

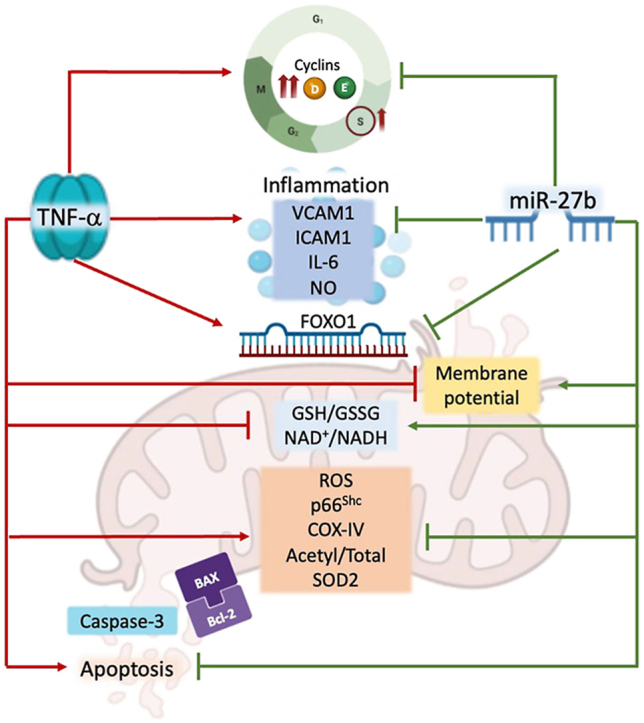
Abstract
MiR-27b is highly expressed in endothelial cells (EC) but its function in this context is poorly characterized. This study aims to investigate the effect of miR-27b on inflammatory pathways, cell cycle, apoptosis, and mitochondrial oxidative imbalances in immortalized human aortic endothelial cells (teloHAEC), human umbilical vein endothelial cells (HUVEC), and human coronary artery endothelial cells (HCAEC) exposed to TNF-α. Treatment with TNF-α downregulates the expression of miR-27b in all EC lines, promotes the activation of inflammatory pathways, induces mitochondrial alteration and reactive oxygen species accumulation, fostering the induction of intrinsic apoptosis. Moreover, miR-27b mimic counteracts the TNF-α-related cytotoxicity and inflammation, as well as cell cycle arrest and caspase-3-dependent apoptosis, restoring mitochondria redox state, function, and membrane polarization. Mechanistically, hsa-miR-27b-3p targets the 3′untranslated regions of FOXO1 mRNA to downregulate its expression, blunting the activation of the Akt/FOXO1 pathway. Here, we show that miR-27b is involved in the regulation of a broad range of functionally intertwined phenomena in EC, suggesting its key role in mitigating mithochondrial oxidative stress and inflammation, most likely through targeting of FOXO1. Overall, results reveal for the first time that miR-27b could represent a possible target for future therapies aimed at improving endothelial health.
Keywords: Endothelial dysfunction, Mitochondria, hsa-miR-27b-3p, Inflammation, Apoptosis
Graphical abstract
Highlights
-
•
Hsa-miR-27b-3p counteracts the TNF-α-induced endothelial dysfunction.
-
•
Hsa-miR-27b-3p opposes the TNF-α-related mitochondrial redox alteration.
-
•
The TNF-α-triggered mitochondrial apoptosis is attenuated by hsa-miR-27b-3p.
-
•
Hsa-miR-27b-3p downregulates FOXO1 mRNA expression blunting the Akt/FOXO1 pathway.
1. Introduction
Redox homeostasis alteration leading to endothelial dysfunction is one of the hallmarks of ageing, cardiovascular diseases, and type 2 diabetes [[1], [2], [3], [4]]. Dysfunctional endothelium is characterized by impaired vascular wall and activated endothelial cells (EC), displaying an unbalanced redox state and the release of prothrombotic and proinflammatory molecules promoting proliferation, migration, and cell interactions [[5], [6], [7], [8]]. As a result, the upregulation of different cytokines, chemokines, adhesion molecules and growth factors by activated EC promotes a chronic proinflammatory state which underlies the progression of cardiovascular disorders [[9], [10], [11]].
Oxidative stress in EC is a pivotal factor contributing to endothelial dysfunction and to the pathogenesis of cardiovascular diseases. A key driver of reactive oxygen species (ROS) accumulation is the aberrant functioning of different ROS-related enzymes, i.e. the mitochondrial electron transport system [[12], [13], [14]]. As a main contributor to cellular ROS burden, mitochondria are players of multiple pathways responsible for excess oxidative stress and low-grade inflammation in EC [[15], [16], [17], [18]]. In particular, the imbalance between mitochondrial ROS overproduction and an inefficient antioxidant response is held to contribute to the development of mitochondria-driven endothelial dysfunction [19,20], and has thus been suggested as a potential target for a variety of pathologycal conditions [21,22].
A large body of evidence suggest that microRNAs (miRNAs) play an active role in inflammation and oxidative stress-related EC dysfunction [15,23,24]. These nucleotide non-coding regulatory RNA molecules are emerging as potential biomarkers, effectors, and targets for vascular disease diagnosis, prognosis, and treatment [25]. MiRNAs regulate target gene expression inhibiting the translation or promoting the degradation of mRNA, and affecting multiple pathophysiological cellular processes [26,27]. MiR-27 is a family of miRNAs highly expressed in EC. In vivo and in vitro studies reported the miR-27b ability to promote angiogenesis, while its inhibition impaired embryonic vessel formation, endothelial cell sprout formation, and decreased the number of perfused vessels [28]. However, the precise functions of miR-27b in EC are largely unknown [29]. As a result, whether the modulation of miR-27 influences key pathways underlying endothelial homeostasis and represents a potential target to improve endothelial health has not been thoroughly explored. Here, we aimed to unveil the effects of miR-27b on EC stressed with TNF-α by evaluating inflammatory pathway, cell cycle/death mechanisms, and mitochondrial oxidative unbalance. We also explored the possible molecular targets of hsa-miR-27b-3p in immortalized human aortic endothelial cells (teloHAEC), human umbilical vein endothelial cells (HUVEC), and human coronary artery endothelial cells (HCAEC).
2. Materials and methods
2.1. Cell culture, transfection, and treatment
TeloHAEC (CRL-4052), HUVEC (PCS-100-010) and HCAEC (PCS-100-020) were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). TeloHAEC and HUVEC cells were grown in vascular cell basal media (PCS-100-030, ATCC, Manassas, VA, USA) supplemented with endothelial cell growth kit-VEGF (PCS-100-041, ATCC, Manassas, VA, USA), while HCAEC cells from a single male donor were grown in EGM-2MV (CC-3202 Lonza, Basel, Switzerland) supplemented with 200 U/mL penicillin and 200 μg/mL of streptomycin. All EC lines were grown as a monolayer under a humidified atmosphere at 37 °C with 5% CO2. Before inflammatory stimulus, EC were transfected with 20 nM mimic miR-27b (hsa-miR-27b miRNA Mimic, MCH01638, Applied Biological Materials, Inc. Richmond, BC, Canada) or miRNA mimic Negative Control (miR-NC, MCH00000, Applied Biological Materials, Inc. Richmond, BC, Canada), in serum- and antibiotic-free medium, using Lullaby (LL70500, OZ Biosciences, Marseille, France) as transfectant reagent. Cells were incubated for 6h, before the addition of FBS for 12h, and then treated with TNF-α. By using qRT-PCR, the overexpression's effectiveness was verified. To induce not only inflammatory state but also determine cell cycle modulation, oxidative stress and cell death induction, EC were exposed up to 50 ng/mL for 8h to recombinant human TNF-α protein (210-TA020, Biotechne, R&D Systems, Inc., Minneapolis, MN, USA), dissolved in phosphate buffered saline (PBS) containing 0.1% bovine serum albumin. Control cells (Ctr) were maintained in complete culture medium with the corresponding volume of Hanks' balanced salt solution (HBSS)-10 mM Hepes.
2.2. Cell viability assessment
EC viability was detected by Cell Counting Kit-8 (CCK-8, Donjindo Molecular Technologies, Inc., Rockville, MD, USA). Following manufacturer's guidance, 10 μL of CCK-8 solution was added to each well and plate incubated for 4h at 37 °C. Absorbance was measured at 450 nm with a microplate reader (model 680, Bio-Rad, Hercules, CA, USA) and viability expressed as % of control. All experiments were performed with n = 4 replicates.
2.3. Lactate dehydrogenase (LDH) release
The release of LDH by EC was assessed by the LDH Assay Kit-WST (CK12, Dojindo Molecular Technologies, Inc., Rockville, MD, USA). According to the manufacturer's protocol, 100 μL of working solution was added to 50 μL of cell suspension. After 30 min incubation, absorbance was measured with a microplate reader (model 680, Bio-Rad, Hercules, CA, USA) at 490 nm, and the % of LDH release measured with the equation: (Test Substance - Low Control)/(High Control - Low Control) × 100. Experiments were carried out with n = 3 replicates.
2.4. Malondialdehyde (MDA) content
Lipid peroxidation was assessed by measuring MDA content with lipid peroxidation assay kit (ab118970, Abcam, Cambridge, UK). According to the manufacturer's instructions, EC were homogenized in 303 μL of MDA lysis solution and 600 μL of thiobarbituric acid (TBA) before 1h of incubation at 95 °C before cooling on ice. The absorbance of the MDA-TBA adduct in supernatant was measured at 532 nm with a microplate reader (model 680, Bio-Rad, Hercules, CA, USA) and total MDA levels, normalized to protein content, calculated by comparing sample absorbance to a standard curve.
2.5. Inflammatory mediators
Levels of ICAM1, VCAM1 and IL-6 were measured using human ELISA kits (ab174445, ab223591, and ab178013, respectively, all from Abcam, Cambridge, UK). According to the manufacturer's protocols, cell culture media were centrifuged at 2000×g for 10 min to remove debris. Antibody cocktails (50 μL) were added to each sample and standard (50 μL) and sealed plate incubated for 1h at room temperature on a plate shaker set to 400 rpm. Each well was washed 3 time with wash buffer, before the addition of development solution (100 μL) and a 10 min incubation in the dark on a plate shaker. The stop solution was added (100 μL), mixed for 1 min on the shaker and 450 nm absorbance recorded with a microplate reader (model 680, Bio-Rad, Hercules, CA, USA). Concentration of ICAM1, VCAM1 and IL-6 in the samples were determined by interpolating the absorbance values against the standard curve.
2.6. Nitric oxide (NO) release
Nitric Oxide assay kit (ab272517, Abcam, Cambridge, UK) was used to quantitate NO production following reduction of nitrate to nitrite, according to manufacturer's protocols. Working solution (200
μL) was added to 100 μL of cell culture media or standard and incubated for 10 min at 60 °C. After a brief centrifugation, supernatants were transferred to a 96-well plate and absorbance detected at 540 nm by a microplate reader (model 680, Bio-Rad, Hercules, CA, USA). The content of NO, reported as μM, was derived by plotting the sample OD against standard concentrations.
2.7. Quantitative real-time PCR
Total RNA was obtained by Total RNA purification kit (17200, Norgen Biotek Corp., Thorold, ON, Canada), following manufacturer's instructions, and quantified by a NanoDrop2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). Reverse transcription from RNA to cDNA was performed with ID3EAL cDNA Synthesis System (1103101, MiRXES, Singapore, Republic of Singapore) and the ID3EAL Individual miRNA RT Primer 1-plex hsa-miR-27b-3p (1103111-HSA0000419A, MiRXES, Singapore, Republic of Singapore) for validation of hsa-miR-27b-3p overexpression and by Tetro cDNA Synthesis Kit (BIO-65043, Meridian Bioscience Inc., Cincinnati, OH, USA) for assessment of FOXO1 mRNA levels, on a thermal cycler SureCycler 8800 (Agilent Technologies, Santa Clara, CA, USA). Quantitative real-time was conducted with CFX96 Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA) with ID3EAL miRNA qPCR Master Mix (1104202, MiRXES, Singapore, Republic of Singapore) and ID3EAL miRNA qPCR hsa-miR-27b-3p (1104101-HSA0000419A, MiRXES, Singapore, Republic of Singapore) primers or with a QuantiTect SYBR Green PCR Kit (204,143, Qiagen, Hilden, Germany) and the following primers: (0.2 μM):
FOXO1 (2308): F-AGACAACGACACATAGCTGG, R-AGGGAGTTGGTGAAAGACATC.
GAPDH (2597): F-GAAGGTGAAGGTCGGAGTC, R-GAAGATGGTGATGGGATTTC.
The relative expression levels were determined by comparing the expression of FOXO1 or hsa-miR-27b-3p to that of GAPDH or U6, using the 2−ΔΔCt method (ΔΔ cycle threshold, Ct = (Ct FOXO1 or hsa-miR-27b-3p - Ct GAPDH or U6) of treated or transfected cells/(Ct FOXO1 or hsa-miR-27b-3p - Ct GAPDH or U6) of control), and data reported as the mean ± SD of n = 3 independent experiments, with each reaction performed in triplicate.
2.8. Cell cycle evaluation
After treatments, EC were incubated for 30 min in a stain buffer containing 50 μg/ml PI, 0.1% sodium citrate, 25 μg/ml RNase A, and 0.1% Triton in PBS. The intracellular DNA content was detected by a BD Accuri C6 (Becton Dickinson, San Jose, CA) cytometer, recording at least 20,000 events for each sample, and analysis carried with FLOWJO V10 software (Williamson Way, Ashland, OR, USA).
2.9. ATP production
The ATP assay kit (ab83355, Abcam, Cambridge, UK) was used to quantify total ATP in EC. Following the supplier's instructions, for each sample 1.0 × 106 cells were homogenized in 100 μL ATP assay buffer and centrifuged at 13,000 g for 5 min at 4 °C. Supernatants were deproteinizated with 1 M ice cold perchloric acid and 2 M KOH and centrifugated at 13,000×g for 15 min at 4 °C. ATP reaction mix (50 μL) was added to 50 μL samples or ATP standard in a 96-well plate. After 30 min incubation protected from light, the optimal output density was measured at 570 nm with a microplate reader (model 680, Bio-Rad, Hercules, CA, USA). The ATP quantity was calculated by plotting absorbance values with the calibration curve.
2.10. GSH/GSSG ratio
The GSSG/GSH quantification kit (G257, Dojindo Molecular Technologies, Tokyo, Japan) was used to obtain GSH and GSSG levels as indicators of oxidative stress. According to the supplier's instructions, after treatments the GSH and GSSG levels were independently evaluated in each sample by 1h incubation with substrate followed by 10 min with enzyme/coenzyme working solution. GSH and GSSG standard solutions were also made up. The absorbance was calculated using a microplate reader model 680 Bio-Rad (Bio-Rad, Hercules, CA, USA) at 405 nm and the quantity of GSH and GSSG in the samples derived by standard calibration curves.
2.11. NAD+/NADH ratio
The NAD+/NADH Assay Kit-WST (N509, Dojindo Molecular Technologies, Tokyo, Japan) was used to detect NAD+ and NADH content in EC, according to manufacturer's protocols. After treatments, 5.0 × 105 cells were extracted with NAD+/NADH extraction buffer. To assess NADH levels, the content of NAD+ was removed by incubation at 60 °C for 1h, while the total amount of NAD+/NADH incubated with working reagent at 37 °C for 1h. The absorbance at 450 nm was detected through a microplate reader model 680 Bio-Rad (Bio-Rad, Hercules, CA, USA). The content of total NAD+/NADH and NADH derived by calibration curves, while NAD+ levels were calculated by subtracting NADH from the total NAD+/NADH levels.
2.12. Mitochondrial analysis
Mitochondrial integrity was assessed with the fluorescent probe MitoTracker Green FM (M7514, Invitrogen, Waltham, MA, USA) Invitrogen), mitochondrial ROS amount was determined with MitoSOX Red Mitochondrial Superoxide Indicator (M36008, Invitrogen, Waltham, MA, USA), and impaired mitochondria fused to lysosomes were identified with Mitophagy Detection Kit (MD01, Dojindo Molecular Technologies, Tokyo, Japan). After treatments, EC were stained for 30 min with 5 μM MitoTracker, MitoSOX or with 100 nM Mtphagy Dye (red) followed by additional 30 min incubation with 1 μM Lyso Dye (green). Cells were imaged on a EVOS FL Cell Imaging System (Thermo Scientific, Rockford, IL, USA) microscope and fluorescence intensities measured by a BD Accuri C6 cytometer (BD Biosciences, San José, CA, USA). At least 20,000 events were recorded for all the samples and analysis conducted with FLOWJO V10 software (WilliamsonWay, Ashland, OR, USA). Treatment with 50 μM menadione (M57405, Sigma Aldrich, St. Louis, MO, USA) for 20 min at 37 °C was used as ROS positive control.
2.13. Mitochondrial membrane potential
Mitochondrial membrane potential was measured with JC-1 stain (MT09, Dojindo Molecular Technologies, Tokyo, Japan), a lipophilic cation fluorescent dye able to accumulate in mitochondria depending to membrane potential. Red fluorescence is related to high mitochondrial membrane potential, while green fluorescent JC-1 to decreased polarization. After treatments, EC were incubated for 1h with 5 μM JC-1 probe at 37 °C in the dark. Mitochondrial pore activity was assessed by Mitochondrial permeability transition pore assay kit (ab239704, Abcam, Cambridge, UK). Cells were incubated in the dark for 30 min at 37 °C in 500 μL wash buffer containing 5 μL staining dye and 5 μL CoCl2. Nuclei were counterstained with 2 μM Hoechst 33342 (ab228551, Abcam, Cambridge, UK), while ionomycin (5 μL) was used as negative control. After both stains, images were performed by a fluorescence microscope (EVOS FL Cell Imaging System, Thermo Scientific, Rockford, IL, USA) and fluorescence detected in detached cells with BD Accuri™ C6. At least 20,000 events were recorded for each sample and results analyzed with FlowJo V10 software (Williamson Way, Ashland, OR, USA).
2.14. Apoptotic cell death
Viable and apoptotic EC were determined by Annexin V Apoptosis detection kit (556547, BD Pharmigen, Franklin Lakes, NJ, USA). After treatments, cells were trypsinized, washed with PBS, and incubated for 30 min in 500 μL binding buffer 1 × containing 2 μL Annexin V-FITC and 2 μL PI (20 μg/mL). The involvement of caspase-3 and mitochondrial depolarization were detected with the Nucview 488 and Mitoview 633 Apoptosis Assay kit (30062, Biotium, Fremont, CA, USA). Staining with MitoView 633 (red) and NucView 488 (green) was performed for 15 min at room temperature, before imaging on a fluorescence microscope EVOS FL Cell Imaging System (Thermo Scientific, Rockford, IL, USA). For both assay, flow cytometry was performed using a BD Accuri C6 cytometer (BD Biosciences, San José, CA, USA) recording at least 20,000 events for each sample and analysis carried out with FlowJo V10 software (Williamson Way, Ashland, OR, USA).
2.15. Bioinformatics analysis
The prediction analysis of hsa-miR-27b-3p target genes has been performed using different engines, as MirDIP, miRDB and TargetScan. Bioinformatics evaluation conducted by mirDIP (https://ophid.utoronto.ca/mirDIP/) recognized FOXO1 as hsa-miR-27b-3p target with high (top 5%) minimum score class and an integrated score value of 0.79. The MiRDB - MicroRNA Target Prediction Database (https://mirdb.org/) identified for hsa-miR-27b-3p two predictive binding sites (position 426–432 and 2000–2006) in FOXO1 3′ UTR region with the target score value of 82. Bioinformatics study by TargetScan 8.0 (https://www.targetscan.org/vert_80/) tool confirmed both positions 426–432 and 2000–2006 as hsa-miR-27b-3p binding sites. This prediction tool ascribed values of 92 as context score percentile and −0.19 as context score to the conserved position 426–432 of FOXO1 3′ UTR and assigned a context score percentile value of 47 with −0.02 as context score for the poor conserved region 2000–2006. Based on these results, validation of FOXO1 as a direct target of hsa-miR-27b-3p has been performed investigating the 426–432 region as the possible functional interaction region.
2.16. Luciferase assay
The FOXO1-related fluorescence was firstly evaluated in EC transfected with fluorescent pEGFP rbFOX1 plasmid (63085, Addgene, Watertown, MA, USA) and Dreamfect 1000 reagent (DF41000, OZ Biosciences, Quimigen, Madrid, Spain), to assess the best transfectant experimental conditions. Dual-luciferase reporter assay was then performed by Firefly & Renilla luciferase single tube assay kit (30081-T, Biotium, Fremont, CA, USA), following manufacturer's protocols, on EC co-transfected with customized luciferase plasmid containing the 350–550 region of the 3′UTR target expression clone for human FOXO1 (CS-HmiT127365-MT06-01, GeneCopoeia, Rockville, MD, USA), and miR-NC or miR-27b mimic. Luciferase activity was detected by Tecan Infinite M200 (Tecan, Männedorf, Switzerland) and the luminescent intensity of Firefly luciferase activity derived by normalization with Renilla luciferase as an internal control. Transfection with control vector, without a 3′ UTR as baseline (CmiT000001-MT06, GeneCopoeia, Rockville, MD, USA) has been performed.
2.17. Cell lysis and western blotting
Cell lysis was conducted for 30 min at 4 °C in buffer containing 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 10 μg/mL aprotinin, leupeptin, and 1 mM phenylmethylsulfonylfluoride in PBS. Proteins (40–60 μg) were separated by SDS-PAGE and transferred to nitrocellulose membranes. Blocking of non-specific binding sites was performed for 1h in 1 × TBS 1% casein blocker (1610782, Bio-Rad, Hercules, CA, USA) and then membranes incubated overnight at 4 °C with specific primary antibodies: anti-BAX (1:500, orb216030, Biorbyt, Cambridge, UK), anti-Bcl-2 (1:500, E-AB-15522, Elabscience Biotechnology Inc., Houston, TX, USA), anti-phospho-Akt (Thr308) (1:1000, 13038, Cell Signaling Technology, Danvers, MA, USA), anti-Akt (1:2000, 2920, Cell Signaling Technology, Danvers, MA, USA), anti-superoxide dismutase 2 (SOD2, 1:2000, sc-137254, Santa Cruz Biotechnology, Dallas, TX, USA), anti-SOD2 (acetyl K68) (1:500, ab137037, Abcam Cambridge, UK), anti-COX-IV (1:2000, MA5-15078, Invitrogen, Waltham, MA, USA), anti-caspase-3 (1:1000, 9662, Cell Signaling Technology, Danvers, MA, USA) anti-cyclin D1 (1:1000, ab134175, Abcam, Cambridge, UK), anti-cyclin E1 (1:1000, ab33911, Abcam, Cambridge, UK); anti-forkhead box O1 (FOXO1A, 1:1000, ab39670, Abcam, Cambridge, UK), anti-p66Shc (1:1000, ab54518, Abcam, Cambridge, UK), anti-α-tubulin (1:5000, E-AB-20036, Elabscience Biotechnology Inc., Houston, TX, USA), anti-actin (1:3000, ab179467, Abcam, Cambridge, UK), and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH, 1:2000, ab9485, Abcam, Cambridge, UK). The peroxidase-conjugated secondary antibodies were incubated for 1h and immunocomplexes revealed by Excellent Chemiluminescent Substrate kit (E-IR-R301, Elabscience Biotechnology Inc., Houston, TX, USA), on dried membranes. The immunoblotting signals were imaged by ChemiDoc Imaging System and Image Lab 6.0.1 software (Bio-Rad Laboratories, Milan, Italy), while densitometric intensities measured with ImageJ software 1.52n (Wayne Rasband, National Institutes of Health, Bethesda, MD, USA). Results are expressed as arbitrary units (AU) where signal of each band has been compared to that of corresponding loading control.
2.18. Statistical analysis
Results are reported as the mean ± standard deviation (SD) of at least 3 independent experiments. GraphPad Prism software version 9.1.2 (GraphPad Software Inc, La Jolla, CA, USA) was used to perform statistical analyses. Significance among the variables was assessed by one-way ANOVA and Tukey's post hoc test. Differences were considered statistically significant with the p-value less than 0.05.
3. Results
3.1. miR-27b opposed the TNF-α-induced cytotoxicity
To investigate the molecular mechanisms that contribute to the etiology of vascular diseases such as diabetes, atherosclerosis and hypertension, we investigated the response of teloHAEC, HUVEC and HCAEC cell lines to the pro-inflammatory cytokine TNF-α, able to generate a robust and reproducible inflammatory response. Moreover, we evaluated the action of the proangiogenic hsa-miR-27b-3p (miR-27b) on the inflammatory state induced by TNF-α. Dose-response revealed that TNF-α (40 ng/mL) was cytotoxic in all cell lines after 8h incubation (p < 0.001) (Fig. S1), with a decrease of hsa-miR-27b-3p (p < 0.05 vs Ctr) (Fig. 1). In light of these results, overexpression experiments with miR-27b mimic were performed.
Fig. 1.

TNF-α and hsa-miR-27b-3p (miR-27b) impact on EC. Hsa-miR-27b-3p (miR-27b) expression, measured by qRT-PCR and normalized with U6 as endogenous control, in (A,B) TeloHAEC, (E,F) HUVEC and (I,J) HCAEC cells treated for 8h with 40 ng/mL TNF-α or transfected with 20 nM mimic Negative Control (miR-NC) and miR-27b mimic (miR-27b). MiR levels are reported as floating bars with line representing the mean ± SD. EC viability and LDH release were evaluated after 8h treatment with 40 ng/mL TNF-α on (C,D) TeloHAEC, (G,H) HUVEC and (K,L) HCAEC transfected or not with miR-NC and miR-27b. Control cells (Ctr) were treated with corresponding volumes of HBSS-10 mM Hepes. Data are expressed as mean ± SD of n = 3 experiments. *p < 0.05 vs. Ctr; ¶p < 0.001 vs. Ctr; §p < 0.001 vs. miR-NC; †p < 0.01 vs. miR-NC+TNF-α.
Analysis by qRT-PCR confirmed the cellular transfection efficacy with mimic miR-27b (p < 0.05) and the absence of cytotoxicity up to 72h (Fig. 1 and S1). Exposure of transfected EC with mimic negative control (miR-NC) to increasing doses of TNF-α also resulted in the induction of cytotoxicity after 8h with 40 ng/mL (p < 0.001) (Fig. S1).
Of interest, miR-27b overexpression for 18h counteracted the EC cytotoxicity and LDH release induced by the inflammatory TNF-α in all EC lines (p < 0.01 vs miR-NC+TNF-α) (Fig. 1).
In teloHAEC cells the TNF-α-induced cytotoxicity correlated with apoptosis (p < 0.001) and S-phase cell cycle accumulation (p < 0.001), underlining the cell cycle-dependent sensitivity of EC to TNF-α (Fig. S2), according to previous studies [30,31]. TNF-α treatment up to 30 ng/ml for 8h did not induce significant effects on cell cycle regulation neither was able to trigger apoptotic or necrotic death phenomena (Fig. S2).
3.2. miR-27b ameliorated the TNF-α-induced inflammation
The miR-27b overexpression was able to counteract the TNF-α-related cell damage, as assessed by the decreased levels of pro-inflammatory mediators, as VCAM1, ICAM1, IL-6, and the restored NO levels (p < 0.01 vs miR-NC+TNF-α) (Fig. 2).
Fig. 2.

TNF-α and miR-27b effects on inflammation. Evaluation of VCAM1, ICAM1, IL-6, and NO levels in (A–D) TeloHAEC, (E–H) HUVEC and (I–L) HCAEC cells exposed for 8h to 40 ng/mL TNF-α or transfected with miR-NC and miR-27b before TNF-α stimulation. Control cells (Ctr) were treated with corresponding volumes of HBSS-10 mM Hepes. Data are expressed as mean ± SD of n = 3 experiments. ¶p < 0.001 vs. Ctr; §p < 0.001 vs. miR-NC; †p < 0.01 vs. miR-NC+TNF-α.
Cells transfected with miR-NC exposed to TNF-α (miR-NC+TNF-α) showed effects comparable to non-transfected cells treated with TNF-α (TNF-α). Therefore, miR-NC+TNF-α treatment was used to induce inflammatory stress in further experiments. Moreover, the response to TNF-α stimulation was highly concordant among immortalized teloHAEC, primary HCAEC and HUVEC cells, suggesting that teloHAEC represents a powerful cellular model to study vascular endothelial cell dysfunction and the underlined molecular mechanism(s).
3.3. miR-27b counteracted the cell cycle arrest induced by TNF-α
In teloHAEC cells, cell cycle analysis showed the capability of miR-27b mimic to counteract the S phase arrest (34.6 ± 2.3% vs 41.6 ± 2.8%) and the concomitant decrease of G1 population (40.8 ± 2.9% vs 25.7 ± 3.1%) caused by TNF-α treatment (p < 0.01 vs miR-NC+TNF-α) (Fig. 3A and B).
Fig. 3.

Cell cycle evaluation. (A,B) Representative cell cycle analysis and cropped blots with relative immunoblotting analysis of (C,D) cyclin D1 and (E,F) cyclin E1 protein levels in teloHAEC cells transfected with miR-NC and miR-27b before TNF-α stimulation. Control cells (Ctr) were dealt with correponding volumes of HBSS-10 mM Hepes. Lane 1 = Ctr; lane 2 = miR-NC; lane 3 = miR-NC+TNF-α; lane 4 = miR-27b; lane 5 = miR-27b+TNF-α; M = molecular weight markers. Western blotting results are expressed as arbitrary units (AU). §p < 0.001 vs. miR-NC; †p < 0.01 vs. miR-NC+TNF-α.
The cytoprotective effect of miR-27b on EC cycle perturbation was accompanied by a reduction of cyclin D1 and E1 expression levels, accumulated during TNF-α exposure (p < 0.01 vs miR-NC+TNF-α) (Fig. 3C–F).
3.4. MiR-27b improved the TNF-α-induced mitochondrial redox stress
Exposure to TNF-α resulted in EC impaired oxidative state (Fig. 4). MiR-27b overexpression decreased the cytokine-induced mitochondrial ROS accumulation and lipid peroxidation (p < 0.01 vs miR-NC+TNF-α) (Fig. 4A–D), as well restored the levels of GSH/GSSG and NAD+/NADH ratios (p < 0.01 vs miR-NC+TNF-α) (Fig. 4E and F). As for the viability and inflammation, the mitochondrial redox response was highly concordant among teloHAEC, HUVEC and HCAEC. In detail, the miR-27b overexpression displayed protective effects in HCAEC and HUVEC cells, counteracting the TNF-α-induced mitochondrial oxidative stress and MDA content, as well as the impaired GSH/GSSG and NAD+/NADH ratios (p < 0.01 vs miR-NC+TNF-α) (Fig. S3).
Fig. 4.

Mitochondrial oxidative stress. (A–C) Representative fluorescent images and FACS analysis of mitochondrial ROS detection, (D) MDA, (E) GSH/GSSG and (F) NAD+/NADH content and cropped blots with relative immunoblotting analysis of (G,H) p66Shc, (I,J) COX-IV, and (K,L) acetylated-SOD2/SOD2 protein levels in teloHAEC transfected with miR-NC and miR-27b before TNF-α stimulation. Control cells (Ctr) were dealt with corresponding volumes of HBSS-10 mM Hepes. Scale bars = 100 μm. Lane 1 = Ctr; lane 2 = miR-NC; lane 3 = miR-NC+TNF-α; lane 4 = miR-27b; lane 5 = miR-27b+TNF-α; M = molecular weight markers. Western blotting results are expressed as arbitrary units (AU). §p < 0.001 vs. miR-NC; †p < 0.01 vs. miR-NC+TNF-α.
In teloHAEC cells miR-27b overexpression also attenuated the expression levels of some oxidative markers, increased during TNF-α exposure (p < 0.001 vs miR-NC). In detail, miR-27b+TNF-α cells showed downregulated p66Shc, COX-IV and acetylated-SOD2/SOD2 protein levels compared to miR-NC+TNF-α (p < 0.01) (Fig. 4G-L). Menadione, a ROS inductor, was also examined as a positive oxidative control (data not shown).
3.5. miR-27b blocked the mitochondrial membrane depolarization induced by TNF-α
Fluorimetric and cytometric assays indicated that miR-27b could oppose the loss of mitochondrial membrane potential induced by TNF-α treatment (Fig. 5). In detail, miR-27b+TNF-α cells showed lower rate of membrane depolarization (p < 0.01 vs miR-NC+TNF-α) (Fig. 5A–C) and an ameliorated mitochondrial permeability of transition pore was observed in teloHAEC, HCAEC, and HUVEC cells (p < 0.01 vs miR-NC+TNF-α) (Fig. 5D and E and Fig. S4).
Fig. 5.

Mitochondrial membrane depolarization. (A–C) Representative fluorescent images and FACS analysis of mitochondrial membrane potential, (D,E) mitochondrial permeability transition pore and (F) ATP content in teloHAEC transfected with miR-NC and miR-27b before TNF-α stimulation. Control cells (Ctr) were treated with corresponding volumes of HBSS-10 mM Hepes. Scale bars = 100 μm. Data are expressed as mean ± SD of n = 3 experiments. §p < 0.001 vs. miR-NC; •p < 0.05 vs. miR-NC+TNF-α; †p < 0.01 vs. miR-NC+TNF-α.
Moreover, in all EC lines miR-27b partly restored the ATP content impaired by TNF-α exposure (p < 0.05 vs miR-NC+TNF-α) (Fig. 5F and Fig. S4).
3.6. miR-27b opposed the TNF-α-induced mitochondrial damage
Mitochondrial perturbation was assessed by different approaches. Due to the fusion of lysosomes and damaged mitochondria, a double staining was used to evaluate the occurrence of mitochondrial injury accompanied by lysosome accumulation, while the fluorescent probe MitoTracker revealed the mitochondrial integrity (Fig. 6). Results showed that TNF-α-induced lysosome accumulation and decreased functional mitochondria (p < 0.001) were ameliorated in miR-27b+TNF-α cells (p < 0.01 vs miR-NC+TNF-α) (Fig. 6A–C).
Fig. 6.

Mitochondrial damage. (A–C) Representative fluorescent images and FACS analysis of mitophagy (red) and lysosome (green) dyes and (D–F) green-stained intact mitochondria in teloHAEC transfected with miR-NC and miR-27b before TNF-α stimulation. Control cells (Ctr) were treated with corresponding volumes of HBSS-10 mM Hepes. Scale bars = 100 μm. Data are expressed as mean ± SD of n = 3 experiments. *p < 0.05 vs. miR-NC; §p < 0.001 vs. miR-NC; †p < 0.01 vs. miR-NC+TNF-α. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Likewise, miR-27b+TNF-α upregulated the green fluorescence related to functional mitochondria compared to miR-NC+TNF-α in all cell lines analyzed (p < 0.01) (Fig. 6D–F and Fig. S5).
3.7. miR-27b counteracted the TNF-α-related mitochondrial apoptosis
Double fluorescent staining showed that TNF-α-induced depletion of mitochondrial polarization was accompanied by caspase-3 activation (p < 0.001 vs miR-NC), while miR-27b+TNF-α attenuated both events (p < 0.01 vs miR-NC+TNF-α) (Fig. 7A–C).
Fig. 7.

Mitochondrial apoptosis. (A–C) Representative fluorescent images and FACS analysis of Nucview 488 and Mitoview 633 staining, (F,G) dot plots and analysis of annexin V-FITC and PI-staining and cropped blots with relative immunoblotting analysis of (D,E) caspase-3 and (H,I) Bax/Bcl-2 protein levels in teloHAEC transfected with miR-NC and miR-27b before TNF-α stimulation. Control cells (Ctr) were treated with corresponding volumes of HBSS-10 mM Hepes. Western blotting results are expressed as arbitrary units (AU). Lane 1 = Ctr; lane 2 = miR-NC; lane 3 = miR-NC+TNF-α; lane 4 = miR-27b; lane 5 = miR-27b+TNF-α; M = molecular weight markers. Cell viability/death was assessed by flow cytometry where at least 10,000 events were acquired. Q1: necrotic cells; Q2: late apoptotic cells; Q3: early apoptotic cells; Q4: viable cells. Scale bars = 100 μm §p < 0.001 vs. miR-NC; •p < 0.05 vs. miR-NC+TNF-α; †p < 0.01 vs. miR-NC+TNF-α.
Immunoblotting analysis validated the restoration of both procaspase and active caspase-3 protein levels in miR-27b+TNF-α cells (p < 0.01 vs miR-NC+TNF-α), increased in miR-NC+TNF-α cells (p < 0.001 vs miR-NC) (Fig. 7D and E). Annexin V/PI measurements revealed that the cytotoxicity induced by TNF-α was accompanied by a reduction of live cells (66.4 ± 2.1% vs. 82.1 ± 3.5% in miR-NC, p < 0.001) and an increase of apoptotic cells (26.4 ± 3.4% vs. 4.8 ± 1.1% in miR-NC, p < 0.001). Moreover, cytokine-mediated apoptotic effects were reduced in miR-27b+TNF-α cells (74.5 ± 2.4% of live cells and 14.5 ± 2.9% of apoptotic cells, p < 0.01) (Fig. 7F and G). Cytofluorimetric results were confirmed in HUVEC and HCAEC cells showing that the TNF-α-related loss of live population (69.1 ± 2.7% vs. 83.1 ± 2.9% in miR-NC and 65.1 ± 3.1% vs. 82.9 ± 2.4% in miR-NC, respectively, p < 0.001) accompanied by apoptotic rate (27.0 ± 3.5% vs. 11.3 ± 1.5% in miR-NC and 31.0 ± 2.9% vs. 8.2 ± 2.4% in miR-NC, respectively, p < 0.001), was counteracted in miR-27b+TNF-α cells (76.7 ± 1.9% and 73.3 ± 2.2 of live cells and 16.0 ± 2.1% and 20.8 ± 2.9% of apoptotic cells, respectively, p < 0.01) (Fig. S6). Finally, the TNF-α-induced apoptotic mechanism was sustained by upregulated Bax/Bcl-2 ratio (p < 0.001 vs miR-NC) which resulted downregulated in miR-27b+TNF-α cells (p < 0.05) (Fig. 7H and I).
3.8. FOXO1 as hsa-miR-27b-3p target
Different bioinformatic tools revealed two predictive binding sequences for hsa-miR-27b-3p within the FOXO1-mRNA 3ʹ-UTR region (Fig. 8A), of which the most conserved (420–426) has been investigated in functional studies (Fig. 8H).
Fig. 8.

MiR-27b-FOXO1 interaction. (A) Alignment of hsa-miR-27b-3p with two regions (426–432, 2000–2006) of FOXO1 3′-UTR from miRNA-mRNA integration analysis using miRNA target prediction tools, as TargetScan and miRDB. (B) Schematic diagram of luciferase reporter plasmid containing 3′-UTR target sequence (region 350–550) of FOXO1. P: promoter, LUC: Luciferase. Representative analysis of (C) mRNA and (D,E) cropped blots with relative immunoblotting analysis of FOXO1 and (F,G) phospho-Akt/Akt protein levels in teloHAEC transfected with miR-NC and miR-27b before TNF-α stimulation. Western blotting results are expressed as arbitrary units (AU). Lane 1 = Ctr; lane 2 = miR-NC; lane 3 = miR-NC+TNF-α; lane 4 = miR-27b; lane 5 = miR-27b+TNF-α; M = molecular weight markers. (H) The relative luciferase activity in teloHAEC co-transfected with luciferase reporter plasmid, containing the region 350–550 of FOXO1 3′-UTR, and miR-NC or miR-27b. Control cells (Ctr) were treated with Corresponding volumes of HBSS-10 mM Hepes. Data are expressed as mean ± SD of n = 3 experiments. *p < 0.05 vs. miR-NC; ‡p < 0.01 vs. miR-NC; §p < 0.001 vs. miR-NC; •p < 0.05 vs. miR-NC+TNF-α; †p < 0.01 vs. miR-NC+TNF-α.
TNF-α exposure induced upregulated FOXO1 mRNA levels (p < 0.05 vs miR-NC), inhibited in both miR-27b overexpression and miR-27b+TNF-α cells (p < 0.05) (Fig. 8C). Immunoblotting analysis confirmed the induction of FOXO1 protein levels under TNF-α treatment (p < 0.001 vs miR-NC) and the inhibition by miR-27b overexpression (p < 0.001 vs miR-NC), even in miR-27b+TNF-α cells (p < 0.01 vs miR-NC+TNF-α) (Fig. 8D and E). Furthermore, stimulation with TNF-α downregulated protein levels of phospho-Akt/Akt (p < 0.001 vs miR-NC), the main regulator of FOXO1, which increased in miR-27b+TNF-α cells (p < 0.01 vs miR-NC+TNF-α) (Fig. 8F and G). To validate the binding prediction on FOXO1 mRNA as a direct target of hsa-miR-27b-3p, a dual-luciferase reporter assay was performed (Fig. 8H). The region containing the conserved 420–426 region of 3′-UTR FOXO1 gene, predicted binding site for hsa-miR-27b-3p, was cloned downstream of luciferase reporter vector (Fig. 8B) and used to co-transfect EC, along with miR-27b. Notably, miR-27b overexpression inhibited the luciferase activity of 3′-UTR FOXO1 (p < 0.01 vs miR-NC) (Fig. 8H), thus confirming the binding between hsa-miR-27b-3p and the FOXO1-mRNA 3ʹ-UTR in 420–426 region, leading to its downregulation.
4. Discussion
Herein, we showed for the first time the endothelial protective role of miR-27b in attenuating the harmful effects of TNF-α, including cell cycle arrest, apoptotic cell death, impaired mitochondrial redox state, and membrane polarization.
Dysfunctional endothelium due to inflammation and oxidative stress shifted endothelial function from vasoprotective to vasoconstrictive, prothrombotic, and proapoptotic profile [32]. The pro-inflammatory cytokine TNF-α is known to promote endothelial dysfunction playing a key role in a plethora of chronic disorders, such as diabetes, hypoxia and atherosclerosis [33,34]. Our results show the ability of TNF-α to induce cytotoxicity by triggering mitochondrial ROS accumulation, decreased permeability of transition pore and ATP production. These events were accompanied by a decreased ratio of reduced (GSH) to oxidized (GSSG) glutathione GSH/GSSG and NAD+/NADH. In turn, TNF-α-induced oxidative stress can promote or sustain EC damage, given the ROS ability to sustain critical intracellular oxidative stress and inflammatory cascade, structural rearrangement, and phenotype transition [35]. ROS are crucial mediators of mitochondrial membrane permeability and proapoptotic Bax protein regulation [17,19,36]. Consistently, our data indicated that the TNF-α-induced mitochondrial redox alteration was accompanied by intrinsic apoptotic cell death induction via caspase-3 upregulation.
Several studies showed that the inflammatory response of EC can be modulated by miRNAs which in turn downregulate target genes via post-translational modifications [23,26]. The role of miRNAs as potential therapeutic agents in EC dysfunction is widely reported [[37], [38], [39]].
TNF-α induces the expression of several miRNAs involved in vascular inflammation by modulating VCAM1, E-selectin and ICAM1, identified and validated as targets of the TNF-α-induced miRNAs [40]. Different miRNAs have been shown to prevent the impairment of endothelium alteration induced by inflammatory cytokines [41], while miR-126-5p and miR-132 are involved in EC proliferation and angiogenesis and miR-29 contributed to the development of pulmonary arterial hypertension via its effects on energy metabolism [42].
Previous evidence relative to the function of hsa-miR-27b in EC are scarce. One report described a protective effect by increasing EC sprouting via the angiogenesis inhibitor semaphorin 6A [28]. Our data showed that miR-27b overexpression was able to reduce the EC dysfunction related to TNF-α treatment by ameliorating oxidative condition and mitochondrial redox state and function. The beneficial effect of miR-27b mimic occurred by increasing the levels of NO and by attenuating the expression of caspase-3 and Bax/Bcl-2 ratio, in line with previous evidence showing the ability of miR-27b to prevent apoptosis in bone marrow-derived progenitor cells by suppressing the Bax/Bcl-2 ratio [43].
MiR-27b-3p clusters with miR-23b and miR-24 within an intron of the chromosome 9 open reading frame 3 (C9orf3) gene on the human chromosome 9q22.32. Of note, this cluster was previously shown to be modulated in patients with type 1 diabetes and to consistently predict its progression [44]. In addition, a recent study showed that glucose tolerance was impaired in mice after ablation of the whole miR-23b/27b/24-1 cluster via glycolytic pathway [45]. On the other hand, few studies explored a possible role for C9orf3, also known as AOPEP, in the setting of diabetes. One study evidence that a specific genetic variant of C9orf3 is associated with the prevalence of hypertension among subjects with type 2 diabetes [46], while another study found it as an hypermethylated gene in human atherosclerotic aorta [47]. However, none of these studies explored a putative mechanism explaining these associations. Thus, additional studies are warranted to explore the role of both the miR-23b/27b/24 cluster and of AOPEP in the context of endothelial damage.
The beneficial outcomes mediated by hsa-miR-27b-3p might be associated, at least in part, with its ability to regulate FOXO1 expression levels. On the other hand, the precise role of FOXO1 in the endothelium is still unclear. FOXO1 signaling is essential to the homeostasis of EC, restricting vascular growth, controlling angiogenesis-related genes and modulating metabolism, through reduction of the metabolic rate and enforcement of endothelial quiescence [48]. FOXO1 activation by the metabolite S-2-hydroxyglutarate induced a potent but reversible cell cycle arrest in EC [49], while siRNA-mediated FOXO1 knockdown in HUVEC significantly increased endothelial migration, tube formation in the Matrigel assay and sprouting activity of endothelial spheroids [50]. Mechanistically, phosphorylation of Akt inactivates FOXO by inducing shuttling from the nuclear localization to the cytoplasm [51]. Enhancement of the Akt/FOXO1 signaling pathway was also reported during hypoxia induced by genetic ablation of IRS2, increasing pulmonary vascular muscularization, proliferation, and perivascular macrophage recruitment [52]. In primary EC, hyperglycaemia suppresses PI3K/Akt activation, while FOXO1 gain-of-function increased inflammation in vitro and in vivo [53]. The functional relationship between miR-27b and FOXO1, already documented in different biological contexts, prompted this study aimed at deepening knowledge on the molecular network involved in the vascular inflammatory state and in the development of cardiovascular diseases. In cardiac microvascular EC, miR-27b-3p-FOXO1 signaling has been found to be associated with inflammasome-induced pyroptosis thus mediating protective effect against ischemia-reperfusion injury [54]. In C2C12 myoblast miR-27b overexpression mimics the effects of resveratrol on mitochondrial function by targeting FOXO1 [55], playing critical roles in the regulation of muscle mass, muscle fiber size, and gene expression of different fiber types [56]. In endometrial adenocarcinoma cell line, miR-27 downregulates the expression of FOXO, whereas miR-27 silencing regulates the invasiveness of cancer cells by inducing FOXO1 overexpression [57,58].
Although the relationship between miRNAs and endothelial dysfunction is increasingly recognized, multiple aspects of miRNA's role in cardiovascular and other diseases still warrant further investigation.
However, we provide the first evidence that hsa-miR-27b-3p might exert protective functions in EC exposed to a pro-inflammatory stressor, suggesting its role as a potential therapeutic target against endothelial dysfunction, now still considered a vital contributor to the pathogenesis of cardiovascular diseases. Indeed, long non-coding RNAs have received increasing attention as potential translational targets, due to their contribution to the molecular dynamics modulating the expression of vascular smooth muscle cells contractile/synthetic gene programs. In this context, a genomic approach and sequencing technologies could deep our knowledge of the complex gene expression regulatory networks mediated by hsa-miR-27b-3p, and favor the rise of innovative therapeutic approaches targeting the non-coding transcriptome in the cardiovascular disease. Further studies are necessary to clarify the relevance of these findings in vivo and to explore whether hsa-miR-27b-3p can represent a target in the prevention or treatment of cardiovascular diseases. Also, the role of FOXO1 in mediating the observed effects and, more broadly, in the context of cardiovascular diseases should be further investigated. Finally, future research might eventually disclose additional targets of miR-27b with a relevance for endothelial health.
5. Conclusions
Our work is the first evidence of the key role of hsa-miR-27b-3p to counteract endothelial dysfunction driven by inflammation. Treatment with TNF-α induced hsa-miR-27b-3p downregulation, while miR overexpression was able to counteract cytokine-induced inflammatory cascade, cell cycle arrest, mitochondrial impairment, loss of membrane potential, and apoptotic cell death. These activities of hsa-miR-27b-3p are likely mediated by FOXO1. Overall, these findings pave the way for further studies on the role of hsa-miR-27b-3p as a potential cardioprotective molecule.
Authors’ contributions
Conceptualization, N.D., F.P., M.L.B., and G.P.; methodology, N.D., E.M., L.M. and F.P.; formal analysis, N.D., E.M., and C.A.; investigation, N.D., E.M., C.S. and F.P.; data curation, N.D., E.M., L.M. and F.P.; writing—original draft preparation, E.M., N.D., M.L.B. and G.P.; writing—review and editing, N.D., E.M, F.P., C.S., R.L.G., M.L.B., R.M., A.C. and G.P.; funding acquisition, M.L.B. All authors have read, edited, and approved the final article.
Funding
This work was supported by PON I&C 2014–2020-TABAREZO-F/200085/03/X45, PON I&C 2014-2020-CAPSULE-F/200016/01_03/X45, and Italian Ministery of Health - Ricerca Corrente to IRCSS Multimedica.
Declaration of competing interest
The authors declare no conflict of interest.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2023.102681.
Contributor Information
Nunzia D'Onofrio, Email: nunzia.donofrio@unicampania.it.
Francesco Prattichizzo, Email: francesco.prattichizzo@multimedica.it.
Elisa Martino, Email: elisa.martino@unicampania.it.
Camilla Anastasio, Email: camilla.anastasio@unicampania.it.
Luigi Mele, Email: luigi.mele@unicampania.it.
Rosalba La Grotta, Email: rosalba.lagrotta@multimedica.it.
Celestino Sardu, Email: celestino.sardu@unicampania.it.
Antonio Ceriello, Email: antonio.ceriello@hotmail.it.
Raffaele Marfella, Email: raffaele.marfella@unicampania.it.
Giuseppe Paolisso, Email: giuseppe.paolisso@unicampania.it.
Maria Luisa Balestrieri, Email: marialuisa.balestrieri@unicampania.it.
Appendix A. Supplementary data
The following are the Supplementary data to this article.
References
- 1.Sardu C., Paolisso P., Sacra C., Mauro C., Minicucci F., Portoghese M., Rizzo M.R., Barbieri M., Sasso F.C., D'Onofrio N., Balestrieri M.L., Calabrò P., Paolisso G., Marfella R. Effects of metformin therapy on coronary endothelial dysfunction in patients with prediabetes with stable angina and nonobstructive coronary artery stenosis: the CODYCE multicenter prospective study. Diabetes Care. 2019;42:1946–1955. doi: 10.2337/dc18-2356. [DOI] [PubMed] [Google Scholar]
- 2.Leung S.W., Vanhoutte P.M. Endothelium-dependent hyperpolarization: age, gender and blood pressure, do they matter? Acta Physiol. 2017;219:108–123. doi: 10.1111/apha.12628. [DOI] [PubMed] [Google Scholar]
- 3.Suryavanshi S.V., Kulkarni Y.A. NF-kappabeta: a potential target in the management of vascular complications of diabetes. Front. Pharmacol. 2017;8:798. doi: 10.3389/fphar.2017.00798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boulanger C.M., Arterioscler Endothelium. Thromb. Vasc. Biol. 2016;36:e26–e31. doi: 10.1161/ATVBAHA.116.306940. [DOI] [PubMed] [Google Scholar]
- 5.Ooi B.K., Chan K.G., Goh B.H., Yap W.H. The role of natural products in targeting cardiovascular diseases via Nrf2 pathway: novel molecular mechanisms and therapeutic approaches. Front. Pharmacol. 2018;9:1308. doi: 10.3389/fphar.2018.01308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carter A.M. Complement activation: an emerging player in the pathogenesis of cardiovascular disease. Sci. Tech. Rep. 2012;2012 doi: 10.6064/2012/402783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park-Windhol C., D’amore P.A. Disorders of vascular permeability. Annu. Rev. Pathol. 2016;11:251–281. doi: 10.1146/annurev-pathol-012615-044506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baghai T.C., Varallo-Bedarida G., Born C., Häfner S., Schüle C., Eser D., Zill P., Manook A., Weigl J., Jooyandeh S., Nothdurfter C., von Schacky C., Bondy B., Rupprecht R. Classical risk factors and inflammatory biomarkers: one of the missing biological links between cardiovascular disease and major depressive disorder. Int. J. Mol. Sci. 2018;19:E1740. doi: 10.3390/ijms19061740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Castro-Ferreira R., Cardoso R., Leite-Moreira A., Mansilha A. The role of endothelial dysfunction and inflammation in chronic venous disease. Ann. Vasc. Surg. 2018;46:380–393. doi: 10.1016/j.avsg.2017.06.131. [DOI] [PubMed] [Google Scholar]
- 10.Haybar H., Shokuhian M., Bagheri M., Davari N., Saki N. Involvement of circulating inflammatory factors in prognosis and risk of cardiovascular disease. J. Mol. Cell. Cardiol. 2019;132:110–119. doi: 10.1016/j.yjmcc.2019.05.010. [DOI] [PubMed] [Google Scholar]
- 11.Zhong S., Li L., Shen X., Li Q., Xu W., Wang X., Tao Y., Yin H. An update on lipid oxidation and inflammation in cardiovascular diseases. Free Radic. Biol. Med. 2019;144:266–278. doi: 10.1016/j.freeradbiomed. [DOI] [PubMed] [Google Scholar]
- 12.Li S., Deng J., Sun D., Chen S., Yao X., Wang N., Zhang J., Gu Q., Zhang S., Wang J., Zhu S., Zhu H., Li H., Xu X., Wei F. FBXW7 alleviates hyperglycemia-induced endothelial oxidative stress injury via ROS and PARP inhibition. Redox Biol. 2022;58 doi: 10.1016/j.redox.2022.102530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Magenta A., Greco S., Capogrossi M.C., Gaetano C., Martelli F. Nitric oxide, oxidative stress, and p66Shc interplay in diabetic endothelial dysfunction. BioMed Res. Int. 2014;2014 doi: 10.1155/2014/193095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Servillo L., D'Onofrio N., Casale R., Cautela D., Giovane A., Castaldo D., Balestrieri M.L. Ergothioneine products derived by superoxide oxidation in endothelial cells exposed to high-glucose. Free Radic. Biol. Med. 2017;108:8–18. doi: 10.1016/j.freeradbiomed.2017.03.009. [DOI] [PubMed] [Google Scholar]
- 15.Giuliani A., Prattichizzo F., Micolucci L., Ceriello A., Procopio A.D., Rippo M.R. Mitochondrial (dys) function in inflammaging: do mitomirs influence the energetic, oxidative, and inflammatory status of senescent cells? Mediat. Inflamm. 2017;2017 doi: 10.1155/2017/2309034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ungvari Z., Tarantini S., Donato A.J., Galvan V., Csiszar A. Mechanisms of vascular aging. Circ. Res. 2018;123:849–867. doi: 10.1161/circresaha.118.311378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y.J., Jin X., Li D., Lu J., Zhang X.N., Yang S.J., Zhao Y.X., Wu M. New insights into vascular aging: emerging role of mitochondria function. Biomed. Pharmacother. 2022;156 doi: 10.1016/j.biopha.2022.113954. [DOI] [PubMed] [Google Scholar]
- 18.Martino E., Balestrieri A., Anastasio C., Maione M., Mele L., Cautela D., Campanile G., Balestrieri M.L., D'Onofrio N. SIRT3 modulates endothelial mitochondrial redox state during insulin resistance. Antioxidants. 2022;11:1611. doi: 10.3390/antiox11081611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amorim J.A., Coppotelli G., Rolo A.P., Palmeira C.M., Ross J.M., Sinclair D.A. Mitochondrial and metabolic dysfunction in ageing and age-related diseases. Nat. Rev. Endocrinol. 2022;18:43–258. doi: 10.1038/s41574-021-00626-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Csiszar A., Labinskyy N., Zhao X., Hu F., Serpillon S., Huang Z., Ballabh P., Levy R.J., Hintze T.H., Wolin M.S., Austad S.N., Podlutsky A., Ungvari Z. Vascular superoxide and hydrogen peroxide production and oxidative stress resistance in two closely related rodent species with disparate longevity. Aging Cell. 2007;6:783–797. doi: 10.1111/j.1474-9726.2007.00339.x. [DOI] [PubMed] [Google Scholar]
- 21.Pollack R.M., Barzilai N., Anghel V., Kulkarni A.S., Golden A., O'Broin P., Sinclair D.A., Bonkowski M.S., Coleville A.J., Powell D., Kim S., Moaddel R., Stein D., Zhang K., Hawkins M., Crandall J.P. Resveratrol improves vascular function and mitochondrial number but not glucose metabolism in older adults. J. Gerontol. A Biol. Sci. Med. Sci. 2017;72:1703–1709. doi: 10.1093/gerona/glx041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Y., Li S., Yin M., Li Y., Chen C., Zhang J., Sun K., Kong X., Chen Z., Qian J. Isorhapontigenin attenuates cardiac microvascular injury in diabetes mellitus via the inhibition of mitochondrial-derived ferroptosis through PRDX2-MFN2-ACSL4 pathways. Diabetes Online ahead of print. 2022:db220553. doi: 10.2337/db22-0553. [DOI] [PubMed] [Google Scholar]
- 23.Kumar S., Kim C.W., Simmons R.D., Jo H. Role of flow-sensitive microRNAs in endothelial dysfunction and atherosclerosis: mechanosensitive athero-miRs. Arterioscler. Thromb. Vasc. Biol. 2014;34:2206–2216. doi: 10.1161/ATVBAHA.114.303425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olivieri F., Prattichizzo F., Giuliani A., Matacchione G., Rippo M.R., Sabbatinelli J., Bonafè M. miR-21 and miR-146a: the microRNAs of inflammaging and age-related diseases. Ageing Res. Rev. 2021;70 doi: 10.1016/j.arr.2021.101374. [DOI] [PubMed] [Google Scholar]
- 25.Prattichizzo F., Matacchione G., Giuliani A., Sabbatinelli J., Olivieri F., de Candia P., De Nigris V., Ceriello A. Extracellular vesicle-shuttled miRNAs: a critical appraisal of their potential as nano-diagnostics and nano-therapeutics in type 2 diabetes mellitus and its cardiovascular complications. Theranostics. 2021;11:1031–1045. doi: 10.7150/thno.51605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leitão A.L., Enguita F.J. A structural view of miRNA biogenesis and function. Noncoding RNA. 2022;8:10. doi: 10.3390/ncrna8010010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ullah M., Ng N.N., Concepcion W., Thakor A.S. Emerging role of stem cell-derived extracellular microRNAs in age-associated human diseases and in different therapies of longevity. Ageing Res. Rev. 2020;57 doi: 10.1016/j.arr.2019.100979. [DOI] [PubMed] [Google Scholar]
- 28.Urbich C., Kaluza D., Frömel T., Knau A., Bennewitz K., Boon R.A., Bonauer A., Doebele C., Boeckel J.N., Hergenreider E., Zeiher A.M., Kroll J., Fleming I., Dimmeler S. MicroRNA-27a/b controls endothelial cell repulsion and angiogenesis by targeting semaphorin 6A. Blood. 2012;119:1607–1616. doi: 10.1182/blood-2011-08-373886. [DOI] [PubMed] [Google Scholar]
- 29.Urbich C., Kuehbacher A., Dimmeler S. Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc. Res. 2008;79:581–588. doi: 10.1093/cvr/cvn156. [DOI] [PubMed] [Google Scholar]
- 30.Rastogi S., Rizwani W., Joshi B., Kunigal S., Chellappan S.P. TNF-α response of vascular endothelial and vascular smooth muscle cells involve differential utilization of ASK1 kinase and p73. Cell Death Differ. 2012;19(2):274–283. doi: 10.1038/cdd.2011.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou P., Lu S., Luo Y., Wang S., Yang K., Zhai Y., Sun G., Sun X. Attenuation of TNF-α-Induced inflammatory injury in endothelial cells by ginsenoside Rb1 via inhibiting NF-κB, JNK and p38 signaling pathways. Front. Pharmacol. 2017;8:464. doi: 10.3389/fphar.2017.00464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Csiszar A., Ungvari Z. Endothelial dysfunction and vascular inflammation in type 2 diabetes: interaction of AGE/RAGE and TNF-alpha signaling. Am. J. Physiol. Heart Circ. Physiol. 2008;295:H475–H476. doi: 10.1152/ajpheart.00644.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taverner K., Murad Y., Yasunaga A.B., Furrer C., Little J., Li I.T.S. The effect of type-2 diabetes conditions on neutrophil rolling adhesion. BMC Res. Notes. 2022;15:355. doi: 10.1186/s13104-022-06248-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abdi Sarabi M., Shiri A., Aghapour M., Reichardt C., Brandt S., Mertens P.R., Medunjanin S., Bruder D., Braun-Dullaeus R.C., Weinert S. Normoxic HIF-1α stabilization caused by local inflammatory factors and its consequences in human coronary artery endothelial cells. Cells. 2022;11:3878. doi: 10.3390/cells11233878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen Z., Xue Q., Cao L., Wang Y., Chen Y., Zhang X., Xiao F., Yang Y., Hayden M.R., Liu Y., Yang K. Toll-like receptor 4 mediated oxidized low-density lipoprotein-induced foam cell formation in vascular smooth muscle cells via Src and Sirt1/3 pathway. Mediat. Inflamm. 2021;2021 doi: 10.1155/2021/6639252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rasola A., Bernardi P. The mitochondrial permeability transition pore and its adaptive responses in tumor cells. Cell Calcium. 2014;56:437–445. doi: 10.1016/j.ceca.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.D'Onofrio N., Sardu C., Paolisso P., Minicucci F., Gragnano F., Ferraraccio F., Panarese I., Scisciola L., Mauro C., Rizzo M.R., Mansueto G., Varavallo F., Brunitto G., Caserta R., Tirino V., Papaccio G., Barbieri M., Paolisso G., Balestrieri M.L., Marfella R. MicroRNA-33 and SIRT1 influence the coronary thrombus burden in hyperglycemic STEMI patients. J. Cell. Physiol. 2020;235:1438–1452. doi: 10.1002/jcp.29064. [DOI] [PubMed] [Google Scholar]
- 38.Cheng M., Yang Z., Qiao L., Yang Y., Deng Y., Zhang C., Mi T. AGEs induce endothelial cells senescence and endothelial barrier dysfunction via miR-1-3p/MLCK signaling pathways. Gene. 2022;851 doi: 10.1016/j.gene.2022.147030. [DOI] [PubMed] [Google Scholar]
- 39.Zhang W., Liu B., Wang Y., Sun Phd L., Liu C., Zhang H., Qin W., Liu J., Han L., Shan W. miR-195-3p/BDNF axis regulates hypoxic injury by targeting P-ERK1/2 expression. Medicine. 2022;101 doi: 10.1097/MD.0000000000031586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suárez Y., Wang C., Manes T.D., Pober J.S. Cutting edge: TNF-induced microRNAs regulate TNF-induced expression of E-selectin and intercellular adhesion molecule-1 on human endothelial cells: feedback control of inflammation. J. Immunol. 2010;184:21–25. doi: 10.4049/jimmunol.0902369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vikram A., Kim Y.R., Kumar S., Li Q., Kassan M., Jacobs J.S., Irani K. Vascular microRNA-204 is remotely governed by the microbiome and impairs endothelium-dependent vasorelaxation by downregulating Sirtuin1. Nat. Commun. 2016;7 doi: 10.1038/ncomms12565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen X., Talati M., P Fessel J., Hemnes A.R., Gladson S., French J., Shay S., Trammell A., Phillips J.A., Hamid R., Cogan J.D., Dawson E.P., Womble K.E., Hedges L.K., Martinez E.G., Wheeler L.A., Loyd J.E., Majka S.J., West J., Austin E.D. Estrogen metabolite 16alpha-hydroxyestrone exacerbates bone morphogenetic protein receptor type II-associated pulmonary arterial hypertension through microRNA-29-mediated modulation of cellular metabolism. Circulation. 2016;133:82–97. doi: 10.1161/CIRCULATIONAHA.115.016133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H., Liu J., Wang Y., Fu Z., Hüttemann M., Monks T.J., Chen A.F., Wang J.M. MiR-27b augments bone marrow progenitor cell survival via suppressing the mitochondrial apoptotic pathway in type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2017;313:E391–E401. doi: 10.1152/ajpendo.00073.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garavelli S., Bruzzaniti S., Tagliabue E., di Silvestre D., Prattichizzo F., Mozzillo E., Fattorusso V., La Sala L., Ceriello A., Puca A.A., Mauri P., Strollo R., Marigliano M., Maffeis C., Petrelli A., Bosi E., Franzese A., Galgani M., Matarese G., de Candia P. Plasma circulating miR-23∼27∼24 clusters correlate with the immunometabolic derangement and predict C-peptide loss in children with type 1 diabetes. Diabetologia. 2020;63:2699–2712. doi: 10.1007/s00125-020-05237-x. [DOI] [PubMed] [Google Scholar]
- 45.Jiang Y.H., Man Y.Y., Liu Y., Yin C.J., Li J.L., Shi H.C., Zhao H., Zhao S.G. Loss of miR-23b/27b/24-1 cluster impairs glucose tolerance via glycolysis pathway in mice. Int. J. Mol. Sci. 2021;22(2):550. doi: 10.3390/ijms22020550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ichikawa M., Konoshita T., Makino Y., Suzuki J., Ishizuka T., Nakamura H. An association study of C9orf3, a novel component of the renin-angiotensin system, and hypertension in diabetes. Sci. Rep. 2020;10(1) doi: 10.1038/s41598-020-73094-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lacey M., Baribault C., Ehrlich K.C., Ehrlich M. Atherosclerosis-associated differentially methylated regions can reflect the disease phenotype and are often at enhancers. Atherosclerosis. 2019;280:183–191. doi: 10.1016/j.atherosclerosis.2018.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilhelm K., Happel K., Eelen G., Schoors S., Oellerich M.F., Lim R., Zimmermann B., Aspalter I.M., Franco C.A., Boettger T., Braun T., Fruttiger M., Rajewsky K., Keller C., Brüning J.C., Gerhardt H., Carmeliet P., Potente M. FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature. 2016;529:216–220. doi: 10.1038/nature16498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Andrade J., Shi C., Costa A.S.H., Choi J., Kim J., Doddaballapur A., Sugino T., Ong Y.T., Castro M., Zimmermann B., Kaulich M., Guenther S., Wilhelm K., Kubota Y., Braun T., Koh G.Y., Grosso A.R., Frezza C., Potente M. Control of endothelial quiescence by FOXO-regulated metabolites. Nat. Cell Biol. 2021;23:413–423. doi: 10.1038/s41556-021-00637-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Potente M., Urbich C., Sasaki K., Hofmann W.K., Heeschen C., Aicher A., Kollipara R., DePinho R.A., Zeiher A.M., Dimmeler S. Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. J. Clin. Invest. 2005;115:2382–2392. doi: 10.1172/JCI23126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim D.H., Bang E., Ha S., Jung H.J., Choi Y.J., Yu B.P., Chung H.Y. Organ-differential roles of Akt/FoxOs axis as a key metabolic modulator during aging. Aging Dis. 2021;12:1713–1728. doi: 10.14336/AD.2021.0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nakahara M., Ito H., Skinner J.T., Lin Q., Tamosiuniene R., Nicolls M.R., Keegan A.D., Johns R.A., Yamaji-Kegan K. The inflammatory role of dysregulated IRS2 in pulmonary vascular remodeling under hypoxic conditions. Am. J. Physiol. Lung Cell Mol. Physiol. 2021;321:L416–L428. doi: 10.1152/ajplung.00068.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sissaoui S., Egginton S., Ting L., Ahmed A., Hewett P.W. Hyperglycaemia up-regulates placental growth factor (PlGF) expression and secretion in endothelial cells via suppression of PI3 kinase-Akt signalling and activation of FOXO1. Sci. Rep. 2021;11 doi: 10.1038/s41598-021-95511-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang B., Sun C., Liu Y., Bai F., Tu T., Liu Q. Exosomal miR-27b-3p derived from hypoxic cardiac microvascular endothelial cells alleviates rat myocardial ischemia/reperfusion injury through inhibiting oxidative stress-induced pyroptosis via Foxo1/GSDMD signaling. Oxid. Med. Cell. Longev. 2022;2022 doi: 10.1155/2022/8215842. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55.Zhou X., Zuo S., Xin W. miR-27b overexpression improves mitochondrial function in a Sirt1-dependent manner. J. Physiol. Biochem. 2015;71(4):753–762. doi: 10.1007/s13105-015-0439-3. [DOI] [PubMed] [Google Scholar]
- 56.Kamei Y., Miura S., Suzuki M., Kai Y., Mizukami J., Taniguchi T., Mochida K., Hata T., Matsuda J., Aburatani H., Nishino I., Ezaki O. Skeletal muscle FOXO1 (FKHR) transgenic mice have less skeletal muscle mass, down-regulated Type I (slow twitch/red muscle) fiber genes, and impaired glycemic control. J. Biol. Chem. 2004;279(39):41114–41123. doi: 10.1074/jbc.M400674200. [DOI] [PubMed] [Google Scholar]
- 57.Myatt S.S., Wang J., Monteiro L.J., Christian M., Ho K.K., Fusi L., Dina R.E., Brosens J.J., Ghaem-Maghami S., Lam E.W. Definition of microRNAs that repress expression of the tumor suppressor gene FOXO1 in endometrial cancer. Cancer Res. 2010;70:367–377. doi: 10.1158/0008-5472.CAN-09-1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mozos A., Catasús L., D'Angelo E., Serrano E., Espinosa I., Ferrer I., Pons C., Prat J. The FOXO1-miR27 tandem regulates myometrial invasion in endometrioid endometrial adenocarcinoma. Hum. Pathol. 2014;45(5):942–951. doi: 10.1016/j.humpath.2013.12.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.