

Abstract
Background:
Lysine-specific histone demethylase 1 (KDM1A/LSD1) regulates multiple cellular functions, including cellular proliferation, differentiation, and DNA repair. KDM1A is overexpressed in squamous cell carcinoma of the skin, and inhibition of KDM1A can suppress cutaneous carcinogenesis. Despite the role of KDM1A in skin and DNA repair, the effect of KDM1A inhibition on cellular ultraviolet (UV) response has not been studied.
Methods:
The ability of KDM1A inhibitor bizine to modify cell death after UVA and UVB exposure was tested in normal human keratinocytes and melanocytes, HaCaT, and FaDu cell lines. KDM1A was also downregulated using shRNA and inhibited by phenelzine in HaCaT and FaDu cells to confirm the role of KDM1A in UVA response. In addition, cellular reactive oxygen species (ROS) changes were assessed by a lipid-soluble fluorescent indicator of lipid oxidation, and ROS related gene regulation using qPCR. During photodynamic therapy (PDT) studies HaCaT and FaDu cells were treated with aminolaevulinic acid (5-ALA) or HPPH (2-(1-Hexyloxyethyl)-2-devinyl pyropheophorbide-a) sodium and irradiated with 0–8J/cm2 red LED light.
Results:
KDM1A inhibition sensitized cells to UVA radiation-induced cell death but not to UVB. KDM1A inhibition increased ROS generation as detected by increased lipid peroxidation and the upregulation of ROS responsive genes. The effectiveness of both ALA and HPPH PDT significantly improved in vitro in HaCaT and FaDu cells after KDM1A inhibition.
Conclusion:
KDM1A is a regulator of cellular UV response, and KDM1A inhibition can improve PDT efficacy.
Keywords: skin, ultraviolet A light (UVA), reactive oxygen species, KDM1A inhibitors, photodynamic therapy (PDT)
Summary Statement
Lysine-specific histone demethylase 1 (KDM1A/LSD1) regulates multiple cellular functions, including cellular proliferation and DNA repair, but its role in modifying ultraviolet light response has not been clear. We found that KDM1A inhibition increases lipid peroxidation and upregulated ROS responsive genes. Moreover, we observed that KDM1A inhibition enhanced the efficacy of photodynamic therapy with both delta-aminolaevulinic acid and the chlorin compound HPPH (2-(1-Hexyloxyethyl)-2-devinyl pyropheophorbide-a). These findings highlight the role of KDM1A as a regulator of cellular UV response and identified a potential novel therapeutic role for KDM1A inhibitors in improving the efficacy and selectivity of PDT.
Introduction:
Ultraviolet radiation (UV) exposure is responsible for most skin cancers[1,2]. Terrestrial UV radiation is divided into two main regions based on wavelength and biological function[3]. UVB (280–315 nm) radiation primarily causes DNA damage including cyclobutane pyrimidine dimers and 6–4 photoproducts and penetrates superficially in the skin[4], while the longer wavelength UVA (315–400 nm) radiation penetrates deeper and exerts most of its biological role through inducing reactive oxygen species (ROS) such as singlet oxygen, hydrogen peroxide and superoxides[5–7]. When cells are exposed to UVA radiation, lipids are peroxidized and other macromolecules also suffer oxidative damage while cellular antioxidants try to mitigate ROS[8,9]. ROS can overwhelm cellular defenses and may also induce DNA damage (8-oxoguanine generation) followed by alteration in gene expression and mutations leading to cancer [8]. Ultimately, if the antioxidant system is completely overwhelmed and cellular macromolecule and organelle damage becomes irreparable, the toxic effects of ROS lead to cell death [10].
Photodynamic therapy (PDT) exploits the potential toxic effect of ROS for the treatment of cancer. PDT involves the administration of a photosensitizer which after exposure to an appropriate wavelength of light, interacts with triplet oxygen (3O2) to form singlet oxygen (1O2) and in lower quantities also generates other ROS like hydroxyl radicals (•OH) and superoxide (O2•−) ions [11]. PDT induces ROS-mediated cell death [11]. PDT is frequently used for the treatment of skin cancers [12]. Aminolevulinic acid (5-ALA), a precursor in heme biosynthesis, was the first FDA-approved photosensitizer for PDT and is used for the treatment of precancerous skin lesions and early skin cancers[13]. Other photosensitizers including synthetic dyes phenothiazinium, squaraine and BODIPY (boron-dipyrromethene), 2-[1-hexyloxyethyl]-2-devinyl pyropheophorbide-a (HPPH), transition metal complexes, and natural products such as hypericin and curcumin are also investigated for the treatment of cancer [14–17]. Since ROS generation is limited by the photosensitizer’s tissue and cellular distribution, PDT is somewhat selective for tumors [18] and does not induce mutations in normal cells.[19] Combining PDT with chemotherapy or targeted therapy could enhance both selectivity and efficacy of treatment. One of the major limiting factors for PDT is the availability of oxygen for ROS generation [20]. Since ROS mediates the cellular and tissue effects of PDT, drugs that could hypersensitize cells to ROS or increase ROS production [14,21] could be employed to improve PDT efficacy.
KDM1A/LSD1 is a ubiquitously expressed transcriptional coregulator, which specifically demethylates mono- and di-methyl residues from H3 on lysine 4 [22]. KDM1A can also demethylate important non-histone proteins including p53, E2F1, DMNT and MYC [23] to regulate normal cellular processes. KDM1A has also been shown to modulate DNA repair. KDM1A is recruited to sites of DNA damage marked by phosphorylated histone H2AX[24] and associates with several factors (53BP1, BRACA1) involved in nucleotide excision repair (NER) [24–26]. KDM1A can also demethylate p53, which is essential to the DNA damage response, impairing its DNA binding ability and association with 53BP1 [25]. Through its enzymatic activity in which hydrogen peroxide is generated [25], KDM1A recruits base excision repair (BER) enzymes, 8-oxoguanine-DNA glycosylase 1 (OGG1) and topoisomerase II (TOP2A) [27] to sites of oxidative DNA damage. In the skin, KDM1A has been described to be a regulator of differentiation[28] and more recently inhibition of KDM1A in the epidermis was suggested to prevent the initiation of squamous cell carcinoma[28]. Based on the role of KDM1A in oxidative DNA damage repair and skin carcinogenesis, in this study we explored KDM1A’s ability to modify UV response and identified KDM1A inhibitors as potential therapeutic agents for increasing PDT efficacy.
Materials and methods
Cell Culture
Cell lines used are HaCaT (human, adult, low calcium, high temperature keratinocytes cells, Addex Biosciences, USA), NHEK (normal human epidermal keratinocytes, neonatal, ATCC, USA), FaDu (human head and neck squamous cell carcinoma, ATCC, USA) and NHEM (normal human epidermal melanocytes, ATCC, USA). All cells were maintained in humidified 5% CO2 incubator at 37 degrees centigrade. HaCaT and FaDu cells were cultured in either DMEM (Corning, NY, USA) or supplemented with 1mM Glutagro supplement (Corning, NY, USA), 1% antibiotic solution (100 mg/L streptomycin, 100 U/ml penicillin) and 10% fetal bovine serum. NHEK and NHEM were grown in dermal cell basal medium (ATCC, USA) supplemented with components of keratinocyte growth kit (ATCC, USA) and melanocyte growth kit (ATCC, USA) respectively, according to manufacturer’s instructions.
UV irradiation
Cells were trypsinized and plated in 96 well (2500 cells per well) or 6 well plates (1*105 cells per well). All drug treatments were performed 24 hours prior to UV exposure. Cells were then irradiated under a thin layer of 1X PBS with calcium and magnesium supplemented with 25mM glucose. The temperature was controlled by placing the plated cells on metal plates that were maintained at 4°C using a water recirculator, PolyScience Refrigerated Recirculating Chiller. This ensured that the temperature of irradiated plates and control plates were at 22–25 °C. PBS was replaced with complete media after irradiation and death was measured using cell titer blue assay 24 hours post UV exposure. Terrestrial UV light was from the Oriel Solar Simulator (Newport Corporation) (Figure S1A). The UVA light source was composed of a bank of 4 UVA light tubes (F8T5 Black Light, 8-Watt UVA fluorescent bulb, Hitachi, Tokyo, Japan) with UV emission peak at 365nm. The UVA device emitted 82% in UVA (315–400nm) and 17.9% in the visible (>400nm) spectrum and negligible UVB and no UVC (Figure S1B). The UVB light source was a bank of 6 UVB light tubes (USHIO G8T5E 7.2W cat# 300318, Ushio America, Cypress, CA) with UV emission peak at 313nm. The UVB device emitted 51% in the UVB (280–315nm) and 47% in the UVA (315–400nm) spectrum and the device showed negligible visible or UVC light emission (Figure S1C). The UVA spectral component of the UVB device had no biologically relevant effect during the experiments. This is due to the approximately 1000-fold higher biological efficacy of the UVB component of the UV spectrum [4].
KDM1A down regulation
KDM1A was downregulated using lentivirus made with plasmids GIPZ Control, KDM1A sh1 clone V3LHS_361041 and KDM1A sh6 clone V2LHS_34926 (Horizon Discovery, Waterbeach, UK). Viruses were prepared using the 293T cell line (Clontech, USA) with packaging plasmids (ΔR) and G-protein vesicular stomatitis virus (VSVG) using JetPrime transfection reagent (PolyPlus transfection, USA) according to the manufacturer’s instructions. For transduction cells were plated in 60- or 100-mm tissue culture dishes and allowed to achieve 60% confluence before adding viral supernatant in the presence of 8 μg/ml polybrene for 24 hours (Sigma, St. Louis, MO). Cells were selected by treatment with puromycin (1ug/ml for FaDu cells and 2ug/ml for HaCaT) for 5 days.
Antioxidant and KDM1A inhibitor treatment
Cells were trypsinized and plated in 96 wells (2500 cells per well) or 6 well plates (1*105) cells per well). The pH of N-acetylcysteine (NAC) (Sigma, St. Louis, MO) stock solution was adjusted to 7.4 with sodium bicarbonate before adding to cells. NAC was added to cells at 2–10mM along with the KDM1A specific inhibitors, bizine or phenelzine (Axon MedChem, BV) for 18 hours prior to UVA exposure.
Cell Titer Blue assay
Cells were plated in 96 well plates at 2500–3000 cells per well and treated with KDM1A inhibitors, bizine or phenelzine the next day. After 24 hours of drug treatment cells were exposed to UVA or UVB. 24 hours after light exposure 6X Cell Titer Blue solution (Promega, Madison, WI, USA) was added into wells to a final 1X concentration and incubated for 3 hours at 3700B0C and 5% CO2. Fluorescence was measured using a 560 nm excitation/590 nm emission filter set. Wells with plain media were used as blank.
Lipid peroxidation assay
Cells were plated in 6-well plates at 1.5×105 per well. Next day cells were treated with bizine or phenelzine at the specified doses. 24 hours after drug treatment cells were exposed to UVA light as usual. Immediately after irradiation, 1X PBS was replaced with 1ml complete media after which BODIPY® 581/591 C11 reagent (Thermofisher Scientific, USA) was added to a final concentration of 10μM according to the manufacturer’s instructions.
Quantitative RT-PCR
Total RNA was isolated using RNeasy Mini Kit (Qiagen Inc, MD, USA) according to the manufacturer’s protocol. cDNA was synthesized from 500 ng total RNA using High-Capacity cDNA Reverse Transcription Kit (Thermofisher Scientific, USA) in a 20μl reaction according to the manufacturer’s protocol. Quantitative reverse-transcription PCR was performed using QS6 PCR System (Applied Biosystems, Carlsbad, CA) using SYBR Green Master Mix (Invitrogen) or TaqMan Universal Master Mix II (Applied Biosystems, Carlsbad, CA). All reactions were performed in triplicates, and the experiments were repeated at least twice. The results are presented as the mean of at least 2 experiments. Primers used for SYBR Green assays: HMOX-1 F- TTCTCCGATGGGTCCTTACACT R- GGCATAAAGCCCTACAGCAACT, KLF-9 F- CTCCGAAAAGAGGCACAAGT R- CGGGAGAACTTTTTAAGGCAGT, SDHA F- TGGGAACAAGAGGGCATCTG R- CCACCACTGCATCAAATTCATG and TaqMan: KDM1A (Thermofisher Scientific, USA) and PGK1 (Thermofisher Scientific, USA).
Immunoblotting
Protein extracts were prepared by lysing cells using 1x lysis buffer (1% SDS, 0.01% Tris-HCl). Protein concentration was estimated using Pierce™ BCA Protein Assay Kit (Thermofisher Scientific, Frederick, MD, USA). Equal amounts of protein were run on 4–20% gradient precast gels and transferred to PVDF membrane. Membranes were blocked with 5% milk and incubated with primary antibodies overnight and one hour with secondary antibodies after which blots were developed using Biorad ChemiDoc™ Imaging Systems (Biorad, Hercules, CA, USA. All antibodies were purchased from Cell Signaling Technologies, Danvers, MA, USA KDM1A (cat no: 2184, final dilution 1:1000) and Lamin A/C (cat no: 2032, final dilution: 1:3000).
Photodynamic therapy
Cells were plated at 2500 cells per well in 96 well plates. 24 hours after cells were plated, they were treated with bizine and the photosensitizer (as below) overnight after which plates were exposed to red LED light (Aktilite-CL128, Galderma, Lausanne, Switzerland; peak wavelength: 630nm; fluence: 70mW/cm2). The Aktilite emission spectrum was collected using a fiber-coupled isotropic probes (IP85; Medlight SA) at the surface of a well-plate connected to a light dosimetry system [29]. The absorption spectrum of HPPH (diluted to 10 μM in PBS containing 10% FBS) was collected using the UV-vis (Cary-60, Agilent Technology). The extinction coefficient was calculated as the absorption divided by the product of the concentration and path length. A total of five emission spectra were collected over a 58-second exposure. The average of these spectra is plotted in Fig S3. Depending on the photosensitizer, protocol was modified as follows. For ALA treatment the cells were treated with 2 or 4mM 5-ALA (5-Aminolevulinic acid hydrochloride Sigma, St. Louis, MO) diluted in media containing 1% FBS. Cells were treated with bizine for 18 hours and then incubated with 5-ALA for 4 hours. 5-ALA was washed out of the plates and replaced with 100μl of phenol red free media and exposed to red light and cell viability was measured 24 hours post light exposure. During HPPH treatment the cells were treated with both bizine and 0.25–1μM of HPPH (2-[1-hexyloxyethyl]-2-devinyl pyropheophorbide-a Medkoo, Morrisville, NC, USA) diluted in complete phenol red free media for 18 hours. Next plates were exposed to red light from Aktilite (Figure S3) and cell viability was measured 24 hours post light exposure using cell titer blue assay.
Statistical methods
Representative graphs are presented from multiple experiments assessing the UVA, UVB and PDT effects. Depicted bars represent mean values (n=2–3). Mann-Whitney-U or T-tests were used when appropriate, and p<0.05 was considered statistically significant.
Results:
KDM1A inhibition modifies UV response and sensitizes cells to UVA light
To understand the ability of KDM1A to modify UV response, we first assessed whether KDM1A inhibition modified cell death after exposure to solar simulated UV light using a KDM1A specific inhibitor, bizine [30] in HaCaT keratinocytes. Out of the six doses of solar simulated UV and bizine treatment only two points showed a significant increase in cell viability.” (Figure 1A). Solar UV light is a combination of UVA and UVB. UVA and UVB have different cellular effects[4], therefore after treatment with predetermined non-toxic doses of bizine (Table S1) we tested the effect of UVA and UVB separately in a panel of cells including normal keratinocytes, melanocytes and squamous cell carcinoma cells. KDM1A inhibition significantly reduced cell viability in all tested cell lines after UVA exposure but not after UVB exposure (Figure 1B, C). KDM1A inhibition using 2 different chemical inhibitors of KDM1A sensitized keratinocytes (NHEKn and HaCaT) and head and neck squamous cell carcinoma cells (FaDu) to UVA radiation in a dose dependent manner (Figure 1D–F). We also downregulated KDM1A using lentivirus expressing 2 different shRNA (sh1 and sh6) specific to KDM1A, in FaDu and HaCaT cells. Reduction in KDM1A mRNA and protein expression was validated using RT-qPCR and western blot (Figure 1G, I). These cells were then exposed to UVA radiation. Both FaDu and HaCaT cells with down regulated KDM1A showed significant dose dependent decrease in cell viability after UVA exposure (Figure 1H, J)
Figure 1:
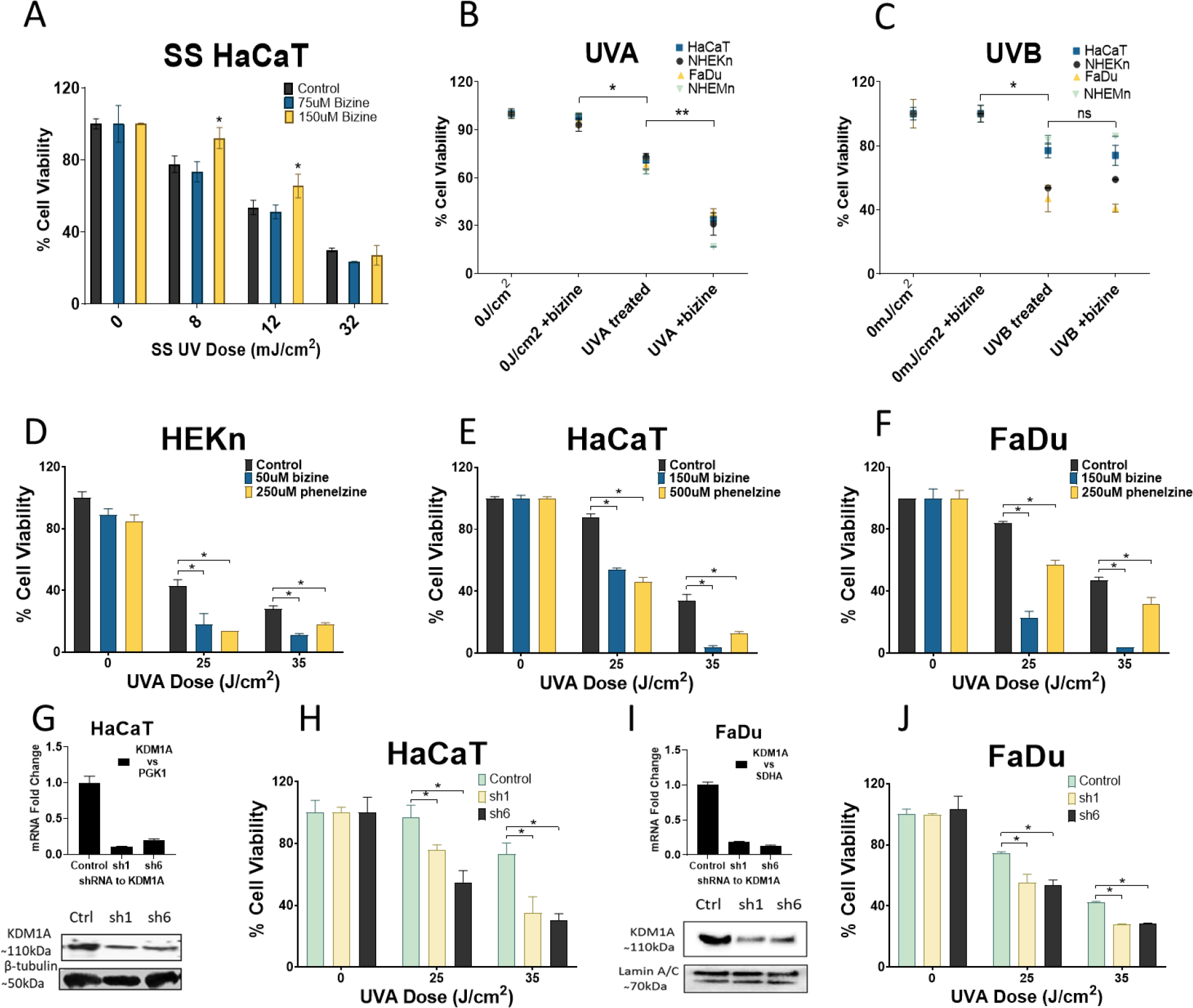
KDM1A inhibition modifies UV response: (A) HaCaT (immortalized keratinocytes) cells were treated with predetermined nontoxic doses of KDM1A inhibitor bizine (75–150μM) and exposed to solar simulated UV light (B,C) A panel of epithelial, melanocytic normal and cancer cells treated for 24 hours with vehicle or bizine (doses in Table S1) and then exposed to UVA-1 or UVB radiation. Cell viability was assessed 24 hours post irradiation KDM1A inhibition sensitizes cells to UVA radiation (D,E,F) Normal human keratinocytes (NHEKn), FaDu (squamous cell carcinoma) and HaCaT cells were treated with treated KDM1A inhibitor bizine and phenelzine and exposed to UVA light (G,I) mRNA expression by RT-qPCR and western blot for KDM1A downregulation using shRNA, sh1 and sh6 in HaCaT and FaDu cells (H,J) Effect of KDM1A downregulation on UVA irradiation. Cell viability is expressed as a percentage of control and was assessed using cell titer blue assay. Bars represent mean ± SD (n=3)
KDM1A inhibition increases in total cellular ROS
KDM1A activity was previously found to be modified by reactive oxygen species (ROS) [31] but the expression of KDM1A after UVA radiation exposure is unknown. To determine whether KDM1A expression is regulated by UVA radiation, HaCaT and FaDu cells were exposed to UVA light. KDM1A expression was not modified by UVA radiation in either cell line and bizine treatment had no significant effect on KDM1A protein levels (Figure 2 A and B). Since KDM1A can generate hydrogen peroxide locally to facilitate increased transcription of genes [25] and KDM1A expression was not modified by UVA exposure we tested whether KDM1A inhibition could modify total ROS levels in our system. We measured changes in ROS levels using fluorescent dye BODIPY® 581/591 C11 to measure lipid peroxidation in HaCaT and FaDu cells. Bizine increased lipid peroxidation after UVA exposure in a dose dependent manner but not at baseline (Figure 2 C and D). We also looked at the expression of several genes commonly regulated by ROS increase after KDM1A inhibition with bizine and UVA exposure in HaCaT cells. Kruppel-like factor 9 (KLF-9), heme oxygenase 1 (HMOX-1) and Phorbol-12-myristate-13-acetate-induced protein 1 (NOXA) mRNA expression was quantified using RT-qPCR. KLF-9, HMOX-1 and NOXA were all upregulated only after UVA exposure in bizine treated cells but not at baseline (Figure 2 E–G). Also, treatment with antioxidant N-acetyl cysteine rescued HaCaT and FaDu cells from bizine induced UVA sensitivity (Figure 2 H and I)
Figure 2:
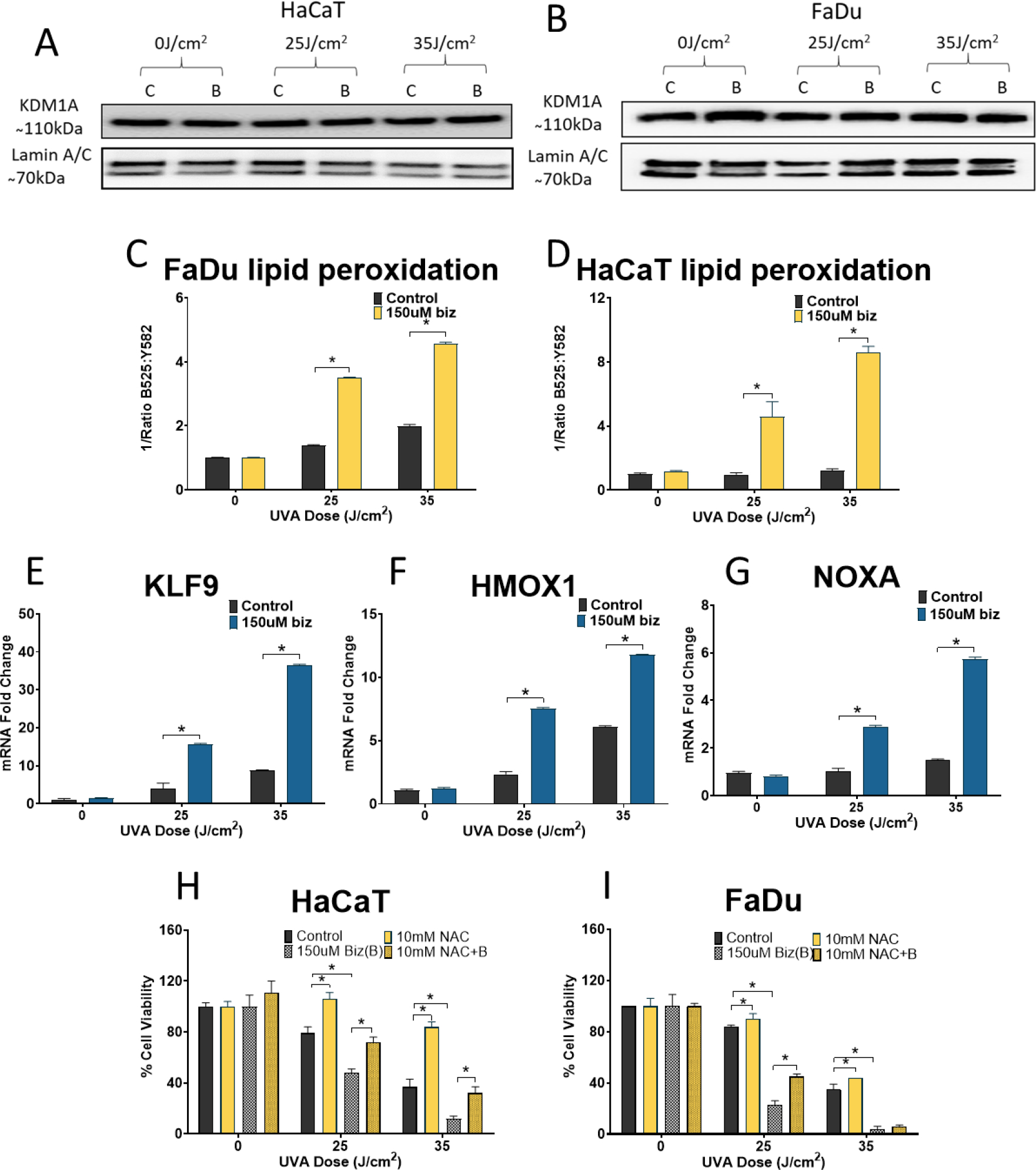
KDM1A expression is not regulated by UVA radiation: (A, B) KDM1A protein level expression compared to normalizer Lamin A/C expression in bizine treated HaCat and FaDu after UVA exposure at doses which in parallel experiments induced significant decrease in cell viability. C-Control, B-Bizine KDM1A inhibition increases lipid peroxidation and mRNA expression of antioxidant enzymes after UVA irradiation : (C, D) Lipid peroxidation by estimating red to green shift in BODIPY® 581/591 C11 measured in HaCaT and FaDu cells treated with bizine and exposed to UVA radiation (E-G) ROS responsive genes KLF-9, HMOX-1 and NOXA expression compared to PGK1 after KDM1A inhibition using bizine UVA exposure. RNA was harvested 4 hours after the addition UVA exposure in HaCaT cells NAC salvaged cells from KDM1A inhibition induced death (H,I) HaCaT and FaDu cells were treated with NAC, bizine or NAC and bizine for 18 hours before exposure to either 2 doses of UVA. Cell viability is expressed as a percentage of control and was assessed using cell titer blue assay. (G-I). Cell viability is expressed as a percentage of control and was assessed using cell titer blue assay. Bars represent mean ± SD (n=3). *: p<0.05
KDM1A inhibition enhances the efficacy of photodynamic therapy
KDM1A has been shown to be highly expressed in cancer which has been linked to poor prognosis in patients [33,34]. Inhibition of KDM1A has also been shown to repress development of squamous cell carcinoma in the skin[28] and from GEODatasets [35,36] we found that KDM1A was overexpressed in patients with basal cell carcinoma and squamous cell carcinoma in skin and esophagus (Figure S2A,B). PDT treatment is used for the treatment of skin cancers [35,36]. During photodynamic therapy (PDT), when a photosensitizer is exposed to light, ROS is released causing cellular damage and cell death. We showed that KDM1A inhibition increases ROS after UVA irradiation. The combination of PDT and KDM1A inhibition could enhance the efficacy and selectivity of the therapy. Therefore, we tested the combination of KDM1A inhibitor with PDT. Bizine treated HaCaT and FaDu cells were subjected to PDT with 5-ALA as the photosensitizer. Both cell lines showed increased sensitivity to 5-ALA PDT after KDM1A inhibition (Figure 3A and B). To rule out a photosensitizer specific effect, HaCaT and FaDu cells were treated with bizine and subjected to PDT using HPPH as photosensitizer. Bizine also enhanced cell sensitivity to HPPH PDT (Figure 3C and D).
Figure 3.
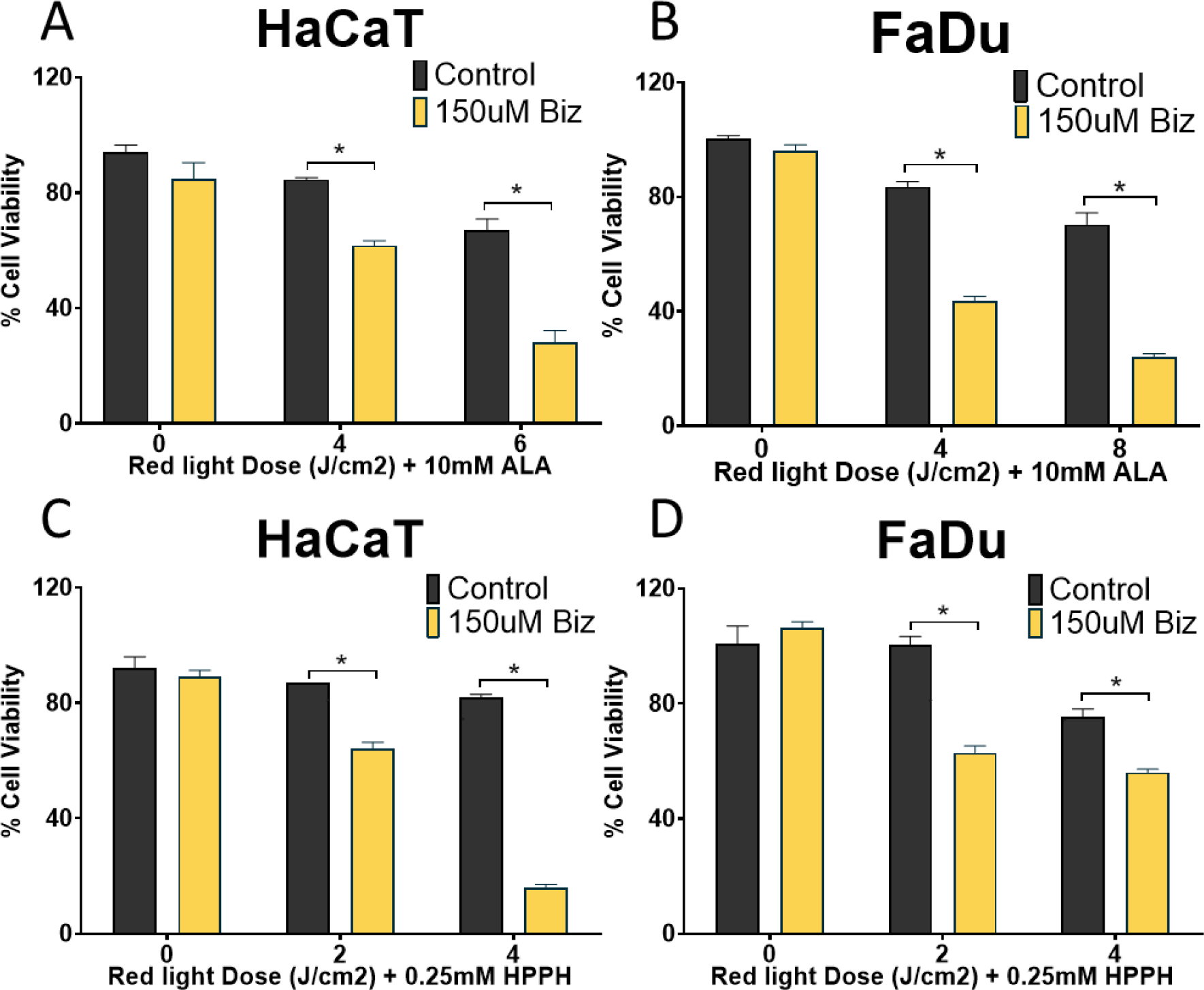
Effect of KDM1A inhibition on 5-ALA and HPPH PDT: (A, B) HaCaT and FaDu cells were treated with bizine and 5-ALA and exposed to red light (635nm) (C, D) HaCaT and FaDu cells treated with bizine and HPPH and exposed to red light (635nm). Cell viability is expressed as a percentage of control and was assessed using cell titer blue assay. Bars represent mean ± SD (n=3) *:p<0.05
Discussion
KDM1A is involved in multiple cellular processes including DNA damage repair, transcriptional regulation and cellular differentiation[24,37,38]. In this paper we focused on a novel role for KDM1A in modifying UVA response. We show that both chemical and genetic inhibition of KDM1A can sensitize cells to UVA radiation (Figure 1B, D–J) through the amplification of ROS as determined by increased lipid peroxidation (Figure 2 C, D) and upregulation of ROS responsive genes (Figure 2E–G). This is an acute effect, with the ROS amplification detected within 30 min of exposure to UVA radiation. Due to the rapidity of response and the effects of KDM1A being mitigated by NAC treatment (Figure 2H, I) the toxic effects of KDM1A inhibition after UVA exposure appears to be directly mediated by ROS. These findings suggested that KDM1A inhibitors may modify PDT efficacy.
PDT treatment is a controllable non-invasive treatment used for precancers and early carcinomas in the skin and head and neck cancers[39,40]. Although PDT is effective it is not completely selective. Combining PDT with chemotherapy or targeted therapy could enhance its effectiveness and selectivity[21,41,42]. We show that the KDM1A inhibitor, bizine significantly increases the efficacy of both 5-ALA and HPPH PDT in vitro (Figure 3). During UVA radiation ROS is amplified throughout the cell while in PDT ROS generation is localized at the photosensitizer [40]. Although, UVA radiation and PDT have different cellular effects we can still enhance PDT using bizine. This also shows the detected increase in ROS after UVA radiation exposure in KDM1A inhibited cells is not a by-product of cell death but the cause of it, which is an important distinction to make when combining any treatment with PDT. KDM1A inhibition has been found to repress invasion and cancer growth in a model of cutaneous squamous cell carcinoma[28]. The upregulation of KDM1A in SCC (Figure S2) and the role of KDM1A in inhibiting SCC growth suggests that KDM1A inhibitors beyond enhancing PDT efficacy, may also enhance the selectivity of PDT.
A limitation of our work is that our studies were performed in vitro on cultured cells, and the effects of KDM1A inhibition and PDT may be different in vivo and in the clinical setting. Both KDM1A and PDT can modify the immune response [43–45], which cannot be assessed with in vitro studies. In actinic keratosis and squamous cell carcinoma, PDT has been described to activate local inflammatory responses stimulating tumor specific cytotoxic T-cells [46] and enhanced immunogenicity of stromal cells[47]. In some cancer models, inhibition of KDM1A has been shown to abrogate PD-1 expression of tumor infiltrating lymphocytes increasing tumor cell clearance[48,49]. It has been recently shown that KDM1A inhibition in combination with checkpoint inhibitors could also be effective in treating melanoma[50]. The above evidence for PDT and KDM1A in immune modulation suggests that in vivo KDM1A inhibition beyond enhancing PDT efficacy may also favorably modify PDT induced immune modulation.
We have identified a novel role for KDM1A in the UVA response. KDM1A inhibition resulted in greatly increased ROS and upregulation of ROS responsive genes. Our study has established a pioneering use for KDM1A inhibitors in enhancing the efficacy of PDT and in the future could be employed to elicit positive outcomes after PDT treatment.
Supplementary Material
ACKNOWLEDGMENT
The authors are grateful for the support and help of Mr. Lawrence Tworek, laser engineer, for helping with light output and spectrum measurements. This work was mainly supported by the Roswell Park Alliance Foundation. The work of Dr. Shafirstein, Dr. Bellnier, and Mr. Tworek was supported by NCI P01 CA55791. The used Genomics Shared Resources at Roswell Park Comprehensive Cancer Center were supported by NCI grant P30CA016056.
Footnotes
Declarations of conflicts of interest: none
BIBLIOGRAPHY
- [1].Kim IY, He YY, Ultraviolet radiation-induced non-melanoma skin cancer: Regulation of DNA damage repair and inflammation, Genes Dis. (2014). 10.1016/j.gendis.2014.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Moon H, Donahue LR, Choi E, Scumpia PO, Lowry WE, Grenier JK, Zhu J, White AC, Melanocyte Stem Cell Activation and Translocation Initiate Cutaneous Melanoma in Response to UV Exposure, Cell Stem Cell. (2017). 10.1016/j.stem.2017.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Holick MF, Biological Effects of Sunlight, Ultraviolet Radiation, Visible Light, Infrared Radiation and Vitamin D for Health, Anticancer Res. 36 (2016) 1345–1356. [PubMed] [Google Scholar]
- [4].Ravanat JL, Douki T, Cadet J, Direct and indirect effects of UV radiation on DNA and its components, J. Photochem. Photobiol. B Biol. (2001). 10.1016/S1011-1344(01)00206-8. [DOI] [PubMed] [Google Scholar]
- [5].McCormick J, Fischer J Pachlatko, A. Eisenstark, Characterization of a cell-lethal product from the photooxidation of tryptophan: hydrogen peroxide, Science (80-. ). (2003). 10.1126/science.1108203. [DOI] [PubMed] [Google Scholar]
- [6].Cunningham ML, Johnson JS, Giovanazzi SM, Peak MJ, PHOTOSENSITIZED PRODUCTION OF SUPEROXIDE ANION BY MONOCHROMATIC (290–405 nm) ULTRAVIOLET IRRADIATION OF NADH and NADPH COENZYMES, Photochem. Photobiol. (1985). 10.1111/j.1751-1097.1985.tb01549.x. [DOI] [PubMed] [Google Scholar]
- [7].Grossweiner LI, Membrane photosensitization by hematoporphyrin and hematoporphyrin derivative., Prog. Clin. Biol. Res. (1984). [PubMed] [Google Scholar]
- [8].Lautier D, Luscher P, Tyrrell RM, Endogenous glutathione levels modulate both constitutive and uva radiation/hydrogen peroxide inducible expression of the human heme oxygenase gene, Carcinogenesis. (1992). 10.1093/carcin/13.2.227. [DOI] [PubMed] [Google Scholar]
- [9].Morlière P, Moysan A, Santus R, Hüppe G, Mazière JC, Dubertret L, UVA-induced lipid peroxidation in cultured human fibroblasts, Biochim. Biophys . Acta (BBA)/Lipids Lipid Metab. (1991). 10.1016/0005-2760(91)90068-S. [DOI] [PubMed] [Google Scholar]
- [10].Danpure HJ, Tyrrell RM, OXYGEN-DEPENDENCE OF NEAR UV (365 NM) LETHALITY AND THE INTERACTION OF NEAR UV AND X-RAYS IN TWO MAMMALIAN CELL LINES, Photochem. Photobiol. (1976). 10.1111/j.1751-1097.1976.tb07238.x. [DOI] [PubMed] [Google Scholar]
- [11].Henderson BW, Dougherty TJ, HOW DOES PHOTODYNAMIC THERAPY WORK?, Photochem. Photobiol. (1992). 10.1111/j.1751-1097.1992.tb04222.x. [DOI] [PubMed] [Google Scholar]
- [12].Cohen DK, Lee PK, Photodynamic Therapy for Non-Melanoma Skin Cancers, Cancers. 8 (2016). 10.3390/cancers8100090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Filho JDV, Andrade CT, Buzza HH, Blanco K, Carbinatto F, Bagnato VS, Allison RR, PDT and emerging therapies for Actinic Keratosis—A resource letter, Photodiagnosis Photodyn. Ther. (2017). 10.1016/j.pdpdt.2016.09.002. [DOI] [PubMed] [Google Scholar]
- [14].Lobel J, MacDonald IJ, Ciesielski MJ, Barone T, Potter WR, Pollina J, Plunkett RJ, Fenstermaker RA, Dougherty TJ, 2-[1-hexyloxyethyl]-2-devinyl pyropheophorbide-a (HPPH) in a nude rat glioma model: Implications for photodynamic therapy, Lasers Surg. Med. 29 (2001) 397. 10.1002/lsm.10001.abs. [DOI] [PubMed] [Google Scholar]
- [15].Garg AD, V Krysko D, Vandenabeele P, Agostinis P, Hypericin-based photodynamic therapy induces surface exposure of damage-associated molecular patterns like HSP70 and calreticulin, Cancer Immunol. Immunother. 61 (2011) 215–221. 10.1007/s00262-011-1184-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Leite DPV, Paolillo FR, Parmesano TN, Fontana CR, Bagnato VS, Effects of photodynamic therapy with blue light and curcumin as mouth rinse for oral disinfection: a randomized controlled trial, Photomed. Laser Surg. 32 (2014) 627–632. 10.1089/pho.2014.3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Li Y, Tan C-P, Zhang W, He L, Ji L-N, Mao Z-W, Phosphorescent iridium(III)-bis-N-heterocyclic carbene complexes as mitochondria-targeted theranostic and photodynamic anticancer agents, Biomaterials. 39 (2015) 95–104. 10.1016/j.biomaterials.2014.10.070. [DOI] [PubMed] [Google Scholar]
- [18].Henderson BW, Dougherty TJ, HOW DOES PHOTODYNAMIC THERAPY WORK?, Photochem. Photobiol. 55 (1992) 145–157. 10.1111/j.1751-1097.1992.tb04222.x. [DOI] [PubMed] [Google Scholar]
- [19].Feruszová J, Imreová P, Bodnárová K, Ševcovicova A, Kyzek S, Chalupa I, Gálová E, Miadoková E, Photoactivated hypericin is not genotoxic, Gen. Physiol. Biophys. (2016). 10.4149/gpb_2015045. [DOI] [PubMed] [Google Scholar]
- [20].Casas A, Di Venosa G, Hasan T, Batlle A, Mechanisms of Resistance to Photodynamic Therapy, Curr. Med. Chem. (2011). 10.2174/092986711795843272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gomez C, Muangnoi C, Sorasitthiyanukarn FN, Wongpiyabovorn J, Rojsitthisak P, Rojsitthisak P, Synergistic effects of photo-irradiation and curcumin-chitosan/alginate nanoparticles on tumor necrosis factor-alpha-induced psoriasis-like proliferation of keratinocytes, Molecules. (2019). 10.3390/molecules24071388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y, Histone demethylation mediated by the nuclear amine oxidase homolog LSD1, Cell. 119 (2004) 941–953. 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- [23].Nicholson TB, Chen T, LSD1 demethylates histone and non-histone proteins, Epigenetics. 4 (2009) 129–132. [DOI] [PubMed] [Google Scholar]
- [24].Mosammaparast N, Kim H, Laurent B, Zhao Y, Lim HJ, Majid MC, Dango S, Luo Y, Hempel K, Sowa ME, Gygi SP, Steen H, Harper JW, Yankner B, Shi Y, The histone demethylase LSD1/KDM1A promotes the DNA damage response, J. Cell Biol. 203 (2013) 457–470. 10.1083/jcb.201302092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Peng B, Wang J, Hu Y, Zhao H, Hou W, Zhao H, Wang H, Liao J, Xu X, Modulation of LSD1 phosphorylation by CK2/WIP1 regulates RNF168-dependent 53BP1 recruitment in response to DNA damage, Nucleic Acids Res. 43 (2015) 5936–5947. 10.1093/nar/gkv528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Palomera-Sanchez Z, Zurita M, Open, repair and close again: Chromatin dynamics and the response to UV-induced DNA damage, DNA Repair (Amst). (2011). 10.1016/j.dnarep.2010.10.010. [DOI] [PubMed] [Google Scholar]
- [27].Li J, Braganza A, Sobol RW, Base excision repair facilitates a functional relationship between Guanine oxidation and histone demethylation, Antioxid. Redox Signal. 18 (2013) 2429–2443. 10.1089/ars.2012.5107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Egolf S, Aubert Y, Doepner M, Anderson A, Maldonado-Lopez A, Pacella G, Lee J, Ko EK, Zou J, Lan Y, Simpson CL, Ridky T, Capell BC, LSD1 Inhibition Promotes Epithelial Differentiation through Derepression of Fate-Determining Transcription Factors, Cell Rep. (2019). 10.1016/j.celrep.2019.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Shafirstein G, Battoo A, Harris K, Baumann H, Gollnick SO, Lindenmann J, Nwogu CE, Photodynamic therapy of non-small cell lung cancer narrative review and future directions, Ann. Am. Thorac. Soc. (2016). 10.1513/AnnalsATS.201509-650FR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Prusevich P, Kalin JH, Ming SA, Basso M, Givens J, Li X, Hu J, Taylor MS, Cieniewicz AM, Hsiao P-Y, Huang R, Roberson H, Adejola N, Avery LB, Casero RA Jr, Taverna SD, Qian J, Tackett AJ, Ratan RR, McDonald OG, Feinberg AP, Cole PA, A selective phenelzine analogue inhibitor of histone demethylase LSD1, ACS Chem. Biol. 9 (2014) 1284–1293. 10.1021/cb500018s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ricq EL, Hooker JM, Haggarty SJ, Activity-dependent Regulation of Histone Lysine Demethylase KDM1A by a Putative Thiol/Disulfide Switch, J. Biol. Chem. 291 (2016) 24756–24767. 10.1074/jbc.M116.734426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chen Y, Yang Y, Wang F, Wan K, Yamane K, Zhang Y, Lei M, Crystal structure of human histone lysine-specific demethylase 1 (LSD1), Proc. Natl. Acad. Sci. U. S. A. 103 (2006) 13956–13961. 10.1073/pnas.0606381103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Schenk T, Chen WC, Göllner S, Howell L, Jin L, Hebestreit K, Klein H-U, Popescu AC, Burnett A, Mills K, Casero RA Jr, Marton L, Woster P, Minden MD, Dugas M, Wang JCY, Dick JE, Müller-Tidow C, Petrie K, Zelent A, Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia, Nat. Med. 18 (2012) 605–611. 10.1038/nm.2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Huang Z, Li S, Song W, Li X, Li Q, Zhang Z, Han Y, Zhang X, Miao S, Du R, Wang L, Lysine-specific demethylase 1 (LSD1/KDM1A) contributes to colorectal tumorigenesis via activation of the Wnt/β-catenin pathway by down-regulating Dickkopf-1 (DKK1) [corrected], PLoS One. 8 (2013) e70077. 10.1371/journal.pone.0070077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hu N, Clifford RJ, Yang HH, Wang C, Goldstein AM, Ding T, Taylor PR, Lee MP, Genome wide analysis of DNA copy number neutral loss of heterozygosity (CNNLOH) and its relation to gene expression in esophageal squamous cell carcinoma, BMC Genomics. (2010). 10.1186/1471-2164-11-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Jee BA, Lim H, Kwon SM, Jo Y, Park MC, Lee IJ, Woo HG, Molecular classification of basal cell carcinoma of skin by gene expression profiling, Mol. Carcinog. (2015). 10.1002/mc.22233. [DOI] [PubMed] [Google Scholar]
- [37].Duquette ML, Kim J, Shi LZ, Berns MW, LSD1 mediated changes in the local redox environment during the DNA damage response, PLoS One. (2018). 10.1371/journal.pone.0201907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Boxer LD, Barajas B, Tao S, Zhang J, Khavari PA, Znf750 interacts with KLF4 and RCOR1, KDM1A, And CTBP1/2 chromatin regulators to repress epidermal progenitor genes and induce differentiation genes, Genes Dev. (2014). 10.1101/gad.246579.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Nowis D, Makowski M, Stokłosa T, Legat M, Issat T, J. Goła̧b, Direct tumor damage mechanisms of photodynamic therapy, Acta Biochim. Pol. 52 (2005) 339–352. [PubMed] [Google Scholar]
- [40].Hopper C, PDT in head and neck cancer, Photodiagnosis Photodyn. Ther. 17 (2017) A38–A39. 10.1016/j.pdpdt.2017.01.085. [DOI] [Google Scholar]
- [41].Lange C, Bednarski PJ, Evaluation for synergistic effects by combinations of photodynamic therapy (PDT) with temoporfin (mTHPC) and Pt(II) complexes carboplatin, cisplatin or oxaliplatin in a set of five human cancer cell lines, Int. J. Mol. Sci. (2018). 10.3390/ijms19103183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Osaki T, Takahashi K, Ishizuka M, Tanaka T, Okamoto Y, Antimalarial drugs enhance the cytotoxicity of 5-aminolevulinic acid-based photodynamic therapy against the mammary tumor cells of mice in vitro, Molecules. (2019). 10.3390/molecules24213891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Mroz P, Castano AP, Wu MX, Kung AL, Hamblin MR, Anti-tumor immune response after photodynamic therapy, Photodyn. Ther. Back to Futur. (2009). 10.1117/12.822994. [DOI] [Google Scholar]
- [44].Reginato E, Wolf P, Hamblin MR, Immune response after photodynamic therapy increases anti-cancer and anti-bacterial effects, World J Immunol. 4 (2014) 1–11. 10.5411/wji.v4.i1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Falk-Mahapatra R, Gollnick SO, Photodynamic Therapy and Immunity: An Update, Photochem. Photobiol. (2020). 10.1111/php.13253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Gellén E, Fidrus E, Péter M, Szegedi A, Emri G, Remenyik É, Immunological effects of photodynamic therapy in the treatment of actinic keratosis and squamous cell carcinoma, Photodiagnosis Photodyn. Ther. (2018). 10.1016/j.pdpdt.2018.10.018. [DOI] [PubMed] [Google Scholar]
- [47].Udartseva OO, Zhidkova OV, Ezdakova MI, Ogneva IV, Andreeva ER, Buravkova LB, Gollnick SO, Low-dose photodynamic therapy promotes angiogenic potential and increases immunogenicity of human mesenchymal stromal cells, J. Photochem. Photobiol. B Biol. (2019). 10.1016/j.jphotobiol.2019.111596. [DOI] [PubMed] [Google Scholar]
- [48].Su S-T, Ying H-Y, Chiu Y-K, Lin F-R, Chen M-Y, Lin K-I, Involvement of histone demethylase LSD1 in Blimp-1-mediated gene repression during plasma cell differentiation, Mol. Cell. Biol. 29 (2009) 1421–1431. 10.1128/MCB.01158-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Bally A, Austin JW, Lu P, Boss J, The histone demethylase LSD1 inhibits PD-1 expression in acute viral infections, J. Immunol. 196 (2016) 129.7–129.7. [Google Scholar]
- [50].Maes T, Mascaró C, Sacilotto N, Lufino MM, Ciceri F, Targeting KDM1A with iadademstat in combination with immunotherapy in an in vivo model of melanoma., J. Clin. Oncol. (2019). 10.1200/jco.2019.37.15_suppl.e14248. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.