Rinaldis, A. , Castellazzi, G. , Calamante, F. , Muhlert, N. , Declan, C. , Donald Tournier, J. , Magenes, G. , D'Angelo, E. , Claudia, A. M. , & Gandini Wheeler‐Kingshott . (2017). Contralateral Cortico‐Ponto‐cerebellar pathways reconstruction in humans in vivo: Implications for reciprocal Cerebro‐cerebellar structural connectivity in motor and non‐motor areas. Scientific Reports, 7(1), 12841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, R. , Steele, C. J. , Chen, A. G. X. , Patel, S. , Devenyi, G. A. , Germann, J. , Tardif, C. L. , & Chakravarty, M. M. (2020). Investigating microstructural variation in the human hippocampus using non‐negative matrix factorization. NeuroImage, 207, 116348. [DOI] [PubMed] [Google Scholar]
- Phillips, J. R. , Hewedi, D. H. , Eissa, A. M. , & Moustafa, A. A. (2015). The cerebellum and psychiatric disorders. Frontiers in Public Health, 3, 66. 10.3389/fpubh.2015.00066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratti, E. , Domoto‐Reilly, K. , Caso, C. , Murphy, A. , Brickhouse, M. , Hochberg, D. , Makris, N. , Cudkowicz, M. E. , & Dickerson, B. C. (2021). Regional prefrontal cortical atrophy predicts specific cognitive‐behavioral symptoms in ALS‐FTD. Brain Imaging and Behavior, 15, 2540–2551. 10.1007/s11682-021-00456-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer, J. D. , Warren, J. D. , Modat, M. , Ridgway, G. R. , Douiri, A. , Rossor, M. N. , Ourselin, S. , & Fox, N. C. (2009). Patterns of cortical thinning in the language variants of frontotemporal lobar degeneration. Neurology, 72(18), 1562–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer, J. D. , Nicholas, J. M. , Cash, D. M. , van Swieten, J. , Dopper, E. , Jiskoot, L. , van Minkelen, R. , Rombouts, S. A. , Cardoso, M. J. , Clegg, S. , Espak, M. , Mead, S. , Thomas, D. L. , de Vita, E. , Masellis, M. , Black, S. E. , Freedman, M. , Keren, R. , MacIntosh, B. J. , … Binetti, G. (2015). Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the genetic frontotemporal dementia initiative (GENFI) study: A cross‐sectional analysis. Lancet Neurology, 14(3), 253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santamaría‐García, H. , Baez, S. , Reyes, P. , Santamaría‐García, J. A. , Santacruz‐Escudero, J. M. , Matallana, D. , Arévalo, A. , Sigman, M. , García, A. M. , & Ibáñez, A. (2017). A lesion model of envy and schadenfreude: Legal, deservingness and moral dimensions as revealed by neurodegeneration. Brain: A Journal of Neurology, 140(12), 3357–3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellami, L. , Bocchetta, M. , Masellis, M. , Cash, D. M. , Dick, K. M. , van Swieten, J. , Borroni, B. , Galimberti, D. , Tartaglia, M. C. , Rowe, J. B. , Graff, C. , Tagliavini, F. , Frisoni, G. , Finger, E. , de Mendonça, A. , Sorbi, S. , Warren, J. D. , Rohrer, J. D. , Laforce, R. , & Genetic FTD Initiative, GENFI . (2018). Distinct neuroanatomical correlates of neuropsychiatric symptoms in the three Main forms of genetic frontotemporal dementia in the GENFI cohort. Journal of Alzheimer's Disease: JAD, 65(1), 147–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha, S. J. , Takada, L. T. , Rankin, K. P. , Yokoyama, J. S. , Rutherford, N. J. , Fong, J. C. , Khan, B. , Karydas, A. , Baker, M. C. , DeJesus‐Hernandez, M. , Pribadi, M. , Coppola, G. , Geschwind, D. H. , Rademakers, R. , Lee, S. E. , Seeley, W. , Miller, B. L. , & Boxer, A. L. (2012). Frontotemporal dementia due to C9ORF72 mutations: Clinical and imaging features. Neurology, 79, 1002–1011. 10.1212/wnl.0b013e318268452e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman, S. M. (2016). Thalamus plays a central role in ongoing cortical functioning. Nature Neuroscience, 19(4), 533–541. [DOI] [PubMed] [Google Scholar]
- Sturm, V. E. , Perry, D. C. , Wood, K. , Hua, A. Y. , Alcantar, O. , Datta, S. , Rankin, K. P. , Rosen, H. J. , Miller, B. L. , & Kramer, J. H. (2017). Prosocial deficits in behavioral variant frontotemporal dementia relate to reward network atrophy. Brain and Behavior, 7(10), e00807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavares, T. P. , Mitchell, D. G. V. , Coleman, K. K. , Coleman, B. L. , Shoesmith, C. L. , Butler, C. R. , Santana, I. , Danek, A. , Gerhard, A. , de Mendonca, A. , Borroni, B. , Tartaglia, M. C. , Graff, C. , Galimberti, D. , Tagliavini, F. , Moreno, F. , Frisoni, G. , Rowe, J. B. , Levin, J. , … GENFI Initiative . (2020). Early symptoms in symptomatic and preclinical genetic frontotemporal lobar degeneration. Journal of Neurology, Neurosurgery, and Psychiatry, 91(9), 975–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeighami, Y. , Fereshtehnejad, S.‐M. , Mahsa, D. , Louis Collins, D. , Postuma, R. B. , Mišić, B. , & Dagher, A. (2017). A clinical‐anatomical signature of Parkinson's disease identified with partial least squares and magnetic resonance imaging. NeuroImage, 190, 69–78. 10.1016/j.neuroimage.2017.12.050 [DOI] [PubMed] [Google Scholar]
- Zhang, Y. , Schuff, N. , An‐Tao, D. , Rosen, H. J. , Kramer, J. H. , Gorno‐Tempini, M. L. , Miller, B. L. , & Weiner, M. W. (2009). White matter damage in frontotemporal dementia and Alzheimer's disease measured by diffusion MRI. Brain: A Journal of Neurology, 132(Pt 9), 2579–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1: Supporting information
Appendix
Data Availability Statement
The data that support the findings of this study are available on request via https://www.genfi.org/study/ or by emailing genfi@ucl.ac.uk. The data are not publicly available due to privacy or ethical restrictions.