

Abstract
A novel doubly cyclopentannulated carbazole which is accessible through a successive π‐expansion of di(1‐naphthylamine) is disclosed. The carbazole moiety is generated in the final step through intramolecular oxidative coupling. The π‐expansion of carbazole resulted in strongly altered optoelectronic and electrochemical properties. The solid‐state structure features an interesting packing motif with alternating face‐to‐face π⋅⋅⋅π and edge‐to‐face C−H⋅⋅⋅π interactions. The experimental findings were corroborated by theoretical calculations.
Keywords: carbazole, cyclopentannulation, electron donor, N-heterocycle, triarylamine
Building big. A symmetrically π‐expanded carbazole incorporating fluoranthene moieties was assembled through a five‐step synthetic route. In comparison to parent N‐phenyl‐substituted carbazole the compound features significantly bathochromically shifted UV‐Vis absorption and emission maxima with considerably increased photoluminescence quantum yield. The π‐expansion results in facilitated oxidation with improved reversibility, while at the same time, the aromaticity of the central carbazole moiety remains unaffected.
Introduction
Carbazole, which was already discovered in 1872 by Graebe and Glaser in a coal tar, [1] is nowadays intensely studied, [2] for example, as an important building block for the materials used in organic electronics, mainly organic lighting devices[ 2b , 3 ] and photovoltaics. [4] In these applications, carbazole serves as an electron‐donating moiety which is mostly linked to other π‐systems through the nitrogen atom or at the 3‐ and 6‐positions. In recent years, efforts were directed towards diverse materials in which the carbazole moiety itself is π‐expanded. For example, benzothieno‐ and benzofuro‐fused carbazole scaffolds were realized and their performance in optoelectronic devices was evaluated. [5] Moreover, related indolo‐ and thienopyrrolo[3,2,1‐jk]carbazoles [6] represent a growing family of versatile electron donors based on ortho‐bridged triphenylamines. [7]
Another type of π‐expanded carbazole was obtained through annulation to all‐carbon polycyclic moieties. [8] In this context, the incorporation of π‐conjugated cyclopentadiene moieties has been established as a particularly potent strategy to modulate the electronic and structural properties of the polycyclic frameworks. [9] This approach has so far resulted in a number of interesting compounds with unusual properties including cyclopentannulated acenes [10] and their nitrogen‐containing counterparts. [11] The cyclopentannulated carbazole derivative 1 was reported by Wang and coworkers to exhibit intense blue fluorescence (Figure 1). [12] The structurally related compound 2 was implemented as a hole transport material with improved stability in OLEDs by Lee and coworkers. [13] Only recently, structurally appealing corannulene‐carbazole hybrids such as 3 were disclosed by Tanaka and coworkers. [14] However, in spite of this indisputable progress π‐expanded carbazoles incorporating cyclopentannulated all‐carbon polycyclic moieties still remain scarce. Such compounds are not only of general interest to study the impact of π‐expansion on the optoelectronic properties of the carbazole moiety, but also as potential optoelectronic materials with tailored characteristics.
Figure 1.

Overview of π‐expanded cyclopentannulated carbazoles reported to date and the title compound described herein.
To address this issue, we report herein the synthesis of a novel symmetrically π‐expanded carbazole with two annulated fluoranthene units. The compound was obtained through a concise synthetic route starting from di(1‐naphthylamine) relying on a 2‐fold Pd‐catalyzed cyclopentannulation followed by oxidative cyclodehydrogenation to form the carbazole core. Comprehensive spectroscopic and electrochemical studies supported by theoretical calculations revealed strongly altered optoelectronic properties when compared to parent carbazole.
Results and Discussion
Di(1‐naphthyl)amine was synthesized according to Venkataraman and coworkers from 1‐naphthylamine and 1‐bromonaphthalene via a Buchwald‐Hartwig amination reaction. [15] Subsequent Cu‐catalyzed Ullmann [16] coupling with 1‐(tert‐butyl)‐4‐iodobenzene provided triarylamine 4 in 70 % yield (Scheme 1). Electrophilic bromination of 4 with N‐bromosuccinimide (NBS) at 0 °C selectively resulted in compound 5 (87 % yield). In the following, a Suzuki‐Miyaura [17] coupling of 5 with boronic acid ester 6 [18] was conducted to afford blue fluorescent compound 7 in 88 % yield. Pd‐catalyzed intramolecular cyclization of 7 provided 8 in nearly quantitative yield as orange solid with yellow fluorescence both in solution and in the solid state.
Scheme 1.

Synthetic route towards symmetrically cyclopentannulated carbazole 9 from di(1‐naphthyl)amine. For model compounds 10 and 11 the yields of the final synthetic steps are shown. NBS=N‐bromosuccinimide, DBU=1,8‐diazabicyclo[5.4.0]undec‐7‐ene, DMA=N,N‐dimethylacetamide.
Final oxidative cyclodehydrogenation to achieve the carbazole moiety in 9 was achieved using FeCl3 and MeNO2 in CH2Cl2. [19] Compound 9 was obtained in 52 % yield as a yellow powder soluble in common organic solvents to give solutions with intense blue fluorescence. Interestingly, green fluorescence was observed in the solid state (see below). In addition, model compounds 10 [17] and 11 [20] were synthesized for further studies (for synthetic details, see the Supporting Information).
Single crystals of 9 suitable for X‐ray crystallographic analysis were grown by slow evaporation of a CH2Cl2/MeOH solution of the compound at room temperature. As shown in Figure 2, the nitrogen center is virtually planar with the C−N−C angles summing to 358.5°. The adjacent phenyl ring is oriented perpendicularly with respect to the polycyclic framework. At the same time, the phenyl is slightly bent from planarity with an angle of 169.1° between the phenyl ring and the center of the pyrrole ring. The C−C and C−N bond lengths within the pyrrole ring range between 1.40 and 1.43 Å, indicating efficient π‐electron delocalization and aromaticity. Moreover, the observed bond lengths are in good agreement with those reported for N‐phenylcarbazole. [21] In the crystal packing the molecules are stacked in a staggered arrangement. In the stack, two molecules interact by face‐to‐face π⋅⋅⋅π interactions with a distance of 3.37 Å between the planar polycyclic frameworks. The distance between the supramolecular dimers within the stack is 4.53 Å. In addition, edge‐to‐face C(sp2)−H⋅⋅⋅π‐interactions [22] between the perpendicularly oriented phenyl moieties and the polycyclic framework of the neighboring molecule are observed (see the Supporting Information). C(sp3)−H⋅⋅⋅π‐interactions were found between the tert‐butyl groups and the π‐systems of the molecules within the neighboring stack. [23]
Figure 2.
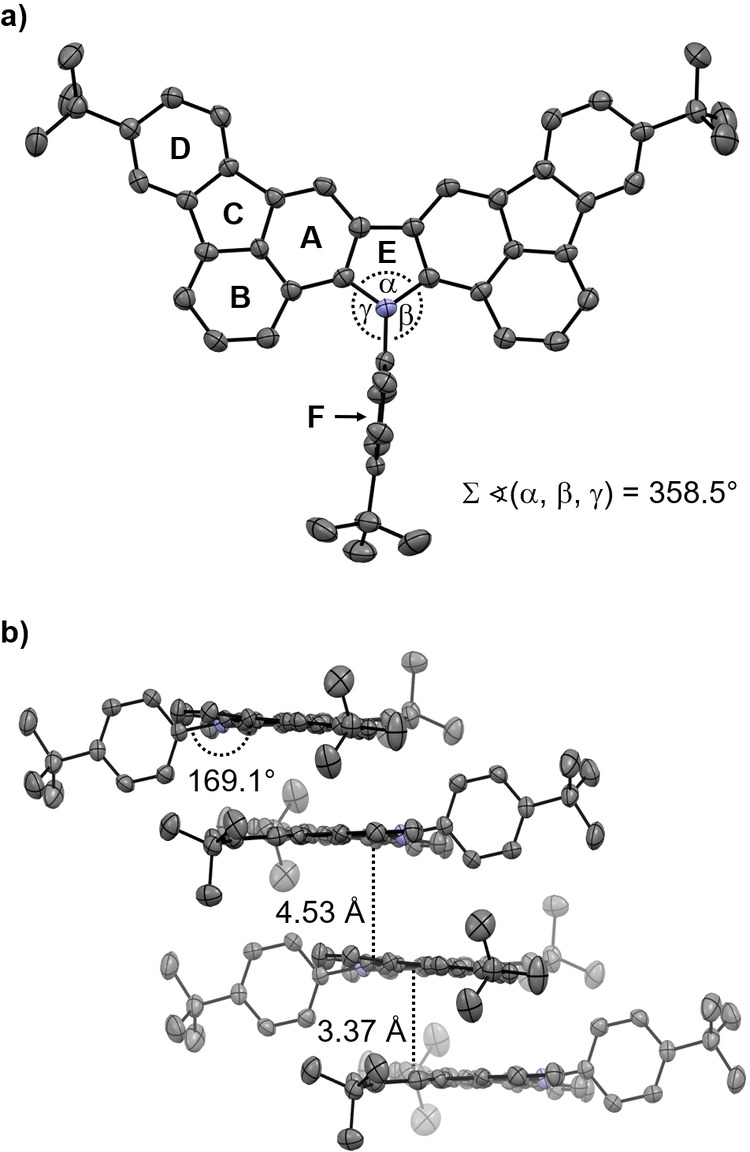
X‐ray crystal structure of 9 (50 % probability level, H‐atoms omitted). a) Top view with angles around the nitrogen atom and assignment of the rings A–F. b) Packing motif with alternating face‐to‐face and edge‐to‐face π⋅⋅⋅π interactions.
To assess the local aromaticity within the polycyclic framework of the π‐expanded carbazole 9 and of its congeners, nucleus independent chemical shift (NICS(X); X=−1, 0, +1) [24] values were calculated by the gauge‐independent atomic orbital (GIAO) [25] method at the B3LYP/6‐31G(d) [26] level of theory. The molecular geometries were optimized at the same level of theory (for further details, see the Supporting Information). The formation of the pyrrole moiety during the final cyclization step of 8 towards 9 exerts basically no influence on the aromatic character within the fluoranthene subunits. Hence, the 6‐membered rings A, B, and D retain their benzenoid nature defined by their Clar sextets with the NICS(+1) values ranging from −10.2 to −8.1 (for compound 8) and from −10.6 to −8.4 (for compound 9) (for the ring labels, see Figure 2a). In contrast, the 5‐membered ring C with the NICS(+1) value close to zero is basically non‐aromatic in both compounds 8 (−0.4) and 9 (−0.5), which is in line with the previous reports. [28] Interestingly, NICS(+1) and NICS(−1) slightly differ for the triarylamine precursor 8, while they are identical for title compound 9, which clearly reflects the planarity of the polycyclic scaffold of the latter. The central pyrrole moiety in 9 (ring E) features a negative NICS(+1) value of −9.2, which nicely corroborates its pronounced aromatic character as already deduced from the structural parameters obtained by X‐ray crystallographic analysis. The fact that for the N‐arylated model compound 11 a comparable value of −8.7 was calculated suggests that the π‐expansion has only a negligible impact on the electronic situation within the pyrrole moiety.
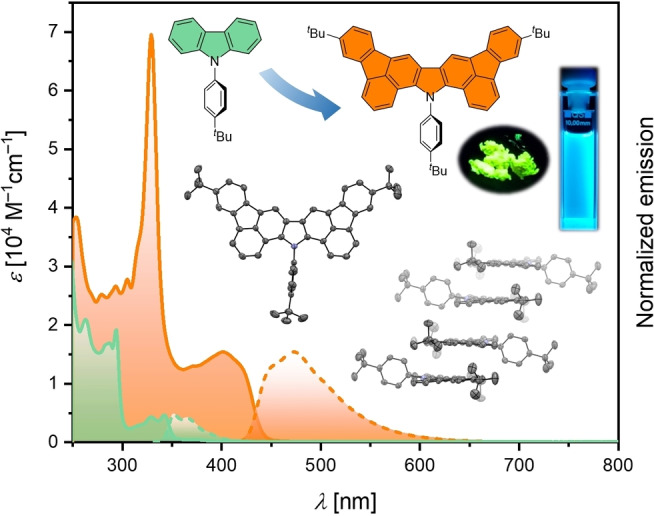
The impact of the successive π‐expansion on the optoelectronic properties was investigated by UV‐Vis absorption and emission spectroscopy in CH2Cl2 at room temperature (Figure 3 and Table 1). Triarylamine 4 and the model compound 10 show a comparable absorption behavior with the longest wavelength absorption maxima (λ max) occurring exclusively in the UV region at 346 and 361 nm, respectively (Table 1). This is not surprising in view of the nearly orthogonal arrangement of the lateral tert‐butylphenyl substituents in 10 (see below). Please note that the model compound 10 was synthesized in order to investigate the isolated influence of the enlarged π‐system without the interference from the electron‐withdrawing chloro substituents in 7 (see the Supporting Information).
Figure 3.

UV‐Vis absorption (solid lines) and emission (dashed lines) spectra of 4, 8, 9, and 10 recorded in CH2Cl2 at room temperature.
Table 1.
Experimental optoelectronic and electrochemical data of compounds 4, 8, 9, 10, and 11 as well as calculated HOMO and LUMO energies.
|
|
λ max [nm][a] |
ϵ [L mol−1 cm−1][a] |
λ ae [nm][a,b] |
E g opt [eV][b] |
λ em [nm][a] |
Stokes Shift [cm−1][c] |
Φ [a,d] |
E ox [V][a] |
HOMO/LUMO [eV][e] |
|---|---|---|---|---|---|---|---|---|---|
|
4 |
346 |
11100 |
389 |
3.19 |
437 |
6018 |
0.13 |
+0.50 |
−4.82/−1.04 |
|
8 |
447 |
16500 |
514 |
2.41 |
553 |
4288 |
0.39 |
+0.37 |
−4.78/−1.76 |
|
9 |
415 |
13800 |
440 |
2.82 |
473 |
2954 |
0.50 |
+0.69 |
−5.12/−1.54 |
|
10 |
361 |
18200 |
408 |
3.04 |
460 |
5961 |
0.34 |
+0.44 |
−4.74/−1.12 |
|
11 |
342 |
4680 |
354 |
3.50 |
352 |
831 |
0.20 |
+0.89 |
−5.27/−0.61 |
[a] Measured at rt in CH2Cl2; Excitation wavelengths: 345 (4), 448 (8), 402 (9), 360 (10), 293 nm (11). [b] Optical band gap E g opt=h c/λ ae. [27] [c] Calculated according to (1/λ em)–(1/λ max). [d] Determined with an integrating sphere. [e] Calculated at the B3LYP/6‐31G(d) level of theory.
A significant bathochromic shift of the absorption maxima λ max at 447 nm is observed for the 2‐fold cyclized compound 8 with the two fluoranthene units. Interestingly, λ max of 9 incorporating the carbazole moiety is found hypsochromically shifted at 415 nm, which is at first glance somewhat unexpected, considering the further expanded π‐system of 9 compared to its triarylamine precursor 8. At the same time, the longest wavelength absorption maximum of the N‐phenyl carbazole model compound 11 occurs with lower intensity at 342 nm (for details, see the Supporting Information), which highlights the impact of the π‐expansion in 9. In general, the absorptions at shorter wavelengths most likely originate from π–π* and n–π* transitions, while the low energy bands may be attributable to HOMO–LUMO transitions.[ 5 , 6 ] All compounds show pronounced fluorescence in CH2Cl2 (Table 1). The most bathochromically shifted emission maximum is observed for the yellow fluorescent triarylamine 8 at 553 nm with a photoluminescence quantum yield (PLQY) of 0.39. The formation of the carbazole moiety in 9 leads to a hypsochromic shift of the emission maximum to 473 nm and an increased PLQY of 0.50. Similar trend is observed for the solid‐state emission of 8 and 9 with the maxima occurring at 567 (PLQY 0.13) and 523 nm (PLQY 0.18), respectively (see Figures S27 and S28 in the Supporting Information).
To investigate the possible reasons behind the unexpected shifts observed in the UV‐Vis absorption spectra, the geometries of molecules 4 and 8–11 were optimized by DFT calculations at the B3LYP/6‐31G(d) level of theory and the energies of the frontier molecular orbitals (FMOs) were estimated. The highest occupied molecular orbitals (HOMOs) of triarylamines 4, 8, and 10 are of comparable energy and are delocalized across all three aryl moieties with a significant contribution of the N‐phenyl substituent (Figure 4 and the Supporting Information). The final cyclization to form the carbazole moiety in 9 is accompanied by an increased stabilization of the HOMO by 0.34 eV compared to precursor 8. This evolution is most likely a result of the bonding interaction of the HOMO across the newly formed pyrrole ring and the adjacent 6‐membered rings in combination with the aromaticity gain due to the additional π‐electron sextet. In contrast, the respective lowest unoccupied molecular orbitals (LUMOs) experience no significant contribution from the N‐phenyl moieties and their energy levels continuously decrease with increased size of the two remaining aryl substituents when going from 4 over 10 to 8. Consequently, triarylamine 8 has the lowest HOMO‐LUMO gap of all examined compounds, which is in agreement with its most bathochromically shifted UV‐Vis absorption. In contrast, the LUMO of the title compound 9 has no contribution of the nitrogen atom and is destabilized by 0.22 eV compared to 8. This results in an overall larger HOMO‐LUMO gap of 9, which is nicely reflected by the hypsochromic shift observed in the UV‐Vis absorption spectrum. Nevertheless, the HOMO‐LUMO gap of 9 is considerably reduced by more than 1.0 eV in comparison to parent N‐aryl carbazole 11, thus highlighting the impact of the π‐expansion.
Figure 4.
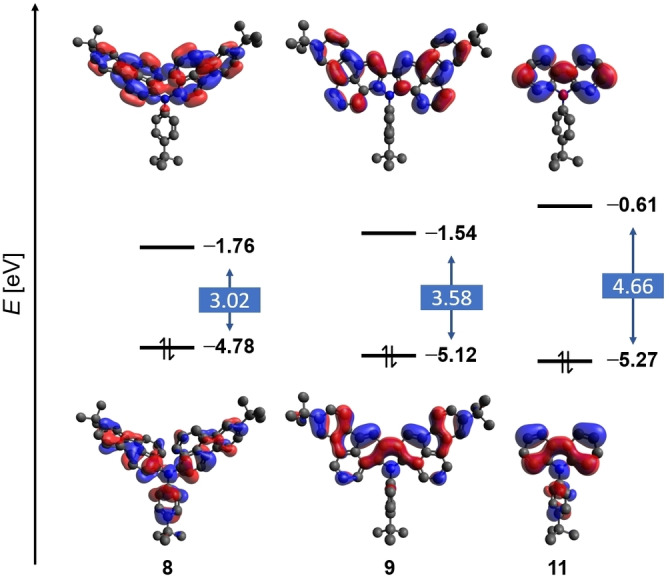
Frontier molecular orbitals of compounds 8, 9, and 11 calculated at the B3LYP/6‐31G(d) level of theory.
The compounds 4 and 8–11 were studied by cyclic voltammetry (CV) in CH2Cl2 with n‐Bu4NPF6 (0.1 m) as an inert supporting electrolyte to determine their redox properties (Figure 5 and Table 1). All compounds show reversible oxidations between +0.37 V (for 8; vs. ferrocene/ferrocenium (Fc/Fc+)) to +0.89 V (for 11). For compound 8 an additional irreversible oxidation is observed at +1.00 V which is most likely located at the fluoranthene moieties. According to literature fluoranthenes are prone to electropolymerization upon oxidation, which explains the observed irreversibility of the second oxidation. [29] Overall, the progressive cathodic shift of the first oxidation step on the way from 4 over 10 to 8 nicely reflects the successive expansion of the respective π‐system, facilitating stabilization of the positive charge upon delocalization. In contrast, a pronounced anodic shift of 320 mV of the first oxidation is encountered when going from triarylamine 8 (+0.37 V) to π‐expanded carbazole 9 (+0.69 V). This finding can be most likely ascribed to the efficient delocalization of the lone pair of the nitrogen to form the π‐electron sextet within the aromatic pyrrole subunit, which is in line with the enhanced HOMO stabilization in 9 (see above). In comparison, the model carbazole compound 11 is irreversibly oxidized at +0.89 V towards the corresponding radical cation which readily dimerizes (see the Supporting Information). [20a] The reversibility of the facilitated oxidation in case of 9 further highlights the beneficial effect of π‐expansion.
Figure 5.
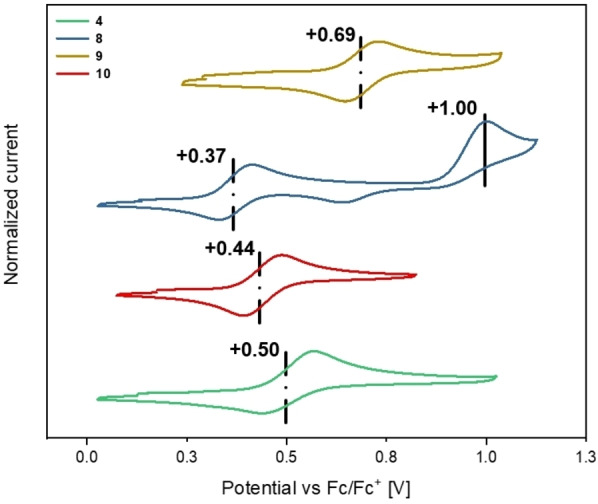
Cyclic voltammetry of compounds 4, 8, 9, and 10 measured in CH2Cl2 at room temperature with n‐Bu4NPF6 as the supporting electrolyte (ν=100 mV s−1, Fc/Fc+).
Conclusion
In summary, a novel π‐expanded carbazole derivative incorporating fluoranthene moieties was prepared through a five‐step procedure from known di(1‐naphthylamine) in an overall yield of 27 %. According to the X‐ray crystallographic analysis, the polycyclic framework of the compound is perfectly planar with the lateral N‐phenyl ring being slightly bent out of planarity. In the solid state the compound forms a staggered packing motif with alternating face‐to‐face π⋅⋅⋅π and edge‐to‐face C−H⋅⋅⋅π interactions. The calculated NICS values indicate that the extent of aromaticity within the pyrrole moiety retains largely unaffected throughout the π‐expansion and is comparable to parent N‐phenyl‐substituted carbazole. At the same time, the π‐expansion leads to pronounced bathochromic shifts both in absorption and emission accompanied by an increase of the photoluminescence quantum yield by a factor of 2.5. The electrochemical data suggests an improved stabilization of the positive charge generated upon oxidation. We believe that these characteristics make the newly established π‐expanded carbazole scaffold highly interesting for the development of functional electron donors upon appropriate functionalization.
Experimental Section
General: All reactions involving oxygen‐ or moisture‐sensitive compounds were carried out in dry reaction vessels under an inert atmosphere of nitrogen using anhydrous solvents and standard Schlenk techniques. Reagents were purchased at reagent grade from commercial suppliers and used without further purification. Di(1‐naphthyl)amine was prepared according to literature. [15] Dry CH2Cl2 was purified by a SPS‐800 (M. Braun). Dry THF was purified by distillation over sodium metal. TLC analysis was performed on aluminum plates coated with 0.20 mm silica gel containing a fluorescent indicator (Macherey‐Nagel, ALUGRAM®, SIL G/UV254). The results were visualized by exposure to ultraviolet light (λ=254 and 366 nm). Column chromatography was performed on silica gel (Macherey‐Nagel, Silica Gel 60 A, 230–400 mesh.
Instrumentation: 1H, 13C, and 11B NMR spectra were recorded on a Bruker Avance III 300 (Bruker, 301 MHz for 1H), a Bruker Avance DRX 300 (Bruker, 300 MHz for 1H, and 75 MHz for 13C), a Bruker Avance III 400 (Bruker, 400 MHz for 1H,101 MHz for 13C, and 128 MHz for 11B), a Bruker Avance III 500 (Bruker, 500 MHz for 1H, and 126 MHz for 13C), a Bruker Avance III 600 (Bruker, 600 MHz for 1H and 151 MHz for 13C), or a Bruker Avance Neo 700 (Bruker, 700 MHz for 1H and 176 MHz for 13C). CDCl3 (Sigma Aldrich, 99.8 %), CD2Cl2 (Sigma Aldrich, 99.5 %), or (CD3)2CO (Sigma Aldrich, 99.5 %) were used to dissolve the samples. Chemical shifts (δ) are reported in ppm and were referenced to the residual solvent signal as an internal reference (CDCl3: 7.26 ppm for 1H, 77.16 ppm for 13C; CD2Cl2: 5.32 ppm for 1H, 53.84 ppm for 13C; (CD3)2CO: 2.05 ppm for 1H, 29.84 ppm for 13C) Coupling constants (J) are given in Hz and the apparent resonance multiplicity is reported as s (singlet), d (doublet), t (triplet), or m (multiplet). All NMR spectra were recorded at rt. Melting points were determined on a Büchi M‐560 melting point apparatus in open capillaries. UV‐Vis absorption spectra were recorded on a Cary 60 UV‐Vis (Agilent) spectrometer and emission spectra on a JASCO FP‐8500 fluorescence spectrometer. IR spectra were recorded on a FT/IR‐4600 (Jasco, ATR mode) spectrometer. Characteristic IR absorptions were reported in cm−1 and labeled as strong (s), medium (m), or weak (w). Mass spectra were obtained from an Accu TOF GCx (Jeol, EI), or an AutoFlex Speed (Bruker, MALDI).
N ‐(4‐tert‐Butylphenyl)‐N‐(naphthalen‐1‐yl)naphthalen‐1‐amine (4): 1‐(tert‐Butyl)‐4‐iodobenzene (0.47 mL, 695 mg, 2.67 mmol), di(1‐naphthyl)amine (200 mg, 0.74 mmol), activated copper (189 mg, 2.97 mmol), and K2CO3 (205 mg, 1.49 mmol) were added to a microwave reaction vessel under N2 atmosphere. The reaction mixture was heated to 200 °C and stirred for 42 h. After cooling to room temperature, the reaction mixture was purified by column chromatography (SiO2, petroleum ether/CH2Cl2 4 : 1) to afford 4 (208 mg, 0.52 mmol 70 %) as a colorless solid. R f=0.62 (SiO2, petroleum ether/CH2Cl2 4 : 1). Mp 125–130 °C. 1H NMR (400 MHz, (CD3)2CO) δ 8.09–8.07 (m, 2H), 7.95 (d, J=8.2 Hz, 2H), 7.76 (d, J=8.2 Hz, 2H), 7.47 (ddd, J=8.1, 6.9, 1.1 Hz, 2H), 7.43–7.39 (m, 2H), 7.33 (ddd, J=8.3, 6.8, 1.2 Hz, 2H), 7.19 (ddd, J=8.9, 7.1, 1.6 Hz, 4H), 6.67–6.63 (m, 2H), 1.26 (s, 9H) ppm. 13C NMR (101 MHz, (CD3)2CO) δ 149.1, 146.2, 144.6, 138.0, 136.3, 131.1, 129.5, 128.7, 127.0, 126.7, 126.3, 125.5, 125.0, 121.4, 34.7, 31.8 ppm. IR (ATR): 3048 (w), 2957 (w), 2901 (w), 2864 (w), 1572 (m), 1502 (m), 1390 (s), 1268 (m), 1016 (m), 832 (m), 770 (s) cm−1. MALDI HRMS (DCTB): calc. for C30H27N: m/z 401.2138 [M+], found 401.2140. UV/Vis (CH2Cl2, rt): λ max (ϵ in L mol−1 cm−1) 266 (20900), 301 (6100), 346 (11100) nm.
4‐Bromo‐N‐(4‐bromonaphthalen‐1‐yl)‐N‐(4‐tert‐butylphenyl)‐naphthalen‐1‐amine (5): Compound 4 (500 mg, 1.25 mmol) was dissolved in CH2Cl2 (100 mL) and cooled to 0 °C. N‐bromosuccinimide (488 mg, 2.74 mmol) was added portionwise over the course of 5 min and the mixture was stirred for 40 min at 0 °C. The reaction mixture was treated with 10 wt.% aq. Na2S2O4 (50 mL) and extracted with CH2Cl2 (3×50 mL). The combined organic layers were washed with saturated aq. NaCl (50 mL) and dried (Na2SO4). The solvent was removed under reduced pressure and the residue was purified by column chromatography (SiO2, petroleum ether/CH2Cl2 8 : 1). Compound 5 (611 mg, 1.09 mmol, 87 %) was obtained as a beige solid. R f=0.85 (SiO2, petroleum ether/CH2Cl2 4 : 1). Mp 216 °C. 1H NMR (300 MHz, CD2Cl2) δ 8.26 (d, J=8.3 Hz, 2H), 8.10 (d, J=8.4 Hz, 2H), 7.65 (d, J=8.1 Hz, 2H), 7.57 (ddd, J=8.4, 6.7, 1.1 Hz, 2H), 7.37 (ddd, J=8.2, 6.7, 1.0 Hz, 2H), 7.22–7.14 (m, 2H), 7.00 (d, J=8.0 Hz, 2H), 6.75–6.66 (m, 2H), 1.27 (s, 9H) ppm. 13C NMR (75 MHz, CD2Cl2) δ 148.0, 145.7, 145.4, 133.7, 131.6, 130.4, 128.1, 127.9, 127.4, 126.4, 125.4, 125.1, 121.6, 119.5, 34.5, 31.5 ppm. IR (ATR): 3068 (w), 3046 (w), 2961 (m), 2866 (w), 1584 (m), 1502 (m), 1452 (m), 1418 (m) 1375 (s), 1263 (s), 1025 (m), 935 (m), 829 (s), 756 (s) cm−1. MALDI HRMS (DCTB): calc. for C30H25Br2N: m/z 557.0348 [M+], found 557.0345. UV/Vis (CH2Cl2, rt): λ max (ϵ in L mol−1 cm−1) 269 (20900), 295 (10600), 359 (14100) nm.
4‐(4‐tert‐Butyl‐2‐chlorophenyl)‐N‐[4‐(4‐tert‐butyl‐2‐chlorophenyl)‐naphthalen‐1‐yl]‐N‐(4‐tert‐butylphenyl)naphthalen‐1‐amine (7): Compounds 5 (500 mg, 0.89 mmol) and 6 (790 mg, 2.68 mmol) were dissolved in ethanol/toluene (1 : 2, vol./vol., 90 mL) and the solution was deoxygenated with N2 for 30 min. [Pd(PPh3)4] (21 mg, 0.02 mmol) was added and the mixture was degassed again for 15 min. A deoxygenated aq. K2CO3 solution (2 m, 11.2 mL, 3.09 g, 22.4 mmol) was added to the reaction mixture, which was heated to 100 °C and stirred for 2 h. After cooling to room temperature, the solvent was removed under reduced pressure. H2O (50 mL) was added to the residue and the aq. phase was extracted with CH2Cl2 (3×50 mL). The combined organic layers were washed with saturated aq. NaCl and dried (Na2SO4). The solvent was removed under reduced pressure and the crude product was purified by column chromatography (SiO2, petroleum ether/CH2Cl2 5 : 1). Compound 7 (570 mg, 0.78 mmol, 88 %) was obtained as a colorless solid. R f=0.62 (SiO2, petroleum ether/CH2Cl2 4 : 1). Mp 251–260 °C. 1H NMR (600 MHz, (CD3)2CO) δ 8.25 (d, J=8.4, 2H), 7.62 (d, J=1.9, 2H), 7.52 (dd, J=8.0, 1.9, 2H), 7.49 (d, J=8.5, 2H), 7.45–7.42 (m, 2H), 7.39‐7.36 (m, 2H), 7.35 (d, J=8.0, 2H), 7.32–7.29 (m, 4H), 7.26–7.23 (m, 2H), 6.75–6.74 (m, 2H), 1.41 (s, 18H), 1.27 (s, 9H) ppm. 13C NMR (151 MHz, (CD3)2CO) δ 153.7, 149.1, 146.1, 146.0, 144.77, 144.75, 137.0, 135.7, 134.4, 134.3, 132.8, 131.0, 128.4, 127.4, 127.3, 127.2, 127.0, 126.8, 125.4, 125.03, 124.96, 124.9, 121.6, 121.5, 35.4, 34.7, 31.7, 31.5 ppm. Within the applied temperature range (295–353 K) the compound occurs in different conformers and consequently the spectrum is reported as empiric enumeration of the observed signals. IR (ATR): 3044 (w), 2959 (m), 2867 (w), 1579 (m) 1508 (s), 1458 (m), 1386 (m), 1261 (m), 828 (m), 760 (s) cm−1. MALDI HRMS (DCTB): calc. for C50H49Cl2N: m/z 733.3237 [M+], found 733.3238. UV‐Vis (CH2Cl2, rt): λ max (ϵ in L mol−1 cm−1) 271 (98800), 357 (62900) nm.
8‐tert‐Butyl‐N‐(8‐tert‐butylfluoranthen‐3‐yl)‐N‐(4‐tert‐butylphenyl)‐fluoranthen‐3‐amine (8): To a high pressure vessel under N2 atmosphere, DMA (40 mL), DBU (4 mL), 7 (400 mg, 544 μmol), and [Pd(PCy3)2Cl2] (281 mg, 381 μmol) were added. The reaction mixture was heated to 180 °C and stirred for 24 h. After cooling to room temperature 2 m aq. HCl (10 mL) and H2O (10 mL) were added and the mixture was passed through a pad of SiO2 (CH2Cl2). The combined organic layers were washed with saturated aq. NaCl (2×50 mL), dried (Na2SO4), and the solvent was removed under reduced pressure. After purification by column chromatography (SiO2, petroleum ether/CH2Cl2 5 : 1) the product 8 (348.7 mg, 527 μmol, 97 %) was obtained as an orange solid. R f=0.48 (SiO2, petroleum ether/CH2Cl2 4 : 1). Mp 237 °C. 1H NMR (700 MHz, CDCl3) δ 7.91 (d, J=1.6, 2H), 7.85 (d, J=6.9, 2H), 7.72 (d, J=7.9, 2H), 7.69 (d, J=7.4, 2H), 7.64 (d, J=8.4, 2H), 7.38 (dd, J=8.0, 1.7, 2H), 7.32 (dd, J=8.3, 7.0, 2H), 7.22–7.20 (m, 4H), 6.99 (d, J=8.7, 2H), 1.43 (s, 18H), 1.31 (s, 9H) ppm. 13C NMR (176 MHz, CDCl3) δ 150.4, 148.2, 146.3, 145.1, 139.7, 137.7, 136.7, 134.8, 133.6, 127.6, 127.1, 126.1, 125.7, 124.9, 124.4, 122.5, 120.7, 120.7, 119.8, 118.6, 35.1, 34.4, 31.7, 31.6 ppm. IR (ATR): 3035 (w), 2953 (m), 2865 (w), 1599 (w) 1509 (m), 1487 (m), 1429 (s), 1362 (m), 1255 (s), 1090 (w), 811 (s) 774 (s) cm−1. MALDI HRMS (DCTB): calc. for C50H47N: m/z 661.3703 [M+], found 661.3705. UV/Vis (CH2Cl2, rt): λ max (ϵ in L mol−1 cm−1) 282 (37200), 298 (49900), 332 (12000), 392 (9400), 447 (16500) nm.
3,13‐Di‐tert‐butyl‐8‐(4‐tert‐butylphenyl)‐8H‐difluoreno[1,9‐ab:9’,1’‐hi]carbazole (9): Under N2 atmosphere, 8 (50.0 mg, 75.5 μmol) was dissolved in dry CH2Cl2 (40 mL) and the solution was cooled to −10 °C. FeCl3 (25.0 mg, 151 μmol) was dissolved in MeNO2 (1 mL) and added dropwise to the reaction mixture over the course of 5 min. The reaction mixture was allowed to warm to room temperature and was stirred for 24 h. The reaction mixture was treated with MeOH (10 mL), washed with saturated aq. NaHCO3 (20 mL) and the combined organic layers were dried over (Na2SO4). The solvent was removed under reduced pressure and the residue was passed through a pad of SiO2 (CH2Cl2). Compound 9 (25.9 mg, 39.2 μmol, 52 %) was obtained as a yellow solid. R f=0.67 (SiO2, petroleum ether/CH2Cl2 3 : 1). Mp 330–362 °C (decomp.). 1H NMR (301 MHz, CDCl3) δ 8.80 (s, 2H), 8.03–7.97 (m, 4H), 7.95 (d, J=6.9 Hz, 2H), 7.83 (d, J=8.5 Hz, 2H), 7.70 (d, J=8.5 Hz, 2H), 7.51 (dd, J=8.0, 1.8 Hz, 2H), 7.40 (dd, J=8.4, 7.0 Hz, 2H), 7.01 (d, J=8.3 Hz, 2H), 1.58 (s, 9H), 1.47 (s, 18H) ppm. 13C NMR (176 MHz, CDCl3) δ 153.9, 149.9, 139.7, 138.6, 137.6, 137.1, 135.6, 132.7, 130.5, 129.0, 128.1, 127.0, 124.9, 122.5, 121.0, 120.5, 119.8, 118.33, 118.30, 113.6, 35.4, 35.2, 31.8, 29.9 ppm. IR (ATR): 3064 (w), 2953 (s), 2923 (s), 2855 (m), 1606 (w), 1510 (m), 1476 (s), 1414 (vs), 1240 (s), 1103 (m), 882 (s), 805 (vs), 746 (s), 655 (vs) cm−1. MALDI HRMS (DCTB): calc. for C50H45N: m/z 659.3547 [M+], found 659.3552. UV‐Vis (CH2Cl2, rt): λ max (ϵ in L mol−1 cm−1) 253 (38400), 279 (25300), 293 (26700), 305 (27800), 329 (69600), 373 (13000), 401 (15400), 415 (13800) nm.
Deposition Numbers 2170907 (for 5), and 2170908 (for 9) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
The generous funding by the Deutsche Forschungsgemeinschaft (DFG)‐Project number 182849149 – SFB 953 is acknowledged. Open Access funding enabled and organized by Projekt DEAL.
A. Vogel, T. Schreyer, J. Bergner, F. Rominger, T. Oeser, M. Kivala, Chem. Eur. J. 2022, 28, e202201424.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Graebe C., Glaser C., Ann. Chem. Pharm. 1872, 163, 343–360. [Google Scholar]
- 2.
- 2a. Lellouche J.-P., Koner R. R., Ghosh S., Rev. Chem. Eng. 2013, 29, 413–437; [Google Scholar]
- 2b. Wex B., Kaafarani B. R., J. Mater. Chem. C 2017, 5, 8622–8653; [Google Scholar]
- 2c. Yin J., Ma Y., Li G., Peng M., Lin W., Coord. Chem. Rev. 2020, 412, 213257; [Google Scholar]
- 2d. Wang G., Sun S., Guo H., J. Med. Chem. 2022, 229, 113999. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Blouin N., Leclerc M., Acc. Chem. Res. 2008, 41, 1110–1119; [DOI] [PubMed] [Google Scholar]
- 3b. Jiang H., Asian J. Org. Chem. 2014, 3, 102–112; [Google Scholar]
- 3c. Ledwon P., Org. Electron. 2019, 75, 105422; [Google Scholar]
- 3d. Krucaite G., Grigalevicius S., Synth. Met. 2019, 247, 90–108. [Google Scholar]
- 4.
- 4a. Li J., Grimsdale A. C., Chem. Soc. Rev. 2010, 39, 2399–2410; [DOI] [PubMed] [Google Scholar]
- 4b. Sathiyan G., Sivakumar E., Ganesamoorthy R., Thangamuthu R., Sakthivel P., Tetrahedron Lett. 2016, 57, 243–252; [Google Scholar]
- 4c. Rodríguez-Seco C., Cabau L., Vidal-Ferran A., Palomares E., Acc. Chem. Res. 2018, 51, 869–880; [DOI] [PubMed] [Google Scholar]
- 4d. Harvey P. D., Sharma G. D., Witulski B., Chem. Lett. 2021, 50, 1345–1355; [Google Scholar]
- 4e. Chen T.-W., Karapala V. K., Chen J.-T., Hsu C.-S., J. Chin. Chem. Soc. 2021, 68, 1186–1196. [Google Scholar]
- 5.
- 5a. Royer H., Joseph D., Prim D., Kirsch G., Synth. Commun. 1998, 28, 1239–1251; [Google Scholar]
- 5b. Gong M.-S., Cha J.-R., Lee C. W., Org. Electron. 2017, 42, 66–74; [Google Scholar]
- 5c. Arai A., Sasabe H., Nakao K., Masuda Y., Kido J., Chem. Eur. J. 2021, 27, 4971–4976. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Wharton S. I., Henry J. B., McNab H., Mount A. R., Chem. Eur. J. 2009, 15, 5482–5490; [DOI] [PubMed] [Google Scholar]
- 6b. Bader D., Fröhlich J., Kautny P., J. Org. Chem. 2020, 85, 3865–3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Hirai M., Tanaka N., Sakai M., Yamaguchi S., Chem. Rev. 2019, 119, 8291–8331; [DOI] [PubMed] [Google Scholar]
- 7b. Schaub T. A., Padberg K., Kivala M., J. Phys. Org. Chem. 2020, 33, e4022. [Google Scholar]
- 8.
- 8a. Han L., Meng X., Ke Y., Ye H., Cui Y., J. Photochem. Photobiol. A 2019, 376, 127–134; [Google Scholar]
- 8b. Wang Y., Jia D., Zeng J., Liu Y., Bu X., Yang X., Org. Lett. 2021, 23, 7740–7745. [DOI] [PubMed] [Google Scholar]
- 9. Liu Y.-H., Perepichka D. F., J. Mater. Chem. C 2021, 9, 12448–12461. [Google Scholar]
- 10.
- 10a. Wood J. D., Jellison J. L., Finke A. D., Wang L., Plunkett K. N., J. Am. Chem. Soc. 2012, 134, 15783–15789; [DOI] [PubMed] [Google Scholar]
- 10b. Bheemireddy S. R., Ubaldo P. C., Rose P. W., Finke A. D., Zhuang J., Wang L., Plunkett K. N., Angew. Chem. Int. Ed. 2015, 54, 15762-15766; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 15988–15992. [Google Scholar]
- 11. Heckershoff R., Maier S., Wurm T., Biegger P., Brödner K., Krämer P., Hoffmann M. T., Eberle L., Stein J., Rominger F., Rudolph M., Freudenberg J., Dreuw A., Hashmi A. S. K., Bunz U. H. F., Chem. Eur. J. 2022, 28, e202104203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xing Y., Hu B., Yao Q., Lu P., Wang Y., Chem. Eur. J. 2013, 19, 12788–12793. [DOI] [PubMed] [Google Scholar]
- 13. Kim S. M., Yun J. H., Han S. H., Lee J. Y., J. Ind. Eng. Chem. 2020, 84, 217–225. [Google Scholar]
- 14. Kise K., Ooi S., Saito H., Yorimitsu H., Osuka A., Tanaka T., Angew. Chem. Int. Ed. 2022, 61, e202112589; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2022, 134, e202112589. [DOI] [PubMed] [Google Scholar]
- 15. Field J. E., Hill T. J., Venkataraman D., J. Org. Chem. 2003, 68, 6071–6078. [DOI] [PubMed] [Google Scholar]
- 16. Ullmann F., Ber. Dtsch. Chem. Ges. 1903, 36, 2382–2384. [Google Scholar]
- 17. Miyaura N., Yanagi T., Suzuki A., Synth. Commun. 1981, 11, 513–519. [Google Scholar]
- 18. Demory E., Blandin V., Einhorn J., Chavant P. Y., Org. Process Res. Dev. 2011, 15, 710–716. [Google Scholar]
- 19.
- 19a. Sarhan A. A. O., Bolm C., Chem. Soc. Rev. 2009, 38, 2730–2744; [DOI] [PubMed] [Google Scholar]
- 19b. Grzybowski M., Skonieczny K., Butenschön H., Gryko D. T., Angew. Chem. Int. Ed. 2013, 52, 9900–9930; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 10084–10115; [Google Scholar]
- 19c. Grzybowski M., Sadowski B., Butenschön H., Gryko D. T., Angew. Chem. Int. Ed. 2020, 59, 2998–3027; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 3020–3050. [Google Scholar]
- 20.
- 20a. Ambrose J. F., Carpenter L. L., Nelson R. F., J. Electrochem. Soc. 1975, 122, 876–894; [Google Scholar]
- 20b. Wei L., Li J., Xue K., Ye S., Jiang H., New J. Chem. 2019, 43, 16629–16638. [Google Scholar]
- 21. Zhang K., Sun Q., Zhang Z., Tang L., Xie Z., Chi Z., Xue S., Zhang H., Yang W., Chem. Commun. 2018, 54, 5225–5228. [DOI] [PubMed] [Google Scholar]
- 22. Salonen L. M., Ellermann M., Diederich F., Angew. Chem. Int. Ed. 2011, 50, 4808–4842; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 4908–4944. [Google Scholar]
- 23.
- 23a. Kohl B., Bohnwagner M. V., Rominger F., Wadepohl H., Dreuw A., Mastalerz M., Chem. Eur. J. 2016, 22, 646–655; [DOI] [PubMed] [Google Scholar]
- 23b. Rösel S., Balestrieri C., Schreiner P. R., Chem. Sci. 2017, 8, 405–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.
- 24a. Schleyer P. v. R., Maerker C., Dransfeld A., Jiao H., van Eikema Hommes N. J. R., J. Am. Chem. Soc. 1996, 118, 6317–6318; [DOI] [PubMed] [Google Scholar]
- 24b. Schleyer P. v. R., Jiao H., van Hommes N. J. R. E., Malkin V. G., Malkina O. L., J. Am. Chem. Soc. 1997, 119, 12669–12670; [Google Scholar]
- 24c. Chen Z., Wannere C. S., Corminboeuf C., Puchta R., Schleyer P. v. R., Chem. Rev. 2005, 105, 3842–3888. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. London F., J. Phys. Radium 1937, 8, 397–409; [Google Scholar]
- 25b. McWeeny R., Phys. Rev. 1962, 126, 1028–1034; [Google Scholar]
- 25c. Ditchfield R., Mol. Phys. 1974, 27, 789–807; [Google Scholar]
- 25d. Wolinski K., Hinton J. F., Pulay P., J. Am. Chem. Soc. 1990, 112, 8251–8260; [Google Scholar]
- 25e. Cheeseman J. R., Trucks G. W., Keith T. A., Frisch M. J., J. Chem. Phys. 1996, 104, 5497–5509. [Google Scholar]
- 26.
- 26a. Hehre W. J., Ditchfield R., Pople J. A., Appl. Phys. Lett. 1972, 56, 2257–2261; [Google Scholar]
- 26b. Hariharan P. C., Pople J. A., Theor. Chim. Acta 1973, 28, 213–222; [Google Scholar]
- 26c. Lee C., Yang W., Parr R. G., Phys. Rev. B Condens. Matter 1988, 37, 785–789; [DOI] [PubMed] [Google Scholar]
- 26d. Becke A. D., Phys. Rev. A 1988, 38, 3098–3100; [DOI] [PubMed] [Google Scholar]
- 26e. Becke A. D., Appl. Phys. Lett. 1993, 98, 5648–5652. [Google Scholar]
- 27. Costa J. C., Taveira R. J., Lima C. F., Mendes A., Santos L. M., Opt. Mater. 2016, 58, 51–60. [Google Scholar]
- 28.
- 28a. Aihara J., Sekine R., Ishida T., J. Phys. Chem. A 2011, 115, 9314–9321; [DOI] [PubMed] [Google Scholar]
- 28b. Alvarez-Ramírez F., Ruiz-Morales Y., J. Chem. Inf. Model. 2020, 60, 611–620. [DOI] [PubMed] [Google Scholar]
- 29. Xu J., Hou J., Zhang S., Xiao Q., Zhang R., Pu S., Wei Q., J. Phys. Chem. B 2006, 110, 2643–2648. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.