

Key words: Macaque, metatranscriptomics, Pentatrichomonas, Tetratrichomonas, Trichomitus, Trichomonads
Abstract
Trichomonads, anaerobic microbial eukaryotes members of the phylum Parabasalia, are common obligate extracellular symbionts that can lead to pathological or asymptomatic colonization of various mucosal surfaces in a wide range of animal hosts. Results from previous in vitro studies have suggested a number of intriguing mucosal colonization strategies by Trichomonads, notably highlighting the importance of interactions with bacteria. However, in vivo validation is currently lacking. A previous metatranscriptomics study into the cause of idiopathic chronic diarrhoea in macaques reported the presence of an unidentified protozoan parasite related to Trichomonas vaginalis. In this work, we performed a reanalysis of the published data in order to identify the parasite species present in the macaque gut. We also leveraged the information-rich metatranscriptomics data to investigate the parasite behaviour in vivo. Our results indicated the presence of at least 3 genera of Trichomonad parasite; Tetratrichomonas, Pentatrichomonas and Trichomitus, 2 of which had not been previously reported in the macaque gut. In addition, we identified common in vivo expression profiles shared amongst the Trichomonads. In agreement with previous findings for other Trichomonads, our results highlighted a relationship between Trichomonads and mucosal bacterial diversity which could be influential in health and disease.
Introduction
Trichomonads are a group of microbial eukaryotes within the phylum Parabasalia, almost all of which are known as obligate mucosal symbionts that colonize a wide range of mammals, birds and reptiles (Malik et al., 2011). Parasitic Trichomonads have no known free-living stages and are assumed to be transmitted almost exclusively by direct contact. The molecular basis of virulence, mucosal colonization (Sommer et al., 2005; de Miguel et al., 2010; Handrich et al., 2019; Martínez-Herrero et al., 2019) and metabolism (Matthews, 1986; Westrop et al., 2017) of Trichomonads has been the subject of extensive in vitro investigation. The vast majority of this work has focused on the human sexually transmitted parasite Trichomonas vaginalis. However, the importance of the proposed mechanisms during colonization of the complex mucosal environment in vivo is unclear. Validation of hypotheses in the natural setting is essential to avoid misinterpretation of results (Bello-Ortí et al., 2015; Marzano et al., 2017). The conservation of genes encoding virulence and mucosal colonization mechanisms across a wider range of Trichomonad species is also largely unknown, necessitating comparative studies.
There is extensive evidence for an interaction between Trichomonads and mucosal bacteria which is influential in health and disease. For example, T. vaginalis infection can induce dysbiotic changes in the urogenital tract (UGT) microbiota (Fichorova et al., 2013). Such results have been validated in vivo in several Trichomonad species and hosts (Wei et al., 2020; Bierlein et al., 2021). Notably, Trichomonas gallinae infection was correlated with changes in the microbiota at local and distant mucosal sites in pigeon squabs (Ji et al., 2020). However, previously, methods have been exclusively limited to 16S profiling, which provides no information on the potential mechanisms underlying parasite–bacteria interactions. T. vaginalis is a phagocytic predator of bacteria (Juliano et al., 1991; Rendon-Maldonado et al., 1998) and fungi (Pereira-Neves and Benchimol, 2007), with some evidence for selective preference of prey species (Juliano et al., 1991). In addition, T. vaginalis can form symbiotic associations with Mycoplasma spp. (Dessì et al., 2019). Functional work is required to determine the contribution of predation, symbiosis or other mechanisms to Trichomonad-induced in vivo microbiota changes.
A recent fecal metatranscriptomics investigation by Westreich et al. (2019) into the cause of idiopathic chronic diarrhoea (ICD) in laboratory macaques revealed the presence of GIT-localized protozoa. The authors stated ‘Protozoans with the most abundant transcripts in the faecal samples from the macaques were Blastocystis sp. and Trichomonas vaginalis’; however, also qualified ‘T. vaginalis was the only species in the reference data set representative of the Trichomonas genus, so it is possible that the particular species with increased gene expressed in macaques with ICD was not T. vaginalis’. Trichomonads do exhibit mucosal and host plasticity (Maritz et al., 2014). For example, T. vaginalis has been detected in the oral cavity (Costello et al., 2017) and respiratory tract (Duboucher et al., 2003). In addition, T. vaginalis is thought to originate from zoonosis of an avian oral parasite (Maritz et al., 2014). However, T. vaginalis is essentially a human UGT parasite. We suggest that misidentification as ‘T. vaginalis’ due to sequence database incompleteness (Watts et al., 2019) is very likely, as there are no reports of T. vaginalis in non-human animals or the GIT. Thus, further investigation into the parasite identity is warranted. Studies conducted on wild macaques did not report the presence of intestinal Trichomonads (Adhikari and Dhakal, 2018), although infection with Pentatrichomonas hominis has been reported in immunocompromised laboratory macaques (Zaragoza et al., 2011) and those suffering from ICD (Laing et al., 2018). Thus, P. hominis is a likely candidate for the parasite infecting the macaques with ICD.
In this work, we utilized the existing fecal metatranscriptomics data from macaques (Westreich et al., 2019) to identify the Trichomonads present, and to investigate their in vivo gene expression. Our results also suggested relationships between Trichomonads and the mucosal microbiota in vivo, with potential implications for the aetiology of ICD.
Materials and methods
Macaque fecal metatranscriptomics data analysis
Full details for experimental methodology used to generate the previously published macaque fecal metatranscriptomics data are available from Westreich et al. (2019). Briefly fecal samples were collected from 12 macaques suffering from ICD and 12 healthy control animals. For 30 days prior to sample collection animals were housed indoors in pairs, separating macaques with ICD and healthy controls. For stool collection, cage pans were placed under the cages of individually housed animals overnight and collected in the morning. Stool samples were used as a proxy for the intestinal mucosal environment. Total RNA was extracted from stool samples and used for cDNA library preparation.
The workflow used to assess macaque fecal metatranscriptomics data is shown in Fig. S1. The metatranscriptomics dataset for macaques was obtained from the NCBI SRA database (Leinonen et al., 2011) under accessions SRX3517701-SRX3517724 (Westreich et al., 2019). There were approximately 95 million paired end reads per animal. The average Phred quality score for all reads did not fall below 30, as assessed using FastQC version 0.11.9 (Andrews, 2018). For quantitative analysis of taxonomic abundance and functional expression, reads derived from rRNA were filtered by alignment to a prokaryotic and eukaryotic rRNA database using SortMeRNA version 4.2.0 (Kopylova et al., 2012). If both reads in a pair aligned, the pair was excluded. Kraken2 version 2.0.8-beta (Wood et al., 2019) with default parameters was used to taxonomically classify reads. The Kraken2 reference database was enriched by including de novo assembled contigs derived from in vitro RNA-Seq data for P. hominis strain PhGII, Tetratrichomonas gallinarum strain M3, and Trichomitus batrachorum strain BUB, kindly provided by Sriram Garg and Sven Gould (Handrich et al., 2019) (Heinrich Heine University, Düsseldorf) to improve the Trichomonad sequence diversity. The NCBI Taxonomy Toolkit version 0.5.0 (Shen and Xiong, 2019) was used to manipulate taxonomy IDs generated by Kraken2.
A de novo assembly was generated from the data using metaSPAdes version 3.13.0 (Nurk et al., 2017). To assess the accuracy of the assembly, reads were aligned to assembled contigs using STAR version 2.7.3a (Dobin et al., 2013). Samtools version 0.1.20 (Li et al., 2009) was used to manipulate alignment files. Overview for the dataset and assembly are presented in Table S1. De novo assembled contigs derived from combined Parabasalia reads from all samples to maximize coverage (available in Data files S1, selected genes, and S2, all genes) were used to examine Parabasalia gene expression. For transcript annotation, assembled parasite contigs were aligned to the T. vaginalis G3 annotated proteins (Carlton et al., 2007) using BLASTx version 2.9.0+ (Altschul et al., 1990). A single top hit for each contig was selected after sorting by, respectively and in priority order, E value and percentage identity, and excluding hits with an E value greater than 1 × 10−10 or a query coverage of less than 70%. Annotated contigs are presented in Table S2 (specific genes used as phylogenetic markers) and Table S3 (all genes).
Phylogenetic analysis
To sequence type parasites, BLASTn version 2.9.0+ (Altschul et al., 1990) was used to identify de novo assembled contigs homologous to Parabasalia genes of interest, with E value, percentage identity and query coverage cut-off values of 1 × 10−10, 88% and 90%, respectively. To broaden the taxonomic sampling for genes of interest, additional homologues were identified by consulting the literature and through the use of online BLAST (Altschul et al., 1990) searches against the NCBI non-redundant protein or nucleotide databases (O'Leary et al., 2016). Alignments were generated using Clustal omega version 1.7 (Sievers et al., 2011) and visually inspected in Seaview version 5.0.4 (Gouy et al., 2010). To improve phylogenetic resolution of parasite sequencing typing, DNA alignments for protein-coding genes were generated. Protein sequences were aligned using Clustal omega version 1.7 (Sievers et al., 2011), and corresponding codon alignments were derived using pal2nal version 14 (Suyama et al., 2006). Poorly aligned sequences were removed, and alignments were trimmed to remove excessive gaps (sites containing a gap for greater than 90–95% of sequences) using TrimAl version 1.2 (Capella-Gutiérrez et al., 2009). Alignments are available in supplementary data files S3–S8. IQ-tree version 1.6.1 (Nguyen et al., 2015) was used to generate maximum likelihood phylogenies, using automatic model selection. Support for tree topology was assessed by computing 1000 bootstrap replicates. iTol (Letunic and Bork, 2019) was used to generate annotated figures from the phylogenies.
Microbial diversity and expression analysis
Microbial diversity analysis was performed using the R packages PhyloSeq version 1.34.0 (McMurdie and Holmes, 2013) and Microbiome version 1.12.0 (Lahti and Shetty, 2017), excluding reads assigned within equivalent or child taxa to animals, viruses or Parabasalia. ANCOM-BC version 1.0.5 (Lin and Peddada, 2020) was used for differential abundance analysis, excluding reads assigned within equivalent or child taxa to animals or plants. SparCC (Friedman and Alm, 2012) was used for microbial correlation analysis, including only bacterial and parabasalid genera representing at least 0.005% of the sequencing library in at least 1 sample. Boostrapped samples (100 replicates) of microbial abundance were used to calculate 2-sided pseudo-P values. Microbial correlation networks were derived from the sparCC results using the R package igraph version 1.2.11 (Csardi and Nepusz, 2006), with edges linking genera sharing a correlation coefficient greater than 0.8 and pseudo P value lower than 0.05. Networks were split into modular components by the Louvain method (Blondel et al., 2008) and Cytoscape version 3.6.1 (Shannon et al., 2003) was used to generate figures and calculate network summary statistics. For functional analysis of microbial transcription, reads assigned within equivalent or child taxa to animals, plants, Parabasalia or viruses were excluded. HUMAnN version 2.8.2 (Franzosa et al., 2018) was used to functionally classify reads at the gene family level by translated alignment to UniRef90 protein families (Suzek et al., 2015). Gene family abundance values were normalized to library size (in counts per million; CPM) prior to assignment to MetaCyc pathways (Caspi et al., 2016) to calculate pathway abundance using HUMAnN version 3.0.0 (Beghini et al., 2021). Pathway abundance values were log2 transformed, with an added pseudocount of 0.01, before differential abundance test by the limma-trend method (Law et al., 2014).
Results
Identity of trichomonads colonizing the macaque gut
We performed an analysis on the unidentified Trichomonads which were reported in published fecal metatranscriptomics data from rhesus macaques with ICD (Westreich et al., 2019). Metatranscriptomes were available for 12 macaques with ICD (Macaques 1–12) and 12 healthy control animals (Macaques 13–24).
We aimed to investigate the identity of Trichomonad parasites reportedly present in this dataset (Westreich et al., 2019). To generate sequences for molecular typing, we generated a de novo assembly of the metatranscriptome. Contig length statistics suggested an overall low degree of assembly, with a mode contig length of 161 bp across all samples, and N50 values ranging from 544 to 1021 bp. Alignment of reads to the assembly indicated no major compositional biases (Fig. S2). Summary statistics for the dataset and assembly are presented in Table S1. The de novo assembly is available in Data file S2.
Due to the low sequence coverage and high complexity of parasite sequences, we utilized the 18S rRNA, actin and elongation factor 1 alpha (EF-1α) loci to identify the Parabasalia colonizing the macaque gut. Amongst all the samples, we assembled 58, 10 and 11 18S rRNA, actin and EF-1α sequences, respectively, which shared greater than 88% sequence identity with reference Parabasalia sequences for at least 90% of their length (Table S2). We assessed the diversity of sequences present by maximum likelihood analysis and identified 10 well-supported clades for the 18S rRNA locus (Fig. S3), 4 for the actin locus (Fig. S4), and 1 for the EF-1α locus (Fig. S5). We generated phylogenies using representative sequences from each clade alongside a range of Parabasalia reference sequences in order to refine the identity of the parasite sequences. Analysis of a single representative sequence from each of the 18S rRNA sequence groups revealed at least 3 major lineages, related to Tetratrichomonas, Pentatrichomonas and Trichomitus spp., with strong bootstrap support only present for the latter (99%; Fig. 1). In contrast, there was strong bootstrap support (99%) for grouping of all identified actin sequences with Tetratrichomonas gallinarum (Fig. 2), and all identified EF-1α sequences with P. hominis (100%; Fig. 3). Integrating these analyses, we inferred that there are likely to be 3 Parabasalia lineages, related to Trichomitus, Tetratrichomonas and Pentatrichomonas, present amongst the macaque fecal samples.
Fig. 1.

Unrooted maximum likelihood phylogeny (GTR model with empirical base frequencies, invariable sites and the discrete gamma model) of Parabasalia-like 18S rRNA sequences from the macaque fecal metatranscriptome, alongside a range of Parabasalia species. Bootstrap values (1000 replicates) greater than 75% are shown on branches in red. Units for tree scale are inferred substitutions per base pair. Macaque-derived sequences (highlighted in orange) are named sequentially according to the animal from which they originated, e.g. Macaque1:1, Macaque1:2. Major lineages of macaque-derived Tetratrichomonas-like, Pentatrichomonas-like and Trichomitus-like sequences are highlighted in red, yellow and blue, respectively. Where available, Genbank accessions (Benson et al., 2015) are shown at the ends of tip labels.
Fig. 2.

Maximum likelihood phylogeny (TIM2e model with equal base frequencies and the discrete gamma model) of Parabasalia-like actin sequences from the macaque fecal metatranscriptome alongside a range of Parabasalia species. Phylogeny is rooted using sequences from Giardia lamblia (accession L29032.1) and Spironucleus salmonicida (accession KI546119.1) as an outgroup (not shown). Bootstrap values (1000 replicates) greater than 75% are shown on branches in red. Units for tree scale are inferred substitutions per base pair. Macaque-derived sequences (highlighted in orange) are named sequentially according to the animal from which they originated, e.g. Macaque1:1, Macaque1:2. The major Tetratrichomonas-like lineage of macaque-derived sequences is highlighted in red. Coloured dots indicate animal host taxa. Where available, Genbank accessions (Benson et al., 2015) are shown at the ends of tip labels.
Fig. 3.

Maximum likelihood phylogeny (TIM2 model allowing unequal base frequencies, with empirical base frequencies, invariable sites and the discrete gamma model) of Parabasalia-like EF-1α sequences from the macaque fecal metatranscriptome, alongside a range of Parabasalia species. Phylogeny is rooted using sequences from Giardia intestinalis (accession HQ179602.1) and Spironucleus barkhanus (accession AB665178.1) as an outgroup (not shown). Bootstrap values (1000 replicates) greater than 75% are shown on branches in red. Units for tree scale are inferred substitutions per base pair. Macaque-derived sequences (highlighted in orange) are named sequentially according to the animal from which they originated, e.g. Macaque1:1, Macaque1:2. The major Pentatrichomonas-like lineage of macaque-derived sequences is highlighted in yellow. Coloured dots indicate animal host taxa. Where available, Genbank accessions (Benson et al., 2015) are shown at the ends of tip labels.
To taxonomically assign the metatranscriptome reads, we included de novo assembled contigs derived from in vitro RNA-Seq analysis of P. hominis, Tetratrichomonas gallinarum and Trichomitus batrachorum (Handrich et al., 2019) in the reference database to improve assignment for sequences derived from the putative parasite genera of interest. In agreement with the phylogenetic results, Trichomitus, Pentatrichomonas and Tetratrichomonas were the 3 most abundant (mean across all samples) parabasalid genera which were identified (Fig. 4A). According to read assignment, Trichomitus was the most abundant individual genus of interest (mean abundance 0.096%), followed by Pentatrichomonas (mean abundance 0.025%) and Tetratrichomonas (mean abundance 0.020%). A substantial number of sequences (mean abundance 0.093%) were identified as parabasalid in origin but could not be assigned to a particular genus. Unidentified parabasalid reads appeared more abundant among animals in which Tetratrichomonas, Pentatrichomonas or Trichomitus classified reads were abundant, likely suggesting that they originated from 1 or more of these genera.
Fig. 4.

Summary of microbial abundances amongst control macaques and this with idiopathic chronic diarrhoea. (A) Parabasalia genera of interest, (B) Phyla excluding the host or Parabasalia (C) Bacteroidetes genera and (D) Firmicutes genera. (B-D) show the abundance of the top 10 most abundant taxa (sum across all samples). Abundances are presented as a percentage of the total sequence library size. The ‘other’ category groups the rest of taxa not shown, and lines separate the subdivisions within these bars. The ‘unclassified’ category represents sequence reads which have been assigned to the relevant taxon of interest for the plot, but not to any specific phylum or genus. Samples are ordered 1–24 from left to right.
In addition, a notable fraction of reads were classified as Trichomonas (mean abundance 0.019%). This most likely reflects the greater representation of Trichomonas whole genome sequences available in the reference database, including T. vaginalis and T. gallinae, whereas the genera of interest were only represented by in vitro RNA-Seq data, which is likely to have an incomplete gene content. However, while it cannot be ruled out that Trichomonas spp. were present amongst the samples, we have focused our analysis on the most likely genera based on the phylogenetic results. Only 2 control macaques showed a total abundance of Parabasalia greater than 0.125%, limiting the statistical power for tests correlating variables with Parabasalia abundance amongst the control animals.
Trichomonad gene expression
We focused on the most abundant putative Parabasalia-derived contigs to explore the most biologically important functions, which are summarized in Table 1. Potential energy-generation pathways included glycolysis, hydrogenosome metabolism, catabolism of GlcNAc, GalNAc, galactose and glucosamine and amino acid catabolism, including the arginine dihydrolase (ADH) pathway. The presence of a putative xanthine dehydrogenase could also indicate catabolism of nucleotides as an additional nutrient source (Wang et al., 2016). Synthesis of glucose and storage as glycogen was suggested by gluconeogenesis and glycogen processing enzymes.
Table 1.
Summary of selected Parabasalia-like contigs of interest derived from the macaque fecal metatranscriptome
| Contig name | Annotation | Pathway/function | T. vaginalis hit % identity (protein level) | Mean abundance (RPM) | Rank (ranked by abundance of all Parabasalia-like contigs |
|---|---|---|---|---|---|
| NODE_1480 | Glutamate dehydrogenase | Amino acid catabolism | 56.1 | 6031 | 1 |
| NODE_1122 | Phosphoenol pyruvate carboxykinase | Gluconeogenesis | 55.9 | 4187 | 3 |
| NODE_1126 | Starch branching enzyme | Starch synthesis | 48.0 | 3211 | 4 |
| NODE_791 | Fructose-1,6-bisphosphate aldolase | Glycolysis | 65.7 | 3006 | 6 |
| NODE_638a | Glyceradehyde-3-phosphate dehydrogenase | 85.3 | 2791 | 8 | |
| NODE_1712 | Phosphoglucomutase/phosphomannomutase | 42.4 | 2136 | 12 | |
| NODE_24 | Pyruvate:ferredoxin oxidoreductase BII | Hydrogenosome metabolism | 74.3 | 875 | 45 |
| NODE_952 | Ornithine carbamoyltransferase family protein | Arginine dihydrolase pathway | 89.8 | 758 | 53 |
| NODE_539 | Malic enzyme | Hydrogenosome metabolism | 83.6 | 289 | 200 |
| NODE_134 | Aldehyde oxidase and xanthine dehydrogenase | Purine catabolism | 82.8 | 281 | 207 |
| NODE_2461 | Glucosamine-6-phosphate isomerase family protein | GlcNAc catabolism | 49.8 | 265 | 222 |
| NODE_2958 | Transketolase family protein | Pentose phosphate pathway | 45.9 | 242 | 239 |
| NODE_418 | Coronin | Phagocytosis | 75.9 | 226 | 252 |
| NODE_1833a | Lysozyme | Bacterial cell wall degradation | 70.6 | 134 | 434 |
| NODE_2008a | Lysozyme | 64.8 | 126 | 462 | |
| NODE_589 | Galactokinase family protein | Galactose metabolism | 65.2 | 71 | 800 |
| NODE_864 | Succinyl-CoA ligase beta-chain | Hydrogenosome metabolism | 78.1 | 60 | 946 |
Annotation for the top T. vaginalis hit was uninformative, and so a lower hit within the top 10 hits was selected.
A number of Parabasalia contigs were annotated with putative lysozyme activity, thus potentially targeting the bacterial microbiota. A maximum likelihood phylogeny was generated to investigate the possibility that the contigs were bacterial in origin (Fig. S6). Results suggested that the lysozyme-like contig NODE_2008 originated from Trichomitus with strong support, and an additional contig NODE_1833 may have originated from Pentatrichomonas or Tetratrichomonas, although this was poorly supported. In addition, we identified a contig with high similarity to T. vaginalis coronin, an actin-binding protein implicated in phagocytosis (Bricheux et al., 2000), thus consistent with parasite phagocytosis targeting microbial or host cells.
Numerous contigs showed strong similarity to T. vaginalis genes previously implicated in pathobiology. Of particular interest for parasite adhesion to host or microbial cells (Handrich et al., 2019), we detected expression of 762 contigs with substantial sequence similarity to T. vaginalis BspA proteins. As the BspA family represent strong candidate LGTs of prokaryotic origin (Handrich et al., 2019), it is likely that this list could include bacterial contigs, due to high similarity between the Trichomonad and bacterial sequences. The list included 70 contigs with greater than 60% sequence identity with the nearest T. vaginalis homologue (by BLASTx). Cysteine peptidases are also implicated in Trichomonas pathobiology (Sommer et al., 2005) and 93 contigs were detected which shared high similarity with T. vaginalis Clan CA, family C1, cathepsin L-like cysteine peptidases. Of particular interest, 17 contigs were close homologues of TvCP39 (locus tag TVAG_298080; mean percentage identity 69%), a secreted cysteine peptidase demonstrated to induce host cell apoptosis (Arroyo et al., 2015).
Trichomonad interactions with the microbiota
To further investigate potential interactions between parabasalid parasites and the microbiota, we examined the taxonomic composition of the samples (Fig. 4). We reproduced a microbiota profile which was in agreement with the previous report (Westreich et al., 2019); Bacteroidetes and Firmicutes were the most abundant phyla (Fig. 4B), the former largely dominated by the genus Prevotella (Fig. 4C) and the latter composed of a diverse range of genera (Fig. 4D). A large proportion of sequences could not be taxonomically classified at the phylum level (mean 66.7% of reads across all samples).
Principal component analysis (PCA) based on the microbial profile showed clear separation between the healthy and ICD groups. The macaques with ICD appeared to resolve into 3 subgroups, potentially indicating distinct microbial communities (Fig. 5). An obvious association between PCA-based clustering and abundance of the parabasalid genera of interest was not clear. However, we tentatively suggest a loose clustering of diseased animals somewhat consistent with Tetratrichomonas abundance (Fig. 5B–D). Of particular interest, a single healthy control animal, macaque 17, clustered amongst the diseased animals. Macaque 17 showed the greatest abundance of total Parabasalia, Trichomitus and Pentatrichomonas of all macaques, and the greatest Tetratrichomonas abundance amongst the macaques with ICD.
Fig. 5.

Principal component analysis (PCA) plot for Aitchison distance based on non-parabaslid microbial abundances amongst macaque fecal samples. Points are shaded according to the centred log ratio normalized abundance values for all Parabasalia, Trichomitus, Pentatrichomonas and Tetratrichomonas, with darker shades indicating greater abundance. Triangle and circular points indicate healthy and diseased animals, respectively.
Our results suggested a possible relationship between parasite abundance and microbiota diversity. Amongst the macaques with ICD, there was a significant positive relationship between Parabasalia abundance and microbial alpha diversity measures (number of observed taxa, Chao1 and Fisher diversity). There was also a significant negative relationship between Parabasalia abundance and Simpson evenness, indicating a more non-uniform distribution of abundance amongst microbial taxa in animals with greater abundance of Parabasalia (Fig. 6). However, this may be restricted to the ICD condition, as a significant relationship between Parabasalia abundance and alpha diversity could not be demonstrated amongst the control macaques (P values derived from linear regression ranged from 0.21 to 0.27), although this is likely to have been influenced by the Parabasalia scarcity amongst the control animals (Fig. 5A). In addition, amongst the full cohort of macaques, using a combined linear model with disease state and Parabasalia abundance as predictors, only disease state showed a significant relationship with the same alpha diversity measures (P value derived from linear regression <0.0001), whereas Parabasalia abundance did not (P value ranged from 0.074 to 0.18). Intriguingly, ICD macaques with greater Parabasalia abundance appeared to more closely resemble control macaques in terms of alpha diversity.
Fig. 6.

Relationship between Parabasalia abundance (normalized by centred log-ratio; clr) and microbial diversity metrics. Observed refers to the total number of observed taxa. Macaques with idiopathic chronic diarrhoea (ICD) and healthy controls are shown in pink and turquoise, respectively. Lines indicate separate linear regressions fitted for the ICD and control groups, and the shaded areas indicate 95% confidence intervals. The significant linear regression P values (<0.05) and corresponding R2 values derived from the ICD sample are indicated next to the corresponding line. The linear regression results for the control macaques were not significant (P value >0.05).
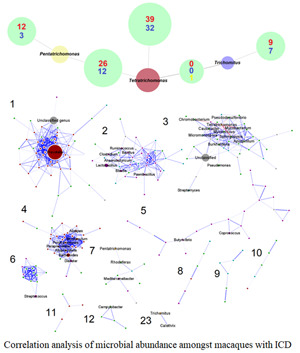
To investigate specific interactions between Parabasalia and bacterial members of the microbiota, we performed an all vs all correlation analysis at the genus level. We focused on the macaques with ICD and included only the most abundant taxa (greater than 0.005% in at least 1 sample). Amongst 358 taxa, with a total of 64 261 possible interactions, our results indicated 11 606 significant abundance correlations between genera. Of the 3 parabasalid genera of interest, Tetratrichomonas showed the greatest number of significant correlations with bacteria (110) followed by Pentatrichomonas (53), and Trichomitus, which showed far fewer significant correlations (17). Tetratrichomonas and Pentatrichomonas substantially overlapped in terms of bacterial genera showing significant positive and negative correlations, possibly indicating shared relationships with bacteria. In contrast, Trichomitus did not share common negative or positive relationships with any bacterial genera with either of the other parabasalid genera (Fig. 7A). Amongst the full complement of significant correlations, Tetratrichomonas stood out as participating in a large number of positive correlations. To investigate this further, we performed a network analysis by linking genera which shared a strong positive correlation. The genera were resolved into 29 connected components (Fig. 7). Of the 12 larger connected components (greater than 3 genera), 4 had a network clustering coefficient of greater than 0.5, indicating the majority of genera correlated with a given genus were also correlated with one another, suggesting well-supported and interdependent networks. Seventeen connected components had fewer than 4 genera, which in total accounted for 41 genera. This overall suggests a complex mixture of interdependent and independent bacterial genera. Tetratrichomonas inhabited the largest connected component of the 3 parabasalid genera of interest (network 3), with a moderate network clustering coefficient of 0.431, indicating a greatest potential interdependence with bacteria. Within network 3, the closeness centrality of Tetratrichomonas was 0.465, the 10th highest in the 41-node network, suggesting a relatively central hub-like position in comparison to most bacterial nodes. In contrast, Pentatrichomonas (network 7) and Trichomitus (network 23) inhabited smaller and more sparsely interconnected components, with 7 and 2 genera, respectively.
Fig. 7.

Correlation analysis of microbial abundance amongst macaques with idiopathic chronic diarrhoea. (A) DiVenn (Sun et al., 2019) figure showing overlap of positive (red) and negative (blue) correlations with bacteria amongst the parabasalid genera of interest. Yellow colour indicates shared correlations for which the direction of correlation differs between the groups. (B) Summary table for the microbial correlation networks. (C) Correlation network of the most abundant microbial genera (greater than 0.005% in at least 1 sample). Edges link nodes (genera) for which abundance was strongly correlated (sharing a significant correlation coefficient greater than 0.8). Node size is scaled to percentage abundance (ignoring the abundance of the ‘unclassified’ group), and labels for genera with less than 0.1% abundance are omitted (except for Parabasalia nodes and their immediate neighbours). Nodes are coloured according to phylum (Actinobacteria; turquoise, Bacteroides; pink, Firmicutes; purple, Proteobacteria; green and Parabasalia; orange) and edge transparency is scaled to the magnitude of the correlation coefficient. Edges linking to Tetratrichomonas are highlighted in red. Connected components are numbered sequentially by decreasing number of nodes and connected components with fewer than 7 nodes are not shown, except those that contain Parabasalia (21 connected components are not shown).
Notably, almost all bacterial genera which showed a significant negative correlation with Tetratrichomonas and Pentatrichomonas were Gram negative, but this pattern did not extend to Trichomitus (Table 2). Pentatrichomonas in particular showed a negative correlation with many bacterial genera reported to contain mucosal inhabitants which are opportunistic pathogens in various host species, including Gemella (Nazik et al., 2018), Moraxella (Goldstein et al., 2009), Mannheimia (Clawson and Murray, 2014) and Aggregatibacter (Karched et al., 2012). Importantly, Tetratrichomonas showed a strong negative correlation with Prevotella, which was the most abundant bacterial genus across all samples. The full list of significant correlations for Tetratrichomonas, Pentatrichomonas and Trichomitus amongst the macaques with ICD is shown in Table S4.
Table 2.
Top significant negative correlations between parabasalid and bacterial genera by sparCC analysis
| Parasite genus | Bacterial genus | Bacterial phylum | Bacterial Gram stain | Correlation coefficient | P value |
|---|---|---|---|---|---|
| Tetratrichomonas | Leeuwenhoekiella | Bacteroidetes | − | −0.824 | 0.00 |
| Algibacter | Bacteroidetes | − | −0.800 | 0.00 | |
| Desulfocapsa | Proteobacteria | − | −0.793 | 0.00 | |
| Veillonella | Firmicutes | − | −0.790 | 0.01 | |
| Desulfitobacterium | Firmicutes | + | −0.784 | 0.00 | |
| Alteromonas | Proteobacteria | − | −0.765 | 0.00 | |
| Emticicia | Bacteroidetes | − | −0.751 | 0.00 | |
| Xenorhabdus | Proteobacteria | − | −0.740 | 0.01 | |
| Prevotella | Bacteroidetes | − | −0.721 | 0.00 | |
| Psychrobacter | Proteobacteria | − | −0.718 | 0.00 | |
| Pentatrichomonas | Basfia | Proteobacteria | − | −0.735 | 0.00 |
| Gemella | Firmicutes | +/− | −0.675 | 0.00 | |
| Moraxella | Proteobacteria | − | −0.639 | 0.03 | |
| Avibacterium | Proteobacteria | − | −0.638 | 0.00 | |
| Acidaminococcus | Firmicutes | − | −0.635 | 0.05 | |
| Roseburia | Firmicutes | + | −0.630 | 0.03 | |
| Aggregatibacter | Proteobacteria | − | −0.617 | 0.04 | |
| Mannheimia | Proteobacteria | − | −0.614 | 0.03 | |
| Chania | Proteobacteria | − | −0.614 | 0.04 | |
| Thalassospira | Proteobacteria | − | −0.610 | 0.05 | |
| Trichomitus | Dickeya | Proteobacteria | − | −0.696 | 0.00 |
| Kocuria | Actinobacteria | +/− | −0.689 | 0.02 | |
| Amycolatopsis | Actinobacteria | + | −0.633 | 0.05 | |
| Aerococcus | Firmicutes | + | −0.614 | 0.03 | |
| Prochlorococcus | Cyanobacteria | − | −0.545 | 0.04 | |
| Phascolarctobacterium | Firmicutes | − | −0.527 | 0.05 | |
| Pseudonocardia | Actinobacteria | + | −0.525 | 0.05 | |
| Desulfosporosinus | Firmicutes | + | −0.508 | 0.05 |
The majority of relationships identified amongst the macaques with ICD were not consistent amongst the control group. Only 1188 out of 11 606 total significant relationships for the macaques with ICD were homodirectionally concordant amongst the control animals, including 2 out of 110 relationships for Tetratrichomonas (Bradyrhizobium and Pentatrichomonas), 5 out of 53 for Pentatrichomonas (Acinetobacter, Janthinobacterium, Mesorhizobium, Streptomyces and Tetratrichomonas) and 2 out of 17 for Trichomitus (Calothrix and Colwellia). This may indicate that the microbial community structure and interdependence was dramatically different between the ICD and control conditions.
We performed a differential abundance analysis between the ICD and control groups in order to investigate any potential impact of the parabasalids on disease aetiology. Interestingly, differential abundance analysis suggested a moderate significantly higher abundance of Tetratrichomonas and Pentatrichomonas (log2 fold differences were 1.62 and 1.80, respectively; adjusted P value <0.001), but not Trichomitus, amongst the macaques with ICD compared with the healthy controls. The original authors ruled out several known common GI pathogens (Bacteria: Campylobacter jejuni, Salmonella, Shigella flexneri and Yersinia enterocolitica and Parasites: Cryptosporidium, Giardia) as the cause of ICD by culture and microscopy-based methods. To complement this, we searched the dataset for potentially pathogenic viral lineage amongst the taxonomic profile. We focused on a selection of 29 potential primate-infecting eukaryotic viruses which we identified by the literature search (Oberste et al., 2007; Handley et al., 2012; Campanini et al., 2013; Janowski et al., 2017; Gao et al., 2018; Zhang et al., 2019b; Smura et al., 2020; Kang et al., 2021) (Fig. S7). Abundance of these viruses was low; total abundance of all 29 viruses was less than 1.6% for all animals, and the highest individual viral abundance was for Simian enterovirus 19, at 0.52%. Notably, we did not identify any significant difference in abundance for any of the viruses comparing between diseased and control animals (Mann–Whitney U test, P value >0.05).
In order to further query the potential influence of parabasalids on the microbiota, we examined the relationship between the HUMAnN2-annotated functional microbial gene expression and parabasalid abundance. The mean-variance relationship of the MetaCyc pathway quantification data is shown in Figure S8. A PCA of the ICD samples based on microbial pathway abundance showed tentative segregation of samples with low and high Tetratrichomonas abundance (Fig. S9). We identified a significant negative relationship between the abundances of 12 MetaCyc pathways and that of Tetratrichomonas amongst the macaques with ICD (Table 3), although the magnitude of the log2 fold changes were relatively small. The strongest relationship was detected for the Superpathway of N-acetylglucosamine, N-acetylmannosamine and N-acetylneuraminate degradation. The majority of functional sequences could not be attributed to a particular microbial species. However, many functions corresponded to likely constitutive bacterial functions such as peptidoglycan synthesis, potentially indicating a negative relationship with bacteria which could not be classified. The analysis did not identify any significant positive relationships between Tetratrichomonas and MetaCyc pathway abundances, and no significant relationships were found for the abundances of both Pentatrichomonas and Trichomitus amongst the macaques with ICD. The significant negative relationships identified for Tetratrichomonas could not be detected amongst the control animals.
Table 3.
Microbial pathways showing a significant relationship with Tetratrichomonas abundance
| MetaCyc ID | Description | Log2 fold changea | Average abundance (CPM) | Species stratification | Stratified abundance | Adjusted P value |
|---|---|---|---|---|---|---|
| GLCMANNANAUT-PWY | Superpathway of N-acetylglucosamine, N-acetylmannosamine and N-acetylneuraminate degradation | −1.28 | 5.19 | NA | 0.04 | |
| P441-PWY | Superpathway of N-acetylneuraminate degradation | −1.12 | 12.5 | NA | 0.03 | |
| PWY-6507 | 4-Deoxy-L-threo-hex-4-enopyranuronate degradation | −0.90 | 16.6 | NA | 0.03 | |
| PWY-7242 | D-fructuronate degradation | −0.76 | 25.4 | NA | 0.04 | |
| GALACTUROCAT-PWY | D-galacturonate degradation I | −0.73 | 21.9 | NA | 0.04 | |
| GALACT-GLUCUROCAT-PWY | Superpathway of hexuronide and hexuronate degradation | −0.65 | 25.7 | NA | 0.04 | |
| GLUCUROCAT-PWY | Superpathway of β-D-glucuronosides degradation | −0.61 | 27.1 | NA | 0.04 | |
| PWY-5686 | UMP biosynthesis | −0.44 | 38.7 | Treponema succinifaciens | 0.014 | 0.04 |
| Bifidobacterium angulatum | 0.018 | |||||
| Coprococcus catus | 0.050 | |||||
| Eubacterium biforme | 0.061 | |||||
| PWY-6386 | UDP-N-acetylmuramoyl-pentapeptide biosynthesis II (lysine-containing) | −0.38 | 41.8 | Bifidobacterium angulatum | 0.0054 | 0.05 |
| Treponema succinifaciens | 0.023 | |||||
| Phascolarctobacterium succinatutens | 0.051 | |||||
| Eubacterium biforme | 0.070 | |||||
| PWY-6385 | Peptidoglycan biosynthesis III (mycobacteria) | −0.37 | 46.9 | Treponema succinifaciens | 0.024 | 0.04 |
| Phascolarctobacterium succinatutens | 0.130 | |||||
| PWY-6387 | UDP-N-acetylmuramoyl-pentapeptide biosynthesis I (meso-diaminopimelate containing) | −0.36 | 53.3 | Bifidobacterium angulatum | 0.0050 | 0.04 |
| Treponema succinifaciens | 0.021 | |||||
| Eubacterium biforme | 0.053 | |||||
| Phascolarctobacterium succinatutens | 0.130 | |||||
| PEPTIDOGLYCANSYN-PWY | Peptidoglycan biosynthesis I (meso-diaminopimelate containing) | −0.35 | 55.9 | Bifidobacterium angulatum | 0.0059 | 0.04 |
| Treponema succinifaciens | 0.024 | |||||
| Eubacterium biforme | 0.051 | |||||
| Phascolarctobacterium succinatutens | 0.13 | |||||
The coefficient resulting from the linear regression fit between pathway and Tetratrichomonas abundance, interpreted as the log2 fold change in pathway abundance per unit of Tetratrichomonas abundance.
Discussion
We aimed to investigate potential relationships between the host, Trichomonads and the mucosal microbiota during health and disease through re-analysis of an existing metatranscriptomics dataset derived from macaque fecal samples. We identified a novel combination of Pentatrichomonas, Tetratrichomonas and Trichomitus parasites in the macaque gut. P. hominis was previously reported in laboratory macaques with ICD (Laing et al., 2018), and simian immunodeficiency virus (Zaragoza et al., 2011). However, to our knowledge, this is the first description of Trichomitus and Tetratrichomonas spp. colonizing the macaque gut. Tetratrichomonas spp. (Cepicka et al., 2006) and Pentatrichomonas spp. (Li et al., 2016; Bastos et al., 2018; Kim et al., 2020) are common GI-inhabitants of mammals. Trichomitus spp. typically infect reptiles and amphibians (Viscogliosi and Müller, 1998; Delgado-Viscogliosi et al., 2000), although mammalian infection has been reported (Dimasuay et al., 2013). Previous reports of macaque-infecting Trichomonads have also included gastric-localized Tritrichomonas spp. (Kondova et al., 2005). It is possible that the intestinal Trichomonad infections represent an artefact resulting from laboratory husbandry, as reports are scarce, and studies on wild macaques did not identify intestinal Trichomonads (Adhikari and Dhakal, 2018; Zhang et al., 2019a). As the animals were at times housed together (Westreich et al., 2019), the possibility of transmission of parasites between the laboratory animals seems likely. Any potential relationship between Trichomonads and diseases such as ICD in macaques has not been reported.
Our results suggested commonality in the expressed functional genes across the Trichomonads. We observed similar energy generation mechanisms for the macaque-infecting parabasalids as have been previously reported for T. vaginalis, demonstrated by a high abundance of transcripts associated with glycolysis, hydrogenosomal metabolism, amino acid catabolism (including the ADH pathway) and glycogen storage and processing (Müller, 1990; Kulda, 1999; Westrop et al., 2017). We also detected BspA expression potentially attributed to the macaque-infecting parabasalids. BspAs are of interest because proteins of this family have demonstrated roles in host adhesion by bacteria as well as adhesion between bacterial cells (Sharma, 2010). Importantly, T. vaginalis BspAs have been implicated in host adhesion in vitro (Handrich et al., 2019). In addition, T. vaginalis BspAs are differentially expressed in response to Mycoplasma symbionts, suggesting a potential role in parasite–bacteria interactions in modulating parasite binding to host cells (Margarita et al., 2022) and possibly binding to members of the microbiota too.
Our results also indicated a potential influential interaction between Trichomonads and microbial diversity in the macaque gut, as has been reported for other hosts and mucosa (El Sayed Zaki et al., 2010; Ji et al., 2020; Wei et al., 2020; Li et al., 2021). Probable coinfection with at least 3 Trichomonad genera complicated accurate abundance estimation due to the portion of sequences which could not be assigned to a specific genus (Watts et al., 2019). Thus, dissection of the relative effects for individual parasites was recalcitrant. This highlights a limitation of observational studies (Cani, 2018). Despite the greater abundance of Trichomitus, our results suggested Tetratrichomonas had the greatest abundance correlation with differences in the microbiota. Tetratrichomonas participated in the greatest number of significant abundance correlations, and was a central node within a densely interconnected microbial positive correlation network. Correlation networks have been effectively utilized to identify keystone species within microbial communities with biological significance (Duran-Pinedo et al., 2011). An overlap in specific relationships between differing Trichomonad spp. and bacteria may be suggested by shared correlations with bacterial abundance between Pentatrichomonas and Tetratrichomonas. In addition, of particular interest, a positive abundance correlation with Prevotella, which we observed for Tetratrichomonas in the macaque gut, has been described for T. vaginalis in the human UGT (Martin et al., 2013; Jarrett et al., 2019). Conserved interactions may result from biochemical features shared amongst the bacteria, supported by our observation that many of the bacteria negatively correlated with Tetratrichomonas and Pentatrichomonas were Gram negative. This is notable because other Trichomonads such as Dientamoeba fragilis (Chan et al., 1993) depend on Gram-negative bacteria for in vitro growth. Microbial interactions identified in this study varied hugely between the diseased and healthy conditions, similarly to previous results from the healthy and diseased human oral microbiota (Duran-Pinedo et al., 2011). This could suggest wholesale changes in community structure between conditions, but may also reflect unreliability in quantifying microbial abundance correlation (Weiss et al., 2016; Matchado et al., 2021). Although only a single sample, 1 control macaque both resembled the ICD macaques in terms of microbial profile, and showed the greatest abundance of Trichomonads, consistent with a potential parasite–bacterial interaction.
Previous studies have suggested the interaction between Trichomonads and the vaginal microbiota is bidirectional. The microbial profile can influence the ability of Trichomonads to colonize the mucosa (Rathod et al., 2011), and the presence of Trichomonad can perturb the microbial profile (Fichorova et al., 2013; Wei et al., 2020). However, the direction of influence between Trichomonads and the microbiota in the macaque gut could not be determined in the absence longitudinal data. In the macaque, Parabasalia expression of potential microbial-targeting genes such as lysozyme could indicate predation, as has been demonstrated for T. vaginalis (Pinheiro et al., 2018). This could provide a mechanistic basis for negative correlations between parasite and microbial abundance. However, we could only reliably attribute lysozyme-encoding transcripts to Trichomitus, whereas Tetratrichomonas was the only Trichomonad genus correlated with bacterial functional expression. Negative correlation of Tetratrichomonas abundance with bacterial degradation pathways for monosaccharides such as GlcNAc and Sia5NAc, potentially derived from mucin glycoproteins (Yurewicz et al., 1987) or microbial cells (Pinheiro et al., 2018), could indicate nutritional competition. This is supported by the detected expression of GlcNAc-targeting glycosyl hydrolases and potentially associated catabolic enzymes by the Trichomonads.
The absence of several known GI bacteria pathogens and microbial parasites was confirmed by culture and microscopy-based methods, and thus may be excluded as causative agents of ICD in the macaques. We performed an additional search for potentially pathogenic viruses amongst the datasets. However, we did not identify any clear differences for any putative host-infecting virus when comparing between diseased and control animals, suggesting viral infection may not be the primary cause of ICD. A greater abundance of reads classified as originating from the Campylobacter genus amongst the animals with ICD was originally reported (Westreich et al., 2019), and so the potential presence of other pathogens in this genus cannot be ruled out. Our results did not establish a causal link between Trichomonads and ICD in macaques. The higher abundance of Pentatrichomonas and Tetratrichomonas could indicate a causal role in disease. High P. hominis abundance in macaques with ICD was previously reported, but not causally liked to disease (Laing et al., 2018). However, abundance of these parasites appeared to promote a more diverse (control-like) microbiota. Trichomonads were positively correlated with microbial diversity, which was also higher in healthy animals, and has been considered characteristic of healthy human gut (Malard et al., 2021). This contrasts with previous work which suggested that the presence of T. gallinae and Tritrichomonas musculis decreases GI microbial diversity (Ji et al., 2020; Wei et al., 2020). Notably, we did not detect any correlation between Trichomonads and the abundance of bacterial genes underlying mucin degradation or fucose utilization, the previously proposed determinants of macaque ICD (Westreich et al., 2019). It is feasible that the ICD state provides a beneficial environment for Trichomonad colonization, within which the parasites exert a disruptive influence. This is supported by the observation that Trichomonad–microbial interactions appeared to be highly dependent on disease state.
Our results revealed a relatively low parasite abundance in the macaque fecal samples, highlighting the need for greater sequencing depth or selective target enrichment (Gaudin and Desnues, 2018) to quantitatively study the parasite transcriptome in vivo. Reference sequences from closely related parasite strains would also have greatly facilitated analysis (Breitwieser et al., 2019). As is typical for a diverse in vivo metatranscriptome (Li et al., 2019), a large proportion of sequences could not be assigned to a specific phylum.
In summary, these metatranscriptomics analyses of Trichomonads in the macaque gut have provided the first in vivo insight into Trichomonad mucosal colonization, which validates numerous in vitro studies (Müller, 1990; Kulda, 1999; Westrop et al., 2017; Handrich et al., 2019). Our findings support previous reports of Trichomonad–microbiota interactions (Ji et al., 2020; Wei et al., 2020; Bierlein et al., 2021), and demonstrate that such interactions vary between parasite species and are highly context-dependent. Longitudinal studies, or those involving experimental Trichomonad infection, could be used to investigate causality and underlying mechanisms in the parasite–microbiota–disease interrelationship.
Acknowledgements
We acknowledge the bioinformatics advice contributed by John Casement at the Bioinformatics Support Unit (Newcastle University, UK). This research also made use of the Rocket High Performance Computing service at Newcastle University.
Supplementary material
For supplementary material accompanying this paper visit https://doi.org/10.1017/S0031182022001688.
click here to view supplementary material
Data availability
This work presents a re-analysis of metatranscriptomics data generated by Westreich et al. (2019), DOI: https://doi.org/10.1186/s40168-019-0664-z. Original data are available from the NCBI SRA database (Leinonen et al., 2011) under accession numbers SRX3517701-SRX3517724. All the supplementary files containing de novo assemblies of the original sequence data and alignments used for phylogenetics are available via figshare (https://figshare.com/s/5d6f50cb71ed2ffc82fb).
Author's contributions
For this paper, Robert P. Hirt contributed to supervision, project administration, funding acquisition, conceptualization, methodology and writing (review and editing). Nicholas P. Bailey contributed to conceptualization, data curation, methodology, formal analysis, validation, data visualization and writing (original draft, review and editing).
Financial support
This work was supported by the Biotechnology and Bioscience Research Council Doctoral Training Partnership for Newcastle, Liverpool and Durham (with Nicholas P. Bailey as the student and Robert P. Hirt as the supervisor; grant number: BB/M011186/1).
Conflict of interest
The authors declare that there are no conflicts of interest.
Ethical standards
No experimental data collection requiring ethical approval was performed during the course of this work.
References
- Adhikari PP and Dhakal P (2018) Prevalence of gastro-intestinal parasites of rhesus macaque (Macaca mulatta Zimmermann, 1780) and hanuman langur (Semnopithecus entellus Dufresne, 1797) in Devghat, Chitwan, Nepal. Journal of Institute of Science and Technology 22, 7. [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW and Lipman DJ (1990) Basic local alignment search tool. Journal of Molecular Biology 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Andrews S (2018) FastQC. Vol. 2018 Babraham bioinformatics. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- Arroyo R, Cárdenas-Guerra RE, Figueroa-Angulo EE, Puente-Rivera J, Zamudio-Prieto O and Ortega-López J (2015) Trichomonas vaginalis cysteine proteinases: iron response in gene expression and proteolytic activity. Biomedical Research International 2015, 946787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastos BF, Brener B, de Figueiredo MA, Leles D and Mendes-de-Almeida F (2018) Pentatrichomonas hominis infection in two domestic cats with chronic diarrhoea. Journal of Feline Medicine and Surgery Open Reports 4, 2055116918774959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beghini F, McIver LJ, Blanco-Míguez A, Dubois L, Asnicar F, Maharjan S, Mailyan A, Manghi P, Scholz M, Thomas AM, Valles-Colomer M, Weingart G, Zhang Y, Zolfo M, Huttenhower C, Franzosa EA and Segata N (2021) Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. Elife 10, e65088. doi: 10.7554/eLife.65088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bello-Ortí B, Howell KJ, Tucker AW, Maskell DJ and Aragon V (2015) Metatranscriptomics reveals metabolic adaptation and induction of virulence factors by Haemophilus parasuis during lung infection. Veterinary Research 46, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DA, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J and Sayers EW (2015) GenBank. Nucleic Acids Research 43, D30–D35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierlein M, Hedgespeth BA, Azcarate-Peril MA, Stauffer SH and Gookin JL (2021) Dysbiosis of fecal microbiota in cats with naturally occurring and experimentally induced Tritrichomonas foetus infection. PLoS One 16, e0246957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondel VD, Guillaume JL, Lambiotte R and Lefebvre E (2008) Fast unfolding of communities in large networks. Journal of Statistical Mechanics – Theory and Experiment 12, P10008. doi: 10.1088/1742-5468/2008/10/p10008. [DOI] [Google Scholar]
- Breitwieser FP, Lu J and Salzberg SL (2019) A review of methods and databases for metagenomic classification and assembly. Briefings in Bioinformatics 20, 1125–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricheux G, Coffe G, Bayle D and Brugerolle G (2000) Characterization, cloning and immunolocalization of a coronin homologue in Trichomonas vaginalis. European Journal of Cell Biology 79, 413–422. [DOI] [PubMed] [Google Scholar]
- Campanini G, Rovida F, Meloni F, Cascina A, Ciccocioppo R, Piralla A and Baldanti F (2013) Persistent human cosavirus infection in lung transplant recipient, Italy. Emerging Infectious Diseases 19, 1667–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cani PD (2018) Human gut microbiome: hopes, threats and promises. Gut 67, 1716–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella-Gutiérrez S, Silla-Martínez JM and Gabaldón T (2009) trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics (Oxford, England) 25, 1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlton JM, Hirt RP, Silva JC, Delcher AL, Schatz M, Zhao Q, Wortman JR, Bidwell SL, Alsmark UC, Besteiro S, Sicheritz-Ponten T, Noel CJ, Dacks JB, Foster PG, Simillion C, Van de Peer Y, Miranda-Saavedra D, Barton GJ, Westrop GD, Müller S, Dessi D, Fiori PL, Ren Q, Paulsen I, Zhang H, Bastida-Corcuera FD, Simoes-Barbosa A, Brown MT, Hayes RD, Mukherjee M, Okumura CY, Schneider R, Smith AJ, Vanacova S, Villalvazo M, Haas BJ, Pertea M, Feldblyum TV, Utterback TR, Shu CL, Osoegawa K, de Jong PJ, Hrdy I, Horvathova L, Zubacova Z, Dolezal P, Malik SB, Logsdon JM, Henze K, Gupta A, Wang CC, Dunne RL, Upcroft JA, Upcroft P, White O, Salzberg SL, Tang P, Chiu CH, Lee YS, Embley TM, Coombs GH, Mottram JC, Tachezy J, Fraser-Liggett CM and Johnson PJ (2007) Draft genome sequence of the sexually transmitted pathogen Trichomonas vaginalis. Science (New York, N.Y.) 315, 207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi R, Billington R, Ferrer L, Foerster H, Fulcher CA, Keseler IM, Kothari A, Krummenacker M, Latendresse M, Mueller LA, Ong Q, Paley S, Subhraveti P, Weaver DS and Karp PD (2016) The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Research 44, D471–D480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepicka I, Hampl V, Kulda J and Flegr J (2006) New evolutionary lineages, unexpected diversity, and host specificity in the parabasalid genus Tetratrichomonas. Molecular Phylogenetics and Evolution 39, 542–551. [DOI] [PubMed] [Google Scholar]
- Chan FT, Guan MX and Mackenzie AM (1993) Application of indirect immunofluorescence to detection of Dientamoeba fragilis trophozoites in fecal specimens. Journal of Clinical Microbiology 31, 1710–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clawson ML and Murray RW (2014) Pathogen variation across time and space: sequencing to characterize Mannheimia haemolytica diversity. Animal Health Research Reviews 15, 169–171. [DOI] [PubMed] [Google Scholar]
- Costello EK, Sun CL, Carlisle EM, Morowitz MJ, Banfield JF and Relman DA (2017) Candidatus Mycoplasma girerdii replicates, diversifies, and co-occurs with Trichomonas vaginalis in the oral cavity of a premature infant. Scientific Reports 7, 3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csardi G and Nepusz T (2006) The igraph software package for complex network research. Vol. Complex Systems pp. 1695. InterJournal.
- Delgado-Viscogliosi P, Viscogliosi E, Gerbod D, Kulda J, Sogin ML and Edgcomb VP (2000) Molecular phylogeny of parabasalids based on small subunit rRNA sequences, with emphasis on the Trichomonadinae subfamily. Journal of Eukaryotic Microbiology 47, 70–75. [DOI] [PubMed] [Google Scholar]
- de Miguel N, Lustig G, Twu O, Chattopadhyay A, Wohlschlegel JA and Johnson PJ (2010) Proteome analysis of the surface of Trichomonas vaginalis reveals novel proteins and strain-dependent differential expression. Molecular and Cellular Proteomics 9, 1554–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessì D, Margarita V, Cocco AR, Marongiu A, Fiori PL and Rappelli P (2019) Trichomonas vaginalis and Mycoplasma hominis: new tales of two old friends. Parasitology 146, 1150–1155. [DOI] [PubMed] [Google Scholar]
- Dimasuay KG, Lavilla OJ and Rivera WL (2013) New hosts of Simplicimonas similis and Trichomitus batrachorum identified by 18S ribosomal RNA gene sequences. Journal of Parasitology Research 2013, 831947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M and Gingeras TR (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics (Oxford, England) 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duboucher C, Noël C, Durand-Joly I, Gerbod D, Delgado-Viscogliosi P, Jouveshomme S, Leclerc C, Cartolano GL, Dei-Cas E, Capron M and Viscogliosi E (2003) Pulmonary coinfection by Trichomonas vaginalis and Pneumocystis sp. as a novel manifestation of AIDS. Human Pathology 34, 508–511. [DOI] [PubMed] [Google Scholar]
- Duran-Pinedo AE, Paster B, Teles R and Frias-Lopez J (2011) Correlation network analysis applied to complex biofilm communities. PLoS One 6, e28438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Sayed Zaki M, Raafat D, El Emshaty W, Azab MS and Goda H (2010) Correlation of Trichomonas vaginalis to bacterial vaginosis: a laboratory-based study. Journal of Infection in Developing Countries 4, 156–163. [DOI] [PubMed] [Google Scholar]
- Fichorova RN, Buck OR, Yamamoto HS, Fashemi T, Dawood HY, Fashemi B, Hayes GR, Beach DH, Takagi Y, Delaney ML, Nibert ML, Singh BN and Onderdonk AB (2013) The Villain Team-Up or how Trichomonas vaginalis and bacterial vaginosis alter innate immunity in concert. Sexually Transmitted Infections 89, 460–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzosa EA, McIver LJ, Rahnavard G, Thompson LR, Schirmer M, Weingart G, Lipson KS, Knight R, Caporaso JG, Segata N and Huttenhower C (2018) Species-level functional profiling of metagenomes and metatranscriptomes. Nature Methods 15, 962–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J and Alm EJ (2012) Inferring correlation networks from genomic survey data. PLOS Computational Biology 8, e1002687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F, Bian L, Hao X, Hu Y, Yao X, Sun S, Chen P, Yang C, Du R, Li J, Zhu F, Mao Q and Liang Z (2018) Seroepidemiology of coxsackievirus B5 in infants and children in Jiangsu province, China. Human Vaccines & Immunotherapeutics 14, 74–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudin M and Desnues C (2018) Hybrid capture-based next generation sequencing and its application to human infectious diseases. Frontiers in Microbiology 9, 2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein E, Murphy TF and Parameswaran GI (2009) Moraxella catarrhalis, a human respiratory tract pathogen. Clinical Infectious Diseases 49, 124–131. [DOI] [PubMed] [Google Scholar]
- Gouy M, Guindon S and Gascuel O (2010) SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Molecular Biology and Evolution 27, 221–224. [DOI] [PubMed] [Google Scholar]
- Handley SA, Thackray LB, Zhao G, Presti R, Miller AD, Droit L, Abbink P, Maxfield LF, Kambal A, Duan E, Stanley K, Kramer J, Macri SC, Permar SR, Schmitz JE, Mansfield K, Brenchley JM, Veazey RS, Stappenbeck TS, Wang D, Barouch DH and Virgin HW (2012) Pathogenic simian immunodeficiency virus infection is associated with expansion of the enteric virome. Cell 151, 253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handrich MR, Garg SG, Sommerville EW, Hirt RP and Gould SB (2019) Characterization of the BspA and Pmp protein family of trichomonads. Parasites & Vectors 12, 406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janowski AB, Krishnamurthy SR, Lim ES, Zhao G, Brenchley JM, Barouch DH, Thakwalakwa C, Manary MJ, Holtz LR and Wang D (2017) Statoviruses, A novel taxon of RNA viruses present in the gastrointestinal tracts of diverse mammals. Virology 504, 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrett OD, Srinivasan S, Richardson BA, Fiedler T, Wallis JM, Kinuthia J, Jaoko W, Mandaliya K, Fredricks DN and McClelland RS (2019) Specific vaginal bacteria are associated with an increased risk of Trichomonas vaginalis acquisition in women. Journal of Infectious Diseases 220, 1503–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji F, Zhang D, Shao Y, Yu X, Liu X, Shan D and Wang Z (2020) Changes in the diversity and composition of gut microbiota in pigeon squabs infected with Trichomonas gallinae. Scientific Reports 10, 19978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliano C, Cappuccinelli P and Mattana A (1991) In vitro phagocytic interaction between Trichomonas vaginalis isolates and bacteria. European Journal of Clinical Microbiology & Infectious Diseases: Official Publication of the European Society of Clinical Microbiology 10, 497–502. [DOI] [PubMed] [Google Scholar]
- Kang HJ, Yoon Y, Lee YP, Kim HJ, Lee DY, Lee JW, Hyeon JY, Yoo JS, Lee S, Kang C, Choi W and Han MG (2021) A different epidemiology of enterovirus A and enterovirus B co-circulating in Korea, 2012–2019. Journal of the Pediatric Infectious Diseases Society 10, 398–407. [DOI] [PubMed] [Google Scholar]
- Karched M, Furgang D, Planet PJ, DeSalle R and Fine DH (2012) Genome sequence of Aggregatibacter actinomycetemcomitans RHAA1, isolated from a rhesus macaque, an Old World primate. Journal of Bacteriology 194, 1275–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Whon TW, Lim MY, Kim YB, Kim N, Kwon MS, Kim J, Lee SH, Choi HJ, Nam IH, Chung WH, Kim JH, Bae JW, Roh SW and Nam YD (2020) The human gut archaeome: identification of diverse haloarchaea in Korean subjects. Microbiome 8, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondova I, Simon MA, Klumpp SA, MacKey J, Widmer G, Domingues HG, Persengiev SP and O'Neil SP (2005) Trichomonad gastritis in rhesus macaques (Macaca mulatta) infected with simian immunodeficiency virus. Veterinary Pathology 42, 19–29. [DOI] [PubMed] [Google Scholar]
- Kopylova E, Noé L and Touzet H (2012) SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics (Oxford, England) 28, 3211–3217. [DOI] [PubMed] [Google Scholar]
- Kulda J (1999) Trichomonads, hydrogenosomes and drug resistance. International Journal for Parasitology 29, 199–212. [DOI] [PubMed] [Google Scholar]
- Lahti L and Shetty S (2017) microbiome R package. Bioconductor. 10.18129/B9.bioc.microbiome [DOI]
- Laing ST, Merriam D, Shock BC, Mills S, Spinner A, Reader R and Hartigan-O'Connor DJ (2018) Idiopathic colitis in rhesus macaques is associated with dysbiosis, abundant enterochromaffin cells and altered T-cell cytokine expression. Veterinary Pathology 55, 741–752. [DOI] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W and Smyth GK (2014) Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biology 15, R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinonen R, Sugawara H and Shumway M and Collaboration INSD (2011) The sequence read archive. Nucleic Acids Research 39, D19–D21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I and Bork P (2019) Interactive Tree of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Research 47, W256–W259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R and Subgroup GPDP (2009) The sequence alignment/map format and SAMtools. Bioinformatics (Oxford, England) 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WC, Ying M, Gong PT, Li JH, Yang J, Li H and Zhang XC (2016) Pentatrichomonas hominis: prevalence and molecular characterization in humans, dogs, and monkeys in Northern China. Parasitology Research 115, 569–574. [DOI] [PubMed] [Google Scholar]
- Li F, Hitch TCA, Chen Y, Creevey CJ and Guan LL (2019) Comparative metagenomic and metatranscriptomic analyses reveal the breed effect on the rumen microbiome and its associations with feed efficiency in beef cattle. Microbiome 7, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Liu ZH, Zhang Z, Bai HH, Zong XN, Wang FJ and Fan LY (2021) Comparative analysis of the vaginal microbiome of Chinese women with Trichomonas vaginalis and mixed infection. Microbial Pathogenesis 154, 104790. doi: 10.1016/j.micpath.2021.104790. [DOI] [PubMed] [Google Scholar]
- Lin H and Peddada SD (2020) Analysis of compositions of microbiomes with bias correction. Nature Communications 11, 3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malard F, Dore J, Gaugler B and Mohty M (2021) Introduction to host microbiome symbiosis in health and disease. Mucosal Immunology 14, 547–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik SB, Brochu CD, Bilic I, Yuan J, Hess M, Logsdon JM and Carlton JM (2011) Phylogeny of parasitic parabasalia and free-living relatives inferred from conventional markers vs Rpb1, a single-copy gene. PLoS One 6, e20774. doi: 10.1371/journal.pone.0020774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margarita V, Bailey NP, Rappelli P, Diaz N, Dessì D, Fettweis JM, Hirt RP and Fiori PL (2022) Two different species of Mycoplasma endosymbionts can influence Trichomonas vaginalis pathophysiology. Mbio 13, e0091822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maritz JM, Land KM, Carlton JM and Hirt RP (2014) What is the importance of zoonotic Trichomonads for human health? Trends in Parasitology 30, 333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DH, Zozaya M, Lillis RA, Myers L, Nsuami MJ and Ferris MJ (2013) Unique vaginal microbiota that includes an unknown Mycoplasma-like organism is associated with Trichomonas vaginalis infection. Journal of Infectious Diseases 207, 1922–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Herrero MDC, Garijo-Toledo MM, González F, Bilic I, Liebhart D, Ganas P, Hess M and Gómez-Muñoz MT (2019) Membrane associated proteins of two Trichomonas gallinae clones vary with the virulence. PLoS One 14, e0224032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzano V, Mancinelli L, Bracaglia G, Del Chierico F, Vernocchi P, Di Girolamo F, Garrone S, Tchidjou Kuekou H, D'Argenio P, Dallapiccola B, Urbani A and Putignani L (2017) “Omic” investigations of protozoa and worms for a deeper understanding of the human gut “parasitome”. PLoS Neglected Tropical Diseases 11, e0005916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matchado MS, Lauber M, Reitmeier S, Kacprowski T, Baumbach J, Haller D and List M (2021) Network analysis methods for studying microbial communities: a mini review. Computational and Structural Biotechnology Journal 19, 2687–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews HM (1986) Carbohydrate utilization by Trichomonas gallinae – effects on growth and metabolic-activity under nongrowth conditions. Journal of Parasitology 72, 170–174. [PubMed] [Google Scholar]
- McMurdie PJ and Holmes S (2013) Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8, e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M (1990) Biochemistry of Trichomonas vaginalis. In Honigberg BM (ed.), Trichomonads Parasitic in Humans. New York: Springer-Verlag, pp. 156. [Google Scholar]
- Nazik S, Cingöz E, Şahin AR and Ateş S (2018) Evaluation of cases with Gemella infection: cross-sectional study. Journal of Infectious Disease and Epidemiology 4, 063. doi: doi.org/ 10.23937/2474-3658/1510063. [DOI] [Google Scholar]
- Nguyen LT, Schmidt HA, von Haeseler A and Minh BQ (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Molecular Biology and Evolution 32, 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk S, Meleshko D, Korobeynikov A and Pevzner PA (2017) metaSPAdes: a new versatile metagenomic assembler. Genome Research 27, 824–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberste MS, Maher K and Pallansch MA (2007) Complete genome sequences for nine simian enteroviruses. Journal of General Virology 88, 3360–3372. [DOI] [PubMed] [Google Scholar]
- O'Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, Astashyn A, Badretdin A, Bao Y, Blinkova O, Brover V, Chetvernin V, Choi J, Cox E, Ermolaeva O, Farrell CM, Goldfarb T, Gupta T, Haft D, Hatcher E, Hlavina W, Joardar VS, Kodali VK, Li W, Maglott D, Masterson P, McGarvey KM, Murphy MR, O'Neill K, Pujar S, Rangwala SH, Rausch D, Riddick LD, Schoch C, Shkeda A, Storz SS, Sun H, Thibaud-Nissen F, Tolstoy I, Tully RE, Vatsan AR, Wallin C, Webb D, Wu W, Landrum MJ, Kimchi A, Tatusova T, DiCuccio M, Kitts P, Murphy TD and Pruitt KD (2016) Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Research 44, D733–D745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira-Neves A and Benchimol M (2007) Phagocytosis by Trichomonas vaginalis: new insights. Biology of the Cell 99, 87–101. [DOI] [PubMed] [Google Scholar]
- Pinheiro J, Biboy J, Vollmer W, Hirt RP, Keown JR, Artuyants A, Black MM, Goldstone DC and Simoes-Barbosa A (2018) The protozoan Trichomonas vaginalis targets bacteria with laterally acquired NlpC/P60 peptidoglycan hydrolases. Mbio 9, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathod SD, Krupp K, Klausner JD, Arun A, Reingold AL and Madhivanan P (2011) Bacterial vaginosis and risk for Trichomonas vaginalis infection: a longitudinal analysis. Sexually Transmitted Diseases 38, 882–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rendon-Maldonado JG, Espinosa-Cantellano M, Gonzalez-Robles A and Martinez-Palomo A (1998) Trichomonas vaginalis: In vitro phagocytosis of lactobacilli, vaginal epithelial cells, leukocytes, and erythrocytes. Experimental Parasitology 89, 241–250. [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Research 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A (2010) Virulence mechanisms of Tannerella forsythia. Periodontology 2000 54, 106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W and Xiong J (2019) TaxonKit: a cross-platform and efficient NCBI taxonomy toolkit. bioRxiv, 513523. doi: 10.1101/513523. [DOI] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD and Higgins DG (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smura T, Blomqvist S, Kolehmainen P, Schuffenecker I, Lina B, Böttcher S, Diedrich S, Löve A, Brytting M, Hauzenberger E, Dudman S, Ivanova O, Lukasev A, Fischer TK, Midgley S, Susi P, Savolainen-Kopra C, Lappalainen M and Jääskeläinen AJ (2020) Aseptic meningitis outbreak associated with echovirus 4 in Northern Europe in 2013–2014. Journal of Clinical Virology: The Official Publication of the Pan American Society for Clinical Virology 129, 104535. [DOI] [PubMed] [Google Scholar]
- Sommer U, Costello CE, Hayes GR, Beach DH, Gilbert RO, Lucas JJ and Singh BN (2005) Identification of Trichomonas vaginalis cysteine proteases that induce apoptosis in human vaginal epithelial cells. Journal of Biological Chemistry 280, 23853–23860. [DOI] [PubMed] [Google Scholar]
- Sun L, Dong S, Ge Y, Fonseca JP, Robinson ZT, Mysore KS and Mehta P (2019) DiVenn: an interactive and integrated web-based visualization tool for comparing gene lists. Frontiers in Genetics 10, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suyama M, Torrents D and Bork P (2006) PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Research 34, W609–W612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzek BE, Wang Y, Huang H, McGarvey PB, Wu CH and Consortium U (2015) UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics (Oxford, England) 31, 926–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viscogliosi E and Müller M (1998) Phylogenetic relationships of the glycolytic enzyme, glyceraldehyde-3-phosphate dehydrogenase, from parabasalid flagellates. Journal of Molecular Evolution 47, 190–199. [DOI] [PubMed] [Google Scholar]
- Wang CH, Zhang C and Xing XH (2016) Xanthine dehydrogenase: an old enzyme with new knowledge and prospects. Bioengineered 7, 395–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts GS, Thornton JE, Youens-Clark K, Ponsero AJ, Slepian MJ, Menashi E, Hu C, Deng W, Armstrong DG, Reed S, Cranmer LD and Hurwitz BL (2019) Identification and quantitation of clinically relevant microbes in patient samples: comparison of three k-mer based classifiers for speed, accuracy, and sensitivity. PLoS Computational Biology 15, e1006863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei YX, Gao J, Kou YB, Meng LY, Zheng XP, Liang M, Sun HX, Liu ZZ and Wang YG (2020) Commensal bacteria impact a protozoan's integration into the murine Gut microbiota in a dietary nutrient-dependent manner. Applied and Environmental Microbiology 86, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss S, Van Treuren W, Lozupone C, Faust K, Friedman J, Deng Y, Xia LC, Xu ZZ, Ursell L, Alm EJ, Birmingham A, Cram JA, Fuhrman JA, Raes J, Sun F, Zhou J and Knight R (2016) Correlation detection strategies in microbial data sets vary widely in sensitivity and precision. The ISME Journal 10, 1669–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westreich ST, Ardeshir A, Alkan Z, Kable ME, Korf I and Lemay DG (2019) Fecal metatranscriptomics of macaques with idiopathic chronic diarrhoea reveals altered mucin degradation and fucose utilization. Microbiome 7, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westrop GD, Wang L, Blackburn GJ, Zhang T, Zheng L, Watson DG and Coombs GH (2017) Metabolomic profiling and stable isotope labelling of Trichomonas vaginalis and Tritrichomonas foetus reveal major differences in amino acid metabolism including the production of 2-hydroxyisocaproic acid, cystathionine and S-methylcysteine. PLoS One 12, e0189072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood DE, Lu J and Langmead B (2019) Improved metagenomic analysis with Kraken 2. Genome Biology 20, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurewicz EC, Matsuura F and Moghissi KS (1987) Structural studies of sialylated oligosaccharides of human midcycle cervical mucin. Journal of Biological Chemistry 262, 4733–4739. [PubMed] [Google Scholar]
- Zaragoza MM, Sankaran-Walters S, Canfield DR, Hung JK, Martinez E, Ouellette AJ and Dandekar S (2011) Persistence of gut mucosal innate immune defenses by enteric α-defensin expression in the simian immunodeficiency virus model of AIDS. Journal of Immunology 186, 1589–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Han S, Liu K, Luo J, Lu J and He H (2019a) Occurrence of selected zoonotic fecal pathogens and first molecular identification of. Biomedical Research International 2019, 2494913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Kataoka M, Doan HY, Ami Y, Suzaki Y, Takeda N, Muramatsu M and Li TC (2019b) Characterization of a novel simian sapelovirus isolated from a cynomolgus monkey using PLC/PRF/5 cells. Scientific Reports 9, 20221. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
For supplementary material accompanying this paper visit https://doi.org/10.1017/S0031182022001688.
click here to view supplementary material
Data Availability Statement
This work presents a re-analysis of metatranscriptomics data generated by Westreich et al. (2019), DOI: https://doi.org/10.1186/s40168-019-0664-z. Original data are available from the NCBI SRA database (Leinonen et al., 2011) under accession numbers SRX3517701-SRX3517724. All the supplementary files containing de novo assemblies of the original sequence data and alignments used for phylogenetics are available via figshare (https://figshare.com/s/5d6f50cb71ed2ffc82fb).