

Abstract
Poor water solubility and low bioavailability of active pharmaceutical ingredients (APIs) are major causes of friction in the pharmaceutical industry and represent a formidable hurdle for pharmaceutical drug development. Drug delivery remains the major challenge for the application of new small‐molecule drugs as well as biopharmaceuticals. The three challenges for synthetic delivery systems are: (i) controlling drug distribution and clearance in the blood; (ii) solubilizing poorly water‐soluble agents, and (iii) selectively targeting specific tissues. Although several polymer‐based systems have addressed the first two demands and have been translated into clinical practice, no targeted synthetic drug delivery system has reached the market. This Review is designed to provide a background on the challenges and requirements for the design and translation of new polymer‐based delivery systems. This report will focus on chemical approaches to drug delivery for systemic applications.
Keywords: Drug Delivery Systems, PEGylation, Polymer–Drug Conjugates, Polymer–Protein Conjugates, Self-Assembled Systems
Over 100 years after Paul Ehrlich's vision of the “magic bullet”, the major challenges in drug delivery remain unchanged: 1) controlling pharmacokinetic and biodistribution drug distribution and clearance in the blood; 2) solubilizing hydrophobic agents, and 3) selectively targeting specific tissues. This Review describes new alternatives for synthetic drug delivery systems for systemic applications.
1. General Aspects and Scope
The ongoing COVID‐19 pandemic has revealed the critical need for safe and effective drug delivery systems, especially for oligonucleotide‐based therapeutics and vaccines. [1] Drug delivery as a field of study traces its origins to the early 1970s, and it is certainly a mature field. But the clinical studies required for drug delivery systems mean that it can take ten to twenty years for a new drug delivery system to gain regulatory approval. Often, many important but incremental improvements are required on a new concept's path to eventual use in humans.
Over 100 years after Paul Ehrlich's vision of the “magic bullet”, [2] the major challenges in drug delivery remain unchanged: (i) controlling drug clearance in the blood; (ii) solubilizing poorly water‐soluble agents, and (iii) selectively targeting specific tissues. Although several synthetic delivery systems have addressed the first two needs and have been translated into clinical practice, no targeted synthetic drug delivery system has reached the market. This report will focus on chemical approaches to drug delivery for systemic applications. It will not address biotechnological approaches of drug delivery, [3] medical devices, [4] oral uptake, [5] or local delivery forms [6] such as sustained release forms that can be found in other reviews. [7] The long‐time paradigm that polyethylene glycol (PEG) is the “gold standard” for biomedical polymers has been challenged over the last few years as several reports on antibodies against PEG in patients were published and several PEGylated biopharmaceuticals received warnings by the regulatory institutions. [8] Therefore, a major effort is required to identify new alternatives for synthetic drug delivery systems for systemic applications by learning from the previously approved systems and taking the open challenge for targeted delivery in humans without the use of biopharmaceuticals, such as antibodies.
2. Polymer Conjugates
2.1. Polymer–Protein Conjugates
Therapeutic biomolecules like antibodies, proteins, and peptides have become an important class of drugs in the 21st century, enabling new treatment options by replacing or inhibiting native proteins or other structural targets. [9] But despite their high specificity, these so‐called biopharmaceuticals display some drawbacks like low solubility and metabolic stability. [10]
Modifying biopharmaceuticals with the synthetic macromolecule poly(ethylene glycol) (PEG) offers a powerful approach for addressing these challenges. PEGylation is the covalent attachment of one or more PEG chains to an active pharmaceutical ingredient (API) or carrier system, and it stands out among the techniques explored so far for delivering therapeutic proteins. Upon conjugation, PEG forms a stable hydration layer that usually consists of two to three water molecules per monomer unit. [11] This hydration layer increases the size of protein drugs, thereby extending their blood circulation time and metabolic stability while reducing their immunogenicity. [12] Moreover, conjugation of PEG to proteins or nanosized objects (i.e., nanoparticles and liposomes) results in the so‐called “stealth effect”, hindering the approach of plasma proteins and macrophages and thereby further increasing circulation time. [8]
The clinical success of this technology is evident: more than 14 PEGylated protein drugs are currently on the market (Table 1), with several more in clinical trials. [13] But despite this success, PEGylation suffers from a number of drawbacks, including a loss of bioactivity and the formation of anti‐PEG antibodies that can elicit immunological reactions or accelerated blood clearance, negating the core benefits of PEGylation.[ 13a , 14 ] Recent research has therefore focused on alternative macromolecules for the chemical conjugation and half‐life extension of therapeutic proteins and biomedical nanosystems.
Table 1.
Regulatory‐approved PEGylated biopharmaceuticals.
|
Trade name |
API |
PEG M w [no. of PEG per protein] |
PEG architecture/PEG linker |
Approval |
Developer |
|---|---|---|---|---|---|
|
Adagen® |
PEG‐adenosine deaminase, Pegademase |
5 kDa [11–17] |
Linear/mPEG‐SS |
1990 |
Enzon |
|
Oncaspar® |
PEG‐asparaginase, Pegaspargase |
5 kDa [69–82] |
Linear/mPEG‐SS |
1994 |
Enzon |
|
PegIntron® |
PEG‐interferon α‐2b |
12 kDa [1] |
Linear/mPEG‐SC |
2000 |
Schering‐Plough/ Enzon |
|
Pegasys® |
PEG‐interferon α‐2a |
40 kDa [1] |
Branched via lysine/mPEG‐NHS |
2001 |
Hoffmann‐La Roche |
|
Neulasta® |
PEG‐granulocyte colony stimulating factor, Pegfilgrastim |
20 kDa [1] |
Linear/mPEG‐propionaldehyde |
2002 |
Amgen |
|
Somavert® |
PEG‐human growth hormone receptor antagonist Pegvisomant |
5 kDa [4–6] |
Linear/mPEG‐NHS |
2003 |
Pfizer |
|
Macugen® |
PEG‐anti‐VEGF aptamer, Pegabtanib |
40 kDa [1] |
Branched via lysine/mPEG‐NHS |
2004 |
Pfizer |
|
Mircera® |
PEG‐erythropoietin beta |
30 kDa [1] |
Linear/mPEG‐NHS |
2007 |
Hoffman‐La Roche |
|
Cimzia® |
PEG‐certolizumab |
40 kDa [1] |
Branched via lysine/mPEG‐maleimide |
2008 |
Nektar/UCB Pharma |
|
Krystexxa® |
PEG‐ericase, pegloticase |
10 kDa [40.8 on avg.] |
Linear/mPEG‐pNPC |
2010 |
Savient |
|
Omontys® |
PEG‐erythropoietin‐mimetic peptide, peginesatide |
40 kDa [1] |
Branched via lysine/mPEG‐NHS |
2012 |
Affymax/ Takeda |
|
Plegridy® |
PEG‐interferon beta‐1a |
20 kDa [1] |
Linear/mPEG‐O2‐propionaldehyde |
2014 |
Biogen |
|
Adynovi®/Adynovate® |
PEG‐recombinant factor VIII Antihemophilic factor |
20 kDa [2 on avg.] |
Branched via glycerol/ mPEG‐NHS |
2015 |
Baxalta |
|
Refixia®/Rebinyn® |
PEG‐recombinant factor IX Antihemophilic factor |
40 kDa [1] |
Linear/conjugation by enzyme |
2017 |
Novo Nordisk |
|
RevcoviTM |
Elapegademase |
5.6 kDa [13] |
Linear/mPEG‐SC |
2018 |
Leadient Biosciences |
|
AsparlasTM |
Calaspargase pegol |
5 kDa [31–39] |
Linear/mPEG‐SC |
2018 |
Servier |
|
PalynzigTM |
PEG‐phenylalanine ammonia‐lyase, pegvaliase‐pqpz |
20 kDa [32–36] |
Linear/mPEG‐NHS |
2018 |
Biomarin |
|
Jivi® |
PEG‐recombinant factor VIII antihemophilic factor |
60 kDa [1] |
Branched/mPEG‐maleimide |
2018 |
Bayer |
Data adapted from Refs. [13, 19] or publicly available sources (EU: European public assessment reports (EPAR) and Summary of Product Characteristics). See text for further references. M w: molecular weight; avg.: average; API: active pharmaceutical ingredient. SC: succinimidyl carbonate; SS: succinimidyl succinate.
The early research on PEGylation, its further development, and PEGylated systems available on the market were comprehensively discussed previously.[ 8 , 15 ] This Review will briefly summarize the different generations of PEG–drug conjugates depending on their bioconjugation chemistry and focus on recent developments as well as future perspectives of PEGylation and PEG‐alternative macromolecules. The earliest research on PEGylation dates to 1977, when Abuchowski and Davies examined its impact on the immunogenicity of the model proteins bovine liver catalase (BLC) and bovine serum albumin (BSA). [16] PEGs used for protein modification generally have a molecular weight between 2 and 40 kDa, display a low dispersity (<1.1), and are typically used as linear polymers or alternatively branched architectures. [17] The monofunctional methoxy‐PEG (mPEG) is typically used to avoid cross‐linking. [18] In addition to its molecular weight, which mostly affects blood circulation time and bioavailability, another key trait of mPEG is its linker chemistry, which plays a crucial role in determining conjugate stability.
The first generation of PEGs are characterized by low molecular weights (<12 kDa) and linear architectures and are mainly used for random PEGylation on the amine group of lysine residues. Both approved first‐generation PEG–protein conjugate drugs, Adagen® and Oncaspar®, employ this conjugation strategy. Furthermore, both were synthesized using an unstable succinimidyl succinate linker (PEG‐SS), which contains an ester bond that is prone to hydrolysis at neutral pH. [20] Unstable linkers like PEG‐SS often serve as a new hapten on the protein surface, enhancing immunogenicity as seen with PEG‐asparaginase. The second generation of PEGylated proteins consist mainly of PEGs with higher molecular weights (>12 kDa) and a more stable linker, and they tend to use branched architectures. These so‐called Y‐shaped PEGs can be synthesized on the base of a lysine core and are more effective than their linear analogs in reducing antigenicity, immunogenicity, and proteolysis.[ 17 , 21 ] The first blockbuster within this group was surely the PEGylated form of IFN‐α2a (Pegasys®), marketed since its 2001 approval by the company Roche. mPEG‐N‐hydroxysuccinimide (mPEG‐NHS), which is used to modify IFN‐α2a, is an improved version of the initially used PEG‐SS, in that it results in a more stable amide linker. [22] The reactivity of mPEG‐NHS can be tuned by adjusting the spacer between the polymer backbone and the reactive NHS group. [23]
The third generation of PEGs aimed to diminish the loss in bioactivity after PEGylation. Therefore, researchers set out to attach PEG to the protein in a more site‐selective manner. The first product using site‐selective PEG chemistry was approved in the year 2002: Pegfilgrastim (Neulasta®) is agranulocyte‐colony stimulating factor (G‐CSF) modified with a 20‐kDa linear PEG at its N‐terminus. [24] Compared to other linker chemistries mentioned above, this strategy enables charge retention in the native protein. [25] Another site‐selective PEGylation approach is to target thiol groups on cysteines. Cysteines are rarely present in proteins; when they are, they tend not to be suitable for conjugation, owing to their hydrophobic nature, they are often located in the active binding site or interior of a protein. However, genetic engineering allows the introduction of cysteines far away from the active site, thereby enabling modification with PEG. [18] Certolizumab Pegol (Cimzia®), approved in 2008, is the first PEGylated protein on the market that uses thiol‐maleimide PEGylation. Cimzia®, an antibody Fab fragment that is directed against tumor necrose factor α (TNF‐α), bears an engineered cysteine modified with a 40‐kDa branched PEG‐maleimide. [26] Another example of this conjugation strategy is PEGylated factor VIII (Jivi®), which in 2018 became the first approved site‐specific PEGylated blood factor. [27]
An even more selective way to conjugate PEG to proteins is by using strain‐promoted or CuI‐catalyzed azide–alkyne cycloaddition (Sp‐AAC, CuAAC). This approach demands genetic engineering of unnatural amino acids into the protein sequence, which can then be conjugated to the polymer in question. [28] However, site‐specific coupling is not always feasible and can lead to low yields, and so this technology remains limited to the lab scale.
Site‐specific PEGylation can also be achieved with enzymes. Usually, the protein moiety to be PEGylated is genetically modified with a tag that allows enzyme‐mediated ligation of the PEG substrate that carries a certain functional group. [29] Typical enzymes used in this approach are sortase [30] and transglutaminase, but others are also possible. [19b] Enzymatic PEGylation was already successfully demonstrated for several biomolecules [31] and even made it onto the market in the form of Rebinyn®/Refixia®, a PEGylated recombinant blood coagulation factor IX from Novo Nordisk that was approved in 2017.
2.1.1. Future Perspectives on PEGylation
With several candidates currently in clinical trials, PEG will remain the preferred macromolecule for the delivery and half‐life modulation of therapeutic proteins for the next several years. [32] PEG is generally considered a safe, non‐toxic excipient, but has recently faced challenges regarding its tendency to incite the formation of antibodies when conjugated to a protein or nanocarrier system. These anti‐PEG antibodies were found to diminish the initial benefits of PEGylation, leading to accelerated blood clearance and sometimes immunological reactions.[ 13a , 33 ] Even in healthy populations that had never had contact with a PEGylated therapeutic, anti‐PEG antibodies were found (up to 72 % were found in 2016 by Yang et al.), presumably due to the widespread presence of PEG in household and cosmetic products.[ 14d , 34 ] Rare anaphylactic reactions reported for BioNTech–Pfizer's recently approved Comirnaty® vaccine against SARS‐CoV‐2 are suggested to be caused by the 2‐kDa PEG moieties on the surface of their nanoparticle formulation. [35] While so far only two PEGylated protein drugs have been removed from the market—Krystexxa® in the EU in 2016, and Omontys® in the EU in 2013, due to hypersensitivity against its drug part—the formation of anti‐PEG antibodies is well‐documented in the product characteristics summaries of many approved PEGylated proteins (publicly accessible files at EMA (EPAR), e.g., for Plegridy®, PalynzigTM or Jivi®), leading also to a special warning about PEG‐related hypersensitivity in the case of Jivi®. Time will tell whether such antibodies emerge as a larger patient safety issue, but PEG‐associated immunogenicity is still generally considered a rare event, with the benefits of PEGylation outweighing its drawbacks. Regarding PEG chemistry, next‐generation PEGylation will most likely aim to improve site‐specific protein conjugation methods to mitigate loss in bioactivity and to enable PEGylated products as potent as their unmodified analogs. Enzymatic PEGylation could be a way to address this issue, though upscaling of the process remains a problem. [19b] Other studies report the synthesis of biodegradable PEG [36] and reduction‐responsive PEGylation, [37] which could suggest a path to preventing the accumulation of larger PEG moieties within organs.
2.1.2. PEG‐Alternative Macromolecules
Several PEG‐alternative macromolecules for half‐life extension and delivery of therapeutic proteins have already reviewed in the literature. [38] Here we focus on approaches involving a chemical conjugation between polymer and protein, necessarily excluding other promising biotechnological half‐life extension strategies like XTENylation, PASylation, and fusion proteins.
2.1.2.1. Polyglycerols (PGs)
Polyglycerols, also termed as polyglycidols, are a class of polyether‐based macromolecules containing side‐chain methyl hydroxy groups (Figure 1). Their high hydrophilicity and water solubility make them attractive for a variety of potential applications, for example as stealth polymers to prevent protein adsorption on surfaces or as responsive scaffolds for drug delivery. [39] PGs are considered to have good biocompatibility profiles and low toxicity. [40] In contrast to PEG, PGs are significantly more hydrophilic as indicated by the water contact angle of their monolayers on a gold surface (34° for PEG vs. 20° for PG). [39a] This also results in a lower unspecific protein binding and an extremely low protein corona of PG‐coated nanoparticles. [41] Additionally, the circulation half‐life of high‐molecular‐weight LPG (100 kDa) was shown to be longer than that of many other linear polymers like PEG, polyvinyl alcohol (PVA), or hydroxypropyl acrylamide (HPMA), suggesting polyglycerols’ great potential for extending the mean residence time of protein therapeutics. [40c] PGs are typically synthesized by anionic ring‐opening polymerization (AROP) of the protected monomer ethoxy ethyl glycidyl ether (EEGE), with various architectures possible, e.g., linear (LPG), hyperbranched (HPG), or dendronized brush‐type (denPG), among others. [42] Different backbone functionalities can be introduced by varying the monomer, but EEGE is the most commonly used. [43]
Figure 1.

Overview of PEG‐alternative macromolecules for protein and drug conjugation: poly(ethylene glycol) (PEG), linear polyglycerol (LPG), poly(2‐oxazoline) (POx), polycarboxybetaines (PCB), polysulfobetaines (PSB), poly(2‐methacryloyloxyethyl phosphorylcholine) (PMPC), polyglutamic acid (PGA), polysarcosine (PSar), poly(N‐(2‐hydroxypropyl) methacrylamide) (PHPMA), and polysialic acid (PSA).
For selective conjugation on proteins, polymers with a monofunctional end group are desired; this configuration can be achieved by using a suitable initiator, for example tetraoctylammonium bromide or azide. [44] Other successfully generated mono‐LPGs include LPG‐NH2, [45] LPG‐SH,[ 39a , 45b , 46 ] LPG‐propargyl, [47] LPG‐cyclooctyne, [42a] and LPG‐vinylsulfonate. [48] The end‐functionalized LPGs can be modified with a short linker or used directly for the attachment to proteins.
Tully et al. used LPG‐aldehyde of various molecular weights (5–40 kDa) for the N‐terminal ligation of the therapeutic protein anakinra by using a reductive amination approach. [49] PEG‐anakinra conjugates of similar molecular weights were synthesized by the same coupling strategy and served as reference material. LPG conjugates displayed a slightly more compact hydrodynamic size compared to PEG analogs of same molecular weight, whereas the in vitro receptor affinity decreased in a molecular‐weight‐dependent manner, with no significant differences observed between PEG and LPG. The terminal half‐life of anakinra modified with a 40 kDa LPG moiety in mice was extended fourfold compared to the native protein, into the same range as its PEG analog of similar molecular weight. The same LPG‐aldehyde compounds were also used for the N‐terminal modification of the protein interleukin‐4. [49]
Another study by our group used LPG‐N3 (10, 20, 40 kDa) for the C‐terminal modification of propargyl‐modified exenatide, a small protein used to treat diabetes mellitus type II. [50] Conjugation was enabled by CuAAC, and PEG variants of similar molecular weights again served as points of comparison. The in vitro bioactivity of exenatide decreased after polymer conjugation, whereas LPG and PEG showed similar behavior before and after conjugation. Moreover, a single injection of a 40 kDa LPG‐exenatide conjugate in diabetic mice caused a significant reduction of blood glucose for up to 72 h, a performance comparable to its PEG analog, confirming the potential of LPG to extend the therapeutic activity of protein drugs. Other studies report LPG conjugates of interferon‐α2a, [42a] lysozyme and bovine serum albumin (BSA), [45a] with BSA also being used for a “grafting‐from” approach. [51]
2.1.2.2. Poly(2‐oxazoline) (POx)
Polyoxazolines (POx) are a promising polymer class with several potential applications in the area of drug delivery (Figure 1). [52] They are generated by cationic ring‐opening polymerization (CROP) and are often termed as pseudo‐polypeptides, as they contain a peptide bond on each of their repeating units. [53] To terminate the polymerization, nucleophiles (e.g., OH−, −NH−, −S−, or −COO−) are added that allow the synthesis of end‐functional POx for selective protein modification. [53b] Additionally, side‐chain functionalities can be introduced by varying the monomer to influence the thermal properties or solubility of POx. The two most studied forms for protein conjugation are 2‐methyl‐2‐oxazoline (PMeOx) and the slightly more hydrophobic 2‐ethyl‐2‐oxazolines (PEtOx), which both display good biocompatibility in vitro. [54] Mero et al. already demonstrated the successful conjugation of PEtOx to the therapeutically relevant protein G‐CSF. Linear PEtOx of 5, 10, and 20 kDa were equipped either with a terminal aldehyde group for chemical N‐terminal ligation or with a terminal amine group for enzymatic transglutaminase (TG)‐mediated conjugation. [55] The PEtOx conjugates displayed somewhat diminished bioactivity in vitro but led to a higher therapeutic activity in vivo, with the PEtOx conjugate synthesized via TG performing slightly better. Other studies describe the conjugation of PEtOx to RNAse, catalase, uricase, insulin, and erythropoietin (EPO) by using PEtOx‐NHS to target lysine residues.[ 53b , 56 ] A site‐specific coupling strategy for PEtOx was exploited by Hauptstein et al., who modified interferon‐α2a with either PEtOx, LPG, or PEG of 10 kDa by using bioorthogonal SpAAC. The respective PEtOx bioconjugates displayed a similar bioactivity in vitro but slightly lower thermal stability than their PEG and LPG analogs. [42a] Another example of the site‐specific coupling of POx can be found in the work of Lühmann et al., who successfully used biorthogonal CuAAC to selectively attach PMeOx (4 kDa) to interleukin‐4. [57]
2.1.2.3. Polyzwitterions (PZIs/Polybetaines)
Polyzwitterions (PZIs), also named polybetaines (Figure 1), are macromolecules that, while neutrally charged overall, feature a positively and a negatively charged functional group on the same monomer. Generated by controlled radical polymerization (RAFT or ATRP), they comprise a poly(meth)acrylic acid or amide backbone along with both a cationic quaternized ammonium group moiety and an anionic moiety consisting of sulfonate, carboxy, or phosphate groups. Several subclasses can be described, including polysulfobetaines (pSB), polycarboxybetaines (pCB), or polyphosphobetaines (pPB). [58] Their strong hydration, high hydrophilicity, and biocompatibility make them a promising polymer class with interesting properties like strong electrostatic interactions and anti‐fouling behavior. [59]
Most PZI–protein conjugates reported in the literature were synthesized by a “grafting‐to” approach, but some of the available studies used an in situ “grafting‐from” method in which the biomolecule is modified with an initiator before polymerization. [60] Hu et al. expressed IFN‐α bearing a short C‐terminal tag, which was used for sortase‐mediated attachment of an initiator for in situ polymerization of poly(2‐methacryloyloxyethyl phosphorylcholine) (PMPC), a derivative of pPB. [60a] Their IFN‐PMPC conjugate with a polymer mass of 57 kDa outperformed the commercial Pegasys® (40 kDa polymer) in terms of in vitro bioactivity but showed comparable circulation time and tumor accumulation in vivo. Sortase can also be used to introduce a small azide linker on the protein, which can be subsequently attached to a polymer‐alkyne as successfully demonstrated for PMPC conjugates of exenatide. [61]
Formation of antipolymer antibodies can be a problem in the case of PEG, as they are often associated with immunological reactions or with accelerated blood clearance of PEGylated proteins and nanocarriers.[ 13a , 62 ] An interesting study by Jiang and co‐workers addressed this issue by quantifying the amount of antipolymer antibodies formed in mice after repeated injections of PEG and pCB conjugates of proteins with varying immunogenicity. [63] The conjugates were generated by conjugating PEG‐SH or pCB‐SH of 5, 10, or 20 kDa to the respective proteins, which had been modified with a short maleimide linker. SPR and ELISA techniques confirmed an increased amount of anti‐PEG‐IgM and ‐IgG antibodies which were correlated to the immunogenicity of the protein moiety, confirming the haptenic character of PEG. In contrast, in the case of the pCB conjugates minimal to virtually no anti‐pCB antibody formation was detected.
2.1.2.4. Polypeptides (PPs)
Polypeptides (PPs) are biodegradable macromolecules that are generated by the polymerization of highly reactive N‐carboxyanhydrides as monomers. [53a] Two main types of PPs are used for protein conjugation: poly‐γ‐glutamic acid (γ‐PGA) and the polypeptoid polysarcosine (PSar, poly(N‐methylglycine); Figure 1).
γ‐PGA is FDA‐approved for use in cosmetics and has already been successfully conjugated to IFN and other biomolecules. [64] A study by Lu and co‐workers pointed out the importance of PGA conformation within IFN‐PGA conjugates impacting various properties like activity or stealth behavior towards the immune system. In short, a “brush‐type” PGA (20 kDa, bearing three ethylene glycol units per monomer) with either unstructured (DL‐PGA) or helical (L‐PGA) conformation was conjugated to IFN's N‐terminus. [64b] Thereby, the rigid, helical L‐PGA‐IFN led to higher antitumor activity in vitro and in vivo and also prevented accelerated blood clearance by anti‐polymer antibodies, which contrasted with the unstructured DL‐PGA‐IFN and a similarly synthesized PEG‐IFN conjugate. Comparable results were found for the protein human growth hormone within the same study.
2.1.2.5. Polysarcosine (PSar)
Polysarcosine (PSar) is a polymer based on the endogenous, non‐proteinogenic amino acid sarcosine and displays stealth properties similar to PEG with, however, a smaller chain flexibility in solution. [65] PSar has already been exploited as a PEG alternative for the surface modification of lipid nanoparticles to deliver genetic material (RNA, DNA), a system comparable to the current SARS‐Cov2 vaccine developed by the company BioNTech. [66] Grafted on liposomes, PSar was able to prevent accelerated blood clearance and exhibited lower anti‐polymer antibody‐formation upon repeated administration than PEG. [67] To our knowledge, the first successful conjugation of PSar to a therapeutic protein was reported by Lu and co‐workers, who synthesized N‐terminal PSar conjugates of IFN. [68] These conjugates showed higher in vitro activity and slower in vivo tumor growth than their respective PEG analogs, while their terminal half‐life was comparable. Furthermore, PSar was better able to prevent the formation of anti‐IFN antibodies upon repeated administration, suggesting an improved immunocamouflage effect over PEG.
2.1.2.6. Polysialic Acid (PSA)
Polysialic acid (PSA) is a highly hydrophilic, linear, and negatively charged macromolecule found in capsules of several Gram‐negative bacteria (e.g. Escherichia coli) and consists of sialic acid moieties linked via α‐glycosidic bonds. [69] It naturally serves as a stealth polymer for bacteria, preventing accelerated blood clearance and detection by the immune system. The various isotypes of PSA strongly impact its circulation time in vivo, where the degree of phospholipid acylation and the type of glycosidic linkage play a major role. [70] Additionally, charge repulsion at the glomerular membrane can diminish PSA elimination and extend its circulation time. [71] PSA displays sufficient stability at physiological pH, but cellular neuraminidases can degrade it and prevent its accumulation in organs. [72] Grafted onto liposomes, PSA was shown to prevent IgM antibody formation and thereby mitigate accelerated blood clearance, a problem often observed with PEGylated liposomes. [73] PSAylation technology is commercialized under the trade name PolyXen™ and is currently offered by the company Xenetic Biosciences (formerly Lipoxen PLC).
For conjugation to proteins, PSA from the E. coli K1 strain (also termed colominic acid, or CA) is mainly used. The latter contains a single vicinal diol at its non‐reducing end, which can be oxidized under mild conditions (e.g. by NaIO4) to generate an aldehyde group for direct protein conjugation or further modification with bifunctional linkers. [74] Others also reported the successful enzymatic conjugation of PSA. [75] The direct N‐terminal attachment of PSA‐aldehyde by reductive amination has already been exploited for a large variety of proteins, including insulin, erythropoietin (EPO), and deoxyribonuclease I, [76] with the two latter even evaluated in clinical trials. [76e] Interesting findings regarding tissue permeability were also obtained: PSAylation of an antibody fragment led to a 30‐fold higher tumor uptake in comparison with its unmodified version. [77]
2.2. Polymer–Drug Conjugates
Polymer–drug conjugates (PDCs), or polymeric prodrugs, are one of the drug delivery tools in nanomedicine in which one or more therapeutic agents are covalently bound to a polymeric carrier. Conjugation of a therapeutic agent to a polymer offers several advantages, including increased blood circulation time, controlled delivery, and improved pharmacokinetics, along with highly improved water solubility, reduced toxicity, and intracellular delivery. This conjugation strategy has mostly been applied for potent anti‐tumor drugs with high cytotoxicity and poor solubility.
For the first PDC, reported by Jatzkewitz in 1955, mescaline was conjugated to a copolymer of N‐vinylpyrrolidone and acrylic acid through a dipeptide spacer. [78] In 1975, Ringsdorf proposed a conceptual framework in which an ideal PDC is defined by the conjugation of a pharmaceutically active agent to a biocompatible polymeric backbone (Figure 2). In this model, additional targeting moieties and water‐solubilizing groups can also be attached to the backbone to improve therapeutic efficiency. [79]
Figure 2.
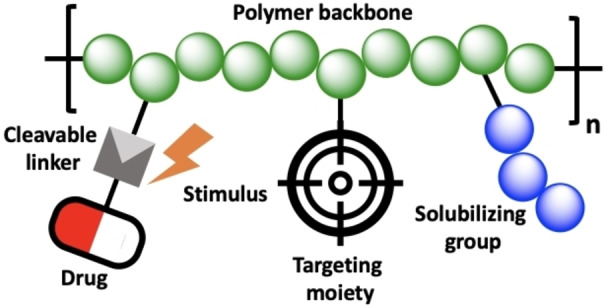
Conceptual scheme of the Ringsdorf model.
The field was then propelled by pioneering work in the late 1970s and early 1980s, when the research of Kopecek and Duncan brought the first progress toward clinical trials. [80] The progress in polymer chemistry since these groundbreaking works has led to a remarkable growth in the number of suitable polymers, with structures that can be tailored to design and optimize delivery systems. The biocompatible polymers with hydrophilic backbones that have been transferred to clinical use include synthetic polymers such as PEG, N‐(2‐hydroxypropyl)methacrylamide copolymers, poly(vinylpyrrolidone), poly(ethyleneimine), and linear polyamidoamines; natural polymers including polyglucose, dextrin, hyaluronic acid, and chitosans; as well as pseudosynthetic polymers such as poly(amino acids), poly(L‐lysine), poly(glutamic acid) (PGA), poly(malic acid), and poly(aspartamides). [81] Although several polymer–drug conjugates are in clinical trials, none has yet entered the market.
The general example provided by Ringsdorf's conceptual framework (Figure 2) has developed into an enormous list of linear PDCs designed and investigated for cancer therapy (Table 2). Therapeutic and targeting agents are linked to a hydrophilic polymeric backbone to enhance their circulation time in the body, increase the system's water solubility, reduce unwanted toxicity, and target the drug to the desired tissues. The chemistry of the linker also plays an important role: it must be stable enough to avoid premature drug release, but it must also facilitate drug release at the site of action in response to a change in pH, the presence of enzymes, or sensitivity to overexpressed molecules in the tumor microenvironment. [82] The drug can be linked to the polymer backbone either by post‐conjugation to the already synthesized polymer or by conjugation to the monomer before polymerization. Conjugation of the drug to the monomer before polymerization offers control over the density of the final conjugated drug, but the risk of interfering with the polymerization process should be considered. [83]
Table 2.
Overview of polymer–drug conjugates in clinical trials.
|
Trade name |
API |
Polymer structure |
Indication |
Trial phase |
Developer |
|---|---|---|---|---|---|
|
PK1 |
Doxorubicin |
Poly(N‐(2‐hydroxypropyl)methacrylate) |
Breast cancer, non‐small‐cell lung cancer, colorectal cancer |
II |
CRC/Pharmacia |
|
PK2 |
Doxorubicin |
Poly(N‐(2‐hydroxypropyl)methacrylate) |
Primary/metastatic liver cancer |
II |
CRC/Pharmacia |
|
AP5280 |
Platinum |
Poly(N‐(2‐hydroxypropyl)methacrylate) |
Various malignancies |
II |
Access Pharmaceuticals |
|
ProLindacTM/AP5346 |
Oxaliplatin |
Poly(N‐(2‐hydroxypropyl)methacrylate) |
Ovarian cancer |
II |
Access Pharmaceuticals |
|
PNU166945 |
Paclitaxel |
Poly(N‐(2‐hydroxypropyl)methacrylate) |
Breast cancer, solid tumors |
I |
Pharmacia |
|
MAG‐CPT, PNU166148 |
Camptothecin |
Poly(N‐(2‐hydroxypropyl)methacrylate) |
Advanced solid malignancies |
I |
Pharmacia |
|
CRLX‐101 |
Camptothecin |
PEG‐cyclodextrin |
Ovarian, peritoneal, and fallopian tube cancer |
II |
Blue link Pharmaceuticals |
|
NKTR‐118 |
Naloxol |
PEG |
Opioid‐induced constipation |
III |
Nektar Therapeutic |
|
NKTR‐102 |
Irinotecan |
PEG |
Breast cancer |
III |
Nektar Therapeutic |
|
NKTR‐262 |
TLR7/TLR8 agonist |
PEG |
Solid tumors |
I/II |
Nektar Therapeutics |
|
NKTR‐105 |
Docetaxel |
PEG |
Solid tumors |
I |
Nektar Therapeutics |
|
Onzeald |
Irinotecan |
PEG |
Breast cancer |
III |
Nektar Therapeutic |
|
Opaxio™/Xyotax™/CT‐2103 |
Paclitaxel |
Polyglutamic acid |
Ovarian cancer, peritoneal cancer, and fallopian tube cancer |
III |
CTI BioPharma |
|
CT‐2106 |
Camptothecin |
Polyglutamic acid |
Colon cancer, ovarian cancer |
I/II |
Cell Therapeutics |
|
OsteoDex |
Alendronate |
Dextran |
Prostate cancer |
II |
DexTechMedical |
|
Somadex |
Somatostatin |
Dextran |
Neuroendocrine tumors |
II |
DexTechMedical |
|
BP‐C1 |
PtII |
Benzo‐polycarbonic acid polymer |
Breast cancer |
II |
Meabco A/S |
|
DFP‐13318 |
SN38 |
PEG |
Solid tumors |
I |
ProLynx |
|
Delimotecan |
T2513 |
Carboxymethyl dextran |
Various malignancies |
I |
Daiichi Pharmaceuticals |
|
EZN‐2208 |
SN38 |
PEG |
Solid tumors |
II |
Enzon |
|
Fleximer®/XMT‐1001 |
Camptothecin |
poly(1‐hydroxymethylethylene hydroxymethylformal) |
Gastric cancer, lung cancer |
I |
Mersana |
|
PROTHECAN |
Camptothecin |
PEG |
Lung cancer |
II |
Enzon |
2.2.1. Poly‐N‐(2‐hydroxypropyl)methacrylamide (PHPMA)
When research began on effective anticancer PDCs, linear copolymers of N‐(2‐hydroxypropyl)methacrylamide (HPMA) were among the most studied water‐soluble polymers due to their excellent biocompatibility and nonimmunogenic properties. Furthermore, the HPMA copolymers used in these first studies were biocompatible in vitro with LEP and HeLa cell lines and did not stimulate antibody formation in vivo. [84] Subsequent clinical studies proved that HPMA copolymer could be tolerated in the human body even at concentrations more than 20 g m−2 without immunogenicity or polymer‐related toxicity. HPMA has been used as an N‐substituted methacrylamide monomer since the α‐carbon substitution and the N‐substituted amide bond ensure the hydrolytic stability of the resulting polymer's side chains. HPMA copolymers offer a high loading capacity, featuring multivalent backbones on which a high number of drugs can be covalently linked. [85] Building on Kopecek and colleagues’ work in the 1990s, the first passive targeting PDC to enter clinical trials was HPMA copolymer‐doxorubin (PK1), consisting of an HPMA copolymer conjugated to doxorubicin (DOX) via a degradable tetrapeptide linker. [86] Shortly after PK1 was investigated in phase I clinical trials, PK2 entered trials as a sister compound for treating liver hepatocytes. PK2 started with the same structure as PK1 and added galactosamine as targeting agent. Although preclinical and phase I clinical studies of PK1 and PK2 demonstrated that the attachment of DOX to HPMA copolymer enhances the plasma circulation half‐life from 5 min to 1 h as compared to free DOX, the studies stopped at phase II due to the lower efficacy compared to an animal study and the lack of efficient tumor accumulation. HPMA copolymer‐paclitaxel (PNU166945) conjugates were developed by linking the drug to a 30‐kDa copolymer via an ester bond with a loading capacity of 5 wt %. [87] PNU166945 was evaluated in phase I clinical trials on just 12 patients. The studies were discontinued due to the serious neurotoxicity of free paclitaxel (PTX) found in rats. It is speculated that after the cleavage of the ester bond in blood circulation, the free PTX can pass through the blood–brain barrier, while it is known that the polymer‐PTX conjugate cannot cross this barrier. [88] HPMA copolymer‐camptothecin (CPT) was developed by esterification of the −OH group of CPT using a glycine residue, followed by conjugating the modified CPT to the polymer through a pendant glycylaminohexanoyl spacer. [89] High bladder toxicity and a lack of apparent antitumor activity, possibly due to the rapid hydrolysis of ester linkage, halted the HPMA‐CPT study in phase I of clinical trials. Two other HPMA copolymer‐drug conjugates, AP5280 (carboplatin) and AP5346 (oxaliplatin) have been investigated in phase II clinical trials for the treatment of various malignancies as well as ovarian cancer. [90] In these formulations platinum derivatives are linked to HPMA copolymers via a glycyl‐phenylalanyl‐leucyl glycine tetrapeptide (GFLG) spacer that is responsive to pH and Cathepsin B. [91] The structure and size of HPMA copolymer‐drug conjugates also play an important role in their antitumor efficiency. Nakumura et al. [92] showed that the starlike HPMA copolymer‐pirarubicin conjugate (400 kDa, 26 nm), based on a PAMAM dendrimer, outperforms linear HPMA copolymer‐pirarubicin conjugate (39 kDa, 8.2 nm) in tumor growth inhibition in S‐180 tumor‐bearing mice; in both systems the drug was conjugated via a hydrazone linker.
2.2.2. Polyglutamic Acid
Together with polyaspartic acid, polyglutamic acid (PGA) is among the most used poly amino acids for drug delivery. PGA features a pendant carboxylate group, which gives the polymer a negative charge and provides functionality for drug conjugation. PGA is usually synthesized either chemically, by polymerization of N‐carboxyanhydride of L‐glutamic acid, or in a biosynthetic route expressed in certain types of bacteria. [93] Its biocompatibility, biodegradability, non‐immunogenic property, and water solubility make PGA a suitable candidate for drug delivery applications. PGA has been widely used for drug delivery in linear [94] and branched structures [95] as a homopolymer or amphiphilic block copolymer that can form micellar aggregations. [96]
The PGA–drug conjugation can be performed in different ways: the anticancer drug can be directly attached to the polymer via an ester bond [97] or a cleavable linker, [98] or in a simpler process PGA can form an ionic complex with a positively charged drug due to its polyanionic feature. [99] Linker chemistry has been shown to have an important effect on the biological activity of conjugates. In a comparative study, the Vicent group discovered that using a small, flexible glycine linker for drug conjugation causes higher in vivo activity as compared to direct attachment or using more bulky linkers; this effect is due to the linker's effect on drug release kinetics, size, secondary structure, and internal arrangement of conjugates. [100] Opaxio™ and CT‐2106 [101] are PGA–drug conjugates currently being evaluated for cancer treatment, in which paclitaxel and camptothecin are linked via ester bonds (Figure 4). OpaxioTM, also known as CT‐2103, Xyotax, and paclitaxel poliglumex, is a PGA‐paclitaxel conjugate developed by CTI BioPharma in which paclitaxel is attached to polyglutamic acid. Preclinical studies showed stability in blood circulation, lower toxicity, and higher tumor accumulation as compared to conventional paclitaxel. OpaxioTM contains 37 wt % paclitaxel linked to polymer, which is cleaved by cathepsin B to release diglutamyl‐paclitaxel. [102] The conjugate has been tested in phase III clinical trials for ovarian and lung cancer treatment, but has not been approved to enter the market, given that paclitaxel poliglumex caused neurotoxicity at a dose of 210 mg m−2. [103]
Figure 4.
Architectures and categories of supramolecular drug delivery systems.
2.3. Dendrimer–Drug Conjugates
In contrast to linear polymers, dendrimers are a class of macromolecules characterized by highly branched and well‐defined architectures. The components of a dendrimer are (i) an initiator core, (ii) generations of repeating units attached to this inner core, and (iii) terminal functional groups attached to the outermost generation. [104] Dendrimers show great potential as drug delivery systems due to their globular structure, high functionality, and controlled size (1–15 nm), along with their rapid cellular uptake, eruption through the endothelial lining and capillary walls, penetration through biological barriers, targetability, and their ability to increase the solubility of hydrophobic drugs. [105] Yet the only dendritic systems to enter clinical trials and reach the market are poly‐L‐lysine (PLL) dendrimers and derivatives (www.starpharma.com) (Table 3). Nevertheless, the dendrimer polyamidoamine (PAMAM) also shows strong potential for application in dendrimer–drug conjugate systems; still the stability of PAMAM dendrimer can be a limiting factor since it can undergo retro‐Michael reactions (β‐eliminations) at high temperature or pH, which may be necessary during the synthesis. [106]
Table 3.
Overview of dendrimer–drug conjugates.
|
Trade name |
API |
Dendritic structure |
Indication |
Trial phase |
Developer |
|---|---|---|---|---|---|
|
Vivagel® |
– |
PEGylated PLL |
Antiviral activity |
Marketed |
Starpharma |
|
DEP® docetaxel |
Docetaxel |
PEGylated PLL |
Lung, prostate cancer |
II |
Starpharma |
|
DEP®CABAZITAXEL |
Cabazitaxel |
PEGylated PLL |
Prostate, ovarian cancer |
I/II |
Starpharma |
|
DEP®IRINOTECAN |
Irinotecan |
PEGylated PLL |
Colorectal, pancreatic cancer |
I/II |
Starpharma |
|
AZD0466 |
AZD4320 |
PEGylated PLL |
Dual Bcl2/xL inhibitor |
II |
Starpharma/AstraZeneca |
|
DEP®GEMCITABINE |
Gemcitabine |
PEGylated PLL |
Pancreatic, lung cancer |
Preclinical |
Starpharma |
2.3.1. Dendritic Poly‐L‐Lysine (PLL)
DEP® docetaxel has progressed furthest in clinical trials among dendrimer–drug conjugates and is currently in phase II (EudraCT number: 2016‐000877‐19). In trials, DEP® docetaxel has been shown to cause less neutropenia and lower excipient toxicity than Taxotere®. [107] Starpharma's first marketed dendrimer is SPL7013 (Vivagel®), a therapeutic dendrimer for preventing HIV and herpes simplex virus (HSV) infections. SPL7013 is a fourth‐generation poly‐L‐lysine dendrimer that contains a divalent benzhydrylamine (BHA) core and 32 naphthalene disulfonic acid groups at the surface, bearing a molecular weight of 16 581 kDa. [108] The end groups provide the dendritic surface with a high anionic charge and impart hydrophobicity. [109] SPL7013 has shown in vitro activity against HIV‐1 clades and HIV‐2 by inhibiting viral attachment and entry. SPL7013 has also demonstrated low toxicity in cervical and colorectal epithelial cell lines and cannot disrupt intercellular tight junctions of polarized epithelial cells. [110]
Patterson et al. reported a dendrimer–drug conjugate in which AZD4320 was chemically conjugated to the free lysines on a PEGylated fifth‐generation poly‐L‐lysine dendrimer with glutarate, thiol diglycolate, and diglycolate as chemical linkers (Figure 3). The dendrimer's molecular weight was approximately 105 kDa, and its loading was reported at 24–30 wt % (25–42 AZD4320 molecules). Its solubility in aqueous buffer is higher than 100 mg ml−1, and it has a hydrodynamic diameter of approximately 10 nm (polydispersity index, PDI<0.2). [111] AZD4320‐dendrimer conjugates significantly improved drug solubility, which would ease intravenous administration. AZD4320 is a potent dual Bcl‐2/Bcl‐xL inhibitor that has shown good efficacy, but its cardiovascular toxicity has prevented its clinical development as a standalone therapeutic. The dendrimer–drug conjugate has shown efficacy and cardiovascular tolerability in preclinical models, allowing it to proceed into clinical development. [111]
Figure 3.

Chemical structure of AZD0466 and dendrimer‐AZD4320 conjugates showing the dendrimer structure of PEGylated PLL and the linkers used. Adapted from Ref. [111].
On the other hand, Fréchet et al. reported a core‐functionalized, symmetrically PEGylated poly‐L‐lysine dendrimer–drug conjugate with a 40 kDa molecular weight that was loaded at 4–6 wt % with polymer‐conjugated camptothecin. [112] Whereas the free polymer‐camptothecin conjugate was eliminated from the blood after 30 min and showed poor tumor accumulation, the dendritic system improved blood circulation half‐life up to 30.9±8.8 h and enhanced tumor uptake to 4.2±2.3 % of the injected dose per g of tissue. [112]
AstraZeneca and the Northern Institute of Cancer Research developed a dendrimer–drug conjugate where a 5th generation poly‐L‐lysine that had been partially modified with a polyoxazoline conjugate bonded to SN‐38, the active metabolite of irinotecan, to improve the therapeutic index of the drug. The system was tested using different linker technologies (ester, primary amine carbamate, and secondary amine carbamate linkers) to compare the pharmacokinetic profiles of these different versions of the system. The conjugates were tested in a SW‐620 mouse xenograft model. The conjugate with an ester linker exhibited a medium release (half‐life of 21 h) and achieved regression of the SW‐620 tumor due to prolonged circulation in the blood and effective administration of the drug. [113]
2.3.2. Dendritic Polyamidoamine (PAMAM)
PAMAM dendrimers are characterized by high amino group density and abundant functional end groups, which mark it as a candidate for drug delivery applications. [114] Yan‐Yan Jiang et al. reported a PEGylated PAMAM (64 surface primary amino groups, molecular weight of 14 kDa) covalently coupled to methotrexate (MTX) via amide linkage. The conjugates showed stability in human plasma and lysosomal media, and when injected in rodents they demonstrated prolonged blood residence time and effective antitumor effects as compared to free MTX. [115]
2.3.3. Recent Developments on PDCs
Recent progress in polymer chemistry has yielded new PDCs with improved pharmacological properties, resulting in several important achievements that have addressed key constraints of these systems. [116] One challenge with many current PDCs is the non‐degradability of the polymer commonly used for this application. This problem has inspired several recent attempts to synthesize degradable polymers by incorporating cleavable moieties into the polymer backbone. [117] Another key point is the drug loading that has been addressed by the use of highly branched polymers and dendrimers as multifunctional platforms, a role where these dendritic polymers excel as an alternative to di‐end‐functional linear polymers such as PEG. [111] Also surface‐charge‐switchable PDCs have been developed that switch charge from negative to positive within the tumor tissue, enabling the transendothelial and transcellular transport of the drug. [118]
3. Supramolecular Drug Delivery Systems
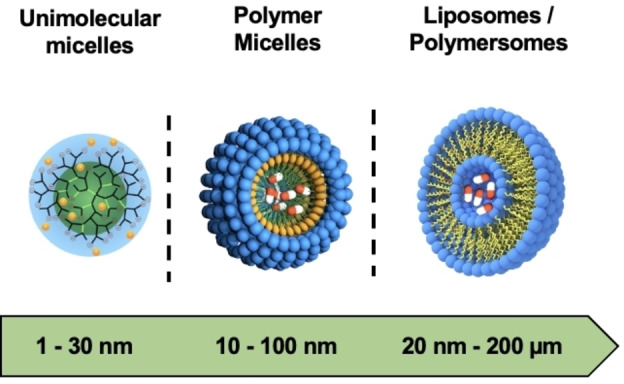
In the previous sections, various polymeric building blocks for the transport of APIs have been described. Depending on the application, carriers with a larger functional surface area and tunable physico‐chemical and mechanical properties are needed to further improve the efficient shielding, transport, and release of sensitive cargo. Here, a wide range of nanocarriers with different sizes, architectures, and surface properties have been developed. These include unimolecular and polymeric micelles as well as liposomes and polymersomes (Figure 4).
3.1. Unimolecular Micelles
Unimolecular systems are single‐molecule micelles having compartments, core and shell, that are covalently bonded. [119] Their design includes various architecture such as dendrimers, dendrimer‐like star polymers, hyperbranched polymers, and dendronized polymers. Unimolecular systems have gained popularity in the field of targeted drug delivery because they show excellent stability to other microenvironment alterations such as temperature, pH, and ionic strength (Figure 5). [120]
Figure 5.
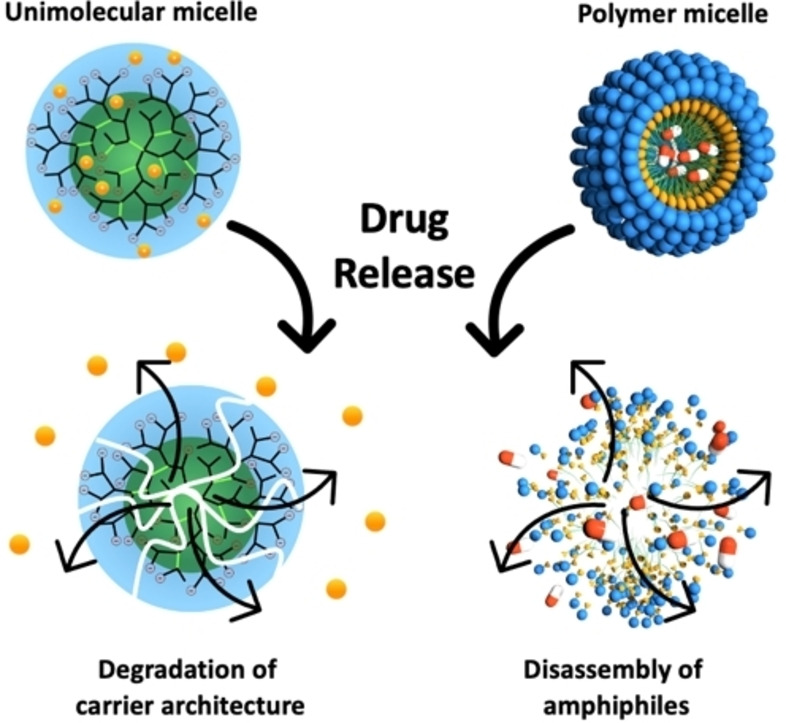
The different physical behavior of unimolecular and polymeric micelles upon degradation and drug release.
A unimolecular micelle is a supramolecular drug delivery system when a guest molecule is physically encapsulated in the system through multivalent interactions as opposed to a monovalent covalent bond. The internal cavities and surface functionalities of dendritic systems make them good candidates for drug delivery. Depending on the structural properties, the drug can be physically encapsulated, through noncovalent interactions, in either the internal cavities of the dendritic core (i.e. endoreceptors) [121] or at the system's multivalent surface (i.e. an exoreceptor). [122]
One of earliest examples of a dendrimer–guest physical interaction is known as the “dendritic box” proposed by Meijer. [121] A modified surface of G5‐PPI dendrimers with Boc‐protected phenylalanine was able to encapsulate guest molecules of different sizes. In this model, the interactions depended on the molecular size of the guest molecule and the physical size of the dendrimer cavities. [123] However, the drawback of this unimolecular micelle is its poor water solubility, as it was designed for organic solvents.
The binding of the guest to the dendrimer core can also be achieved via hydrophobic, electrostatic, or hydrogen‐bonding interactions. Fréchet et al. reported a unimolecular micelle that is a dendritic network of polyaryl ether with carboxylate surface groups. The system showed the capacity to dissolve pyrene, an non‐polar molecule, in water. [124] In this case, the concentration of dendrimers to dissolved pyrene was proportional, and the host–guest interactions were arbitrated through π–π interactions between the aryl ether, rich in electrons, and the aromatic guest molecule. [125] Michell and co‐workers developed an approach for encapsulating acidic aromatic antibacterial compounds that are responsive to a lower pH value. These dendrimers are derived from PAMAM by modifying the amines of the surface with a glycerol derivative, resulting in a water‐soluble system. It is assumed that the host–guest complex is formed through acid–base interactions and hydrogen bonding between the inner tertiary amine core and the acidic substrate. Table 4 summarizes some of the advanced unimolecular drug delivery systems.
Table 4.
Overview of unimolecular drug delivery systems.
|
API |
Polymer structure |
Polymer architecture |
Application |
Trial phase |
Developer |
Reference |
|---|---|---|---|---|---|---|
|
Rhenium‐188 complex |
Polylysine |
Dendrimer |
Liver cancer therapy |
I |
Yang et al. |
|
|
Paclitaxel |
PEI‐C18‐hPG and hPG‐C10‐PEG |
Derivatized hyperbranched polyglycerols |
Intravesical therapy for bladder cancer |
– |
Burt et al. |
|
|
Dehydroepiandosterone (DHEA) |
PAMAM‐PVL‐ PEG‐Cy5.5/CTB |
Multi‐arm block copolymer |
Loss of retinal ganglion cells (glaucoma) |
– |
Guo et al. |
|
|
1,2‐Diaminocyclohexaneplatinum(II) |
PAM‐PGlub‐PEG |
Multi‐arm block copolymer |
Lung cancer therapy |
– |
Mei et al. |
|
|
AB3 |
PAMAM–PVL– PEG–OCH3/Cy5/KE108 |
Multi‐arm block copolymer |
Therapy for medullary thyroid cancer |
– |
Chen et al. |
|
|
Thailandepsin‐A |
H40‐PLA‐PEG‐OCT |
Multi‐arm block copolymer |
Neuroendocrine cancer therapy |
– |
Chen et al. |
|
|
Doxorubicin |
β‐CD‐(PCLPAEMA‐ PPEGMA)21 |
21‐arm star‐like triblock polymer |
Tumor therapy |
– |
Zhang et al. |
|
|
Sunitinib |
hPG‐PCL sulfates |
Sulfated hyperbranched polyglycerols |
Anti‐tumor activity |
– |
Haag et al. |
Imdendrim is a drug delivery system developed by the French Association for the Advancement of Medical Research and is currently in clinical trials (NCT03255343). Belhadj‐Tahar et al. proposed this 5th‐generation poly‐L‐lysine dendrimer, mixed with “nitro‐imidazole‐methyl‐1,2,3‐triazol‐methyl‐di‐(2‐pycolyl) amine” and loaded with [188re]rhenium nitro‐imidazole ligand, as an in situ cancer treatment for solid tumors unresponsive to conventional therapy. [126] The objectives of the trials are to evaluate the drug delivery system's efficacy and safety upon in situ introduction and to compare it to conventional liver cancer treatment methods. Preliminary results showed the safety and efficiency of Imdendrim after a patient diagnosed with stage IV adenocarcinoma of the descending colon received 50 mCi of the drug. The patient was discharged one week after the treatment and examined regularly. After 30 days, the patient showed a standardized uptake value (SUV) of 1.7 (pre‐/post treatment ratio) and was considered a good responder. [126]
Even though no other unimolecular micellar system has reached clinical trials, several systems have proven their effectiveness in vivo. Brooks et al. developed unimolecular micelles, based on derivatives of hyperbranched polyglycerols, [127] and Brut et al. encapsulated paclitaxel as mucoadhesive agents against non‐muscle‐invasive bladder cancer in a unimolecular system. [128] The active pharmaceutical ingredient was encapsulated using the solvent evaporation method and tested in vivo on nude mice with orthotopic KU7‐luc tumors. The results showed that the unimolecular system was not only well tolerated in vivo, but was also significantly more effective than the free drug in reducing orthotopic tumor growth. [128]
On the other hand, Guo et al. reported a unimolecular micellar system formed from a single multi‐arm star amphiphilic block copolymer of poly(amidoamine)‐polyvalerolactone‐poly(ethylene glycol) to target glaucoma, a common blinding disease characterized by loss of retinal ganglion cells (RGCs). The unimolecular micelles were conjugated with cholera toxin B domain (CTB) to target RGCs and Cy5.5 for tracking the system. The system further encapsulated S1R agonist dehydroepiandrosterone (DHEA) to exploit the RGC‐protective sigma‐1 receptor (S1R). The drug delivery system was then injected intraocularly into mice, where it was proven that the system accumulated at the RGC layer and protected it for at least 14 days. [129]
Mei et al. reported the increased half‐life and stability of a unimolecular micelle in comparison to its diblock copolymer micelle equivalent. This unimolecular micelle consisted of a hydrophilic, biodegradable, PEGylated dendritic block copolymer (generation 3 PAMAM, polyglutamic acid). [130]
Chen et al. have also developed two systems that showed great stability and promising results when tested in vivo. Their first unimolecular system was formed from a hyperbranched core (Boltorn® H40) and approximately 25 amphiphilic polylactide‐poly(ethylene glycol) block copolymer arms (H40‐PLA‐PEG). The system was further functionalized to target somatostatin receptors in neuroendocrine (NE) cancers. Then, thailandepsin‐A (TDP‐A) was encapsulated in the system and tested on NE‐cancer‐bearing nude mice. In vivo studies revealed that the unimolecular micelles had greater anticancer efficacy as compared to TDP‐A alone. [131] The second unimolecular micelle was devised to target medullary thyroid cancer (MTC). It was used to encapsulate an API (AB3) that can effectively inhibit MTC but suffers from poor aqueous solubility and stability and is rapidly cleared from the body, unable to target tumors. [132]
The unimolecular micelle developed by Zhang et al., which consists of a multifunctional nanocarrier for disease‐site targeting and controlled release, can also function as an imaging agent for both diagnostics and targeted therapy. The system is formed by a 21‐arm star‐like triblock polymer of β‐cyclodextrin‐poly(caprolactone)‐{poly(2‐aminoethylmethacrylate) poly[poly(ethylene glycol)‐methyl ether methacrylate)]}21, or in short, β‐CD‐PCL‐(PAEMA‐PPEGMA)21. The synthesis was performed using ROP and ATRP techniques, and the resulting unimolecular micelles showed high colloidal stability. They successfully entrapped 60 % of the starting amount of doxorubicin (DOX). [133]
Recently, our group reported a 100‐gram‐scale synthesis of a unimolecular drug delivery system based on biodegradable polyglycerol sulfates. Its biodegradability came from the caprolactone units integrated in the polymeric backbones of the hyperbranched structure. [134] The catalytically driven synthesis was achieved on a 100‐gram scale. The system was proven to increase the solubility of sunitinib, a multi‐targeted receptor tyrosine kinase inhibitor that was approved by the FDA for the treatment of renal cell carcinoma and Imatinib‐resistant gastrointestinal stromal tumors. This unimolecular system showed superior performance when tested in mice against an A431 tumor xenograft model.[ 134 , 135 ]
3.2. Micelles and Polymer Micelles
Taxol® is an anticancer formulation of Paclitaxel with Cremophor EL®, approved by the FDA in 1992 and based on self‐assembled micellar drug delivery system. Taxol® is widely used for treating ovarian and breast cancer. The high hydrophobicity of most chemotherapeutics requires dropwise intravenous injection over several hours. During infusion, the excipients in the formulation leach plasticizers from the tubes, altering the material's properties and causing undesired side reactions. [136] Furthermore, solubilization enhancers are associated with toxicity and allergic reactions in patients. [137] Although Cremophor EL® and Tween® 80 are amphiphiles, they immediately disassemble upon injection, leading to fast clearance from the bloodstream and causing high drug accumulation in healthy tissue. One way to overcome this issue is by substituting polymeric surfactants for low‐molecular‐weight ones. Due to the increased interfacial energy derived from their larger insoluble polymer segments, the in vivo stability of polymeric micelles is significantly higher than those based on low‐molecular‐weight surfactants. This extends circulation times, increases drugs’ half‐lives, and boosts accumulation, e.g. for cancer therapeutics in tumors. Furthermore, polymeric micelles can effectively reduce required therapeutic dosages and administration intervals. The retention time of the intravenously injected particles is prolonged by the lack of lymphatic drainage.[ 138 , 165 ] Micelles exhibit a more dynamic character than nanosized drug delivery systems, which are mostly “static” systems like the polymer conjugates described in Section 2. Consequently, they can release their cargo at the targeted site more easily than polymer–drug conjugates. However, these dynamics can also favor undesired leaching events. Micelles in aggregation always exist in an equilibrium between free unimers and formed micelles that is controlled by the critical micelle concentration (CMC).
The stability of polymeric micelles, a crucial factor in determining the circulation time of injected particles, can be controlled by varying the chain length and density of the hydrophilic shell‐forming polymer. [139] Furthermore, larger polymer particles circulate longer in the blood than smaller ones. [140] The longer the circulation time in the bloodstream, the higher the accumulation in tumor tissue as tailored by the EPR effect. Nonetheless, the response of injected particles to their surrounding biological environment is still a largely unresolved question. [141] The formation of a biomolecular layer around injected particles has been shown to alter their pharmacokinetic properties, [142] adversely influencing their stability [143] and affecting their outer composition. [144] The particle's surface charge determines its ability to penetrate cell membranes and its internalization into the cytosol. [145] Negatively charged particles show prolonged circulation times, whereas positive charges result in particles’ rapid sequestration in the spleen and liver. [146] Once internalized into the cytosol, a drug's release can be selectively triggered by activation of stimuli‐sensitive bonds within the polymer structure. [147] It was also found that the drug loading of polymeric micelles can influence their shape and conformation and might change the biological response that they elicit. [148] A significant challenge in designing block copolymer micelles for biomedical applications is the interplay between these multiple factors that influence the pharmaceutical efficacy of the system. The characterization and evaluation of polymeric micelles is a key point in their development. [149] As a rule of thumb, for micellar systems to be translated into clinical applications, the following criteria must be met: (i) the self‐assembled systems must be in equilibrium, and dilution upon injection must not lead to quick disaggregation; (ii) the carrier system must deliver its cargo to the site of action with no leaching into the bloodstream; (iii) once the loaded carrier reaches its destination, the drug must be released selectively; and (iv) nonspecific interactions with the reticuloendothelial system (RES) and other systems within the body must be avoided.
Two frequently used pathways have been developed for the synthesis of amphiphilic block copolymers. The simpler one is the use of mPEG‐OH or mPEG‐NH2 as a macroinitiator for the polymerization of cyclic monomers such as lactide, glycolide, caprolactone, NCAs, or epoxide‐based monomers (Figure 6). As this approach is limited to cyclic monomers, another strategy was used to enable the creation of functional block copolymers from vinyl‐containing monomers. This process couples mPEG‐OH to ABCPA, leading to a bifunctional mPEG2‐ABCPA azoinitiator used for free radical polymerizations of mostly HPMA‐based monomers. Besides the access of this approach to functional block copolymers, the problems of removal of free homopolymers which is hard to achieve by purification methods, such as precipitation, dialysis, or size‐exclusion chromatography.
Figure 6.

Overview of synthetic approaches towards amphiphilic block copolymers and the most frequently used monomers.
In the 1990s, Kabanov's group developed A‐B‐A triblock DOX‐loaded Pluronic® micelles based on PEO‐b‐PPO‐b‐PEO (Pluronic F127: EO100‐PO65‐EO100, Pluronic L61: EO2‐PO30‐EO2). [150] The final micelles are 30 nm in size with 8.2 wt % DOX. SP1049 C was the first polymeric micelle drug formulation to enter clinical trials, in 1999; preclinical studies of this drug had revealed pharmacokinetics like those of DOX. [151] Its rapid dissociation consequently led to comparable toxicity following intravenous administration of free DOX formulations. SP1049 C is being evaluated in phase II clinical trials for the treatment of multi‐resistant cancer. However, its low stability remains an unresolved drawback.
Around the same time, Kataoka and co‐workers described one of the first A‐B diblock micellar drug delivery systems, based on PEG‐b‐poly(α,β‐aspartic acid‐DOX) block copolymers (NK911). [152] DOX was covalently conjugated to the polymer backbone via amide bonds (Figure 7). As the amide bond is highly hydrolytically stable, DOX was physically loaded into the micelles. The conjugated DOX enhances the stability of formed micelles via π–π stacking. It also serves as an agglomerate interacting with loaded DOX, leading to a carrier system with reduced CMC, low leakage, and prolonged circulation time. The formed micelle is 40 nm in size with a drug loading of 17 wt % (drug/polymer). [153] In 2001, this system went into clinical trials, where its investigation kicked off the era of exploring micelles for clinical use, making it the first true breakthrough in this field. In clinical studies, this system showed a higher plasma value (area under the curve or AUC) than that of free DOX. Although NK911 showed lower stability, its smaller‐sized micelles exerted better tumor cell uptake than DOXIL®, with a liposome size of 100–150 nm. The DOX conjugated covalently to the polymer backbone did not exercise any antitumor activity, likely due to slowly degrading amide bonds. This failure spotlighted the obligatory requirement for both cohesive forces in the core to enhance micellar stability and cleavable bonds in the polymer backbone to enable drug release. [154] For instance, in subsequently designed systems, the drug was covalently connected by either hydrazone or ester bonds (see NC‐6300 [155] and NK012 [156] ), making the systems degradable in the acidic environment of cancerous tissue. Furthermore, the polymer backbone was modified to increase hydrophobicity by introducing aromatic moieties to the polymer structure (see NK105 [157] ).
Figure 7.

Overview of polymer–drug conjugate block copolymer micelles that reached clinical trials.
Besides the developments based on mPEG‐b‐poly(amino acid) micelles and mPEG‐b‐poly(HPMA‐Lac), other systems have emerged, such as those based on polyethers (Pluronics®, SP1049 C) [151b] and mPEG‐b‐poly(esters) (mPEG‐b‐p(PLA) (Genexol®, Nanoxel®). [158] So far, the only approved polymeric micelles are Genexol® and Nanoxel®, both based on mPEG‐b‐p(PLA) and loaded with the taxane‐derived drugs Paclitaxel and Docetaxel, respectively. Genexol® was approved in South Korea in 2006, whereas Nanoxel® entered clinics in 2007. Both formulations are comparable in acting as solubilizing enhancers of the hydrophobic drugs, but due to their low in vivo stability, neither drug offers tailored release kinetics or increased drug retention. [159]
To date, 11 polymeric micelle formulations have been translated into drugs undergoing advanced clinical trials. Some candidates currently under investigation have demonstrated promising outcomes in treating different types of cancer. Table 5 summarizes all polymeric micelles that have reached clinical trials and are still under active clinical investigation. The journey and status of all other systems excluded is discussed elsewhere in detail.[ 151c , 160 ]
Table 5.
Overview of polymeric micelle drug formulations in clinical trials. Source: https://clinicaltrials.gov (date: April 17, 2022).
|
Trade name |
API |
Polymer structure |
Drug loading type |
Trial phase/ NCT number |
Indication |
Developer |
|---|---|---|---|---|---|---|
|
NK012 |
7‐ethyl‐10‐hydroxy‐CPT (SN‐38) |
mPEG‐b‐p(Glu‐SN‐38) |
Covalent via ester bond |
Completed (phase II) NCT00951054 |
Triple‐negative breast cancer |
Nippon Kayaku, Japan |
|
NC‐6300 |
Epirubicin (4′‐epimer of DOX) |
mPEG‐b‐p(Asp‐Epirubicin)(Asp‐ben) |
Covalent via hydrazone bond |
Ongoing (phase I/II) NCT03168061 |
Advanced solid tumors; advanced, metastatic, or unresectable soft‐tissue sarcoma |
NanoCarrier, Japan |
|
| ||||||
|
Nanoxel® |
Docetaxel |
mPEG‐b‐p(PLA) |
Physical via hydrophobic interactions |
Approved in India in 2007 |
Several cancers |
Samyang Biopharmaceuticals Corporation, South Korea |
|
Genexol® |
Paclitaxel |
mPEG‐b‐p(PLA) |
Physical via hydrophobic interactions |
Approved in South Korea in 2006 |
Metastatic breast cancer |
Samyang Biopharmaceuticals Corporation, South Korea |
|
| ||||||
|
SP1049 C |
Doxorubicin |
PEO‐b‐PPO‐b‐PEO, Pluronic® F127: EO100‐PO65‐EO100, L61: EO2‐PO30‐EO2 |
Physical via hydrophobic interactions |
Completed (phase II) Not listed at clinicaltrials.gov |
Gastrointestinal, colorectal, and non‐small‐cell lung cancers |
Supratek Pharma, Canada |
|
| ||||||
|
CriPec634 |
Docetaxel derivatized with methacrylate |
mPEG‐b‐p(HPMAm‐Lac) |
Core‐crosslinked micelles with chemically loaded (crosslinked) drug |
Completed (phase II) NCT03742713 |
Platinum‐resistant ovarian cancer |
Cristal Therapeutics, Netherlands |
3.3. Liposomes and Polymersomes
3.3.1. Liposomes
The pioneering work in developing liposomal vesicles for drug delivery goes back to the 1970s, when Gregoriadis established the concept of drug entrapment in liposomes. [161] The first demonstration of improved in vivo activity of a liposomal drug in an animal model was reported a few years later. [162]
Two decades after Gregoriadis’ initial concept, liposomes had overcome their initial problems, with two drug formulations being approved. First, a special high‐pressure formulation allowed the amphiphilic drug amphotericin to be inserted into the membrane of liposomes. This liposomal formulation, AmBisome®, is used to treat serious fungal infections with clinical success and reduced toxicity.[ 161b , 163 ]
Another breakthrough was the addition of PEGylated lipids, [16a] which can increase liposomes’ circulation time in vivo. This small but important lipid additive established the “stealth” effect that transformed the cytotoxic drug doxorubicin into a longer‐circulating anti‐tumor agent with lower cardiotoxicity. [164] The first clinical trials of liposomal drugs were performed in 1989, and 1994 saw the first human studies demonstrating the longer circulation of PEGylated liposomes loaded with doxorubicin, which later became DOXIL®, the first nanomedicine approved by the FDA.[ 164 , 165 ] Since then, PEGylated liposomes have addressed various complex formulation problems to enhance circulation time, and they are currently applied to drug delivery in about 15 commercial drugs and many more in clinical studies. A number of adaptations to the lipid formulations (i.e. cationic lipids for RNA and DNA), as well as other smaller innovations, have contributed to the success that led Alnylam to consider them for siRNA delivery in a clinical trial. [166] As applied to COVID‐19 vaccines in technologies developed by Arbutus Biopharma for Moderna and BioNTech/Pfizer, liposomal mRNA formulations have saved many thousands of lives during the current pandemic, and they are currently being adapted to other unsolved problems such as malaria and tumor vaccinations.[167][168]
3.3.2. Polymersomes
Yet liposomes’ lipid membranes limit their mechanical and thermal stability, a drawback that has been addressed by their polymer analogs. “Polymersome” refers to synthetic vesicles consisting of a hydrophobic bilayer membrane and an aqueous lumen. In the decades since the first polymersome was formed [21] and six different morphologies [169] were observed in the 1990s, extensive studies of these vesicles as carriers for a wide array of therapeutic drugs, enzymes, peptides, and nucleotides have attracted increasing interest. In contrast to polymer micelles, polymersomes can encapsulate hydrophilic species in an aqueous interior and hydrophobic compounds within the membrane. Polymersomes are self‐assembled from amphiphilic block copolymers in diverse architectures. With their capacity for customizable polymer chains, the polymersomes’ size, [170] morphology, [171] and stimuli‐responsiveness [172] can be further tuned by adjusting the molecular weight [173] of the block copolymer and by modifying the membrane structure. Compared to liposomes with reproduced biological lipids, polymersomes display a compact membrane with improved thickness, mechanical stability, and chemical variety. Polymersomes composed of PEG block have shown great inherent potential due to their enhanced blood circulation time and low off‐target accumulation, owing in particular to stability improvements from reversibly crosslinking the polymer bilayers. [174] Furthermore, their external surfaces can be easily modified by targeting ligands, [175] and researchers have reported a growing number of studies on polymersomes designed to respond to various stimuli. [176]
Considering the significant impact of polymersome size on intracellular uptake efficiency, recent decades have seen the development of multiple strategies to control the size of polymersomes. In general, the size of a polymersome depends on the packing parameters of its amphiphilic polymer chains during self‐assembly in water phase. [177] Avenues that have been explored for controlling polymersome size include polymer chain length, the mixing rate of organic solvent into water phase, and post‐extrusion and sonication processes. The correlation between the morphology of assemblies and the geometry of BCPs was first established by Ahmed and co‐workers. [178] Both the size and morphology of vesicles formed from BCPs are crucially determined by the mass or volume fraction of the hydrophilic block in BCPs (fA). For instance, in self‐assembled systems consisting of PEG with high hydrophilic interaction, vesicular structure is optimized when fPEG is within the range of 10–40 %, [179] and the diameter of the polymersome core increases from 9.6 nm to 10.6 nm as the fPEG is reduced to 10 %. [173] External shear forces can also affect the diameter of a polymersome. In contrast to liposomes, polymersomes possess thicker membranes with excellent mechanical performance and tunable chemical properties. The great number of biocompatible polymers offer practically infinite potential to chemically modify the polymeric membrane to optimize polymersome’ stability and selectivity and thereby enhance circulation time and biocompatibility.
PEGylated liposomes that can minimize both electrostatic and hydrophobic interactions with proteins constitute a breakthrough in 20th century drug carrier development. [180] Compared to PEGylated liposomes, polymersomes fabricated from PEG of similar molecular weight achieved a twofold increase in circulation time, to 20–30 h. [173] Nevertheless the potential immunogenic aspects of PEG have to be considered (see Section 2.1).
Besides improved stability by introducing a PEG chain, polymersomes formulated from amphiphilic BCPs composed of PEG also exhibit notably enhanced membrane permeability. Based on the discussion about PEGylated lipids’ improved membrane permeability over natural liposomes, vesicles formulated from BCPs containing PEG blocks have been developed as precisely controllable delivery systems for payloads such as drugs, proteins, and nucleotides, though hydrodynamic repulsion by the PEG chain continues to make low efficiency a challenge in protein encapsulation into polymersomes. However, polymersomes assembled from the triblock copolymer dextran‐b‐PCL (DEX‐b‐PCL) have been reported by Zhang and colleagues. This polymersome's symmetric membrane results in permeabilization and the synchronized release of the cargo molecule, erythropoietin (EPO). [181] Discher and co‐workers’ pioneering study on polymersomes based on BCPs like PEG‐PCL, PEG‐PLA, and PEG‐PBD for siRNA delivery [182] offered a clear view of polymeric vesicles’ potential role in the field of gene delivery. In cNGQ peptide‐directed polymersomes co‐self‐assembled from biodegradable poly(ethylene glycol)‐b‐poly(trimethylene carbonate‐co‐dithiolane trimethylene carbonate)‐b‐polyethylenimine (PEG‐P(TMC‐DTC)‐PEI) asymmetric triblock copolymer and cNGQ‐PEG‐P(TMC‐DTC) diblock copolymer, Zhong's group demonstrated both the efficient loading and uptake of siRNA by A549 lung cancer cells in vivo. [183] Scott conceived of stimuli‐responsive polymersomes based on PEG‐PPS for the endocytic delivery of antigen protein or TLR agonist adjuvants to dendritic cells to induce immune response; the model antigen ovalbumin, encapsulated and released in vitro, was successful in activating dendritic cells from the spleen and in stimulating the priming of T‐cells. [184]
3.3.3. Stimuli‐Responsive Polymersomes
The tumor microenvironment's reliable extracellular pH range of 6.5–6.9, as compared to the 7.4 pH of blood under normal metabolism, inspired the development and study of pH‐responsive polymersomes. The sustained and controllable release of polymersomes formed from PEG‐PCL has been observed for release periods ranging from 20 to 200 h. [185] In contrast, polymersomes bearing bonds that are cleavable in response to pH change exhibit rapid release. pH‐sensitive linkers including acetal, ester, and amine groups are extensively incorporated into BCPs.
Secondly, the accumulation of the natural reducing agent glutathione in tumor sites offers a critical access point for constructing redox‐sensitive polymersomes for drug delivery. Polymersomes formed from amphiphilic block copolymers connected by a disulfide bond show a fast in vitro release of 10 min or less. Reduction‐cleavable polymersomes formulated from triblock copolymer pPEGMA‐PCL‐SS‐PCL‐pPEGMA (poly(polyethylene glycol methacrylate)‐poly(caprolactone)‐SS‐poly(caprolactone)‐poly‐(polyethylene methacrylate)), customized with folate and trastuzumab ligands on the membrane, exhibits effective delivery of DOX to breast cancer cell lines BT474 and MCF‐7 as compared to nontargeted polymersomes. During in vivo studies on mice, ≈85 % tumor regression was observed without any significant cardiotoxicity, as compared to only 40 % tumor inhibition of free DOX treatment. [186]
4. Future Directions for Supramolecular Drug Delivery Systems
4.1. Application of the Core‐Crosslinking Strategy
In vivo targeted drug delivery by conventional polymeric carriers may fall short due to adverse circulation kinetics, which hinder accumulation at the tumor site. Interactions with blood components may cause undesired aggregation or disassembly of the polymeric drug delivery systems. For example, interaction between plasma proteins and the hydrophobic part of micelles can influence carrier stability by affecting e.g. the sensitive CMC equilibrium between unimers and micelles and thus induce a rapid, premature release of the encapsulated drug. These drawbacks can be overcome by an increase in stability achieved by crosslinking the polymeric systems to improve their pharmacokinetic profile and optimize their circulation performance in vivo. More importantly, the design of crosslinking processes requires synchronized stabilization and the retention of intrinsic properties. Initial studies on physical stabilization and chemical crosslinking of the core, shell, and intermediate layers have allowed researchers to find techniques that enable the creation of stable, biodegradable systems that can release their therapeutic cargo at the target site.
Besides non‐covalent crosslinked micelles (e.g. those formed by π–π stacking, hydrogen bonding, dipole interactions, or host–guest interactions) [187] chemical crosslinking can be ensured by using radical or photo(thermal) polymerization techniques. [188] In addition, bifunctional groups on the side‐ or end‐group of the copolymers can be used to introduce biodegradable groups on the micellar system. Rijcken and co‐workers developed a platform based on the initial research on mPEG‐b‐p(HEMA‐Lac) copolymers, where the methacrylated blocks were thermally crosslinked. Hydrolysis of the lactate and cleavage of the incorporated ester bond made them biodegradable. [189] The further developed formulation CriPec634 completed a phase II study with docetaxel on platinum‐resistant ovarian cancer (Table 5). Besides that, dithiolane‐crosslinked micelles can be used to introduce dynamic and reversible crosslinks between the micellar polymer chains. [190]
For polymersomes, crosslinking between lipid membranes is considered a unique means of stabilizing them against post‐injection dilution and interaction with the blood complex, thus prolonging their circulation period and enabling the translation into clinical use.[ 188 , 191 ] Recently developed techniques such as introducing bifunctional crosslinker to BCPs, usage of monomers with bifunctional groups undergoing self‐polymerization, as well as thiol–ene click chemistry have been demonstrated to improve polymersome stability. By introducing dithiolane‐functionalized carbonates to the polymer backbone, which undergo ring‐opening polymerization, self‐crosslinking behavior was observed in reduction‐sensitive polymersomes formed from PEG‐P(TMC‐DTC). [192] Accordingly, specific drugs can be actively loaded and released via thiol–disulfide exchange. Furthermore, bifunctional crosslinkers like cysteamine with stimuli moieties have been exploited for combined one‐step crosslinking and functionalization. For instance, Liu and co‐workers reported a light‐regulated “traceless” crosslinking strategy. Here, through amidation among primarily decayed amines by UV stimulation, prominent vesicle crosslinking associated with bilayer hydrophobicity‐to‐hydrophilicity transition, occurs upon self‐assembly. [193]
To enhance the surface areas of nanocarriers, crosslinking techniques can be used for the formation of other types of architectures such as nanosized hydrogels (i.e., nanogels). These highly hydrated polymeric networks display suitable inherent properties for applications as drug delivery systems, including biocompatibility as well as a large surface area for further functionalization, and a hydrophilic interior network which renders them particularly interesting for the encapsulation, transport, and release of sensitive biomolecules as payload. Methods that can be used for the formation of nanogels include emulsion techniques, nanoprecipitation, or nanolithography and they all rely on similar crosslinking strategies as for the previously discussed carrier systems. However, enhanced carrier stability is concomitant with the need to ensure safe release of the loaded API at the desired site of action. For this, the stimuli for the cleavage of the respective release exploit the biochemical microenvironment and physico‐chemical features of the targeted site of action (i.e., gradients in pH and redox potential, overexpression of enzymes and marker molecules, or ionic strength). [194]
4.2. Targeted Supramolecular Drug Delivery Systems
Besides the development of new polymer architectures designed to enhance the stability and loading capacities of polymer materials, the significance of targeting ligands has been a subject of considerable research effort. Click and biorthogonal reactions have emerged as powerful tools to attach multiple targeting moieties to functional polymer materials. [195] As already discussed in Section 2.1 (polymer‐conjugates), the biorthogonality and biocompatibility of a system's coupling reactions is crucial in order for it to be applicable in nanomedicine. However, as micelles and liposomes take on greater roles in the field of targeting polymeric materials, another point for consideration is the possibility that interactions of the ligand or linker structure may adversely affect the nanoparticle′s stability (Figure 8). [196]
Figure 8.

Overview of synthetic approaches for the introduction of targeting ligands onto amphiphilic block copolymers.
4.2.1. Delivery of Oligonucleotides: DNA‐ and RNA‐based Materials
The COVID‐19 pandemic has revealed the essential need for safe and suitable nanomedicine delivery systems, e.g., for oligonucleotide‐based materials.[ 1 , 197 ]
Polymersomes engineered with surface‐based targeting moieties including peptides, protein, and antibodies play an essential role in the effective delivery of therapeutic agents. Correspondingly, desired functional end groups on the hydrophilic blocks indicate the possibility of various post modifications. For instance, polymersomes formed from the block copolymer polyethylene glycol‐polytrimethylene carbonate‐polydithiolane trimethylene carbonate (PEG‐P(TMC)‐P(DTC)) functionalized with cNGQ peptide and loaded with doxorubicin show great binding ability to A549 lung cancer cells in vitro and significantly reduced tumor volume in vivo. [192] A cooperative study by Kataoka and Zhong, based on the superb selectivity and efficiency of small interfering RNA (siRNA), has exemplified the induction of highly potent and sequence‐specific gene silencing on α3β1‐integrin‐overexpressing A549 lung cancer cells. [183] In 1995, Kataoka and co‐workers discovered the mixing two oppositely charged amphiphilic block copolymers results in narrowly distributed micelles, termed polyion complex (PIC) micelles. [198] Typically, PICs precipitate under charge‐neutral conditions, but in the case of PIC micelles, the hydrophilic shell enhances the system's solubility. Their findings paved the way for the investigation of PIC micelles for delivering new classes of charged (macro‐)molecules, such as proteins, nucleic acids, negatively charged photosensitizers, pDNA, and mRNA. PIC micelles have been intensively evaluated for photodynamic therapy and for protein and gene delivery.
4.2.2. Charge‐Reversal Polymer Architectures: Delivery of Proteins and Drugs upon Charge Manipulation
PEGylation is one attractive concept for overcoming the main hurdles to delivering protein therapeutics; another is their integration in PIC micelles. These micelles typically consist of a hydrophilic segment and a charged block copolymer that interacts with the oppositely charged target protein; a protein‐complexing micelle is thereby formed, with the protein incorporated into the micellar core. A recent study demonstrated the ability of PEG‐polycation block copolymers to form well‐defined PIC micelles with different proteins through electrostatic interactions. [199] The low charge density of most proteins inhibits the formation of stable complexes under physiological conditions; PIC micelle complexes tend to dissociate under the harsh conditions found in vivo. [200] One frequently applied strategy, strengthening the electrotactic interaction between protein and polymer, is accomplished via “charge conversion” by installing pH‐degradable charged moieties. For instance, an excess of positive charges is consumed by introducing carboxylic acid moieties and vice versa. To prevent in vivo dissociation, protein‐loaded PIC micelles were developed with pH‐sensitive linkers between the amine in the protein and a maleic anhydride derivative in the polymer backbone (Figure 9). [201]
Figure 9.

The concept of charge conversion of biomacromolecules to improve their therapeutic efficacy.
Despite numerous attempts in nanomedicine technology to improve drug accumulation in tumor tissue, several unsolved obstacles remain, including tightly packed tumor cells and high interstitial fluid pressure, which results in limited molecular diffusion and extravasation into tumor tissues. Even oxygen molecules can only diffuse up to 200 μm from the vasculature. [202] It is known that positively charged or cationized nanocarriers can effectively enhance in‐tumor accumulation by inducing transcytosis and facilitating penetration across multiple cell layers. [203] However, polycationic polymers are well known for nonspecific cellular uptake, in vivo toxicity, and opsonization‐induced rapid clearance from blood circulation, problems not seen with neutral or negatively charged nanocarriers. [146] To address these limitations, pH‐ and enzyme‐triggered charge‐reversal polymer–drug conjugates have been developed that are neutral or slightly anionic in the bloodstream but generate positive charge in the tumor microenvironment. Shen and colleagues developed polyamides in which the amide groups are neighbored by carboxylic acid. Such amide groups can hydrolyze under acidic conditions and regenerate amine groups carrying positive charge, leading to enhanced cellular uptake and localization in the cell nucleus.[ 118b , 204 ] Taking advantage of γ‐glutamyl transpeptidase enzyme, which is overexpressed on the cell membrane, an enzyme‐activatable polymer‐camptothecin conjugate was synthesized. The enzyme cleaves the γ‐glutamyl moieties to generate positively charged primary amine groups. The resulting cationic conjugate can effectively penetrate tumor tissues via caveolae‐mediated endocytosis and transcytosis, which enables transendothelial and transcellular transport. [118a] These achievements might enable the successful translation of charge‐reversal polymer–drug conjugates into effective therapies.
4.3. Targeted Nanoparticle Drug Delivery Systems
Nanoparticle drug delivery systems have been clinically investigated since the late 1980s. Most of these systems are passively transported in the body by fluid movement. Over the past two decades, actively targeted generations of nanoparticles have reached the preclinical stage of drug development. With the availability of clinically proven antibodies against a variety of tumor‐cell‐specific membrane receptors, antibody‐linked nanoparticles were investigated for targeted drug delivery. However, due to the complexity of the pharmaceutical formulations, only a very few formulations entered clinical phase I/II studies. Specifically, Synergene Therapeutics is developing both SGT‐53, a cationic liposome, modified with an anti‐transferrin receptor antibody fragment, that encapsulates the wild‐type p53 sequence for the treatment of glioblastoma, solid tumors, and pancreatic cancer, and SGT‐94, an RB94 plasmid DNA inside a liposome decorated with an anti‐transferrin receptor antibody fragment. BIND‐014 (BIND Therapeutics) represents the first polymer‐based targeted nanoparticulate drug delivery system. Here, a PSMA‐targeted 2‐(3‐((S)‐5‐amino‐1‐carboxypentyl)ureido)pentanedioic acid (ACUPA) and docetaxel‐containing PEG‐PLGA or PLA‐PEG particles entered clinical Phase I trials for the treatment of PSMA‐positive prostate cancers; non‐small‐cell lung, cervical, head and neck cancers; and KRAS‐positive lung cancers. [205]
Beyond targeted nanoparticulate polymers and liposomes, targeting peptides represent another synthetic drug delivery system of future interest. In contrast to antibodies and targeted nanoparticulate systems, peptides may deliver more effector molecules to the target and take them deeper into the disease tissue. The advent of advanced peptide discovery platforms has enabled today's extremely successful screening of peptides for high‐affinity binding to extracellular and cell membrane drug targets. In addition to screening for high target affinity, peptides can be chemically modified to fine‐tune their pharmacokinetic behavior, chemical resistance, proteolysis stability, and physicochemical properties. With respect to the short half‐life of peptides, two different chemical synthesis strategies are applied to slow their clearance from the bloodstream: modification of the chemical structure by cyclization, peptide stapling, or amino acid substitution; and chemical conjugation to larger molecules such as PEG. Altogether, peptides are perfectly suited for the targeted delivery of therapeutic and diagnostic cargos. They also offer a multitude of chemical approaches to tune all the relevant properties of the final peptide–drug conjugate. Peptides’ attractiveness for the targeted delivery of therapeutic effector molecules relies on their potential to achieve a high target‐to‐blood drug concentration within a short time. While ADCs may take one week or more to be eliminated from blood circulation, PDCs can achieve high target‐to‐blood ratios within hours after intravenous application, provided that the affinity of the targeting peptide is in the low nanomolar range. Peptides are therefore well‐suited for the targeted delivery of therapeutic effector molecules with a very small therapeutic window.
Despite this attractive chemical profile, only a few PDCs are in the clinical space today. Lutetium‐containing 177Lu‐dotatate (LUTATHERA, Advanced Accelerator Applications USA, Inc.), a radiolabeled somatostatin analog for treating somatostatin‐receptor‐positive gastroenteropancreatic neuroendocrine tumors (GEP‐NETs), was approved by the FDA in 2018. The development of radiolabeled somatostatin ligands was a long‐term endeavor that began with the first description of somatostatin more than 45 years ago. [206] Many diagnostic as well as therapeutic somatostatin ligands have been developed. [207]
Most recently, 3B Pharmaceuticals published the first clinical results for Lutetium 177Lu‐PDC in targeting the neurotensin‐1 receptor in pancreatic cancer . [208] FAP‐2286, another PDC for radiotherapy that targets fibroblast activation protein (FAP), is in early clinical development (NCT 04939610).
PDCs for targeting homing receptors like RGD or NGR represent another area of drug development. [209] Most PDCs target the tripeptide Arg‐Gly‐Asp (RGD) acid sequence because of its high affinity and specificity for integrin αvβ3, which is overexpressed in many tumors.
5. Conclusion
Drug development is a lengthy, complex, and expensive endeavor bearing a high degree of uncertainty at every step of the process. Poor water solubility of active pharmaceutical ingredients (APIs) is a major cause of attrition in the pharmaceutical industry and represents a formidable hurdle for pharmaceutical drug development. With the advent of today's drug development algorithms, such as high‐capacity screening and combinatorial chemistry, the number of poorly water‐soluble drugs is sharply increasing. Advanced technologies for drug delivery are needed to ensure that tomorrow's cutting‐edge therapeutics will be bioavailable and effective in patients’ bodies. The most recent advances in polymeric material design have enabled dedicated drug formulations even in the case of critical physicochemical drug properties. New polymer materials need to be tested in clinical trials and approved by medical authorities. Because of breakthroughs in the chemical synthesis of biocompatible polymers and in physicochemical characterization methods, polymers for tailored drug delivery solutions are now fully established in both scientific research and drug development. A growing universe of synthetic polymer architectures and sophisticated drug formulation methods are now available to address the complex demands of drug adsorption, blood kinetics, metabolism, and finally clearance from the blood. Nevertheless, more polymer drug delivery systems need to be clinically evaluated; however, it is difficult for individual research groups to finance the further development steps such as GMP production, long‐term stability tests, and clinical studies, after finishing initial evaluations in various in vitro and in vivo experiments. On the other hand, the clinical development of new polymers is not the primary goal of pharmaceutical companies, as their research capabilities are primarily focused on the identification and validation of novel drug pharmacophores. Future drug development in industry needs to fully implement most recent findings in polymer chemistry, pharmaceutical technology, and translational research to deliver novel solutions and enable successful clinical translation. However, funding of public–private partnerships (e.g. between research institutions and companies) is required along with early address development risks and regulatory hurdles to push new drug delivery systems into early clinical trials. In addition, new drug delivery solutions will focus on target‐specific drug accumulation in diseased organs, tissues, and cells, and will enable stimulus‐controlled drug release. Finally, with respect to the economic and regulatory requirements of the pharmaceutical industry, less complex drug delivery solutions are needed to facilitate their integration into the early development process.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
From top left to bottom left: G. Ma, R. Haag, M. Cherri, V. Ahmadi, M. Tully, D. Braatz, E. Mohammadifar, M. Dimde, F. Reisbeck, M. Schirner. Vahid Ahmadi completed his doctoral study at Freie Universität Berlin in 2021, studying functional polymers with defined architecture for biointerface interactions under the supervision of Rainer Haag. His current postdoctoral research is focused on developing polymer architectures with antiviral properties within a start‐up project NoVirall (EXIST‐Forschungstransfer). Daniel Braatz received his master′s degree in chemistry at the Freie Universität Berlin in 2019. He is currently pursuing his doctoral degree there under the supervision of Rainer Haag and Matthias Ballauff. His research is dedicated to the development of amphiphilic dendritic block copolymer micelles and their systematic characterization and application. Mariam Cherri served two years as a process engineer and technical consultant in the energy industrial sector. Currently, she is a doctoral candidate in the group of Rainer Haag at Freie Universität Berlin, focusing her research on the synthesis of functional polymeric architectures, their characterization, scalability, and systemic applications. Mathias Dimde received his PhD in Chemistry at Freie Universität Berlin. From 2017 to 2021, he was a postdoctoral fellow in a joint project in the group of Rainer Haag and Uwe Schedler (PolyAn GmbH). He is currently working in the Research Center of Electron Microscopy (FZEM) in the Ludwig group and coordinating the Core Facility BioSupraMol at Freie Universität Berlin. His primary research interests are in the development of functional carrier systems for diagnostics and therapy and studying their structural organization and supramolecular assemblies. Rainer Haag is Professor of Organic and Macromolecular Chemistry at Freie Universität Berlin. Since 2021, he has been spokesperson of the Collaborative Research Center SFB 1449 “Dynamic Hydrogels at Biological Interfaces”. His research focuses on biodegradable and multivalent macromolecules, supramolecular architectures, nanotransporters for drug delivery, and sustainable polymer syntheses. In start‐up‐oriented teaching, he won the 2014 teaching award at Freie Universität Berlin with his project “Translation of Project Ideas.” Together with the company Dendropharm, he received the Innovation Award Berlin‐Brandenburg in 2016. In 2019 he was elected as a member of the German Academy of Science and Engineering (acatech). In 2022 he was awarded the ERC Advanced Grant. Guoxin Ma studied polymer science and engineering at Sichuan University and Freie Universität Berlin. Currently she is a doctoral candidate under the supervision of Rainer Haag at Freie Universität Berlin. Her research mainly focuses on synthesis and biomedical applications of multifunctional polymer materials. Ehsan Mohammadifar completed his PhD in chemistry at the University of Tehran (2018). Currently he is working as a postdoc in in the group of Rainer Haag at Freie Universität Berlin with a focus on the synthesis and modification of biocompatible polymers for biomedical applications. Felix Reisbeck received his PhD in chemistry from Freie Universität Berlin with a research focus on the synthesis of various architectures of polymers for biomedical applications (Haag group). Further research interests include polymers in optoelectronics (Fraunhofer IAP). Currently he co‐leads a start‐up concept on the development of novel stem cell matrices. Michael Schirner received his MD from the Friedrich‐Schiller‐University Jena. He is board certified for pharmacology and toxicology and since 2016 he has been a visiting professor for polymer science at the Freie Universität Berlin. His research interests are novel therapeutics in oncology and inflammatory diseases, medical device technologies for disease diagnosis, and novel drug delivery technologies. Before 2015, he worked for 25 years in research and early development functions in the pharmaceutical and medical device industry. Michael Schirner is the successful founder of three start‐up companies in the fields of medical device technology, biotechnology, and research diagnostics, and is also advisor to universities and companies for early technology transfer. Michael Tully studied pharmacy at the Ludwig‐Maximilians University in Munich. During his doctoral studies in the group of Rainer Haag, he focused on the synthesis and characterization of PEG‐alternative polymer bioconjugates for the extended circulation half‐life of biopharmaceuticals. Currently he is based in Turku, Finland.

Acknowledgements
The authors acknowledge funding from the German Ministry of Science and Education within the project “Next‐PEG”, German Science Foundation (DFG) within the SFB 1449 and Sino‐German center on targeted drug delivery. We thank Benjamin Allen (Boston) for careful editing and proofreading of this review. Open Access funding enabled and organized by Projekt DEAL.
D. Braatz, M. Cherri, M. Tully, M. Dimde, G. Ma, E. Mohammadifar, F. Reisbeck, V. Ahmadi, M. Schirner, R. Haag, Angew. Chem. Int. Ed. 2022, 61, e202203942; Angew. Chem. 2022, 134, e202203942.
References
- 1. Sadeghi I., Byrne J., Shakur R., Langer R., J. Controlled Release 2021, 331, 503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ehrlich P., in The Collected Papers of Paul Ehrlich (Ed.: Himmelweit F.), Pergamon, Oxford, 1960, pp. 106–117. [Google Scholar]
- 3.
- 3a. Alvarez-Erviti L., Seow Y., Yin H., Betts C., Lakhal S., Wood M. J. A., Nat. Biotechnol. 2011, 29, 341–345; [DOI] [PubMed] [Google Scholar]
- 3b. Cavarretta I. T., Altanerova V., Matuskova M., Kucerova L., Culig Z., Altaner C., Mol. Ther. 2010, 18, 223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Maloney J. M., Uhland S. A., Polito B. F., Sheppard N. F., Pelta C. M., Santini J. T., J. Controlled Release 2005, 109, 244–255. [DOI] [PubMed] [Google Scholar]
- 5. Kohli K., Chopra S., Dhar D., Arora S., Khar R. K., Drug Discovery Today 2010, 15, 958–965. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Sullivan S. P., Koutsonanos D. G., del Pilar Martin M., Lee J. W., Zarnitsyn V., Choi S.-O., Murthy N., Compans R. W., Skountzou I., Prausnitz M. R., Nat. Med. 2010, 16, 915–920; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Lee J. W., Park J.-H., Prausnitz M. R., Biomaterials 2008, 29, 2113–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brambilla D., Luciani P., Leroux J.-C., J. Controlled Release 2014, 190, 9–14. [DOI] [PubMed] [Google Scholar]
- 8. Knop K., Hoogenboom R., Fischer D., Schubert U. S., Angew. Chem. Int. Ed. 2010, 49, 6288–6308; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 6430–6452. [Google Scholar]
- 9. Szymkowski D. E., Curr. Opin. Drug Discovery Dev. 2005, 8, 590–600. [PubMed] [Google Scholar]
- 10.
- 10a. Strohl W. R., BioDrugs 2015, 29, 215–239; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Caliceti P., Veronese F. M., Adv. Drug Delivery Rev. 2003, 55, 1261–1277; [DOI] [PubMed] [Google Scholar]
- 10c. Kontermann R. E., Expert Opin. Biol. Ther. 2016, 16, 903–915. [DOI] [PubMed] [Google Scholar]
- 11. Daley K. R., Kubarych K. J., J. Phys. Chem. B 2017, 121, 10574–10582. [DOI] [PubMed] [Google Scholar]
- 12. Veronese F. M., Pasut G., Drug Discovery Today 2005, 10, 1451–1458. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Kozma G. T., Shimizu T., Ishida T., Szebeni J., Adv. Drug Delivery Rev. 2020, 154–155, 163–175; [DOI] [PubMed] [Google Scholar]
- 13b. Ramos-de-la-Peña A. M., Aguilar O., Int. J. Pept. Res. Ther. 2020, 26, 333–348. [Google Scholar]
- 14.
- 14a. Armstrong J. K., Hempel G., Koling S., Chan L. S., Fisher T., Meiselman H. J., Garratty G., Cancer 2007, 110, 103–111; [DOI] [PubMed] [Google Scholar]
- 14b. Mima Y., Hashimoto Y., Shimizu T., Kiwada H., Ishida T., Mol. Pharmaceutics 2015, 12, 2429–2435; [DOI] [PubMed] [Google Scholar]
- 14c. Sundy J. S., Ganson N. J., Kelly S. J., Scarlett E. L., Rehrig C. D., Huang W., Hershfield M. S., Arthritis Rheum. 2007, 56, 1021–1028; [DOI] [PubMed] [Google Scholar]
- 14d. Yang Q., Jacobs T. M., McCallen J. D., Moore D. T., Huckaby J. T., Edelstein J. N., Lai S. K., Anal. Chem. 2016, 88, 11804–11812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Olson R. A., Korpusik A. B., Sumerlin B. S., Chem. Sci. 2020, 11, 5142–5156; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Chen C., Ng D. Y. W., Weil T., Prog. Polym. Sci. 2020, 105, 101241. [Google Scholar]
- 16.
- 16a. Abuchowski A., McCoy J. R., Palczuk N. C., van Es T., Davis F. F., J. Biol. Chem. 1977, 252, 3582–3586; [PubMed] [Google Scholar]
- 16b. Abuchowski A., van Es T., Palczuk N. C., Davis F. F., J. Biol. Chem. 1977, 252, 3578–3581. [PubMed] [Google Scholar]
- 17. Veronese F. M., Caliceti P., Schiavon O., J. Bioact. Compat. Polym. 1997, 12, 196–207. [Google Scholar]
- 18. Roberts M. J., Bentley M. D., Harris J. M., Adv. Drug Delivery Rev. 2012, 64, 116–127. [Google Scholar]
- 19.
- 19a. Turecek P. L., Bossard M. J., Schoetens F., Ivens I. A., J. Pharm. Sci. 2016, 105, 460–475; [DOI] [PubMed] [Google Scholar]
- 19b. Dozier J. K., Distefano M. D., Int. J. Mol. Sci. 2015, 16, 25831–25864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Carter M. C., Meyerhoff M. E., J. Immunol. Methods 1985, 81, 245–257. [DOI] [PubMed] [Google Scholar]
- 21. Monfardini C., Schiavon O., Caliceti P., Morpurgo M., Harris J. M., Veronese F. M., Bioconjugate Chem. 1995, 6, 62–69. [DOI] [PubMed] [Google Scholar]
- 22. Zalipsky S., Barany G., Polym. Prepr. Am. Chem. Soc. Div. Polym. Chem. 1986, 27, 1–2. [Google Scholar]
- 23. Harris J. M., Kowzlowski A., US Patent 5672662, 1997.
- 24. Molineux G., Curr. Pharm. Des. 2004, 10, 1235–1244. [DOI] [PubMed] [Google Scholar]
- 25. Kinstler O., Molineux G., Treuheit M., Ladd D., Gegg C., Adv. Drug Delivery Rev. 2002, 54, 477–485. [DOI] [PubMed] [Google Scholar]
- 26. Nesbitt A., Fossati G., Bergin M., Stephens P., Stephens S., Foulkes R., Brown D., Robinson M., Bourne T., Inflamm. Bowel Dis. 2007, 13, 1323–1332. [DOI] [PubMed] [Google Scholar]
- 27. Mei B., Pan C., Jiang H., Tjandra H., Strauss J., Chen Y., Liu T., Zhang X., Severs J., Newgren J., Chen J., Gu J. M., Subramanyam B., Fournel M. A., Pierce G. F., Murphy J. E., Blood 2010, 116, 270–279. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a. Tamshen K., Wang Y., Jamieson S. M. F., Perry J. K., Maynard H. D., Bioconjugate Chem. 2020, 31, 2179–2190; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28b. Wu L., Chen J., Wu Y., Zhang B., Cai X., Zhang Z., Wang Y., Si L., Xu H., Zheng Y., Zhang C., Liang C., Li J., Zhang L., Zhang Q., Zhou D., J. Controlled Release 2017, 249, 84–93; [DOI] [PubMed] [Google Scholar]
- 28c. Lühmann T., Jones G., Gutmann M., Rybak J. C., Nickel J., Rubini M., Meinel L., ACS Biomater. Sci. Eng. 2015, 1, 740–746. [DOI] [PubMed] [Google Scholar]
- 29. Rashidian M., Dozier J. K., Distefano M. D., Bioconjugate Chem. 2013, 24, 1277–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Popp M. W., Antos J. M., Grotenbreg G. M., Spooner E., Ploegh H. L., Nat. Chem. Biol. 2007, 3, 707–708. [DOI] [PubMed] [Google Scholar]
- 31.
- 31a. Popp M. W., Dougan S. K., Chuang T. Y., Spooner E., Ploegh H. L., Proc. Natl. Acad. Sci. USA 2011, 108, 3169–3174; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31b. da Silva Freitas D., Mero A., Pasut G., Bioconjugate Chem. 2013, 24, 456–463; [DOI] [PubMed] [Google Scholar]
- 31c. Grigoletto A., Mero A., Yoshioka H., Schiavon O., Pasut G., J. Drug Targeting 2017, 25, 856–864; [DOI] [PubMed] [Google Scholar]
- 31d. Hui X., Cao W., Zhang D., Ge W., Li S., Li Y., Shengwu Gongcheng Xuebao 2020, 36, 750–762. [Google Scholar]
- 32. Swierczewska M., Lee K. C., Lee S., Expert Opin. Emerging Drugs 2015, 20, 531–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.
- 33a. Wenande E., Garvey L. H., Clin. Exp. Allergy 2016, 46, 907–922; [DOI] [PubMed] [Google Scholar]
- 33b. Sellaturay P., Nasser S., Ewan P., J. Allergy Clin. Immunol. 2021, 9, 670–675. [DOI] [PubMed] [Google Scholar]
- 34.
- 34a. Richter A. W., Akerblom E., Int. Arch. Allergy Appl. Immunol. 1984, 74, 36–39; [DOI] [PubMed] [Google Scholar]
- 34b. Armstrong J. K., in PEGylated Protein Drugs:Basic Science and Clinical Applications, Springer, Berlin, 2009. [Google Scholar]
- 35. Sellaturay P., Nasser S., Islam S., Gurugama P., Ewan P. W., Clin. Exp. Allergy 2021, 51, 861–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cai Q., Li X., Zhu W., Macromolecules 2020, 53, 2177–2186. [Google Scholar]
- 37. Chen J., Zhao M., Feng F., Sizovs A., Wang J., J. Am. Chem. Soc. 2013, 135, 10938–10941. [DOI] [PubMed] [Google Scholar]
- 38.
- 38a. Qi Y., Chilkoti A., Curr. Opin. Chem. Biol. 2015, 28, 181–193; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38b. Hoang Thi T. T., Pilkington E. H., Nguyen D. H., Lee J. S., Park K. D., Truong N. P., Polymers 2020, 12, 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.
- 39a. Siegers C., Biesalski M., Haag R., Chemistry 2004, 10, 2831–2838; [DOI] [PubMed] [Google Scholar]
- 39b. Deng Y., Saucier-Sawyer J. K., Hoimes C. J., Zhang J., Seo Y. E., Andrejecsk J. W., Saltzman W. M., Biomaterials 2014, 35, 6595–6602; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39c. Khandare J., Calderón M., Dagia N. M., Haag R., Chem. Soc. Rev. 2012, 41, 2824–2848; [DOI] [PubMed] [Google Scholar]
- 39d. Calderon M., Quadir M. A., Sharma S. K., Haag R., Adv. Mater. 2010, 22, 190–218. [DOI] [PubMed] [Google Scholar]
- 40.
- 40a. Kainthan R. K., Hester S. R., Levin E., Devine D. V., Brooks D. E., Biomaterials 2007, 28, 4581–4590; [DOI] [PubMed] [Google Scholar]
- 40b. Kainthan R. K., Janzen J., Levin E., Devine D. V., Brooks D. E., Biomacromolecules 2006, 7, 703–709; [DOI] [PubMed] [Google Scholar]
- 40c. Imran ul-haq M., Lai B. F., Chapanian R., Kizhakkedathu J. N., Biomaterials 2012, 33, 9135–9147. [DOI] [PubMed] [Google Scholar]
- 41. Bewersdorff T., Vonnemann J., Kanik A., Haag R., Haase A., Int. J. Nanomed. 2017, 12, 2001–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.
- 42a. Pouyan P., Nie C., Bhatia S., Wedepohl S., Achazi K., Osterrieder N., Haag R., Biomacromolecules 2021, 22, 2298; [DOI] [PubMed] [Google Scholar]
- 42b. Gheybi H., Sattari S., Bodaghi A., Soleimani K., Dadkhah A., Adeli M., in Engineering of Biomaterials for Drug Delivery Systems, Elsevier, Amsterdam, 2018, pp. 103–171. [Google Scholar]
- 43. Thomas A., Muller S. S., Frey H., Biomacromolecules 2014, 15, 1935–1954. [DOI] [PubMed] [Google Scholar]
- 44.
- 44a. Gervais M., Brocas A.-L., Cendejas G., Deffieux A., Carlotti S., Macromolecules 2010, 43, 1778–1784; [Google Scholar]
- 44b. Gervais M., Brocas A.-L., Cendejas G., Deffieux A., Carlotti S., Macromol. Symp. 2011, 308, 101–111; [Google Scholar]
- 44c. Gervais A. M. L., Carlotti S., Deffieux A., Macromolecules 2009, 42, 2395–2400. [Google Scholar]
- 45.
- 45a. Wurm F., Dingels C., Frey H., Klok H. A., Biomacromolecules 2012, 13, 1161–1171; [DOI] [PubMed] [Google Scholar]
- 45b. Weinhart M., Grunwald I., Wyszogrodzka M., Gaetjen L., Hartwig A., Haag R., Chem. Asian J. 2010, 5, 1992–2000. [DOI] [PubMed] [Google Scholar]
- 46. Bej R., Achazi K., Haag R., Ghosh S., Biomacromolecules 2020, 21, 3353–3363. [DOI] [PubMed] [Google Scholar]
- 47. Thomas A., Niederer K., Wurm F., Frey H., Polym. Chem. 2014, 5, 899–909. [Google Scholar]
- 48. Haamann D., Keul H., Klee D., Möller M., Macromolecules 2010, 43, 6295–6301. [Google Scholar]
- 49. Tully M., Dimde M., Weise C., Pouyan P., Licha K., Schirner M., Haag R., Biomacromolecules 2021, 22, 1406–1416. [DOI] [PubMed] [Google Scholar]
- 50. Tully M., Wedepohl S., Kutifa D., Weise C., Licha K., Schirner M., Haag R., Eur. J. Pharm. Biopharm. 2021, 164, 105–113. [DOI] [PubMed] [Google Scholar]
- 51. Spears B. R., Waksal J., McQuade C., Lanier L., Harth E., Chem. Commun. 2013, 49, 2394–2396. [DOI] [PubMed] [Google Scholar]
- 52.
- 52a. Hoogenboom R., Angew. Chem. Int. Ed. 2009, 48, 7978–7994; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 8122–8138; [Google Scholar]
- 52b. Lorson T., Lubtow M. M., Wegener E., Haider M. S., Borova S., Nahm D., Jordan R., Sokolski-Papkov M., Kabanov A. V., Luxenhofer R., Biomaterials 2018, 178, 204–280. [DOI] [PubMed] [Google Scholar]
- 53.
- 53a. Barz M., Luxenhofer R., Zentel R., Vicent M. J., Polym. Chem. 2011, 2, 1900–1918; [Google Scholar]
- 53b. Viegas T. X., Bentley M. D., Harris J. M., Fang Z., Yoon K., Dizman B., Weimer R., Mero A., Pasut G., Veronese F. M., Bioconjugate Chem. 2011, 22, 976–986. [DOI] [PubMed] [Google Scholar]
- 54.
- 54a. Bauer M., Schroeder S., Tauhardt L., Kempe K., Schubert U. S., Fischer D., J. Polym. Sci. Part A 2013, 51, 1816–1821; [Google Scholar]
- 54b. Bauer M., Lautenschlaeger C., Kempe K., Tauhardt L., Schubert U. S., Fischer D., Macromol. Biosci. 2012, 12, 986–998. [DOI] [PubMed] [Google Scholar]
- 55. Mero A., Fang Z., Pasut G., Veronese F. M., Viegas T. X., J. Controlled Release 2012, 159, 353–361. [DOI] [PubMed] [Google Scholar]
- 56. Viegas T. X., Fang Z., Yoon K., Weimer R., Dizman B., in Engineering of Biomaterials for Drug Delivery Systems. Beyond Polyethylene Glycol. (Ed.: Parambath A.), Woodhead Publishing Series in Biomaterials, Cambridge, 2018, pp. 173–198. [Google Scholar]
- 57. Lühmann T., Schmidt M., Leiske M. N., Spieler V., Majdanski T. C., Grube M., Hartlieb M., Nischang I., Schubert S., Schubert U. S., Meinel L., ACS Biomater. Sci. Eng. 2017, 3, 304–312. [DOI] [PubMed] [Google Scholar]
- 58. Kudaibergenov S., Jaeger W., Laschewsky A., in Supramolecular Polymers Polymeric Betains Oligomers, Springer, Berlin, 2006, pp. 157–224. [Google Scholar]
- 59.
- 59a. Shao Q., Jiang S., Adv. Mater. 2015, 27, 15–26; [DOI] [PubMed] [Google Scholar]
- 59b. Yang W., Zhang L., Wang S., White A. D., Jiang S., Biomaterials 2009, 30, 5617–5621; [DOI] [PubMed] [Google Scholar]
- 59c. Jiang S., Cao Z., Adv. Mater. 2010, 22, 920–932. [DOI] [PubMed] [Google Scholar]
- 60.
- 60a. Hu J., Wang G., Zhao W., Gao W., J. Controlled Release 2016, 237, 71–77; [DOI] [PubMed] [Google Scholar]
- 60b. Bhattacharjee S., Liu W., Wang W. H., Weitzhandler I., Li X., Qi Y., Liu J., Pang Y., Hunt D. F., Chilkoti A., ChemBioChem 2015, 16, 2451–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pang Y., Liu J., Qi Y., Li X., Chilkoti A., Angew. Chem. Int. Ed. 2016, 55, 10296–10300; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 10452–10456. [Google Scholar]
- 62. Zhang P., Sun F., Liu S., Jiang S., J. Controlled Release 2016, 244, 184–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li B., Yuan Z., Hung H. C., Ma J., Jain P., Tsao C., Xie J., Zhang P., Lin X., Wu K., Jiang S., Angew. Chem. Int. Ed. 2018, 57, 13873–13876; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14069–14072. [Google Scholar]
- 64.
- 64a. Hou Y., Lu H., Bioconjugate Chem. 2019, 30, 1604–1616; [DOI] [PubMed] [Google Scholar]
- 64b. Hou Y., Zhou Y., Wang H., Sun J., Wang R., Sheng K., Yuan J., Hu Y., Chao Y., Liu Z., Lu H., ACS Cent. Sci. 2019, 5, 229–236; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64c. Hou Y., Zhou Y., Wang H., Wang R., Yuan J., Hu Y., Sheng K., Feng J., Yang S., Lu H., J. Am. Chem. Soc. 2018, 140, 1170–1178; [DOI] [PubMed] [Google Scholar]
- 64d. Wang H., Hou Y., Hu Y., Dou J., Shen Y., Wang Y., Lu H., Biomacromolecules 2019, 20, 3000–3008. [DOI] [PubMed] [Google Scholar]
- 65.
- 65a. Bleher S., Buck J., Muhl C., Sieber S., Barnert S., Witzigmann D., Huwyler J., Barz M., Suss R., Small 2019, 15, 1904716; [DOI] [PubMed] [Google Scholar]
- 65b. Weber B., Birke A., Fischer K., Schmidt M., Barz M., Macromolecules 2018, 51, 2653–2661; [Google Scholar]
- 65c. Settanni G., Schafer T., Muhl C., Barz M., Schmid F., Comput. Struct. Biotechnol. J. 2018, 16, 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nogueira S. S., Schlegel A., Maxeiner K., Weber B., Barz M., Schroer M. A., Blanchet C. E., Svergun D. I., Ramishetti S., Peer D., Langguth P., Sahin U., Haas H., ACS Appl. Nano Mater. 2020, 3, 10634–10645. [Google Scholar]
- 67. Son K., Ueda M., Taguchi K., Maruyama T., Takeoka S., Ito Y., J. Controlled Release 2020, 322, 209–216. [DOI] [PubMed] [Google Scholar]
- 68. Hu Y., Hou Y., Wang H., Lu H., Bioconjug. Chem. 2018, 29, 2232-2238.. [DOI] [PubMed] [Google Scholar]
- 69. Colley K. J., Kitajima K., Sato C., Crit. Rev. Biochem. Mol. Biol. 2014, 49, 498–532. [DOI] [PubMed] [Google Scholar]
- 70. Encapsulation and Controlled Release, Woodhead Publishing Series in Food, Science, Technology and Nutrition (Eds.: Gregoriadis G., McCormack B., Karsa D. R., Stephenson R. A.), Elsevier, Amsterdam, 2005, pp. 75–85. [Google Scholar]
- 71. Therapeutic Proteins: Strategies to Modulate Their Plasma Half-Lives (Eds.: Constantinou A., Chen C., Deonarain M. P., Kontermann R.), Wiley-VCH, Weinheim, 2012, pp. 95–115. [Google Scholar]
- 72. Siekmann J., Turecek P. L., in Polymer-Protein Conjugates, Elsevier, Amsterdam, 2020, pp. 455–469. [Google Scholar]
- 73. Han X., Zhang T., Liu M., Song Y., Liu X., Deng Y., Coatings 2020, 10(9), 834. [Google Scholar]
- 74. Gregoriadis G., Jain S., Papaioannou I., Laing P., Int. J. Pharm. 2005, 300, 125–130. [DOI] [PubMed] [Google Scholar]
- 75. Lindhout T., Iqbal U., Willis L. M., Reid A. N., Li J., Liu X., Moreno M., Wakarchuk W. W., Proc. Natl. Acad. Sci. USA 2011, 108, 7397–7402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.
- 76a. Fernandes A. I., Gregoriadis G., Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1996, 1293, 90–96; [DOI] [PubMed] [Google Scholar]
- 76b. Fernandes A. I., Gregoriadis G., Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1997, 1341, 26–34; [DOI] [PubMed] [Google Scholar]
- 76c. Fernandes A. I., Gregoriadis G., Int. J. Pharm. 2001, 217, 215–224; [DOI] [PubMed] [Google Scholar]
- 76d. Jain S., Hreczuk-Hirst D. H., McCormack B., Mital M., Epenetos A., Laing P., Gregoriadis G., Biochim. Biophys. Acta Gen. Subj. 2003, 1622, 42–49; [DOI] [PubMed] [Google Scholar]
- 76e. Meng H., Jain S., Lockshin C., Shaligram U., Martinez J., Genkin D., Hill D. B., Ehre C., Clark D., H. Hoppe IV , Recent Pat. Drug Delivery Formulation 2018, 12, 212–222. [DOI] [PubMed] [Google Scholar]
- 77. Constantinou A., Epenetos A. A., Hreczuk-Hirst D., Jain S., Wright M., Chester K. A., Deonarain M. P., Bioconjugate Chem. 2009, 20, 924–931. [DOI] [PubMed] [Google Scholar]
- 78. Jatzkewitz H., Z. Naturforsch. B 1955, 10, 27–31. [Google Scholar]
- 79. Ringsdorf H., J. Polym. Sci. Part C 1975, 51, 135–153. [Google Scholar]
- 80. Duncan R., Seymour L. W., O'Hare K. B., Flanagan P. A., Wedge S., Hume I. C., Ulbrich K., Strohalm J., Subr V., Spreafico F., Grandi M., Ripamonti M., Farao M., Suarato A., J. Controlled Release 1992, 19, 331–346. [Google Scholar]
- 81. Duncan R., Nat. Rev. Drug Discovery 2003, 2, 347–360. [DOI] [PubMed] [Google Scholar]
- 82.
- 82a. Ekladious I., Liu R., Zhang H., Foil D. H., Todd D. A., Graf T. N., Padera R. F., Oberlies N. H., Colson Y. L., Grinstaff M. W., Chem. Sci. 2017, 8, 8443–8450; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82b. Namgung R., Mi Lee Y., Kim J., Jang Y., Lee B.-H., Kim I.-S., Sokkar P., Rhee Y. M., Hoffman A. S., Kim W. J., Nat. Commun. 2014, 5, 3702; [DOI] [PubMed] [Google Scholar]
- 82c. Rades N., Achazi K., Qiu M., Deng C., Haag R., Zhong Z., Licha K., J. Controlled Release 2019, 300, 13–21; [DOI] [PubMed] [Google Scholar]
- 82d. Shen S., Xu X., Lin S., Zhang Y., Liu H., Zhang C., Mo R., Nat. Nanotechnol. 2021, 16, 104–113. [DOI] [PubMed] [Google Scholar]
- 83. Feng Q., Tong R., Bioeng. Transl. Med. 2016, 1, 277–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Korčáková L., Paluska E., Hašková V., Kopeček J., Z. Immunitaetsforsch. Exp. Klin. Immunol. 1976, 151, 219–223. [PubMed] [Google Scholar]
- 85. Kopeček J., Kopečková P., Adv. Drug Delivery Rev. 2010, 62, 122–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Vasey P. A., Kaye S. B., Morrison R., Twelves C., Wilson P., Duncan R., Thomson A. H., Murray L. S., Hilditch T. E., Murray T., Burtles S., Fraier D., Frigerio E., Cassidy J., Clin. Cancer Res. 1999, 5, 83–94. [PubMed] [Google Scholar]
- 87. Meerum Terwogt J. M., ten Bokkel Huinink W. W., Schellens J. H., Schot M., Mandjes I. A., Zurlo M. G., Rocchetti M., Rosing H., Koopman F. J., Beijnen J. H., Anti-Cancer Drugs 2001, 12, 315–323. [DOI] [PubMed] [Google Scholar]
- 88. Yang J., Kopeček J., Curr. Opin. Colloid Interface Sci. 2017, 31, 30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Schoemaker N. E., van Kesteren C., Rosing H., Jansen S., Swart M., Lieverst J., Fraier D., Breda M., Pellizzoni C., Spinelli R., Porro M. G., Beijnen J. H., Schellens J. H. M., ten Bokkel Huinink W. W., Br. J. Cancer 2002, 87, 608–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Rademaker-Lakhai J. M., Terret C., Howell S. B., Baud C. M., de Boer R. F., Pluim D., Beijnen J. H., Schellens J. H. M., Droz J.-P., Clin. Cancer Res. 2004, 10, 3386–3395. [DOI] [PubMed] [Google Scholar]
- 91.
- 91a. Parveen S., Arjmand F., Tabassum S., RSC Adv. 2019, 9, 24699–24721; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91b. Nowotnik D. P., Cvitkovic E., Adv. Drug Delivery Rev. 2009, 61, 1214–1219. [DOI] [PubMed] [Google Scholar]
- 92. Nakamura H., Koziolová E., Etrych T., Chytil P., Fang J., Ulbrich K., Maeda H., Eur. J. Pharm. Biopharm. 2015, 90, 90–96. [DOI] [PubMed] [Google Scholar]
- 93. Ogunleye A., Bhat A., Irorere V. U., Hill D., Williams C., Radecka I., Microbiology 2015, 161, 1–17. [DOI] [PubMed] [Google Scholar]
- 94. Manocha B., Margaritis A., J. Nanomater. 2010, 2010, 780171. [Google Scholar]
- 95. Pu Y., Chang S., Yuan H., Wang G., He B., Gu Z., Biomaterials 2013, 34, 3658–3666. [DOI] [PubMed] [Google Scholar]
- 96. Li C., Adv. Drug Delivery Rev. 2002, 54, 695–713. [DOI] [PubMed] [Google Scholar]
- 97. Li C., Yu D.-F., Newman R. A., Cabral F., Stephens L. C., Hunter N., Milas L., Wallace S., Cancer Res. 1998, 58, 2404–2409. [PubMed] [Google Scholar]
- 98. Bae Y., Nishiyama N., Fukushima S., Koyama H., Yasuhiro M., Kataoka K., Bioconjugate Chem. 2005, 16, 122–130. [DOI] [PubMed] [Google Scholar]
- 99. Duro-Castano A., Sousa-Herves A., Armiñán A., Charbonnier D., Arroyo-Crespo J. J., Wedepohl S., Calderón M., Vicent M. J., J. Controlled Release 2021, 332, 10–20. [DOI] [PubMed] [Google Scholar]
- 100. Arroyo-Crespo J. J., Deladriere C., Nebot V. J., Charbonnier D., Masiá E., Paul A., James C., Armiñán A., Vicent M. J., Adv. Funct. Mater. 2018, 28, 1800931. [Google Scholar]
- 101. Homsi J., Simon G. R., Garrett C. R., Springett G., De Conti R., Chiappori A. A., Munster P. N., Burton M. K., Stromatt S., Allievi C., Angiuli P., Eisenfeld A., Sullivan D. M., Daud A. I., Clin. Cancer Res. 2007, 13, 5855–5861. [DOI] [PubMed] [Google Scholar]
- 102. Shaffer S. A., Baker-Lee C., Kennedy J., Lai M. S., de Vries P., Buhler K., Singer J. W., Cancer Chemother. Pharmacol. 2007, 59, 537–548. [DOI] [PubMed] [Google Scholar]
- 103. Paz-Ares L., Ross H., O'Brien M., Riviere A., Gatzemeier U., Von Pawel J., Kaukel E., Freitag L., Digel W., Bischoff H., García-Campelo R., Iannotti N., Reiterer P., Bover I., Prendiville J., Eisenfeld A. J., Oldham F. B., Bandstra B., Singer J. W., Bonomi P., Br. J. Cancer 2008, 98, 1608–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.
- 104a. Tomalia D. A., Baker H., Dewald J., Hall M., Kallos G., Martin S., Roeck J., Ryder J., Smith P., Polym. J. 1985, 17, 117–132; [Google Scholar]
- 104b. Frey H., Haag R., Rev. Mol. Biotechnol. 2002, 90, 257–267. [DOI] [PubMed] [Google Scholar]
- 105.
- 105a. Jain N. K., Gupta U., Expert Opin. Drug Metab. Toxicol. 2008, 4, 1035–1052; [DOI] [PubMed] [Google Scholar]
- 105b. Wooley K. L., Fréchet J. M. J., Hawker C. J., Polymer 1994, 35, 4489–4495. [Google Scholar]
- 106. Astruc D., Boisselier E., Ornelas C., Chem. Rev. 2010, 110, 1857–1959. [DOI] [PubMed] [Google Scholar]
- 107.Starpharma.
- 108.
- 108a. McCarthy T. D., Karellas P., Henderson S. A., Giannis M., O'Keefe D. F., Heery G., Paull J. R., Matthews B. R., Holan G., Mol. Pharmaceutics 2005, 2, 312–318; [DOI] [PubMed] [Google Scholar]
- 108b. Gong E., Matthews B., McCarthy T., Chu J., Holan G., Raff J., Sacks S., Antiviral Res. 2005, 68, 139–146. [DOI] [PubMed] [Google Scholar]
- 109. Tyssen D., Henderson S. A., Johnson A., Sterjovski J., Moore K., La J., Zanin M., Sonza S., Karellas P., Giannis M. P., Krippner G., Wesselingh S., McCarthy T., Gorry P. R., Ramsland P. A., Cone R., Paull J. R., Lewis G. R., Tachedjian G., PLoS One 2010, 5, e12309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.
- 110a. Price C. F., Tyssen D., Sonza S., Davie A., Evans S., Lewis G. R., Xia S., Spelman T., Hodsman P., Moench T. R., Humberstone A., Paull J. R., Tachedjian G., PLoS One 2011, 6, e24095; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110b. Dezzutti C. S., James V. N., Ramos A., Sullivan S. T., Siddig A., Bush T. J., Grohskopf L. A., Paxton L., Subbarao S., Hart C. E., Antimicrob. Agents Chemother. 2004, 48, 3834–3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Patterson C. M., Balachander S. B., Grant I., Pop-Damkov P., Kelly B., McCoull W., Parker J., Giannis M., Hill K. J., Gibbons F. D., Hennessy E. J., Kemmitt P., Harmer A. R., Gales S., Purbrick S., Redmond S., Skinner M., Graham L., Secrist J. P., Schuller A. G., Wen S., Adam A., Reimer C., Cidado J., Wild M., Gangl E., Fawell S. E., Saeh J., Davies B. R., Owen D. J., Ashford M. B., Commun. Biol. 2021, 4, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Fox M. E., Guillaudeu S., Fréchet J. M. J., Jerger K., Macaraeg N., Szoka F. C., Mol. Pharmaceutics 2009, 6, 1562–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. England R. M., Hare J. I., Barnes J., Wilson J., Smith A., Strittmatter N., Kemmitt P. D., Waring M. J., Barry S. T., Alexander C., Ashford M. B., J. Controlled Release 2017, 247, 73–85. [DOI] [PubMed] [Google Scholar]
- 114. Xu Z., Wang Y., Ma Z., Wang Z., Wei Y., Jia X., Polym. Chem. 2016, 7, 715–721. [Google Scholar]
- 115. Jiang Y.-Y., Tang G.-T., Zhang L.-H., Kong S.-Y., Zhu S.-J., Pei Y.-Y., J of Drug Targ. 2010, 18, 389–403. [DOI] [PubMed] [Google Scholar]
- 116. Ekladious I., Colson Y. L., Grinstaff M. W., Nat. Rev. Drug Discovery 2019, 18, 273–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.
- 117a. Suksiriworapong J., Taresco V., Ivanov D. P., Styliari I. D., Sakchaisri K., Junyaprasert V. B., Garnett M. C., Colloids Surf. B 2018, 167, 115–125; [DOI] [PubMed] [Google Scholar]
- 117b. Tsai F.-T., Wang Y., Darensbourg D. J., J. Am. Chem. Soc. 2016, 138, 4626–4633. [DOI] [PubMed] [Google Scholar]
- 118.
- 118a. Zhou Q., Shao S., Wang J., Xu C., Xiang J., Piao Y., Zhou Z., Yu Q., Tang J., Liu X., Gan Z., Mo R., Gu Z., Shen Y., Nat. Nanotechnol. 2019, 14, 799–809; [DOI] [PubMed] [Google Scholar]
- 118b. Zhou Z., Shen Y., Tang J., Fan M., Van Kirk E. A., Murdoch W. J., Radosz M., Adv. Funct. Mater. 2009, 19, 3580–3589; [Google Scholar]
- 118c. Lee Y., Fukushima S., Bae Y., Hiki S., Ishii T., Kataoka K., J. Am. Chem. Soc. 2007, 129, 5362–5363; [DOI] [PubMed] [Google Scholar]
- 118d. Tangsangasaksri M., Takemoto H., Naito M., Maeda Y., Sueyoshi D., Kim H. J., Miura Y., Ahn J., Azuma R., Nishiyama N., Miyata K., Kataoka K., Biomacromolecules 2016, 17, 246–255. [DOI] [PubMed] [Google Scholar]
- 119. Xu J., Luo S., Shi W., Liu S., Langmuir 2006, 22, 989–997. [DOI] [PubMed] [Google Scholar]
- 120.
- 120a. Lukowiak M. C., Thota B. N., Haag R., Biotechnol. Adv. 2015, 33, 1327–1341; [DOI] [PubMed] [Google Scholar]
- 120b. Savić R., Azzam T., Eisenberg A., Maysinger D., Langmuir 2006, 22, 3570–3578; [DOI] [PubMed] [Google Scholar]
- 120c. Gong J., Chen M., Zheng Y., Wang S., Wang Y., J. Controlled Release 2012, 159, 312–323. [DOI] [PubMed] [Google Scholar]
- 121. Jansen J. F., de Brabander-van den Berg E. M., Meijer E. W., Science 1994, 266, 1226–1229. [DOI] [PubMed] [Google Scholar]
- 122. Zeng F., Zimmerman S. C., Chem. Rev. 1997, 97, 1681–1712. [DOI] [PubMed] [Google Scholar]
- 123. Jansen J. F. G. A., Meijer E. W., De Brabander - Van Den Berg E. M. M., Macromol. Symp. 1996, 102, 27–33. [Google Scholar]
- 124. Hawker C. J., Frechet J. M. J., J. Am. Chem. Soc. 1990, 112, 7638–7647. [Google Scholar]
- 125. Hawker C. J., Wooley K. L., Fréchet J. M. J., J. Chem. Soc. Perkin Trans. 1 1993, 1287–1297. [Google Scholar]
- 126. Belhadj-Tahar H., Chen A., Jia Y., Wu S., Sadeg N., Zhao H., Li C., Gu G., Gao Y., Yang G., Am. J. Clin. Oncol. 2018, 36, e15569–e15569. [Google Scholar]
- 127. Kainthan R. K., Mugabe C., Burt H. M., Brooks D. E., Biomacromolecules 2008, 9, 886–895. [DOI] [PubMed] [Google Scholar]
- 128. Mugabe C., Hadaschik B. A., Kainthan R. K., Brooks D. E., So A. I., Gleave M. E., Burt H. M., BJU Int. 2009, 103, 978–986. [DOI] [PubMed] [Google Scholar]
- 129. Zhao L., Chen G., Li J., Fu Y., Mavlyutov T. A., Yao A., Nickells R. W., Gong S., Guo L.-W., J. Controlled Release 2017, 247, 153–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Liu G., Gao H., Zuo Y., Zeng X., Tao W., Tsai H. I., Mei L., ACS Appl. Mater. Interfaces 2017, 9, 112–119. [DOI] [PubMed] [Google Scholar]
- 131. Jaskula-Sztul R., Xu W., Chen G., Harrison A., Dammalapati A., Nair R., Cheng Y., Gong S., Chen H., Biomaterials 2016, 91, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Jaskula-Sztul R., Chen G., Dammalapati A., Harrison A., Tang W., Gong S., Chen H., J. Mater. Chem. B 2017, 5, 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Lin W., Zhang X., Qian L., Yao N., Pan Y., Zhang L., Biomacromolecules 2017, 18, 3869–3880. [DOI] [PubMed] [Google Scholar]
- 134. Cherri M., Ferraro M., Mohammadifar E., Quaas E., Achazi K., Ludwig K., Grötzinger C., Schirner M., Haag R., ACS Biomater. Sci. Eng. 2021, 7, 2569–2579. [DOI] [PubMed] [Google Scholar]
- 135. Mohammadifar E., Zabihi F., Tu Z., Hedtrich S., Nemati Kharat A., Adeli M., Haag R., Polym. Chem. 2017, 8, 7375–7383. [Google Scholar]
- 136. Faouzi M. E. A., Dine T., Luyckx M., Brunet C., Mallevais M.-L., Goudaliez F., Gressier B., Cazin M., Kablan J., Cazin J. C., J. Pharm. Biomed. Anal. 1995, 13, 1363–1372. [DOI] [PubMed] [Google Scholar]
- 137. Gelderblom H., Verweij J., Nooter K., Sparreboom A., Eur. J. Cancer 2001, 37, 1590–1598. [DOI] [PubMed] [Google Scholar]
- 138. Maeda H., Wu J., Sawa T., Matsumura Y., Hori K., J. Controlled Release 2000, 65, 271–284. [DOI] [PubMed] [Google Scholar]
- 139. Kanapathipillai M., Brock A., Ingber D. E., Adv. Drug Delivery Rev. 2014, 79–80, 107–118. [DOI] [PubMed] [Google Scholar]
- 140. Wang J., Mao W., Lock L. L., Tang J., Sui M., Sun W., Cui H., Xu D., Shen Y., ACS Nano 2015, 9, 7195–7206. [DOI] [PubMed] [Google Scholar]
- 141.
- 141a. Achazi K., Haag R., Ballauff M., Dernedde J., Kizhakkedathu J. N., Maysinger D., Multhaup G., Angew. Chem. Int. Ed. 2021, 60, 3882–3904; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 3926–3950; [Google Scholar]
- 141b. Schöttler S., Landfester K., Mailänder V., Angew. Chem. Int. Ed. 2016, 55, 8806–8815; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 8950–8959. [Google Scholar]
- 142. Bertrand N., Grenier P., Mahmoudi M., Lima E. M., Appel E. A., Dormont F., Lim J.-M., Karnik R., Langer R., Farokhzad O. C., Nat. Commun. 2017, 8, 777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Lu J., Owen S. C., Shoichet M. S., Macromolecules 2011, 44, 6002–6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.
- 144a. Alberg I., Kramer S., Schinnerer M., Hu Q., Seidl C., Leps C., Drude N., Möckel D., Rijcken C., Lammers T., Diken M., Maskos M., Morsbach S., Landfester K., Tenzer S., Barz M., Zentel R., Small 2020, 16, 1907574; [DOI] [PubMed] [Google Scholar]
- 144b. Tenzer S., Docter D., Kuharev J., Musyanovych A., Fetz V., Hecht R., Schlenk F., Fischer D., Kiouptsi K., Reinhardt C., Landfester K., Schild H., Maskos M., Knauer S. K., Stauber R. H., Nat. Nanotechnol. 2013, 8, 772–781. [DOI] [PubMed] [Google Scholar]
- 145. Cho E. C., Xie J., Wurm P. A., Xia Y., Nano Lett. 2009, 9, 1080–1084. [DOI] [PubMed] [Google Scholar]
- 146. Nel A. E., Mädler L., Velegol D., Xia T., Hoek E. M. V., Somasundaran P., Klaessig F., Castranova V., Thompson M., Nat. Mater. 2009, 8, 543–557. [DOI] [PubMed] [Google Scholar]
- 147.
- 147a. Kamaly N., Yameen B., Wu J., Farokhzad O. C., Chem. Rev. 2016, 116, 2602–2663; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147b. Wong P. T., Choi S. K., Chem. Rev. 2015, 115, 3388–3432. [DOI] [PubMed] [Google Scholar]
- 148. Stenzel M. H., Angew. Chem. Int. Ed. 2021, 60, 2202–2206; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 2230–2234. [Google Scholar]
- 149. Ghezzi M., Pescina S., Padula C., Santi P., Del Favero E., Cantù L., Nicoli S., J. Controlled Release 2021, 332, 312–336. [DOI] [PubMed] [Google Scholar]
- 150. Venne A., Li S., Mandeville R., Kabanov A., Alakhov V., Cancer Res. 1996, 56, 3626. [PubMed] [Google Scholar]
- 151.
- 151a. Alakhov V., Klinski E., Li S., Pietrzynski G., Venne A., Batrakova E., Bronitch T., Kabanov A., Colloids Surf. B 1999, 16, 113–134; [Google Scholar]
- 151b. Danson S., Ferry D., Alakhov V., Margison J., Kerr D., Jowle D., Brampton M., Halbert G., Ranson M., Br. J. Cancer 2004, 90, 2085–2091; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151c. Hwang D., Ramsey J. D., Kabanov A. V., Adv. Drug Delivery Rev. 2020, 156, 80–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.
- 152a. Masayuki Y., Mizue M., Noriko Y., Teruo O., Yasuhisa S., Kazunori K., Shohei I., J. Controlled Release 1990, 11, 269–278; [Google Scholar]
- 152b. Yokoyama M., Inoue S., Kataoka K., Yui N., Okano T., Sakurai Y., Makromol. Chem. 1989, 190, 2041–2054. [Google Scholar]
- 153. Cabral H., Kataoka K., J. Controlled Release 2014, 190, 465–476. [DOI] [PubMed] [Google Scholar]
- 154. Bae Y., Fukushima S., Harada A., Kataoka K., Angew. Chem. Int. Ed. 2003, 42, 4640–4643; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 4788–4791. [Google Scholar]
- 155. Harada M., Bobe I., Saito H., Shibata N., Tanaka R., Hayashi T., Kato Y., Cancer Sci. 2011, 102, 192–199. [DOI] [PubMed] [Google Scholar]
- 156. Koizumi F., Kitagawa M., Negishi T., Onda T., Matsumoto S.-I., Hamaguchi T., Matsumura Y., Cancer Res. 2006, 66, 10048. [DOI] [PubMed] [Google Scholar]
- 157. Hamaguchi T., Matsumura Y., Suzuki M., Shimizu K., Goda R., Nakamura I., Nakatomi I., Yokoyama M., Kataoka K., Kakizoe T., Br. J. Cancer 2005, 92, 1240–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Kim S. C., Kim D. W., Shim Y. H., Bang J. S., Oh H. S., Kim S. W., Seo M. H., J. Controlled Release 2001, 72, 191–202. [DOI] [PubMed] [Google Scholar]
- 159. Sun X., Wang G., Zhang H., Hu S., Liu X., Tang J., Shen Y., ACS Nano 2018, 12, 6179–6192. [DOI] [PubMed] [Google Scholar]
- 160.
- 160a. He H., Liu L., Morin E. E., Liu M., Schwendeman A., Acc. Chem. Res. 2019, 52, 2445–2461; [DOI] [PubMed] [Google Scholar]
- 160b. Mi P., Miyata K., Kataoka K., Cabral H., Adv. Therapeutics 2021, 4, 2000159. [Google Scholar]
- 161.
- 161a. Gregoriadis G., Ryman B. E., Biochem. J. 1971, 124, 58P; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161b. Saravolatz L. D., Bern C., Adler-Moore J., Berenguer J., Boelaert M., den Boer M., Davidson R. N., Figueras C., Gradoni L., Kafetzis D. A., Ritmeijer K., Rosenthal E., Royce C., Russo R., Sundar S., Alvar J., Arch. Clin. Infect. Dis. 2006, 43, 917–924. [DOI] [PubMed] [Google Scholar]
- 162. Kobayashi T., Tsukagoshi S., Sakurai Y., Gann 1975, 66, 719–720. [PubMed] [Google Scholar]
- 163. Proffitt D. R., Bhalla M., Gossweiler R., Midgett J., Psychon. Bull. Rev. 1995, 2, 409–428. [DOI] [PubMed] [Google Scholar]
- 164. Barenholz Y., J. Controlled Release 2012, 160, 117–134. [DOI] [PubMed] [Google Scholar]
- 165. Gabizon A., Catane R., Uziely B., Kaufman B., Safra T., Cohen R., Martin F., Huang A., Barenholz Y., Cancer Res. 1994, 54, 987–992. [PubMed] [Google Scholar]
- 166. Semple S. C., Akinc A., Chen J., Sandhu A. P., Mui B. L., Cho C. K., Sah D. W., Stebbing D., Crosley E. J., Yaworski E., Hafez I. M., Dorkin J. R., Qin J., Lam K., Rajeev K. G., Wong K. F., Jeffs L. B., Nechev L., Eisenhardt M. L., Jayaraman M., Kazem M., Maier M. A., Srinivasulu M., Weinstein M. J., Chen Q., Alvarez R., Barros S. A., De S., Klimuk S. K., Borland T., Kosovrasti V., Cantley W. L., Tam Y. K., Manoharan M., Ciufolini M. A., Tracy M. A., de Fougerolles A., MacLachlan I., Cullis P. R., Madden T. D., Hope M. J., Nat. Biotechnol. 2010, 28, 172–176. [DOI] [PubMed] [Google Scholar]
- 167. Mallory K. L., Taylor J. A., Zou X., Waghela I. N., Schneider C. G., Sibilo M. Q., Punde N. M., Perazzo L. C., Savransky T., Sedegah M., Dutta S., Janse C. J., Pardi N., Lin P. J. C., Tam Y. K., Weissman D., Angov E., npj Vaccines 2021, 6, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.BionTech, 2022.
- 169. Zhang L., Eisenberg A., Science 1995, 268, 1728–1731. [DOI] [PubMed] [Google Scholar]
- 170.
- 170a. Bleul R., Thiermann R., Maskos M., Macromolecules 2015, 48, 7396–7409; [Google Scholar]
- 170b. Thiermann R., Mueller W., Montesinos-Castellanos A., Metzke D., Löb P., Hessel V., Maskos M., Polymer 2012, 53, 2205–2210. [Google Scholar]
- 171. Habel J., Ogbonna A., Larsen N., Schulte L., Almdal K., Hélix-Nielsen C., J. Polym. Sci. Part B 2016, 54, 699–708. [Google Scholar]
- 172.
- 172a. Chen W., Meng F., Cheng R., Zhong Z., J. Controlled Release 2010, 142, 40–46; [DOI] [PubMed] [Google Scholar]
- 172b. Fang Y., Yang W., Cheng L., Meng F., Zhang J., Zhong Z., Acta Biomater. 2017, 64, 323–333; [DOI] [PubMed] [Google Scholar]
- 172c. Gao H., Shi W., Freund L. B., Proc. Natl. Acad. Sci. USA 2005, 102, 9469–9474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Photos P. J., Bacakova L., Discher B., Bates F. S., Discher D. E., J. Controlled Release 2003, 90, 323–334. [DOI] [PubMed] [Google Scholar]
- 174. Zou Y., Fang Y., Meng H., Meng F., Deng C., Zhang J., Zhong Z., J. Controlled Release 2016, 244, 326–335. [DOI] [PubMed] [Google Scholar]
- 175. Zhong Y., Dimde M., Stöbener D., Meng F., Deng C., Zhong Z., Haag R., ACS Appl. Mater. Interfaces 2016, 8, 27530–27538. [DOI] [PubMed] [Google Scholar]
- 176. Gu W., Meng F., Haag R., Zhong Z., J. Controlled Release 2021, 329, 676–695. [DOI] [PubMed] [Google Scholar]
- 177. Nagarajan R., Langmuir 2002, 18, 31–38. [Google Scholar]
- 178. Discher D. E., Ahmed F., Annu. Rev. Biomed. Eng. 2006, 8, 323–341. [DOI] [PubMed] [Google Scholar]
- 179. Lee H., Larson R. G., Biomacromolecules 2016, 17, 1757–1765. [DOI] [PubMed] [Google Scholar]
- 180. Papahadjopoulos D., Allen T., Gabizon A., Mayhew E., Matthay K., Huang S., Lee K., Woodle M., Lasic D., Redemann C., Proc. Natl. Acad. Sci. USA 1991, 88, 11460–11464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Zhang Y., Wu F., Yuan W., Jin T., J. Controlled Release 2010, 147, 413–419. [DOI] [PubMed] [Google Scholar]
- 182. Kim Y., Tewari M., Pajerowski J. D., Cai S., Sen S., Williams J., Sirsi S., Lutz G., Discher D. E., J. Controlled Release 2009, 134, 132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Zou Y., Zheng M., Yang W., Meng F., Miyata K., Kim H. J., Kataoka K., Zhong Z., Adv. Mater. 2017, 29, 1703285. [DOI] [PubMed] [Google Scholar]
- 184. Scott E. A., Stano A., Gillard M., Maio-Liu A. C., Swartz M. A., Hubbell J. A., Biomaterials 2012, 33, 6211–6219. [DOI] [PubMed] [Google Scholar]
- 185. Ahmed F., Discher D. E., J. Controlled Release 2004, 96, 37–53. [DOI] [PubMed] [Google Scholar]
- 186. Kumar A., Lale S. V., Mahajan S., Choudhary V., Koul V., ACS Appl. Mater. Interfaces 2015, 7, 9211–9227. [DOI] [PubMed] [Google Scholar]
- 187. Lin M., Dai Y., Xia F., Zhang X., Mater. Sci. Eng. C 2021, 119, 111626. [DOI] [PubMed] [Google Scholar]
- 188.
- 188a. Talelli M., Barz M., Rijcken C. J., Kiessling F., Hennink W. E., Lammers T., Nano Today 2015, 10, 93–117; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188b. O′Reilly R. K., Hawker C. J., Wooley K. L., Chem. Soc. Rev. 2006, 35, 1068–1083. [DOI] [PubMed] [Google Scholar]
- 189. Rijcken C. J., Snel C. J., Schiffelers R. M., van Nostrum C. F., Hennink W. E., Biomaterials 2007, 28, 5581–5593. [DOI] [PubMed] [Google Scholar]
- 190. Liu Y., van Steenbergen M. J., Zhong Z., Oliveira S., Hennink W. E., van Nostrum C. F., Macromolecules 2020, 53, 7009–7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Li Y., Xiao K., Zhu W., Deng W., Lam K. S., Adv. Drug Delivery Rev. 2014, 66, 58–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Zou Y., Meng F., Deng C., Zhong Z., J. Controlled Release 2016, 239, 149–158. [DOI] [PubMed] [Google Scholar]
- 193. Wang X., Liu G., Hu J., Zhang G., Liu S., Angew. Chem. Int. Ed. 2014, 53, 3138–3142; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3202–3206. [Google Scholar]
- 194.
- 194a. Mekuria S. L., Ouyang Z., Song C., Rodrigues J., Shen M., Shi X., Bioconjugate Chem. 2022, 33, 87–96; [DOI] [PubMed] [Google Scholar]
- 194b. Gruber A., Navarro L., Klinger D., Adv. Mater. Interfaces 2020, 7, 1901676; [Google Scholar]
- 194c. Jiang Y., Chen J., Deng C., Suuronen E. J., Zhong Z., Biomaterials 2014, 35, 4969–4985; [DOI] [PubMed] [Google Scholar]
- 194d. Oehrl A., Schötz S., Haag R., Colloid Polym. Sci. 2020, 298, 719–733; [Google Scholar]
- 194e. Gao L., Zabihi F., Ehrmann S., Hedtrich S., Haag R., J. Controlled Release 2019, 300, 64–72; [DOI] [PubMed] [Google Scholar]
- 194f. Zhang X., Malhotra S., Molina M., Haag R., Chem. Soc. Rev. 2015, 44, 1948–1973. [DOI] [PubMed] [Google Scholar]
- 195. Taiariol L., Chaix C., Farre C., Moreau E., Chem. Rev. 2022, 122, 340–384 [DOI] [PubMed] [Google Scholar]
- 196. Srinivasarao M., Low P. S., Chem. Rev. 2017, 117, 12133–12164. [DOI] [PubMed] [Google Scholar]
- 197. Germain M., Caputo F., Metcalfe S., Tosi G., Spring K., Åslund A. K. O., Pottier A., Schiffelers R., Ceccaldi A., Schmid R., J. Controlled Release 2020, 326, 164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Harada A., Kataoka K., Macromolecules 1995, 28, 5294–5299. [Google Scholar]
- 199.
- 199a. Kim A., Miura Y., Ishii T., Mutaf O. F., Nishiyama N., Cabral H., Kataoka K., Biomacromolecules 2016, 17, 446–453; [DOI] [PubMed] [Google Scholar]
- 199b. Lee Y., Ishii T., Cabral H., Kim H. J., Seo J.-H., Nishiyama N., Oshima H., Osada K., Kataoka K., Angew. Chem. Int. Ed. 2009, 48, 5309–5312; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 5413–5416; [Google Scholar]
- 199c. Lee Y., Ishii T., Kim H. J., Nishiyama N., Hayakawa Y., Itaka K., Kataoka K., Angew. Chem. Int. Ed. 2010, 49, 2552–2555; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 2606–2609. [Google Scholar]
- 200. Harada A., Kataoka K., J. Am. Chem. Soc. 1999, 121, 9241–9242. [Google Scholar]
- 201. Tao A., Huang G. L., Igarashi K., Hong T., Liao S., Stellacci F., Matsumoto Y., Yamasoba T., Kataoka K., Cabral H., Macromol. Biosci. 2020, 20, 1900161. [DOI] [PubMed] [Google Scholar]
- 202.
- 202a. Matsumoto Y., Nichols J. W., Toh K., Nomoto T., Cabral H., Miura Y., Christie R. J., Yamada N., Ogura T., Kano M. R., Matsumura Y., Nishiyama N., Yamasoba T., Bae Y. H., Kataoka K., Nat. Nanotechnol. 2016, 11, 533–538; [DOI] [PubMed] [Google Scholar]
- 202b. Flessner M. F., Choi J., Credit K., Deverkadra R., Henderson K., Clin. Cancer Res. 2005, 11, 3117–3125; [DOI] [PubMed] [Google Scholar]
- 202c. Minchinton A. I., Tannock I. F., Nat. Rev. Cancer 2006, 6, 583–592. [DOI] [PubMed] [Google Scholar]
- 203.
- 203a. Miura S., Suzuki H., Bae Y. H., Nano Today 2014, 9, 695–704; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203b. Syvänen S., Edén D., Sehlin D., Biochem. Biophys. Res. Commun. 2017, 493, 120–125. [DOI] [PubMed] [Google Scholar]
- 204. Xu P., Van Kirk E. A., Zhan Y., Murdoch W. J., Radosz M., Shen Y., Angew. Chem. Int. Ed. 2007, 46, 4999–5002; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 5087–5090. [Google Scholar]
- 205. Anselmo A. C., Mitragotri S., Bioeng. Transl. Med. 2019, 4, e10143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Brazeau P., Vale W., Burgus R., Ling N., Butcher M., Rivier J., Guillemin R., Science 1973, 179, 77–79. [DOI] [PubMed] [Google Scholar]
- 207. Kwekkeboom D. J., Kam B. L., van Essen M., Teunissen J. J., van Eijck C. H., Valkema R., de Jong M., de Herder W. W., Krenning E. P., Endocr.-Relat. Cancer 2010, 17, R53–R73. [DOI] [PubMed] [Google Scholar]
- 208. Baum R. P., Singh A., Schuchardt C., Kulkarni H. R., Klette I., Wiessalla S., Osterkamp F., Reineke U., Smerling C., J. Nucl. Med. 2018, 59, 809–814. [DOI] [PubMed] [Google Scholar]
- 209. Battistini L., Bugatti K., Sartori A., Curti C., Zanardi F., Eur. J. Org. Chem. 2021, 2506–2528. [Google Scholar]