

Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal central nervous system neurodegenerative disease. Despite intense research, current ALS management remains suboptimal, from diagnosis to prognosis. Recognition of ALS phenotypic heterogeneity, global central nervous system dysfunction, genetic architecture, and development of novel diagnostic criteria are clarifying the spectrum of clinical presentation and facilitating diagnosis. Insights into ALS pathophysiology, identification of disease biomarkers and modifiable risks, along with new predictive models, scales, and scoring systems, and a clinical trial pipeline of mechanism-based therapies are changing the prognostic landscape. Although most recent advances have yet to translate to patient benefit, the view of ALS as a complex syndrome is already having tangible effects in the clinic. This review will outline these recent insights and discuss the status of ALS management for the general neurologist, along with future prospects, which may improve care and outcomes for ALS patients.
Keywords: Amyotrophic lateral sclerosis, biomarker, clinical overlap, diagnosis, epidemiology, exposome, genetic architecture, motor neuron disease, pathophysiology, phenotype, prognosis, scoring, staging
Introduction
Amyotrophic lateral sclerosis (ALS), a fatal central nervous system neurodegenerative disease, can be difficult to recognize, especially in the early stages. The disease is rare and more common illnesses are considered before ALS, frequently delaying diagnosis. However, the lifetime risk of ALS is approximately 1 in 350, though limited life expectancy reduces the prevalence.(1) Recent recognition of phenotypic heterogeneity and ALS as a complex syndrome that frequently includes behavioral deficits, may help physicians better recognize ALS earlier in the disease course. Development of new diagnostic criteria and identification of genetic risk factors could also expedite the diagnostic process.(2) Regarding prognosis, our clearer understanding of the multisystem nature of ALS including cognitive dysfunction and behavioral changes, has important ramifications for caregiving support and end of life decision making. Moreover, newly developed predictive models, scales, and scoring systems can give ALS patients and their physicians a clearer idea of their disease course.(2) Finally, advances in our understanding of disease pathophysiology are leading to mechanism-based and potentially disease-modifying therapies, currently in clinical trials. This review will outline these topics and current clinical practice for ALS, along with research advances, which may facilitate future improvements in diagnosis and prognosis for ALS patients.
ALS epidemiology
ALS incidence rises with age and is highest around 60 to 79 years,(3, 4) although variation can occur by ancestral background.(5) Some studies show stable incidence over the past two or three decades,(1) whereas others report a possible increase.(6, 7) Changes in perceived incidence may arise from improved diagnosis or changes in reporting standards over time, advocating the construction of well-curated population registries. It is unclear whether ALS incidence has changed in the past couple of decades, although it is anticipated to grow with an aging population.(8) ALS prevalence is also expected to increase due to an aging population in addition to improved management, which supports increased life expectancy.(8, 9) Still, ALS remains a relatively rare disease. Standardized global ALS incidence by meta-analysis is only 1·68 per 100,000 person-years of follow-up but varies by region.(10) In populations of predominantly European descent, such as Europe and North America, incidence is slightly higher than the global average and ranges from 1·71 to 1·89 per 100,000 and may even be higher within population-based studies.(11) Asian populations have lower incidences, varying from 0·73 per 100,000 in South Asia to 0·94 per 100,000 in West Asia, whereas Oceania universally has among the highest incidence rates.(7, 10) ALS incidence also varies by sex with an overall standardized male-to-female ratio of 1·35, which is affected by age of onset.(12) Genetics also plays a role; heritability is higher in mother-daughter pairs(1) whereas the most common known ALS risk gene, C9orf72, lowers onset age in males versus females.(13) Thus, ALS arises from complex interrelationships between age, sex, and genetics,(14) which has implications for preclinical and clinical research and clinical trials.
ALS clinical presentation
ALS phenotypic heterogeneity
ALS presents as a combination of upper (UMN) and lower motor neuron (LMN) dysfunction affecting the bulbar, cervical, thoracic, and/or lumbar segments.(2) This leads to progressive weakness of voluntary skeletal muscles involved in limb movement, swallowing (dysphagia), speaking (dysarthria), and respiratory function, with different clinical presentations (Panel 1). Sphincter and extraocular muscles are classically spared, although autonomic dysfunction in ALS is increasingly recognized, e.g., urinary urgency and incontinence.(15) Clinical weakness spreads contralaterally and rostrally and caudally, most often in an anatomically contiguous manner. A recent survey of ALS patients found that 85% had focal onset in one body segment, which progressed to the contralateral side and then to adjacent anatomical segments.(16) Disease spread to non-contiguous segments was less common.
Panel 1. Definitions of ALS motor signs and phenotypes.
Lower motor neurons (LMN): Brainstem cranial motor nerve nuclei or anterior horn cells; LMN dysfunction is characterized by muscle weakness, atrophy, and fasciculations.
Upper motor neurons (UMN): Betz cells in layer 5 of the primary motor cortex; UMN dysfunction is characterized by increase and pathologic reflexes (including Hoffmann, Babinski, snout), pathologic spread of reflexes, preserved reflexes in a weak limb, and/or spasticity.
Bulbar ALS: Phenotype presents with weakness starting in the muscles controlling speaking and swallowing. Both LMN and UMN signs are present.
Pseudobulbar palsy: A nonclassical subset of bulbar onset, characterized by prominent bulbar features, predominantly from UMN signs, which slowly spread to limbs.
Pseudobulbar affect (PBA): Uncontrollable emotional outbursts, including laughing, crying, and excessive yawning.
Classical ALS: Phenotype presents with muscle weakness starting in the limbs. Both LMN and UMN signs are present.
Cervical onset ALS: A subset of classical ALS with weakness commencing in the upper limbs, especially hand weakness.
Lumbar onset ALS: A subset of classical ALS with weakness commencing in the lower limbs, especially foot drop.
Flail arm: Prominent LMN dysfunction initially causing proximal > distal muscle weakness in the arms. Unlike progressive muscular atrophy, patients with flail arm do manifest progressive UMN dysfunction. This entity may also be referred to as brachial amyotrophic diplegia.
Flail leg: LMN dysfunction causing muscle weakness in the legs. Unlikely progressive muscular atrophy, this phenotype does not generalize or generalizes very slowly.
Primary lateral sclerosis*: UMN dysfunction causing weakness in muscles controlling limbs, swallowing, and speaking. Less commonly causes respiratory dysfunction.
Pyramidal: Like PLS but additionally eventually exhibiting LMN signs.
Progressive muscular atrophy*: LMN dysfunction causing weakness in muscles controlling limbs, swallowing, speaking, and respiratory function.
Respiratory onset: LMN and UMN dysfunction causing weakness commencing in the respiratory muscles.
Hemiplegic: Predominantly UMN dysfunction causing muscle weakness in one side of the body.
Cachexia: Unexplained weight and muscle loss.
*This review considers primary lateral sclerosis and progressive muscular atrophy are on the spectra of ALS phenotypes, although they may also be considered as separate clinical entities.
ALS presents as multiple phenotypes (Figure 1A–B; Appendix Table 1). Bulbar onset and spinal (cervical, lumbar) onset ALS are the most common presentations, each constituting about a quarter to a third of cases, with less frequent manifestations of flail arm and leg, primary lateral sclerosis, progressive muscular atrophy, respiratory onset, and hemiplegic presentations.12,13 This review considers primary lateral sclerosis and progressive muscular atrophy are on the spectra of ALS phenotypes, although they may also be considered as separate clinical entities. Age, sex, and genetics contribute to ALS phenotypes. Older female patients may more commonly develop bulbar onset ALS, younger males classical ALS, younger males and females pure UMN diseases, males flail arm variant, older males flail leg variant and respiratory onset.(14) Specific genetic mutations favor certain phenotypes (see “ALS genetic architecture” section). One recent study of German and Chinese registries suggest that phenotypes may vary globally.(18) German ALS patients have an older onset age (66.6 years), larger proportion of bulbar onset (35.9%), and smaller male-to-female ratio (1.33) versus Chinese patients (53.2 years onset age, 22.8% bulbar, 1.51 male-to-female ratio).(18) Consensus phenotyping between registries would advance our knowledge of age, sex, genetics, and racial/ethnic contributions to phenotypes.
Figure 1. ALS phenotypic variation and spectrum with FTD.
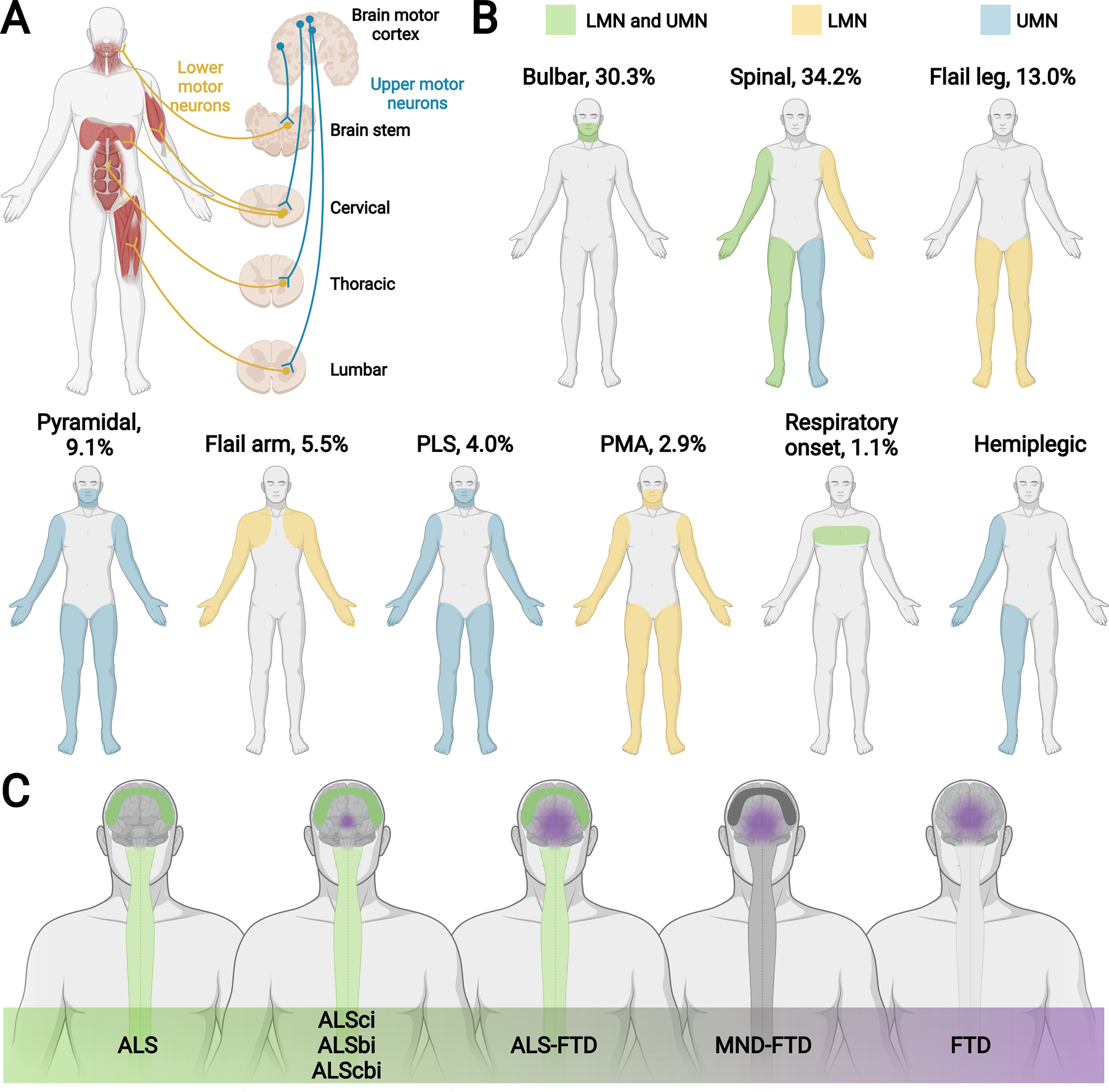
(A) Schematic showing upper motor neurons (UMN; blue), which relay signals from the motor cortex to the lower motor neurons (LMN; yellow, i.e., cranial motor nerve nuclei in the brainstem and anterior horn cells in the spinal cord), which relay signals to the muscles. Motor neurons connecting within the brain stem innervate, among other muscles, cranial muscles. Initial UMN and LMN degeneration in the brain stem are linked to bulbar onset ALS. Motor neurons connecting within the cervical region of the spinal cord innervate, among other muscles, upper limb and respiratory muscles. Motor neurons connecting within the thoracic and lumbar regions of the spinal cord innervate, among other muscles, accessory respiratory, abdominal, and lower limb muscles. Initial UMN and LMN degeneration in the cervical and lumbar regions are linked to spinal onset ALS. (B) ALS patients can present with signs of UMN (blue), LMN (yellow), and combined UMN + LMN (green) dysfunction. Most common ALS phenotypic presentations are bulbar and classical spinal limb onset (cervical, lumbar). Less common ALS phenotypic presentations are flail leg, pyramidal, flail arm, primary lateral sclerosis (PLS), progressive muscular atrophy (PMA), respiratory onset, and hemiplegic. Proportion of various ALS phenotypes shown in the figure as the percentage (%) of a total representative ALS population.(14, 17) Pyramidal is predominantly UMN, as shown in the figure, but still exhibits some LMN signs, differentiating it from PLS. See Appendix Table 1 for more information. (C) ALS occurs on a continuum with FTD. ALS is on one end of the spectrum and presents with pure motor signs from UMN + LMN neurodegeneration (green, spinal cord and motor cortex degeneration). FTD is on the other end of the spectrum and presents with behavioral and/or cognitive deficits from frontotemporal neurodegeneration (purple frontotemporal lobe degeneration). After pure ALS are ALS patients not meeting FTD criteria, defined as ALS cognitive impairment (ALSci), ALS behavioral impairment (ALSbi), and ALS cognitive and behavioral impairment (ALScbi) (green, spinal cord and motor cortex degeneration; small purple sphere of frontotemporal lobe degeneration). Next are ALS patients meeting FTD criteria, defined as ALS-FTD (green, spinal cord and motor cortex degeneration; purple frontotemporal lobe degeneration). Patients on the remainder of the continuum have FTD but do not meet the criteria for ALS. Patients still exhibiting evidence of motor neuron disease (MND) with FTD are defined as MND-FTD (dark grey, spinal cord and motor cortex degeneration; purple frontotemporal lobe degeneration) and patients with no MND signs have FTD (purple, frontotemporal lobe degeneration).
ALS cognitive and behavioral changes
Classically, ALS was predominantly considered a disease of motor dysfunction, e.g., dysarthria, dysphagia, weakness of upper and/or lower limb muscles. However, cognitive and behavioral changes, which can occur early in the disease course,(19, 20) are now recognized to occur in 35 to 50% of ALS patients.(21, 22) Individuals with ALS experience loss of normal language and executive function, i.e., poor working memory, inhibition, and fluency. Typically, more long-term memory and spatial domains remain intact.(21) Other behavioral changes include apathy, irritability, disregard for hygiene, and eating habit changes. Approximately 15% of ALS cases meet the diagnostic criteria for frontotemporal dementia (FTD).(20, 23) Furthermore, depression, anxiety, and sleep disruptions occur in ALS(24) along with pseudobulbar affect, which causes emotional lability.16
These cognitive and behavioral changes support the concept that ALS is a global neurodegenerative disease along the same continuum as FTD (Figure 1C). Transactive response DNA binding protein (TDP)-43 proteinopathy, an almost universal pathological hallmark of ALS, is present in ~97% of cases and ~50% of FTD cases. Mild deficits in executive function, language, and fluency have 100% specificity for TDP-43 pathology in non-motor brain regions corresponding to these domains.(25) Certain patient characteristics, such as C9orf72 status(26, 27) and bulbar onset,(27) are strong determinants of cognitive impairment and may help the physician and patient to anticipate this complication. Furthermore, cognitive dysfunction and behavioral abnormalities may be prognostic of disease stage.(21) In a report of 146 ALS patients, cognition worsened in 30% of cases after 6-months, even among patients that were initially normal.(22) Those patients with cognitive decline had a more rapid clinical progression and shorter survival. Network analyses of brain MRIs demonstrate widespread disruption of motor and extra-motor networks that correspond with ALS phenotypes. Specifically, abnormal structural connectivity correlates with motor impairment, whereas disrupted functional connectivity aligns with changes in cognition and behavior.(28)
Collectively, this new understanding of ALS as a multi-system disorder underscores the importance of managing cognitive decline and neuropsychological problems, e.g., depression, dysfunctional sleep, apathy and irritability.(24) Importantly, when cognitive symptoms emerge, care teams should engage early with patients and their families to inquire about end-of-life care preferences to ensure the patient plays an active role in these important conversations.
ALS diagnosis
ALS criteria
ALS patients are unlikely to encounter a neurologist early in the diagnostic journey;(29, 30) therefore, there should be a low threshold for neurological referral when patients present with progressive dysarthria, dysphagia, limb weakness, or neuromuscular respiratory failure. The ALS Association “thinkALS” tool encourages early neurological referral to avoid unnecessary procedures, begin patients on disease-modifying treatments, and fast-track patient enrollment into clinical trials.(31) Additional clues indicating a diagnosis of ALS include unexplained weight loss, pseudobulbar affect, changes in cognition or executive functioning, and a family history of ALS or other neurodegenerative diseases. Clinical features that do not support ALS include sensory, sphincter, and autonomic nervous system dysfunction and anterior visual pathway abnormalities. A detailed neurological examination should identify signs of UMN and LMN dysfunction in bulbar, cervical, thoracic, and/or lumbosacral segments (Panel 2).
Panel 2. ALS diagnosis.
Clinical history: Symptoms (e.g., weakness, time course) and family history of ALS or other neurodegenerative diseases.
Neurological examination: Signs of UMN and LMN dysfunction in bulbar, cervical, thoracic, and/or lumbosacral segments, e.g., hand weakness (split hand), foot drop. Unexplained weight loss, cognition or executive functioning dysfunction, and pseudobulbar affect are additional signs.
Electrodiagnostic testing: Nerve conduction studies and needle electromyography to confirm LMN signs.
Laboratory testing: Serology should be normal except for elevated creatine phosphokinase levels, which can also lead to abnormal liver function tests.
Magnetic resonance imaging (MRI): Imaging the spinal cord by MRI is essential to rule out more common differential diagnoses, e.g., disc herniation, cord compression.
Criteria: Most neurologists employ the revised El Escorial criteria.(32)
Classifies ALS patients as possible, probable, probable laboratory supported, and definite, based on clinical presentation and electrodiagnostic findings.
The revised El Escorial criteria are:
(A) Presence of:
(A:1) LMN signs by clinical, electrodiagnostic testing, or neuropathologic exam,
(A:2) UMN signs by clinical examination, and
(A:3) progressive symptom or sign spread within a region or to other regions, as determined by history or exam,
With:
(B) Absence of:
(B:1) electrodiagnostic or pathological evidence of other diseases explaining LMN and/or UMN signs, and
(B:2) neuroimaging evidence of other diseases explaining the observed clinical and electrodiagnostic signs.
The diagnostic categories are:
Clinically definite: Clinical evidence of (1) UMN + LMN signs in the bulbar and two spinal regions OR (2) UMN + LMN signs in three spinal regions.
Clinically probable: Clinical evidence of UMN + LMN signs in at least two regions with UMN signs rostral to LMN signs.
Clinically probable – laboratory supported: Clinical evidence of (1) UMN + LMN signs in one region or UMN signs alone in one region AND (2) LMN by electrodiagnostic criteria in at least two regions.
Clinically possible: Clinical evidence of (1) UMN + LMN in one region OR (2) UMN signs in two or more regions OR (3) LMN signs are rostral to UMN signs.
Clinical history and neurological examination are accompanied by serological and electrodiagnostic testing. ALS patients have normal serology, except for elevated creatine phosphokinase levels in some cases. Other abnormal serologies call into question an ALS diagnosis (see “Differential diagnosis and ALS overlap syndromes” section). Nerve conduction studies exclude sensory nerve involvement and motor nerve conduction block and needle electromyography confirm LMN involvement, with the provision that testing of distal muscles and muscles in the involved clinical segment have the highest sensitivity.(33, 34) Most neurologists still use the revised El Escorial criteria to subclassify ALS, which categorizes patients as possible, probable, probable laboratory supported, and definite ALS, depending on clinical presentation and electromyography findings. The revised El Escorial criteria are outlined in Panel 2, since they are most widely used.(32)
Regarding advances in diagnostic criteria for ALS, the Gold Coast Criteria were recently proposed to simplify and potentially replace the revised El Escorial and improve inter-rater reliability (Appendix Table 2).(35) The Gold Coast criteria are primarily based on clinical presentation, although they do not consider cognitive changes, which the authors noted were covered by the 2017 Strong criteria.(36) Gold Coast classifies patients as having or not having ALS, streamlining diagnostic certainty and eliminating confusion to patients and their relatives from El Escorial terminology. A comparison of the sensitivity and specificity of the various criteria reveal that Gold Coast criteria are the most sensitive while El Escorial are the most specific (Appendix Table 2). Additionally, the revised El Escorial Criteria provide information that the Gold Coast criteria do not, such as the distribution of clinical segmental involvement, which is important for stratifying disease severity in ALS patients. Although the revised El Escorial presently remain the mainstay of ALS diagnosis, the field may be slowly moving towards simpler criteria, such as the Gold Coast.
Overall, early diagnosis of ALS is important. Educational efforts for physicians most likely to encounter ALS patients during initial symptom onset are essential to support prompt recognition of the disease with timely initiation of treatment. As simplified diagnostic criteria become more universally accepted, we anticipate that more practitioners will recognize and treat ALS early in the disease course.
ALS cognitive assessment
While not part of formal ALS diagnostic criteria, it is essential to evaluate cognition and behavior in ALS patients, despite potentially further fatiguing individuals undergoing long and complex clinical visits. Assessments of cognitive and behavioral impairment are essential as they relate to prognosis and progression rate, and, thus, inform clinical management.(21, 22) Assessment of cognitive impairment in ALS patients should include multiple cognitive domains (e.g., executive and language dysfunction, social cognition).(37) Behavioral impairment (e.g., apathy, disinhibition, loss of empathy, compulsive behavior, etc) also affects the well-being of patients and family-members and requires evaluation.
Some ALS patients are diagnosed with FTD (ALS-FTD), as defined by the Neary(38) or Rascovsky(39) criteria. For patients not meeting formal FTD criteria, the revised Strong criteria define ALS patients with cognitive dysfunction as “ALS cognitive impairment” (ALSci), with behavioral problems as “ALS behavioral impairment” (ALSbi) or with both, as “ALS combined cognitive behavioral deficits” (ALScbi) (Appendix Table 3).(36) Several assessment batteries can classify these changes. The Edinburgh Cognitive and Behavioral ALS Screen (ECAS) is a validated, multidomain assessment tool developed for ALS patients, which can be administered by neuropsychological and non-neuropsychological professionals.(37) ECAS, available in 23 languages, covers the largest number of ALS-specific cognitive or behavioral assessment scales. Incorporating ECAS into ALS management has a positive impact on the quality of care, by stimulating end-of-life care discussions, referrals to other services, and identifying caregiver support needs.(40)
The ALS Cognitive Behavioral Screen, available in three languages, can also identify cognitive and behavioral impairment and FTD, in ALS patients.(37) The ALS-FTD Questionnaire (ALS-FTD-Q) is completed by healthcare professionals or caregivers to assess behavioral changes in ALS patients.(37) ALS-FTD-Q, translated into nine languages, identifies patients with behavioral variant FTD. The Beaumont Behavioral Inventory is a new screening tool for evaluating behavioral impairment in ALS patients and may be more sensitive than ALS-FTD-Q.(37)
Overall, it is important to recognize that cognitive symptoms are a manifestation of ALS, and properly identifying these symptoms improves disease management, counseling, and prognostication. Since cognitive symptoms may change with disease progression, it is critical to regularly assess them to best care for the patient. Future directions include standardizing cognitive assessments for in-clinic screening, determining whether neuropsychologists should become part of the regular multidisciplinary team, and developing evidence-based treatments for cognitive impairment in ALS.
ALS genetic architecture
ALS is presently classified as either familial or sporadic. Familial ALS, which constitutes 10 to 15% of cases, is defined as inheritance among family members of ALS and associated syndromes, e.g., FTD.(41) About 70% of familial cases have mutations within known ALS genes. Sporadic ALS, which constitutes the remaining approximately 85% of cases, is defined as disease arising in patients lacking a family history of ALS. About 15% of sporadic ALS patients harbor “private” pathogenic mutations to known ALS genes, i.e., mutations limited to a single individual, hence lacking a family history of ALS.(41) There is no known cause in the remaining 85% of sporadic ALS cases. Apparently sporadic cases harboring low penetrant mutations and belonging to small families or having incomplete or poor knowledge of family history may in fact be familial ALS. Thus, familial ALS may be underreported and represent closer to 20% of cases.(42),(43) As genetic testing becomes more widely implemented and potential candidate therapies more targeted, it may become useful to drop the familial versus sporadic dichotomy of ALS in favor of genetically confirmed versus non-genetically confirmed ALS, i.e., presence versus lack of an ALS mutation underpinning molecular subclassification of disease.
ALS genetic architecture is highly complex and largely based on monogenic inheritance of rare variants, i.e., single disease-causing genes (Figure 2A).(44) To date, over 40 ALS-associated genes have been identified,(45, 46) which vary in frequency, mode of inheritance (mostly dominant, rarely recessive), and penetrance (Figure 2B; Appendix Table 4). The most common and penetrant mutations are C9orf72, TARDBP, SOD1, and FUS,(45) although the frequency of genetic subtypes does vary by population ancestry.(47) Some ALS genes are not necessarily disease-inducing, but rather confer an increased risk of developing ALS, e.g., ANG, ATXN2, DCTN1.(45) Importantly, uncertainty remains on the relevance of some identified genes to ALS, which require further confirmation and replication efforts.(48) Consortia of ALS genetics experts can curate and maintain an up-to-date list of ALS genes as evidence emerges,(49) facilitating clinical translation for genetic testing. Since ALS genetic architecture is complex, it is advisable that specialist ALS centers perform genetic testing to avoid over diagnosing or missing genetic ALS. It is also important to recognize that genetic testing in ALS might not identify rare pathogenic variants, i.e., allele frequency less than 1%.
Figure 2. ALS genetic architecture.
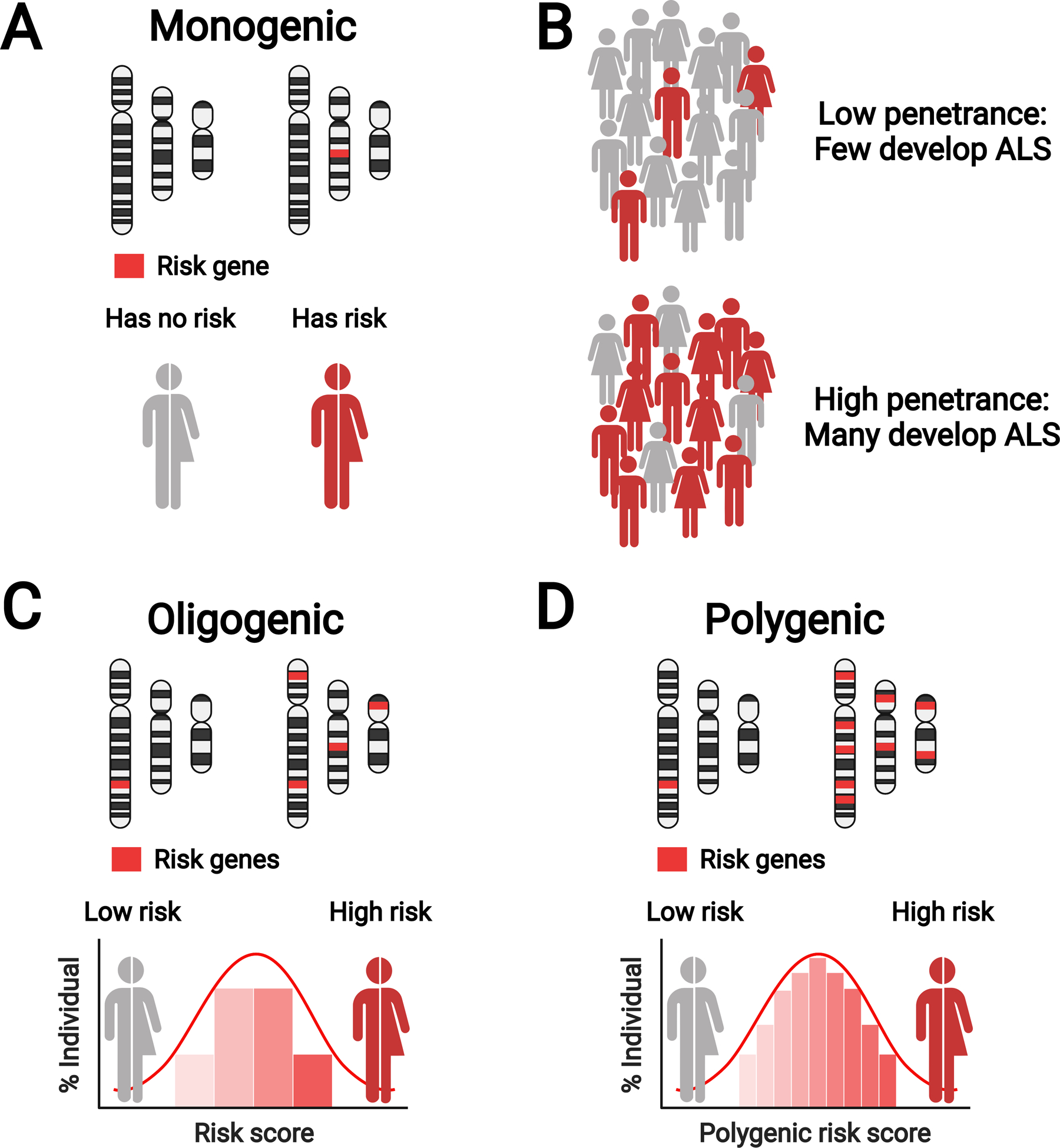
ALS genetics is characterized by monogenic, oligogenic, and polygenic risk; image featuring only three representative chromosomes (within each panel, chromosomes on the left for healthy person, on the right for person with ALS). (A) Monogenic inheritance in ALS, characterized by inheritance of a single gene. (B) ALS genes are not fully penetrant and pathogenicity of certain variants is uncertain. Left: In a population of gene carriers, low penetrance variants lead to a low frequency of ALS onset (red figures). Right: In a population of gene carriers, high penetrance variants lead to a high frequency of ALS onset (red figures). (C) Oligogenic inheritance in ALS, characterized by inheritance of several genes (four shown in the figure). (D) Polygenic inheritance in ALS, characterized by inheritance of many genes (nine shown in the figure). Created with BioRender.com. Adapted, with permission, from Goutman et al. The Lancet Neurology, 2022.
In addition to primary monogenic inheritance in ALS, interest in the impact on oligogenic and polygenic inheritance on disease risk has also gained traction. Several studies highlight that oligogenic inheritance, meaning a trait or disease controlled by inheritance of several genes, may have a role in ALS risk and/or disease progression (Figure 2C).(50, 51) Genetic screening identified a subset of sporadic ALS patients harboring two or more variants in ALS genes; these patients were more likely to have earlier onset disease versus patients harboring one or no variants.(50, 51) Polygenic inheritance, arising from inheritance of multiple genetic variants, is also a component of ALS genetic architecture (Figure 2D).(52, 53) Analysis of ALS genetic profiles identified shared polygenic risk of ALS with traits and single nucleotide polymorphisms correlated with smoking status, physical activity, cognitive performance, and educational attainment,(52) as well as obesity-related traits,(52, 53) particularly hyperlipidemia. Our growing knowledge of ALS genetic architecture is due in great part to large collaborative projects, which are driving discovery in this relatively rare disease, such as the ALS Sequencing Consortium,(54) International ALS Genomics Consortium,(55) Genomic Translation for ALS Care Consortium,(54) Answer ALS Foundation,(54) and Project MinE.(54) We anticipate that these consortia will continue to bear fruit and foster further investigation.
Importantly, ALS is also characterized by incomplete heritability, meaning genetics does not fully account for all disease burden. Estimates vary but most studies report heritability of 45 to 50% in ALS dyads, driven in large part by rare genetic variants.(1) However, heritability estimates can be as high as 66% in some dyad comparisons and as low as 37% in patients lacking a known genetic risk.(1) In addition to rare variants,(56) several additional factors can account for “missing heritability” in ALS, such as alterations in the non-coding genome, structural variants,(57) epigenetic changes,(58) and environmental factors.(59) The contribution of the environment has led to the “gene-time-environment” hypothesis of ALS,(60) which proposes that an interaction of genes and environment over time causes ALS through a multistep process.(61) An evolving body of evidence demonstrates that the environment does impact ALS risk and progression in a gene-dependent manner (see “Environmental exposure in ALS” section).
As therapeutics that target certain genetic forms of ALS become a possibility, genetic testing for all patients with ALS will likely become standard practice. Genetic treatment paradigms will increase the need for classifying and assessing genetic variants in ALS. Additionally, partnership with genetic counselors will expand to facilitate discussions of these complex results with patients and their families.(62)
Differential diagnosis and overlap syndromes
General physicians and even specialist neurologists, may not initially recognize an ALS diagnosis in a patient with ALS symptoms due to overlap of disease presentation with other conditions. Thus, classical differential diagnosis based on clinical presentation is an important element of the diagnostic process in ALS (Figure 3; Appendix Table 5).
Figure 3. ALS differential diagnosis.
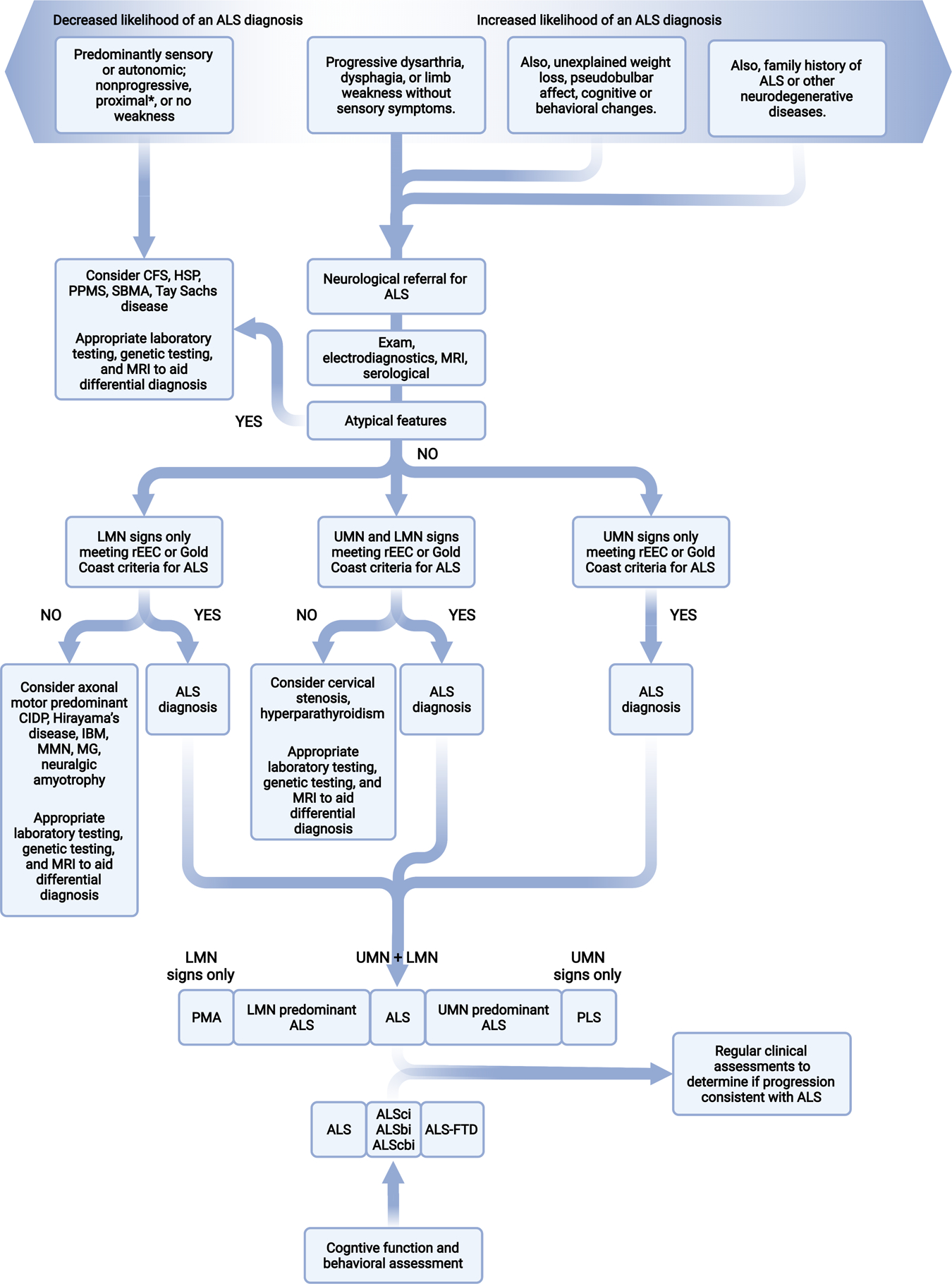
Differential diagnosis, represented here by a flowchart for the classical process using symptoms and signs, is central to the diagnostic process in ALS. At minimum, individuals suspected of ALS will undergo physical and neurological exams, electrodiagnostic assessment, MRI of involved regions, and relevant serological testing. This figure is based on a summary of potential differential diagnoses for diseases more common or as common as ALS is outlined in Appendix Table 3. Overlap of known ALS genes with other diseases and syndromes also occurs and is outlined in Appendix Table 4. CFS, cramp-fasciculation syndrome; CIDP, chronic inflammatory demyelinating polyneuropathy; HSP, hereditary spastic paraparesis; IBM, inclusion body myositis; LMN, lower motor neuron; MMN, multifocal motor neuropathy; MG, myasthenia gravis; PPMS primary progressive multiple sclerosis; rEEC, revise El Escorial criteria; SBMA, spinobulbar muscular atrophy; UMN, upper motor neuron. *Several potential differential diagnoses present with proximal weakness and should be considered along with flail arm ALS, which also presents with proximal greater than distal upper extremity weakness. Thus, check for increased proximal reflexes on exam and neurogenic motor unit action potentials on electromyography.
Diseases more common than ALS are often considered and thoroughly evaluated first, which ultimately delays an ALS diagnosis. Conditions that most commonly mimic ALS include multifocal motor neuropathy with conduction block, axonal motor predominant chronic inflammatory demyelinating polyneuropathy, spinobulbar muscular atrophy, and inclusion body myositis.(63) Simultaneous cervical nerve root and spinal cord compression by disc herniations, tumors or malformations may cause combined LMN signs in the arms and UMN in the legs, and be misdiagnosed as classical ALS.(63) UMN dominant ALS or primary lateral sclerosis may be confused with hereditary spastic paraplegias or primary progressive multiple sclerosis. Additional, but rare, differential diagnoses include hyperparathyroidism and hexosaminidase A-/B-deficiency.(63) Since some of these conditions are treatable, it is important to rule out these possibilities.
In conjunction with clinical presentation, genetic testing is increasingly utilized to explain disease cause and predict family risk. Risk ALS genes can cause other syndromes or phenocopy alternative neurodegenerative diseases (Appendix Table 4). C9orf72 expansions, the most common ALS gene, are linked to movement disorders(64, 65) and phenocopy Huntington’s disease in patients lacking huntingtin (HTT) expansions.(66) Conversely, ALS patients may harbor HTT repeat expansions simultaneously with TDP-43 inclusions.(67) Thus, patients may present with atypical ALS delaying diagnosis. Additional ALS genes overlap with other syndromes and an improved understanding of the complexity of genotype-phenotype relationships will expedite ALS diagnosis. Finally, ALS is associated with neuropsychiatric illnesses, such as psychosis and suicidal ideation;(68, 69) thus, clinicians should obtain comprehensive detailed family history, not just of ALS, but of neurodegenerative and neuropsychiatric illnesses.
ALS risk, progression, and pathophysiology
Identifying factors that increase ALS risk and progression is central to patient diagnosis and care. Genetics are a major ALS risk factor (see “ALS Genetic Architecture”; Appendix Table 4). For instance, C9orf72 expansions are penetrant and confer high ALS risk, and are also associated with bulbar onset(14) and a decreased survival (70) in some studies. However, there are genetic mutations that confer risk but do not impact progression; therefore, risk and progression can be independent processes and factors influencing either or both are an active area of research.(71) It is increasingly recognized that a patient’s cumulative environmental lifetime exposures, known as the exposome, can also confer ALS risk and may accelerate disease progression.(72) Independent of whether risk is secondary to genetics and/or the exposome, a knowledge of ALS pathophysiology will promote the development of novel treatment and prevention strategies, such as genetic therapies for asymptomatic carriers of highly penetrant pathogenic mutations.(73)
Molecular pathomechanisms in ALS
In ALS, pathological processes arise from toxic gain-of-function or loss-of-function mutations to the approximately 40 ALS genes known to date. Toxicity also occurs from aggregates of both wild-type and mutant proteins, which is a universal pathological feature in sporadic and familial ALS.(74) Pathophysiological processes broadly fall into four major categories, impaired RNA metabolism, altered proteostasis/ autophagy, cytoskeletal/ trafficking defects, and mitochondrial dysfunction.(75) Several ALS genes, including C9orf72, TARDBP, and FUS, impair RNA metabolism. Aggregation of the DNA/RNA binding proteins, TDP-43 and FUS, into inclusions impairs their normal function, causing broad changes to transcription and RNA processing. TDP-43, among several other ALS genes, also dysregulates proteostasis and autophagy by preventing the clearance of damaged proteins. Multiple mutant ALS genes, such as tubulin alpha 4a (TUBA4A) and profilin 1 (PFN1), induce cytoskeletal and/or tubulin defects, blocking axonal trafficking. Mitochondrial dysfunction, as triggered by SOD1, is a central ALS characteristic, which also increases oxidative stress.
Although much progress has been made, the full molecular underpinnings of ALS pathophysiology are incompletely understood. In addition to the major abovementioned processes, TDP-43 and SOD1 aggregates also transfer from cell-to-cell in prion-like transmission,(76, 77) which would propagate ALS pathology. TARDBP, FUS, and a handful of other genes, are linked to dysfunctional DNA repair in ALS; for instance, loss of nuclear TDP-43 induces accumulation of double-stranded DNA breaks,(78) which would compromise genome stability. TDP-43 aggregates,(79) mutant FUS,(80) and C9orf72 repeat expansions(81) also impair nucleocytoplasmic transport, the shuttling of cargo between the nucleus and cytoplasm.(79) Dipeptide repeat proteins derived from mis-translated C9orf72 expansion transcripts are neurotoxic and may promote heterochromatin anomalies(82) and TDP-43 aggregation.(83)
Central and peripheral inflammatory mechanisms are important contributors to ALS,(84) both in the context of specific genetic mutations(85–87) and likely as a consequence of the general disease process in sporadic disease.(88, 89) In ALS, changes occur in specific immune cell levels,(88, 89) their activation state,(88) and cytokine production.(86, 87) Importantly, immune system involvement in ALS is double-edged; a protective initial response is overcome by a destructive cytotoxic phase.(84) Hypermetabolism is also a broad ALS characteristic,(90) both dependent and independent of ALS mutations, and metabolomics investigations(91) could shed light on the specific molecular changes that underscore disease progression. Pathways related to ALS genes, inflammation, hypermetabolism and other continued insights into the pathological mechanisms underlying ALS provide an essential knowledge base for ALS therapeutic development (see “Novel ALS treatment approaches” section) and ALS prevention strategies.
Environmental exposure in ALS
The gene-time-environment hypothesis of ALS suggests that genetic susceptibility, age-related cellular damage, and a burden of environmental exposures combine to trigger ALS.(60) Several lines of evidence support this model. First, genetic variants do not fully account for ALS.(92) Second, population-based modeling of ALS indicates that disease occurs in a multi-step process,(61) even in patients with highly penetrant monogenic mutations, e.g., mutant SOD1.(93) Third, a growing body of research supports the association of environmental exposures with disease risk, with a new focus on the ALS exposome.(59)
The ALS exposome is defined as the cumulative lifetime impact of environmental exposures, including lifestyle factors. Since the exposome involves exposures throughout a patient’s lifespan, multiple study designs are needed to interrogate its role in ALS. Many case-control studies have explored the relationship between occupational, residential, and avocational environmental risk factors on ALS risk. While studies leveraging population-based registries would provide a higher level of evidence, studies based on retrospective cohorts show reassuringly consistent results (Appendix Table 6).
Of exposures with documented relevance to ALS, plasma persistent organic pollutants(94) and blood metals(95),(96) correlate with disease risk and shortened survival.(72) Lifestyle factors associate with ALS risk, including higher cigarette pack-years, a lower current body mass index and lifetime alcohol consumption.(97) Some relationships are dependent on C9orf72 status,(97) demonstrating an interaction between genes and environment. Physical activity as an ALS risk is supported by several studies,(97, 98) including analysis of the National Football League players.(99) Military service is also a recurring theme in ALS risk assessments.(100)
There are important unanswered questions relating to the ALS exposome. Are there periods of greater susceptibility to exposure throughout life, which increase ALS risk? Will it be possible to adopt a preventative approach to ALS if modifiable risks are identified? Prospective studies using well curated population registries and biorepositories can help answer these questions and are a future goal of the field.(59)
ALS prognosis
ALS prognosis is dependent on disease progression. Currently, clinicians monitor ALS progression using the ALS functional rating score-revised (ALSFRS-R), a multidomain assessment, which also serves as the gold standard for primary efficacy outcomes in clinical trials.(101) Respiratory function, which is a domain of the ALSFRS-R, provides prognostic information.(102) One shortcoming of the ALSFRS-R is that certain subscores increase with symptom improvement despite continued underlying disease progression.(101, 103) The Rasch-Built Overall ALS Disability Scale (ROADS) was designed to specifically capture functional decline arising from the underlying disease course,(103) thereby overcoming the limitations of the ALSFRS-R. The ROADS currently awaits clinical validation prior to widespread adoption.
New staging paradigms have also been developed to inform prognosis. Patients assessed with these tools, the King’s(104) and ALS Milano-Torino Staging (ALS-MiToS),(105) consistently progress along stages, which are associated with decreasing median survival (Figure 4A–B). The King’s is more sensitive early in the disease course, the ALS-MiToS later in the disease course.(106, 107) Neither staging system is yet in widespread clinical use.
Figure 4. ALS risk and prognosis.
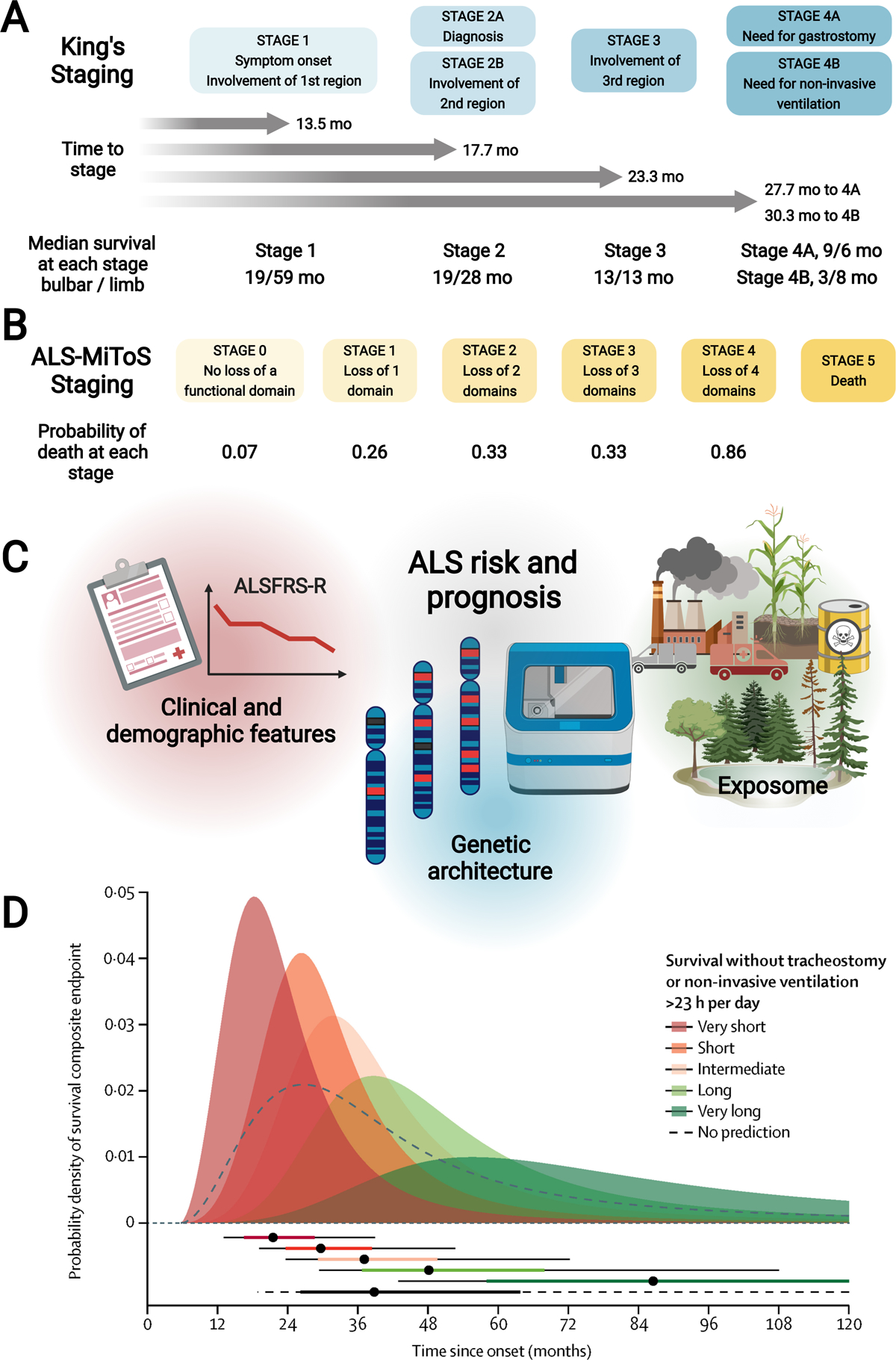
(A) King’s staging with four stages indicated (1, 2A/B, 3, 4A/B; blue); time to progress to stages and median survival at each stage (in months) are also annotated. (B) ALS-MiToS staging with six stages indicated (0, 1, 2, 3, 4, 5; orange); staging based on four functional domains from the ALSFRS-R: (i) movement (walking/self-care; ALSFRS-R question 6 or 8); (ii) swallowing (ALSFRS-R question 3); (iii) communicating (ALSFRS-R questions 1 and 4), and (iv) breathing (ALSFRS-R question 10 or 12). Intensifying color indicates progression along stages for both King’s and ALS-MiToS. (C) Schematic overview of factors that affect ALS risk (onset) and prognosis, which include clinical and demographic features (e.g., age at onset, segment onset, ALSFRS-R progression rate, forced vital capacity, FTD), genetic architecture (e.g., rapidly progressive SOD1A5V, slowly progressive DCTN1 mutations), and exposome (e.g., environmental exposures). (D) ENCALS prediction model of ALS prognosis, represented, with permission, from Westeneng, The Lancet Neurology, 2018.(70) The model leverages 8 clinical predictors to the composite endpoint (survival without tracheostomy or non-invasive ventilation >23 hours per day): age at onset, time to diagnosis, ALSFRS-R progression rate, forced vital capacity, bulbar onset, definite ALS by revised El Escorial criteria, FTD, and C9orf72 repeat expansion. Top: The model defines five survival groups: very short (red; predicted median survival [MS] 17·7 months), short (orange; predicted MS 25·3 months), intermediate (light orange; predicted MS 32·2 months), long (light green; predicted MS 43·7 months), and very long (predicted MS green; 91·0 months). The dashed black line represents MS without employing the ENCALS prediction model, which is overly optimistic for ALS patients classified to the very short and short survival groups, i.e., they end up with less time, and overly pessimistic for patients classified to long and very long groups, i.e., they end up with more time. Bottom: Horizontal bars have dots to represent median times to composite outcome, thick lines to represent probability interquartile range, thin lines to represent 10 to 90% probability intervals to composite outcome. Created, in part, with BioRender.com.
Although median survival in ALS is only 2 to 4 years, there is a broad distribution of individual patient survival, affecting both the clinician’s ability to discuss and the patient’s ability to understand, disease prognosis. This is attributable to various factors that influence ALS survival (Figure 4C), such as clinical and demographic features (e.g., age at onset, site of onset, presence of FTD), genetic architecture (e.g., rapidly progressive SOD1A5V, slowly progressive DCTN1 mutations; Appendix Table 5), and the exposome (e.g., environmental exposures). The European Network for the Cure of ALS (ENCALS) model was created to predict personalized survival (defined as survival without tracheostomy or non-invasive ventilation >23 hours/day) based on eight parameters: onset age, time to diagnosis, ALSFRS-R progression rate, forced vital capacity, bulbar onset, definite ALS by revised El Escorial criteria, FTD, and C9orf72 repeat expansion (Figure 4D).(70) Although not in routine clinical use, the ENCALS prediction tool can potentially benefit patients by giving them a more accurate perspective of life expectancy.
Overall, accurate prognostication of the clinical course of ALS remains in its infancy since even predictions by the best models retain uncertainty. Thus, clinical care teams should advise patients and their families on the anticipated disease course and range of expected symptoms, with the caveat that these predictions can vary with each patient. Variation of disease phenotypes even within the same family attests to this unpredictability. Finally, although clinical staging methods provide useful metrics for comparing participant stages in clinical research populations, their use in the clinic remains to be determined.
ALS treatment
As ALS remains incurable, treatment is focused on using disease modifying therapies and maximizing quality of life. The American Academy of Neurology, the European Federation of Neurological Societies, the United Kingdom National Institute for Health and Care Excellence (NICE)(108) and ALS Canada(109) have published evidence-based and expert consensus guidelines for managing ALS, and supportive multidisciplinary care improves survival and quality-of-life for ALS patients (Table 1).(110) The two medications with approval in some countries for slowing ALS are riluzole and edaravone. Riluzole, an anti-glutamate agent, improves ALS patient survival in clinical trials and post-marketing analyses, but whether this prolongation occurs at all stages of ALS or just at advanced disease stages remains a topic of debate.(116, 117) The antioxidant edaravone for 6 months showed some efficacy in post hoc analysis of the first phase 3 trial for participants meeting the criteria of definite or probable ALS (El Escorial and revised Airlie House diagnostic criteria), disease duration less than 24 months, FVC >80%, and ALSFRS-R subscale scores all >2.(121) The trial was repeated prospectively using this defined patient population,(118, 122) and again reported edaravone slowed disease progression. However, this trial design may lack generalizability to the broader ALS patient population and post-marketing analyses raise questions about edaravone’s safety and benefits.(119, 120) Thus, edaravone use remains controversial and has not obtained worldwide approval. A combination of dextromethorphan and quinidine is approved in the United States for managing symptoms of pseudobulbar affect.(123) This drug is not marketed in all countries and alternative and more cost-effective treatments are available. Non-invasive ventilation also improves ALS survival and quality of life.(124) For this reason, patients with ALS should be regularly monitored for respiratory symptoms and undergo the appropriate respiratory assessments such as overnight oximetry or measures for blood gas partial pressure of CO2, blood bicarbonate levels, vital capacity, or maximum inspiratory pressure to confirm if they qualify for non-invasive ventilation.(125)
Table 1.
ALS management
| Indication | Treatments and interventions* |
|---|---|
| Disease-modifying treatments | |
| Disease progression | Only two drugs with regulatory approval are available, riluzole and edaravone. They are of marginal efficacy and only in select populations and merely lengthen survival by a few months;(116–118) however, even within select populations, the efficacy of edaravone is contested.(119, 120) |
| Symptomatic management | |
| Comprehensive care | A multidisciplinary clinic plans the comprehensive, multidisciplinary care needed to manage symptoms in ALS patients. Care spans managing respiration and oral symptoms (speech, swallowing) nutrition and gastrointestinal symptoms, pain and symptoms secondary to muscle loss, and cognition, mood and behavioral changes. |
| Respiratory and oral symptoms | |
| Bronchial secretions | Cease provoking medications. Administer mucolytics if the patient exhibits sufficient cough flow, which includes N-acetylcysteine, anticholinergic bronchodilator, β-receptor antagonist and nebulized saline, furosemide, and guaifenesin. Mechanical or non-pharmacological approaches are also available, which include manual assisted cough, mechanical insufflator-exsufflator, portable home suction device, and room humidifier. Additionally, patients are encouraged to remain hydrated or drink pineapple or papaya juice to break up secretions. |
| Dysarthria | Evaluate speech-language regularly and identify language impairments. Provide assistive communication tools, such as electronic writing, voice banking, and voice amplification devices. |
| Dyspnea | Options include elevating the head of the bed, use of a hospital bed for elevation, noninvasive ventilation, and invasive tracheostomy ventilation. |
| Sialorrhea | Administer anticholinergics, such as amitriptyline, atropine ophthalmic drops, glycopyrrolate, scopolamine patch. If sialorrhea is refractory to anticholinergics, botulinum toxin injections, external beam radiation therapy, and surgery may be considered. A portable suction device is a less aggressive approach. Dark grape juice and ginger tea are reported to decrease saliva production. |
| Nutrition and gastrointestinal symptoms | |
| Constipation | Increase fluid and fiber intake or adjust enteral nutrition. Administer an osmotic or stimulant laxative. Increase physical activity. |
| Dysphagia | Evaluate speech-language pathology regularly and provide swallowing technique education. Gastrostomy feeding tube placement by percutaneous endoscopic gastrostomy, radiologically inserted gastrostomy, or per-oral image-guided gastrostomy. Ideally, gastrostomy placement should occur early in the disease course prior to significant weight loss. |
| Nutrition | Dietary evaluation by a nutritionist and modify food and fluid consistency accordingly. Administer appetite stimulants and/or high-protein/high-caloric supplements. |
| Pain and symptoms secondary to muscle loss | |
| Cramps | Administer magnesium, levetiracetam, mexiletine, or quinidine (controversial). Non-pharmacological approaches span physiotherapy, exercise, massage, and hydrotherapy. Hydration with tonic water. |
| Joint pain | Administer analgesics, including acetaminophen and nonsteroidal anti-inflammatory drugs. Non-pharmacological approaches span physical therapy, especially to limit joint contractures, and repositioning, pressure relief, mechanical support for weak limbs. |
| Spasticity | Administer spasmolytic drugs (tizanidine, baclofen, intrathecal baclofen pump), benzodiazepines, dantrolene, or botulinum toxin injections. Tetrahydrocannabinol is a novel direction. Non-pharmacological approaches include stretching and range of motion physical therapy. |
| Venous thromboembolism | Administer anticoagulants. Non-pharmacological preventive approaches span physiotherapy, limb elevation, and compression stockings. |
| Weakness | To address upper limb weakness, occupational therapy for adaptive equipment and hand splints. To address lower limb weakness, physical therapy, which includes transfer and gait assessments. Fall prevention. Various aids, such as ankle-foot orthoses, Hoyer lift, walker, transport wheelchair, and powered wheelchair. |
| Mood | |
| Depression and anxiety | Administer antidepressants or anxiolytics. Non-pharmacological approaches span psychotherapy and counseling. |
| Fatigue | Address related symptoms (e.g., sleep disturbances, depression, cramps, pain, respiratory distress). Encourage patients to conserve energy. |
| Insomnia | Address related symptoms (e.g., depression, cramps, pain, respiratory distress). Monitor sleep hygiene and address the role of noninvasive ventilation. |
| Pseudobulbar affect | Administer tricyclic antidepressants, selective serotonin reuptake inhibitors (fluvoxamine, citalopram), or dextromethorphan-quinidine. Educate patients that these are ALS symptoms. |
| Care planning | |
| Cognitive dysfunction | Encourage early discussions regarding treatment goals while the patient still retains decision-making capacity. Educate family and caregivers. Respite care. |
| Palliative and end-of-life care | Administer benzodiazepine and/or opioids for symptomatic dyspnea or intractable pain. Encourage early conversations with patients, family members, and caregivers regarding treatment choices. Palliative care referral can occur early in the disease course with hospice care at end of life. |
Sources: EFNS Task Force on Diagnosis and Management of Amyotrophic Lateral Sclerosis,(62) The American Academy of Neurology,(111–113) the United Kingdom National Institute for Health and Care Excellence (NICE),(108) ALS Canada(109) and other Canadian guidelines,(114) and Bradley and Daroff’s Neurology in Clinical Practice.(115) These therapies, in most cases, represent good clinical practice as few clinical trials involving patients with ALS exist to provide a robust evidence base for these interventions.
Gastrostomy is also an effective therapy for supporting nutrition and is likely of greater benefit when established earlier in the disease course. Gastrostomy tubes can be inserted using percutaneous endoscopic gastrostomy, radiologically inserted gastrostomy, and per-oral image-guided gastrostomy placement with similar mortality.(126) Factors that associated with a poor outcome following gastrostomy include use of non-invasive ventilation > 16 hours per day, older age, body mass index <20 kg/m2 and recurrent accumulation of airway secretions.(127) High calorie nutrition has also been investigated for treating ALS(90) and post-hoc analysis of a phase 3 trial suggests that it might be helpful for rapidly progressing patients,(128) though confirmatory trials are needed.
Additional treatments are outlined in Table 1. ALS patients may also contemplate alternative and off label treatments, often found on the internet. ALS Untangled (https://www.alsuntangled.com/), was conceived to provide a systematic review of unproven treatments. ALS care guidelines encourage providers to have an open dialogue about the use and risks of these treatments, especially as some can carry medical or financial risk.
Emerging directions in ALS
Novel ALS treatment approaches
Recognition of ALS heterogeneity, genetics, and a deeper understanding of pathophysiology bring new treatment approaches to the ALS community. These span new trial designs to address heterogeneity, genetic therapies, immune-targeting agents against inflammation, and stem cells to enrich the CNS environment.
New trial designs:
New ALS clinical trials can leverage a basket design of targeted agents against phenotypically- or genetically-defined participant populations (see Genetic therapies section).(129, 130) Novel platform trial paradigms simultaneously evaluate multiple therapies in distinct arms against a single placebo group, lowering the number of required participants and shortening trial duration.(129) Adaptive designs can further shorten trial duration by response-adaptive randomization, which increases participant allocation to more promising arms.(129) Several major trials with novel compounds and treatment approaches are currently underway (Appendix Table 7).
Genetic therapies:
There is growing consensus that gene therapy is a promising avenue in ALS. One strategy is silencing toxic gain-of-function genes by targeting mRNA and pre-mRNA using antisense oligonucleotides (ASOs). The first clinical trial of the SOD1 ASO, BIIB067, demonstrated safety, evidence of target engagement, and promising trends in exploratory secondary outcome measures.(131) However, the phase 3 clinical trial did not meet its primary efficacy outcome of slowing disease progression as measured by the ALSFRS-R, though cerebrospinal fluid (CSF) SOD1 protein levels and neurofilament levels were significantly decreased.(132) A new approach is earlier intervention with BIIB067 during the pre-symptomatic phase of disease in mutant SOD1 carriers (NCT04856982; Appendix Table 7). Clinical trials are also underway of ASOs targeting other autosomal dominant gain-of-function mutations, including C9orf72, FUS, and ATAXN2.(133)
Antibodies:
Monoclonal antibodies against mutant C9orf72 and TDP-43 are in preclinical development.(134) Several clinical trials have also been launched, but besides demonstrating safety, none were effective, e.g., tocilizumab, ozanezumab.(134) A few antibody candidates are still in the clinical trial pipeline, including AP-101 against SOD1 aggregates (NCT05039099), ANX005 against C1q protein (NCT04569435), and AT-1501 against CD40L protein (NCT04322149; Appendix Table 7).
Immune-targeting:
New anti‐inflammatory therapies targeting the immune system are also in the clinical pipeline (Appendix Table 7). Phase 1/2 clinical trial results report that low-dose IL-2 is well tolerated and immunologically effective in increasing regulatory T cell numbers, although its effect on ALS progression is still being evaluated in a phase 2b/3 trial (MIROCALS).(135) Autologous infusion of expanded Treg cells in a small patient cohort slowed disease progression.(136) Masitinib, a tyrosine kinase inhibitor, reduces microglial activation and showed promise in a phase 2/3 trial.(137) These reports underscore the feasibility of immune-targeting drugs as ALS candidate therapies.
Stem cells:
Stem cells offer the unique opportunity to simultaneously target multiple dysregulated pathways while providing CNS neurotrophic support.(138) They can derive from diverse sources, e.g., mesenchymal stem cells, neural progenitor cells (Appendix Table 7), each offering distinct advantages and disadvantages.(138) A recent meta-analysis concluded that adult stem cells are safe and well tolerated;(139) however, apart from a possible transient positive effect, trials have failed to demonstrate long-lasting efficacy from stem cells.
Novel diagnostic ALS biomarkers
There is an urgent need for ALS biomarkers to expedite diagnosis, particularly in atypical phenotypes, and enable improved prognosis of disease course. Biomarkers can also refine clinical trial participant stratification, facilitate the estimation of progression rates, monitor target engagement, and detect early potential treatment effects.
Neurofilaments:
CSF and plasma neurofilaments are well-characterized and promising fluid biomarkers. Elevated CSF and plasma neurofilament light chain levels correlate with shorter survival, more aggressive disease phenotypes, and presence of C9orf72 expansion.(140–142) Plasma neurofilaments are also elevated up to five years prior to disease onset in sporadic and familial ALS cases,(143, 144) and indicate phenoconversion in clinically asymptomatic mutant SOD1 carriers.(143) Recent clinical trials support their use as pharmacodynamic markers of ALS progression.(131, 145)
Brain imaging:
While routine magnetic resonance tomography (MRI) cannot diagnose ALS, MRI with quantitative analysis of fluid-attenuated inversion recovery (FLAIR) can identify increased corticospinal tract and corpus callosum intensities in ALS patients.(146) More advanced structural and functional MRI techniques are not yet in routine clinical practice but may provide new diagnostic biomarkers. Examples include diffusion tensor imaging (DTI)(147, 148) and multimodal(147, 149) approaches, such as quantitative susceptibility mapping to detect iron-related motor cortex changes and connectome analyses of motor- and non-motor networks. T1-weighted imaging and DTI detects abnormalities (cortical and subcortical atrophy, white matter changes), already present in presymptomatic C9orf72 repeat expansion carriers.(150) While not a disease-specific biomarker, positron emission tomography using tracers to quantify brain metabolism ([18F]-fluorodeoxyglucose) or glial activation ([11C]-PBR28) provides new insights into disease mechanisms and may prove useful as pharmacodynamic indices in future clinical trials.(151, 152)
Neurophysiological:
Neurophysiological markers of disease-associated changes are currently available. Spectral electroencephalogram mapping reveals brain connectivity changes in ALS, which correlate with MRI findings and could become useful, cost-effective markers of cortical network disruption.(153, 154) Magnetoencephalography shows enhanced connectivity during ALS progression.(155)
Cortical motor neuronal hyperexcitability can sometimes be detected by routine transcranial magnetic stimulation (TMS); however, more often, refined techniques such as threshold-tracking TMS measuring short-interval intracortical inhibition and intracortical facilitation are necessary to detect subclinical UMN involvement.(156) Cortical hyperexcitability across ALS phenotypes distinguishes ALS from non-ALS disorders, correlates with clinically affected body regions,(157) disease spread,(157) and cognitive dysfunction.(158) TMS may also have a role in prognosis, with increased cortical hyperexcitability associated with longer disease duration(159) and cortical inexcitability with poorer clinical trajectory.(160) Change in short-interval intracortical inhibition was the primary endpoint in a phase 2 ALS trial of retigabine, a potassium channel activator, demonstrating the potential of neurophysiological outcome measures as pharmacodynamic disease markers.(161)
LMN degeneration can be quantified by the non-invasive motor unit index (MUNIX), which correlates with the number of functioning motor units.(156) MUNIX detects motor unit decline already in clinically unaffected muscle groups and can monitor motor unit loss over time. When used as an outcome measure in clinical trials, MUNIX requires thorough rater qualification to ensure reliability.(162)
Conclusions
ALS remains difficult to diagnose and manage. This is due to heterogenous ALS presentation and phenotype, and symptom and sign overlap with other illnesses. Earlier on in the diagnostic process, physicians should refer patients presenting with progressive dysarthria, dysphagia, limb weakness, or respiratory failure to a neurologist. This aligns with suggestions by advocate groups, as they lobby to help patients seek early treatment and enroll in clinical trials. Unfortunately, effective disease-modifying drugs are lacking, and treatment revolves around multidisciplinary care to manage symptoms and aid end-of-life planning.
Research into improved diagnostic and prognostic tools could expedite diagnosis and give patients a better understanding of their disease course. Thus, we anticipate future directions in clinical ALS management will move towards simpler diagnostic criteria, such as the Gold Coast, and widespread genetic testing. Research will evaluate whether newly developed scoring, staging, and predictive tools will give patients meaningful and accurate insight into their anticipated clinical trajectory. Pathophysiology research and novel trial designs are developing rational, targeted candidates, which are passing through the clinical testing pipeline more efficiently. We anticipate that these research efforts will translate into improved outcomes for current and future patients with ALS.
Search strategy and selection criteria
We searched PubMed for English language articles from September 15th, 2021, to October 5th, 2021, and then again January 2022, with the terms, in addition to “amyotrophic lateral sclerosis”: Epidemiology section: “epidemiology”. ALS clinical presentation section: “phenotype”. ALS diagnosis section: “diagnostic”, “diagnosis”, “cognition”, and “cognitive”. ALS genetic architecture section: “GWAS,” “genetic”, “risk”, “oligogenic”, “polygenic”, “heritability”. Differential diagnosis section: “mimic”, “GWAS” combined with every ALS gene in turn. ALS risk, progression, and pathophysiology section: “pathophysiology”, “mechanism”, “nucleocytoplasmic transport”, “cell-to-cell transmission”, “immune system”, “exposure”, “environment”, “pollutant”, “toxin”, “metals”, “traffic”. ALS prognosis: “prognosis”, “scoring”, “scaling”, “staging”. ALS treatment: “multidisciplinary care”, “riluzole”, “edaravone”, “non-invasive ventilation”, “gastrostomy”. Emerging directions in ALS: “gene therapy”, “antisense oligonucleotide, “antibody,” “immune,” “clinical trial”, “neurofilaments”, “imaging”, “PET”, “connectome”, “EEG”, “hyperexcitability”. The search focused on articles published from Jan 1st, 2017, to Jan 31st, 2022, though seminal older articles were also considered. We also included articles from the authors’ personal reference lists. Selected articles were based on relevance to this review. Additionally, we searched clinicaltrials.gov for “amyotrophic lateral sclerosis”.
Supplementary Material
Acknowledgments
SAG and ELF receive funding from the National ALS Registry/CDC/ATSDR (1R01TS000289; R01TS000327); National ALS Registry/CDC/ATSDR CDCP-DHHS-US (CDC/ATSDR 200-2013-56856); NIEHS K23ES027221; NIEHS R01ES030049; NINDS R01NS127188; NINDS R01NS120926; ALS Association 20-IIA-532; NeuroNetwork for Emerging Therapies, the NeuroNetwork Therapeutic Discovery Fund, the Peter R. Clark Fund for ALS Research, the Sinai Medical Staff Foundation, Scott L. Pranger, University of Michigan. LM research is partly funded by the AGING Project for Department of Excellence at the Department of Translational Medicine (DIMET), Università del Piemonte Orientale, Novara, Italy. PJS receives funding from the National Institute for Health Research (NIHR), including for the NIHR Sheffield Biomedical Research Centre, UK Medical Research Council, LifeArc, Motor Neurone Disease Association, MyName’5 Doddie Foundation, the Darby Rimmer Foundation, the Nick Smith Foundation, Fight MND, EU Innovative Medicines Initiative, EU Innovative Training Network, and EU Horizon 2020. SG is supported by Japan Agency for Medical Research and Development, AMED under Grant Number JP21wn0425009h0001, JP21ak0101111h0003, JP21ak0101124h0002, JP21ek0109492h0002.
Footnotes
Declaration of interests
ELF has a patent US20200253977A1 issued. SAG reports personal fees from Biogen, ITF Pharma, and Watermark, outside the submitted work; in addition, SAG has a patent US20200253977A1 issued. SP reports grants from the German Neuromuscular Society, the German-Israeli Foundation for Scientific Research and Development (GIF), and personal fees from Cytokinetics, Desitin Pharma, Italfarmaco, Biogen, Roche, and Zambon outside the submitted work. LM declared no conflicts of interest. MGS declared no conflicts of interest. PJS reports consultancy and advisory board membership with Biogen, Benevolent AI, QurALIS, Quell, and Aclipse Therapeutics, outside the submitted work. GS reports personal fees from Mitsubishi Tanabe Pharma Corporation, Cyberdyne, Biogen Japan, Takeda Pharmaceutical, Nihon Pharmaceutical, and Teijin Pharma, outside the submitted work.
References
- 1.Ryan M, Heverin M, McLaughlin RL, Hardiman O. Lifetime Risk and Heritability of Amyotrophic Lateral Sclerosis. JAMA Neurol. 2019;76(11):1367–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goutman SA, Hardiman O, Al-Chalabi A, Chió A, Savelieff MG, Kiernan MC, et al. Recent advances in the diagnosis and prognosis of amyotrophic lateral sclerosis. Lancet Neurol. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marin B, Fontana A, Arcuti S, Copetti M, Boumédiene F, Couratier P, et al. Age-specific ALS incidence: a dose-response meta-analysis. Eur J Epidemiol. 2018;33(7):621–34. [DOI] [PubMed] [Google Scholar]
- 4.Mehta P, Kaye W, Raymond J, Punjani R, Larson T, Cohen J, et al. Prevalence of Amyotrophic Lateral Sclerosis - United States, 2015. MMWR Morb Mortal Wkly Rep. 2018;67(46):1285–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luna J, Diagana M, Ait Aissa L, Tazir M, Ali Pacha L, Kacem I, et al. Clinical features and prognosis of amyotrophic lateral sclerosis in Africa: the TROPALS study. J Neurol Neurosurg Psychiatry. 2019;90(1):20–9. [DOI] [PubMed] [Google Scholar]
- 6.Feigin VL, Vos T, Alahdab F, Amit AML, Bärnighausen TW, Beghi E, et al. Burden of Neurological Disorders Across the US From 1990–2017: A Global Burden of Disease Study. JAMA Neurol. 2021;78(2):165–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu L, Liu T, Liu L, Yao X, Chen L, Fan D, et al. Global variation in prevalence and incidence of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol. 2020;267(4):944–53. [DOI] [PubMed] [Google Scholar]
- 8.Arthur KC, Calvo A, Price TR, Geiger JT, Chio A, Traynor BJ. Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat Commun. 2016;7:12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gowland A, Opie-Martin S, Scott KM, Jones AR, Mehta PR, Batts CJ, et al. Predicting the future of ALS: the impact of demographic change and potential new treatments on the prevalence of ALS in the United Kingdom, 2020–2116. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20(3–4):264–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marin B, Boumediene F, Logroscino G, Couratier P, Babron MC, Leutenegger AL, et al. Variation in worldwide incidence of amyotrophic lateral sclerosis: a meta-analysis. Int J Epidemiol. 2017;46(1):57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Longinetti E, Fang F. Epidemiology of amyotrophic lateral sclerosis: an update of recent literature. Curr Opin Neurol. 2019;32(5):771–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fontana A, Marin B, Luna J, Beghi E, Logroscino G, Boumédiene F, et al. Time-trend evolution and determinants of sex ratio in Amyotrophic Lateral Sclerosis: a dose-response meta-analysis. J Neurol. 2021;268(8):2973–84. [DOI] [PubMed] [Google Scholar]
- 13.Murphy NA, Arthur KC, Tienari PJ, Houlden H, Chio A, Traynor BJ. Age-related penetrance of the C9orf72 repeat expansion. Sci Rep. 2017;7(1):2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chio A, Moglia C, Canosa A, Manera U, D’Ovidio F, Vasta R, et al. ALS phenotype is influenced by age, sex, and genetics: A population-based study. Neurology. 2020;94(8):e802–e10. [DOI] [PubMed] [Google Scholar]
- 15.Fang T, Jozsa F, Al-Chalabi A. Nonmotor Symptoms in Amyotrophic Lateral Sclerosis: A Systematic Review. Int Rev Neurobiol. 2017;134:1409–41. [DOI] [PubMed] [Google Scholar]
- 16.Walhout R, Verstraete E, van den Heuvel MP, Veldink JH, van den Berg LH. Patterns of symptom development in patients with motor neuron disease. Amyotroph Lateral Scler Frontotemporal Degener. 2018;19(1–2):21–8. [DOI] [PubMed] [Google Scholar]
- 17.Chiò A, Calvo A, Moglia C, Mazzini L, Mora G. Phenotypic heterogeneity of amyotrophic lateral sclerosis: a population based study. J Neurol Neurosurg Psychiatry. 2011;82(7):740–6. [DOI] [PubMed] [Google Scholar]
- 18.Rosenbohm A, Liu M, Nagel G, Peter RS, Cui B, Li X, et al. Phenotypic differences of amyotrophic lateral sclerosis (ALS) in China and Germany. J Neurol. 2018;265(4):774–82. [DOI] [PubMed] [Google Scholar]
- 19.Beeldman E, Govaarts R, de Visser M, Klein Twennaar M, van der Kooi AJ, van den Berg LH, et al. Progression of cognitive and behavioural impairment in early amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 91 2020. p. 779–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pender N, Pinto-Grau M, Hardiman O. Cognitive and behavioural impairment in amyotrophic lateral sclerosis. Curr Opin Neurol. 2020;33(5):649–54. [DOI] [PubMed] [Google Scholar]
- 21.Crockford C, Newton J, Lonergan K, Chiwera T, Booth T, Chandran S, et al. ALS-specific cognitive and behavior changes associated with advancing disease stage in ALS. Neurology. 2018;91(15):e1370–e80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bersano E, Sarnelli MF, Solara V, Iazzolino B, Peotta L, De Marchi F, et al. Decline of cognitive and behavioral functions in amyotrophic lateral sclerosis: a longitudinal study. Amyotroph Lateral Scler Frontotemporal Degener. 2020;21(5–6):373–9. [DOI] [PubMed] [Google Scholar]
- 23.Ringholz GM, Appel SH, Bradshaw M, Cooke NA, Mosnik DM, Schulz PE. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology. 2005;65(4):586–90. [DOI] [PubMed] [Google Scholar]
- 24.Nicholson K, Murphy A, McDonnell E, Shapiro J, Simpson E, Glass J, et al. Improving symptom management for people with amyotrophic lateral sclerosis. Muscle Nerve. 2018;57(1):20–4. [DOI] [PubMed] [Google Scholar]
- 25.Gregory JM, McDade K, Bak TH, Pal S, Chandran S, Smith C, et al. Executive, language and fluency dysfunction are markers of localised TDP-43 cerebral pathology in non-demented ALS. J Neurol Neurosurg Psychiatry. 2020;91(2):149–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iazzolino B, Peotta L, Zucchetti JP, Canosa A, Manera U, Vasta R, et al. Differential Neuropsychological Profile of Patients With Amyotrophic Lateral Sclerosis With and Without C9orf72 Mutation. Neurology. 2021;96(1):e141–e52. [DOI] [PubMed] [Google Scholar]
- 27.Yang T, Hou Y, Li C, Cao B, Cheng Y, Wei Q, et al. Risk factors for cognitive impairment in amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2021;92(7):688–93. [DOI] [PubMed] [Google Scholar]
- 28.Basaia S, Agosta F, Cividini C, Trojsi F, Riva N, Spinelli EG, et al. Structural and functional brain connectome in motor neuron diseases: A multicenter MRI study. Neurology. 2020;95(18):e2552–e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williams JR, Fitzhenry D, Grant L, Martyn D, Kerr DA. Diagnosis pathway for patients with amyotrophic lateral sclerosis: retrospective analysis of the US Medicare longitudinal claims database. BMC Neurol. 2013;13:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Falcão de Campos C, Gromicho M, Uysal H, Grosskreutz J, Kuzma-Kozakiewicz M, Oliveira Santos M, et al. Delayed Diagnosis and Diagnostic Pathway of ALS Patients in Portugal: Where Can We Improve? Front Neurol. 2021;12:761355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. https://www.als.org/thinkals/thinkals-tool.
- 32.Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(5):293–9. [DOI] [PubMed] [Google Scholar]
- 33.Babu S, Pioro EP, Li J, Li Y. Optimizing muscle selection for electromyography in amyotrophic lateral sclerosis. Muscle Nerve. 2017;56(1):36–44. [DOI] [PubMed] [Google Scholar]
- 34.Shayya L, Babu S, Pioro EP, Li J, Li Y. Distal Predominance of Electrodiagnostic Abnormalities in Early-Stage Amyotrophic Lateral Sclerosis. Muscle Nerve. 2018;58(3):389–95. [DOI] [PubMed] [Google Scholar]
- 35.Shefner JM, Al-Chalabi A, Baker MR, Cui LY, de Carvalho M, Eisen A, et al. A proposal for new diagnostic criteria for ALS. Clinical neurophysiology : official journal of the International Federation of Clinical Neurophysiology. 2020;131(8):1975–8. [DOI] [PubMed] [Google Scholar]
- 36.Strong MJ, Abrahams S, Goldstein LH, Woolley S, McLaughlin P, Snowden J, et al. Amyotrophic lateral sclerosis - frontotemporal spectrum disorder (ALS-FTSD): Revised diagnostic criteria. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(3–4):153–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gosselt IK, Nijboer TCW, Van Es MA. An overview of screening instruments for cognition and behavior in patients with ALS: selecting the appropriate tool for clinical practice. Amyotroph Lateral Scler Frontotemporal Degener. 2020;21(5–6):324–36. [DOI] [PubMed] [Google Scholar]
- 38.Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51(6):1546–54. [DOI] [PubMed] [Google Scholar]
- 39.Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(Pt 9):2456–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hodgins F, Mulhern S, Abrahams S. The clinical impact of the Edinburgh Cognitive and Behavioural ALS Screen (ECAS) and neuropsychological intervention in routine ALS care. Amyotroph Lateral Scler Frontotemporal Degener. 2020;21(1–2):92–9. [DOI] [PubMed] [Google Scholar]
- 41.Goutman SA, Hardiman O, Al-Chalabi A, Chió A, Savelieff MG, Kiernan MC, et al. Emerging insights into the complex genetics and pathophysiology of amyotrophic lateral sclerosis. Lancet Neurol. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Al-Chalabi A, Lewis CM. Modelling the effects of penetrance and family size on rates of sporadic and familial disease. Hum Hered. 2011;71(4):281–8. [DOI] [PubMed] [Google Scholar]
- 43.Ryan M, Heverin M, Doherty MA, Davis N, Corr EM, Vajda A, et al. Determining the incidence of familiality in ALS: A study of temporal trends in Ireland from 1994 to 2016. Neurol Genet. 2018;4(3):e239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cady J, Allred P, Bali T, Pestronk A, Goate A, Miller TM, et al. Amyotrophic lateral sclerosis onset is influenced by the burden of rare variants in known amyotrophic lateral sclerosis genes. Ann Neurol. 2015;77(1):100–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chia R, Chio A, Traynor BJ. Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol. 2018;17(1):94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goutman SA, Chen KS, Paez-Colasante X, Feldman EL. Emerging understanding of the genotype-phenotype relationship in amyotrophic lateral sclerosis. Handbook of clinical neurology. 2018;148:603–23. [DOI] [PubMed] [Google Scholar]
- 47.Zou ZY, Zhou ZR, Che CH, Liu CY, He RL, Huang HP. Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2017;88(7):540–9. [DOI] [PubMed] [Google Scholar]
- 48.Gregory JM, Fagegaltier D, Phatnani H, Harms MB. Genetics of Amyotrophic Lateral Sclerosis. Current Genetic Medicine Reports. 2020;8(4):121–31. [Google Scholar]
- 49.Rehm HL, Berg JS, Brooks LD, Bustamante CD, Evans JP, Landrum MJ, et al. ClinGen--the Clinical Genome Resource. N Engl J Med. 2015;372(23):2235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McCann EP, Henden L, Fifita JA, Zhang KY, Grima N, Bauer DC, et al. Evidence for polygenic and oligogenic basis of Australian sporadic amyotrophic lateral sclerosis. J Med Genet. 2020. [DOI] [PubMed] [Google Scholar]
- 51.Shepheard SR, Parker MD, Cooper-Knock J, Verber NS, Tuddenham L, Heath P, et al. Value of systematic genetic screening of patients with amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2021;92(5):510–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bandres-Ciga S, Noyce AJ, Hemani G, Nicolas A, Calvo A, Mora G, et al. Shared polygenic risk and causal inferences in amyotrophic lateral sclerosis. Annals of neurology. 2019;85(4):470–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li C, Ou R, Wei Q, Shang H. Shared genetic links between amyotrophic lateral sclerosis and obesity-related traits: a genome-wide association study. Neurobiol Aging. 2021;102:211 e1–e9. [DOI] [PubMed] [Google Scholar]
- 54.Nicolas A, Kenna KP, Renton AE, Ticozzi N, Faghri F, Chia R, et al. Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron. 2018;97(6):1268–83 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Saez-Atienzar S, Bandres-Ciga S, Langston RG, Kim JJ, Choi SW, Reynolds RH, et al. Genetic analysis of amyotrophic lateral sclerosis identifies contributing pathways and cell types. Sci Adv. 2021;7(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dekker AM, Diekstra FP, Pulit SL, Tazelaar GHP, van der Spek RA, van Rheenen W, et al. Exome array analysis of rare and low frequency variants in amyotrophic lateral sclerosis. Sci Rep. 2019;9(1):5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Theunissen F, Flynn LL, Anderton RS, Mastaglia F, Pytte J, Jiang L, et al. Structural Variants May Be a Source of Missing Heritability in sALS. Front Neurosci. 2020;14:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang S, Cooper-Knock J, Weimer AK, Shi M, Moll T, Marshall JNG, et al. Genome-wide identification of the genetic basis of amyotrophic lateral sclerosis. Neuron. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goutman SA, Feldman EL. Voicing the Need for Amyotrophic Lateral Sclerosis Environmental Research. JAMA Neurol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Al-Chalabi A, Hardiman O. The epidemiology of ALS: a conspiracy of genes, environment and time. Nature reviews Neurology. 2013;9(11):617–28. [DOI] [PubMed] [Google Scholar]
- 61.Vucic S, Higashihara M, Sobue G, Atsuta N, Doi Y, Kuwabara S, et al. ALS is a multistep process in South Korean, Japanese, and Australian patients. Neurology. 2020;94(15):e1657–e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Andersen PM, Abrahams S, Borasio GD, de Carvalho M, Chio A, Van Damme P, et al. EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS)--revised report of an EFNS task force. Eur J Neurol. 2012;19(3):360–75. [DOI] [PubMed] [Google Scholar]
- 63.Goutman SA. Diagnosis and Clinical Management of Amyotrophic Lateral Sclerosis and Other Motor Neuron Disorders. Continuum (Minneap Minn). 2017;23(5, Peripheral Nerve and Motor Neuron Disorders):1332–59. [DOI] [PubMed] [Google Scholar]
- 64.Estevez-Fraga C, Magrinelli F, Hensman Moss D, Mulroy E, Di Lazzaro G, Latorre A, et al. Expanding the Spectrum of Movement Disorders Associated With C9orf72 Hexanucleotide Expansions. Neurol Genet. 2021;7(2):e575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cooper-Knock J, Frolov A, Highley JR, Charlesworth G, Kirby J, Milano A, et al. C9ORF72 expansions, parkinsonism, and Parkinson disease: a clinicopathologic study. Neurology. 2013;81(9):808–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hensman Moss DJ, Poulter M, Beck J, Hehir J, Polke JM, Campbell T, et al. C9orf72 expansions are the most common genetic cause of Huntington disease phenocopies. Neurology. 2014;82(4):292–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dewan R, Chia R, Ding J, Hickman RA, Stein TD, Abramzon Y, et al. Pathogenic Huntingtin Repeat Expansions in Patients with Frontotemporal Dementia and Amyotrophic Lateral Sclerosis. Neuron. 2021;109(3):448–60 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.O’Brien M, Burke T, Heverin M, Vajda A, McLaughlin R, Gibbons J, et al. Clustering of Neuropsychiatric Disease in First-Degree and Second-Degree Relatives of Patients With Amyotrophic Lateral Sclerosis. JAMA Neurol. 2017;74(12):1425–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Devenney EM, Ahmed RM, Halliday G, Piguet O, Kiernan MC, Hodges JR. Psychiatric disorders in C9orf72 kindreds: Study of 1,414 family members. Neurology. 2018;91(16):e1498–e507. [DOI] [PubMed] [Google Scholar]
- 70.Westeneng HJ, Debray TPA, Visser AE, van Eijk RPA, Rooney JPK, Calvo A, et al. Prognosis for patients with amyotrophic lateral sclerosis: development and validation of a personalised prediction model. The Lancet Neurology. 2018;17(5):423–33. [DOI] [PubMed] [Google Scholar]
- 71.Wang MD, Little J, Gomes J, Cashman NR, Krewski D. Identification of risk factors associated with onset and progression of amyotrophic lateral sclerosis using systematic review and meta-analysis. Neurotoxicology. 2017;61:101–30. [DOI] [PubMed] [Google Scholar]
- 72.Goutman SA, Boss J, Patterson A, Mukherjee B, Batterman S, Feldman EL. High plasma concentrations of organic pollutants negatively impact survival in amyotrophic lateral sclerosis. Journal of neurology, neurosurgery, and psychiatry. 2019;90(8):907–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Benatar M, Wuu J, McHutchison C, Postuma RB, Boeve BF, Petersen R, et al. Preventing amyotrophic lateral sclerosis: insights from pre-symptomatic neurodegenerative diseases. Brain. 2022;145(1):27–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mackenzie IR, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010;9(10):995–1007. [DOI] [PubMed] [Google Scholar]
- 75.Nguyen HP, Van Broeckhoven C, van der Zee J. ALS Genes in the Genomic Era and their Implications for FTD. Trends Genet. 2018;34(6):404–23. [DOI] [PubMed] [Google Scholar]
- 76.Pokrishevsky E, Grad LI, Cashman NR. TDP-43 or FUS-induced misfolded human wild-type SOD1 can propagate intercellularly in a prion-like fashion. Sci Rep. 2016;6:22155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sackmann C, Sackmann V, Hallbeck M. TDP-43 Is Efficiently Transferred Between Neuron-Like Cells in a Manner Enhanced by Preservation of Its N-Terminus but Independent of Extracellular Vesicles. Front Neurosci. 2020;14:540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mitra J, Guerrero EN, Hegde PM, Liachko NF, Wang H, Vasquez V, et al. Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc Natl Acad Sci U S A. 2019;116(10):4696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chou CC, Zhang Y, Umoh ME, Vaughan SW, Lorenzini I, Liu F, et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat Neurosci. 2018;21(2):228–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lin YC, Kumar MS, Ramesh N, Anderson EN, Nguyen AT, Kim B, et al. Interactions between ALS-linked FUS and nucleoporins are associated with defects in the nucleocytoplasmic transport pathway. Nat Neurosci. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature. 2015;525(7567):129–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang YJ, Guo L, Gonzales PK, Gendron TF, Wu Y, Jansen-West K, et al. Heterochromatin anomalies and double-stranded RNA accumulation underlie C9orf72 poly(PR) toxicity. Science. 2019;363(6428). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cook CN, Wu Y, Odeh HM, Gendron TF, Jansen-West K, Del Rosso G, et al. C9orf72 poly(GR) aggregation induces TDP-43 proteinopathy. Sci Transl Med. 2020;12(559). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Beers DR, Appel SH. Immune dysregulation in amyotrophic lateral sclerosis: mechanisms and emerging therapies. Lancet Neurol. 2019;18(2):211–20. [DOI] [PubMed] [Google Scholar]
- 85.Coque E, Salsac C, Espinosa-Carrasco G, Varga B, Degauque N, Cadoux M, et al. Cytotoxic CD8(+) T lymphocytes expressing ALS-causing SOD1 mutant selectively trigger death of spinal motoneurons. Proc Natl Acad Sci U S A. 2019;116(6):2312–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McCauley ME, O’Rourke JG, Yáñez A, Markman JL, Ho R, Wang X, et al. C9orf72 in myeloid cells suppresses STING-induced inflammation. Nature. 2020;585(7823):96–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yu CH, Davidson S, Harapas CR, Hilton JB, Mlodzianoski MJ, Laohamonthonkul P, et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell. 2020;183(3):636–49.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Murdock BJ, Famie JP, Piecuch CE, Raue KD, Mendelson FE, Pieroni CH, et al. Natural killer cells associate with amyotrophic lateral sclersois in a sex- and age-dependent manner. JCI Insight. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Murdock BJ, Zhou T, Kashlan SR, Little RJ, Goutman SA, Feldman EL. Correlation of Peripheral Immunity With Rapid Amyotrophic Lateral Sclerosis Progression. JAMA neurology. 2017;74(12):1446–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Guillot SJ, Bolborea M, Dupuis L. Dysregulation of energy homeostasis in amyotrophic lateral sclerosis. Curr Opin Neurol. 2021;34(5):773–80. [DOI] [PubMed] [Google Scholar]
- 91.Goutman SA, Guo K, Savelieff MG, Patterson A, Sakowski SA, Habra H, et al. Metabolomics identifies shared lipid pathways in independent amyotrophic lateral sclerosis cohorts. Brain. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.McLaughlin R, Vajda A, Hardiman O. Heritability of amyotrophic lateral sclerosis: Insights from disparate numbers. JAMA neurology. 2015. [DOI] [PubMed] [Google Scholar]
- 93.Chio A, Mazzini L, D’Alfonso S, Corrado L, Canosa A, Moglia C, et al. The multistep hypothesis of ALS revisited: The role of genetic mutations. Neurology. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Su FC, Goutman SA, Chernyak S, Mukherjee B, Callaghan BC, Batterman S, et al. Association of Environmental Toxins With Amyotrophic Lateral Sclerosis. JAMA Neurol. 2016;73(7):803–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Peters S, Broberg K, Gallo V, Levi M, Kippler M, Vineis P, et al. Blood Metal Levels and Amyotrophic Lateral Sclerosis Risk: A Prospective Cohort. Ann Neurol. 2021;89(1):125–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Figueroa-Romero C, Mikhail KA, Gennings C, Curtin P, Bello GA, Botero TM, et al. Early life metal dysregulation in amyotrophic lateral sclerosis. Ann Clin Transl Neurol. 2020;7(6):872–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Westeneng HJ, van Veenhuijzen K, van der Spek RA, Peters S, Visser AE, van Rheenen W, et al. Associations between lifestyle and amyotrophic lateral sclerosis stratified by C9orf72 genotype: a longitudinal, population-based, case-control study. Lancet Neurol. 2021;20(5):373–84. [DOI] [PubMed] [Google Scholar]
- 98.Julian TH, Glascow N, Barry ADF, Moll T, Harvey C, Klimentidis YC, et al. Physical exercise is a risk factor for amyotrophic lateral sclerosis: Convergent evidence from Mendelian randomisation, transcriptomics and risk genotypes. EBioMedicine. 2021;68:103397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Daneshvar DH, Mez J, Alosco ML, Baucom ZH, Mahar I, Baugh CM, et al. Incidence of and Mortality From Amyotrophic Lateral Sclerosis in National Football League Athletes. JAMA Netw Open. 2021;4(12):e2138801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.McKay KA, Smith KA, Smertinaite L, Fang F, Ingre C, Taube F. Military service and related risk factors for amyotrophic lateral sclerosis. Acta Neurol Scand. 2021;143(1):39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.van Eijk RPA, de Jongh AD, Nikolakopoulos S, McDermott CJ, Eijkemans MJC, Roes KCB, et al. An old friend who has overstayed their welcome: the ALSFRS-R total score as primary endpoint for ALS clinical trials. Amyotroph Lateral Scler Frontotemporal Degener. 2021;22(3–4):300–7. [DOI] [PubMed] [Google Scholar]
- 102.Pirola A, De Mattia E, Lizio A, Sannicolò G, Carraro E, Rao F, et al. The prognostic value of spirometric tests in Amyotrophic Lateral Sclerosis patients. Clin Neurol Neurosurg. 2019;184:105456. [DOI] [PubMed] [Google Scholar]
- 103.Fournier CN, Bedlack R, Quinn C, Russell J, Beckwith D, Kaminski KH, et al. Development and Validation of the Rasch-Built Overall Amyotrophic Lateral Sclerosis Disability Scale (ROADS). JAMA neurology. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Roche JC, Rojas-Garcia R, Scott KM, Scotton W, Ellis CE, Burman R, et al. A proposed staging system for amyotrophic lateral sclerosis. Brain : a journal of neurology. 2012;135(Pt 3):847–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chiò A, Hammond ER, Mora G, Bonito V, Filippini G. Development and evaluation of a clinical staging system for amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2015;86(1):38–44. [DOI] [PubMed] [Google Scholar]
- 106.Fang T, Al Khleifat A, Stahl DR, Lazo La Torre C, Murphy C, Young C, et al. Comparison of the King’s and MiToS staging systems for ALS. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(3–4):227–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Luna J, Couratier P, Lahmadi S, Lautrette G, Fontana A, Tortelli R, et al. Comparison of the ability of the King’s and MiToS staging systems to predict disease progression and survival in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2021:1–9. [DOI] [PubMed] [Google Scholar]
- 108. https://www.nice.org.uk/guidance/ng42 [
- 109. https://www.cmaj.ca/content/cmaj/192/46/E1453.full.pdf.
- 110.Klavžar P, Koritnik B, Leonardis L, Dolenc Grošelj L, Kirbiš M, Ristić Kovačič S, et al. Improvements in the multidisciplinary care are beneficial for survival in amyotrophic lateral sclerosis (ALS): experience from a tertiary ALS center. Amyotroph Lateral Scler Frontotemporal Degener. 2020;21(3–4):203–8. [DOI] [PubMed] [Google Scholar]
- 111.Miller RG, Jackson CE, Kasarskis EJ, England JD, Forshew D, Johnston W, et al. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73(15):1227–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Miller RG, Jackson CE, Kasarskis EJ, England JD, Forshew D, Johnston W, et al. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: drug, nutritional, and respiratory therapies (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73(15):1218–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Miller RG, Brooks BR, Swain-Eng RJ, Basner RC, Carter GT, Casey P, et al. Quality improvement in neurology: amyotrophic lateral sclerosis quality measures: report of the quality measurement and reporting subcommittee of the American Academy of Neurology. Neurology. 2013;81(24):2136–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shoesmith C, Abrahao A, Benstead T, Chum M, Dupre N, Izenberg A, et al. Canadian best practice recommendations for the management of amyotrophic lateral sclerosis. Cmaj. 2020;192(46):E1453–e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fearon C, Murray B, Mitsumoto H. Disorders of Upper and Lower Motor Neurons, Chapter 97. In: Jankovic, Joseph, Mazziotta CJ, Pomeroy LS, et al. , editors. Bradley and Daroff’s Neurology in Clinical Practice. 8th ed: Elsevier; 2022. p. 1535–67.e5. [Google Scholar]
- 116.Fang T, Al Khleifat A, Meurgey JH, Jones A, Leigh PN, Bensimon G, et al. Stage at which riluzole treatment prolongs survival in patients with amyotrophic lateral sclerosis: a retrospective analysis of data from a dose-ranging study. Lancet Neurol. 2018;17(5):416–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Andrews JA, Jackson CE, Heiman-Patterson TD, Bettica P, Brooks BR, Pioro EP. Real-world evidence of riluzole effectiveness in treating amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2020;21(7–8):509–18. [DOI] [PubMed] [Google Scholar]
- 118.Shefner J, Heiman-Patterson T, Pioro EP, Wiedau-Pazos M, Liu S, Zhang J, et al. Long-term edaravone efficacy in amyotrophic lateral sclerosis: Post-hoc analyses of Study 19 (MCI186–19). Muscle Nerve. 2020;61(2):218–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lunetta C, Moglia C, Lizio A, Caponnetto C, Dubbioso R, Giannini F, et al. The Italian multicenter experience with edaravone in amyotrophic lateral sclerosis. J Neurol. 2020;267(11):3258–67. [DOI] [PubMed] [Google Scholar]
- 120.Witzel S, Maier A, Steinbach R, Grosskreutz J, Koch JC, Sarikidi A, et al. Safety and Effectiveness of Long-term Intravenous Administration of Edaravone for Treatment of Patients With Amyotrophic Lateral Sclerosis. JAMA Neurol. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.A post-hoc subgroup analysis of outcomes in the first phase III clinical study of edaravone (MCI-186) in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(sup1):11–9. [DOI] [PubMed] [Google Scholar]
- 122.Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017;16(7):505–12. [DOI] [PubMed] [Google Scholar]
- 123.Brooks BR, Thisted RA, Appel SH, Bradley WG, Olney RK, Berg JE, et al. Treatment of pseudobulbar affect in ALS with dextromethorphan/quinidine: a randomized trial. Neurology. 2004;63(8):1364–70. [DOI] [PubMed] [Google Scholar]
- 124.Radunovic A, Annane D, Rafiq MK, Brassington R, Mustfa N. Mechanical ventilation for amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst Rev. 2017;10(10):Cd004427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Niedermeyer S, Murn M, Choi PJ. Respiratory Failure in Amyotrophic Lateral Sclerosis. Chest. 2019;155(2):401–8. [DOI] [PubMed] [Google Scholar]
- 126.López-Gómez JJ, Ballesteros-Pomar MD, Torres-Torres B, Pintor-De la Maza B, Penacho-Lázaro MA, Palacio-Mures JM, et al. Impact of Percutaneous Endoscopic Gastrostomy (PEG) on the Evolution of Disease in Patients with Amyotrophic Lateral Sclerosis (ALS). Nutrients. 2021;13(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hesters A, Amador MDM, Debs R, Le Forestier N, Lenglet T, Pradat PF, et al. Predictive factors for prognosis after gastrostomy placement in routine non-invasive ventilation users ALS patients. Sci Rep. 2020;10(1):15117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ludolph AC, Dorst J, Dreyhaupt J, Weishaupt JH, Kassubek J, Weiland U, et al. Effect of High-Caloric Nutrition on Survival in Amyotrophic Lateral Sclerosis. Ann Neurol. 2020;87(2):206–16. [DOI] [PubMed] [Google Scholar]
- 129.Kiernan MC, Vucic S, Talbot K, McDermott CJ, Hardiman O, Shefner JM, et al. Improving clinical trial outcomes in amyotrophic lateral sclerosis. Nature reviews Neurology. 2021;17(2):104–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.van Eijk RPA, Nikolakopoulos S, Roes KCB, Kendall L, Han SS, Lavrov A, et al. Innovating Clinical Trials for Amyotrophic Lateral Sclerosis: Challenging the Established Order. Neurology. 2021;97(11):528–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Miller T, Cudkowicz M, Shaw PJ, Andersen PM, Atassi N, Bucelli RC, et al. Phase 1–2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. The New England journal of medicine. 2020;383(2):109–19. [DOI] [PubMed] [Google Scholar]
- 132.Mullard A ALS antisense drug falters in phase III. Nat Rev Drug Discov. 20. England 2021. p. 883–5. [DOI] [PubMed] [Google Scholar]
- 133.Amado DA, Davidson BL. Gene therapy for ALS: A review. Mol Ther. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Poulin-Brière A, Rezaei E, Pozzi S. Antibody-Based Therapeutic Interventions for Amyotrophic Lateral Sclerosis: A Systematic Literature Review. Front Neurosci. 2021;15:790114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Camu W, Mickunas M, Veyrune JL, Payan C, Garlanda C, Locati M, et al. Repeated 5-day cycles of low dose aldesleukin in amyotrophic lateral sclerosis (IMODALS): A phase 2a randomised, double-blind, placebo-controlled trial. EBioMedicine. 2020;59:102844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Thonhoff JR, Beers DR, Zhao W, Pleitez M, Simpson EP, Berry JD, et al. Expanded autologous regulatory T-lymphocyte infusions in ALS: A phase I, first-in-human study. Neurol Neuroimmunol Neuroinflamm. 2018;5(4):e465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mora JS, Genge A, Chio A, Estol CJ, Chaverri D, Hernández M, et al. Masitinib as an add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: a randomized clinical trial. Amyotroph Lateral Scler Frontotemporal Degener. 2020;21(1–2):5–14. [DOI] [PubMed] [Google Scholar]
- 138.Goutman SA, Savelieff MG, Sakowski SA, Feldman EL. Stem cell treatments for amyotrophic lateral sclerosis: a critical overview of early phase trials. Expert Opin Investig Drugs. 2019;28(6):525–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Morata-Tarifa C, Azkona G, Glass J, Mazzini L, Sanchez-Pernaute R. Looking backward to move forward: a meta-analysis of stem cell therapy in amyotrophic lateral sclerosis. NPJ Regen Med. 2021;6(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Huang F, Zhu Y, Hsiao-Nakamoto J, Tang X, Dugas JC, Moscovitch-Lopatin M, et al. Longitudinal biomarkers in amyotrophic lateral sclerosis. Ann Clin Transl Neurol. 2020;7(7):1103–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Poesen K, De Schaepdryver M, Stubendorff B, Gille B, Muckova P, Wendler S, et al. Neurofilament markers for ALS correlate with extent of upper and lower motor neuron disease. Neurology. 2017;88(24):2302–9. [DOI] [PubMed] [Google Scholar]
- 142.Benatar M, Zhang L, Wang L, Granit V, Statland J, Barohn R, et al. Validation of serum neurofilaments as prognostic and potential pharmacodynamic biomarkers for ALS. Neurology. 2020;95(1):e59–e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Benatar M, Wuu J, Andersen PM, Lombardi V, Malaspina A. Neurofilament light: A candidate biomarker of presymptomatic amyotrophic lateral sclerosis and phenoconversion. Annals of neurology. 2018;84(1):130–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Bjornevik K, O’Reilly EJ, Molsberry S, Kolonel LN, Le Marchand L, Paganoni S, et al. Prediagnostic Neurofilament Light Chain Levels in Amyotrophic Lateral Sclerosis. Neurology. 2021;97(15):e1466–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Dorst J, Schuster J, Dreyhaupt J, Witzel S, Weishaupt JH, Kassubek J, et al. Effect of high-caloric nutrition on serum neurofilament light chain levels in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 91. England 2020. p. 1007–9. [DOI] [PubMed] [Google Scholar]
- 146.Fabes J, Matthews L, Filippini N, Talbot K, Jenkinson M, Turner MR. Quantitative FLAIR MRI in Amyotrophic Lateral Sclerosis. Acad Radiol. 2017;24(10):1187–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Weidman EK, Schweitzer AD, Niogi SN, Brady EJ, Starikov A, Askin G, et al. Diffusion tensor imaging and quantitative susceptibility mapping as diagnostic tools for motor neuron disorders. Clin Imaging. 2019;53:6–11. [DOI] [PubMed] [Google Scholar]
- 148.Welton T, Maller JJ, Lebel RM, Tan ET, Rowe DB, Grieve SM. Diffusion kurtosis and quantitative susceptibility mapping MRI are sensitive to structural abnormalities in amyotrophic lateral sclerosis. Neuroimage Clin. 2019;24:101953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Acosta-Cabronero J, Machts J, Schreiber S, Abdulla S, Kollewe K, Petri S, et al. Quantitative Susceptibility MRI to Detect Brain Iron in Amyotrophic Lateral Sclerosis. Radiology. 2018;289(1):195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bertrand A, Wen J, Rinaldi D, Houot M, Sayah S, Camuzat A, et al. Early Cognitive, Structural, and Microstructural Changes in Presymptomatic C9orf72 Carriers Younger Than 40 Years. JAMA Neurol. 2018;75(2):236–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.D’Hulst L, Van Weehaeghe D, Chio A, Calvo A, Moglia C, Canosa A, et al. Multicenter validation of [(18)F]-FDG PET and support-vector machine discriminant analysis in automatically classifying patients with amyotrophic lateral sclerosis versus controls. Amyotroph Lateral Scler Frontotemporal Degener. 2018;19(7–8):570–7. [DOI] [PubMed] [Google Scholar]
- 152.Alshikho MJ, Zurcher NR, Loggia ML, Cernasov P, Reynolds B, Pijanowski O, et al. Integrated magnetic resonance imaging and [(11) C]-PBR28 positron emission tomographic imaging in amyotrophic lateral sclerosis. Ann Neurol. 2018;83(6):1186–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Dukic S, McMackin R, Buxo T, Fasano A, Chipika R, Pinto-Grau M, et al. Patterned functional network disruption in amyotrophic lateral sclerosis. Hum Brain Mapp. 2019;40(16):4827–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Nasseroleslami B, Dukic S, Broderick M, Mohr K, Schuster C, Gavin B, et al. Characteristic Increases in EEG Connectivity Correlate With Changes of Structural MRI in Amyotrophic Lateral Sclerosis. Cereb Cortex. 2019;29(1):27–41. [DOI] [PubMed] [Google Scholar]
- 155.Sorrentino P, Rucco R, Jacini F, Trojsi F, Lardone A, Baselice F, et al. Brain functional networks become more connected as amyotrophic lateral sclerosis progresses: a source level magnetoencephalographic study. Neuroimage Clin. 2018;20:564–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Huynh W, Dharmadasa T, Vucic S, Kiernan MC. Functional Biomarkers for Amyotrophic Lateral Sclerosis. Front Neurol. 2018;9:1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Menon P, Yiannikas C, Kiernan MC, Vucic S. Regional motor cortex dysfunction in amyotrophic lateral sclerosis. Ann Clin Transl Neurol. 2019;6(8):1373–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Higashihara M, Pavey N, van den Bos M, Menon P, Kiernan MC, Vucic S. Association of Cortical Hyperexcitability and Cognitive Impairment in Patients With Amyotrophic Lateral Sclerosis. Neurology. 2021;96(16):e2090–e7. [DOI] [PubMed] [Google Scholar]
- 159.Menon P, Higashihara M, van den Bos M, Geevasinga N, Kiernan MC, Vucic S. Cortical hyperexcitability evolves with disease progression in ALS. Ann Clin Transl Neurol. 2020;7(5):733–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Dharmadasa T, Howells J, Matamala JM, Simon NG, Burke D, Vucic S, et al. Cortical inexcitability defines an adverse clinical profile in amyotrophic lateral sclerosis. Eur J Neurol. 2021;28(1):90–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Wainger BJ, Macklin EA, Vucic S, McIlduff CE, Paganoni S, Maragakis NJ, et al. Effect of Ezogabine on Cortical and Spinal Motor Neuron Excitability in Amyotrophic Lateral Sclerosis: A Randomized Clinical Trial. JAMA Neurol. 2021;78(2):186–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Neuwirth C, Braun N, Claeys KG, Bucelli R, Fournier C, Bromberg M, et al. Implementing Motor Unit Number Index (MUNIX) in a large clinical trial: Real world experience from 27 centres. Clin Neurophysiol. 2018;129(8):1756–62. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.