

Abstract
Human enteric adenovirus species F (HAdV-F) is a leading cause of childhood diarrhoeal deaths. The genomic analysis would be key to understanding transmission dynamics, potential drivers of disease severity, and vaccine development. However, currently, there are limited HAdV-F genomic data globally. Here, we sequenced and analysed HAdV-F from stool samples collected in coastal Kenya between 2013 and 2022. The samples were collected at Kilifi County Hospital in coastal Kenya from children <13 years of age who reported a history of three or more loose stools in the previous 24 hours. The genomes were analysed together with the data from the rest of the world by phylogenetic analysis and mutational profiling. Types and lineages were assigned based on phylogenetic clustering consistent with the previously described criteria and nomenclature. Participant clinical and demographic data were linked to genotypic data. Of ninety-one cases identified using real-time Polymerase Chain Reaction, eighty-eight near-complete genomes were assembled, and these were classified into HAdV-F40 (n = 41) and HAdV-F41 (n = 47). These types co-circulated throughout the study period. Three and four distinct lineages were observed for HAdV-F40 (Lineages 1–3) and HAdV-F41 (Lineages 1, 2A, 3A, 3C, and 3D). Types F40 and F41 coinfections were observed in five samples and F41 and B7 in one sample. Two children with F40 and 41 coinfections were also infected with rotavirus and had moderate and severe diseases as defined using the Vesikari Scoring System, respectively. Intratypic recombination was found in four HAdV-F40 sequences occurring between Lineages 1 and 3. None of the HAdV-F41 cases had jaundice. This study provides evidence of extensive genetic diversity, coinfections, and recombination within HAdV-F40 in a rural coastal Kenya that will inform public health policy, vaccine development that includes the locally circulating lineages, and molecular diagnostic assay development. We recommend future comprehensive studies elucidating on HAdV-F genetic diversity and immunity for rational vaccine development.
Keywords: human adenovirus F, Kenya, recombination, F40/41
Background
Human enteric adenovirus species F (HAdV-F) is a leading cause of paediatric viral diarrhoea and deaths globally (Institute for Health Metrics and Evaluation 2021). In 2019, a total of 83,491 deaths from HAdV-F were recorded globally (95 per cent confidence interval (CI): 43,914–143,867) in children below 14 years of age (Institute for Health Metrics and Evaluation 2021). Clinical isolates of HAdV-F can be classified into two types (40 and 41) based on genetic differences (Kidd et al. 1984; Rafie et al. 2021). In Kenya, the positivity rate of HAdV-F40/41 in diarrhoeal stools from hospitalized under 13-year olds was estimated at 5.8 per cent (95 per cent CI: 3.2–9.6) and 7.3 per cent (95 per cent CI: 5.2–10.1) pre- and post-rotavirus vaccine introduction, respectively (Agoti et al. 2022). Cases were observed in all years studied, with peaks in dry months (Lambisia et al. 2020).
HAdV-F40/41 are double-stranded deoxyribonucleic acid (DNA) viruses with short and long fibre proteins distinct from other adenoviruses (A-G). The genomes of HAdV-F40 and HAdV-41 are approximately 35,000 base pairs (Hung-Yueh et al. 1994). Previously, there have been studies of HAdV-F in Ethiopia, Albania, China, India, and Brazil following which the researchers have reported a higher prevalence in HAdV-F41 cases compared to HAdV-F40 (La Rosa et al. 2015; Gelaw, Pietsch, and Liebert 2019; Tahmasebi et al. 2020; Chandra et al. 2021; Tang et al. 2022). Coinfections between F40 and F41 and F41 and C5 have been reported in Brazil and Ethiopia (Gelaw, Pietsch, and Liebert 2019; Tahmasebi et al. 2020). However, there are limited data on the clinical implications among individuals with HAdV-F coinfections.
The emergence of new adenovirus strains due to recombination has previously been described in adenovirus species B, C, and D (Lukashev et al. 2008; Singh et al. 2013; Dehghan et al. 2019; Yang et al. 2019). Intertypic recombination has not been reported in HAdV-F types, but intratypic recombination in HAdV-F41 Lineage 3B sequences has been shown to occur around the short fibre region (Götting et al. 2022). Since early 2022, HAdV-F41 has been associated with severe paediatric hepatitis, either due to triggering a dysregulated immune response that led to liver injury or through an unknown coinfection-mediated mechanism (Gutierrez Sanchez et al. 2022; World Health Organization—WHO 2022).
Understanding the genomic diversity among HAdV-F types is key to the optimization of molecular diagnostic assays, vaccine development, tracking global spread, and linking viral variation with disease severity. However, despite HAdV-F being among the top viral causes of diarrhoea, its genomic diversity is poorly understood due to the limited genomic data globally (∼120 as of September 2022). Notably, in Africa, there are only ten type 40 and 41 complete genomes, all collected from South Africa.
Here, we aimed to investigate the molecular epidemiology and diversity of HAdV-F40/41 at the coastal Kenyan utilizing ninety-one HAdV-F-positive samples collected between January 2013 and May 2022 from children admitted to the Kilifi County Hospital with diarrhoea.
Methods
Study site and population
The samples were collected as part of a prospective hospital-based rotavirus surveillance study at Kilifi County Hospital paediatric ward in Kilifi, Kenya (Agoti et al. 2022). The target population was children below 13 years who presented with three or more loose stools in a 24-hour period. The samples were collected at the hospital, then transported, and stored at KEMRI-Wellcome Trust Research Programme at −80°C.
Ethical consideration
An informed written consent was obtained from each child’s parent/guardian before sample collection. The study protocol was approved by the Scientific and Ethics Review Unit at Kenya Medical Research Institute, Kenya (SERU#CGMRC/113/3624).
Laboratory methods
Extraction and screening
A total of ninety-one real-time positive stool samples were retrieved from a −80°C storage. These included positives in samples collected between January and December 2013 and January 2016 and May 2022. The positives had been identified either by conventional real-time reverse transcription polymerase chain reaction (RT-PCR) approach or by custom TaqMan Array cards as previously described (Lambisia et al. 2020; Agoti et al. 2022). Briefly, the samples were extracted from 0.2 g of the specimen (or 200 µl if liquid) using the QIAamp Fast DNA Stool Mini kit (Qiagen, Manchester, UK) as per the manufacturer’s instructions and screened using the TaqMan Fast Virus 1-Step Master Mix and adenovirus 40/41 specific primers (forward primer: 5’-CACTTAATGCTGACACGGGC-3’; probe: ‘FAM-TGCACCTCTTGGACTAGT-MGBNFQ’; and Reverse primer: 5’-ACTGGATAGAGCTAGCGGGC-3’). The thermocycling conditions were 95°C for 20 seconds and 35 cycles of 94°C for 15 seconds and 60°C for 30 seconds (Agoti et al. 2022). No cut-off in the cycle threshold value was used during the sample selection. In the HAdV-F positives, Group A rotavirus was screened using a conventional real-time RT-PCR approach or by custom TaqMan Array cards as previously described (Lambisia et al. 2020; Agoti et al. 2022).
Whole-genome sequencing
DNA Amplification
Total nucleic acids were amplified using the Q5® Hot Start High-Fidelity 2X Master Mix (NEB) kit. The master mix was prepared as follows: Q5® Hot Start High-Fidelity 2X Master Mix (6.25 μl), H2O (3 μl), Primer pool (1/2/3/4) (2 μl), and total nucleic acids (1.25 μl). The primers were designed by the Quick group using the ‘Jackhammer’ approach and divided the adenovirus genome into ninety-two amplicons of 1,200 base pairs (bp) amplified in four pools (https://github.com/quick-lab/HAdV/blob/main/HAdV-F41/v1.0/HAdV-F41_2000jh.primer.bed) (Mailis et al. 2022). The reaction was then incubated on a thermocycler using the following conditions: 98°C for 30 seconds followed by 35 cycles of 98°C for 15 seconds and 65°C for 5 minutes.
Library preparation and sequencing
Oxford Nanopore Technologies
Library preparation was performed using the SQK-LSK109 ligation kit with EXP-NBD196 barcoding kit as previously described (https://www.protocols.io/view/ncov-2019-sequencing-protocol-v3-locost-bp2l6n26rgqe/v3) (Quick 2020). Briefly, the amplicons were end-repaired, barcoded using EXP-NBD196 native barcoding expansion kit, and pooled into one tube, and adapters ligated to the library and the final library sequenced using the FLOW-MIN106D R9.4.1 flow cell on the GridION platform (Oxford Nanopore Technologies [ONT]).
Illumina Miseq
Samples that were classified as F41 based on the ONT data (described in the next section) were resequenced on the Illumina Miseq platform. An aliquot of the amplicons obtained in the DNA amplification section was used to generate libraries using an Illumina library preparation kit as recommended by the manufacturer. Briefly, the amplicons were tagmented, indexed, and amplified. The libraries were then normalized, pooled, and sequenced as paired-end reads (2*250 bp).
Consensus genome generation and typing
Consensus HAdV-F40/41 genomes using ONT sequence data were generated using a modified primer scheme with an adenovirus reference (NC_001454) in the ARTIC field bioinformatics pipeline (https://github.com/artic-network/fieldbioinformatics) with a minimum read depth of 20x. The fast5 reads were basecalled and demultiplexed as the run progressed on the GridION machine with Guppy v.6.1.5. The reads in FASTQ format were filtered based on the length (1,200 bp) and then mapped to the NC_001454 reference genome using minimap2 v.2.2.4. Variant calling and pre-consensus genome generation were performed using bcftools v.1.10.2, and then, polishing and final consensus generation were performed using medaka v.1.0.3 as previously described (Mailis et al. 2022).
MetaSPAdes v3.13.2 was used to de novo assemble the Illumina reads (https://github.com/ablab/spades). The reads were trimmed using QUASR v.7.03 to remove low-quality reads, adapters, and primer sequences. The reads were then assembled using default MetaSPAdes parameters. The genomes generated from the scaffolds were then used as references, and the raw reads were mapped against them to check the integrity of the genomes and generate final consensus sequences.
BLASTN was used to identify the closest match for each genome in the standard nucleotide nr/nt database on National Center for Biotechnology Information (https://blast.ncbi.nlm.nih.gov/). All the genomes were checked for the presence of all expected HAdV-F coding sequence and annotated using Geneious Prime® 2022.2.1.
Phylogenetic analysis
Only HAdV sequences with a genome coverage of ≥80 per cent were utilized for phylogenetic analysis. There was a significant correlation between genome coverage and cycle threshold value for HAdV-F40, unlike for HAdV-F41 (Supplementary Fig. S1). The sequences were compared against 111 F41 and 5 F40 genomes on GenBank as of 20 September 2022 with ≥80 per cent genome coverage (Supplementary Table S1). Multiple sequence alignments were generated using mafft v7.487 (https://mafft.cbrc.jp/alignment/software/). The alignments were used to generate maximum likelihood (ML) phylogenies with 1,000 bootstraps on IQTREE v2.1.3 (http://www.iqtree.org/) using the GTR model. The ML phylogenies were visualized using the package ‘ggtree’ v2.4.2 (https://github.com/YuLab-SMU/ggtree) in R (https://www.r-project.org/). F40/41 types were assigned based on the closest hit on BLASTN and the clustering of the genome on the ML tree. F41 lineages were assigned based on previously established clusters and new lineages proposed for the newly observed clusters. The F40 lineages were defined based on the clustering on the ML trees and the single nucleotide polymorphisms observed in the alignments.
Recombination analysis
Recombination was checked using the RDP4 software and included all the detection methods, i.e. RDP, GENECONV, Bootscan, Maxchi, Chimaera, SiSscan, PhylPro, LARD, and 3seq (Martin et al. 2015). Simplot++ (https://github.com/Stephane-S/Simplot_PlusPlus) was used to show similarity plots using a window and step size of 500 bp and 100 bp, respectively (Samson, Lord, and Makarenkov 2022). Snipit was used to visualize the mutations relative to a reference sequence (https://github.com/aineniamh/snipit).
Statistical analysis
All statistical analyses were performed using R version 4.1.1 (R Core Team 2019). Comparisons among groups were made using χ2 statistics, and a P value of <0.05 was considered statistically significant. Vesikari Clinical Severity Scoring System Manual was used to assess the disease severity by taking into account parameters including duration and episodes of diarrhoea and vomiting, dehydration, fever, and treatment status (Lewis 2011). The disease severity categories were mild, moderate, and severe for scores of <7, 7–10, and ≥11, respectively (Lewis 2011).
Results
Diarrhoeal cases and HAdV-F types in Kenya
A total of 3,741 diarrhoeal cases were documented across the years studied, with peaks observed between July and September (Fig. 1A). Out of 1,667 screened diarrhoeal cases, 91 (5.5 per cent) were positive for HAdV-F. Healthcare workers’ strikes interrupted sample collection in 2013, 2016, and 2017, and the coronavirus disease 2019 (COVID-19) pandemic affected the sample collection in 2020 and early 2021 (Khagayi et al. 2020). Seventy-eight newly generated HAdV-F genomes were classified into either single type 40 (n = 36, 43 per cent) or 41 (n = 42, 51 per cent) samples using BLASTN and phylogenetic clustering on trees (Fig. 1B). Five samples had a coinfection of F40 and 41 types, and one sample had a coinfection of F41 and B7 types. The coinfections were investigated and confirmed by de novo assembly and investigation of reads from both ONT and Illumina platforms. Among the five participants with the F40 and 41 coinfections, three had the severe diarrhoeal disease (Table 1). Furthermore, two participants with F40 and 41 coinfections were also positive for rotavirus and had moderate and severe diarrhoeal diseases (Table 1).
Figure 1.
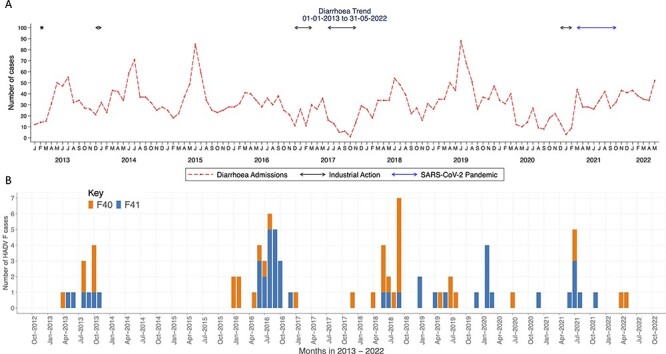
The temporal distribution of diarrhoeal cases and HAdV-F40/41 cases in children under the age of 13 years in Kilifi, Kenya. (A) A line graph showing the monthly number of diarrhoeal cases in Kilifi, Kenya, from January 2013 to May 2022. The y-axis represents the number of diarrhoeal cases, and the x-axis represents months. (B) The monthly human adenovirus F40 and F41 cases observed in Kilifi, Kenya, from January to December 2013 and January 2016 to May 2022. The y-axis shows the absolute number of HAdV-F40 and HAdV-F41 cases and the x-axis shows the time in months.
Table 1.
The demographic characteristics of human F40/41 in children under the age of 13 years in Kilifi, Kenya.
| F40 (n = 36) | F41 (n = 42) | Coinfection (n = 5) | Total (n = 83) | P value | |
|---|---|---|---|---|---|
| Gender | – | – | – | – | 0.05 |
| Female | 21 (58%) | 13 (31%) | 2 (40%) | 36 (43%) | |
| Male | 15 (42%) | 29 (69%) | 3 (60%) | 47 (57%) | |
| Age (months) | – | – | – | – | – |
| Median interquartile range (IQR) | 13.2 (8.2–21.6) | 15.2 (9.0–19.6) | 17.8 (16.3–21.6) | 15.2 (8.8–21.2) | – |
| Age strata (months) | – | – | – | – | 0.2 |
| <12 | 16 (45%) | 13 (31%) | 0 (0%) | 29 (35%) | |
| 12–23 | 14 (39%) | 21 (50%) | 4 (80%) | 39 (47%) | |
| 24–59 | 3 (8%) | 7 (17%) | 1 (20%) | 11 (13%) | |
| >60 | 3 (8%) | 1 (2%) | 0 (0%) | 4 (5%) | |
| Disease severity | – | – | – | – | 0.8 |
| Moderate | 17 (47%) | 22 (52%) | 2 (40%) | 41 (49%) | |
| Severe | 19 (53%) | 20 (48%) | 3 (60%) | 42 (51%) | |
| Outcome | – | – | – | – | 0.4 |
| Alive | 30 (83%) | 38 (90%) | 5 (100%) | 73 (88%) | – |
| Dead | 6 (17%) | 4 (10%) | 0 (0%) | 10 (12%) | – |
| Rotavirus coinfection | – | – | – | – | 0.06 |
| Positive | 2 (6%) | 6 (14%) | 2 (40%) | 10 (12%) | – |
| Negative | 34 (94%) | 34 (81%) | 3 (60%) | 71 (86%) | – |
| No data | 0 (0%) | 2 (5%) | 0 (0%) | 2 (2%) |
There were no significant differences in gender, age strata, outcome, and disease severity between the cases infected with either HAdV-F40 or HAdV-41 (P > 0.05) (Table 1). HAdV-F40/41 and rotavirus coinfections were detected in ten samples (12 per cent). Of these, six were coinfections with type F41(n = 6). One child with HAdV-F41 and rotavirus coinfection had severe dehydration and was unconscious at the time of admission and eventually died; the other children were treated and discharged. A total of ten HAdV-F cases succumbed due to either gastroenteritis and sepsis (n = 3) or gastroenteritis and a respiratory illness (n = 3) or gastroenteritis only (n = 3) or gastroenteritis and human immunodeficiency virus immunosuppression (n = 1). None of the children had jaundice as one of their illness symptoms at the time of admission.
Diversity of HAdV-F in coastal Kenya
To determine the diversity of HAdV-F40/41 in coastal Kenya, we combined the coastal sequences with a global dataset (n = 118) to give context to the newly generated genomes. We observed the clustering patterns of the coastal Kenya sequences on the global ML phylogenetic trees and identified lineages within HAdV-F40/41. We also analysed the mutation profiles in each protein relative to a reference genome to identify synonymous and non-synonymous mutations within and between the lineages.
HAdV-F40
Five major clusters were observed from the global phylogenetic tree (Fig. 2). The synonymous and non-synonymous substitutions relative to the HAdV-F reference (NC_001454.1) that were characteristics for each cluster are summarized in Supplementary Table S2 and Supplementary Fig. S2. Each of these clusters was assigned as a lineage. The Kenyan sequences clustered with Lineages 1, 2, and 3, with one global sequence observed in Lineage 1 from Bangladesh (Fig. 2A). Lineages 4 and 5 were purely non-Kenyan sequences with Lineage 4 comprising genomes from South Africa. Varied genetic divergence was observed between Lineages 1–4 (Supplementary Fig. S4). The per cent nucleotide similarity was 99.52 per cent (nucleotide differences (nd) = 163), 99.95 per cent (nd = 14), 99.76 per cent (nd = 80), and 99.89 per cent (nd = 37) for Lineages 1–4, respectively, and 99.06 per cent among all the five lineages.
Figure 2.
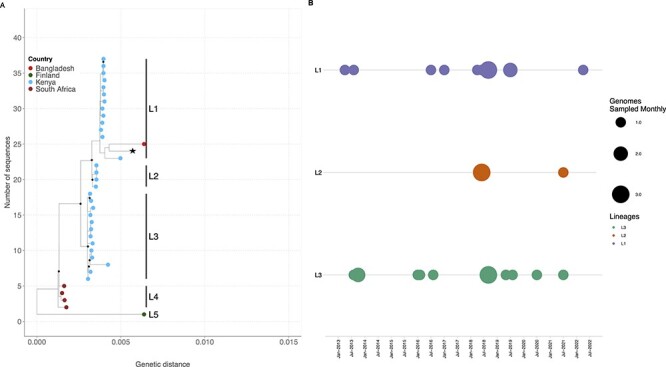
The ML phylogenetic tree showing the genetic relatedness of HAdV-F40 genotypes when using the whole-genome sequence. The tips are coloured by country. The nodes with a bootstrap support of ≥80 are shown by a black diamond. (A) The prototype sequence (L19443.1) is shown by an asterisk next to the tip label. (B) The temporal distribution of HAdV-F40 lineages in sequenced samples from January to December 2013 and January 2016 to May 2022.
No amino acid differences were observed in the hexon gene of the Kenyan sequences among Lineages 1–3 (Supplementary Table S2). However, within the long fibre protein, Lineage 2 sequences had an additional S81T amino acid mutation that was not observed in Lineages 1 and 3, while Lineage 3 lacked the V120I mutation observed in Lineages 1 and 2. Within Lineage 1, sequence MN_968817.1 (Dhaka, BGD) and OP581380.1 (Kenya) contained additional mutations compared to the other sequences, lacked synonymous substitutions observed within the short and long fibre proteins and had a higher genetic distance compared to other Lineage 1 sequences (Supplementary Fig. S2). The mutation profile for the five lineages showed that Lineage 2 contained polymorphic sites that contained either Lineage 1 or 3 mutation profiles, suggesting intertypic recombination (Supplementary Fig. S2). Lineages 1 and 3 were in circulation throughout the study period with Lineage 2 observed in 2018 and 2021 (Fig. 2B).
HAdV-F41
Five lineages, 1, 2A, 2B, 3A, and 3B, had been previously defined (Götting et al. 2022). The Kilifi HAdV-F41 sequences clustered with global sequences characterized as Lineage 1 (n = 1), Lineage 2A (n = 14), and Lineage 3A (n = 4). Eleven sequences clustered within Lineage 3 but neither with previously defined Lineage 3A or 3B. We propose these sequences be assigned new HAdV-F41 Lineages 3C and 3D (Fig. 3A). Non-synonymous and synonymous substitutions relative to sequence KF303070.1 (NY/2010, within Lineage 2A) characteristic for each lineage were identified in multiple proteins (Supplementary Fig. S3 and Supplementary Table S3). Many of the amino acid differences were observed in the hexon, 100K, E3-19.4K, E3-31.6K, E3-14.5K, short and long fibre, and E4-orf6 proteins (Supplementary Table S3). The varied genetic divergence among the lineages has been shown in Supplementary Fig. S5.
Figure 3.
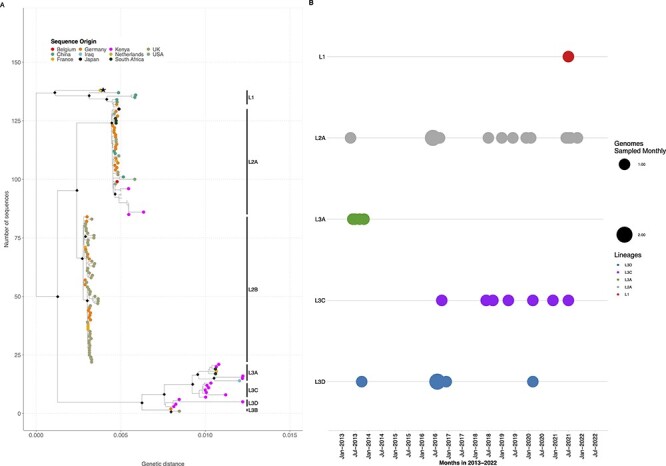
The ML phylogenetic trees showing the diversity within the HAdV-F41 genotypes when using the whole-genome sequence. The tips are coloured by country. The nodes with a bootstrap support of ≥80 are shown by a black diamond. (A) The prototype sequence (DQ315364.2) is shown by an asterisk next to the tip label. (B) The temporal distribution of HAdV-F41 lineages in sequenced samples from January to December 2013 and January 2016 to May 2022.
In the Kenyan sequences, Lineage 1 was only observed in 2021, Lineage 3A in 2013, Lineage 3D in 2013 and 2016, and Lineage 3C from 2016 to 2021 (Fig. 3B). Notably, Lineage 2A was persistent across the study period. The per cent nucleotide identity for the newly proposed lineages is 99.87 per cent (nd = 44) and 99.85 per cent (nd = 50) for Lineages 3C and 3D, respectively. The per cent nucleotide identity for the other lineages, 1, 2A, 2B, and 3A, has been previously described and ranges from 99.6 per cent to 99.9 per cent (Götting et al. 2022). Within-lineage diversity for each of the lineages is shown in Supplementary Fig. S6.
The diversity observed within HAdV-F41 was confirmed by sequencing the samples on both the ONT and Illumina Miseq platforms. The Illumina Miseq platform yielded a higher number of raw reads per sample and subsequently better genome coverage compared to ONT (Supplementary Fig. S7). Amplicon dropout was observed in the E1B 55K, E2B, hexon, and 100k regions, especially in the ONT data due to low read depth (<20). However, some HAdV-F41 genomes from the Illumina Miseq reads had enough depth to avoid the amplicon dropout and did not show any difference during clustering and lineage assignment in the phylogenetic tree when compared to their respective genomes with the amplicon dropout from ONT reads.
Recombination analysis
To investigate the possibility of recombination among the HAdV-F40 Lineage 2 genomes based on the observed mutation profiles and clustering on the phylogenetic tree, we ran a recombination analysis using the RDP4 software. Two sequences were flagged as recombinants between HAdV-F40 Lineage 1 (major parent) and Lineage 3 (minor parent). The recombination was significant by RDP (P = 0.00055), GENECONV (P = 0.0179), Maxchi (P = 0.006), Chimaera (P = 0.003), SiSscan (P = 0.039), and 3seq (P = 0.000013). The mutation profile and similarity plot of the recombinant sequences show the switching of polymorphisms between Lineage 1 and 3 sequences (Supplementary Figs S2 and S6).
Discussion
We report on the genomic epidemiology of HAdV-F40/41 types and recombination in viruses detected in hospitalized children under 13 years of age on the Kenya coast. The observed genomic diversity has not been observed elsewhere globally. HAdV-F40/41 types co-circulated similar to reports from other countries (La Rosa et al. 2015; Gelaw, Pietsch, and Liebert 2019; Tahmasebi et al. 2020; Tang et al. 2022). In previous studies, the proportion of F40 in sequenced HAdV-F-positive samples was lower compared to F41 (3–36 per cent vs. 11–97 per cent). This may be partly due to the higher genetic diversity in the F41 type that is hypothesized to enable it to outcompete F40 potentially via immune escape (La Rosa et al. 2015; Gelaw, Pietsch, and Liebert 2019; Tahmasebi et al. 2020; Chandra et al. 2021; Tang et al. 2022). A similar finding of lower F40 diversity was observed in our study. However, the proportion of HAdV-F-positive samples with a single type F40 infection was 41 per cent, which is higher than the reports from other regions.
A coinfection between HAdV-40 and HAdV-41 has only previously been reported in Brazil (one sample) (Tahmasebi et al. 2020). We observed coinfections between F40 and 41 types in five individuals who had moderate-to-severe disease (none were fatal), suggesting that coinfections do not necessarily lead to worse outcomes. Notably, reference-based genome assembly from reads in samples with coinfections generated genomes that have an anomalous phylogenetic placement, which we deconvoluted by de novo assembly (result not shown). Therefore, scientists and clinicians working in high adenovirus transmission settings should have a high suspicion of coinfection when they see strains that appear to be F40 x 41 recombinants and consider de novo assembly pipelines to pick up these coinfections.
This study contributes to the number of both HAdV-F40 and HAdV-F41 genomes available on the public sequence database (GenBank) fourfold. Previously, only six near-complete genomes were available for HAdV-F40, and our study contributes forty-one additional genomes. The Kenyan HAdV-F40 sequences clustered into three major clusters that we defined as Lineages 1–3 and only one global sequence (MN968817.1-Dhaka) clustered within Lineage 1. The other two lineages are purely Kenyan lineages not previously observed elsewhere (Götting et al. 2022). Over the 10-year period studied here, the co-circulation of F40 Lineages 1 and 3 shows the sustained diversity of HAdV-F strains in coastal Kenya. The Kenyan HAdV-F41 genomes clustered within three previously defined lineages, 1, 2A, and 3A, with none of the sequences clustering within Lineages 2B and 3B, which are mainly from Europe (Götting et al. 2022). Two additional clusters composed exclusively of Kenyan sequences were observed within Lineage 3, and these were not observed elsewhere globally. We have designated these two clusters as Lineages 3C and 3D. Interestingly, we observe lineage replacement within the Lineage 3 subtypes over time. Co-circulation and replacement of multiple HAdV-F41 strains in different genome type clusters based on the fibre gene have also been reported in India between 2013 and 2020 (Banerjee et al. 2017; Chandra et al. 2021).
HAdV-F40 Lineage 2 sequences were recombinants between Lineage 1 and Lineage 3 sequences, and their mutation profile showed shared polymorphic sites with either of the parent lineages. The co-circulation of Lineages 1 and 3 HAdV-F40 viruses could have led to the emergence of F40 recombinants. Recombination in HAdV-F40 suggests that fragment sequencing is not the best method for genotyping and lineage assignment as the recombination events may miscue the visualization of the evolutionary relationships depending on the fragment sequenced.
None of the children in this study had jaundice at the time of admission between 2013 and 2022. Previous studies early in 2022 have reported human adenovirus infections among children with acute hepatitis, and further studies in coastal Kenya should investigate such cases in the future (Gutierrez Sanchez et al. 2022).
The study had two limitations. First, the genomes had amplicon dropouts, meaning portions of the genome were missed and redesigning the primers will help recover complete genomes. Finally, samples from 2014 and 2015 were not screened and tested; hence, some key information on the diversity of HAdV-F may have been missed from that period.
In conclusion, this study shows that there is a high genetic diversity of both HAdV-F40 and HAdV-F41 in co-circulation in Kenya, coupled with intratypic recombination events that may have led to the emergence of new lineages. The observed genetic diversity and recombination events necessitate the need for continuous genomic surveillance to track HAdV-F strains, to inform policy and future HAdV-F studies, and to develop vaccines that match what is locally circulating. In addition, the study highlights the importance of storing samples and using infrastructure from other projects, as this helps fill the knowledge gaps in the genetic diversity of pathogens through whole-genome sequencing across the African continent.
Supplementary Material
Acknowledgements
We acknowledge all participants and their parents/guardians for their contribution to the study samples and members of the Virus Epidemiology and Control Research Group (http://virec-group.org/) for their input in the study. For the purpose of Open Access, the authors have applied a CC-BY public copyright licence to any author-accepted manuscript version arising from this submission.
Contributor Information
Arnold W Lambisia, Kenya Medical Research Institute-Wellcome Trust Research Programme, PO Box 230-80108, Kilifi, Kenya.
Timothy O Makori, Kenya Medical Research Institute-Wellcome Trust Research Programme, PO Box 230-80108, Kilifi, Kenya.
Martin Mutunga, Kenya Medical Research Institute-Wellcome Trust Research Programme, PO Box 230-80108, Kilifi, Kenya.
Robinson Cheruiyot, Kenya Medical Research Institute-Wellcome Trust Research Programme, PO Box 230-80108, Kilifi, Kenya.
Nickson Murunga, Kenya Medical Research Institute-Wellcome Trust Research Programme, PO Box 230-80108, Kilifi, Kenya.
Joshua Quick, Institute of Microbiology and Infection, School of Biosciences, University of Birmingham, Birmingham B15 2TT, UK.
George Githinji, Kenya Medical Research Institute-Wellcome Trust Research Programme, PO Box 230-80108, Kilifi, Kenya; Department of Biochemistry and Biotechnology, Pwani University, PO Box 195-80108, Kilifi, Kenya.
D James Nokes, Kenya Medical Research Institute-Wellcome Trust Research Programme, PO Box 230-80108, Kilifi, Kenya; School of Life Sciences and Zeeman Institute for Systems Biology and Infectious Disease Epidemiology Research (SBIDER), University of Warwick, Coventry CV4 7AL, UK.
Charlotte J Houldcroft, Department of Genetics, University of Cambridge, Downing Street, Cambridge CB2 3EH, UK.
Charles N Agoti, Kenya Medical Research Institute-Wellcome Trust Research Programme, PO Box 230-80108, Kilifi, Kenya; School of Public Health, Pwani University, PO Box 195-80108, Kilifi, Kenya.
Data availability
The genomes were then deposited to GenBank with the following accession numbers: OP581345–OP581409. The corresponding strain identifier, lineage, and HAdV type are summarized in Supplementary Table S4. The epidemiological data are available on the VEC dataverse (https://doi.org/10.7910/DVN/AMP10S).
Supplementary data
Supplementary data are available at Virus Evolution online.
Funding
This study was funded by The Wellcome Trust [102975 and 203077]. C.N.A. was supported by the Initiative to Develop African Research Leaders (IDeAL) through the DELTAS Africa Initiative [DEL-407 15-003]. The DELTAS Africa Initiative is an independent funding scheme of the African Academy of Sciences (AAS)’s Alliance for Accelerating Excellence in Science in Africa and is supported by the New Partnership for Africa’s Development Planning and Coordinating Agency (NEPAD Agency). C.J.H. was supported by funding from the Department of Genetics, University of Cambridge. J.Q. was supported by funding from the UK Research and Innovation body. The views expressed in this report are those of the authors and not necessarily those of the AAS, the NEPAD Agency, the Wellcome, the UK Research and Innovation (UKRI), the University of Cambridge, and the UK government.
Conflict of interest:
None declared.
References
- Agoti C. N. et al. (2022) ‘Differences in Epidemiology of Enteropathogens in Children Pre- and Post-Rotavirus Vaccine Introduction in Kilifi, Coastal Kenya’, Gut Pathogens, 14: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A. et al. (2017) ‘Molecular Characterization of Enteric Adenovirus Genotypes 40 and 41 Identified in Children with Acute Gastroenteritis in Kolkata, India During 2013–2014’, Journal of Medical Virology, 89: 606–14. [DOI] [PubMed] [Google Scholar]
- Chandra P. et al. (2021) ‘Genetic Characterization and Phylogenetic Variations of Human Adenovirus‐F Strains Circulating in Eastern India During 2017–2020’, Journal of Medical Virology, 93: 6180–90. [DOI] [PubMed] [Google Scholar]
- Dehghan S. et al. (2019) ‘A Zoonotic Adenoviral Human Pathogen Emerged through Genomic Recombination among Human and Nonhuman Simian Hosts’, Journal of Virology, 93: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelaw A., Pietsch C., and Liebert U. G. (2019) ‘Genetic Diversity of Human Adenovirus and Human Astrovirus in Children with Acute Gastroenteritis in Northwest Ethiopia’, Archives of Virology, 164: 2985–93. [DOI] [PubMed] [Google Scholar]
- Götting J. et al. (2022) ‘Molecular Phylogeny of Human Adenovirus Type 41 Lineages’, BioRxiv, 2022: 05.30.493978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez Sanchez L. H. et al. (2022) ‘A Case Series of Children with Acute Hepatitis and Human Adenovirus Infection’, The New England Journal of Medicine, 387: 620–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung-Yueh Y. et al. (1994) ‘Human Adenovirus Type 41 Contains Two Fibers’, Virus Research, 33: 179–98. [DOI] [PubMed] [Google Scholar]
- Institute for Health Metrics and Evaluation . (2021), Default Results Are Global All-Cause Deaths and DALYs for 2019 with Trends Since 1990. <https://vizhub.healthdata.org/gbd-results/> accessed 27 Sep 2022.
- Khagayi S. et al. (2020) ‘Effectiveness of Monovalent Rotavirus Vaccine against Hospitalization with Acute Rotavirus Gastroenteritis in Kenyan Children’, Clinical Infectious Diseases, 70: 2298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd A. H. et al. (1984) ‘Genome Variants of Human Adenovirus 40 (Subgroup F)’, Journal of Medical Virology, 14: 235–46. [DOI] [PubMed] [Google Scholar]
- Lambisia A. W. et al. (2020) ‘Epidemiological Trends of Five Common Diarrhea-Associated Enteric Viruses Pre- and Post-Rotavirus Vaccine Introduction in Coastal Kenya’, Pathogens, 9: 660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Rosa G. et al. (2015) ‘Genetic Diversity of Human Adenovirus in Children with Acute Gastroenteritis, Albania, 2013–2015’, BioMed Research International, 2015: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis K. (2011), Vesikari Clinical Severity Scoring System Manual. Path, May, 1–50. <https://www.path.org/publications/files/VAD_vesikari_scoring_manual.pdf> accessed 27 Sep 2022.
- Lukashev A. N. et al. (2008) ‘Evidence of Frequent Recombination among Human Adenoviruses’, Journal of General Virology, 89: 380–8. [DOI] [PubMed] [Google Scholar]
- Mailis M. et al. (2022) ‘Enteric Adenovirus F41 Genetic Diversity Comparable to pre-COVID-19 Era_ Validation of a Multiplex Amplicon-MinION Sequencing Method’ OSF-Preprints 1–23.
- Martin D. P. et al. (2015) ‘RDP4: Detection and Analysis of Recombination Patterns in Virus Genomes’, Virus Evolution, 1: 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick J. (2020), nCoV-2019 Sequencing Protocol V2 (Gunit). Protocols.Io. < 10.17504/protocols.io.bdp7i5rn> accessed 27 Sep 2022. [DOI]
- Rafie K. et al. (2021) ‘The Structure of Enteric Human Adenovirus 41—A Leading Cause of Diarrhea in Children’, Science Advances, 7: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . (2019), R: A Language and Environment for Statistical Computing. (Vienna, Austria: ) <https://www.r-project.org/> accessed 27 Sep 2022. [Google Scholar]
- Samson S., Lord É., and Makarenkov V. (2022) ‘SimPlot++: A Python Application for Representing Sequence Similarity and Detecting Recombination’, Bioinformatics, 38: 3118–20. [DOI] [PubMed] [Google Scholar]
- Singh G. et al. (2013) ‘Homologous Recombination in E3 Genes of Human Adenovirus Species D’, Journal of Virology, 87: 12481–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahmasebi R. et al. (2020) ‘Viral Gastroenteritis in Tocantins, Brazil: Characterizing the Diversity of Human Adenovirus F Through Next-Generation Sequencing and Bioinformatics’, Journal of General Virology, 101: 1280–8. [DOI] [PubMed] [Google Scholar]
- Tang X. et al. (2022) ‘Molecular Epidemiology of Human Adenovirus, Astrovirus, and Sapovirus among Outpatient Children with Acute Diarrhea in Chongqing, China, 2017–2019’, Frontiers in Pediatrics, 10: 2017–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization—WHO . (2022), Multi-Country – Acute, severe hepatitis of unknown origin in children, <https://www.who.int/emergencies/disease-outbreak-news/item/2022-DON376> accessed 27 Sep 2022.
- Yang J. et al. (2019) ‘Human Adenovirus Species C Recombinant Virus Continuously Circulated in China’, Scientific Reports, 9: 9781. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The genomes were then deposited to GenBank with the following accession numbers: OP581345–OP581409. The corresponding strain identifier, lineage, and HAdV type are summarized in Supplementary Table S4. The epidemiological data are available on the VEC dataverse (https://doi.org/10.7910/DVN/AMP10S).