

Abstract
Background
Antiangiogenic tyrosine kinase inhibitors (TKIs) provide one of the few therapeutic options for effective treatment of hepatocellular carcinoma (HCC). However, patients with HCC often develop resistance toward antiangiogenic TKIs, and the underlying mechanisms are not understood. The aim of this study was to determine the mechanisms underlying antiangiogenic TKI resistance in HCC.
Methods
We used an unbiased proteomic approach to define proteins that were responsible for the resistance to antiangiogenic TKIs in HCC patients. We evaluated the prognosis, therapeutic response, and serum insulin‐like growth factor‐binding protein‐1 (IGFBP‐1) levels of 31 lenvatinib‐treated HCC patients. Based on the array of results, a retrospective clinical study and preclinical experiments using mouse and human hepatoma cells were conducted. Additionally, in vivo genetic and pharmacological gain‐ and loss‐of‐function experiments were performed.
Results
In the patient cohort, IGFBP‐1 was identified as the signaling molecule with the highest expression that was inversely associated with overall survival. Mechanistically, antiangiogenic TKI treatment markedly elevated tumor IGFBP‐1 levels via the hypoxia‐hypoxia inducible factor signaling. IGFBP‐1 stimulated angiogenesis through activation of the integrin α5β1‐focal adhesion kinase pathway. Consequently, loss of IGFBP‐1 and integrin α5β1 by genetic and pharmacological approaches re‐sensitized HCC to lenvatinib treatment.
Conclusions
Together, our data shed light on mechanisms underlying acquired resistance of HCC to antiangiogenic TKIs. Antiangiogenic TKIs induced an increase of tumor IGFBP‐1, which promoted angiogenesis through activating the IGFBP‐1‐integrin α5β1 pathway. These data bolster the application of a new therapeutic concept by combining antiangiogenic TKIs with IGFBP‐1 inhibitors.
Keywords: hepatocellular carcinoma, hypoxia, IGFBP‐1, lenvatinib, molecular targeting, resistance, tyrosine kinase inhibitors
Abbreviations
- HCC
Hepatocellular carcinoma
- TME
Tumor microenvironment
- TKI
Tyrosine kinase inhibitor
- PFS
Progression‐free survival
- OS
Overall survival
- IGFBP‐1
Insulin‐like growth factor‐binding protein‐1
- CT
Computed tomography
- FBS
Fetal bovine serum
- HUVEC
Human umbilical vein endothelial cells
- EGM‐2
Endothelial cell growth medium‐2
- shRNA
Short hairpin RNA
- KD
Knockdown
- PBS
Phosphate‐buffered saline
- HIF
Hypoxia‐inducible factor
- GAPDH
Glyceraldehyde‐3‐phosphate dehydrogenase
- RIPA
Radioimmunoprecipitation assay
- SDS‐PAGE
Sodium dodecyl sulfate‐polyacrylamide gel electrophoresis
- HRP
Horseradish peroxidase
- IGF‐1
Insulin‐like growth factor‐1
- hr
Human recombinant
- VEGF
Vascular endothelial growth factor
- FGF
Fibroblast growth factor
- FAK
Focal adhesion kinase
- DEG
Differentially expressed gene
- CPM
Counts per million
- LogFC
Log‐fold change
- DAVID
Database for Annotation, Visualization and Integrated Discovery
- PCNA
Proliferating cell nuclear antigen
- PA
Proliferation/apoptosis
- ANOVA
Analysis of variance
- CA9
Carbonic anhydrase 9
- ERK
Extracellular signal‐regulated kinase
- FOXO1
Forkhead box O1
1. BACKGROUND
Hepatocellular carcinoma (HCC) is the most common hepatic malignancy and the third leading cause of cancer‐related death worldwide [1]. Tumor cells construct self‐sufficient structures with surrounding non‐malignant cells, including endothelial cells, known as the tumor microenvironment (TME), for their survival and proliferation [2]. Antiangiogenic tyrosine kinase inhibitors (TKIs) such as sorafenib and lenvatinib are TME‐targeting drugs that have provided apparent survival benefits to patients with advanced HCC [3, 4]. Additionally, our previous studies have shown the clinical utility of these agents; however, patients’ prognoses remain poor [5, 6, 7, 8]. Approximately 80%–90% of patients treated with antiangiogenic TKIs developed adverse events of any grade, and more than half of these patients were forced to stop therapy due to severe adverse events [9]. Antiangiogenic TKI dose reduction or discontinuation is known to promote tumor metastasis or recurrence, which closely associates with poor prognosis [10]. The main therapeutic action of antiangiogenic TKIs is the induction of severe hypoxia in the TME through tumor vascular density depletion [11]. However, hypoxia in the TME can aggravate cytokine expression and induce mutations and epithelial‐mesenchymal transition, resulting in acquired resistance to antiangiogenic TKIs [5, 12‐15]. Nonetheless, the potential mechanisms of acquired resistance to antiangiogenic TKIs remain unclear.
Here, we hypothesized that tumor‐derived proteins induced by hypoxia confered resistance of HCC to antiangiogenic TKIs. We conducted a retrospective analysis on patients with advanced HCC treated with antiangiogenic TKIs and in vitro and in vivo analyses to determine the mechanisms underlying antiangiogenic TKI resistance in hepatoma cells.
2. MATERIALS AND METHODS
2.1. Patient selection
Cohort 1: We retrospectively evaluated 31 patients with advanced HCC who were treated with lenvatinib at Kurume University Hospital (Kurume City, Fukuoka, Japan) and Iwamoto Internal Clinic (Kitakyushu City, Fukuoka, Japan) between March 2018 and April 2019 for exploring the candidate proteins associated with therapeutic response to antiangiogenic TKI treatment and evaluating its ability as a therapeutic/prognostic biomarker. Cohort 2: We retrospectively evaluated 3 patients who underwent hepatectomy after lenvatinib treatment and 9 patients who underwent hepatectomy without prior treatment at Kurume University Hospital between July 2019 and December 2020 for evaluating the effect of lenvatinib treatment on IGFBP‐1 expression in resected tumors.
The patient inclusion criteria were as follows: (1) with histologically or radiologically diagnosed HCC; (2) who were 18 years of age or older; (3) with Eastern Cooperative Oncology Group performance status ≤1; (4) with life expectancy ≥12 weeks (without ≥50% liver occupation and/or obvious portal vein invasion into the trunk). The exclusion criteria were as follows: (1) with brain metastases or spinal cord compression; (2) with poorly controlled or refractory hepatic encephalopathy (West Haven criteria; grade ≥3); (3) with previous gastrointestinal hemorrhage in the month prior to the beginning of the study and/or high risk of bleeding.
2.2. Therapeutic assessments for lenvatinib treatment
Patients received oral lenvatinib (Eisai Co., Ltd., Tokyo, Japan) at a dose of 12 mg/day (for body weight ≥60 kg) or 8 mg/day (for body weight <60 kg). Therapeutic response was evaluated using imaging tests according to the modified Response Evaluation Criteria in Solid Tumors guidelines [16], and progression‐free survival (PFS) and overall survival (OS) after lenvatinib treatment initiation were determined. The receiver operating characteristic curve method for the prediction of long OS (≥median OS for all patients) was used to determine cutoffs for the serum level of insulin‐like growth factor‐binding protein (IGFBP)‐1 before and after lenvatinib treatment as well as changes in IGFBP‐1 levels.
2.3. Definition of computed tomography (CT) value and changes in the CT value
In all patients, tumors could be confirmed in the arterial phase; thus, the intratumoral CT value was measured in this phase. To avoid partial volume calculation errors, a region of interest in each tumor was selected to achieve the best tumor visualization, with a maximum circular or oval area within the tumor. To exclude the influence of the environment during the examination, the CT value of the tumor was corrected with the CT value of the aorta in the same slice. The formula for calculating changes in the CT value is shown in Supplementary Figure S1A. Estimated tumor volume was calculated as 0.52 × length (mm) × width (mm)2.
2.4. Cell lines and culture conditions
The mouse hepatoma cell lines Hep‐55.1C and Hepa1‐6 were purchased from Cell Lines Service GmbH (Oppenheim, Germany) and American Type Culture Collection (ATCC) (Manassas, VA, USA), respectively. Regarding human hepatoma cell lines, HuH7 (JCRB0403) and HLF (JCRB0405) were purchased from the Japanese Collection of Research Bioresource (JCRB) Cell Bank (Tokyo, Japan), HepG2 and Hep3B were purchased from ATCC, and KYN‐2 was originally established and maintained at Department of Pathology, Kurume University School of Medicine (Kurume, Fukuoka, Japan) [17]. These tumor cells were maintained in Dulbecco's modified eagle's medium (Gibco by Invitrogen Cell Culture Co., Auckland, New Zealand) supplemented with 10% heat‐inactivated (56°C, 30 min) fetal bovine serum (FBS; Biowest, Nuaille, France), 100 U/mL penicillin, and 100 mg/mL streptomycin (Nacalai Tesque, Inc., Kyoto, Japan) in a humidified atmosphere containing 5% CO2 at 37°C. Human umbilical vein endothelial cells (HUVECs) were purchased from CAMBREX Bio Science Walkersville, Inc. (Walkersville, MD, USA) and maintained in endothelial cell growth medium‐2 (EGM‐2; Clonetics, CA, USA) supplemented with 5% heat‐inactivated FBS in a humidified atmosphere containing 5% CO2 at 37°C. All cell lines used in our study were authenticated via short tandem repeat profiling and checked for mycoplasma contamination using the mycoplasma removal kit (#88101‐2, DS Pharma Biomedical, Osaka, Japan).
2.5. IGFBP‐1 gene silencing in vitro
Lentiviral transduction particles containing short hairpin RNA (control GFP shRNA, #sc‐108084 and IGFBP‐1 shRNA, # sc‐39584‐V) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Stable IGFBP‐1‐knockdown (KD) HuH7 and HepG2 cell lines were selected using 1 μg/mL puromycin (Sigma‐Aldrich by Merck KGaA, Darmstadt, Germany).
2.6. Establishment of the HCC xenograft mouse models and animal experiments
Mice were purchased from Kyudo KK (Fukuoka, Japan). In total, 5 × 106 Hep‐55.1C cells were suspended in phosphate‐buffered saline (PBS) and subcutaneously inoculated into the flank regions of 5‐week‐old female C57BL/6J mice to establish the Hep‐55.1C subcutaneous mouse model; 2 × 105 Hep‐55.1C cells were suspended in PBS and orthotopically inoculated into the left lobe of the liver of 5‐week‐old female C57BL/6J mice to establish the Hep‐55.1C orthotopic mouse model.
Five‐week‐old female nude mice (BALB/c nu/nu) were housed in specific pathogen‐free conditions. In total, 5 × 106 human tumor cells (HuH7, HepG2, or KYN‐2) were suspended in PBS and subcutaneously inoculated into the flank regions of the nude mice to establish the HuH7, HepG2, or KYN‐2 subcutaneous mouse model. Similarly, stable IGFBP‐1‐KD HuH7 or HepG2 cells were subcutaneously inoculated into the nude mice to establish the IGFBP‐1‐KD HuH7 or HepG2 subcutaneous mouse model.
When the estimated tumor volume reached 150‐200 mm3 in the subcutaneous mouse models or 2 weeks after transplantation in the orthotopic model, the tumor‐bearing mice were allocated to different treatments (5‐6 mice per group). The treatments [lenvatinib, anti‐IGFBP‐1 neutralizing antibody (#MA5‐23727; Thermo Fisher Scientific), and ATN‐161] were initiated. Detailed treatment data in each group are given in Supplementary Table S1. The tumor volumes were measured every 2 days. Mice were euthanized by cervical dislocation under anesthesia using isoflurane and pentobarbital at 2 weeks or indicated points after treatment initiation, and the tumors were resected and evaluated.
2.7. Evaluation of serum angiogenesis‐related proteins
The relative expression of 55 angiogenesis‐related proteins in serum samples from patients in cohort 1 was detected using the Human Angiogenesis Array Kit (#ARY007, R&D Systems, Minneapolis, MN, USA) according to the manufacturer's instructions. Serum IGFBP‐1 concentration in 31 patients (cohort 1) was measured using an ELISA kit (#DGB100, R&D Systems). The serum IGFBP‐1 concentration in mice was measured using the Igfbp1 (Mouse) ELISA kit (#KA3054, Abnova, Taipei, Taiwan, China), via a colorimetric assay according to the manufacturer's instructions.
2.8. Hypoxia experiments
Various O2 concentrations (1%, 2%, and 20%) were applied to a humidified atmosphere containing 5% CO2 at 37°C using a CO2/multigas incubator (#APM‐30D, ASTEC Co. Ltd., Fukuoka, Japan). HepG2, HuH7, Hep‐55.1C, KYN‐2, Hep3B, and Hepa1‐6 cells were seeded onto plates at a density of 1 × 106 cells/well and incubated for 24 h. After overnight incubation at 37°C in the serum‐free medium, the medium was changed and incubated for 48 h under normoxia or hypoxia conditions.
For the hypoxia‐inducible factor (HIF) inhibitory experiment, we used YC‐1 (#AG‐CR‐0120; AdipoGen Life Science, San Diego, CA, USA), an HIF‐1α and ‐2α inhibitor. Cells (HepG2, HuH7, or Hep‐55.1C) were seeded onto plates at a density of 1 × 106 cells/well and incubated for 24 h. After overnight incubation at 37°C in the serum‐free medium, the medium was changed and incubated for 24 h under normoxia condition with 5 μmol/L YC‐1. After incubation, protein and RNA levels of IGFBP‐1, HIF‐1α, HIF‐2α, and tubulin in these cells were measured by reverse transcription‐quantitative real‐time PCR and Western blotting, respectively.
2.9. Reverse transcription‐quantitative real‐time PCR
The extracted RNA from cultured cells or resected mouse tissues (liver or tumor), using an Isogen kit (Nippon Gene Co., Ltd, Tokyo, Japan), was reversely transcribed into cDNA, and quantitative real‐time PCR was performed to detect the mRNA levels of IGFBP‐1. Single‐strand cDNA was synthesized using a high‐capacity RNA‐to‐cDNA kit (Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA). The thermocycling parameters were as follows: 2 min at 50 °C and 10 min at 95 °C, followed by 45 cycles of 15 s at 95 °C and 1 min at 60 °C. Relative quantification of gene expression was performed according to the 2−ΔΔCT method [18] using StepOne Software 2.0 (Applied Biosystems). Glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was used as the reference for the normalization of target gene expression data. The primers used were purchased from TaqMan™ (Applied Biosystems, Foster City, CA, USA) and were as follows: GAPDH, Hs02758991_g1 (#4331182); Gapdh, Mm99999915_g1 (#4331182); IGFBP‐1, Hs00236877_m1 (#4331182); and Igfbp1, Mm005154_m1 (#4331182).
2.10. Western blotting
Resected mice tissues (normal liver or tumor) and cells (hepatoma or endothelial cells) were homogenized in radioimmunoprecipitation assay (RIPA) buffer (Thermo Fisher Scientific) containing 1% protease inhibitor cocktail (Sigma‐Aldrich) and 1% phosphatase inhibitor cocktail (Thermo Fisher Scientific). The protein concentration was determined using the detergent compatible protein assay kit (Bio‐Rad Laboratories, Inc., Berkeley, CA, USA). Equal amounts of protein (20 μg) were loaded per lane onto 10% or 8% sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE), and the proteins were electrophoresed and electrotransferred onto the FluoroTrans membrane (Pall Life Sciences, Port Washington, NY, USA). Following protein transfer, the membranes were incubated with the primary antibodies as described in Supplementary Table S2. Visualization of protein signals was achieved with the horseradish peroxidase (HRP)‐conjugated secondary antibodies [anti‐rabbit IgG, HRP‐conjugated whole Ab (from donkey) (#NA934, 1:10,000, GE Healthcare Life Sciences, Marlborough, MA, USA) or anti‐mouse IgG, HRP‐linked F(ab’)2 fragment (from sheep) (#NA9310, 1:10,000, GE Healthcare Life Sciences)]. Relative amounts of protein were calculated based on the luminescent signals in each sample using Multigauge software (version 3.11, Fujifilm).
2.11. Cell proliferation assay
Cells were seeded onto 96‐well plates at a density of 500 cells/well in sextuplicate and incubated in 100 μL culture medium for 24 h. After overnight incubation at 37°C in the serum‐free medium, the medium was changed. Cell proliferation at various time points (baseline, 24 h, and 48 h) was determined using Cell Count Reagent SF (Nacalai Tesque) according to the manufacturer's protocol.
2.12. Tube formation assay
Tube formation assays were performed using the μ‐Slide Angiogenesis kit (#81506, Ibidi GmbH, Gräfelfing, Germany) according to the manufacturer's protocol. EGM‐2 without hydrocortisone and insulin‐like growth factor‐1 (IGF‐1) were used in these assays. For each assay, HUVECs were resuspended in media as follows: (1) medium supplemented with human recombinant IGFBP‐1 (hrIGFBP‐1; #871‐B1; R&D Systems) at different concentrations (0, 4, 10, and 40 nmol/L); (2) medium supplemented with 2% FBS and 10 nmol/L hrIGFBP‐1 with antiangiogenic TKIs, including 3 μmol/L lenvatinib (#CS‐0109; ChemScene, Monmouth Junction, NJ, USA), 3 μmol/L sorafenib (#CS‐0164; ChemScene), 1 μmol/L fruquintinib [#S5667; Selleck, Houston, TX, USA; a vascular endothelial growth factor (VEGF) receptor‐1, ‐2, and ‐3 inhibitor], or 1 μmol/L AZD4547 [#CS‐0971; ChemScene; a fibroblast growth factor (FGF) receptor‐1, ‐2, and ‐3 inhibitor]; (3) medium supplemented with 10 nmol/L hrIGFBP‐1 and ATN‐161 (#S8454; Selleck; an integrin α5β1 inhibitor) or focal adhesion kinase (FAK) inhibitor 14 (#14485; Cayman Chemical, Ann Arbor, MI, USA) at different concentrations (0, 3, and 10 μmol/L). All cell suspensions were incubated for 12 h before staining with Calcein AM Solution (1:1,000, #NV011, Fujifilm, Tokyo, Japan). The tubes were counted using computer‐assisted imaging software (Adobe Photoshop CC 2021 version 22.2.0, Adobe Systems, Inc., San Jose, CA, USA).
2.13. Total RNA isolation and RNA sequencing
The total RNA was isolated from the cultured HUVECs, which were stimulated with/without 10 nmol/L hrIGFBP‐1 for 3 h, using TRIzol Reagent (Thermo Fisher Scientific) and purified using SV Total RNA Isolation System (Promega, Madison, WI, USA) according to the manufacturer's instructions. RNA samples were quantified using a ND‐1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA), and the quality was confirmed with a TapeStation (Agilent Technologies, Inc, Santa Clara, CA, USA). The sequencing libraries were prepared from 200 ng of RNA with MGIEasy rRNA Depletion Kit and MGIEasy RNA Directional Library Prep Set (MGI Tech Co., Ltd., Shenzhen, Guangdong, China) according to the manufacturer's instructions. The libraries were sequenced on the DNBSEQ‐G400 FAST Sequencer (MGI Tech) with a paired‐end 150 nt strategy.
2.14. Sequencing data analysis
Quality trimming and adapter clipping of the read data were performed using Trimmomatic version 0.38 [19]. Trimmed reads were mapped to the transcript in the reference human hg38 using Bowtie2 aligner within RSEM [20]. The abundance estimation of genes and isoforms with RSEM generated basic count data (expected counts). We used edgeR program to detect differentially expressed genes (DEGs) [21], and obtained normalized counts per million (CPM) values, log‐fold changes (logFC), and P values. We then established the criteria for DEGs: P value ≤ 0.05 and logFC ≥ 1. The heat map of DEGs was generated by MeV software [22]. We used a hierarchical clustering method to sort the genes. The color indicated the distance from the median of each row. The distance metric was “Pearson correlation”, and the linkage method was “average linkage clustering”. To identify the significantly over‐represented gene ontology categories and significantly enriched pathways, we used tools and data provided by the Database for Annotation, Visualization and Integrated Discovery (DAVID) [23].
2.15. Immunohistochemical staining
Paraffin‐embedded tumor tissue sections from mice models and human samples (5‐μm‐thick) were boiled for 30 min in high‐pH target retrieval solution for antigen retrieval and subsequently incubated with primary antibodies and then with secondary antibodies. These antibodies are listed in Supplementary Table S3. Nuclei were stained with DAPI (#H‐1200, Vector Laboratories, Inc., Burlingame, CA, USA) for counterstaining. Immunoreactivity was visualized using anti‐goat IgG HRP‐conjugated antibody (#HAF109, R&D Systems) and Liquid DAB+ Substrate Chromogen System (#K3468, DAKO Japan, Kyoto, Japan). For quantification of IGFBP‐1‐positive cells (in 4‐7 random fields per section), CD31‐positive tumor microvessels (in 5‐10 random fields per section), and CA9‐positive areas (in 4‐6 random fields per section) were counted at a magnification of ×200. The proliferative and apoptosis indices were defined as the percentage of tumor cells with nuclei positive for proliferating cell nuclear antigen (PCNA) and cleaved caspase‐3 staining per 1,000 neoplastic cells, respectively, counted in 5 fields with 200 tumor cells. The proliferation/apoptosis (PA) index was calculated as the ratio of the proliferative index to the apoptosis index. All slides were examined using a confocal microscope (BZ‐X700, Keyence Corporation, Osaka, Japan). Quantitative analyses were performed with Adobe Photoshop CC 2021 version 22.2.0.
2.16. Statistical analyses
All data are expressed as means ± standard error of the mean (SEM). The strength of a linear association between two variables was measured by the Pearson correlation coefficient. Differences between two groups were examined for statistical significance using an unpaired Student's t‐test, and differences among multiple groups were examined using one‐way analysis of variance (ANOVA), followed by a Fisher's least significant difference test. Receiver operating characteristic curve analysis was also performed to evaluate the cutoff value and area under the curve. Kaplan‐Meier curves were constructed to determine OS and PFS in patients with different IGFBP‐1 levels before and after lenvatinib treatment. P values less than 0.05 were considered statistically significant. Data analysis was performed using JMP Pro 15.0 software (JMP, Tokyo, Japan).
3. RESULTS
3.1. Comparison between tumor CT value and serum IGFBP‐1 in antiangiogenic TKI treatment
To detect therapeutic biomarkers of antiangiogenic TKIs in HCC, we first assessed the changes in serum levels of angiogenesis‐related proteins from several patients (cohort 1) with advanced HCC who were treated with lenvatinib, a first‐line antiangiogenic TKI. Figure 1A shows a representative array of data from a patient who responded well to lenvatinib treatment. Among the 55 evaluated angiogenesis‐related proteins, the levels of IGFBP‐1, pentraxin 3, leptin, a disintegrin and metalloproteinase with thrombospondin motif 1, and persephin were significantly changed by more than 50% compared to serum levels before the treatment. Levels of representative angiogenic factors such as VEGF and angiopoietin‐2 were not significantly increased. IGFBP‐1 showed the greatest elevation after lenvatinib treatment compared to other proteins (Figure 1A, Supplementary Figure S1B). In another patient with HCC treated with regorafenib, a second‐line antiangiogenic TKI approved for HCC, IGFBP‐1 level was also elevated by more than 50% compared to the baseline (Supplementary Figure S1C). To validate these findings, we investigated serum IGFBP‐1 levels before and after lenvatinib treatment in 31 patients with advanced HCC (cohort 1). The baseline characteristics of these patients are summarized in Supplementary Table S4. The objective response rate was 35.5%, and the median OS and PFS of these patients were 11.1 and 6.2 months, respectively (Supplementary Figure S1D‐E). Notably, serum IGFBP‐1 levels in almost all patients were elevated under lenvatinib treatment (Figure 1B). The change in serum IGFBP‐1 level was negatively correlated with CT value in the target lesion (Figure 1C). The estimated baseline tumor volume and TNM stage were not associated with serum IGFBP‐1 levels in the clinical data (Supplementary Figure S1F‐G). To corroborate these findings, we collected and immunohistochemically evaluated HCC samples from 3 patients who underwent hepatectomy after lenvatinib treatment and 9 who underwent hepatectomy without prior treatment (cohort 2). Patient characteristics of cohort 2 and HCC samples are listed in Supplementary Table S5. Consistent with the results of clinical cohort 1, lenvatinib significantly increased IGFBP‐1 expression in human HCC samples, which were obtained from hepatectomy after lenvatinib treatment (Figure 1D). In addition, lenvatinib‐treated tumors showed a hypoxic state (carbonic anhydrase 9; CA9 staining) with a marked reduction in tumor vessels (CD31 staining). A significant decrease in tumor cell proliferation and an increase in cell apoptosis were observed in lenvatinib‐treated tumors, leading to a decrease in the PA index (Supplementary Figure S1H).
FIGURE 1.
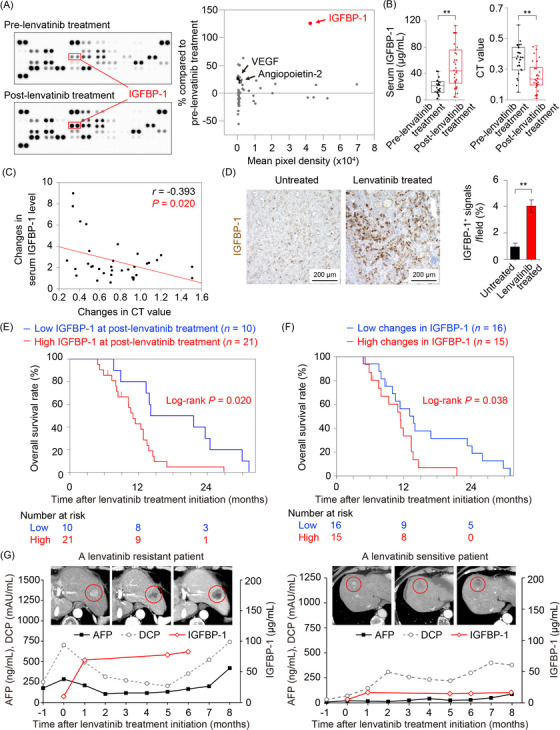
Changes in serum protein levels and CT values with lenvatinib treatment, and association of serum IGFBP‐1 levels with overall survival in patients with HCC. (A) A comprehensive evaluation of angiogenesis‐related proteins before and after lenvatinib treatment (left). The expression levels and rate of change in expression of these proteins after lenvatinib treatment (right). (B) Serum IGFBP‐1 level (left) and CT value (right) before and after lenvatinib treatment in cohort 1 (n = 31; **P < 0.01 vs. pre‐lenvatinib treatment, Student's t‐test). (C) Changes in serum IGFBP‐1 levels were negatively correlated with changes in the CT value in cohort 1 (n = 31; P = 0.020, Pearson correlation coefficient). (D) Lenvatinib treatment significantly increased IGFBP‐1 expression in human HCC samples (**P < 0.01 vs. untreated, Student's t‐test). (E‐F) Kaplan‐Meier curves estimating the overall survival of patients with HCC according to high vs. low serum IGFBP‐1 levels (cutoff: 28.99 μg/mL) after lenvatinib treatment (E), and high vs. low changes in IGFBP‐1 levels (cutoff: 2.054) (F). (G) Representative clinical course in a patient with advanced HCC who developed acquired resistance to lenvatinib therapy and a patient who did not develop the resistance. The enhanced computed tomography images at baseline (left), 1 month (middle), and 6 months (right) after lenvatinib treatment initiation are presented. Red circles indicate the HCC lesion. Data are presented as mean ± SEM.
Abbreviations: HCC, hepatocellular carcinoma; IGFBP‐1, insulin‐like growth factor‐binding protein‐1; VEGF, vascular endothelial growth factor; CT, computed tomography; AFP, alpha‐fetoprotein; DCP, des‐gamma carboxy prothrombin.
3.2. Prognostic biomarker in lenvatinib treatment
We assessed the association between the level of IGFBP‐1 and the prognosis of patients with HCC treated with lenvatinib. We found that the baseline serum IGFBP‐1 levels were not associated with the prognosis (Supplementary Figure S1I). Surprisingly, patients with high serum IGFBP‐1 levels (cutoff: >28.99 μg/mL; area under the curve, 0.723) after lenvatinib treatment had significantly worse prognosis than those with low serum IGFBP‐1 levels (Figure 1E). Moreover, median OS was markedly shortened in the patients with highly elevated IGFBP‐1 levels (cutoff: increased by more than 2.054 times; area under the curve, 0.673) under lenvatinib treatment, when compared to those with slight elevation in IGFBP‐1 level (Figure 1F). A representative time course of changing serum IGFBP‐1 levels and radiological findings of patients treated with lenvatinib are shown in Figure 1G. Before the administration of lenvatinib, an enhanced lesion was detected in hepatic segment 2. The baseline serum IGFBP‐1 level was low. After administration of lenvatinib, arterial blood flow to the target lesion was remarkably decreased and serum IGFBP‐1 level was elevated. After consecutive administration of lenvatinib, the tumor size gradually increased (maximum tumor diameter increased from 15 to 28 mm) with an improvement in arterial blood flow (Figure 1G). In accordance with tumor growth, serum IGFBP‐1 level was further increased. These findings indicated that lenvatinib‐induced elevation in IGFBP‐1 level could be a potential biomarker to detect the poor prognosis of patients with HCC.
3.3. Association between lenvatinib resistance and IGFBP‐1 expression in a mouse xenograft model
To clarify the role of the elevated IGFBP‐1 level after lenvatinib treatment, we conducted xenograft experiments. The basal expression of IGFBP‐1 in five human hepatoma cell lines was assessed. Among them, HepG2 (a human HCC cell line with high IGFBP‐1 expression) showed the highest IGFBP‐1 expression (Figure 2A). Interestingly, IGFBP‐1‐high HCC (HepG2) xenografts showed significant resistance toward lenvatinib treatment (Figure 2B). Consistent with our clinical findings, tumor IGFBP‐1 levels were significantly elevated after lenvatinib treatment in IGFBP‐1‐high HCC (HepG2) xenografts (Figure 2C). Immunohistochemical analyses were conducted to detect changes in the TME after administration of lenvatinib (Figure 2D‐E). Although a decrease in the number of proliferative cells was detected, there was no increase in the number of apoptotic cells in IGFBP‐1‐high HCC (HepG2) xenografts treated with lenvatinib. The microvessel density (CD31 staining) showed a decreasing trend, and hypoxia (CA9 staining) was elevated in IGFBP‐1‐high HCC (HepG2) xenografts treated with lenvatinib (Figure 2D‐E).
FIGURE 2.
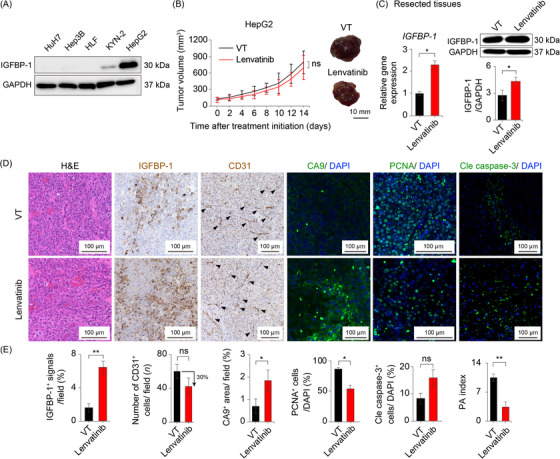
Lenvatinib treatment for the IGFBP‐1‐high HCC xenograft mouse models. (A) Western blotting analysis of IGFBP‐1 protein levels in five cultured human hepatoma cell lines. (B‐E) Assessments in the subcutaneous IGFBP‐1‐high HCC (HepG2) xenograft mouse model. (B) Tumor volume measurements (left panel) and representative macrographs of the tumor (right panel) (6 mice per group). (C) Quantifications of the gene (left) and protein (right) expression levels of IGFBP‐1 in the resected tissues (*P < 0.05 vs. VT, Student's t‐test). (D) Representative micrographs of H&E staining and immunohistochemistry in the VT and lenvatinib groups. (E) Quantification of IGFBP‐1‐positive cells (4‐7 random fields per section), CD31‐positive tumor microvessels (5‐10 random fields per section), CA9‐positive areas (4‐6 random fields per section), PCNA‐positive cells, and cleaved caspase‐3‐positive cells (*P < 0.05, **P < 0.01 vs. VT, Student's t‐test). Data are presented as mean ± SEM.
Abbreviations: IGFBP‐1, insulin‐like growth factor‐binding protein‐1; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; ns, no significance; VT, vehicle treatment; CA9, carbonic anhydrase 9; PCNA, proliferating cell nuclear antigen; Cle caspase‐3, cleaved caspase‐3; PA, proliferation/apoptosis.
3.4. Therapeutic effects of lenvatinib in HCC xenograft mouse models
Next, we subcutaneously implanted HuH7, a human HCC cell line with low IGFBP‐1 expression (hereafter, IGFBP‐1‐low human HCC; Figure 2A), in nude mice (BALB/c nu/nu), and Hep‐55.1C, a mouse HCC cell line with low IGFBP‐1 expression (hereafter, IGFBP‐1‐low mouse HCC; Supplementary Figure S2A) in immunocompetent C57BL/6 mice. Tumor growth was significantly inhibited by lenvatinib in IGFBP‐1‐low human HCC (HuH7) and mouse HCC (Hep‐55.1C) models (Figure 3A‐B). Tumor IGFBP‐1 levels were significantly increased in the lenvatinib group (Figure 3C‐D), and serum IGFBP‐1 levels were also elevated in the lenvatinib group (Figure 3E). Sorafenib, another first‐line antiangiogenic TKI, also showed a similar effect on mouse HCC xenografts (Figure 3D‐E). Antiangiogenic TKIs significantly induced hypoxia (CA9 staining) in the TME of both IGFBP‐1‐low human HCC (HuH7) and mouse HCC (Hep‐55.1C) models (Figure 3F‐G). Consistently, markedly increased IGFBP‐1‐positive areas were seen in the antiangiogenic TKI‐treated group (Figure 3F‐G). The expression of IGFBP‐1 in the liver was not increased in the antiangiogenic TKI group (Supplementary Figure S2B). The proliferative rate of tumor cells as determined by PCNA was largely decreased by lenvatinib treatment (Figure 3F‐G). Importantly, the number of apoptotic cells as measured using cleaved caspase‐3 was substantially increased in lenvatinib‐treated HCC, leading to significantly greater PA index in both IGFBP‐1‐low human and mouse HCC models (Figure 3F‐G). Similar findings were observed in the lenvatinib‐treated orthotopic HCC mouse model (Supplementary Figure S2C‐G). These data show that antiangiogenic TKI transcriptionally and translationally induces IGFBP‐1 in HCC tumors. Taken together, the expression level of IGFBP‐1 in hepatoma cell lines was significantly associated with the therapeutic efficacy of antiangiogenic TKI.
FIGURE 3.
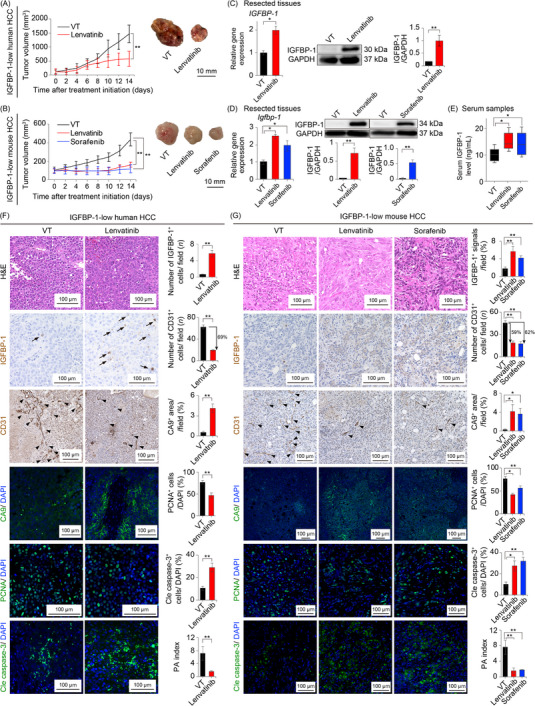
Antiangiogenic TKI treatment in the IGFBP‐1‐low HCC xenograft mouse models. (A‐B) Tumor volume measurements (left panel) and representative macrographs of the tumor (right panel; 5‐6 mice per group) in the subcutaneous IGFBP‐1‐low human HCC (HuH7) xenograft mouse model (A) and those in the subcutaneous IGFBP‐1‐low mouse HCC (Hep‐55.1C) xenograft mouse model (B), respectively. (C‐D) Quantification of the gene (left) and protein (right) expression levels of IGFBP‐1 in the resected tissues from IGFBP‐1‐low human HCC (HuH7) (C) and mouse HCC (Hep‐55.1C) xenograft mouse models (D), respectively (*P < 0.05, **P < 0.01 vs. VT, Student's t‐test or one‐way ANOVA). (E) Serum IGFBP‐1 levels in the subcutaneous IGFBP‐1‐low mouse HCC (Hep‐55.1C) xenograft mouse model (*P < 0.05 vs. VT, one‐way ANOVA). (F‐G) Representative micrographs of tumor tissues with H&E staining from IGFBP‐1‐low human HCC (HuH7) (F) and mouse HCC (Hep‐55.1C) xenograft mouse models (G) after indicated treatments. Arrows indicate IGFBP‐1‐positive cells, and arrowheads indicate CD31‐positive tumor microvessels. Quantification of IGFBP‐1‐positive cells (4‐7 random fields per section), CD31‐positive tumor microvessels (5‐10 random fields per section), CA9‐positive areas (4‐6 random fields per section), PCNA‐positive cells, and cleaved caspase‐3‐positive cells in the IGFBP‐1‐low human HCC (HuH7) (F) and mouse HCC (Hep‐55.1C) tumors (G) (*P < 0.05, **P < 0.01 vs. VT, Student's t‐test or one‐way ANOVA). Data are presented as means ± SEM.
Abbreviations: IGFBP‐1, insulin‐like growth factor‐binding protein‐1; HCC, hepatocellular carcinoma; VT, vehicle treatment; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; CA9, carbonic anhydrase 9; PCNA, proliferating cell nuclear antigen; Cle caspase‐3, cleaved caspase‐3; PA, proliferation/apoptosis.
3.5. Hypoxia promoted tumor IGFBP‐1 expression
To investigate the mechanism of IGFBP‐1 level elevation, we employed in vitro assessments. Although lenvatinib treatment downregulated the expression level of IGFBP‐1 in HUVECs or HCC (HepG2 and Hep‐55.1C) cells (Figure 4A, Supplementary Figure S3), hypoxia significantly induced IGFBP‐1 expression in all hepatoma cell lines (Figure 4B, Supplementary Figure S4). Promoted tumor IGFBP‐1 expression by hypoxia stimulation was suppressed by addition of YC‐1, a HIF‐1α and ‐2α inhibitor, in both mouse and human HCC cell lines (Figure 4C‐E). These data indicated that tumor IGFBP‐1 expression was upregulated by the activation of HIF‐1α and ‐2α, which was caused by TKI‐induced hypoxia in the TME.
FIGURE 4.
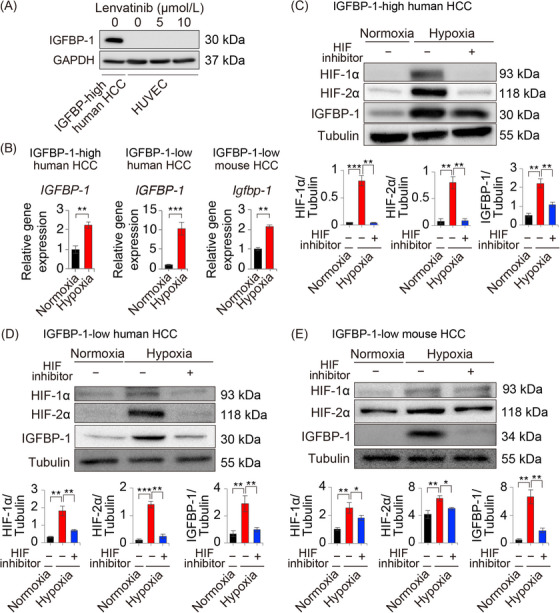
Tumor IGFBP‐1 level is increased by molecular targeted agent‐induced upregulation of HIF‐1α and ‐2α expression. (A) IGFBP‐1 expression in HUVECs under 24 h lenvatinib stimulation. (B) Quantification of the IGFBP‐1 gene expression under 24 h of normoxia (20% O2) or hypoxia (1% O2) in IGFBP‐1‐high human HCC (HepG2)/IGFBP‐1‐low human HCC (HuH7) and IGFBP‐1‐low mouse HCC (Hep‐55.1C) cells (n = 4, **P < 0.01, ***P < 0.001 vs. normoxia, Student's t‐test). (C‐E) Quantification of the IGFBP‐1 protein expression under 24 h of hypoxia condition with/without 5 μmol/L of HIF inhibitor stimulation in HepG2 (D), HuH7 (E), and Hep‐55.1C cells (F) (n = 4, *P < 0.05, **P < 0.01, and ***P < 0.001).
Abbreviations: IGFBP‐1, insulin‐like growth factor‐binding protein‐1; HUVEC, human umbilical vein endothelial cells; HIF, hypoxia‐inducible factor; HCC, hepatocellular carcinoma.
3.6. Functional analysis of IGFBP‐1 in antiangiogenic TKI resistance
Concerning functional insights, we evaluated whether tumor‐secreted IGFBP‐1 acted in an autocrine and/or paracrine manner to participate in tumor cell survival and proliferation. We performed proliferation assays using hrIGFBP‐1 stimulation in IGFBP‐1‐low human HCC (HuH7), IGFBP‐1‐high human HCC (HepG2) cells, and HUVECs under normoxic and hypoxic conditions. We found that hrIGFBP‐1 did not accelerate tumor cell proliferation, but significantly accelerated the proliferation of HUVECs (Figure 5A). Moreover, this effect was well‐preserved even under exposure to lenvatinib (Figure 5B). To further investigate the function of IGFBP‐1 in angiogenesis, we performed a tube formation assay with hrIGFBP‐1 stimulation. The number of tubes was significantly increased with hrIGFBP‐1 stimulation (Figure 5C). Interestingly, hrIGFBP‐1 promoted tube formation under exposure to lenvatinib or sorafenib (Figure 5D).
FIGURE 5.
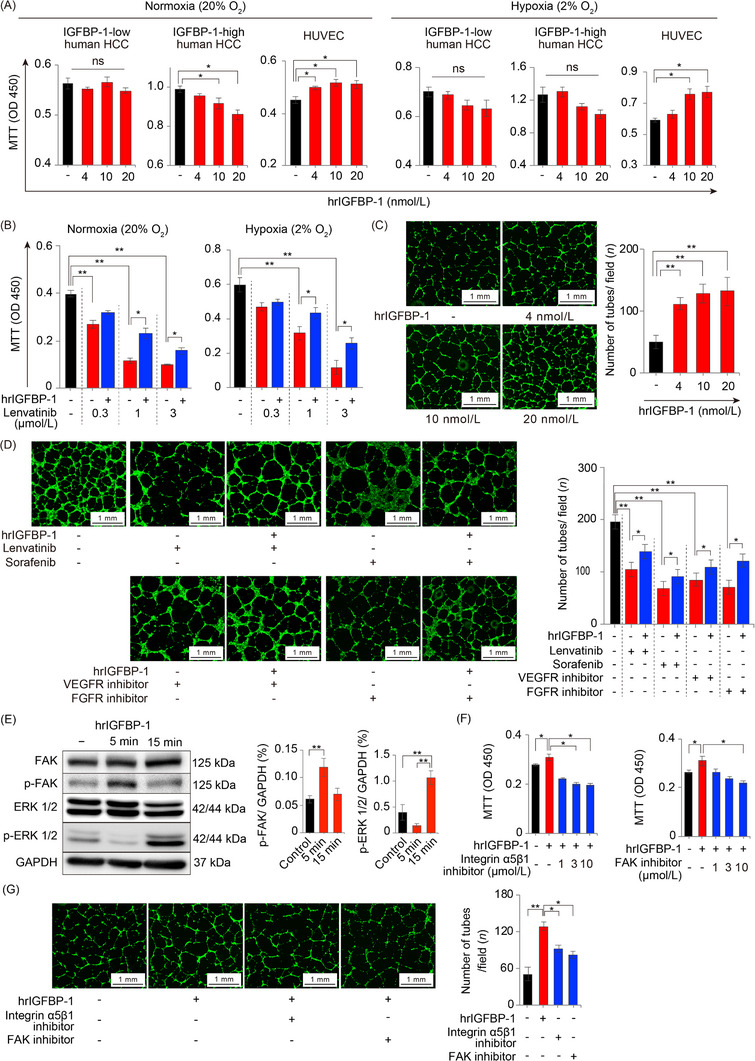
Functions of IGFBP‐1 on tumor cells and endothelial cells. (A) The effect of hrIGFBP‐1 on the proliferation of IGFBP‐1‐low human HCC (HuH7), IGFBP‐1‐high human HCC (HepG2) cells and HUVECs under normoxia and hypoxia for 48 h (n = 6 per group; *P < 0.05 vs. control, one‐way ANOVA). (B) The effect of hrIGFBP‐1 (10 nmol/L) on the proliferation of HUVECs in normoxia (20% O2; left) or hypoxia (2% O2; right) under lenvatinib exposure for 48 h (n = 6 per group; *P < 0.05, **P < 0.01, one‐way ANOVA). (C) Quantification of the tube formation numbers stimulated with hrIGFBP‐1 (n = 3 per group) (**P < 0.001 vs. control, one‐way ANOVA). (D) The HUVEC angiogenesis (left) and proliferation (right) under exposure to antiangiogenic TKIs and 10 nmol/L hrIGFBP‐1 (*P < 0.05, **P < 0.01, one‐way ANOVA). (E) p‐FAK level is upregulated with 5 min hrIGFBP‐1 (10 nmol/L) stimulation and p‐ERK level is upregulated by 15 min hrIGFBP‐1 (10 nmol/L) stimulation. (F) The accelerated proliferation of HUVECs by hrIGFBP‐1 (10 nmol/L) stimulation is reduced by the integrin α5β1 inhibitor or FAK inhibitor (n = 6 per group). (G) Representative micrographs of the cavities (left) and quantification of the number of tubes in the cavities (right). Angiogenesis promoted by 10 nmol/L hrIGFBP‐1 is significantly reduced by the integrin α5β1 inhibitor (3 μmol/L) or FAK inhibitor (10 μmol/L) (n = 3 per group; *P < 0.05, one‐way ANOVA). Data are presented as means ± SEM.
Abbreviations: IGFBP‐1, insulin‐like growth factor‐binding protein‐1; HCC, hepatocellular carcinoma; hrIGFBP‐1, human recombinant IGFBP‐1; HUVEC, human umbilical vein endothelial cells; TKI, tyrosine kinase inhibitor; DMSO, dimethyl sulfoxide; VEGF, vascular endothelial growth factor; FGF, fibroblast growth factor; FAK, focal adhesion kinase; ERK, extracellular signal‐regulated kinase; p‐FAK, phospho‐FAK; p‐ERK, phospho‐ERK; VT, vehicle treatment; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; ns, no significance.
To clarify which signaling pathway played a key role in promoting tube formation under antiangiogenic TKI exposure, we performed tube formation assays using fruquintinib (a VEGFR‐1, ‐2, and ‐3 inhibitor) or AZD4547 (an FGF receptor‐1, ‐2, and ‐3 inhibitor). hrIGFBP‐1 promoted endothelial cell proliferation and tube formation even under the blockade of VEGF or FGF signaling (Figure 5D). These results suggested that IGFBP‐1 was able to induce tumor angiogenesis in a VEGF‐ and FGF‐independent manner.
3.7. IGFBP‐1 promoted angiogenesis through the integrin α5β1/FAK/ERK pathway
To characterize the molecular signaling pathway in endothelial cells, we treated HUVECs with hrIGFBP‐1 and then performed RNA sequencing analysis. From the gene ontology enrichment analysis, 12 pathways were significantly enriched in hrIGFBP‐1‐stimulated HUVECs, such as “angiogenesis,” “positive regulation of fibroblast proliferation,” “positive regulation of endothelial cell proliferation,” and “blood vessel development,” compared to untreated HUVECs (Supplementary Table S6). Pathway analysis revealed that hrIGFBP‐1 increased cell signaling including Ras signaling, phosphatidylinositol‐3 kinase/protein kinase B, Janus kinase/signal transducer and activator of transcription, Wnt, and transforming growth factor‐β signaling (Supplementary Figure S5). A previous study reported that IGFBP‐1 promoted cell proliferation through the integrin α5β1/FAK/extracellular signal‐regulated kinase (ERK) cascade [24]. Addition of hrIGFBP‐1 markedly activated FAK phosphorylation after 5 min of stimulation, followed by a marked increase in ERK phosphorylation after 15 min of stimulation (Figure 5E). Strikingly, the effects of IGFBP‐1 on proliferation and angiogenesis were markedly inhibited by either integrin α5β1 or FAK inhibitors (Figure 5F‐G). Moreover, either integrin α5β1 or FAK inhibitors suppressed the proliferation and angiogenesis of HUVECs independent of lenvatinib treatment (Supplementary Figure S6). These findings suggested that IGFBP‐1 promoted HUVEC proliferation and tube formation through the integrin α5β1/FAK/ERK signaling cascade. Therefore, IGFBP‐1 independently increased tube formation under the blockade of VEGF and FGF signaling.
3.8. Genetic ablation of IGFBP‐1 sensitized HCC to lenvatinib treatment
To determine the role of IGFBP‐1 in antiangiogenic TKI resistance, we generated IGFBP‐1‐KD HCC in both IGFBP‐1‐low (HuH7) and IGFBP‐1‐high (HepG2) human HCC models (Supplementary Figure S7A‐C). IGFBP‐1‐low HCC (HuH7) xenografts became more sensitized to lenvatinib by IGFBP‐1 KD (Figure 6A). After lenvatinib treatment, tumor IGFBP‐1 expression was markedly lower in IGFBP‐1‐KD HepG2 xenografts than in IGFBP‐1‐WT HepG2 xenografts (Supplementary Figure S7D). Strikingly, lenvatinib‐resistant IGFBP‐1‐high HCC (HepG2) xenografts also became sensitized to lenvatinib by IGFBP‐1 KD (Figure 6B). Lenvatinib‐treated IGFBP‐1‐KD tumors showed a marked reduction in the microvessel density (CD31 staining) and were extremely hypoxic (CA9 staining). In addition, a significant decrease in tumor cell proliferation and an increase in cell apoptosis were observed in the lenvatinib‐treated IGFBP‐1 KD groups, leading to a decrease in the PA index (Figure 6C‐H). Thus, we uncovered that reducing IGFBP‐1 expression increased the sensitivity of HCC to lenvatinib by inhibiting its effect on angiogenesis.
FIGURE 6.
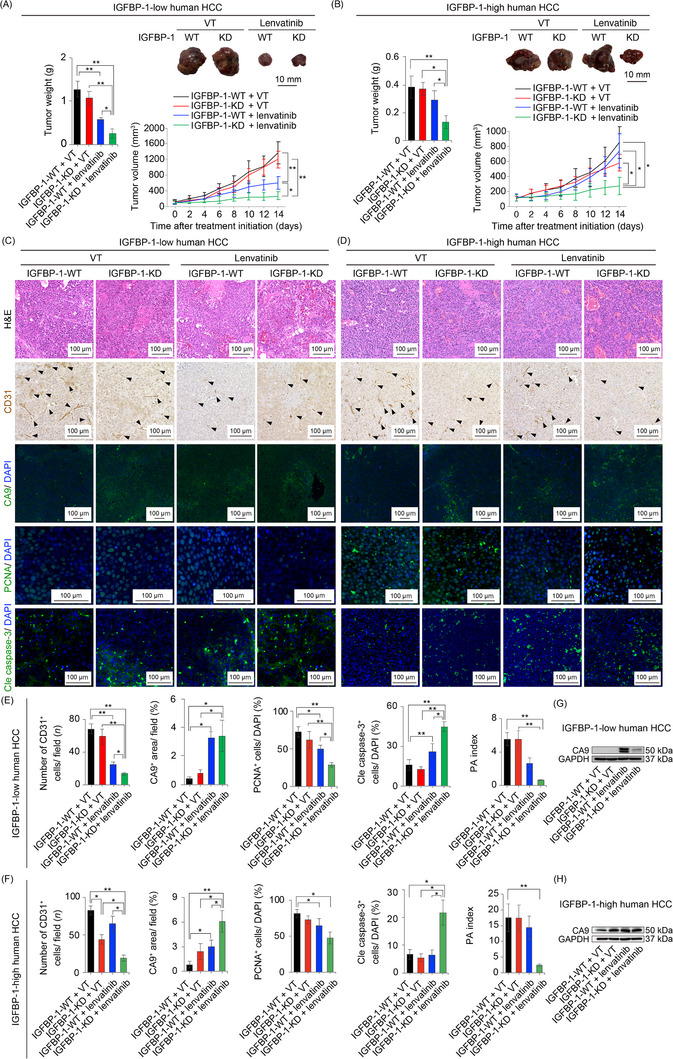
Genetic loss‐of‐function experiment of IGFBP‐1. (A‐B) Quantification of tumor weight (left panel), representative macrographs (right upper panel), and tumor volume measurements (right lower panel) in the IGFBP‐1‐low (HuH7; A) and subcutaneous IGFBP‐1‐high (HepG2; B) human HCC xenograft mouse model (5‐6 mice per group). (C‐D) Representative micrographs of H&E staining, CD31‐positive tumor microvessels, CA9‐positive areas, PCNA‐positive cells, and cleaved caspase‐3‐positive cells in the IGFBP‐1‐low (HuH7; C) and IGFBP‐1‐high (HepG2; D) human HCCs in each treatment group. Arrowheads indicate CD31‐positive tumor microvessels in the tumor microenvironment. (E, F) Quantification of CD31‐positive tumor microvessels (5‐10 random fields per section), CA9‐positive areas (4‐6 random fields per section), PCNA‐positive cells, and cleaved caspase‐3‐positive cells in the IGFBP‐1‐low (HuH7; E) and IGFBP‐1‐high (HepG2; F) tumors (*P < 0.05, **P < 0.01, one‐way ANOVA). (G‐H) Western blotting analysis of CA9 protein levels in the IGFBP‐1‐low (HuH7; G) and IGFBP‐1‐high tumors (HepG2; H). Data are presented as means ± SEM.
Abbreviations: IGFBP‐1, insulin‐like growth factor‐binding protein‐1; HCC, hepatocellular carcinoma; WT, wild type; KD, knockdown; CA9, carbonic anhydrase 9; PCNA, proliferating cell nuclear antigen; Cle caspase‐3, cleaved caspase‐3; PA, proliferation/apoptosis.
3.9. Pharmacological IGFBP‐1 blockade sensitized HCC to lenvatinib treatment
To determine whether the IGFBP‐1/integrin α5β1 signal is a potential therapeutic target in combination with antiangiogenic TKI treatment, we employed a pharmacological IGFBP‐1 signaling inactivation approach using an anti‐IGFBP‐1 neutralizing antibody or an integrin α5β1 inhibitor, a receptor for IGFBP‐1, in combination with lenvatinib in IGFBP‐1‐high HCC (HepG2) (Figure 7A). Notably, the addition of anti‐IGFBP‐1 neutralizing antibody or integrin α5β1 inhibitor contributed to a significant reduction in the growth of lenvatinib‐resistant HCC (Figure 7B). The combination of lenvatinib with the anti‐IGFBP‐1 neutralizing antibody or integrin α5β1 inhibitor significantly inhibited tumor angiogenesis compared with lenvatinib monotherapy, which promoted significant hypoxia in the tumor (Figure 7C‐D). Similar to the genetic loss‐of‐function experiments, a significant decrease in tumor cell proliferation and an increase in cell apoptosis were observed after the combination therapy, leading to a decrease in the PA index, compared with lenvatinib monotherapy (Figure 7E‐F). Although IGFBP‐1‐high HCC (HepG2) xenografts were originally resistant to lenvatinib treatment, we demonstrated that the addition of IGFBP‐1 or integrin α5β1 blockade reversed lenvatinib resistance. Considering that HepG2 is a hepatoblastoma cell line, we performed similar in vivo experiments using KYN‐2, which was an IGFBP‐1‐high human HCC cell line (Figure 2A), to match clinical practice. Remarkably, the combination treatment of anti‐IGFBP‐1 antibody or integrin α5β1 inhibitor with lenvatinib contributed to a significant tumor reduction in the subcutaneous KYN‐2 xenograft mouse model (Supplementary Figure S8A‐B). Again, a significant decrease in tumor cell proliferation and increase in cell apoptosis were observed in the combination treatment groups, leading to a decrease in the PA index, compared with the lenvatinib monotherapy group (Supplementary Figure S8C‐D). These data supported that the combination therapy of antiangiogenic TKIs and IGFBP‐1 blockade could be a promising treatment for antiangiogenic TKI‐resistant HCCs.
FIGURE 7.
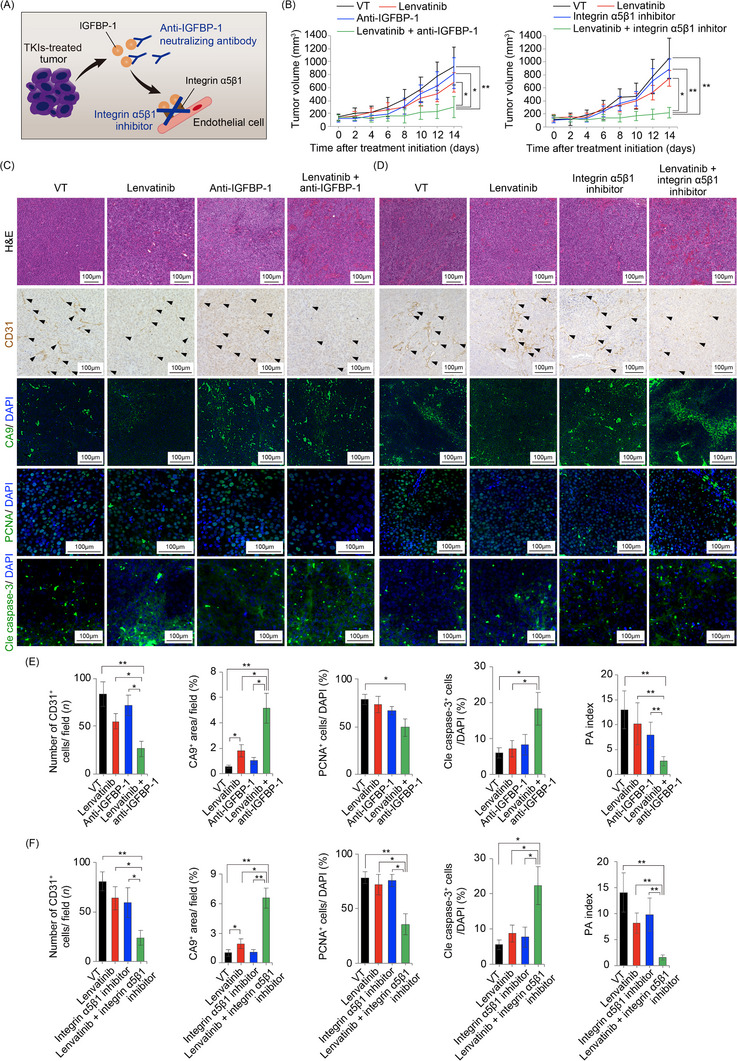
Pharmacological loss‐of‐function experiments using anti‐IGFBP‐1 neutralizing antibody and integrin α5β1 inhibitor. (A) A diagram showing potential pharmacological targets (anti‐IGFBP‐1 neutralizing antibodies or integrin α5β1 inhibitors) in antiangiogenic TKI treatment. (B) Tumor volume measurements of subcutaneous IGFBP‐1‐high human HCC (HepG2) xenograft mouse model at the indicated time points in each indicated treatment group (5‐6 mice per group). (C‐D) Representative micrographs of H&E staining, CD31‐positive tumor microvessels, CA9‐positive areas, PCNA‐positive cells, and cleaved caspase‐3‐positive cells. Arrowheads indicate CD31‐positive tumor microvessels in the tumor microenvironment. (E‐F) Quantification of CD31‐positive tumor vessels (5‐10 random fields per section), CA9‐positive areas (4‐6 random fields per section), PCNA‐positive cells, and cleaved caspase‐3‐positive cells (*P < 0.05, **P < 0.01, one‐way ANOVA). Data are presented as means ± SEM.
Abbreviations: IGFBP‐1, insulin‐like growth factor‐binding protein‐1; CA9, carbonic anhydrase 9; PCNA, proliferating cell nuclear antigen; Cle caspase‐3, cleaved caspase‐3; PA, proliferation/apoptosis.
3.10. Additional therapeutic effects of IGFBP‐1 signaling blockades on long‐term lenvatinib‐treated HCC xenograft mouse model
In the IGFBP‐1‐low human HCC (HuH7) xenograft mouse model experiment, two weeks of lenvatinib treatment elevated IGFBP‐1 expression in the xenografted HCC tissues; however, the tumors were still sensitive to lenvatinib (Figure 3). Therefore, to increase clinical relevance, additional experimentation with long‐term lenvatinib administration in the subcutaneous IGFBP‐1‐low HCC (HuH7) xenograft mouse model was conducted. When the tumor growth was accelerated, combination therapies were initiated (Supplementary Figure S9A‐B). Notably, a combination of anti‐IGFBP‐1 antibody or integrin α5β1 inhibitor with lenvatinib contributed to a significant reduction even in the lenvatinib‐resistant HCC (Supplementary Figure S9A‐B). Strikingly, continued lenvatinib treatment until HCC acquired resistance to lenvatinib led to an inverse in the IGFBP‐1 expression levels in the xenografted HCC (Supplementary Figure S9C).
4. DISCUSSION
In this study, we uncovered that serum IGFBP‐1 elevation could potentially be a biomarker of poor prognosis for patients with advanced HCC after antiangiogenic TKI treatment. in vitro and in vivo experiments demonstrated that antiangiogenic TKI‐induced hypoxia increased IGFBP‐1 expression through activation of HIF‐1α and ‐2α. Tumor‐derived IGFBP‐1 induced tumor angiogenesis by activating integrin α5β1/FAK/ERK signaling, even when VEGF and FGF signaling were inhibited. Furthermore, in vivo IGFBP‐1‐targeted genetic and pharmacological loss‐of‐function experiments revealed that IGFBP‐1 played an important role in antiangiogenic TKI resistance. These data bolster the application of the combination therapy using antiangiogenic TKIs and IGFBP‐1 blockade for effectively treating HCC.
Approval of antiangiogenic TKIs has contributed significantly to survival benefits in patients with advanced HCC [25], but the maximum benefits from these drugs have not been attained owing to the lack of validated therapeutic/prognostic biomarkers. Although several serum proteins such as VEGF, FGF, IGF‐1, and angiopoietin‐2 have been demonstrated as prognostic biomarkers for patients with HCC who received antiangiogenic TKI treatments, most were evaluated only at baseline [3, 26‐28]. Furthermore, the expression of these proteins was very low and hence not suitable for evaluation. Recently, a study has shown that a high serum IGFBP‐1 level was a potential diagnostic marker of high accuracy for upper gastrointestinal cancers [29]. Another study has shown that low IGFBP‐1 levels in HCC tissues were associated with poor prognosis in patients with HCC who had undergone hepatic resection [30]. It has been reported that early changes in FGF‐19 and angiopoietin‐2 levels could be predictive biomarkers of clinical response to lenvatinib in patients with HCC [28], and an increase in circulating FGF‐23 level at 15 days after the initiation of lenvatinib treatment could be a therapeutic biomarker for patients with thyroid cancer [31]. Contrastingly, our findings revealed that serum IGFBP‐1 levels at 4 weeks after the initiation of lenvatinib treatment were associated with poor prognosis. Additionally, the results from a clinical case (Figure 1G) in which IGFBP‐1 expression was measured over time showed that the level of IGFBP‐1 was further elevated even after the patients became resistant to lenvatinib. Furthermore, we have uncovered that antiangiogenic TKI‐induced IGFBP‐1 level elevation was involved in the acquisition of antiangiogenic TKI resistance and poor prognosis in patients with HCC.
IGFBP‐1 is known to confer spatial and temporal regulation of IGF‐1 bioavailability. It modulates migratory and/or proliferative responses in different types of cells in an IGF‐1‐independent manner, predominantly through interaction between the RGD (Arg‐Gly‐Asp) motif within its C‐terminal domain and integrin α5β1 [24]. Several studies have demonstrated that IGFBP‐1 initiated morphological changes in tumor cells, such as proliferation, epithelial‐mesenchymal transition, and acquisition of motility [32, 33, 34, 35]. However, hrIGFBP‐1 did not regulate proliferation in hepatoma cells in the present study. It has been reported that IGFBP‐1 in the liver tissues has a survival‐promoting function by antagonizing the proapoptotic actions of p53 in the mitochondria [36]. Therefore, these findings suggest that IGFBP‐1 secreted from tumor cells may not be involved in the survival or proliferation of hepatoma cells in an autocrine manner.
In the present study, the elevation in serum IGFBP‐1 levels at 4 weeks after initiating lenvatinib treatment reflected intratumoral ischemia, indicating that these patients responded well to lenvatinib; however, high serum IGFBP‐1 levels at that time were associated with poor prognosis. Concerning these contrasting findings, we thought that tumor cells acquired resistance to antiangiogenic TKIs through severe hypoxia in the TME and escaped this condition by elevation in the expression of IGFBP‐1 as the downstream protein of HIF‐1α and ‐2α signaling [24, 37, 38]. Consistent with the previous report that hypoxia stimulation increased IGFBP‐1 expression in HepG2 cells [39], we demonstrated that antiangiogenic TKIs indirectly increased IGFBP‐1 levels in HCC cell lines. The underlying mechanism of this effect was that antiangiogenic TKIs induced severe tumor hypoxia due to the inhibition of tumor angiogenesis‐activated HIF‐1α and ‐2α. We observed that IGFBP‐1 enhanced angiogenesis and HUVEC proliferation via the integrin α5β1/FAK/ERK signaling cascade, and these effects were well‐preserved even under antiangiogenic TKI exposure. These findings support that IGFBP‐1 contributed to antiangiogenic TKI resistance in tumors. in vitro analyses clarified that the hypoxic responsiveness of HIF‐1α‐ and ‐2α‐mediated IGFBP‐1 level elevation differed depending on the cell type. It is known that forkhead box O1 (FOXO1), one of the factors that activate the promoter of IGFBP‐1, encodes a transcription factor that regulates metabolic homeostasis in the presence of oxidative stress and that FOXO1 gene was altered in 1.11% [e.g., FOXO1 mutation (0.85%), FOXO1 loss (0.30%), FOXO1 amplification (0.17%), FOXO1‐FGFR1 fusion (0.29%), and FOXO1 fusion (0.11%)] of patients with malignant solid tumors [40, 41]. Thus, it is suggested that upstream genetic alterations regulating IGFBP‐1 expression may lead to differences in cytoplasmic changes in response to hypoxic stimuli. Therefore, this variation might reflect the differences in antiangiogenic TKI resistance in clinical cases. It is known that once IGFBP‐1 forms a complex with BAK, the formation of a proapoptotic p53/BAK complex and apoptosis induction are impaired [36]. Consistent with the study, in vitro experiments using endothelial cells and gene ontology enrichment analysis revealed that IGFBP‐1 enriched “negative regulation of apoptosis.” Thus, it is also possible that IGFBP‐1 may be involved in lenvatinib resistance by affecting not only angiogenesis but also antiapoptotic effect under antiangiogenic TKIs.
The combination of atezolizumab and bevacizumab has recently been approved as the first‐line HCC treatment showing superiority in OS and PFS over sorafenib treatment [42]. Although antiangiogenic TKIs have provided enormous survival benefits to patients with HCC [25, 43], devising optimal treatment strategies using systemic chemotherapy for HCC is becoming increasingly complex. Although various antiangiogenic TKIs have been approved for HCC treatment, the general therapeutic action of antiangiogenic TKIs involves inducing the TME into severe hypoxia via its antiangiogenic effects [44]. We discovered that IGFBP‐1 expression, upregulated by hypoxia in tumors, was involved in the acquisition of antiangiogenic TKI resistance, through which tumors exerted antiangiogenic TKI‐independent neoangiogenic effects. Thus, in antiangiogenic TKI‐centered systemic chemotherapy for HCC, antiangiogenic TKI‐induced elevation in serum IGFBP‐1 level might be useful as an early therapeutic biomarker for poor prognosis. Moreover, the present study proposed that combining antiangiogenic TKIs and an IGFBP‐1 inhibitor could be a promising treatment strategy for HCC because it might suppress the acquired resistance to antiangiogenic TKIs in tumors.
Nonetheless, this study has several limitations. It was a retrospective study with a limited number of patients with advanced HCC treated with lenvatinib. Regarding the exploration of target proteins, we used a limited number of protein assays (covering only 55 angiogenesis‐related proteins). For a more comprehensive and objective analysis, multiplex immunoassays, such as xMAP® (Luminex Corporation, Austin, TX, USA) or O‐link proximity extension assay (Olink Proteomics AB, Uppsala, Sweden), should be conducted in the future. Additionally, we only evaluated a few antiangiogenic TKIs approved for HCC treatment. Lastly, lenvatinib‐induced elevation in serum IGFBP‐1 level was evaluated at only one time point after treatment initiation. In the future, a prospective study with a large number of cases treated with several antiangiogenic TKIs and antibody preparations, including atezolizumab and bevacizumab, is warranted for evaluating IGFBP‐1 as a prognostic biomarker for HCC.
5. CONCLUSIONS
We showed that antiangiogenic TKI‐induced hypoxia increased the expression of IGFBP‐1, which was involved in the resistance to antiangiogenic TKI treatments in patients with advanced HCC. IGFBP‐1 could be a biomarker of poor prognosis in patients with advanced HCC after antiangiogenic TKI therapy. Furthermore, genetic or pharmacological suppression of IGFBP‐1 activity along with exposure to antiangiogenic TKIs exerted additional antitumor effects, indicating a promising treatment strategy for patients with advanced HCC.
DECLARATIONS
AUTHOR CONTRIBUTIONS
H.S., H.I., and T.S. (Takahiro Seki) designed and performed the experiments, analyzed the data, and drafted the manuscript. T.N. (Takashi Niizeki), M.N. (Masahito Nakano), S.S., T.S. (Tomotake Shirono), Y.N., N.K., M.S., and R.K. were responsible for the acquisition and interpretation of data. T.N. (Toru Nakamura), A.M., T.S. (Takahiko Sakaue), and T.T. (Toshimitsu Tanaka) were responsible for technical or material support. Y.I. performed Western blotting analysis and quantitative real‐time PCR analysis. K.M., M.N. (Masamichi Nakayama), and T.Y. participated in discussions. H.Y., Y.C., H.K., and T.T. (Takuji Torimura) were responsible for editing the manuscript and supervision.
CONFLICT OF INTEREST STATEMENT
The authors have declared that no conflict of interest exists.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This retrospective study was performed in accordance with the principles of the 1975 Declaration of Helsinki and approved by the Institutional Review Board at Kurume University (Ethical code: 19726). The tissue samples were obtained with written informed consent from each patient. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed, and all animal experiments were approved by the ethical committee of the Kurume University School of Medicine (Ethical code: 2020‐056).
CONSENT FOR PUBLICATION
Not applicable.
FINANCIAL SUPPORT STATEMENT
H.S. was partly supported by a grant from the Ishibashi Foundation for the Promotion of Science. H.I. was partly granted by the Takeda Science Foundation, the Shinnihon Foundation of Advanced Medical Treatment Research, the Kurume University Branding Project, the Yasuda Medical Foundation, and the JSPS KAKENHI grant.
Supporting information
SUPPORTING INFORMATION
ACKNOWLEDGEMENTS
The Authors appreciate the technical assistance from Cell Innovator Co., Ltd., Fukuoka, Japan.
Suzuki H, Iwamoto H, Seki T, Nakamura T, Masuda A, Sakaue T, et al. Tumor‐derived insulin‐like growth factor‐binding protein‐1 contributes to resistance of hepatocellular carcinoma to tyrosine kinase inhibitors. Cancer Communications. 2023;43:415–434. 10.1002/cac2.12411
DATA AVAILABILITY STATEMENT
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2021;71:209‐49. [DOI] [PubMed] [Google Scholar]
- 2. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 2012;21:309‐22. [DOI] [PubMed] [Google Scholar]
- 3. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008;359:378‐90. [DOI] [PubMed] [Google Scholar]
- 4. Kudo M, Finn RS, Qin S, Han KH, Ikeda K, Piscaglia F, et al. Lenvatinib versus sorafenib in first‐line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non‐inferiority trial. Lancet 2018;391:1163‐73. [DOI] [PubMed] [Google Scholar]
- 5. Iwamoto H, Suzuki H, Shimose S, Niizeki T, Nakano M, Shirono T, et al. Weekends‐Off Lenvatinib for Unresectable Hepatocellular Carcinoma Improves Therapeutic Response and Tolerability toward Adverse Events. Cancers (Basel) 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shimose S, Kawaguchi T, Tanaka M, Iwamoto H, Miyazaki K, Moriyama E, et al. Lenvatinib prolongs the progression‐free survival time of patients with intermediate‐stage hepatocellular carcinoma refractory to transarterial chemoembolization: A multicenter cohort study using data mining analysis. Oncol Lett 2020;20:2257‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nakano M, Kuromatsu R, Niizeki T, Okamura S, Iwamoto H, Shimose S, et al. Immunological inflammatory biomarkers as prognostic predictors for advanced hepatocellular carcinoma. ESMO Open 2021;6:100020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nakano M, Kuromatsu R, Niizeki T, Okamura S, Iwamoto H, Shimose S, et al. Primary Treatment with Molecular‐Targeted Agents for Hepatocellular Carcinoma: A Propensity Score‐matching Analysis. Hepatol Commun 2020;4:1218‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kudo M, Ikeda M, Takayama T, Numata K, Izumi N, Furuse J, et al. Safety and efficacy of sorafenib in Japanese patients with hepatocellular carcinoma in clinical practice: a subgroup analysis of GIDEON. J Gastroenterol 2016;51:1150‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang Y, Zhang Y, Iwamoto H, Hosaka K, Seki T, Andersson P, et al. Discontinuation of anti‐VEGF cancer therapy promotes metastasis through a liver revascularization mechanism. Nat Commun 2016;7:12680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rapisarda A, Melillo G. Role of the VEGF/VEGFR axis in cancer biology and therapy. Adv Cancer Res 2012;114:237‐67. [DOI] [PubMed] [Google Scholar]
- 12. Tovar V, Cornella H, Moeini A, Vidal S, Hoshida Y, Sia D, et al. Tumour initiating cells and IGF/FGF signalling contribute to sorafenib resistance in hepatocellular carcinoma. Gut 2017;66:530‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tang W, Chen Z, Zhang W, Cheng Y, Zhang B, Wu F, et al. The mechanisms of sorafenib resistance in hepatocellular carcinoma: theoretical basis and therapeutic aspects. Signal Transduct Target Ther 2020;5:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late‐stage pancreatic islet tumors. Cancer Cell 2005;8:299‐309. [DOI] [PubMed] [Google Scholar]
- 15. Iwamoto H, Abe M, Yang Y, Cui D, Seki T, Nakamura M, et al. Cancer Lipid Metabolism Confers Antiangiogenic Drug Resistance. Cell Metab 2018;28:104‐117 e105. [DOI] [PubMed] [Google Scholar]
- 16. Lencioni R, Llovet JM. Modified RECIST (mRECIST) assessment for hepatocellular carcinoma. Semin Liver Dis 2010;30:52‐60. [DOI] [PubMed] [Google Scholar]
- 17. Yano H, Maruiwa M, Murakami T, Fukuda K, Ito Y, Sugihara S, et al. A new human pleomorphic hepatocellular carcinoma cell line, KYN‐2. Acta Pathol Jpn 1988;38:953‐66. [DOI] [PubMed] [Google Scholar]
- 18. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods 2001;25:402‐8. [DOI] [PubMed] [Google Scholar]
- 19. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30:2114‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA‐Seq data with or without a reference genome. BMC Bioinformatics 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26:139‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, et al. TM4: a free, open‐source system for microarray data management and analysis. Biotechniques 2003;34:374‐8. [DOI] [PubMed] [Google Scholar]
- 23. Dennis G, Jr., Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 2003;4:P3. [PubMed] [Google Scholar]
- 24. Slater T, Haywood NJ, Matthews C, Cheema H, Wheatcroft SB. Insulin‐like growth factor binding proteins and angiogenesis: from cancer to cardiovascular disease. Cytokine Growth Factor Rev 2019;46:28‐35. [DOI] [PubMed] [Google Scholar]
- 25. Kobayashi K, Ogasawara S, Takahashi A, Seko Y, Unozawa H, Sato R, et al. Evolution of Survival Impact of Molecular Target Agents in Patients with Advanced Hepatocellular Carcinoma. Liver Cancer 2022;11:48‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Llovet JM, Pena CE, Lathia CD, Shan M, Meinhardt G, Bruix J, et al. Plasma biomarkers as predictors of outcome in patients with advanced hepatocellular carcinoma. Clin Cancer Res 2012;18:2290‐300. [DOI] [PubMed] [Google Scholar]
- 27. Shao YY, Hsu CH, Cheng AL. Predictive biomarkers of sorafenib efficacy in advanced hepatocellular carcinoma: Are we getting there? World J Gastroenterol 2015;21:10336‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chuma M, Uojima H, Numata K, Hidaka H, Toyoda H, Hiraoka A, et al. Early Changes in Circulating FGF19 and Ang‐2 Levels as Possible Predictive Biomarkers of Clinical Response to Lenvatinib Therapy in Hepatocellular Carcinoma. Cancers (Basel) 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu YW, Chen H, Hong CQ, Chu LY, Yang SH, Huang LS, et al. Serum IGFBP‐1 as a potential biomarker for diagnosis of early‐stage upper gastrointestinal tumour. EBioMedicine 2020;51:102566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dai B, Ruan B, Wu J, Wang J, Shang R, Sun W, et al. Insulin‐like growth factor binding protein‐1 inhibits cancer cell invasion and is associated with poor prognosis in hepatocellular carcinoma. Int J Clin Exp Pathol 2014;7:5645‐54. [PMC free article] [PubMed] [Google Scholar]
- 31. Tahara M, Schlumberger M, Elisei R, Habra MA, Kiyota N, Paschke R, et al. Exploratory analysis of biomarkers associated with clinical outcomes from the study of lenvatinib in differentiated cancer of the thyroid. Eur J Cancer 2017;75:213‐21. [DOI] [PubMed] [Google Scholar]
- 32. Kodama S, Yamazaki Y, Negishi M. Pregnane X Receptor Represses HNF4alpha Gene to Induce Insulin‐Like Growth Factor‐Binding Protein IGFBP1 that Alters Morphology of and Migrates HepG2 Cells. Mol Pharmacol 2015;88:746‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thomas D, Radhakrishnan P. Role of Tumor and Stroma‐Derived IGF/IGFBPs in Pancreatic Cancer. Cancers (Basel) 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang X, Yee D. Insulin‐like growth factor binding protein‐1 (IGFBP‐1) inhibits breast cancer cell motility. Cancer Res 2002;62:4369‐75. [PubMed] [Google Scholar]
- 35. Ammoun S, Schmid MC, Zhou L, Ristic N, Ercolano E, Hilton DA, et al. Insulin‐like growth factor‐binding protein‐1 (IGFBP‐1) regulates human schwannoma proliferation, adhesion and survival. Oncogene 2012;31:1710‐22. [DOI] [PubMed] [Google Scholar]
- 36. Leu JI, George DL. Hepatic IGFBP1 is a prosurvival factor that binds to BAK, protects the liver from apoptosis, and antagonizes the proapoptotic actions of p53 at mitochondria. Genes Dev 2007;21:3095‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ferry RJ, Jr ., Katz LE, Grimberg A, Cohen P, Weinzimer SA. Cellular actions of insulin‐like growth factor binding proteins. Horm Metab Res 1999;31:192‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hoefflin R, Harlander S, Schafer S, Metzger P, Kuo F, Schonenberger D, et al. HIF‐1alpha and HIF‐2alpha differently regulate tumour development and inflammation of clear cell renal cell carcinoma in mice. Nat Commun 2020;11:4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu Q, Xu Z, Mao S, Chen W, Zeng R, Zhou S, et al. Effect of hypoxia on hypoxia inducible factor‐1alpha, insulin‐like growth factor I and vascular endothelial growth factor expression in hepatocellular carcinoma HepG2 cells. Oncol Lett 2015;9:1142‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim JJ, Fazleabas AT. Uterine receptivity and implantation: the regulation and action of insulin‐like growth factor binding protein‐1 (IGFBP‐1), HOXA10 and forkhead transcription factor‐1 (FOXO‐1) in the baboon endometrium. Reprod Biol Endocrinol 2004;2:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Consortium APG. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov 2017;7:818‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N Engl J Med 2020;382:1894‐905. [DOI] [PubMed] [Google Scholar]
- 43. Iwamoto H, Shimose S, Noda Y, Shirono T, Niizeki T, Nakano M, et al. Initial Experience of Atezolizumab Plus Bevacizumab for Unresectable Hepatocellular Carcinoma in Real‐World Clinical Practice. Cancers (Basel) 2021;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Weinstein IB, Joe AK. Mechanisms of disease: Oncogene addiction–a rationale for molecular targeting in cancer therapy. Nat Clin Pract Oncol 2006;3:448‐57. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPORTING INFORMATION
Data Availability Statement
The data supporting the findings of this study are available from the corresponding author upon reasonable request.