

SUMMARY
The high genetic diversity of hepatitis C virus (HCV) complicates effective vaccine development. We screened a cohort of 435 HCV-infected individuals and found that 2%–5% demonstrated outstanding HCV-neutralizing activity. From four of these patients, we isolated 310 HCV antibodies, including neutralizing antibodies with exceptional breadth and potency. High neutralizing activity was enabled by the use of the VH1-69 heavy-chain gene segment, somatic mutations within CDRH1, and CDRH2 hydrophobicity. Structural and mutational analyses revealed an important role for mutations replacing the serines at positions 30 and 31, as well as the presence of neutral and hydrophobic residues at the tip of the CDRH3. Based on these characteristics, we computationally created a de novo antibody with a fully synthetic VH1-69 heavy chain that efficiently neutralized multiple HCV genotypes. Our findings provide a deep understanding of the generation of broadly HCV-neutralizing antibodies that can guide the design of effective vaccine candidates.
Graphical Abstract
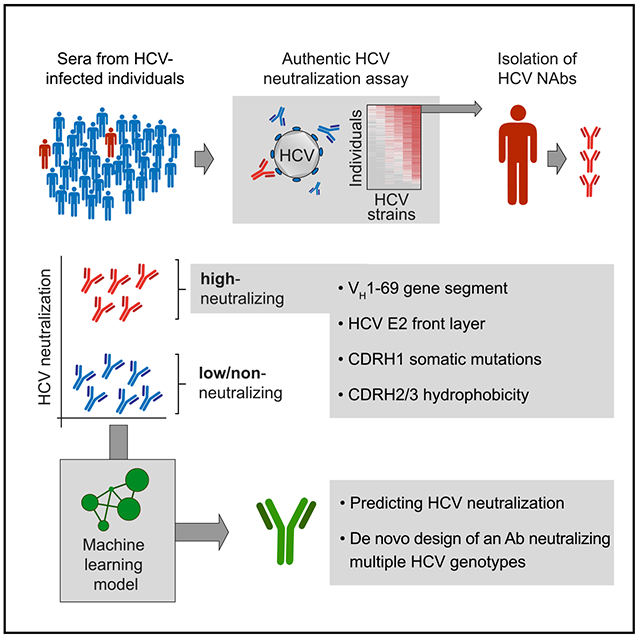
In brief
Broadly neutralizing antibodies (bNAbs) can protect from HCV infection, but little is known about their development and specific characteristics. Weber, Potthoff et al. isolated potent HCV bNAbs from individuals with exceptional HCV antibody responses and determined the genetic requirements for high neutralizing activity. Based on these data, they computationally designed a de novo antibody that efficiently neutralized multiple HCV genotypes.
INTRODUCTION
Hepatitis C virus (HCV) infection can cause progressive liver fibrosis, liver cirrhosis, and hepatocellular carcinoma (Freeman et al., 2001), resulting in almost 400,000 deaths/year (Thomas, 2019). Treatment with direct-acting antivirals (DAAs) has revolutionized HCV therapy, with success rates exceeding 95% (Luna et al., 2019). However, due to a high fraction of missed diagnoses and high treatment costs, less than 10% of HCV-infected individuals worldwide are effectively treated. Moreover, successfully treated individuals are not protected from re-infection (Bailey et al., 2019; Roingeard and Beaumont, 2020; Thomas, 2019). Therefore, the development of an effective HCV vaccine is of critical clinical need.
Neutralizing antibodies (NAbs) can protect from HCV infection in animal models and are therefore of the utmost interest for immunization strategies (Kinchen et al., 2018a). HCV NAbs target two envelope glycoproteins, E1 and E2 (Farci et al., 1996; Osburn et al., 2014). Due to the extraordinary genetic diversity of HCV, with 7 major genotypes that differ in 30% of their amino acid sequence and exceed the diversity of HIV-1 (Bailey et al., 2019), a successful vaccine would have to elicit antibodies with broad neutralizing activity (i.e., broadly neutralizing antibodies, bNAbs). In recent years, several HCV bNAbs were identified, most of which target the CD81 binding site on E2 that is necessary for host cell entry (Bailey et al., 2017; Clayton et al., 2002; Colbert et al., 2019; Law et al., 2008; Merat et al., 2016, 2019). Together with T cells, NAbs play an important role in the spontaneous clearance of HCV infection (Kinchen et al., 2018b; Logvinoff et al., 2004; Osburn et al., 2014; Pestka et al., 2007). In animal models, treatment with HCV bNAbs achieved protection from HCV infection as well as abrogation of established infection (de Jong et al., 2014; Keck et al., 2016; Law et al., 2008; Morin et al., 2012). On a molecular level, HCV bNAbs are characterized by a preference for the use of gene segment VH1-69 (Chen et al., 2019) and a low to average rate of somatic mutations (5%–14% and 1%–9% for the immunoglobulin heavy and light chains, respectively; Bailey et al., 2017). The existence of bNAbs with high germline (GL) identity has raised hopes for a broadly protective HCV vaccine. However, the first HCV vaccine trials have led to mostly strain-specific humoral immune responses both in non-human primates and humans (Bailey et al., 2019; Chen et al., 2020; Choo et al., 1994; Kinchen et al., 2018a; Ströh and Krey, 2020). Therefore, a better understanding of the natural HCV antibody response is required, particularly of antibody features leading to broad neutralization.
Research on bNAbs targeting HIV-1 has benefited from strategies in which patient cohorts were screened for individuals with exceptional serum neutralization (i.e., “elite neutralizers”), followed by single B cell isolation and antibody cloning (Burton et al., 2012; Klein et al., 2013b; Scheid et al., 2009; Schommers et al., 2020). In an analogous approach, we identified HCV elite neutralizers and defined characteristic properties that are critical for mediating broad and potent neutralizing activity. By processing mutational and structural information, we built a machine learning model that was able to predict and to design a de novo E2-specific VH1-69 antibody that efficiently neutralized HCV. Our findings provide a detailed understanding of features that drive HCV-neutralizing activity and support HCV vaccine strategies that aim to induce VH1-69 HCV NAbs (Bailey et al., 2017; Chen et al., 2019, 2021).
RESULTS
HCVcc screening identified 5% of HCV-infected individuals with outstanding neutralizing activity
To identify individuals with broad and potent HCV antibody responses, we collected sera from 435 HCV patients at multiple sites in Germany (Figure 1A). Most of these individuals were chronically infected (>96%), and samples were mainly collected before starting DAA therapy (Figure S1A). The majority of individuals were male (71%) and had a median age of 49 years. Study participants were mainly infected with HCV genotypes 1 (56%) and 3 (21%), with drug use being the predominant risk factor (43%, Figures 1B–1D). Polyclonal immunoglobulin G (pIgG) was isolated from the sera and screened for neutralizing activity using an authentic HCV cell culture (HCVcc) assay (Figure 1E; Koutsoudakis et al., 2006; Wakita et al., 2005). Neutralization was tested against 6 distinct HCVcc strains (Bankwitz et al., 2021) covering 4 major genotypes (1, 2, 4, 5): 1bCon (Con1), 1bJ4 (J4/JFH1), 2a (J6/JFH1), 2b (J8/JFH1), 4a (ED43/JFH1), and 5a (SA13/JFH1). Based on data from immunoglobulin G (IgG) isotype controls, we defined reduction of infection by at least 40% as meaningful virus neutralization (Figure S1B). HCV strains showed different sensitivities to pIgG neutralization. Infection with genotype 5a was inhibited (>40% reduction) by 92% of all samples, whereas infection with genotype 2a was inhibited by only 16% of all samples (Figure 1E). pIgGs that neutralized more resistant strains (2a, 1b con) were also able to neutralize more sensitive strains (4a, 5a), but not vice versa. Ranking of the pIgG samples by average neutralization across all tested genotypes identified 21 of 435 analyzed individuals neutralizing HCV with exceptional breadth and potency (5%; Figure 1E). These so called elite neutralizers exhibited activity against all 6 tested HCVcc strains. Neither the HCV genotype that the individuals were infected with (Figure 1F), nor gender (Figure S1C), or age (Figure S1D) significantly impacted average neutralization. However, pIgGs from individuals who had been infected for more than 20 years showed more robust neutralizing activity than pIgGs from recently infected subjects (p = 0.0028, Figure 1G). pIgGs from individuals co-infected with HIV neutralized HCV less efficiently (p = 0.0042, Figure S1E). Average neutralization was calculated directly from the measured % neutralization values. An alternative calculation method in which we first set values of 40% and below to zero led to almost identical statistics (Figure S5). We concluded that the antibody response against HCV varies substantially between infected individuals, with a small fraction of individuals having elite HCV-neutralizing activity.
Figure 1. HCVcc screening identified 5% of HCV-infected individuals with outstanding “neutralizing activity”.
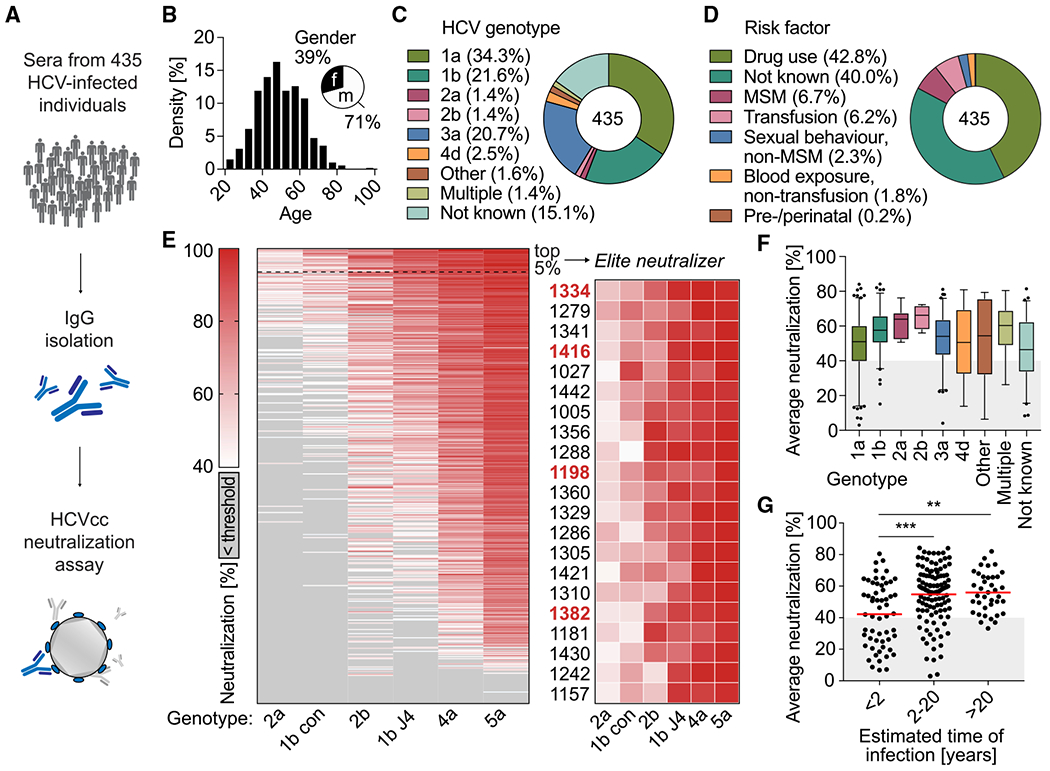
(A) pIgG from 435 HCV-infected individuals was screened for neutralizing activity in an HCVcc assay.
(B) Age and gender distribution of HCV-infected individuals.
(C) HCV genotypes that individuals were infected with.
(D) Self-reported most likely risk factor for HCV infection.
(E) Left panel: screening of pIgG for neutralizing activity against authentic HCV virions. Huh 7.5 cells were infected with 6 different HCVcc strains (columns) carrying a Renilla luciferase reporter gene, in the presence of 300 μg/mL pIgG from HCV-infected individuals (rows). HCV neutralization was determined by reduction of luciferase activity in Huh 7.5 cells relative to cells infected with virus alone. Sorted by row means (average neutralization). Medians of triplicate measurements. Screening was performed once. Right panel: enlarged heatmap showing IDs of individuals with average neutralization in the top 5% range and neutralization of all HCVcc strains. Red IDs: individuals who later donated peripheral blood mononuclear cells.
(F) Influence of the HCV genotype on average neutralization of pIgG samples. Lines, boxes, and whiskers indicate medians, 25%–75%, and 5%–95% percentiles, respectively.
(G) Influence of estimated time of HCV infection on average neutralization. Data are plotted for 209 individuals. No data on time of infection were available for 226 individuals. Lines indicate means.**p ≤ 0.01, ***p ≤ 0.001; two-tailed Mann-Whitney test with Bonferroni correction. See also Figure S1.
HCV elite neutralizer B cell responses are polyclonal and VH1-69 biased
In order to examine the antibody response in HCV elite neutralizers more closely, we collected peripheral blood from 4 selected individuals (Table S1) and isolated single HCV-reactive B cells by flow cytometry. 0.1%–0.8% of IgG+ B cells reacted with fluorescently labeled HCV E2 core protein (Kong et al., 2013) of genotype 2b. In total, 1,887 E2-reactive B cells were isolated by flow cytometry (Figures 2A and S2A; Table S2), of which 1,519 productive heavy-chain sequences were obtained that passed quality control (346–457 per individual). Sequence analysis revealed a polyclonal response, mainly of the IgG1 subclass, with 32%–83% clonally related sequences per individual and not more than 6–15 sequences belonging to the same B cell clone (mean clone size: 3; Figures 2B and S2B; Table S2).
Figure 2. HCV elite neutralizer B cell repertoires are polyclonal and VH1-69 biased.
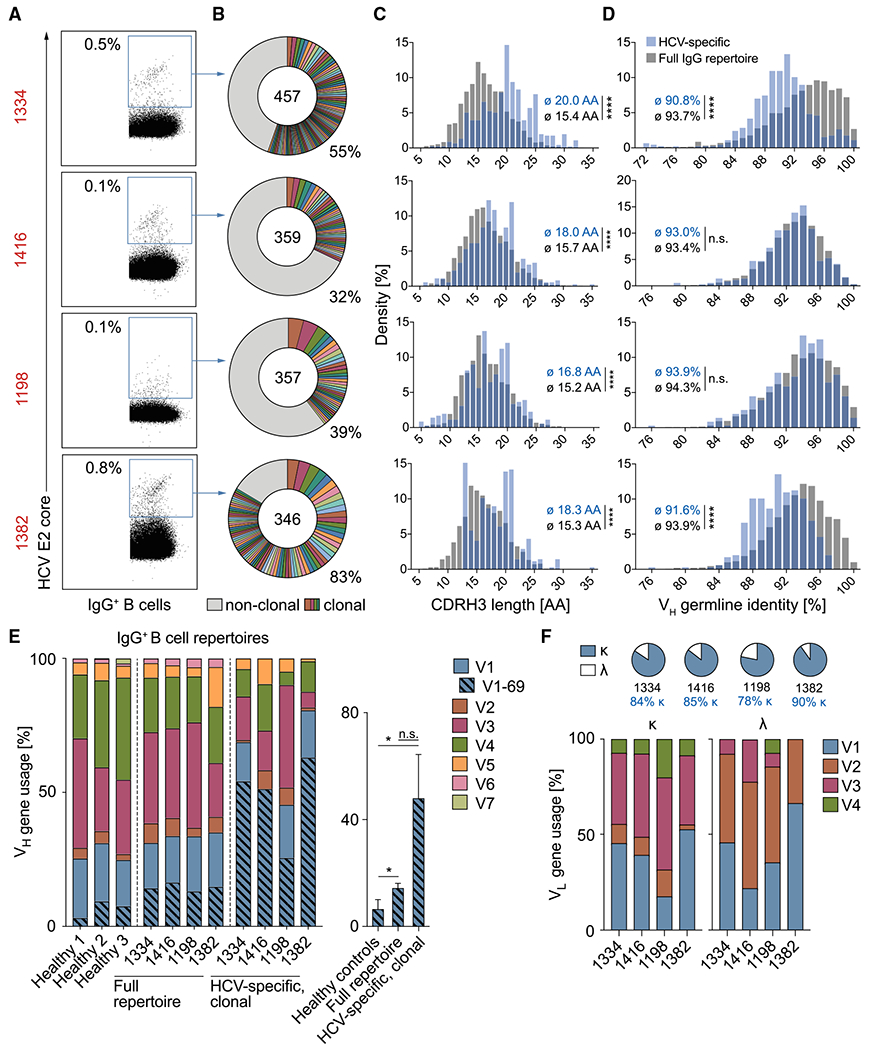
(A) Exemplary dot plots of IgG+ B cell analysis (see also Figure S2A). Numbers in red are IDs of HCV-infected individuals (see also Table S1). Numbers in plots indicate percent frequencies of E2 core-reactive B cells within IgG+ B cells (see also Table S2). Sorting was performed once.
(B) Clonal relationship of E2 core-reactive B cells. Clones are shown in different colors. Numbers of productive heavy-chain sequences are depicted in the chart center. Clone sizes are proportional to the total number of productive heavy-chains per clone.
(C–E) Frequencies of CDR3 length (C), V gene germline identity (D), as well as V gene segments (E) of productive heavy-chain sequences obtained from E2 core-reactive B cells compared with NGS reference data from full IgG+ B cell repertoires of the same individuals. Reference data from three randomly selected healthy donors in (E) are also included in a previous publication (Kreer et al., 2020b). Right panel of (E): quantification of VH1-69 heavy-chain frequencies. Means ± SDs are plotted. Not significant (n.s.), p > 0.05, *p ≤ 0.05, **p ≤ 0.01, ****p ≤ 0.0001; two-tailed Mann-Whitney test with Bonferroni correction (C and D) or two-tailed t test with Bonferroni correction (E).
(F) Characteristics of amplified light chains.
Compared with the general IgG+ B cell repertoires of the same 4 individuals obtained by next-generation sequencing (NGS), HCV-reactive B cells carried longer heavy-chain complementarity-determining regions 3 (CDRH3s; mean difference of 3 amino acids, p < 0.0001 for each individual; Figures 2C and S2C) and had slightly more VH gene somatic mutations (difference in mean GL identity of 0.4%–2.9%, p = 0.079–<0.0001 for each individual; Figures 2D and S2D). HCV-reactive B cells utilized different VH gene segments, however, VH1-69 was 3.3-fold overrepresented in clonal HCV-specific B cells compared with full IgG+ B cell repertoires of the same individuals (p = 0.064) and 7.3-fold overrepresented compared with IgG+ B cell repertoires of healthy donors (p = 0.022; Figure 2E). From 310 corresponding light chains, 84% were κ, utilizing mainly VK1 and VK3 family gene segments (Figure 2F) with moderate somatic mutation (94.8% mean V gene GL identity; Figure S2D). Taken together, E2 core-reactive B cell repertoires from HCV elite neutralizers are polyclonal and show no gross deviation from the normal human B cell repertoire except for a bias toward use of the VH gene segment 1-69 and slightly longer CDRH3s.
Potent HCV neutralization requires VH1-69 and E2 front-layer targeting
To generate a panel of monoclonal antibodies from HCV elite neutralizers, we cloned 310 heavy- and light-chain pairs into expression vectors and produced them as IgG1 isotypes in HEK293-6E cells (Table S3). 218 antibodies bound to E2 core protein in ELISA, whereas no binding to E2 could be detected among 92 antibodies (Figure S3A). Binding affinities were similar to those of the HCV bNAb AR3C (Law et al., 2008), which was previously identified by phage display (EC50 in the range of 0.1 μg/mL; Figures S3A and S3B). ELISA binding was detected for 94% of VH1–69 antibodies and for less than half (49%) of non-VH1-69 antibodies (Figure S3C). No binding was detected for VH1–69 control antibodies reactive against HIV-1, ruling out unspecific binding of VH1–69 antibodies with the HCV E2 core protein (Figure S3D).
To identify HCV antibodies with high neutralizing breadth and potency, we screened all antibodies for neutralization of authentic HCV virions in an HCVcc assay. Of 310 tested antibodies, 39 (13%; range: 8.5%–23% per individual) neutralized all 6 HCVcc strains (>40% inhibition at 50 μg/mL; Figure 3A). Based on average neutralization values, the antibodies could be divided into two groups: (1) potent NAbs with an average neutralization above 40% and (2) low/non-NAbs with average neutralization below 40% (98 and 212 antibodies, respectively; Figures 3A and 3B). The two groups markedly differed by VH gene usage: although only 26% of low/non-NAbs utilized VH1-69, 90% of potent neutralizers were encoded by VH1-69 (Figure 3C). Of note, use of VH1-69 is a feature of previously described HCV bNAbs binding to a structural component of E2 referred to as the front layer (Ströh and Krey, 2020). The E2 front layer consists mainly of a β-strand and a short α-helix. Together with the CD81-binding loop, it forms the hydrophobic CD81-binding site of E2 (Kong et al., 2013; Tzarum et al., 2018). We performed an ELISA using a modified E2 protein in which selected amino acids of the front layer were replaced by alanine residues or, as a control, the same protein with an intact front layer. As a result, 96.6% of potent NAbs depended on an intact front layer, whereas binding of low/non-NAbs was in most cases front-layer independent (96.5%; Figure 3D). We conclude that the presence of both the usage of the VH1-69 gene segment and E2 front-layer dependency is a hallmark of potent HCV-neutralizing activity among human antibodies. VH1-69 E2 antibodies that are front-layer independent show no or only low neutralizing activity (Figure 3E).
Figure 3. B cells of HCV elite neutralizers encode highly broad and potent bNAbs.
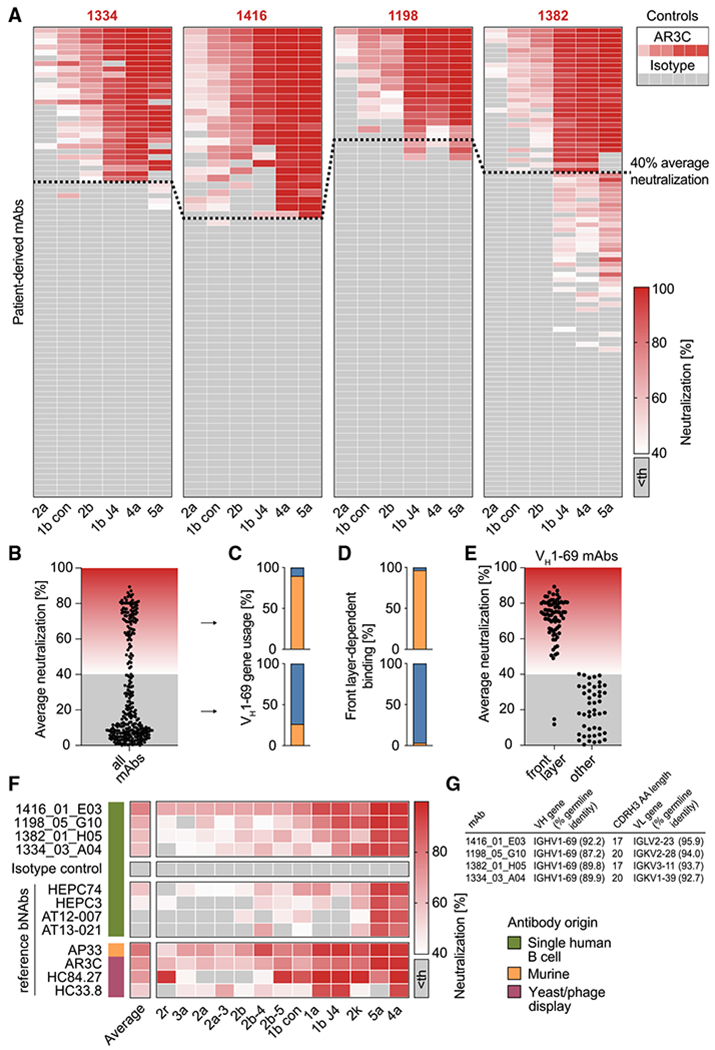
(A) 310 antibodies isolated from HCV elite neutralizers were screened at a concentration of 50 μg/mL for neutralizing activity against HCV as in Figure 1E. Medians of triplicate measurements. Screening was performed once. Antibodies with an average neutralization above 40% (indicated by a dashed line) were defined as potent neutralizers. Th, threshold.
(B) Distribution of average neutralization for all patient-derived antibodies from (A).
(C) Percentage of antibodies utilizing the VH gene segment 1-69 among potent (upper panel) or low/non-neutralizers (lower panel).
(D) Percentage of antibodies binding the E2 protein in a front-layer-dependent manner within all antibodies binding the E2 protein. Determined by ELISA with either wildtype or front layer knockout E2 protein of strain 1a157.
(E) Average neutralization as in (B) for VH1-69 antibodies that bind E2 in either a front layer-dependent or a front layer-independent way.
(F) Four top hits from the neutralization screen in (A) along with a set of reference HCV-specific antibodies were re-tested for neutralizing activity against an extended panel of 13 HCVcc strains, including those used for the first screen. Medians of triplicate measurements, representative of 3 independent experiments.
(G) Genetic properties of the 4 isolated HCV bNAbs assayed in (F). Germline identity is indicated on nucleotide level.
B cells of HCV elite neutralizers encode highly broad and potent bNAbs
From 310 obtained antibodies, we identified 4 antibodies (1416_01_E03, 1198_05_G10, 1382_01_H05, 1334_03_A04) with exceptional neutralizing activity against 6/6 tested HCVcc strains (Figures 3F and 3G). We tested these bNAbs against an extended HCVcc panel and compared their neutralization potencies with 8 reference HCV-specific bNAbs (Figure 3F). The extended HCVcc panel covered 13 strains representing the major genotypes 1–5 and 9 distinct subtypes (Bankwitz et al., 2021). Four of the reference bNAbs originated from single human B cells with the original pairing of heavy- and light chains (HEPC74, HEPC3; Bailey et al., 2017; and AT12-007, AT-13-021; Merat et al., 2016), one is of murine origin (AP33; Clayton et al., 2002), and 3 were identified by screening combinatorial display libraries (AR3C, HC84.27, HC33.8; Keck et al., 2012, 2013). 1416_01_E03 showed the best activity by neutralizing all HCV strains (13/13) with 77% average neutralization (range: 65%–98%; 50 μg/mL) and substantially exceeded all human reference antibodies (Figure 3F; average neutralization and breadth: HEPC74; 57% (12/13), HEPC3 45% (8/13), AT12-007 39% (6/13), AT13-021 35% (5/13)). 1198_05_G10, 1382_01_H05, and 1334_03_A04 inhibited infection of 12, 13, and 12 HCVcc strains by average 65%, 60%, and 59%, respectively. Only phage-display-derived bNAb AR3C, as well as AP33, a bNAb of murine origin, showed similarly high activity. Some VH1-69 bNAbs against HIV-1 (e.g., 4E10) are polyreactive, meaning that they also react with multiple unrelated antigens (Haynes et al., 2005a), but the identified HCV bNAbs did not show any signs of auto- or polyreactivity (Figures S3E and S3F). Thus, the identification of HCV elite neutralizers enabled us to identify bNAbs with extraordinary potency and breadth, targeting all HCV genotypes and subtypes tested.
Crystal structures of 1198_05_G10-E2ecto and 1382_01_H05-E2ecto complexes reveal a distinct binding mode of human HCV bNAbs
To verify the epitope specificity of bNAbs 1198_05_G10 and 1382_01_H05, we determined their crystal structures in complex with the E2 ectodomain (E2ecto; Figure 4; Table S4). In line with our ELISA data, both antibodies bind to the conserved epitope in the E2 front layer (Figure 4A). Although superposition of the two complexes revealed that 1198_05_G10 and 1382_01_H05 bind E2ecto in slightly different orientations (Figure 4A), CDRH3 loops of both bNAbs contacted the same antigenic region on the E2 surface (Figure 4B). As found for other front-layer-specific bNAbs, both antibodies contact E2ecto primarily by CDRH3 residues, burying 462 or 532 Å2 (42% or 48% of the total Fab buried surface area, BSA) for 1198_05_G10 and 1382_01_H05, respectively (Figure 4C; Table S5). However, in contrast to other front-layer-specific bNAbs in which the light chains do not play a role in the interaction with E2 envelope (Flyak et al., 2018; Kong et al., 2013; Tzarum et al., 2019), 1198_05_G10 and 1382_01_H05 light chains accounted for 25% and 21% (280 or 232 Å2) of the total BSA (Figure 4C), respectively.
Figure 4. Crystal structures of 1198_05_G10-E2ecto and 1382_01_H05-E2ecto complexes reveal a distinct binding mode of human HCV bNAbs.
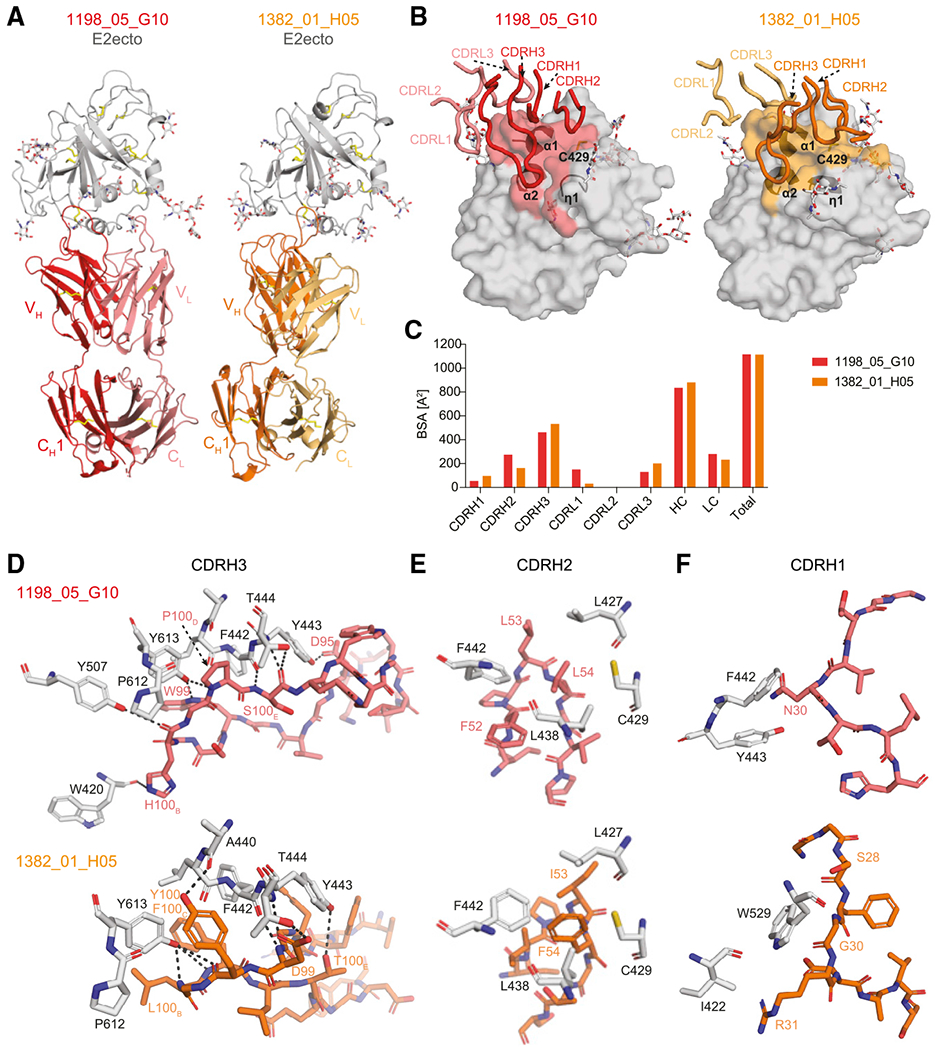
(A) Crystal structures of the 1198_05_G10-E2ecto and 1382_01_H05-E2ecto complexes. E2ecto is shown as a cartoon representation with N-glycans highlighted as sticks and disulfide bonds shown as yellow sticks. Fabs are shown as cartoons with 1198_05_G10-HC colored in red; 1198_05_G10-LC—light red, 1382_01_H05-HC—orange, and 1382_01_H05-LC—yellow. The structures were superimposed on E2 proteins.
(B) Comparison of 1198_05_G10 and 1382_01_H05 CDR loop positions and corresponding E2 epitopes. Epitopes on the E2 front-layer surface were defined as residues in E2 containing an atom within 4 Å of the bound Fab. The positions of three α1-helices and Cys429 residue are indicated.
(C) Comparison of buried surface areas (BSAs).
(D) Interactions of CDRH3 loops with E2ecto. Potential hydrogen bonds are shown as black dashed lines and residues at the interface are indicated. Hydrogen bonds for the 3.2 Å 1382_01 _H05-E2ecto structure should be considered tentative.
(E) Interactions of Fab CDRH2 loops with E2ecto. Residues at the interface are indicated.
(F) Interactions of Fab CDRH1 loops with E2ecto. Residues at the interface are indicated.
Although 1198_05_G10, 1382_01_H05, as well as other front-layer-specific VH1-69 antibodies (e.g., AR3X and HEPC74) show similar binding footprints on the E2 surface (Figure 5; Flyak et al., 2018, 2020), we observed different approach angles of the HCV bNAbs 1198_05_G10 and 1382_01_H05. Both antibodies CDRH3s’ contact the hydrophobic groove formed by three α-helices: short η1-helix in the front-layer N terminus (residues 421–425), helix α1 (residues 438–443) C-terminus, and back layer α2 helix (613–617; Figures 4B and 4D; Table S5). All eight putative hydrogen bonds with the E2 glycoprotein are formed exclusively by 1198_05_G10 and 1382_01_H05 CDRH3 loops, with at least half of hydrogen bonds centered around two E2 residues: Thr444 (C-terminal end of α1-helix) and Tyr613 (α2 helix in the back layer of E2; Figure 4D).
Figure 5. 1198_05_G10 and 1382_01_H05 share a similar binding mode with vaccine-induced bNAbs.
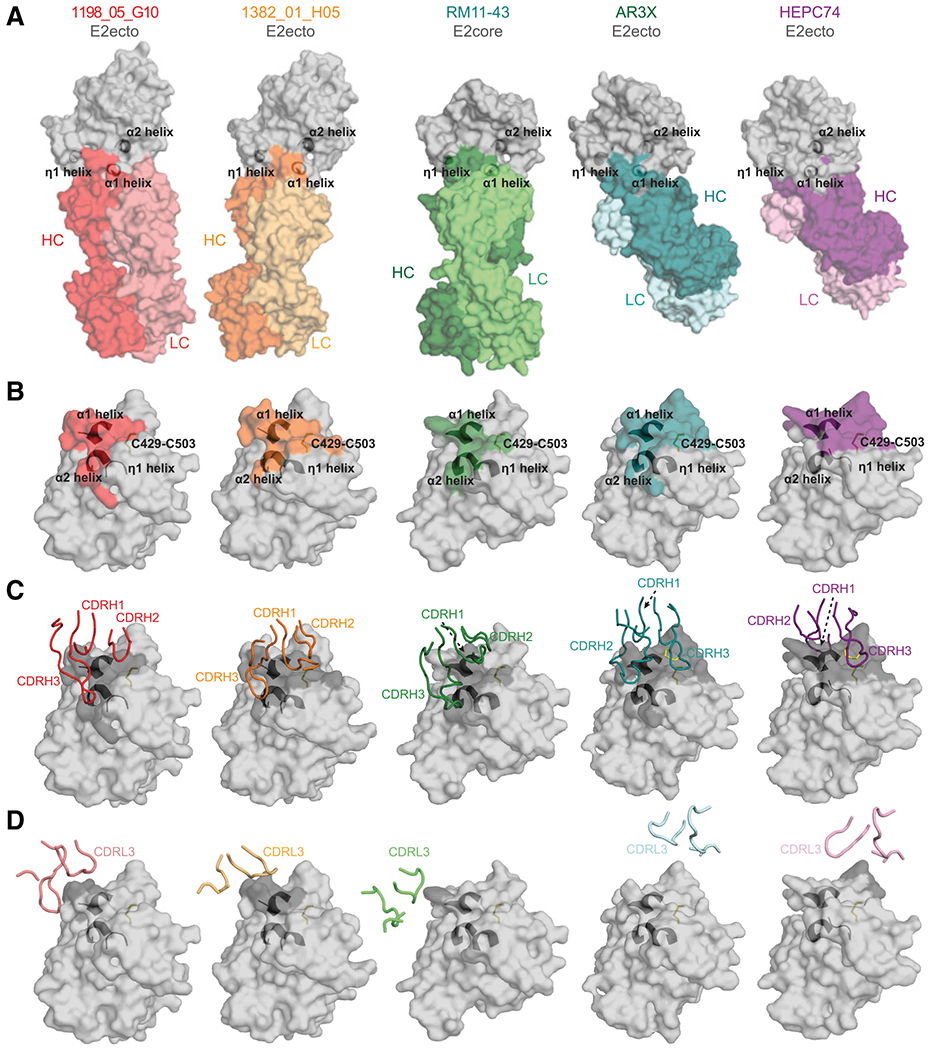
(A) Surface representations of 1198_05_G10-E2, 1382_01_H05-E2, and other bNAb-E2 crystal structures. The location of α1-, α2-, and η1-helices in E2 are indicated by black cartoon representations. The structures were superimposed on E2 proteins.
(B) Comparison of 1198_05_G10, 1382_01_H05, RM11-43, AR3X, and HEPC74 epitopes. Epitopes on the E2 front layer (surface representation) were defined as residues in E2 containing an atom within 4 Å of the bound Fab. The location of α1-, α2-, and η1-helices in E2 are indicated by black cartoon representations and the C429–C503 disulfide bond is indicated by yellow sticks.
(C) CDRH loops mapped onto the E2 surface. HC interacting residues are colored in gray on the E2 surface.
(D) CDRL loops mapped onto the E2 surface. LC interacting residues are colored in gray on the E2 surface.
A unique feature of VH1-69 antibodies is the presence of hydrophobic residues at the tip of the CDRH2 loop that facilitates interactions with hydrophobic epitopes. In contrast to some HCV bNAbs that utilize their CDRH2 tips to interact with a hydrophobic groove formed by three α-helixes (AR3C, HEPC3, HEPC74; Flyak et al., 2018; Kong et al., 2013; Tzarum et al., 2019), the hydrophobic residues of 1198_05_G10 and 1382_01_H05 CDRH2s interact with the E2 front-layer near Cys429 and the α1-helix (Figures 4B and 4E), a region of E2, which is recognized by CDRH3 residues of previously described front-layer-specific HCV bNAbs. Differently positioned CDRH1 loops for both bNAbs play a minor role in the E2-Fab-binding interface by making contacts with the E2 front layer (both 1198_05_G10 and 1382_01_H05) and the CD81-binding loop (only 1382_01_H05; Figure 4F). In summary, 1198_05_G10 and 1382_01_H05 bind the E2 front layer with approach angles and CDR interactions distinct from previously identified HCV bNAbs.
1198_05_G10 and 1382_01_H05 share a similar binding mode with vaccine-induced bNAbs
Recently, two bNAbs (RM2-01 and RM11-43) were isolated from macaques immunized with an HCV vaccine candidate (Chiron recombinant E1E2 complex; Chen et al. 2021). Both macaque antibodies utilized the VH1.36 gene segment, a macaque ortholog to human VH1-69 (94.8% nucleotide identity). Structural comparison with known human HCV bNAbs indicated that RM2-01 and RM11-43 recognized overlapping epitopes. However, both antibodies displayed a unique binding mode in which the CDRH3 loops reach out to the E2 back layer (Chen et al., 2021), a feature not yet described for human HCV bNAbs. Interestingly, structural analyses of 1198_05_G10 and 1382_01_H05 revealed the same binding mode as for RM2-01 and RM11-43 (Figure 5A; Chen et al. 2021). All four antibodies showed a similar binding footprint (Figure 5B; Chen et al., 2021), utilizing their CDRH3s to bind the hydrophobic pocket formed between three E2 α-helixes. Moreover, their hydrophobic CDRH2s bind to the E2 front layer near cysteine 429 (Cys429;Figures 4B and 5C) with a minimal impact of their light chains on the E2-Fab-binding interface (Figure 5D). The binding mode of 1198_05_G10 and 1382_01_H05 is distinct from other human bNAbs that predominantly utilize a CDRH3 ß-hairpin with or without disulfide motif to recognize front-layer epitopes near Cys429 (HEPC74- and AR3C-like bNAbs; Flyak et al., 2018; Kong et al., 2013; Tzarum et al., 2019). Notably, the human bNAb AR3X isolated from a chronically infected individual (Flyak et al., 2020) mimics the CDRH3-interaction of 1198_05_G10 and 1382_01_H05 with E2 by utilizing an ultralong insertion in its CDRH2 to reach out to the α2-helix in the back layer (Figure 5). In summary, the HCV bNAbs 1198_05_G10 and 1382_01_H05 display a distinct binding mode similar to bNAbs induced by an HCV vaccine in macaques. We conclude that immunization of human subjects with an E1E2 complex may induce bNAbs resembling those isolated from elite neutralizers.
VH1-69 CDRH1 somatic mutations are critical for broad HCV neutralization
We then investigated the genetic characteristics of VH1-69 antibodies from HCV elite neutralizers. Although various mutations have been described at an individual HCV antibody level (Bailey et al., 2017; Chen et al., 2019; Flyak et al., 2018; Tzarum et al., 2019), we investigated the impact of somatic mutations in a sample of 12 VH1-69 bNAbs (Figure 6A). To this end, we reverted the heavy-chain of all 12 VH1-69 antibodies to GL. Reverted antibodies showed a marked reduction in neutralizing potency and breadth, dropping from 87% (average breadth of all 12 HCV bNAbs; range: 67%–100%) to 25% (range: 0%–67%). Thus, all VH1-69 antibodies tested require VH somatic mutations for broad HCV-neutralizing activity. To narrow down whether mutations in a defined region are of particular importance, we reverted the CDRH1, CDRH2, and heavy-chain framework region 3 (FWRH3) separately. For none of the antibodies, reversions of a single region (CDRH1, CDRH2, or FWRH3) were sufficient to abolish HCV-neutralizing activity. However, reverting the CDRH1 significantly reduced HCV neutralization (60%–87% breadth; Figure 6A). Next, we reverted single somatic mutations within the CDRH1. Although the majority of changes showed no effect, 4 out of 8 antibodies were highly sensitive to reverting position 30 or 31 back to the GL-encoded serine. These antibodies lost neutralizing activity against one to several HCVcc strains by up to 58%, 78%, 91%, and 94% (Figures 6A and 6B). In contrast, re-insertion of single mutations into the VH GL-reverted antibodies did not substantially restore their HCV-neutralizing capacity (Figure S4A). However, re-insertion of all CDRH1 somatic mutations at once led to a significant improvement in neutralizing activity (57% instead of 25% of HCVcc strains neutralized; Figures 6C and S4B). Thus, distinct somatic mutations within the VH1-69 gene, particularly within the CDRH1, facilitate broad HCV neutralization.
Figure 6. VH1-69 CDRH1 somatic mutations are critical for broad HCV neutralization.
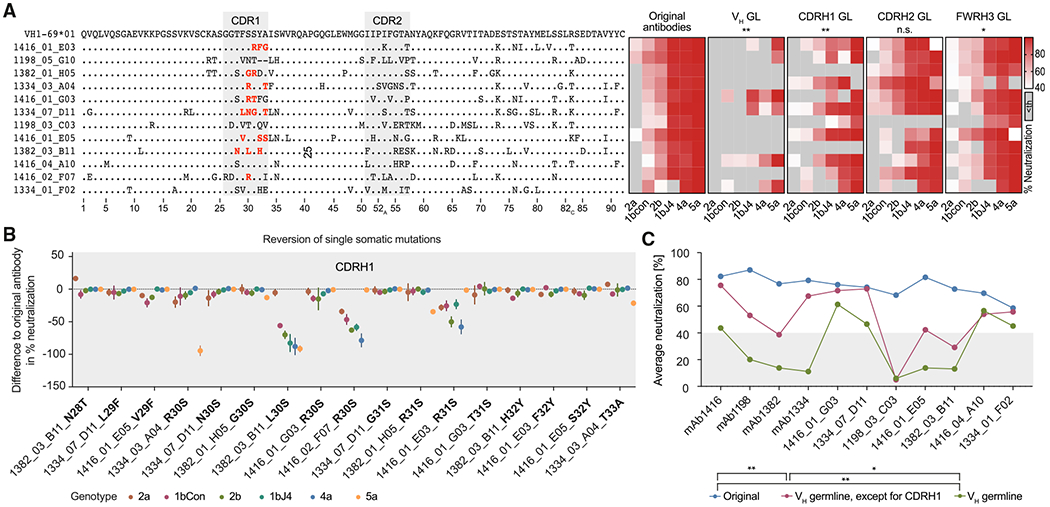
(A) Left panel: alignment of amino acid sequences of 12 HCV antibodies to VH1-69*01 (K73 is encoded by the VH1-69*06 germline allele and therefore is not necessarily a somatic mutation). Right panel: effect of partial germline-reversion on HCVcc neutralization at an antibody concentration of 50 μg/mL. Medians of triplicate measurements. Th, threshold.
(B) Effect of single VH1-69 amino acid germline-reversions on HCVcc neutralization. The reverted mutations are also highlighted in (A) in red. Means ± SD of triplicate measurements.
(C) HCVcc average neutralization (6-virus panel) by antibodies with VH gene segments reverted to germline, except for the CDRH1.
Medians of triplicate measurements. Representative of 3 (A) or 2 (B and C) independent experiments. Not significant (n.s.), *p ≤ 0.05, **p ≤ 0.01; Wilcoxon matched-pairs signed rank test with Bonferroni correction. Average neutralization values across all 6 tested HCVcc strains for each mutated antibody were compared pairwise with average neutralization values of corresponding original antibodies (A) or among each other (C). GL, germline. See also Figure S4.
Potent HCV-neutralizing activity requires a pattern of genetic features and can be predicted
Efficient neutralization of HCV was only achieved by the fraction of E2 core-reactive VH1-69 antibodies targeting the E2 front layer (Figure 3). Therefore, we asked whether common sequence features separate antibodies into potent and non-/low HCV neutralizers. To this end, we used a machine learning approach. First, amino acid sequences of the VH gene segments of all produced VH1–69 antibodies were assigned to the two groups of potent (n = 88) and low/non-neutralizers (n = 56; Figure 3E). This dataset was used to train a support vector machine that provided a classification on the basis of VH amino acid sequences with an average accuracy of 78% as determined by 10-fold cross-validation. The model’s accuracy could be further increased to 87% by considering full VHDHJH amino acid sequences. In both models, distinct somatic mutations within the VH1-69 gene segment contributed to classify high neutralizer. Consistent with our experimental data, somatic mutations within the CDRH1 played an important role, in particular mutations changing the serines at positions 30 and 31 (Figure 7A). Interestingly, there were also somatic mutations determining non-/low neutralizers, for example affecting the CDRH2 germline amino acids F54 and A57 (Figure 7A). Although beneficial mutations, such as S30R or S31L, were associated with higher average neutralization, unfavorable mutations, such as F54S or A57E, were significantly associated with poor average neutralization (Figure 7B). Moreover, in the VHDHJH-based model, CDRH3 loops with neutral or hydrophobic amino acids at the tip contributed to the classification as a potent neutralizer, whereas hydrophilic amino acids in the CDRH3 tip contributed to the classification as a non-/low neutralizer (Figure 7A). This is in line with our structural data showing that CDRH3s of potent neutralizers contact a hydrophobic groove in the E2 protein.
Figure 7. Potent HCV-neutralizing activity requires a pattern of genetic features and can be predicted.
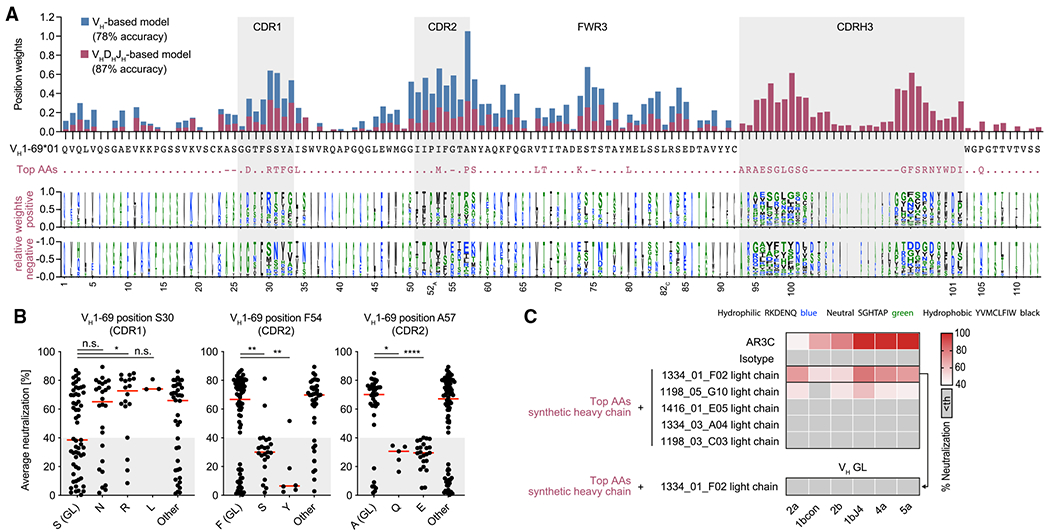
(A) Top panel: total weights of VH1-69-amino-acid positions contributing to the classification as an efficient neutralizer in VH-based or VHDHJH-based machine learning models. The VH1-69*01 germline amino acids are shown in black and the most impactful amino acids for the VHDHJH-based model (top AAs) are shown in violet. Bottom panel: logo plot with letter heights representing relative weights of amino acids at each position for positive or negative classification of the encoded antibodies as efficient neutralizers. Letter widths represent absolute weights. Insertions are omitted for clarity.
(B) Average neutralization values of antibodies with indicated amino acid sequence features in the HCVcc screen shown in Figure 3A. Lines indicate medians.
(C) HCVcc neutralization by antibodies with a synthetic heavy-chain amino acid sequence as depicted in (A) in violet. Medians of triplicate measurements. Representative of 2 independent experiments. Th, threshold.
Not significant (n.s.), p > 0.05, *p ≤ 0.05, **p ≤ 0.01, ****p ≤ 0.0001; two-tailed Mann-Whitney test with Bonferroni correction. GL, germline.
In order to validate model-based predictions for potent HCV-neutralizing activity, we derived a fully synthetic heavy-chain sequence where each position was filled with the amino acid having the strongest weight for the classification as a potent HCV NAb (Figure 7A). This de novo sequence carried 17 somatic mutations and a CDRH3 that had less than 50% identity with any of the CDRH3s of the 1,519 sorted and analyzed HCV-reactive B cells. Pairing this sequence with different light chains from HCV NAbs resulted in identifying a de novo broadly HCV NAb (6/6 tested HCVcc strains, Figure 7C). Like naturally occurring HCV antibodies, the synthetic heavy-chain antibody lost its neutralizing capacity when somatic mutations in the VH gene segment were reverted to GL (Figure 7C), demonstrating the importance of distinct VH1-69 somatic mutations for high HCV-neutralizing activity. We conclude that distinct mutational patterns exist that predict high HCV-neutralizing activity for VH1-69 antibodies. This has direct consequences to inform vaccine-mediated strategies to induce HCV NAbs.
DISCUSSION
Despite the development of efficient treatment with DAAs, HCV infection remains a major public health challenge. Most likely, HCV will not be brought under global control until a protective vaccine is developed. The genetic diversity of HCV has posed a problem for vaccine development. However, HCV NAbs can protect from HCV infection in animal models and a better understanding of their characteristics and mode of induction is urgently needed (Bailey et al., 2019; Kinchen et al., 2018a).
Previously, human bNAbs against HCV were isolated from single individuals using combinatorial display libraries or B cell immortalization (Bailey et al., 2017; Clayton et al., 2002; Colbert et al., 2019; Law et al., 2008; Merat et al., 2016, 2019). More recently, use of large-scale donor screening, as well as the development of single B cell isolation and cloning methods, has led to the identification of numerous very broad and potent human antibodies targeting infectious pathogens (Burton et al., 2012; Ehrhardt et al., 2019; Gieselmann et al., 2021; Kinchen et al., 2018b; Klein et al., 2013a; Kreer et al., 2020b; Liu et al., 2020; Pinto et al., 2020; Scheid et al., 2009; Schommers et al., 2020; Suryadevara et al., 2021). Building on these experiences, we screened HCV-infected individuals and identified elite neutralizers from which we isolated single E2 core-specific B cells and produced a large panel of HCV antibodies. This strategy led us to identify exceptionally broad NAbs, including bNAb 1416_01_E03, which is, to the best of our knowledge, the broadest naturally occurring HCV NAb isolated so far.
We used authentic HCV virions (HCVcc) instead of the more widely used HCV pseudoparticle (HCVpp) assay to determine antibody-mediated neutralization. Authentic HCV virions have a unique structure: they incorporate lipoproteins to form lipo-viro-particles. This impacts host cell entry and might mediate immune evasion (Perin et al., 2016; Sainz et al., 2012; Wrensch et al., 2018). Although there is a correlation between HCVpp and HCVcc neutralization (Kinchen and Bailey, 2018), the differences between HCVpp and HCVcc may impact virus-antibody interplay. The HCVcc assay is thus a more physiological approach (Bankwitz et al., 2021).
Among all the isolated HCV antibodies, VH1-69 was the most prevalent gene segment. VH1-69 antibodies could be divided into two groups: 61% (n = 88) that bound the front layer and neutralized HCV at high or very high activities and 39% (n = 56) that bound the E2 core protein elsewhere and carried no or only low HCV-neutralizing activity. This substantially extends previous observations in which E2-binding VH1-69 antibodies were classified as potent neutralizing in general (Bailey et al., 2017; Chen et al., 2020; Tzarum et al., 2019). Interestingly, HCV E2-reactive VH1–69 NAbs had characteristic sequence properties that distinguish them from low/non-neutralizing E2-reactive VH1–69 antibodies found in the same patients. These included CDRH1 mutations, such as S30R or S31L, as well as CDRH3 hydrophobicity. In addition, other mutations, such as F54S in the CDRH2, were associated with low/non-neutralizing activity. Crystal structure analyses of the two HCV bNAbs 1198_05_G10 and 1382_01_H05 revealed how these features are linked to efficient HCV neutralization. Both CDRH3 and CDRH2 make hydrophobic contacts with E2. Notably, whereas CDRH1 plays a minor role in direct E2 interaction, somatic mutations, such as serine replacement at position 30 or 31, are likely to avoid clashes between CDRH1 and the CD81-binding loop (e.g., by W529) of E2. Based on these findings, high versus low/non-neutralizing activity of VH1-69 antibodies could be predicted with 87% accuracy by using heavy-chain sequence data in a machine learning model. Notably, we were able to computationally derive a fully synthetic heavy-chain that could be used to produce an HCV cross-NAb, demonstrating the existence of a mutational pattern driving HCV neutralization.
Identifying genetic features and distinct somatic mutations is critical for developing effective vaccine design strategies. Although we demonstrated that distinct CDRH1 mutations are critical for HCV-neutralizing activity, a combination of mutations is required in VH1-69 antibodies to achieve broad and potent HCV neutralization. A previously described human-origin HCV bNAb targeting the E2 front layer, HEPC3, also relies on a combination of multiple VH1-69 somatic mutations, including critical mutations within the CDRH1 and dispensable mutations within the CDRH2 (Bailey et al., 2017). The previously identified E2 front-layer-specific VH1-69 bNAbs HEPC3, HEPC74, and AR3C carry CDRH3s of 17-, 18-, and 18-amino-acid length, respectively. Antibodies with exceptional neutralizing activity that we isolated from HCV elite neutralizers carried CDRH3s of similar lengths (17–20 amino acids). There are also VH1-69 bNAbs against HIV-1, such as 4E10, that carry a similarly long hydrophobic CDRH3 (18 amino acids for 4E10; Cardoso et al., 2005; Chen et al., 2019; Irimia et al., 2016). Interestingly, 4E10 and some other HIV bNAbs using VH1-69 are polyreactive. This can complicate the induction of such antibodies because they are more likely to be removed during B cell development (Haynes et al., 2005a, 2005b; Kelsoe and Haynes, 2017). In contrast, the HCV bNAbs isolated in this study did not show any signs of auto- or polyreactivity.
Our findings lead to the question of whether HCV bNAbs like those isolated from elite neutralizers can be induced by a future vaccine. Interestingly, structural analyses of the isolated antibodies demonstrated that they bind the E2 protein in a similar way to antibodies elicited in macaques by immunization with a recombinant E1E2 complex (Chen et al., 2021). Moreover, these macaque antibodies utilized a homolog of VH1-69, with critical CDRH1 somatic mutations, including mutation of germline-encoded S31, analogous to HCV bNAbs that we isolated in our study. Thus, broadly protective HCV antibodies are not only characterized by VH1-69 usage but also require a characteristic CDRH mutational pattern that seems to be inducible by an HCV vaccine. Sequential immunization (Escolano et al., 2016) would be one potential approach to expand suitable B cell clones and drive their maturation.
Limitations of the study
An important question regarding HCV immunity is why some individuals can clear the virus naturally. With our chronically infected cohort of HCV-infected individuals, we were not able to address this question. In addition, we were not able to study the development of broad neutralization in the interplay with viral resistance during the course of chronic infection. HCV-specific B cells were sorted using a fluorescently labeled bait protein (E2 core). With a different bait (e.g., E1-E2 heterodimer), we might have identified different HCV-specific B cells. We have made best efforts to cover the global diversity of HCV strains, but HCV-neutralizing breadth was determined by a selection of representative HCV strains that may not fully cover the genetic diversity of HCV.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Florian Klein (florian.klein@uk-koeln.de).
Materials availability
Antibodies will be made available by the lead contact upon request with a Material Transfer Agreement for non-commercial usage.
Data and code availability
Nucleotide sequences of all antibodies have been deposited at Genbank. NGS data have been deposited at the Sequence Read Archive (SRA). Coordinates for atomic models have been deposited at the Protein Data Bank (PDB). The data are publicly available as of the date of publication and accession numbers are listed in the key resources table.
All original code has been deposited at Zenodo and is publicly available as of the date of publication. The Digital Object Identifier (DOI) is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Human CD20-Alexa Fluor 700 (Clone 2H7) | BD Biosciences | Cat#560631; RRID: AB_1727447 |
| Goat Anti-Human IgG-HRP | Southern Biotech | Cat#2040-05; RRID: AB_2795644 |
| HCV Antibody AP33 | Clayton et al., 2002 | N/A |
| HCV Antibody AR3C | Law et al., 2008 | N/A |
| HCV Antibody AT12-007 | Merat et al., 2016 | N/A |
| HCV Antibody AT13-021 | Merat et al., 2016 | N/A |
| HCV Antibody HC33.8 | Keck et al., 2013 | N/A |
| HCV Antibody HC84.27 | Keck et al., 2012 | N/A |
| HCV Antibody HEPC-3 | Bailey et al., 2017 | N/A |
| IgG1 isotype control antibody MGO-53 | Wardemann et al., 2003 | N/A |
| Monoclonal anti-HCV E2 patient-derived antibodies | This paper | N/A |
| Monoclonal anti-HIV patient-derived antibodies | This paper | N/A |
| PE Mouse Anti-human IgG (clone G18-145) | BD Biosciences | Cat#555787; RRID: AB_396122 |
| StrepMAB-Classic Oyster 645 conjugate | IBA Lifesciences | Cat#2-1555-050; RRID: |
| HIV-1 Antibody 4E10 | Laboratories of P.W.H.I. Parren and D. Burton; The Scripps Research Institute, La Jolla, California | N/A |
| Bacterial and virus strains | ||
| E. coli DH5α | Thermo Fisher | Cat#18263012 |
| HCVcc strain “1b Con” (Con1/1b/R2a ) | Bankwitz et al., 2021 | Gen Bank (E1/E2 sequence): AJ238799.1 |
| HCVcc strain “1b J4” (J4/1b/R2a ) | Bankwitz et al., 2021 | Gen Bank (E1/E2 sequence): AF054247.1 |
| HCVcc strain “2a” (JcR-2a ) | Bankwitz et al., 2021 | Gen Bank (E1/E2 sequence): NC_009823.1 |
| HCVcc strain “2b” (J8/2b/R2a ) | Bankwitz et al., 2021 | Gen Bank (E1/E2 sequence): JQ745651.1 |
| HCVcc strain “4a” (ED43/4a/R2a ) | Bankwitz et al., 2021 | Gen Bank (E1/E2 sequence): NC_009825.1 |
| HCVcc strain “5a” (SA13/5a/R2a ) | Bankwitz et al., 2021 | Gen Bank (E1/E2 sequence): MH427311.1 |
| HCVcc strain “1a.H77” (H77c/1a/R2a ) | Bankwitz et al., 2021 | Gen Bank (E1/E2 sequence): NC_038882.1 |
| HCVcc strain “2a-3” (2a-3/2a/R2a ) | Bankwitz et al., 2021 | Gen Bank (E1/E2 sequence): KM361734.1 |
| HCVcc strain “2b-4” (2b-4/2b/R2a ) | Bankwitz et al., 2021 | Gen Bank (E1/E2 sequence): KM361730.1 |
| HCVcc strain “2b-5” (2b-5/2b/R2a ) | Bankwitz et al., 2021 | Gen Bank (E1/E2 sequence): KM361731.1 |
| HCVcc strain “2k” (2k/2k/R2a ) | Bankwitz et al., 2021 | Gen Bank (E1/E2 sequence): AB031663.1 |
| HCVcc strain “2r” (2r/2r/R2a ) | Bankwitz et al., 2021 | Gen Bank (E1/E2 sequence): JF735115.1 |
| HCVcc strain “3a” (S52/3a/R2a ) | Bankwitz et al., 2021 | Gen Bank (E1/E2 sequence): GU814264.1 |
| Chemicals, peptides, and recombinant proteins | ||
| ABTS solution | Thermo Fisher | Cat#002024 |
| Adenosine triphosphate | Roche (Sigma Aldrich) | Cat#000000011140965001 |
| Bovine Serum Albumin Fraction V (BSA) | Carl Roth | Cat#8076.3 |
| Branched Polyethylenimine, 25 kDa | Sigma Aldrich | Cat#408727; CAS: 9002-98-6 |
| Cardiolipin | Sigma Aldrich | Cat#C0563 |
| Colenterazine | PJK Biotech | Cat#102171 |
| Cytidine triphosphate | Roche (Sigma Aldrich) | Cat#000000011140922001 |
| DAPI | Thermo Fisher | Cat#D1306; CAS: 581-88-4 |
| DMEM, high glucose | Gibco (Thermo Fisher) | Cat#41965039 |
| DMSO | Sigma Aldrich | Cat#D2650; CAS: 67-68-5 |
| dNTP Mix | Thermo Fisher | Cat#R1122 |
| DTT | Sigma Aldrich | Cat#GE17-1318-01 CAS: 3483-12-3 |
| Fetal bovine serum (FBS) | Sigma Aldrich | Cat#F9665 |
| Fetal bovine serum (FBS), HCVcc assay tested | Capricorn Scientific | Cat#FBS-11A, Lot#CP16-1515 |
| FreeStyle Expression Medium | Thermo Fisher | Cat#12338001 |
| Guanosine triphosphate | Roche (Sigma Aldrich) | Cat#000000011140957001 |
| HEPES | Gibco (Thermo Fisher) | Cat#15630-080 |
| Histopaque-1077 | Sigma Aldrich | Cat#H8889 |
| Human insulin | Sigma Aldrich | Cat#I9278 |
| KLH | Sigma Aldrich | Cat#H8283 |
| L-Glutamine | Gibco (Thermo Fisher) | Cat#25030024 |
| LPS from E. coli | Sigma Aldrich | Cat#L2637 |
| MEM NEAA | Gibco (Thermo Fisher) | Cat#11140-050 |
| NOVA Lite Hep-2 ANA Kit | Inova Diagnostics / Werfen | Cat#066708100 |
| Penicillin-Streptomycin | Gibco (Thermo Fisher) | Cat#15140122 |
| Platinum Taq Green Hot Start DNA Polymerase | Thermo Fisher | Cat#11966034 |
| Protein G Sepharose 4 Fast Flow | GE Life Sciences | Cat#17061805 |
| Q5 Hot Start High Fidelity DNA Polymerase | NEB | Cat#M0493L |
| Recombinant HCV E2 protein pT1056 (GT 2b) | This paper | N/A |
| RNaseOUT | Thermo Fisher | Cat#10777019 |
| Rnasin | Promega | Cat#N2515 |
| RQ1 RNase-free DNase I | Promega | Cat#M6101 |
| Spermidine | Sigma Aldrich | Cat#S2626-1G CAS: 124-20-9 |
| SuperScript IV Reverse Transcriptase | Thermo Fisher | Cat#18090050 |
| T4 DNA Polymerase | New England Biolabs | Cat#M0203L |
| T7 RNA Polymerase | Promega | Cat#P2075 |
| Uridine triphosphate | Roche (Sigma Aldrich) | Cat#000000011140949001 |
| Critical commercial assays | ||
| CD19 MicroBeads, human | Miltenyi Biotec | Cat#130-050-301 |
| EZ-Link sulfo-NHS-biotin | Thermo Fisher | A39256 |
| RNA cleanup kit | Machery Nagel | Cat#740948.5 |
| Deposited data | ||
| HCV antibody heavy and light chain sequences | This paper | GenBank: OL704862 - OL705481 |
| NGS data of IgG repertoires | This paper | NCBI SRA: SAMN23561202- SAMN23561205 |
| 1198_05_G10-E2ecto structure | This paper | PDB: 7RFB |
| 1382_01_H05-E2ecto structure | This paper | PDB: 7RFC |
| Matlab code for machine learning | This paper | DOI: 10.5281/zenodo.5713270 |
| Experimental models: Cell lines | ||
| HEK 293-6E cell line | National Research | NRC file 11565 |
| Council Canada (NRC) | ||
| Huh 7.5 cell line | Blight et al., 2002 | RRID: CVCL_7927 |
| Huh 7.5.1 cell line | Zhong et al., 2005 | RRID: CVCL_E049 |
| Experimental models: Organisms/strains | ||
| None | N/A | N/A |
| Oligonucleotides | ||
| Random Hexamer Primer | Thermo Fisher | Cat#SO142 |
| Single cell PCR Primers | Kreer et al., 2020a | N/A |
| SLIC heavy chain reverse primer (GGGTGCCAGGGGGAAGACCGATGGGCCCTTGGTCGAGGC) | Kreer et al., 2020b | N/A |
| SLIC kappa chain reverse primer (CTCATCAGATGGCGGGAAGATGAAGACAGATGGTGCAGCCACCGTACG) | Kreer et al., 2020b | N/A |
| SLIC lambda chain reverse primer (GAAGCTCCTCACTCGAGGGYGGGAACAGAGTG) | Kreer et al., 2020b | N/A |
| VH1-69 genotyping primer fwd (AGGAAGGGATCCTGGTTT) | Adapted from Pappas et al. (2014) | N/A |
| VH1-69 genotyping primer rev (GATGTGGGTTTTCACACTGTGT) | Adapted from Pappas et al. (2014) | N/A |
| Recombinant DNA | ||
| Human antibody expression vector IgG1 | Tiller et al., 2008 | Avaliable through Addgene as plasmid #80795 |
| Human antibody expression vector Ig kappa | Tiller et al., 2008 | Avaliable through Addgene as plasmid #80796 |
| Human antibody expression vector Ig lambda | Tiller et al., 2008 | Avaliable through Addgene as plasmid #99575 |
| Plasmids encoding HCVcc strains (see section Bacterial and Virus Strains for details) | Bankwitz et al., 2021 | N/A |
| Software and algorithms | ||
| Adobe Illustrator CC 2018 | Adobe | NA |
| FlowJo 10.5.3 | FlowJo, LLC | NA |
| Geneious R10 and Geneious Prime | Geneious | RRID: SCR_010519 |
| IgBlast | National Library of Medicine; Ye et al., 2013 | RRID: SCR_002873 |
| Prism 7 | GraphPad | RRID: SCR_002798 |
| Python 3.6.8 | Python Software Foundation; https://www.python.org/ | RRID: SCR_008394 |
| Other | ||
| Amicon MWCO 30 kDa | Merck Millipore | Cat#Z677108 |
| FACSAria Fusion | BD | N/A |
| Microscope DMI3000 B | Leica | N/A |
| Pierce High Sensitivity Streptavidin-HRP | Thermo Fisher | Cat#21130 |
EXPERIMENTAL MODELS AND SUBJECT DETAILS
HCV-infected individuals and sample collection
Samples were obtained under study protocols 11-312 and 16-054 (approved by the Ethics Commission of the Medical Faculty of the University of Cologne) as well as study protocol 017/16 (approved by the Ethics Commission of the Medical Faculty of the University of Bonn). All participants provided written informed consent. Serum was collected from patients (71% male, median age 49 years; see Figure 1B) treated at the Clinic for Gastroenterology, Department I of Internal Medicine, University Hospital of Bonn, Germany, or enrolled in the PEPSI study (Sirkoski et al., 2013) at hospitals or as outpatients in the German cities of Bielefeld, Bonn, Cologne, Dortmund, Dresden, Duisburg, Düsseldorf, Iserlohn, Lüdenscheid, Munich, Osnabrück, and Wuppertal. For the donation of peripheral blood mononuclear cells (PBMC), patients were invited to the University Hospital of Bonn or Cologne.
Cell lines
HEK 293-6E cells (National Research Council Canada, file 11565) were maintained in FreeStyle Expression Medium (Thermo Fisher) containing 0.2 % Penicillin-Streptomycin (Gibco) in a shaking incubator at 37°C and 5% CO2. Huh 7.5 (Blight et al., 2002) and Huh 7.5.1 cells (Zhong et al., 2005) were maintained in DMEM (Gibco) supplemented with 2 mM L-glutamine (Gibco), 10% FBS (Capricorn), 1% Penicillin-Streptomycin (Gibco) and 1% MEM NEAA (Gibco) at 37°C and 5% CO2. HEK 293-6E cells are female. Huh 7.5 and Huh 7.5.1 cells are male.
METHOD DETAILS
Isolation of polyclonal serum IgG (pIgG)
Serum samples were heat-inactivated at 56°C for 40 min and then incubated with Protein G Sepharose (GE Life Sciences) overnight at 4°C. pIgG was eluted by adding 0.1 M glycine at pH 3.0 onto the Protein G Sepharose in chromatography columns. Eluate was buffered with 1/10 volume of 1 M Tris, pH 8.0. Subsequently, Amicon 30 kDa spin membranes (Millipore) were used to perform buffer exchange to PBS (Gibco) and to adjust pIgG concentration. pIgG concentration was measured using a Nanodrop (A280). IgG samples were stored at 4°C.
Virus particle production
HCV particles were produced as previously published (Koutsoudakis et al., 2006; Wakita et al., 2005). Plasmids containing a T7-promoter, a Renilla luciferase gene, and genomes of the previously described (Bankwitz et al., 2021) HCVcc strains 1bCon (Con1), 1bJ4 (J4/JFH1), 2a (J6/JFH1), 2b (J8/JFH1), 4a (ED43/JFH1), and 5a (SA13/JFH1) were used as templates for in vitro transcription with T7 RNA polymerase (Promega) in 80 mM HEPES pH 7.5, 12 mM MgCl2, 3.125 mM (each) ribonucleotides (Roche), 2 mM spermidine (Sigma Aldrich), 40 mM DTT, and 1 U/μL RNasin (Promega) for 4 h at 37°C. DNA was digested with DNase (Promega) for 30 min at 37°C. RNA was purified using an RNA cleanup kit (Machery Nagel). RNA was electroporated at 975 μF and 270 V into 6 x 105 Huh 7.5.1 cells (Zhong et al., 2005), suspended in 400 μL Cytomix buffer (120 mM KCl, 0.15 mM CaCl2, 10 mM K2HPO4/KH2PO4, 25 mM HEPES, 2 mM EGTA, 5 mM MgCl2, pH 7.6). Cells were then cultured in DMEM (Gibco) supplemented with 2 mM L-glutamine (Gibco), 10% FBS (Capricorn), 1% Penicillin-Streptomycin (Gibco) and 1% MEM NEAA (Gibco) at 37°C and 5% CO2. Supernatant was harvested after 48, 72, and 96 h, pooled, aliquoted, and stored at −80°C until further use.
HCVcc neutralization assay
Neutralization assays were performed as previously described (Koutsoudakis et al., 2006; Wakita et al., 2005). In brief, Huh 7.5 cells (Blight et al., 2002) were seeded at a density of 104 cells per well in a 96-well plate (Falcon) in 200 μl supplemented DMEM (see above) and incubated for 24 h at 37°C and 5% CO2. Equal volumes of viral supernatant and antibodies (final concentration of 50 μg/mL) or purified pIgG (final concentration of 300 μg/mL) were mixed and co-incubated at 37°C for 30-60 min and 40 μL was added to precultured Huh 7.5 cells, from which the medium had been aspirated. After 4 h of culture, 160 μL supplemented DMEM (see above) was added to each well. After 3 days of culture, supernatant was removed and cells were washed with PBS (Gibco) and lysed by adding 35 μL ddH2O and freezing at −80°C. 20 μL of the thawed lysate was transferred to a white microtiter assay plate (Berthold). 60 μL of 0.424 μg/mL coelenterazine (PJK Biotech) in PBS was added to each well in a plate reader (Tri Star, Berthold). Renilla luminescence was determined (counting time 0.1 sec) after a reaction delay of 5 sec and shaking (1 sec). Luminescence was quantified relative to wells infected with virus alone (at least 4 control wells on the same assay plate) to obtain % neutralization values. For example, if the median luminescence of the virus-only control wells on a given plate was 10,000 arbitrary units (AU), then ≥ 10,000 AU luminescence of a well on the same plate would indicate 0% neutralization; 2,000 AU luminescence would indicate 80% neutralization; and 0 AU luminescence would indicate 100% neutralization. Besides virus-only control wells, each plate contained one control well with a published HCV bNAb (AR3C) and one control well with an isotype control antibody (MGO-53). For each independent experiment, % neutralization values were determined in technical triplicates. As a measure of neutralization breadth, we determined the fraction of HCVcc strains neutralized. For a strain to be considered neutralized, we chose a cutoff neutralization value of 40% based on our experience that neutralization values of the isotype control were routinely smaller than 20% and never exceeded 40% (see also Figure S1B).
Expression and purification of E2 constructs
An E2 core fragment (residues 412 – 645; pT1056) of isolate UKNP2.5.1 lacking HVR2 with asparagines constituting N-linked glycosylation sites 4 and 9 mutated to aspartic acid was produced as bait for B cell isolation in Drosophila S2 cells as described before (Krey et al., 2010) and purified from the supernatant using a StrepTactin Superflow column (IBA) followed by gel filtration chromatography using a Superdex 200 Increase 10/300 GL column (GE Healthcare). For ELISA experiments to determine front layer-dependent E2 binding, His-tagged E2ecto protein (residues 384 – 643) was expressed by transiently transfecting Expi293F cells (National Research Council of Canada) and purified from clarified supernatants using a HisTrap FF column (GE Healthcare) followed by SEC on a Superdex 200 Increase 10/300 GL column (GE Healthcare) to separate monomeric E2ecto proteins from oligomeric species. 1a157 E2ecto ΔFRLY contains mutations T425A, L427A, N428A, S432A, G436A, W437A, G530A, and D535A that disable E2 front layer epitopes. For structural studies, the His-tag was removed from an expression vector encoding a strain 1b09 E2 ectodomain.
B cell isolation
Blood was drawn into syringes pre-filled with heparin. PBMCs were isolated using density gradient separation medium (Histopaque, Sigma Aldrich) according to the manufacturer’s instructions and stored at −150°C in 10% DMSO (Sigma-Aldrich) and 90% (v/v) FBS (Sigma-Aldrich). B cells were obtained from PBMCs by magnetic separation using CD19 Microbeads (Miltenyi Biotec). B cells were stained with anti-human CD20-Alexa Fluor 700 (BD), anti-human IgG-PE (BD), DAPI (Thermo Fisher), recombinant HCV E2 core protein pT1056 (245 ng/μL), and StrepMAB-Classic Oyster 645 conjugate (IBA Lifesciences). For single-cell sorting of HCV-reactive B cells, DAPI−CD20+IgG+pT1056+ cells were sorted into 96-well plates containing 4 μL of lysis buffer per well consisting of 0.5x PBS (Gibco), 0.5 U/μL RNAsin (Promega), 0.5 U/μL RNaseOUT (Thermo Fisher), and 10 mM DTT using a FACSAria Fusion cell sorter (BD). To obtain IgG B cells for next-generation sequencing, 2 x 105 DAPI−CD20+IgG+ cells were sorted into FBS (Sigma Aldrich), which was subsequently removed by centrifugation. Sorted cells were stored at −80°C.
Antibody heavy- and light chain gene amplification and sequence analysis
Single cell amplification of antibody heavy and light chain genes was done as previously published (Ehrhardt et al., 2019; Gieselmann et al., 2021; Kreer et al., 2020a, 2020b; Schommers et al., 2020). Cells were lysed, followed by reverse transcription of RNA into cDNA using Superscript IV reverse transcriptase (Thermo Fisher) and random hexamer primers (Invitrogen) in the presence of RNasin (Promega) and RNaseOUT (Thermo Fisher). cDNA was used as template for heavy and light chain amplification through semi-nested PCR using PlatinumTaq HotStart DNA polymerase (Thermo Fisher) with V gene-specific forward primer mixes (Kreer et al., 2020a), published reverse primers (Tiller et al., 2008), and KB extender. PCR products were analyzed by gel electrophoresis and Sanger sequencing. Sequences that passed quality control (chromatograms with mean Phred scores of at least 28 and length of at least 240 nucleotides) were annotated with IgBLAST (Ye et al., 2013) and trimmed to the variable region (FWR1 to the end of the J gene). In a second quality control step, sequences were excluded when their variable regions contained more than 15 base calls with Phred scores below 16, frame shifts, or stop codons. For each HCV-infected individual, B cells were grouped into clones with same V, D, and J gene usage and at least 75% amino acid identity within their CDRH3s as previously described (Kreer et al., 2020b).
Next generation sequencing and evaluation of IgG+ B cell repertoires
For reference IgG repertoires, we generated cDNA from 2x105 CD20+/IgG+ B cells by 5’ rapid amplification of cDNA ends (5’RACE) and performed 2x300 bp Illumina sequencing followed by sequence analysis with an in-house pipeline, as described previously (Ehrhardt et al., 2019). Briefly, data processing comprised an initial filtering of raw-reads for a mean Phred score of 25 and a minimal read length of 250 base pairs. Sequences were then grouped by an 18 nucleotide unique molecular identifier (UMI), which was added during 5’RACE, as well as an additional molecular identifier (MID) of 18 nucleotides (covering the CDR3) to exclude wrongly assigned sequences. Sequences that were not assigned to a UMI/MID group (single reads) were excluded from further analyses. For all other groups, alignments were performed with Clustal Omega (Sievers et al., 2011) and consensus sequences were built with quality-weighted base call frequencies. The two 300 bp consensus reads were assembled with the pRESTO toolkit (Vander Heiden et al., 2014) and sequences were annotated with IgBLAST (Ye et al., 2013). Only productive sequences (i.e., without frame-shift or stop-codons) were used for repertoire analyses.
Cloning and production of monoclonal antibodies
PCR products obtained in the first round of semi-nested single-cell PCR (described above) were used as templates for PCR amplification with specific forward- and reverse primers for the respective V- and J-regions with expression vector overhangs (Kreer et al., 2020b; Tiller et al., 2008). Analogous V(D)J segments encoding mutated antibody variants or published control antibodies, flanked by expression vector overhangs, were synthesized as double-stranded DNA fragments (IDT DNA). PCR products or DNA fragments were cloned into human antibody expression vectors (IgG1 heavy-, kappa-, or lambda chain) by sequence- and ligation-independent cloning (SLIC) assembly as previously published (Von Boehmer et al., 2016). 293-6E cells (National Research Council Canada) were co-transfected with plasmids encoding heavy- and light chains using 25 kDa branched polyethylenimine (PEI, Sigma Aldrich). 293-6E cells were cultured for antibody production at 37°C and 6% CO2 in FreeStyle 293 Expression Medium (Gibco) containing 0.2% penicillin/streptomycin (Gibco). After 7 days, supernatants were harvested by centrifugation and incubated with Protein G Sepharose (GE Life Sciences) overnight at 4°C. Antibodies were eluted by adding 0.1 M glycine at pH 3.0 onto the Protein G Sepharose in chromatography columns. Eluate was buffered with 1/10 volume of 1 M Tris, pH 8.0. Subsequently, Amicon 30 kDa spin membranes (Millipore) were used to perform buffer exchange to PBS (Gibco) and to adjust concentrations. Antibody concentrations were measured using a Nanodrop (A280). Antibodies were stored at 4°C.
Control antibodies directed against HIV-1 were isolated as part of an as yet unpublished study in our laboratory. Single B cells directed against a stabilized HIV envelope glycoprotein were sorted as previously published (Schommers et al., 2020) and monoclonal antibodies were cloned and produced as described above.
ELISA analysis to determine antibody binding activity to HCV E2 protein or polyreactivity
96-well ELISA plates (Corning) were coated with 2.5 μg/mL HCV E2 protein in PBS overnight at 4°C. For polyreactivity ELISA (Haynes et al., 2005a; Wardemann et al., 2003), plates were coated with 10 μg/mL human insulin (Sigma) or 10 μg/mL Lipopolysaccharide from E. Coli (Sigma) or 10 μg/mL Keyhole limpet hemocyanin (Sigma) in PBS overnight at room temperature; or plates were coated with 10 μg/mL bovine cardiolipin (Sigma) in pure ethanol overnight at room temperature. For coating with cardiolipin, plates were allowed to dry out. After washing six times with PBS-T (PBS, 0.05% v/v Tween-20), plates were blocked with 200 μL of 2% (w/v) bovine serum albumin (BSA, Carl Roth) in PBS, and incubated for 90 min at room temperature. Plates were then washed as before and horseradish peroxidase (HRP)-conjugated goat anti-human IgG antibody (Southern Biotech) was added (diluted 1:1000 in 2% w/v BSA/PBS) and incubated for 60 min at room temperature. Plates were then washed as before and ABTS solution (Thermo Fisher) was added. Readout was performed at an absorbance of 415 nm with subtraction of 695 nm background using a plate reader (Tecan Sunrise).
Hep-2 assay
Monoclonal antibodies were tested at 100 μg/mL in PBS using the NOVA Lite HEp-2 ANA Kit (Inova Diagnostics) including a kit-supplied high control according to the manufacturer’s instructions. Images were acquired using a DMI3000 B microscope (Leica).
Expression and purification of E2-Fab complexes
1198_05_G10 Fab-1b09 E2ecto and 1382_01_H05 Fab-1b09 E2ecto complexes for structural studies were produced in Expi293F cells in the presence of 5 μM kifunensine (Sigma) by co-transfecting expression vectors encoding His-tagged Fab and untagged E2ecto to allow isolation of stable Fab-E2 complexes (Flyak et al., 2018). Protein complexes were purified from supernatants using Ni-NTA chromatography on HisTrap HP column (GE Healthcare) followed by SEC on a Superdex 200 Increase 10/300 GL column (GE Healthcare).
Crystallization, data collection and structure determinations
Commercially-available screens (Hampton Research and Molecular Dimensions) were used to screen initial crystallization conditions by vapor diffusion in sitting drops. 1198_05_G10-E2ecto crystals were grown using 0.2 μL of protein complex in TBS and 0.2 μL of mother liquor (0.1 M ammonium acetate, 0.1 M BIS-TRIS pH 5.5, 17% PEG 10,000) and cryoprotected in Al’s oil. 1382_01_H05-E2ecto crystals were grown using 0.2 μL of protein complex in TBS and 0.2 μL of mother liquor (0.03 M citric acid, 0.07 M BIS-TRIS propane pH 7.6, 20% PEG 3,350) and cryoprotected in mother liquor supplemented with 25% (v/v) glycerol. X-ray diffraction data from cryopreserved crystals were collected at the Stanford Synchrotron Radiation Lightsource on beamline 12–2 using a PILATUS 6M detector. Images were processed and scaled using iMosflm (Battye et al., 2011) and Aimless as implemented in the CCP4 software suite (Evans and Murshudov, 2013). Structures were solved by molecular replacement using the HEPC74 (PDB 6MEH) and 1b09 HCV E2ecto (PDB 6MEI) structures as search models. The models were refined and validated using Phenix.refine (Adams et al., 2010). Iterative manual model building and corrections were performed using Coot (Emsley and Cowtan, 2004). Glycans were initially interpreted and modeled using Fo – Fc maps calculated with model phases contoured at 2σ, followed by 2Fo – Fc simulated annealing composite omit maps generated in Phenix in which modeled glycans were omitted to remove model bias (Adams et al., 2010). The quality of the final models was examined using MolProbity (Chen et al., 2010).
Models were superimposed and figures rendered using the PyMOL molecular visualization system (Version 2.1, Schrödinger, LLC). Buried surface areas (BSAs) were determined using the PDBePISA web-based interactive tool (Krissinel and Henrick, 2007). Potential hydrogen bonds were assigned using criteria of a distance of <4.0 Å and an A-D-H angle of >90°, and the maximum distance allowed for a van der Waals interaction was 4.0 Å. Rmsd calculations were done in PyMOL following pairwise Cα alignments. Antibody residues were numbered according to the Kabat numbering scheme, and ImMunoGeneTics (IMGT) definitions of CDRs were used throughout the paper (Kabat, 1991; Lefranc et al., 2003).
Reversion of VH1-69 somatic mutations to the germline sequence
First, we determined germline VH1-69 allelic variants by sequencing of genomic DNA. For this, a fragment of the VH1-69 gene was amplified as previously published (Pappas et al., 2014) from PBMC genomic DNA. PCR products were cloned into TOPO vectors. At least 8 clones per individual were sequenced by Sanger sequencing. To revert VH1-69 somatic mutations to germline, sequences were replaced by those of the most similar VH1-69 allelic variant from the global ImMunoGeneTics (IMGT) database up until FWR3 (Lefranc et al., 2015). CDRs were defined according to the IMGT scheme (Lefranc et al., 2003).
Machine learning
Our machine learning problem was a classification task and stated as follows:
Given VH gene segment amino acid sequences and the HCV neutralization efficacy (good/bad) of the corresponding antibodies, build a model which predicts the HCV neutralization efficacy of antibodies encoded by any VH gene segment sequence.
Since this is a binary classification task and the number of training samples was not large enough to build accurate deep learning models, we trained an SVM (Cortes and Vapnik, 1995), which is known to perform well for small to medium sized problems. We made use of the Statistics and Machine Learning toolbox of MATLAB 2019b to train the SVM. We encoded the amino acid sequences with binary encoding and used a linear kernel, since polynomial kernels of degree two and three showed similar performance and linear kernels allow for a more direct explainability. We determined the best parameters (C=1) by 10-fold cross-validation with C ε {10−5, 10−4,… 105}.
In this study we were especially interested in the feature importance, the impact of amino acids at their position on the classifier. Therefore, we determined the weights of the features from the trained SVM: First we retrieved the Lagrange multipliers, which are the coefficients from the dual problem. Second, we computed the weight vector by summing up the product of the Lagrange multiplier, label and binary encoding at each position. In the course of this we can distinguish between positive and negative weights. We end up with one vector resembling the impact of each amino acid at each position.
QUANTIFICATION AND STATISTICAL ANALYSIS
Flow cytometry analysis and quantifications were done with FlowJo10. Statistical analyses were done with GraphPad Prism (v9), Microsoft Excel for Mac (v14.7.3), Python (v3.6.8), and R (v3.6.0). The logo plot in Figure 7A was created with WebLogo 3.
Supplementary Material
Highlights.
Screening identified individuals with exceptional HCV Ab responses
Isolated broad and potent HCV-neutralizing Abs from elite neutralizers
Identified genetic antibody features associated with potent HCV neutralization
Designed de novo HCV Ab by machine learning that neutralized multiple HCV genotypes
ACKNOWLEDGMENTS
We thank all study participants who devoted time to our research; members of the Klein, Pietschmann, Pfeifer, Krey, and Bjorkman Laboratories for continuous support and helpful discussions, Maike Schlotz and Carola Ruping for help with sample processing, and Henning Gruell for valuable discussions. We thank the Caltech Protein Expression Center for help with protein expression and the Caltech Molecular Observatory for assistance with structural studies.
This work was funded by grants from the European Research Council (ERC-STG-639961 to F.K.), the German Center for Infection Research (DZIF TTU 05.817 and TTU 05.821 to F.K., T.P., T.K., and T.W.) and the German Research Foundation (DFG) (CRC 1310 to F.K.). T.P. and T.K. are funded by the DFG under Germany’s Excellence Strategy (EXC 2155 “RESIST” — project ID 39087428). This research was also supported by the U.S. National Institutes of Health (NIH) (NIH grant R01 AI127469 to P.J.B.) and (NIH grant K99 AI153465 to A.I.F.) (content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH) and the Molecular Observatory at Caltech supported by the Gordon and Betty Moore Foundation. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences (contract no DE-AC02-76SF00515). The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by NIHGMS (P41GM103393).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.immuni.2021.12.003.
DECLARATION OF INTERESTS
Reported antibodies are in the process of being patented.
REFERENCES
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JR, Barnes E, and Cox AL (2019). Approaches, progress, and challenges to hepatitis C vaccine development. Gastroenterology 156, 418–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JR, Flyak AI, Cohen VJ, Li H, Wasilewski LN, Snider AE, Wang S, Learn GH, Kose N, Loerinc L, et al. (2017). Broadly neutralizing antibodies with few somatic mutations and hepatitis C virus clearance. JCI Insight 2, e92872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankwitz D, Bahai A, Labuhn M, Doepke M, Ginkel C, Khera T, Todt D, Ströh LJ, Dold L, Klein F, et al. (2021). Hepatitis C reference viruses highlight potent antibody responses and diverse viral functional interactions with neutralising antibodies. Gut 70, 1734–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battye TGG, Kontogiannis L, Johnson O, Powell HR, and Leslie AGW (2011). iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr 67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blight KJ, McKeating JA, and Rice CM (2002). Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol 76, 13001–13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton DR, Poignard P, Stanfield RL, and Wilson IA (2012). Broadly neutralizing antibodies present new prospects to counter highly antigenically diverse viruses. Science 337, 183–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso RMF, Zwick MB, Stanfield RL, Kunert R, Binley JM, Katinger H, Burton DR, and Wilson IA (2005). Broadly neutralizing anti-HIV antibody 4E10 recognizes a helical conformation of a highly conserved fusion-associated motif in gp41. Immunity 22, 163–173. [DOI] [PubMed] [Google Scholar]
- Chen F, Nagy K, Chavez D, Willis S, McBride R, Giang E, Honda A, Bukh J, Ordoukhanian P, Zhu J, et al. (2020). Antibody responses to immunization With HCV envelope glycoproteins as a baseline for B-cell-based vaccine development. Gastroenterology 158, 1058–1071, e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Tzarum N, Lin X, Giang E, Velázquez-Moctezuma R, Augestad EH, Nagy K, He L, Hernandez M, Fouch ME, et al. (2021). Functional convergence of a germline-encoded neutralizing antibody response in rhesus macaques immunized with HCV envelope glycoproteins. Immunity 54, 781–796, e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Tzarum N, Wilson IA, and Law M (2019). V H 1–69 antiviral broadly neutralizing antibodies: genetics, structures, and relevance to rational vaccine design. Curr. Opin. Virol 34, 149–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo QL, Kuo G, Ralston R, Weiner A, Chien D, Van Nest G, Han J, Berger K, Thudium K, Kuo C, et al. (1994). Vaccination of chimpanzees against infection by the hepatitis C virus. Proc. Natl. Acad. Sci. USA 91, 1294–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton RF, Owsianka A, Aitken J, Graham S, Bhella D, and Patel AH (2002). Analysis of antigenicity and topology of E2 glycoprotein present on recombinant hepatitis C virus-like particles. J. Virol 76, 7672–7682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbert MD, Flyak AI, Ogega CO, Kinchen VJ, Massaccesi G, Hernandez M, Davidson E, Doranz BJ, Cox AL, Crowe JE, and Bailey JR (2019). Broadly neutralizing antibodies targeting new sites of vulnerability in hepatitis C virus E1E2. J. Virol 93, e02070–e02018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes C, and Vapnik V (1995). Support-vector networks. Mach. Learn 20, 273–297. [Google Scholar]
- de Jong YP, Dorner M, Mommersteeg MC, Xiao JW, Balazs AB, Robbins JB, Winer BY, Gerges S, Vega K, Labitt RN, et al. (2014). Broadly neutralizing antibodies abrogate established hepatitis C virus infection. Sci. Transl. Med 6, 254ra129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrhardt SA, Zehner M, Krähling V, Cohen-Dvashi H, Kreer C, Elad N, Gruell H, Ercanoglu MS, Schommers P, Gieselmann L, et al. (2019). Polyclonal and convergent antibody response to Ebola virus vaccine rVSV-ZEBOV. Nat. Med 25, 1589–1600. [DOI] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Escolano A, Steichen JM, Dosenovic P, Kulp DW, Golijanin J, Sok D, Freund NT, Gitlin AD, Oliveira T, Araki T, et al. (2016). Sequential immunization elicits broadly neutralizing anti-HIV-1 antibodies in Ig knockin mice. Cell 166, 1445–1458, e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans PR, and Murshudov GN (2013). How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr 69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farci P, Shimoda A, Wong D, Cabezon T, De Gioannis D, Strazzera A, Shimizu Y, Shapiro M, Alter HJ, and Purcell RH (1996). Prevention of hepatitis C virus infection in chimpanzees by hyperimmune serum against the hypervariable region 1 of the envelope 2 protein. Proc. Natl. Acad. Sci. USA 93, 15394–15399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flyak AI, Ruiz S, Colbert MD, Luong T, Crowe JE, Bailey JR, and Bjorkman PJ (2018). HCV broadly neutralizing antibodies use a CDRH3 disulfide motif to recognize an E2 glycoprotein site that can be targeted for vaccine design. Cell Host Microbe 24, 703–716, e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flyak AI, Ruiz SE, Salas J, Rho S, Bailey JR, and Bjorkman PJ (2020). An ultralong CDRH2 in HCV neutralizing antibody demonstrates structural plasticity of antibodies against E2 glycoprotein. Elife 9, e53169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman AJ, Dore GJ, Law MG, Thorpe M, Von Overbeck J, Lloyd AR, Marinos G, and Kaldor JM (2001). Estimating progression to cirrhosis in chronic hepatitis C virus infection. Hepatology 34, 809–816. [DOI] [PubMed] [Google Scholar]
- Gieselmann L, Kreer C, Ercanoglu MS, Lehnen N, Zehner M, Schommers P, Potthoff J, Gruell H, and Klein F (2021). Effective high-throughput isolation of fully human antibodies targeting infectious pathogens. Nat. Protoc 16, 3639–3671. [DOI] [PubMed] [Google Scholar]
- Haynes BF, Fleming J, St Clair EW, Katinger H, Stiegler G, Kunert R, Robinson J, Scearce RM, Plonk K, Staats HF, et al. (2005a). Cardiolipin polyspecific autoreactivity in two broadly neutralizing HIV-1 antibodies. Science 308, 1906–1908. [DOI] [PubMed] [Google Scholar]
- Haynes BF, Moody MA, Verkoczy L, Kelsoe G, and Alam SM (2005b). Antibody polyspecificity and neutralization of HIV-1: a hypothesis. Hum. Antibodies 14, 59–67. [PMC free article] [PubMed] [Google Scholar]
- Irimia A, Sarkar A, Stanfield RL, and Wilson IA (2016). Crystallographic identification of lipid as an integral component of the epitope of HIV broadly neutralizing antibody 4E10. Immunity 44, 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabat EA (1991). Sequences of Proteins of Immunological Interest (United States Department of Health and Human Services. Public Health Service, National Institutes of Health). [Google Scholar]
- Keck Z-Y, Xia J, Wang Y, Wang W, Krey T, Prentoe J, Carlsen T, Li AY-J, Patel AH, Lemon SM, et al. (2012). Human monoclonal antibodies to a novel cluster of conformational epitopes on HCV E2 with resistance to neutralization escape in a genotype 2a isolate. PLoS Pathog 8, e1002653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck Z, Wang W, Wang Y, Lau P, Carlsen THR, Prentoe J, Xia J, Patel AH, Bukh J, and Foung SKH (2013). Cooperativity in virus neutralization by human monoclonal antibodies to two adjacent regions located at the amino terminus of hepatitis C virus E2 glycoprotein. J. Virol 87, 37–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck Z-Y, Wang Y, Lau P, Lund G, Rangarajan S, Fauvelle C, Liao GC, Holtsberg FW, Warfield KL, Aman MJ, et al. (2016). Affinity maturation of a broadly neutralizing human monoclonal antibody that prevents acute hepatitis C virus infection in mice. Hepatology 64, 1922–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsoe G, and Haynes BF (2017). Host controls of HIV broadly neutralizing antibody development. Immunol. Rev 275, 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinchen VJ, and Bailey JR (2018). Defining breadth of hepatitis C virus neutralization. Front. Immunol 9, 1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinchen VJ, Cox AL, and Bailey JR (2018a). Can broadly neutralizing monoclonal antibodies lead to a hepatitis C virus vaccine? Trends Microbiol 26, 854–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinchen VJ, Zahid MN, Flyak AI, Soliman MG, Learn GH, Wang S, Davidson E, Doranz BJ, Ray SC, Cox AL, et al. (2018b). Broadly neutralizing antibody mediated clearance of human hepatitis C virus infection. Cell Host Microbe 24, 717–730, e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein F, Diskin R, Scheid JF, Gaebler C, Mouquet H, Georgiev IS, Pancera M, Zhou T, Incesu RB, Fu BZ, et al. (2013a). Somatic mutations of the immunoglobulin framework are generally required for broad and potent HIV-1 neutralization. Cell 153, 126–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein F, Mouquet H, Dosenovic P, Scheid JF, Scharf L, and Nussenzweig MC (2013b). Antibodies in HIV-1 vaccine development and therapy. Science 341, 1199–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L, Giang E, Nieusma T, Kadam RU, Cogburn KE, Hua Y, Dai X, Stanfield RL, Burton DR, Ward AB, et al. (2013). Hepatitis C virus E2 envelope glycoprotein core structure. Science 342, 1090–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsoudakis G, Kaul A, Steinmann E, Kallis S, Lohmann V, Pietschmann T, and Bartenschlager R (2006). Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J. Virol 80, 5308–5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreer C, Döring M, Lehnen N, Ercanoglu MS, Gieselmann L, Luca D, Jain K, Schommers P, Pfeifer N, and Klein F (2020a). openPrimeR for multiplex amplification of highly diverse templates. J. Immunol. Methods 480, 112752. [DOI] [PubMed] [Google Scholar]
- Kreer C, Zehner M, Weber T, Ercanoglu MS, Gieselmann L, Rohde C, Halwe S, Korenkov M, Schommers P, Vanshylla K, et al. (2020b). Longitudinal isolation of potent near-germline SARS-CoV-2-neutralizing antibodies from COVID-19 patients. Cell 182, 843–854, e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krey T, D’Alayer J, Kikuti CM, Saulnier A, Damier-Piolle L, Petitpas I, Johansson DX, Tawar RG, Baron B, Robert B, et al. (2010). The disulfide bonds in glycoprotein E2 of hepatitis C virus reveal the tertiary organization of the molecule. PLoS Pathog 6, e1000762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, and Henrick K (2007). Inference of macromolecular assemblies from crystalline state. J. Mol. Biol 372, 774–797. [DOI] [PubMed] [Google Scholar]
- Law M, Maruyama T, Lewis J, Giang E, Tarr AW, Stamataki Z, Gastaminza P, Chisari FV, Jones IM, Fox RI, et al. (2008). Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med 14, 25–27. [DOI] [PubMed] [Google Scholar]
- Lefranc M-P, Giudicelli V, Duroux P, Jabado-Michaloud J, Folch G, Aouinti S, Carillon E, Duvergey H, Houles A, Paysan-Lafosse T, et al. (2015). IMGT®, the international ImMunoGeneTics information system® 25 years on. Nucleic Acids Res 43, D413–D422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefranc M-P, Pommié C, Ruiz M, Giudicelli V, Foulquier E, Truong L, Thouvenin-Contet V, and Lefranc G (2003). IMGT unique numbering for immunoglobulin and T cell receptor variable domains and Ig superfamily V-like domains. Dev. Comp. Immunol 27, 55–77. [DOI] [PubMed] [Google Scholar]
- Liu L, Wang P, Nair MS, Yu J, Rapp M, Wang Q, Luo Y, Chan JFW, Sahi V, Figueroa A, et al. (2020). Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature 584, 450–456. [DOI] [PubMed] [Google Scholar]
- Logvinoff C, Major ME, Oldach D, Heyward S, Talal A, Balfe P, Feinstone SM, Alter H, Rice CM, and McKeating JA (2004). Neutralizing antibody response during acute and chronic hepatitis C virus infection. Proc. Natl. Acad. Sci. USA 101, 10149–10154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna JM, Saeed M, and Rice CM (2019). Taming a beast: lessonsfrom the domestication of hepatitis C virus. Curr. Opin. Virol 35, 27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merat SJ, Bru C, van de Berg D, Molenkamp R, Tarr AW, Koekkoek S, Kootstra NA, Prins M, Ball JK, Bakker AQ, et al. (2019). Cross-genotype AR3-specific neutralizing antibodies confer long-term protection in injecting drug users after HCV clearance. J. Hepatol 71, 14–24. [DOI] [PubMed] [Google Scholar]
- Merat SJ, Molenkamp R, Wagner K, Koekkoek SM, Van De Berg D, Yasuda E, Böhne M, Claassen YB, Grady BP, Prins M, et al. (2016). Hepatitis C virus broadly neutralizing monoclonal antibodies isolated 25 years after spontaneous clearance. PLoS One 11, e0165047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin TJ, Broering TJ, Leav BA, Blair BM, Rowley KJ, Boucher EN, Wang Y, Cheslock PS, Knauber M, Olsen DB, et al. (2012). Human monoclonal antibody HCV1 effectively prevents and treats HCV infection in chimpanzees. PLoS Pathog 8, e1002895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osburn WO, Snider AE, Wells BL, Latanich R, Bailey JR, Thomas DL, Cox AL, and Ray SC (2014). Clearance of hepatitis C infection is associated with the early appearance of broad neutralizing antibody responses. Hepatology 59, 2140–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappas L, Foglierini M, Piccoli L, Kallewaard NL, Turrini F, Silacci C, Fernandez-Rodriguez B, Agatic G, Giacchetto-Sasselli I, Pellicciotta G, et al. (2014). Rapid development of broadly influenza neutralizing antibodies through redundant mutations. Nature 516, 418–422. [DOI] [PubMed] [Google Scholar]
- Perin PM, Haid S, Brown RJP, Doerrbecker J, Schulze K, Zeilinger C, von Schaewen M, Heller B, Vercauteren K, Luxenburger E, et al. (2016). Flunarizine prevents hepatitis C virus membrane fusion in a genotype-dependent manner by targeting the potential fusion peptide within E1. Hepatology 63, 49–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestka JM, Zeisel MB, Bläser E, Schürmann P, Bartosch B, Cosset FL, Patel AH, Meisel H, Baumert J, Viazov S, et al. (2007). Rapid induction of virus-neutralizing antibodies and viral clearance in a single-source outbreak of hepatitis C. Proc. Natl. Acad. Sci. USA 104, 6025–6030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D, Park YJ, Beltramello M, Walls AC, Tortorici MA, Bianchi S, Jaconi S, Culap K, Zatta F, De Marco A, et al. (2020). Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature 583, 290–295. [DOI] [PubMed] [Google Scholar]
- Roingeard P, and Beaumont E (2020). Hepatitis C vaccine: 10 good reasons for continuing. Hepatology 71, 1845–1850. [DOI] [PubMed] [Google Scholar]
- Sainz B, Barretto N, Martin DN, Hiraga N, Imamura M, Hussain S, Marsh KA, Yu X, Chayama K, Alrefai WA, et al. (2012). Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptoras a new hepatitis C virus entry factor. Nat. Med 18, 281–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheid JF, Mouquet H, Feldhahn N, Seaman MS, Velinzon K, Pietzsch J, Ott RG, Anthony RM, Zebroski H, Hurley A, et al. (2009). Broad diversity of neutralizing antibodies isolated from memory B cells in HIV-infected individuals. Nature 458, 636–640. [DOI] [PubMed] [Google Scholar]
- Schommers P, Gruell H, Abernathy ME, Tran MK, Dingens AS, Gristick HB, Barnes CO, Schoofs T, Schlotz M, Vanshylla K, et al. (2020). Restriction of HIV-1 escape by a highly broad and potent neutralizing antibody. Cell 180, 471–489, e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirkoski AM, Sierra S, Qurishi N, Bagel B, Schelhorn ES, Lengauer T, Erhardt A, Goeser T, Esser S, Häussinger D, et al. (2013). 1204 The Pepsi project: HCV resistance screening and prediction of pi-containing therapy outcome. J. Hepatol 58, S489–S490. [Google Scholar]
- Ströh LJ, and Krey T (2020). HCV glycoprotein structure and implications for B-cell vaccine development. Int. J. Mol. Sci 21, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suryadevara N, Shrihari S, Gilchuk P, VanBlargan LA, Binshtein E, Zost SJ, Nargi RS, Sutton RE, Winkler ES, Chen EC, et al. (2021). Neutralizing and protective human monoclonal antibodies recognizing the N-terminal domain of the SARS-CoV-2 spike protein. Cell 184, 2316–2331, e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DL (2019). Global elimination of chronic hepatitis. N. Engl. J. Med 380, 2041–2050. [DOI] [PubMed] [Google Scholar]
- Tiller T, Meffre E, Yurasov S, Tsuiji M, Nussenzweig MC, and Wardemann H (2008). Efficient generation of monoclonal antibodies from single human B cells by single cell RT-PCR and expression vector cloning. J. Immunol. Methods 329, 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzarum N, Giang E, Kong L, He L, Prentoe J, Augestad E, Hua Y, Castillo S, Lauer GM, Bukh J, et al. (2019). Genetic and structural insights into broad neutralization of hepatitis C virus by human VH 1–69 antibodies. Sci. Adv 5, eaav1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzarum N, Wilson IA, and Law M (2018). The neutralizing face of hepatitis C virus E2 envelope glycoprotein. Front. Immunol 9, 1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden JA, Yaari G, Uduman M, Stern JNH, O’Connor KC, Hafler DA, Vigneault F, and Kleinstein SH (2014). pRESTO: a toolkit for processing high-throughput sequencing raw reads of lymphocyte receptor repertoires. Bioinformatics 30, 1930–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Boehmer L, Liu C, Ackerman S, Gitlin AD, Wang Q, Gazumyan A, and Nussenzweig MC (2016). Sequencing and cloning of antigen-specific antibodies from mouse memory B cells. Nat. Protoc 11, 1908–1923. [DOI] [PubMed] [Google Scholar]
- Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Kräusslich H-G, Mizokami M, et al. (2005). Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med 11, 791–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, and Nussenzweig MC (2003). Predominant autoantibody production by early human B cell precursors. Science 301, 1374–1377. [DOI] [PubMed] [Google Scholar]
- Wrensch F, Crouchet E, Ligat G, Zeisel MB, Keck Z-Y, Foung SKH, Schuster C, and Baumert TF (2018). Hepatitis C virus (HCV)-apolipoprotein interactions and immune evasion and their impact on HCV vaccine design. Front. Immunol 9, 1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Ma N, Madden TL, and Ostell JM (2013). IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res 41, W34–W40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, and Chisari FV (2005). Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 102, 9294–9299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Nucleotide sequences of all antibodies have been deposited at Genbank. NGS data have been deposited at the Sequence Read Archive (SRA). Coordinates for atomic models have been deposited at the Protein Data Bank (PDB). The data are publicly available as of the date of publication and accession numbers are listed in the key resources table.
All original code has been deposited at Zenodo and is publicly available as of the date of publication. The Digital Object Identifier (DOI) is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Human CD20-Alexa Fluor 700 (Clone 2H7) | BD Biosciences | Cat#560631; RRID: AB_1727447 |
| Goat Anti-Human IgG-HRP | Southern Biotech | Cat#2040-05; RRID: AB_2795644 |
| HCV Antibody AP33 | Clayton et al., 2002 | N/A |
| HCV Antibody AR3C | Law et al., 2008 | N/A |
| HCV Antibody AT12-007 | Merat et al., 2016 | N/A |
| HCV Antibody AT13-021 | Merat et al., 2016 | N/A |
| HCV Antibody HC33.8 | Keck et al., 2013 | N/A |
| HCV Antibody HC84.27 | Keck et al., 2012 | N/A |
| HCV Antibody HEPC-3 | Bailey et al., 2017 | N/A |
| IgG1 isotype control antibody MGO-53 | Wardemann et al., 2003 | N/A |
| Monoclonal anti-HCV E2 patient-derived antibodies | This paper | N/A |
| Monoclonal anti-HIV patient-derived antibodies | This paper | N/A |
| PE Mouse Anti-human IgG (clone G18-145) | BD Biosciences | Cat#555787; RRID: AB_396122 |
| StrepMAB-Classic Oyster 645 conjugate | IBA Lifesciences | Cat#2-1555-050; RRID: |
| HIV-1 Antibody 4E10 | Laboratories of P.W.H.I. Parren and D. Burton; The Scripps Research Institute, La Jolla, California | N/A |
| Bacterial and virus strains | ||
| E. coli DH5α | Thermo Fisher | Cat#18263012 |
| HCVcc strain “1b Con” (Con1/1b/R2a ) | Bankwitz et al., 2021 | Gen Bank (E1/E2 sequence): AJ238799.1 |
| HCVcc strain “1b J4” (J4/1b/R2a ) | Bankwitz et al., 2021 | Gen Bank (E1/E2 sequence): AF054247.1 |
| HCVcc strain “2a” (JcR-2a ) | Bankwitz et al., 2021 | Gen Bank (E1/E2 sequence): NC_009823.1 |
| HCVcc strain “2b” (J8/2b/R2a ) | Bankwitz et al., 2021 | Gen Bank (E1/E2 sequence): JQ745651.1 |
| HCVcc strain “4a” (ED43/4a/R2a ) | Bankwitz et al., 2021 | Gen Bank (E1/E2 sequence): NC_009825.1 |
| HCVcc strain “5a” (SA13/5a/R2a ) | Bankwitz et al., 2021 | Gen Bank (E1/E2 sequence): MH427311.1 |
| HCVcc strain “1a.H77” (H77c/1a/R2a ) | Bankwitz et al., 2021 | Gen Bank (E1/E2 sequence): NC_038882.1 |
| HCVcc strain “2a-3” (2a-3/2a/R2a ) | Bankwitz et al., 2021 | Gen Bank (E1/E2 sequence): KM361734.1 |
| HCVcc strain “2b-4” (2b-4/2b/R2a ) | Bankwitz et al., 2021 | Gen Bank (E1/E2 sequence): KM361730.1 |
| HCVcc strain “2b-5” (2b-5/2b/R2a ) | Bankwitz et al., 2021 | Gen Bank (E1/E2 sequence): KM361731.1 |
| HCVcc strain “2k” (2k/2k/R2a ) | Bankwitz et al., 2021 | Gen Bank (E1/E2 sequence): AB031663.1 |
| HCVcc strain “2r” (2r/2r/R2a ) | Bankwitz et al., 2021 | Gen Bank (E1/E2 sequence): JF735115.1 |
| HCVcc strain “3a” (S52/3a/R2a ) | Bankwitz et al., 2021 | Gen Bank (E1/E2 sequence): GU814264.1 |
| Chemicals, peptides, and recombinant proteins | ||
| ABTS solution | Thermo Fisher | Cat#002024 |
| Adenosine triphosphate | Roche (Sigma Aldrich) | Cat#000000011140965001 |
| Bovine Serum Albumin Fraction V (BSA) | Carl Roth | Cat#8076.3 |
| Branched Polyethylenimine, 25 kDa | Sigma Aldrich | Cat#408727; CAS: 9002-98-6 |
| Cardiolipin | Sigma Aldrich | Cat#C0563 |
| Colenterazine | PJK Biotech | Cat#102171 |
| Cytidine triphosphate | Roche (Sigma Aldrich) | Cat#000000011140922001 |
| DAPI | Thermo Fisher | Cat#D1306; CAS: 581-88-4 |
| DMEM, high glucose | Gibco (Thermo Fisher) | Cat#41965039 |
| DMSO | Sigma Aldrich | Cat#D2650; CAS: 67-68-5 |
| dNTP Mix | Thermo Fisher | Cat#R1122 |
| DTT | Sigma Aldrich | Cat#GE17-1318-01 CAS: 3483-12-3 |
| Fetal bovine serum (FBS) | Sigma Aldrich | Cat#F9665 |
| Fetal bovine serum (FBS), HCVcc assay tested | Capricorn Scientific | Cat#FBS-11A, Lot#CP16-1515 |
| FreeStyle Expression Medium | Thermo Fisher | Cat#12338001 |
| Guanosine triphosphate | Roche (Sigma Aldrich) | Cat#000000011140957001 |
| HEPES | Gibco (Thermo Fisher) | Cat#15630-080 |
| Histopaque-1077 | Sigma Aldrich | Cat#H8889 |
| Human insulin | Sigma Aldrich | Cat#I9278 |
| KLH | Sigma Aldrich | Cat#H8283 |
| L-Glutamine | Gibco (Thermo Fisher) | Cat#25030024 |
| LPS from E. coli | Sigma Aldrich | Cat#L2637 |
| MEM NEAA | Gibco (Thermo Fisher) | Cat#11140-050 |
| NOVA Lite Hep-2 ANA Kit | Inova Diagnostics / Werfen | Cat#066708100 |
| Penicillin-Streptomycin | Gibco (Thermo Fisher) | Cat#15140122 |
| Platinum Taq Green Hot Start DNA Polymerase | Thermo Fisher | Cat#11966034 |
| Protein G Sepharose 4 Fast Flow | GE Life Sciences | Cat#17061805 |
| Q5 Hot Start High Fidelity DNA Polymerase | NEB | Cat#M0493L |
| Recombinant HCV E2 protein pT1056 (GT 2b) | This paper | N/A |
| RNaseOUT | Thermo Fisher | Cat#10777019 |
| Rnasin | Promega | Cat#N2515 |
| RQ1 RNase-free DNase I | Promega | Cat#M6101 |
| Spermidine | Sigma Aldrich | Cat#S2626-1G CAS: 124-20-9 |
| SuperScript IV Reverse Transcriptase | Thermo Fisher | Cat#18090050 |
| T4 DNA Polymerase | New England Biolabs | Cat#M0203L |
| T7 RNA Polymerase | Promega | Cat#P2075 |
| Uridine triphosphate | Roche (Sigma Aldrich) | Cat#000000011140949001 |
| Critical commercial assays | ||
| CD19 MicroBeads, human | Miltenyi Biotec | Cat#130-050-301 |
| EZ-Link sulfo-NHS-biotin | Thermo Fisher | A39256 |
| RNA cleanup kit | Machery Nagel | Cat#740948.5 |
| Deposited data | ||
| HCV antibody heavy and light chain sequences | This paper | GenBank: OL704862 - OL705481 |
| NGS data of IgG repertoires | This paper | NCBI SRA: SAMN23561202- SAMN23561205 |
| 1198_05_G10-E2ecto structure | This paper | PDB: 7RFB |
| 1382_01_H05-E2ecto structure | This paper | PDB: 7RFC |
| Matlab code for machine learning | This paper | DOI: 10.5281/zenodo.5713270 |
| Experimental models: Cell lines | ||
| HEK 293-6E cell line | National Research | NRC file 11565 |
| Council Canada (NRC) | ||
| Huh 7.5 cell line | Blight et al., 2002 | RRID: CVCL_7927 |
| Huh 7.5.1 cell line | Zhong et al., 2005 | RRID: CVCL_E049 |
| Experimental models: Organisms/strains | ||
| None | N/A | N/A |
| Oligonucleotides | ||
| Random Hexamer Primer | Thermo Fisher | Cat#SO142 |
| Single cell PCR Primers | Kreer et al., 2020a | N/A |
| SLIC heavy chain reverse primer (GGGTGCCAGGGGGAAGACCGATGGGCCCTTGGTCGAGGC) | Kreer et al., 2020b | N/A |
| SLIC kappa chain reverse primer (CTCATCAGATGGCGGGAAGATGAAGACAGATGGTGCAGCCACCGTACG) | Kreer et al., 2020b | N/A |
| SLIC lambda chain reverse primer (GAAGCTCCTCACTCGAGGGYGGGAACAGAGTG) | Kreer et al., 2020b | N/A |
| VH1-69 genotyping primer fwd (AGGAAGGGATCCTGGTTT) | Adapted from Pappas et al. (2014) | N/A |
| VH1-69 genotyping primer rev (GATGTGGGTTTTCACACTGTGT) | Adapted from Pappas et al. (2014) | N/A |
| Recombinant DNA | ||
| Human antibody expression vector IgG1 | Tiller et al., 2008 | Avaliable through Addgene as plasmid #80795 |
| Human antibody expression vector Ig kappa | Tiller et al., 2008 | Avaliable through Addgene as plasmid #80796 |
| Human antibody expression vector Ig lambda | Tiller et al., 2008 | Avaliable through Addgene as plasmid #99575 |
| Plasmids encoding HCVcc strains (see section Bacterial and Virus Strains for details) | Bankwitz et al., 2021 | N/A |
| Software and algorithms | ||
| Adobe Illustrator CC 2018 | Adobe | NA |
| FlowJo 10.5.3 | FlowJo, LLC | NA |
| Geneious R10 and Geneious Prime | Geneious | RRID: SCR_010519 |
| IgBlast | National Library of Medicine; Ye et al., 2013 | RRID: SCR_002873 |
| Prism 7 | GraphPad | RRID: SCR_002798 |
| Python 3.6.8 | Python Software Foundation; https://www.python.org/ | RRID: SCR_008394 |
| Other | ||
| Amicon MWCO 30 kDa | Merck Millipore | Cat#Z677108 |
| FACSAria Fusion | BD | N/A |
| Microscope DMI3000 B | Leica | N/A |
| Pierce High Sensitivity Streptavidin-HRP | Thermo Fisher | Cat#21130 |