

Abstract
Recent therapeutic strategies for hemophilia include long-term therapeutic gene expression using adeno-associated virus (AAV) and rebalancing therapy via the downregulation of anticoagulant pathways. However, these approaches have limitations in immune responses or insufficiency to control acute bleeding. Thus, we developed a therapeutic strategy for hemophilia B by a combined rebalancing and human factor 9 (hF9) gene knockin (KI) using a lipid nanoparticle (LNP) and AAV. Antithrombin (AT; Serpin Family C Member 1 [Serpinc1]) was selected as the target anticoagulation pathway for the gene KI. First, the combined use of LNP-clustered regularly interspaced short palindromic repeats (CRISPR) and AAV donor resulted in 20% insertions or deletions (indels) in Serpinc1 and 67% reduction of blood mouse AT concentration. Second, hF9 coding sequences were integrated into approximately 3% of the target locus. hF9 KI yielded approximately 1,000 ng/mL human factor IX (hFIX) and restored coagulation activity to a normal level. LNP-CRISPR injection caused sustained AT downregulation and hFIX production up to 63 weeks. AT inhibition and hFIX protein-production ability could be maintained by the proliferation of genetically edited hepatocytes in the case of partial hepatectomy. The co-administration of AAV and LNP showed no severe side effects except random integrations. Our results demonstrate hemophilia B therapy by a combination of rebalancing and hF9 KI using LNP and AAV.
Keywords: MT: Oligonucleotides: Therapies and Applications, antithrombin, CRISPR-Cas9, hemophilia B, in vivo knockin, lipid nanoparticle
Graphical abstract
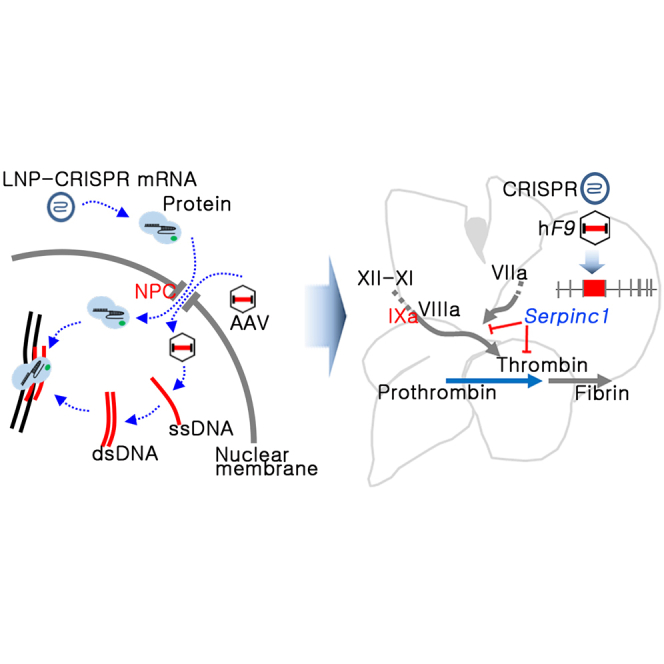
Yeom and colleagues established a gene editing-mediated therapeutic platform for hemophilia B. The platform combines existing concepts of anticoagulant pathway rebalancing and therapeutic gene KI using viral and non-viral vectors and produced the restoration of coagulation activity for the long term.
Introduction
Hemophilia is a genetic disorder involving a coagulation factor deficiency, along with spontaneous and excessive bleeding associated with surgery or trauma.1 Hemophilia B is caused by a deficiency of coagulation factor IX (FIX),2 and its prevalence is 1 in 25,000 male births. Various hemophilia treatments have been developed, but there are still unmet needs in terms of efficacy, safety, ease of treatment, and cost.3
General treatment for hemophilia involves the use of recombinant proteins, but repeated administration is required. Recently, recombinant coagulation factors with extended half-lives have been developed. However, the reported maximum half-life of recombinant FIX is still approximately 100 h, insufficient to solve the limitation.4 Therefore, advanced therapies are being developed to overcome the short-term therapeutic effect of recombinant clotting factors. Adeno-associated virus (AAV)-mediated gene therapy has reduced bleeding events and prolonged blood coagulation factor synthesis for several years.5,6,7 AAV has been used for gene therapies because of its high tissue-specific tropism and efficient transduction potential. However, the immune response to its capsid and the possibility of random integration are safety issues that need to be considered.8,9
Alternatively, rebalancing therapy via downregulation of three representative anticoagulant pathways, namely those of tissue factor pathway inhibitor (TFPI), antithrombin (AT), and activated protein C, has been attempted.10 Inhibition of gene expression using small interfering RNA (siRNA) or monoclonal antibody (mAb) was applied for rebalancing. A therapeutic AT siRNA (such as fitusiran)11,12 and a mAb against TFPI (such as concizumab)13 showed hemostatic effects on hemophilia, with or without inhibitors. Various types of coagulation disorders can be treated by rebalancing against AT through weekly or monthly subcutaneous injections.10 However, as shown in clinical trials for AT siRNA, rebalancing might be insufficient to control acute bleeding.14 Therefore, it needs to be combined with another therapeutic method.
To this end, therapeutic gene knockin (KI) is considered a next-generation strategy for restoring protein synthesis ability.15 The clinical trial was terminated for in vivo human factor 9 gene (hF9) KI using zinc finger nuclease (ClinicalTrials.gov: NCT02695160). In vivo genome editing is carried out by inducing breakage in the targeted DNA using a nuclease and then knocking the therapeutic gene into the target site. The clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated protein 9 (Cas9) technology has made targeted gene editing easier.16 However, the difficulty in delivering CRISPR-Cas9 and donor templates to target cells and low in vivo KI efficiency continue to pose challenges to gene editing.17 To improve the in vivo KI efficiency, the strategy of knocking a therapeutic gene into a locus with high expression can be adopted. For the development of in vivo therapeutic gene KI, CRISPR-Cas9 and donor templates should be delivered into the nucleus of target cells. AAV could deliver nucleic acid templates across the nuclear pore complex, making it a suitable tool for donor delivery for in vivo KI.18 Recent studies have used AAVs for CRISPR-Cas9 and donor template delivery in vivo, but they exhibited low KI efficiencies.19,20 Furthermore, AAV-packed CRISPR-Cas9 could induce long-term expression as an episomal persistence,21 and this property might cause long-lasting double-stranded DNA breakage (DSB) and off-target effects. In contrast, lipid nanoparticles (LNPs) are rapidly activated and degraded within 1–4 days after in vivo transfection.22 Our previous work has demonstrated safe and highly efficient in vivo gene targeting with LNP-incorporated CRISPR-Cas9.23
We attempted to establish a strategy for hemophilia treatment by combining rebalancing anticoagulation pathways and therapeutic gene KI. Hemophilia B was selected as a candidate disease to prove this concept. F9 is a small-sized gene encoding coagulation FIX in hepatocytes and is easy to pack with AAV. After selecting the target locus among the rebalancing-related genes, hF9 was knocked into this locus in vivo using a hybrid system of LNP-packed CRISPR (LNP-CRISPR) and AAV-packed hF9 donor templates. Here, we demonstrate that recovery from hemophilia is possible through a combination of rebalancing therapy and therapeutic gene KI in a mouse model.
Results
Serpin Family C Member 1 was selected as the target locus for therapeutic gene KI
Since we intended to knock the therapeutic gene into the locus of an anticoagulant pathway gene, it was necessary to select the optimal target locus (Figure 1A). Expression of liver tissue-derived Serpin Family C Member 1 (Serpinc1; encoding AT), Tfpi, and Protein C was compared, and Serpinc1 was found to show a significantly higher expression than the others (p < 0.001) (Figure 1B). Therefore, Serpinc1 was selected as the target locus for hF9 KI for inducing higher therapeutic gene expression, even under a low KI frequency.
Figure 1.

Serpinc1 was selected as the target locus for hF9 gene knockin (KI)
(A) Candidate rebalancing genes for therapeutic hF9 KI (blue letters: anticoagulant genes, red line: inhibition signal, blue and gray arrows: coagulation cascade). (B) Relative gene expression of Serpin Family C Member 1 gene (Serpinc1), tissue factor pathway inhibitor (Tfpi), and Protein C in the liver tissue (wild-type [WT]: n = 4). Each gene expression was normalized to that of Gapdh. Each dot represents data from an individual mouse. Data are presented as mean ± standard error of mean, calculated using one-way ANOVA (∗∗∗p < 0.001).
AAV-trap strategy with a bidirectional AAV donor efficiently induced hF9 KI
Since we previously developed a highly efficient LNP-CRISPR for AT targeting,23 LNP-packed single guide RNA (sgRNA) and Cas9 mRNA (LNP-CRISPR) targeting the third exon of Serpinc1 locus were used in this study. As the next step, we needed to select a strategy for in vivo hF9 KI. Recently, we reported a dual AAV introduction strategy with hF9 KI into apolipoprotein C3 (Apoc3) for hemophilia B therapy.18 In this study, AAV8-packed Campylobacter jejuni Cas9 was applied to induce targeted DSB. In addition, three different AAV8-packed donors for homologous recombination (HR), homology-independent targeted integration (HITI), and AAV trap were prepared to compare KI efficiency in different targeted integration strategies. The AAV trap with a bidirectional strategy induced the highest hFIX blood concentration.18 Similarly, target gene integration using bidirectional AAV has been applied to other KI studies of therapeutic genes.24,25,26 Therefore, we used the bidirectional AAV donor, which can trap the endogenous Serpinc1 promoter signal after integration into the KI locus (Figure 2A). The LNP-CRISPR and AAV donor were transfected into a primary hepatocyte or mouse hepatocyte cell line (AML12), and hF9 KI and hFIX production were confirmed in cells treated with LNP-CRISPR and AAV donor simultaneously (Figure 2B). This observation suggested that the AAV-trap strategy with the bidirectional AAV donor could be applied for in vivo hF9 KI through the combined use of LNP-CRISPR.
Figure 2.

In vivo human F9 gene KI into the Serpinc1 locus decreased antithrombin concentration in the blood
(A) Brief schematic representation of in vivo human (h)F9 KI into the third exon of Serpinc1 (thunder symbol: DSB site, blue letters: sgRNA binding sequences, red letters: protospacer adjacent motif [PAM] sequences; SA, splicing acceptor; hF9, coding sequence of hF9; pA, polyadenylation; LNP-CRISPR; lipid nanoparticle-packed SpCas9 mRNA and sgRNA). (B) Polymerase chain reaction (PCR) for KI and enzyme-linked immunosorbent assay (ELISA) for human factor IX (hFIX) was conducted after hF9 KI into the Serpinc1 locus using a primary hepatocyte and AML12 cell line (n = 4). (C) Brief information on experimental groups and injection schedule of genetic materials. (D) Identification of indel by T7E1 and calculation of indel rate using Sanger sequencing and algorithm-based analysis. (E) Mouse antithrombin (AT) concentration in the blood of different treatment groups, measured using ELISA. Each dot represents data from an individual mouse. Data are presented as mean ± standard error of mean, calculated using unpaired one-way ANOVA (∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.000).
Combined use of LNP-packaged CRISPR-Cas9 and AAV donor decreased at concentration efficiently
We expected that AAV donor transduction before LNP-CRISPR injection could reduce the dose of genetic material and provide the time for delivering the donor template in the nucleus before DSB formation by LNP-CRISPR. Hence, transgene expression after AAV transduction was analyzed weekly using AAV8 luciferase and hairless mice. Since high luciferase expression was observed at 2–3 weeks after AAV transduction (Figure S1), we decided on 2 weeks as a time of AAV transduction for donor preparation. We injected AAV donor and LNP-CRISPR into F9Mut mice after a 2 week interval (Figure 2C). LNP-CRISPR-injected groups (F9Mut-LNP and F9Mut-AAV-LNP) resulted in approximately 22% and 27% insertion or deletion (indel) formation in the liver, respectively, but other mouse groups (wild type [WT], F9Mut, and F9Mut-AAV) did not show any evidence of indel formation (Figure 2D). In addition, there was a difference in the reduction in blood AT concentration between F9Mut-LNP and F9Mut-AAV-LNP mice, at 34% and 67%, respectively, compared with that in the F9Mut control group. It has been reported that when DSB occurred by AAV-packed SpCas9, the AAV sequence integration in the DSB site was up to 47%, but AAV integration at the off-target site did not increase.26 Thus, we reasoned that the frequency of error-free DNA repair or non-frameshift deletion might be reduced by AAV-donor integration into the DSB site, which led to a relatively reduced AT concentration of F9Mut-AAV-LNP compared with F9Mut-LNP (Figure 2E). Thus, this implied that the combined use of LNP-CRISPR and AAV donor was effective for inducing rebalancing through potent AT targeting.
Therapeutic hF9 was knocked into the targeted Serpinc1 locus
The combination strategy aims to induce rebalancing and hFIX expression simultaneously. This study applied two different delivery vectors, which could lead to target-organ-specific hF9 KI by overlapping the biodistribution of these vectors. The biodistributions of AAV8 were evaluated using AAV8-delivered luciferase, and AAV8 shows high delivery efficiency to the liver (Figure S1). The hF9 AAV-donor level in the liver was highest among other organs (Figure S2). Consistently, indels were observed only in the liver (Figure S2), leading to liver-specific KI of the therapeutic gene. Since AT targeting in the liver was confirmed, the production of hFIX needed to be verified. hF9 integration into the liver tissue was analyzed 10 weeks after AAV-donor transduction (Figure 2C). Targeted hF9 integration was observed only in the group co-transfected with LNP-CRISPR and AAV donor (F9Mut-AAV-LNP). Sanger sequencing results revealed that partial sequences of the inverted terminal repeat (ITR) were fused into the target Serpinc1 locus (Figure 3A). Genomic DNA (gDNA)-based quantitative PCR showed amplification of the KI sequence solely in F9Mut-AAV-LNP mice (Figure 3B). In addition, detectable KI was only observed in the liver (Figure S2). hF9 KI yielded approximately 1,000 ng/mL hFIX in plasma (Figure 3C). Active hFIX in plasma was detected to be high in the WT and AAV-LNP groups, supporting the therapeutic effect of the AAV-LNP treatment (Figure 3D). The sequencing-based analysis reported possibly fragmented or full-length AAV integration,26 but hFIX production suggested that full hF9 coding sequences were knocked in in the Serpinc1 exon region. Therapeutic hF9 KI was confirmed by detecting hFIX expression in hepatocytes (Figures 3E and S3). Further, low levels of hFIX were detected in the F9Mut-AAV group (Figures 3C and 3E). In brief, combined use of AAV donor and LNP-CRISPR efficiently induced targeted hF9 integration by ITR sequence fusion into the Serpinc1 locus and produced active plasma hFIX.
Figure 3.

Knocked in hF9 produced hFIX in hepatocytes
(A) PCR and Sanger sequencing-based genotyping for analyzing KI and its integration pattern. Primers were designed for both sides of the KI-expected regions. Sequencing was conducted using 10 randomly selected clones from each KI-positive amplicon of F9Mut-AAV-LNP (red bar: KI sequence, mSerpinc1: endogenous sequence of Serpinc1; ITR, inverted terminal repeat; SA, splicing acceptor; yellow bar: unexpected indel). (B) Relative KI rates, as analyzed using quantitative PCR in the 3′ site of the expected KI locus. The KI rate was normalized to that of the F9Mut-AAV-LNP group. (C) hFIX concentration in the blood, measured using ELISA (n = 4 per group). Each dot represents data from an individual mouse. Data are presented as mean ± standard error of mean, calculated using one-way ANOVA. (∗∗p < 0.01 and ∗∗∗∗p < 0.0001. (D) Active hFIX in the blood was measured using a FIX activity assay kit (Fluorometric) (n = 4 per group). (E) Immunofluorescence images showing liver hFIX expression (100×). Yellow scale bar: 200 μm.
Combination of rebalancing and therapeutic gene KI restored coagulation activity
Since each AT-targeting and hF9 KI could exert therapeutic effects in hemophilia B mice,18,23 the improvement of blood coagulation activity by F9Mut-AAV-LNP was analyzed. The results of activated partial thromboplastin time (aPTT) analysis revealed a marked improvement in coagulation activity in the F9Mut-AAV-LNP group. The F9Mut-AAV group showed an improvement in coagulation activity as well. Moreover, restored coagulation activity was expected in F9Mut-LNP mice by rebalancing as a result of reduced Serpinc1 expression and blood AT concentration (Figure 2E).11 However, as per the results of the aPTT analysis, the enhancement in coagulation activity was not seen in the F9Mut-LNP group (Figure 4A). This might have resulted from a limitation of the aPTT analysis, which can evaluate only the initiation phase of coagulation.27
Figure 4.

In vivo hF9 gene KI into the Serpinc1 locus restored coagulation activity
(A) Coagulation activity of different groups analyzed based on aPPT (n = 7–8 per group). (B) Thrombin-generation potential, analyzed using a calibrated automated thrombogram (n = 3 per group). Each dot represents data from an individual mouse. Data are presented as mean ± standard error of mean, calculated using one-way ANOVA. (∗∗∗p < 0.001, ∗∗∗∗p < 0.0001).
Alternatively, the calibrated automated thrombogram (CAT) could evaluate the entire coagulation process, which could better reflect bleeding and thrombotic risks that occurred in vivo.27 Therefore, further CAT analysis was conducted to analyze the thrombin-formation ability. As expected, the F9Mut-AAV-LNP group showed thrombin-generation activity similar to that of the WT mice with regard to the peak time and peak height. Peak height refers to the maximum level of thrombin generated, and peak time refers to the time for reaching peak height. Both indexes represent the restoration of coagulation ability in the F9Mut-AAV-LNP group. The F9Mut-AAV and F9Mut-LNP groups also showed improved thrombin-generation activity (Figure 4B). Summarizing the results of aPTT and CAT analyses, blood coagulation activity can be restored to a level almost comparable to that of WT through combined therapeutic gene KI (hF9 in this case) and AT level reduction.
Prolonged therapeutic protein expression was observed after in vivo gene editing
A goal of CRISPR-mediated gene editing is to bring about sustainable therapeutic effects. WT mice were used to analyze the long-term reduced AT and hFIX synthesis with repeated blood sampling. Two weeks after AAV donor and LNP-CRISPR injection, AT and hFIX concentrations in the blood were measured every 4 weeks (Figure 5A). A single LNP-CRISPR injection decreased blood AT concentration to approximately 35% in control mice and was maintained for 20 weeks. In addition, a combination of AAV donor and LNP-CRISPR significantly lowered the blood AT concentration compared with the LNP-CRISPR control (p = 0.003). This observation was consistent with results from the F9Mut mice (Figure 2E) and indicated that the AAV-donor template decreases the rate of precise repair or non-frameshift indels. Similarly, blood hFIX concentrations were increased to more than 500–700 ng/mL after AAV and LNP injection and were stably maintained for 20 weeks. An additional analysis at 63 weeks after LNP-CRISPR injection revealed sustained AT downregulation and hFIX production. Increased hFIX concentration was observed in the AAV- and LNP-injected groups; thus, we reasoned that hF9 was integrated into the Serpinc1 locus and that gradually increased Serpinc1 expression according to age would affect hF9 gene expression. Although AAV therapy is known to overcome preexisting inhibitors,28 we used WT mice for repeated blood sampling; therefore, short-term therapeutic effects against the inhibitor could not be analyzed in this study. In addition, there was a low hFIX level in the AAV-donor control group (Figure 5B).
Figure 5.

In vivo hF9 KI induced a long-term and sustained therapeutic effect
(A) Brief schematic representation of long-term analysis of hFIX and AT concentrations in blood. AAV donor and LNP-CRISPR were injected at 2 week intervals via the intravenous route, and blood was collected every 4 weeks (red triangle: time point of blood collection). (B) Long-term concentration of AT and hFIX in the blood, measured using ELISA (n = 4 per group). (C) Information on PHx and blood sampling. Each number represents an individual lobe (1: right median lobe, 2: left median lobe, 3: left lobe, 4-1: right lobe, 4-2: right renal fossa, and 5: caudate lobe). White lobes indicate those removed by PHx. Red triangles indicate the time points of blood sampling. (D) Comparison of the shape and weight of the liver of control and PHxed mice 20 weeks from the surgery. The liver weight was normalized by body weight. (E and F) Concentrations of AT and hFIX in the blood were measured (blue arrow: time point of PHx; n = 4 per group). (G) Immunofluorescence images for hFIX and Ki67 expression in control and PHx groups (400×). White triangles indicate Ki67 and hFIX co-expressed cells. Yellow scale bar: 50 μm. (H) Indel rate in Serpinc1 locus, analyzed using Sanger sequencing and algorithm analysis, and relative hF9 KI rate, analyzed using quantitative PCR (control: n = 3, PHx: n = 4). Data are presented as mean ± standard error of mean, calculated using unpaired Student’s t test. (ns, not significant, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001).
The therapeutic effect was sustained even under partial hepatectomy
We assumed that a sustained therapeutic effect of hF9 KI into the Serpinc1 locus might be obtained by direct division of the genetically edited cells. Since 2/3 partial hepatectomy (PHx) facilitates hepatocyte hyperplasia,29 2/3 PHx after in vivo therapeutic gene KI could be a useful tool for analyzing clonal expansion. Thus, 2/3 PHx was conducted after co-injection of AAV-donor and LNP-CRISPR. Following this, blood was collected from these mice every 2 weeks (Figure 5C). Twenty weeks after PHx, the liver mass was restored to 88% of that of control mice (4.56% vs. 4% of liver to body weight in control vs. PHx), and the size of the remaining right and caudate lobes after PHx was increased (Figure 5D). Next, the concentrations of AT and hFIX were measured using plasma. First, there was no decrease in blood AT concentration by PHx in mice without AT targeting, but mice with in vivo AT targeting showed a rapid decrease in blood AT concentration to approximately 55% of the control after PHx and gradually recovered afterward (Figure 5E). We interpreted that the AT concentration was affected by the reduction in liver mass by additional PHx upon AT inhibition by gene targeting. Second, hFIX concentration in the blood was not different between the control and PHx groups (Figure 5F). This observation indicated that the plasma concentration of hFIX after in vivo KI was not affected by the decrease in liver mass caused by PHx, similar to previous reports.30,31 hFIX and Ki67 co-expressed cells were observed in liver tissue from mice with PHx (Figure 5G). Ki67 is a marker for proliferating cells and is poorly expressed in quiescent cells.32 The co-expression of FIX-Ki67 in cells is indicative of hF9 KI hepatocytes undergoing differentiation. In particular, there was no significant difference in the indel and KI rates in the hepatocytes of the control and PHx groups (Figure 5H). A previous PHx study reported that most of the residual hepatocytes were differentiated to induce mass recovery.33 Therefore, we reasoned that AT inhibition and hFIX protein-production ability can be maintained by the proliferation of genetically edited hepatocytes in the case of PHx.
The AAV-LNP hybrid system did not exert severe side effects
In the CRISPR-Cas-mediated therapeutic gene KI approach, safety issues should be assessed in depth. LNP is a safe delivery tool for genetic materials,34 and we reported the safety of LNP-CRISPR for AT targeting.23 In addition, the expression of proinflammatory genes such as interferon-γ, tumor necrosis factor α, and interleukin 1β was analyzed using liver tissues, and there was no significant difference between groups (Figure S4). Similarly, we assessed the possibility of liver toxicity by measuring alanine aminotransferase (ALT) and aspartate aminotransferase (AST) concentrations in the blood after combined use of AAV and LNP; results indicated the absence of any remarkable long-term side effects (Figure 6A). In addition, there were no remarkable clinical symptoms in mice after AAV or LNP injection.
Figure 6.

Combined in vivo KI using AAV donor and LNP-CRISPR did not exert severe side effects
(A) Blood samples from long-term screening (samples same as those mentioned in Figure 5B) were subjected to an enzyme-linked immunosorbent assay for measuring alanine aminotransferase (ALT) and aspartate aminotransferase (AST) concentrations (blue arrow: timing of the final delivery of genetic materials). (B) Whole-genome sequencing was conducted to identify hF9 integration (F9Mut or control: n = 1, F9Mut-AAV-LNP group: n = 3). (C) Rate of on-target indel and KI and the ratio of KI/indel. Each dot indicates data from individual mice. Data are presented as mean ± standard error of mean. (D) Identification of the off-target integration pattern. Off-target soft-clipped read: detected soft-clipped read containing KI sequence and off-target sequence.
Most of all, this study used CRISPR-Cas9 and AAV, and thus off-target DSB and random integration by AAV donor were critical issues. Off-target DSB analysis can be performed through in silico off-target site selection and targeted deep sequencing, but random integration analysis can be conducted using whole-genome sequencing (WGS). We performed WGS with more than 30× resolution using liver gDNA from one control (F9Mut) and three KI (F9Mut-AAV-LNP) mice (Table S1). After aligning WGS reads to the KI-expected gene map, soft-clipped reads with KI sequences and host genomic sequences were extracted. Next, the KI position and rate were calculated by alignment to the mouse genome assembly GRCm39 (mm39) (Figure 6B). The indel rate at the on-target site was approximately 20% (Figure 6C). Moreover, WGS analysis revealed that hF9 coding sequences were integrated into approximately 3% of the target locus in hepatocytes. When the KI/indel rate was calculated for each individual, KI appeared to occur in approximately 15% (3/20) of the indels (Figure 6C).
Next, off-target DSB was analyzed. There were no detectable indels in the ten off-target candidate sites selected by Digenome-seq or harboring the three base-pair mismatches selected through in silico analysis (Table S2).23 Although WGS with 30–40× resolution could not detect every random integration, random integration was observed in analyzing the soft-clipped reads at three genomic sites. The soft-clipped reads and predicted sequence of the random integration site showed no similarity to the sgRNA sequence of the on-target site (Figure 6D; Table S3). This suggests that the bidirectional AAV donor was integrated by itself rather than incorporated at the DSB site induced by LNP-CRISPR. Two of the three detected random integration sites were identified on a CpG island that was associated with frequent AAV integration.35 However, the off-target soft-clipped read sequence also contained an “NGG” sequence; therefore, DSB induced by LNP-CRISPR could not be ruled out. It is still unclear whether bidirectional AAV facilitates random integration. AAV random integration could induce genotoxicity such as hepatocellular carcinoma (HCC).36 Thus, we analyzed the occurrence of HCC in the liver after 26 months of AAV and LNP treatment, and there was no evidence of HCC development (Figure S5). Briefly, the combined use of LNP-CRISPR and AAV bidirectional donors did not induce a remarkable inflammatory response. In addition, AAV integration was observed in approximately 20% of DSB sites, which resulted in hF9 coding sequence integration in approximately 3% of total hepatocytes.
Discussion
This study demonstrates an efficient therapeutic strategy for hemophilia by combining the existing concepts of anticoagulant pathway rebalancing and therapeutic gene KI. Co-transfection with AAV donor and LNP-CRISPR resulted in a long-term, sufficient coagulation activity restoration by AT inhibition and hFIX production in a hemophilia mouse model.
Among the anticoagulant pathway components, Protein C might not be suitable for gene targeting because of its antiinflammatory and antiapoptotic function.37 Further, since TFPI is synthesized in endothelial cells or platelets, targeting efficiency using LNP or AAV is expected to be low.38 In contrast, Serpinc1 (AT) has been proven to be an efficient target for restoring blood coagulation ability by reducing its gene expression through a clinical trial.39 Nevertheless, the lack of hemostatic effects on acute bleeding remains a challenge. In this regard, it was confirmed that knocking the therapeutic gene into a targeted locus of the rebalancing gene may produce the desired coagulation effect.
The liver produces various proteins and is considered the primary target organ for AAV- and non-viral-vector-mediated gene therapy.40,41 Therefore, we reasoned that therapeutic gene KI in the endogenous chromosome of hepatocytes is an efficient approach. The first consideration for in vivo gene KI is to choose a strategy between HR and non-homologous end joining (NHEJ)-mediated KI. HR may be preferred because it can induce precise KI. However, HR is a pathway dependent on the G2/S phase of the cell cycle,42 and hepatocytes are predominantly in a quiescent state.43 Thus, it is challenging to ensure highly efficient KI in hepatocytes. As an alternative, cell cycle-independent NHEJ-mediated KI can be expected to have relatively high in vivo KI efficiencies and could be more suitable when integration of the therapeutic sequence itself is more important than precise gene editing. We applied bidirectional AAV-donor-mediated integration, and it seemed to incorporate intact coding therapeutic genes along with a partial ITR sequence into the DSB site. Although the underlying mechanism is still uncertain, the bidirectional AAV-donor strategy may be applicable to in vivo KI of various genes.
The second consideration is the method of introduction of the donor template and nuclease into hepatocytes. Classically, the dual-AAV strategy was applied in such studies on atherosclerosis, hemophilia A, and hemophilia B.30,44,45 However, a high dose of AAV transduction in humans can cause severe liver toxicity and death.46 In this regard, the AAV-LNP hybrid strategy can reduce the dose of the delivery tool by separating the viral and non-viral vectors. The LNP is suitable for delivering CRISPR-Cas9 mRNA and sgRNA into cells in vivo.47 In a previous study, LNP-CRISPR showed a high potential to induce DSB in vivo.48 Consistent with this finding, in this study, LNP-CRISPR for Serpinc1 also resulted in a DSB rate of 25%. However, the LNP delivers DNA or RNA to the cytoplasm,49 which does not satisfy one of the most important requirements in KI: the delivery of donor template to the nucleus at the time of DSB. Donor nucleotides are difficult to move to the nucleus via the nuclear pore complex,50 and in vivo hepatocytes are in a quiescent state with a nucleus membrane. A recent study reported that conjugating a 3NF sequence to the plasmid could enhance intranuclear delivery,51 but intranuclear delivery using LNPs has not yet been reported. In targeted integration, the amount and preexistence of donor templates around the DSB site are critical,52 and AAV would be the best candidate for delivering donor templates to the nucleus of the target cell.53,54 AAV requires a certain period to be enriched in vivo and can be maintained in the form of double-stranded DNA (dsDNA) for a long time within hepatocytes.55 We suggest that delivery of the AAV donor and LNP-CRISPR for an interval of a few weeks can help improve KI efficiency and minimize toxicity.
The most significant advantage of the rebalancing approach is its applicability in treating all types of hemophilia, with or without inhibitors.12 The combined strategy utilizing LNP-CRISPR and AAV donor restored the thrombin-generation capacity to normal and led to improved aPTT results. This strongly supports the positive effect of combining rebalancing and therapeutic gene KI. Moreover, the AAV and LNP combination approach used in this study resulted in a high KI efficiency, thus showing its high potential for use. Although our results might be confined to a mouse model, stable inhibition of AT and hFIX production was sustained for more than a year through a single AAV-donor and LNP-CRISPR treatment. Therefore, this combined strategy of in vivo hepatocyte gene editing would not require repeated injection. In addition, considering the aPTT result, hF9 KI has a stronger effect on controlling acute bleeding than AT inhibition. Moreover, our combined strategy would be beneficial (1) as a low-dose in vivo KI strategy or (2) when inhibitors are a concern.
A therapeutic approach should secure safety. It is known that the capsid of AAV elicits a humoral immune response. This study utilized AAV serotype 8, which has a lower seroprevalence in humans.56 Thus, an advantage in reducing the neutralizing effect in clinical trials is expected compared with other AAV serotypes. In addition, AAV and CRISPR-Cas9 could activate a T cell response57,58; therefore, repeated delivery of genetic materials would trigger a strong immune response. Previously, dual AAV with Staphylococcus aureus Cas9 and HR donor template was applied to the F9 of hemophilia B mouse and presented a therapeutic effect.59 In contrast, we observed therapeutic effects for up to 63 weeks using single administration of LNP-CRISPR transfection and AAV-donor transduction. Although the humoral and T cell immune response could not be avoided, a single administration with intervals could minimize the side effects caused by an immune response. Consistently, stable ALT and AST concentrations would suggest no significant or continuous hepatocyte damage in the liver. This study confirmed the occurrence of random integration, which seemed to be unavoidable in AAV use. AAV random integration potentially leads to the development of HCC under liver injury.36 However, this study did not find any evidence of HCC in AAV-transduced mice. The combination use of AAV and LNP combination has advantages over the use of dual AAV in therapeutic gene KI. First, the overall AAV dose could be decreased, reducing the chance of AAV-mediated immune response and AAV-random integration. Second, the rapid biodegradability of LNPs is beneficial for minimizing the total amount of DSB and off-target effects by shorter CRISPR-Cas9 expression time.
Combined treatment of fitusiran and coagulation factors in patients with hemophilia A could result in thrombotic adverse events.60 Consequently, the clinical trial protocol for fitusiran was recently adjusted to keep the AT level between 15% and 30% to prevent thrombotic events observed at less than 15%61; an adjustment of dosage is required during a combined use with recombinant coagulation factors. This study revealed an approximately 33% AT level induced by single-time LNP-CRISPR injection and a sustained therapeutic effect via the differentiation of the gene-edited hFIX-producing cells. This is a potential advantage but could also cause irreversible adverse events when too many cells are edited. In both strategies of AT targeting and hF9 KI, a high dose would increase both the therapeutic effect and the risk. Therefore, we suggest that our combined strategy could induce complementary therapeutic effects and lower the dose. Further in vivo studies of the dose, injection number, and interval for reaching the target AT level would be needed to broaden the scope of this study.
In conclusion, this study confirmed the restoration of coagulation activity resulting from combination strategy of AT targeting and hF9 KI. A high rate of in vivo therapeutic gene KI was achieved with the use of an AAV and LNP hybrid, and the therapeutic effect seemed to be sustained for 63 weeks. In addition, the AAV-LNP combination could be beneficial for reducing the AAV dose compared with that employed in the dual-AAV strategy and could serve as a basis for developing gene editing-based advanced therapeutic strategies for protein deficiency-related genetic disorders.
Materials and methods
Quantitative reverse transcription PCR
Total RNA was extracted from liver tissues of WT mice using Trizol reagent (Thermo Fisher Scientific, Waltham, MA, USA), and cDNA was synthesized using a cDNA synthesis kit (Thermo Fisher Scientific). Quantitative reverse transcription PCR (qRT-PCR) was performed using PowerUp SYBR Green Master Mix and the StepOnePlus Real-Time PCR System (both from Thermo Fisher Scientific). All reactions were conducted in triplicate, and the average value of each sample was used for further analyses. The expression of target gene was normalized to that of Gapdh. The primer sequences used in this study were obtained from the primer bank62 or in silico design and are listed in Table S4.
LNP preparation
LNPs were synthesized using the Nano Assemblr instrument (Precision Nanosystems, Vancouver, BC, Canada) as previously reported.23,63 Streptococcus pyogenes (SpCas9) mRNA (TriLink Biotechnologies, San Diego, CA, USA) and highly modified sgRNA (Axolabs, Kulmbach, Germany)64 were dissolved in 10 mM citrate at a 1:1 weight ratio. The lipid components (ionizable lipid, dioleoylphosphatidylethanolamine, cholesterol, and PEG-lipid at a 26.5:20:52:1.5 M ratio) were dissolved in ethanol. The final weight ratio of ionizable lipid to RNA was 10:1, and LNPs were formulated by microfluidic mixing of ionizable lipid and RNA at a 12 mL/min flow rate. Then, the produced LNPs were dialyzed against 1× phosphate-buffered saline using dialysis cassettes with a 3,500 molecular weight cut-off (Life Technologies, Carlsbad, CA, USA). Next, LNPs were characterized by dynamic light scattering, polydispersity index, and zeta potential (Table S5). The encapsulation efficiency was calculated by Quanti-iTTM Ribogreen Assay (Life Technologies).
Recombinant AAV preparation
The AAV vector for the donor was designed to be bidirectional (ITR-splicing acceptor [SA]-hF9-pA-pA-hF9-SA-ITR: AAV donor) and prepared via DNA synthesis.18 Next, 70 μg each AAV vector, helper plasmid, and AAV8-specific rep/cap plasmid was co-transfected into 1 × 108 HEK293T cells using calcium phosphate. Three days after co-transfection, the viral particles were collected and purified through cesium chloride centrifugation. After titration using qRT-PCR, the AAV was stored at −80°C until use. The AAV vector and recombinant AAV were produced using a commercial service (Vigene, Rockville, MD, USA).
In vivo luciferase detection on systemic AAV transduction
The AAV vector for luciferase expression (ITR-CMV promoter-luciferase-pA-ITR: AAV-luciferase) was prepared, and recombinant AAV was produced. Next, 2 × 1010 vg/kg AAV8 luciferase was diluted in 200 μL sterilized saline and injected via the tail vein in hairless mice. The intensity of luciferase expression in the liver region was determined using Lumina caliper IVIS III (PerkinElmer, Waltham, MA, USA) after injecting 150 mg/kg D-luciferin (PerkinElmer) under anesthesia. The luciferase intensities of each mouse were compared with the total normalized flux (photons/s/cm2/radian) at the same exposure condition.
In vitro AAV transduction and LNP transfection into hemophilia B mice
Primary hepatocytes were isolated from a C57BL/6 (B6, WT) mouse. Briefly, the mouse was anesthetized, and the liver was collected after transcardiac perfusion using PBS. The liver tissue was minced using a microscissor in DPBS, followed by filtration of the cell suspension using a 70 μm cell strainer (Corning Life Sciences, Tewksbury, MA, USA). After washing by centrifugation and resuspension, hepatocytes were isolated. Next, 2 × 105 primary hepatocytes and mouse hepatocyte cell line (alpha mouse liver 12 [AML12]) were seeded into a 24-well culture plate and cultured for 24 h. Next, 3 × 1010 viral genome AAV particles or 1 mg LNP was added. The cells were harvested 3 days after treatment and used for gDNA and protein preparation. hF9 KI and hFIX production were analyzed by PCR and enzyme-linked immunosorbent assay (ELISA).
Indel analysis and sanger sequencing
Primer sets covering the on-target sites were designed, and PCR was conducted using gDNA from liver tissues. PCR amplicons were used for indel analysis using T7E1 analysis or algorithm-based indel prediction. For T7E1 analysis, PCR amplicons were subjected to heteroduplex formation by gradually decreasing the annealing temperature; T7 endonuclease (New England Biolabs, Ipswich, MA, USA) was added to the amplicons, and the mixture was incubated for 1 h at 37°C. The cuts observed in the DNA bands upon electrophoresis were considered an indication of the presence of indels. In algorithm-based indel prediction, Sanger sequencing reads were subjected to the “inference of CRISPR edit” tool, and the indel rate was calculated.65 For KI pattern analysis, primer sets were selected from Serpinc1 and the incorporated hF9 gene, respectively. Then, PCR and Sanger sequencing were conducted, and sequence reads were aligned to the Serpinc1 gene. The primer sequences used in this study were obtained from the primer bank62 or in silico design and are listed in Table S4.
In vivo AAV transduction and LNP transfection
C57BL/6-F9em1 (F9Mut) mice were generated by CRISPR-Cas9-mediated gene targeting, and their hemophilia B coagulopathies were confirmed by aPTT analysis.23 Since hemophilia B is an X-linked recessive genetic disorder, only male mice were subjected to in vivo AAV transduction. Seven-to-eight-week-old WT and F9Mut mice were randomly divided into five groups. AAV donor (2 × 1013 vg/kg) or LNP-CRISPR (1.2 mpk) were mixed with warm saline up to a total volume of 500 μL and injected intravenously. LNP-CRISPR was injected 2 weeks after AAV-donor injection in the AAV and LNP combined group. All mice were euthanized 10 weeks after AAV transduction, while their blood and organ tissue samples were collected. For long-term analysis of hFIX or AT blood concentration, 7-week-old male WT mice were subjected to AAV-donor or LNP-CRISPR injection, and 50 μL blood was repeatedly collected from the tail vein monthly. This study was approved by the Institutional Animal Care and Use Committee of the Seoul National University (SNU-200715-2).
ELISA
Concentrations of mouse AT, human FIX, active FIX assay kit, aspartate aminotransferase (AST), and ALT were measured using a commercial ELISA kit (Abcam, Waltham, MA, USA) according to the manufacturer’s instructions. Detailed information on the used ELISA kits and reaction conditions are listed in Table S6.
Immunofluorescence analysis
After anesthetization using 2.5% avertin, prewarmed phosphate-buffered saline and 4% paraformaldehyde were perfused via the heart. Next, the collected liver tissues were subjected to precipitation in 15% and 30% sucrose for 6 h and 1 day and then embedded in an optimal cutting temperature compound (Sakura Finetek, Tokyo, Japan) with gradual freezing to −80°C. Cryo-sections were prepared at 15 μm thick using a Cryostat microtome (Leica, Wetzler, Germany). Immunofluorescence analysis for hFIX and Ki67 detection was conducted by incubating the cryo-sections with target protein reactive primary antibodies at 4°C overnight. This was followed by incubation with a secondary antibody, and reactive protein expressions were detected using the Cytation 5 Cell Imaging Multi-mode Reader (BioTek, Winooski, VT, USA). Information on the antibodies and conditions used in this study is provided in the Table S7.
aPPT analysis
After anesthetizing mice, 450 μL fresh blood was collected from the inferior vena cava using a syringe prefilled with 50 μL 3.2% sodium citrate solution (Medicago, Durham, NC, USA), and plasma was collected after centrifugation. For aPTT analysis, a mixture of 30 μL plasma and 30 μL aPTT reagent (Thermo Fisher Scientific) was added to each well of a 96-well microplate, which was then incubated at 37°C for 5 min. Next, 30 μL calcium chloride (CaCl2; 26 μM) was added to each well. The absorbance was measured at 405 nm wavelength every 10 s for 8 min after the first 10 s shaking using Tecan Sunrise Microplate Reader (Tecan, Männedorf, Switzerland). ΔOD (OD value of each time point – OD value from 10 s before) was calculated, and the time point with the highest ΔOD value was designated as the aPTT.
CAT analysis
Thrombin-generation activity was measured using the Technothrombin TGA kit (Diapharma, West Chester Township, OH, USA). A mixture of 40 μL diluted plasma, 10 μL reagent C low buffer, and 50 μL substrate was added to a single well, and fluorescence was measured for 120 min at 1 min intervals. Fluorescence measurements were performed in a 96-well plate using the Cytation 5 cell imaging multi-mode plate reader (BioTek). The thrombin-generation curves, peak height, and peak time were determined using the software provided by the manufacturer.
PHx
A 2/3 PHx was conducted as previously reported.66 Briefly, 8-week-old B6 male mice were anesthetized using isoflurane, and the abdominal cavity was opened by a midline abdominal incision after disinfecting the skin using 70% ethanol. The xiphoid process was lifted upward and backward using a stuck rubber band, and the falciform ligament or membrane was cut using curved microsurgery scissors. The median and left lateral lobes were removed using scissors after knotting using 4-0 silk thread. Subsequently, the abdomen was closed by suturing.
WGS
Low-quality bases below Q20 were trimmed using the Sickle software (v.1.3.3). High-quality reads were then aligned to the reference sequence using Burrows-Wheeler Aligner (BWA) (v.0.7.17), and duplicate reads were further removed using Picard tools (v.1.98). To determine the potential off-target sites, we predicted up to 3 bp mismatches in silico (www.rgenome.net). Next, to identify therapeutic gene insertion sites, the extracted soft-clipped nucleotides were mapped using BWA. Soft-clipped nucleotides were determined using a Smith-Waterman-like scoring scheme in the BWA software. The hF9 insertion sites were inferred by analyzing the mapped patterns in the soft-clipped sequences. Finally, hF9 insertion was confirmed by manual inspection of the candidate site using the IGV software v.2.9.4.
Statistical analysis
Statistical analysis was performed using unpaired Student’s t test and one-way ANOVA in GraphPad Prism (v.5.02, GraphPad, San Diego, CA, USA). p values under 0.05 were designated as statistically significant.
Acknowledgments
This work was supported by grants from the National Research Foundation (nos. NRF- 2021R1I1A2044117, 2017M3A9B4061409, 2018M3A9H3020844, and 2020R1A2C2004364), the Korea Institute of Toxicology (1711133848), and the Ministry of Food and Drug Safety (21173MFDS562). We are grateful to Theragen Bio for assistance with the WGS experiment and analysis.
Author contributions
J.H.L. and S.C.Y. wrote the manuscript. J.H.L., J.P.H., D.W.S., G.S.L., Y.L., B.S.C., and M.K. conducted the experiments. D.W.S., S.K., and H.L. analyzed the data. S.C.Y. conceived and designed the experiments.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2023.03.008.
Supplemental information
Data availability statement
The data of the current study are available from the corresponding author on reasonable request.
References
- 1.Mehta P., Reddivari A.K.R. StatPearls: Treasure Island (FL); 2021. Hemophilia. [Google Scholar]
- 2.Castaman G., Matino D. Hemophilia A and B: molecular and clinical similarities and differences. Haematologica. 2019;104:1702–1709. doi: 10.3324/haematol.2019.221093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mannucci P.M. Hemophilia therapy: the future has begun. Haematologica. 2020;105:545–553. doi: 10.3324/haematol.2019.232132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Konkle B.A., Coffin D., Pierce G.F., Clark C., George L., Iorio A., Mahlangu J., Naccache M., O'Mahony B., Peyvandi F., et al. World federation of hemophilia gene therapy registry. Haemophilia. 2020;26:563–564. doi: 10.1111/hae.14015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pasi K.J., Rangarajan S., Mitchell N., Lester W., Symington E., Madan B., Laffan M., Russell C.B., Li M., Pierce G.F., Wong W.Y. Multiyear follow-up of AAV5-hFVIII-SQ gene therapy for hemophilia A. N. Engl. J. Med. 2020;382:29–40. doi: 10.1056/NEJMoa1908490. [DOI] [PubMed] [Google Scholar]
- 6.Miesbach W., Meijer K., Coppens M., Kampmann P., Klamroth R., Schutgens R., Tangelder M., Castaman G., Schwäble J., Bonig H., et al. Gene therapy with adeno-associated virus vector 5-human factor IX in adults with hemophilia B. Blood. 2018;131:1022–1031. doi: 10.1182/blood-2017-09-804419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nathwani A.C., Reiss U.M., Tuddenham E.G.D., Rosales C., Chowdary P., McIntosh J., Della Peruta M., Lheriteau E., Patel N., Raj D., et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 2014;371:1994–2004. doi: 10.1056/NEJMoa1407309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colella P., Ronzitti G., Mingozzi F. Emerging issues in AAV-mediated in vivo gene therapy. Mol. Ther. Methods Clin. Dev. 2018;8:87–104. doi: 10.1016/j.omtm.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nguyen G.N., Everett J.K., Kafle S., Roche A.M., Raymond H.E., Leiby J., Wood C., Assenmacher C.A., Merricks E.P., Long C.T., et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat. Biotechnol. 2021;39:47–55. doi: 10.1038/s41587-020-0741-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weyand A.C., Pipe S.W. New therapies for hemophilia. Blood. 2019;133:389–398. doi: 10.1182/blood-2018-08-872291. [DOI] [PubMed] [Google Scholar]
- 11.Sehgal A., Barros S., Ivanciu L., Cooley B., Qin J., Racie T., Hettinger J., Carioto M., Jiang Y., Brodsky J., et al. An RNAi therapeutic targeting antithrombin to rebalance the coagulation system and promote hemostasis in hemophilia. Nat. Med. 2015;21:492–497. doi: 10.1038/nm.3847. [DOI] [PubMed] [Google Scholar]
- 12.Ragni M.V. Targeting antithrombin to treat hemophilia. N. Engl. J. Med. 2015;373:389–391. doi: 10.1056/NEJMcibr1505657. [DOI] [PubMed] [Google Scholar]
- 13.Chowdary P., Lethagen S., Friedrich U., Brand B., Hay C., Abdul Karim F., Klamroth R., Knoebl P., Laffan M., Mahlangu J., et al. Safety and pharmacokinetics of anti-TFPI antibody (concizumab) in healthy volunteers and patients with hemophilia: a randomized first human dose trial. J. Thromb. Haemostasis. 2015;13:743–754. doi: 10.1111/jth.12864. [DOI] [PubMed] [Google Scholar]
- 14.Peters R., Harris T. Advances and innovations in haemophilia treatment. Nat. Rev. Drug Discov. 2018;17:493–508. doi: 10.1038/nrd.2018.70. [DOI] [PubMed] [Google Scholar]
- 15.Porteus M.H. Knock-in editing: it functionally corrects. Blood. 2016;127:2507–2509. doi: 10.1182/blood-2016-03-703181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilbie D., Walther J., Mastrobattista E. Delivery aspects of CRISPR/Cas for in vivo genome editing. Acc. Chem. Res. 2019;52:1555–1564. doi: 10.1021/acs.accounts.9b00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee J.H., Oh H.K., Choi B.S., Lee H.H., Lee K.J., Kim U.G., Lee J., Lee H., Lee G.S., Ahn S.J., et al. Genome editing-mediated knock-in of therapeutic genes ameliorates the disease phenotype in a model of hemophilia. Mol. Ther. Nucleic Acids. 2022;29:551–562. doi: 10.1016/j.omtn.2022.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He X., Zhang Z., Xue J., Wang Y., Zhang S., Wei J., Zhang C., Wang J., Urip B.A., Ngan C.C., et al. Low-dose AAV-CRISPR-mediated liver-specific knock-in restored hemostasis in neonatal hemophilia B mice with subtle antibody response. Nat. Commun. 2022;13:7275. doi: 10.1038/s41467-022-34898-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gaj T., Staahl B.T., Rodrigues G.M.C., Limsirichai P., Ekman F.K., Doudna J.A., Schaffer D.V. Targeted gene knock-in by homology-directed genome editing using Cas9 ribonucleoprotein and AAV donor delivery. Nucleic Acids Res. 2017;45:e98. doi: 10.1093/nar/gkx154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Naso M.F., Tomkowicz B., Perry W.L., 3rd, Strohl W.R. Adeno-associated virus (AAV) as a vector for gene therapy. BioDrugs. 2017;31:317–334. doi: 10.1007/s40259-017-0234-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pardi N., Tuyishime S., Muramatsu H., Kariko K., Mui B.L., Tam Y.K., Madden T.D., Hope M.J., Weissman D. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Contr. Release. 2015;217:345–351. doi: 10.1016/j.jconrel.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han J.P., Kim M., Choi B.S., Lee J.H., Lee G.S., Jeong M., Lee Y., Kim E.A., Oh H.K., Go N., et al. In vivo delivery of CRISPR-Cas9 using lipid nanoparticles enables antithrombin gene editing for sustainable hemophilia A and B therapy. Sci. Adv. 2022;8:eabj6901. doi: 10.1126/sciadv.abj6901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hon-Ren Huang, CM-E, P. Bialek, C. Wang, G. Gong, S. Hartfort, R. Sattler, D. White, K. Lai, D. Chalothorn (2019). CRISPR/Cas9-mediated targeted insertion of human f9 achieves therapeutic circulating protein levels in mice and nonhuman primates. American Society of Gene and Cell Therapy 22nd Meeting.
- 25.Butterfield J.S.S., Hege K.M., Herzog R.W., Kaczmarek R. A molecular revolution in the treatment of hemophilia. Mol. Ther. 2020;28:997–1015. doi: 10.1016/j.ymthe.2019.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanlon K.S., Kleinstiver B.P., Garcia S.P., Zaborowski M.P., Volak A., Spirig S.E., Muller A., Sousa A.A., Tsai S.Q., Bengtsson N.E., et al. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun. 2019;10:4439. doi: 10.1038/s41467-019-12449-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duarte R.C.F., Ferreira C.N., Rios D.R.A., Reis H.J.D., Carvalho M.D.G. Thrombin generation assays for global evaluation of the hemostatic system: perspectives and limitations. Rev. Bras. Hematol. Hemoter. 2017;39:259–265. doi: 10.1016/j.bjhh.2017.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perrin G.Q., Herzog R.W., Markusic D.M. Update on clinical gene therapy for hemophilia. Blood. 2019;133:407–414. doi: 10.1182/blood-2018-07-820720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marongiu F., Marongiu M., Contini A., Serra M., Cadoni E., Murgia R., Laconi E. Hyperplasia vs hypertrophy in tissue regeneration after extensive liver resection. World J. Gastroenterol. 2017;23:1764–1770. doi: 10.3748/wjg.v23.i10.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang L., Yang Y., Breton C.A., White J., Zhang J., Che Y., Saveliev A., McMenamin D., He Z., Latshaw C., et al. CRISPR/Cas9-mediated in vivo gene targeting corrects hemostasis in newborn and adult factor IX-knockout mice. Blood. 2019;133:2745–2752. doi: 10.1182/blood.2019000790. [DOI] [PubMed] [Google Scholar]
- 31.Wang Q., Zhong X., Li Q., Su J., Liu Y., Mo L., Deng H., Yang Y. CRISPR-Cas9-Mediated in vivo gene integration at the albumin locus recovers hemostasis in neonatal and adult hemophilia B mice. Mol. Ther. Methods Clin. Dev. 2020;18:520–531. doi: 10.1016/j.omtm.2020.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun X., Bizhanova A., Matheson T.D., Yu J., Zhu L.J., Kaufman P.D. Ki-67 contributes to normal cell cycle progression and inactive X heterochromatin in p21 checkpoint-proficient human cells. Mol. Cell Biol. 2017;37 doi: 10.1128/MCB.00569-16. 005699-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duncan A.W., Dorrell C., Grompe M. Stem cells and liver regeneration. Gastroenterology. 2009;137:466–481. doi: 10.1053/j.gastro.2009.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Let’s talk about lipid nanoparticles. Nat. Rev. Mater. 2021;6:99. [Google Scholar]
- 35.Miller D.G., Trobridge G.D., Petek L.M., Jacobs M.A., Kaul R., Russell D.W. Large-scale analysis of adeno-associated virus vector integration sites in normal human cells. J. Virol. 2005;79:11434–11442. doi: 10.1128/JVI.79.17.11434-11442.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dalwadi D.A., Torrens L., Abril-Fornaguera J., Pinyol R., Willoughby C., Posey J., Llovet J.M., Lanciault C., Russell D.W., Grompe M., Naugler W.E. Liver injury increases the incidence of HCC following AAV gene therapy in mice. Mol. Ther. 2021;29:680–690. doi: 10.1016/j.ymthe.2020.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Toltl L.J., Austin R.C., Liaw P.C. Activated protein C modulates inflammation, apoptosis and tissue factor procoagulant activity by regulating endoplasmic reticulum calcium depletion in blood monocytes. J. Thromb. Haemostasis. 2011;9:582–592. doi: 10.1111/j.1538-7836.2010.04177.x. [DOI] [PubMed] [Google Scholar]
- 38.Maroney S.A., Mast A.E. Expression of tissue factor pathway inhibitor by endothelial cells and platelets. Transfus. Apher. Sci. 2008;38:9–14. doi: 10.1016/j.transci.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pasi K.J., Rangarajan S., Georgiev P., Mant T., Creagh M.D., Lissitchkov T., Bevan D., Austin S., Hay C.R., Hegemann I., et al. Targeting of antithrombin in hemophilia A or B with RNAi therapy. N. Engl. J. Med. 2017;377:819–828. doi: 10.1056/NEJMoa1616569. [DOI] [PubMed] [Google Scholar]
- 40.Cunningham S.C., Alexander I.E. AAV-mediated gene delivery to the mouse liver. Methods Mol. Biol. 2019;1937:213–219. doi: 10.1007/978-1-4939-9065-8_12. [DOI] [PubMed] [Google Scholar]
- 41.Witzigmann D., Kulkarni J.A., Leung J., Chen S., Cullis P.R., van der Meel R. Lipid nanoparticle technology for therapeutic gene regulation in the liver. Adv. Drug Deliv. Rev. 2020;159:344–363. doi: 10.1016/j.addr.2020.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mao Z., Bozzella M., Seluanov A., Gorbunova V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle. 2008;7:2902–2906. doi: 10.4161/cc.7.18.6679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Furchtgott L.A., Chow C.C., Periwal V. A model of liver regeneration. Biophys. J. 2009;96:3926–3935. doi: 10.1016/j.bpj.2009.01.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao H., Li Y., He L., Pu W., Yu W., Li Y., Wu Y.T., Xu C., Wei Y., Ding Q., et al. In vivo AAV-CRISPR/Cas9-Mediated gene editing ameliorates atherosclerosis in familial hypercholesterolemia. Circulation. 2020;141:67–79. doi: 10.1161/CIRCULATIONAHA.119.042476. [DOI] [PubMed] [Google Scholar]
- 45.Chen H., Shi M., Gilam A., Zheng Q., Zhang Y., Afrikanova I., Li J., Gluzman Z., Jiang R., Kong L.J., Chen-Tsai R.Y. Hemophilia A ameliorated in mice by CRISPR-based in vivo genome editing of human Factor VIII. Sci. Rep. 2019;9:16838. doi: 10.1038/s41598-019-53198-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.High-dose AAV gene therapy deaths. Nat. Biotechnol. 2020;38:910. doi: 10.1038/s41587-020-0642-9. [DOI] [PubMed] [Google Scholar]
- 47.Qiu M., Glass Z., Chen J., Haas M., Jin X., Zhao X., Rui X., Ye Z., Li Y., Zhang F., et al. Lipid nanoparticle-mediated codelivery of Cas9 mRNA and single-guide RNA achieves liver-specific in vivo genome editing of Angptl3. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2020401118. e2020401118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wei T., Cheng Q., Min Y.-L., Olson E.N., Siegwart D.J. Systemic nanoparticle delivery of CRISPR-Cas9 ribonucleoproteins for effective tissue specific genome editing. Nat. Commun. 2020;11:3232. doi: 10.1038/s41467-020-17029-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sinegra A.J., Evangelopoulos M., Park J., Huang Z., Mirkin C.A. Lipid nanoparticle spherical nucleic acids for intracellular DNA and RNA delivery. Nano Lett. 2021;21:6584–6591. doi: 10.1021/acs.nanolett.1c01973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Salman H., Zbaida D., Rabin Y., Chatenay D., Elbaum M. Kinetics and mechanism of DNA uptake into the cell nucleus. Proc. Natl. Acad. Sci. USA. 2001;98:7247–7252. doi: 10.1073/pnas.121067698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Le Guen Y.T., Pichon C., Guégan P., Pluchon K., Haute T., Quemener S., Ropars J., Midoux P., Le Gall T., Montier T. DNA nuclear targeting sequences for enhanced non-viral gene transfer: an in vitro and in vivo study. Mol. Ther. Nucleic Acids. 2021;24:477–486. doi: 10.1016/j.omtn.2021.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Han J.P., Chang Y.J., Song D.W., Choi B.S., Koo O.J., Yi S.Y., Park T.S., Yeom S.C. High homology-directed repair using mitosis phase and nucleus localizing signal. Int. J. Mol. Sci. 2020;21:3747. doi: 10.3390/ijms21113747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang D., Tai P.W.L., Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019;18:358–378. doi: 10.1038/s41573-019-0012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gao J., Bergmann T., Zhang W., Schiwon M., Ehrke-Schulz E., Ehrhardt A. Viral vector-based delivery of CRISPR/Cas9 and donor DNA for homology-directed repair in an in vitro model for canine hemophilia B. Mol. Ther. Nucleic Acids. 2019;14:364–376. doi: 10.1016/j.omtn.2018.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang J., Xie J., Lu H., Chen L., Hauck B., Samulski R.J., Xiao W. Existence of transient functional double-stranded DNA intermediates during recombinant AAV transduction. Proc. Natl. Acad. Sci. USA. 2007;104:13104–13109. doi: 10.1073/pnas.0702778104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Perocheau D.P., Cunningham S., Lee J., Antinao Diaz J., Waddington S.N., Gilmour K., Eaglestone S., Lisowski L., Thrasher A.J., Alexander I.E., et al. Age-related seroprevalence of antibodies against AAV-LK03 in a UK population cohort. Hum. Gene Ther. 2019;30:79–87. doi: 10.1089/hum.2018.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ertl H.C.J. T cell-mediated immune responses to AAV and AAV vectors. Front. Immunol. 2021;12:666666. doi: 10.3389/fimmu.2021.666666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Crudele J.M., Chamberlain J.S. Cas9 immunity creates challenges for CRISPR gene editing therapies. Nat. Commun. 2018;9:3497. doi: 10.1038/s41467-018-05843-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ohmori T., Nagao Y., Mizukami H., Sakata A., Muramatsu S.-i., Ozawa K., Tominaga S.-i., Hanazono Y., Nishimura S., Nureki O., Sakata Y. CRISPR/Cas9-mediated genome editing via postnatal administration of AAV vector cures haemophilia B mice. Sci. Rep. 2017;7:4159. doi: 10.1038/s41598-017-04625-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Machin N., Ragni M.V. An investigational RNAi therapeutic targeting antithrombin for the treatment of hemophilia A and B. Hematol. Res. Rev. 2018;9:135–140. doi: 10.2147/JBM.S159297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nogami K., Shima M. Current and future therapies for haemophilia-Beyond factor replacement therapies. Br. J. Haematol. 2023;200:23–34. doi: 10.1111/bjh.18379. [DOI] [PubMed] [Google Scholar]
- 62.Wang X., Spandidos A., Wang H., Seed B. PrimerBank: a PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic Acids Res. 2012;40:D1144–D1149. doi: 10.1093/nar/gkr1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim M., Jeong M., Hur S., Cho Y., Park J., Jung H., Seo Y., Woo H.A., Nam K.T., Lee K., Lee H. Engineered ionizable lipid nanoparticles for targeted delivery of RNA therapeutics into different types of cells in the liver. Sci. Adv. 2021;7:eabf4398. doi: 10.1126/sciadv.abf4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yin H., Song C.Q., Suresh S., Wu Q., Walsh S., Rhym L.H., Mintzer E., Bolukbasi M.F., Zhu L.J., Kauffman K., et al. Structure-guided chemical modification of guide RNA enables potent non-viral in vivo genome editing. Nat. Biotechnol. 2017;35:1179–1187. doi: 10.1038/nbt.4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tim Hsiau D.C., Rossi N., Maures T., Waite K., Yang J., Joshi S., Reed K., Holden K., Enzmann B.L., Stoner R. Inference of CRISPR edits from sanger trace data. bioRxiv. 2019 doi: 10.1101/251082. Preprint at. [DOI] [PubMed] [Google Scholar]
- 66.Mitchell C., Willenbring H. A reproducible and well-tolerated method for 2/3 partial hepatectomy in mice. Nat. Protoc. 2008;3:1167–1170. doi: 10.1038/nprot.2008.80. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data of the current study are available from the corresponding author on reasonable request.