

Abstract
Background
Paroxetine is the most potent inhibitor of the reuptake of serotonin of all selective serotonin reuptake inhibitors (SSRIs) and has been studied in many randomised controlled trials (RCTs). However, these comparative studies provided contrasting findings and systematic reviews of RCTs have always considered the SSRIs as a group, and evidence applicable to this group of drugs might not be applicable to paroxetine alone. The present systematic review assessed the efficacy and tolerability profile of paroxetine in comparison with tricyclics (TCAs), SSRIs and newer or non‐conventional agents.
Objectives
1. To determine the efficacy of paroxetine in comparison with other anti‐depressive agents in alleviating the acute symptoms of Major Depressive Disorder.
2. To review acceptability of treatment with paroxetine in comparison with other anti‐depressive agents.
3. To investigate the adverse effects of paroxetine in comparison with other anti‐depressive agents.
Search methods
We searched the Cochrane Depression, Anxiety and Neurosis Review Group's Specialized Register (CCDANCTR, to 30 September 2012), which includes relevant randomised controlled trials from the following bibliographic databases: The Cochrane Library (all years), EMBASE (1974 to date), MEDLINE (1950 to date) and PsycINFO (1967 to date). Reference lists of relevant papers and previous systematic reviews were handsearched. Pharmaceutical companies marketing paroxetine and experts in this field were contacted for supplemental data.
Selection criteria
All randomised controlled trials allocating participants with major depression to paroxetine versus any other antidepressants (ADs), both conventional (such as TCAs, SSRIs) and newer or non‐conventional (such as hypericum). For trials which had a cross‐over design, only results from the first randomisation period were considered.
Data collection and analysis
Two review authors independently checked eligibility and extracted data using a standard form. Data were then entered in RevMan 5.2 with a double‐entry procedure. Information extracted included study and participant characteristics, intervention details, settings and efficacy, acceptability and tolerability measures.
Main results
A total of 115 randomised controlled trials (26,134 participants) were included. In 54 studies paroxetine was compared with older ADs, in 21 studies with another SSRI, and in 40 studies with a newer or non‐conventional antidepressant other than SSRIs. For the primary outcome (patients who responded to treatment), paroxetine was more effective than reboxetine at increasing patients who responded early to treatment (Odds Ratio (OR): 0.66, 95% Confidence Interval (CI) 0.50 to 0.87, number needed to treat to provide benefit (NNTb) = 16, 95% CI 10 to 50, at one to four weeks, 3 RCTs, 1375 participants, moderate quality of evidence), and less effective than mirtazapine (OR: 2.39, 95% CI 1.42 to 4.02, NNTb = 8, 95% CI 5 to 14, at one to four weeks, 3 RCTs, 726 participants, moderate quality of evidence). Paroxetine was less effective than citalopram in improving response to treatment (OR: 1.54, 95% CI 1.04 to 2.28, NNTb = 9, 95% CI 5 to 102, at six to 12 weeks, 1 RCT, 406 participants, moderate quality of evidence). We found no clear evidence that paroxetine was more or less effective compared with other antidepressants at increasing response to treatment at acute (six to 12 weeks), early (one to four weeks), or longer term follow‐up (four to six months). Paroxetine was associated with a lower rate of adverse events than amitriptyline, imipramine and older ADs as a class, but was less well tolerated than agomelatine and hypericum. Included studies were generally at unclear or high risk of bias due to poor reporting of allocation concealment and blinding of outcome assessment, and incomplete reporting of outcomes.
Authors' conclusions
Some possibly clinically meaningful differences between paroxetine and other ADs exist, but no definitive conclusions can be drawn from these findings. In terms of response, there was a moderate quality of evidence that citalopram was better than paroxetine in the acute phase (six to 12 weeks), although only one study contributed data. In terms of early response to treatment (one to four weeks) there was moderate quality of evidence that mirtazapine was better than paroxetine and that paroxetine was better than reboxetine. However there was no clear evidence that paroxetine was better or worse compared with other antidepressants at increasing response to treatment at any time point. Even if some differences were identified, the findings from this review are better thought as hypothesis forming rather than hypothesis testing and it would be reassuring to see the conclusions replicated in future trials. Finally, most of included studies were at unclear or high risk of bias, and were sponsored by the drug industry. The potential for overestimation of treatment effect due to sponsorship bias should be borne in mind.
Plain language summary
Paroxetine versus other anti‐depressive agents for depression
Major depression is a severe mental illness characterised by a persistent and unreactive low mood and loss of all interest and pleasure, usually accompanied by a range of symptoms such as appetite change, sleep disturbance, fatigue, loss of energy, poor concentration, inappropriate guilt and morbid thoughts of death. Although medication and psychological treatments are both effective for major depression, antidepressant drugs remain the mainstay of treatment in moderate to severe major depression. However, head‐to‐head comparisons of such drugs provide contrasting findings as to whether they are effective.
This review of the research on the effect of an antidepressant drug called paroxetine was conducted to shed light on the field of drug treatment for depression. In September 2012 we searched, in a wide ranging way, for all the useful studies (randomised controlled trials) which had been completed which compared paroxetine with any other antidepressant in treating people with depression. One hundred and fifteen studies were included in this review, with a total of 26,134 people. We grouped the studies according to the types of drug they compared paroxetine against; we then analysed the combined findings of these groups of studies.
For the primary outcome (number of people who responded to treatment) paroxetine was more effective than reboxetine, but less effective than mirtazapine (in the early phase: one to four weeks follow‐up) and probably citalopram (at endpoint: six weeks follow‐up). There was some evidence that paroxetine is less well tolerated than agomelatine and St John's Wort, as more patients allocated to paroxetine experienced at least some side effects (though this finding for St John's Wort was only based on one study).
In conclusion, some possibly meaningful differences between paroxetine and other antidepressants exist, but no definitive concluions can be drawn due to the limited number of studies per comparison. In addition, most of included studies were sponsored by the drug industry, which means they might potentially have overestimated the effect of paroxetine. Therefore, the results of this review should be interpreted with caution.
Summary of findings
Summary of findings for the main comparison. Paroxetine compared with older ADs for depression.
| Paroxetine compared with older ADs for depression | ||||||
| Patient or population: patients with depression Settings: in‐ and out‐patients Intervention: paroxetine Comparison: older ADs | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Older ADs | Paroxetine | |||||
| Failure to respond at endpoint (6‐12 weeks) | 416 per 1000 | 426 per 1000 (396 to 455) | OR 1.04 (0.92 to 1.17) | 4647 (34 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to respond at 1‐4 weeks | 670 per 1000 | 416 per 1000 (328 to 509) | OR 0.90 (0.61 to 1.33) | 526 (4 studies) | ⊕⊕⊝⊝ low1 | |
| Failure to respond at 16‐24 weeks | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| Failure to remit at endpoint | 550 per 1000 | 365 per 1000 (180 to 601) | OR 1.23 (0.49 to 3.07) | 401 (3 studies) | ⊕⊕⊝⊝ low1,2 | |
| SMD at endpoint | The mean SMD at endpoint in the intervention groups was 0.01 standard deviations higher (0.08 lower to 0.09 higher) | 4745 (35 studies) | ⊕⊕⊕⊝ moderate1 | This effect approaches zero. | ||
| Failure to complete ‐ any cause ‐ | 312 per 1000 | 276 per 1000 (249 to 303) | OR 0.84 (0.73 to 0.96) | 6810 (44 studies) | ⊕⊕⊕⊝ moderate1 | |
| Participants with at least some Side Effects | 707 per 1000 | 606 per 1000 (565 to 650) | OR 0.64 (0.54 to 0.77) | 6132 (42 studies) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; SMD: standardized mean difference; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Blinding stated but not tested. No information on randomisation procedures and allocation concealment. 2 I squared 79%.
Summary of findings 2. Paroxetine compared with agomelatine for depression.
| Paroxetine compared with agomelatine for depression | ||||||
| Patient or population: patients with depression Settings: in‐ and out‐patients Intervention: paroxetine Comparison: agomelatine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Agomelatine | Paroxetine | |||||
| Failure to respond at endpoint (6‐12 weeks) Follow‐up: 6‐12 weeks | 394 per 1000 | 449 per 1000 (337 to 567) | OR 1.25 (0.78 to 2.01) | 284 (1 study) | ⊕⊕⊕⊝ moderate1 | |
| Failure to respond at 1‐4 weeks | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| Failure to respond at 16‐24 weeks | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| Failure to remit at endpoint HDRS Follow‐up: 6‐12 weeks | 701 per 1000 | 748 per 1000 (637 to 834) | OR 1.27 (0.75 to 2.14) | 284 (1 study) | ⊕⊕⊕⊝ moderate1 | |
| SMD at endpoint HDRS Follow‐up: 6‐12 weeks | The mean SMD at endpoint in the intervention groups was 0.18 standard deviations lower (0.38 lower to 0.02 higher) | 1074 (4 studies) | ⊕⊕⊕⊝ moderate1 | The point estimate of the effect size corresponds to a small effect according to Cohen 1992. | ||
| Failure to complete ‐ any cause ‐ | 234 per 1000 | 232 per 1000 (148 to 343) | OR 0.99 (0.57 to 1.71) | 284 (1 study) | ⊕⊕⊕⊝ moderate1 | |
| Participants with at least some Side Effects | 547 per 1000 | 659 per 1000 (545 to 758) | OR 1.60 (0.99 to 2.59) | 284 (1 study) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; SMD: standardized mean difference; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Blinding stated but not tested. No information on randomisation procedures and allocation concealment.
Summary of findings 3. Paroxetine compared with amisulpride for depression.
| Paroxetine compared with amisulpride for depression | ||||||
| Patient or population: patients with depression Settings: in‐ and out‐patients Intervention: paroxetine Comparison: amisulpride | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Amisulpride | Paroxetine | |||||
| Failure to respond at endpoint (6‐12 weeks) | 254 per 1000 | 181 per 1000 (109 to 281) | OR 0.65 (0.36 to 1.15) | 277 (1 study) | ⊕⊕⊕⊝ moderate1 | |
| Failure to respond at 1‐4 weeks | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| Failure to respond at 16‐24 weeks | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| Failure to remit at endpoint | 268 per 1000 | 237 per 1000 (152 to 348) | OR 0.85 (0.49 to 1.46) | 277 (1 study) | ⊕⊕⊕⊝ moderate1 | |
| SMD at endpoint | The mean SMD at endpoint in the intervention groups was 0.13 standard deviations lower (0.37 lower to 0.10 higher) | 272 (1 study) | ⊕⊕⊕⊝ moderate1 | The point estimate of the effect size corresponds to a small effect according to Cohen 1992. | ||
| Failure to complete ‐ any cause ‐ | See comment | See comment | Not estimable | 0 (0) |
See comment | No trial reported this outcome. |
| Participants with at least some Side Effects | Study population | OR 1.49 (0.89 to 2.50) | 277 (1 study) | ⊕⊕⊕⊝ moderate1 | ||
| 261 per 1000 | 345 per 1000 (239 to 469) | |||||
| Moderate | ||||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; SMD: standardized mean difference; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Blinding stated but not tested. No information on randomisation procedures and allocation concealment.
Summary of findings 4. Paroxetine compared with aprepitant for depression.
| paroxetine compared with aprepitant for depression | ||||||
| Patient or population: patients with depression Settings: in‐ and out‐patients Intervention: paroxetine Comparison: aprepitant | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Aprepitant | Paroxetine | |||||
| Failure to respond at endpoint (6‐12 weeks) | See comment | See comment | See comment | 0 (0) | See comment | No trial reported this outcome. |
| Failure to respond at 1‐4 weeks | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| Failure to respond at 16‐24 weeks | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| Failure to remit at endpoint | 444 per 1000 | 557 per 1000 (464 to 647) | OR 1.57 (1.08 to 2.29) | 807 (4 studies) | ⊕⊕⊕⊕ high | |
| SMD at endpoint | The mean SMD at endpoint in the intervention groups was 0.00 standard deviations higher (0.39 lower to 0.39 higher) | 102 (1 study) | ⊕⊕⊕⊕ high | This correspond to no treatment effect. | ||
| Failure to complete ‐ any cause ‐ | 282 per 1000 | 375 per 1000 (230 to 548) | OR 1.53 (0.76 to 3.09) | 143 (1 study) | ⊕⊕⊕⊝ moderate1 | |
| Participants with at least some Side Effects | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; SMD: standardized mean difference; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Blinding stated but not tested. No information on randomisation procedures and allocation concealment.
Summary of findings 5. Paroxetine compared with bupropion for depression.
| Paroxetine compared with bupropion for depression | ||||||
| Patient or population: patients with depression Settings: in‐ and out‐patients Intervention: paroxetine Comparison: bupropion | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Bupropion | Paroxetine | |||||
| Failure to respond at endpoint (6‐12 weeks) | 292 per 1000 | 231 per 1000 (110 to 424) | OR 0.73 (0.3 to 1.79) | 100 (1 study) | ⊕⊕⊕⊝ moderate1 | |
| Failure to respond at 1‐4 weeks | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| Failure to respond at 16‐24 weeks | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| Failure to remit at endpoint | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| SMD at endpoint | The mean SMD at endpoint in the intervention groups was 0.04 standard deviations lower (0.38 lower to 0.3 higher) | 132 (1 study) | ⊕⊕⊕⊝ moderate1 | This effect approaches zero. | ||
| Failure to complete ‐ any cause ‐ | 188 per 1000 | 212 per 1000 (126 to 338) | OR 1.16 (0.62 to 2.2) | 240 (2 studies) | ⊕⊕⊕⊝ moderate1 | |
| Participants with at least some Side Effects | 913 per 1000 | 944 per 1000 (819 to 984) | OR 1.60 (0.43 to 5.92) | 140 (1 study) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; SMD: standardized mean difference; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Blinding stated but not tested. No information on randomisation procedures and allocation concealment.
Summary of findings 6. Paroxetine compared with citalopram for depression.
| Paroxetine compared with citalopram for depression | ||||||
| Patient or population: patients with depression Settings: in‐ and out‐patients Intervention: paroxetine Comparison: citalopram | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Citalopram | Paroxetine | |||||
| Failure to respond at endpoint (6‐12 weeks) | 507 per 1000 | 613 per 1000 (517 to 701) | OR 1.54 (1.04 to 2.28) | 406 (1 study) | ⊕⊕⊕⊝ moderate | |
| Failure to respond at 1‐4 weeks | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| Failure to respond at 16‐24 weeks | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| Failure to remit at endpoint | See comment | See comment | Not estimable | See comment | No trial reported this outcome. | |
| SMD at endpoint | The mean SMD at endpoint in the intervention groups was 0.16 standard deviations lower (0.44 lower to 0.11 higher) | 201 (1 study) | ⊕⊕⊕⊝ moderate1 | The point estimate of the effect size corresponds to a small effect according to Cohen 1992. | ||
| Failure to complete ‐ any cause ‐ | 208 per 1000 | 206 per 1000 (138 to 296) | OR 0.99 (0.61 to 1.6) | 406 (1 study) | ⊕⊕⊕⊝ moderate1 | |
| Participants with at least some Side Effects | Study population | OR 0.74 (0.46 to 1.21) | 406 (1 study) | ⊕⊕⊕⊝ moderate1 | ||
| 821 per 1000 | 773 per 1000 (679 to 848) | |||||
| Moderate | ||||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; SMD: standardized mean difference; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Blinding stated but not tested. No information on randomisation procedures and allocation concealment.
Summary of findings 7. Paroxetine compared with duloxetine for depression.
| Paroxetine compared with duloxetine for depression | ||||||
| Patient or population: patients with depression Settings: in‐ and out‐patients Intervention: paroxetine Comparison: duloxetine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Duloxetine | Paroxetine | |||||
| Failure to respond at endpoint (6‐12 weeks) | 459 per 1000 | 431 per 1000 (373 to 492) | OR 0.89 (0.70 to 1.14) | 1821 (6 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to respond at 1‐4 weeks | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| Failure to respond at 16‐24 weeks | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| Failure to remit at endpoint | 592 per 1000 | 587 per 1000 (537 to 633) | OR 0.98 (0.80 to 1.19) | 1821 (6 studies) | ⊕⊕⊕⊝ moderate1 | |
| SMD at endpoint | The mean SMD at endpoint in the intervention groups was 0.04 standard deviations higher (0.06 lower to 0.15 higher) | 1481 (6 studies) | ⊕⊕⊕⊝ moderate1 | This effect approaches zero. | ||
| Failure to complete ‐ any cause ‐ | 237 per 1000 | 232 per 1000 (193 to 277) | OR 0.97 (0.77 to 1.23) | 1821 (6 studies) | ⊕⊕⊕⊝ moderate1 | |
| Participants with at least some Side Effects | 700 per 1000 | 654 per 1000 (599 to 702) | OR 0.81 (0.64 to 1.01) | 1870 (6 studies) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; SMD: standardized mean difference; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Blinding stated but not tested. No information on randomisation procedures and allocation concealment.
Summary of findings 8. Paroxetine compared with escitalopram for depression.
| Paroxetine compared with escitalopram for depression | ||||||
| Patient or population: depression Settings: in‐ and out‐patients Intervention: paroxetine Comparison: escitalopram | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Escitalopram | Paroxetine | |||||
| Failure to respond at endpoint (6‐12 weeks) | 312 per 1000 | 336 per 1000 (256 to 427) | OR 1.12 (0.76 to 1.65) | 784 (2 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to respond at 1‐4 weeks | See comment | See comment | Not estimable | 0 (0) | No trial reported this outcome. | |
| Failure to respond at 16‐24 weeks | Study population | OR 1.36 (0.87 to 2.12) | 459 (1 study) | ⊕⊕⊕⊝ moderate1 | ||
| 194 per 1000 | 247 per 1000 (173 to 338) | |||||
| Moderate | ||||||
| Failure to remit at endpoint | 407 per 1000 | 441 per 1000 (292 to 604) | OR 1.15 (0.6 to 2.22) | 784 (2 studies) | ⊕⊕⊝⊝ low1,2 | |
| SMD at endpoint | The mean SMD at endpoint in the intervention groups was 0.05 standard deviations higher (0.26 lower to 0.36 higher) | 772 (2 studies) | ⊕⊕⊝⊝ low1,3 | This effect approaches zero. | ||
| Failure to complete ‐ any cause ‐ | 156 per 1000 | 213 per 1000 (124 to 340) | OR 1.47 (0.77 to 2.79) | 784 (2 studies) | ⊕⊕⊕⊝ moderate1 | |
| Participants with at least some Side Effects | Study population | OR 1.28 (0.86 to 1.91) | 454 (1 study) | ⊕⊕⊕⊝ moderate1 | ||
| 668 per 1000 | 720 per 1000 (634 to 794) | |||||
| Moderate | ||||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; SMD: standardized mean difference; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Blinding stated but not tested. No information on randomisation procedures and allocation concealment. 2 I squared 81% 3 I squared 78%
Summary of findings 9. Paroxetine compared with fluoxetine for depression.
| Paroxetine compared with fluoxetine for depression | ||||||
| Patient or population: patients with depression Settings: in‐ and out‐patients Intervention: paroxetine Comparison: fluoxetine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Fluoxetine | Paroxetine | |||||
| Failure to respond at endpoint (6‐12 weeks) | 386 per 1000 | 555 per 1000 (332 to 436) | OR 1.98 (0.79 to 1.23) | 2418 (11 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to respond at 1‐4 weeks | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| Failure to respond at 16‐24 weeks | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| Failure to remit at endpoint | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| SMD at endpoint | The mean SMD at endpoint in the intervention groups was 0.04 standard deviations higher (0.05 lower to 0.12 higher) | 2109 (9 studies) | ⊕⊕⊕⊝ moderate1 | This effect approaches zero. | ||
| Failure to complete ‐ any cause ‐ total drop out rate | 325 per 1000 | 336 per 1000 (300 to 372) | OR 1.05 (0.89 to 1.23) | 2798 (13 studies) | ⊕⊕⊕⊝ moderate1 | |
| Participants with at least some Side Effects | 774 per 1000 | 763 per 1000 (703 to 814) | OR 0.94 (0.69 to 1.28) | 2255 (9 studies) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; SMD: standardized mean difference; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Blinding stated but not tested. No information on randomisation procedures and allocation concealment.
Summary of findings 10. Paroxetine compared with fluvoxamine for depression.
| Paroxetine compared with fluvoxamine for depression | ||||||
| Patient or population: patients with depression Settings: in‐ and out‐patients Intervention: paroxetine Comparison: fluvoxamine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Fluvoxamine | Paroxetine | |||||
| Failure to respond at endpoint (6‐12 weeks) | 466 per 1000 | 510 per 1000 (386 to 629) | OR 1.19 (0.72 to 1.94) | 261 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to respond at 1‐4 weeks | Study population | OR 0.71 (0.41 to 1.24) | 281 (3 studies) | ⊕⊕⊕⊝ moderate1 | ||
| 769 per 1000 | 703 per 1000 (577 to 805) | |||||
| Moderate | ||||||
| Failure to respond at 16‐24 weeks | See comment | See comment | Not estimable | 0 (0) | No trial reported this outcome. | |
| Failure to remit at endpoint | 752 per 1000 | 703 per 1000 (571 to 807) | OR 0.78 (0.44 to 1.38) | 261 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
| SMD at endpoint | The mean SMD at endpoint in the intervention groups was 0.09 standard deviations higher (0.43 lower to 0.6 higher) | 58 (1 study) | ⊕⊕⊕⊝ moderate1 | This effect approaches zero. | ||
| Failure to complete ‐ any cause ‐ | 293 per 1000 | 285 per 1000 (183 to 416) | OR 0.96 (0.54 to 1.72) | 261 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
| Participants with at least some Side Effects | 609 per 1000 | 636 per 1000 (395 to 822) | OR 1.12 (0.42 to 2.97) | 261 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; SMD: standardized mean difference; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Blinding stated but not tested. No information on randomisation procedures and allocation concealment.
Summary of findings 11. Paroxetine compared with hypericum for depression.
| Paroxetine compared with hypericum for depression | ||||||
| Patient or population: patients with depression Settings: in‐ and out‐patients Intervention: paroxetine Comparison: hypericum | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Hypericum | Paroxetine | |||||
| Failure to respond at endpoint (6‐12 weeks) | 312 per 1000 | 420 per 1000 (301 to 550) | OR 1.60 (0.95 to 2.69) | 251 (1 study) | ⊕⊕⊕⊝ moderate1 | |
| Failure to respond at 1‐4 weeks | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| Failure to respond at 16‐24 weeks | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| Failure to remit at endpoint | 512 per 1000 | 659 per 1000 (538 to 762) | OR 1.84 (1.11 to 3.06) | 251 (1 study) | ⊕⊕⊕⊝ moderate1 | |
| SMD at endpoint | The mean SMD at endpoint in the intervention groups was 0.35 standard deviations higher (0.09 to 0.6 higher) | 244 (1 study) | ⊕⊕⊕⊝ moderate1 | The point estimate of the effect size corresponds to a small effect according to Cohen 1992. | ||
| Failure to complete ‐ any cause ‐ | 136 per 1000 | 230 per 1000 (134 to 366) | OR 1.90 (0.98 to 3.67) | 251 (1 study) | ⊕⊕⊕⊝ moderate1 | |
| Participants with at least some Side Effects | 552 per 1000 | 762 per 1000 (650 to 846) | OR 2.60 (1.51 to 4.46) | 251 (1 study) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; SMD: standardized mean difference; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Blinding stated but not tested. No information on randomisation procedures and allocation concealment.
Summary of findings 12. Paroxetine compared with milnacipran for depression.
| Paroxetine compared with milnacipran for depression | ||||||
| Patient or population: patients with depression Settings: in‐ and out‐patients Intervention: paroxetine Comparison: milnacipran | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Milnacipran | Paroxetine | |||||
| Failure to respond at endpoint (6‐12 weeks) | 423 per 1000 | 405 per 1000 (302 to 519) | OR 0.93 (0.59 to 1.47) | 302 (1 study) | ⊕⊕⊕⊝ moderate1 | |
| Failure to respond at 1‐4 weeks | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| Failure to respond at 16‐24 weeks | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| Failure to remit at endpoint | Study population | OR 0.92 (0.57 to 1.49) | 302 (1 study) | ⊕⊕⊕⊝ moderate1 | ||
| 671 per 1000 | 652 per 1000 (538 to 753) | |||||
| Moderate | ||||||
| SMD at endpoint | The mean SMD at endpoint in the intervention groups was 0.05 standard deviations lower (0.28 lower to 0.18 higher) | 299 (1 study) | ⊕⊕⊕⊝ moderate1 | This effect approaches zero. | ||
| Failure to complete ‐ any cause ‐ | 172 per 1000 | 195 per 1000 (122 to 323) | OR 1.17 (0.67 to 2.3) | 343 (2 studies) | ⊕⊕⊕⊝ moderate1 | |
| Participants with at least some Side Effects | 772 per 1000 | 700 per 1000 (581 to 795) | OR 0.69 (0.41 to 1.15) | 302 (1 study) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; SMD: standardized mean difference; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Blinding stated but not tested. No information on randomisation procedures and allocation concealment.
Summary of findings 13. Paroxetine compared with mirtazapine for depression.
| Paroxetine compared with mirtazapine for depression | ||||||
| Patient or population: patients with depression Settings: in‐ and out‐patients Intervention: paroxetine Comparison: mirtazapine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Mirtazapine | Paroxetine | |||||
| Failure to respond at endpoint (6‐12 weeks) | 509 per 1000 | 554 per 1000 (483 to 625) | OR 1.20 (0.90 to 1.61) | 766 (4 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to respond at 1‐4 weeks | 735 per 1000 | 869 per 1000 (797 to 918) | OR 2.39 (1.42 to 4.02) | 726 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to respond at 16‐24 weeks | Study population | OR 1.41 (0.81 to 2.48) | 726 (3 studies) | ⊕⊕⊕⊝ moderate1 | ||
| 735 per 1000 | 869 per 1000 (797 to 918) | |||||
| Moderate | ||||||
| Failure to remit at endpoint | 597 per 1000 | 692 per 1000 (626 to 753) | OR 1.52 (1.13 to 2.06) | 766 (4 studies) | ⊕⊕⊕⊝ moderate1 | |
| SMD at endpoint | The mean SMD at endpoint in the intervention groups was 0.33 standard deviations higher (0.08 to 0.58 higher) | 246 (1 study) | ⊕⊕⊕⊝ moderate1 | The point estimate of the effect size corresponds to a small effect according to Cohen 1992. | ||
| Failure to complete ‐ any cause ‐ | 306 per 1000 | 357 per 1000 (286 to 434) | OR 1.26 (0.91 to 1.74) | 726 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
| Participants with at least some Side Effects | 743 per 1000 | 756 per 1000 (687 to 813) | OR 1.07 (0.76 to 1.50) | 726 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; SMD: standardized mean difference; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Blinding stated but not tested. No information on randomisation procedures and allocation concealment.
Summary of findings 14. Paroxetine compared with nefazodone for depression.
| Paroxetine compared with nefazodone for depression | ||||||
| Patient or population: patients with depression Settings: in‐ and out‐patients Intervention: paroxetine Comparison: nefazodone | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Nefazodone | Paroxetine | |||||
| Failure to respond at endpoint (6‐12 weeks) | 450 per 1000 | 202 per 1000 (54 to 506) | OR 0.31 (0.07 to 1.25) | 40 (1 study) | ⊕⊕⊕⊝ moderate1 | |
| Failure to respond at 1‐4 weeks | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| Failure to respond at 16‐24 weeks | Study population | OR 0.64 (0.17 to 2.38) | 40 (1 study) | ⊕⊕⊕⊝ moderate1 | ||
| 400 per 1000 | 299 per 1000 (102 to 613) | |||||
| Moderate | ||||||
| Failure to remit at endpoint | Not estimable | 0 (0) | ||||
| SMD at endpoint | The mean SMD at endpoint in the intervention groups was 0.12 standard deviations lower (0.37 lower to 0.14 higher) | 235 (2 studies) | ⊕⊕⊕⊝ moderate1 | The point estimate of the effect size corresponds to a small effect according to Cohen 1992. | ||
| Failure to complete ‐ any cause ‐ | 250 per 1000 | 150 per 1000 (35 to 464) | OR 0.53 (0.11 to 2.60) | 40 (1 study) | ⊕⊕⊕⊝ moderate1 | |
| Participants with at least some Side Effects | 838 per 1000 | 781 per 1000 (638 to 879) | OR 0.69 (0.34 to 1.40) | 206 (1 study) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; SMD: standardized mean difference; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Blinding stated but not tested. No information on randomisation procedures and allocation concealment.
Summary of findings 15. Paroxetine compared with reboxetine for depression.
| Paroxetine compared with reboxetine for depression | ||||||
| Patient or population: patients with depression Settings: in‐ and out‐patients Intervention: paroxetine Comparison: reboxetine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Reboxetine | Paroxetine | |||||
| Failure to respond at endpoint (6‐12 weeks) | 500 per 1000 | 451 per 1000 (398 to 505) | OR 0.82 (0.66 to 1.02) | 1369 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to respond at 1‐4 weeks | 846 per 1000 | 784 per 1000 (733 to 827) | OR 0.66 (0.5 to 0.87) | 1375 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to respond at 16‐24 weeks | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| Failure to remit at endpoint | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| SMD at endpoint | The mean SMD at endpoint in the intervention groups was 0.10 standard deviations lower (0.21 lower to 0 higher) | 1291 (3 studies) | ⊕⊕⊕⊝ moderate1 | The point estimate of the effect size corresponds to a small effect according to Cohen 1992. | ||
| Failure to complete ‐ any cause ‐ | 277 per 1000 | 230 per 1000 (164 to 313) | OR 0.78 (0.51 to 1.19) | 1375 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
| Participants with at least some Side Effects | 849 per 1000 | 859 per 1000 (806 to 899) | OR 1.08 (0.74 to 1.58) | 1375 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; SMD: standardized mean difference; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Blinding stated but not tested. No information on randomisation procedures and allocation concealment.
Summary of findings 16. Paroxetine compared with sertraline for depression.
| Paroxetine compared with sertraline for depression | ||||||
| Patient or population: patients with depression Settings: in‐ and out‐patients Intervention: paroxetine Comparison: sertraline | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Sertraline | Paroxetine | |||||
| Failure to respond at endpoint (6‐12 weeks) | 292 per 1000 | 340 per 1000 (269 to 422) | OR 1.25 (0.89 to 1.77) | 618 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to respond at 1‐4 weeks | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| Failure to respond at 16‐24 weeks | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| Failure to remit at endpoint | 577 per 1000 | 562 per 1000 (478 to 647) | OR 0.94 (0.67 to 1.34) | 545 (2 studies) | ⊕⊕⊕⊝ moderate1 | |
| SMD at endpoint | The mean SMD at endpoint in the intervention groups was 0.13 standard deviations lower (0.34 lower to 0.07 higher) | 353 (1 study) | ⊕⊕⊕⊝ moderate1 | The point estimate of the effect size corresponds to a small effect according to Cohen 1992. | ||
| Failure to complete ‐ any cause ‐ | 338 per 1000 | 411 per 1000 (210 to 648) | OR 1.37 (0.52 to 3.6) | 426 (2 studies) | ⊕⊕⊕⊝ moderate1 | |
| Participants with at least some Side Effects | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; SMD: standardized mean difference; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Blinding stated but not tested. No information on randomisation procedures and allocation concealment.
Summary of findings 17. Paroxetine compared with tianeptine for depression.
| Paroxetine compared with tianeptine for depression | ||||||
| Patient or population: patients with depression Settings: in‐ and out‐patients Intervention: paroxetine Comparison: tianeptine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Tianeptine | Paroxetine | |||||
| Failure to respond at endpoint (6‐12 weeks) | 370 per 1000 | 379 per 1000 (294 to 473) | OR 1.04 (0.71 to 1.53) | 648 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
| Failure to respond at 1‐4 weeks | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| Failure to respond at 16‐24 weeks | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| Failure to remit at endpoint | 727 per 1000 | 638 per 1000 (324 to 863) | OR 0.66 (0.18 to 2.36) | 44 (1 study) | ⊕⊕⊕⊝ moderate1 | |
| SMD at endpoint | The mean SMD at endpoint in the intervention groups was 0.04 standard deviations higher (0.12 lower to 0.2 higher) | 586 (3 studies) | ⊕⊕⊕⊝ moderate1 | This effect approaches zero. | ||
| Failure to complete ‐ any cause ‐ | 174 per 1000 | 235 per 1000 (136 to 375) | OR 1.46 (0.75 to 2.85) | 648 (3 studies) | ⊕⊕⊕⊝ moderate1 | |
| Participants with at least some Side Effects | 427 per 1000 | 484 per 1000 (398 to 570) | OR 1.26 (0.89 to 1.78) | 604 (2 studies) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; SMD: standardized mean difference; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Blinding stated but not tested. No information on randomisation procedures and allocation concealment.
Summary of findings 18. Paroxetine compared with trazodone for depression.
| Paroxetine compared with trazodone for depression | ||||||
| Patient or population: patients with depression Settings: in‐ and out‐patients Intervention: paroxetine Comparison: trazodone | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Trazodone | Paroxetine | |||||
| Failure to respond at endpoint (6‐12 weeks) | 127 per 1000 | 94 per 1000 (30 to 260) | OR 0.71 (0.21 to 2.41) | 108 (1 study) | ⊕⊕⊕⊝ moderate1 | |
| Failure to respond at 1‐4 weeks | Study population | OR 0.54 (0.25 to 1.19) | 108 (1 study) | ⊕⊕⊕⊝ moderate1 | ||
| 691 per 1000 | 547 per 1000 (358 to 727) | |||||
| Moderate | ||||||
| Failure to respond at 16‐24 weeks | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| Failure to remit at endpoint | 309 per 1000 | 322 per 1000 (174 to 516) | OR 1.06 (0.47 to 2.38) | 108 (1 study) | ⊕⊕⊕⊝ moderate1 | |
| SMD at endpoint | The mean SMD at endpoint in the intervention groups was 0.08 standard deviations lower (0.46 to 0.30 lower) | 108 (1 study) | ⊕⊕⊕⊝ moderate1 | This effect approaches zero. | ||
| Failure to complete ‐ any cause ‐ | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| Participants with at least some Side Effects | 345 per 1000 | 264 per 1000 (137 to 450) | OR 0.68 (0.30 to 1.55) | 108 (1 study) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; SMD: standardized mean difference; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Blinding stated but not tested. No information on randomisation procedures and allocation concealment.
Summary of findings 19. Paroxetine compared with venlafaxine for depression.
| Paroxetine compared with venlafaxine for depression | ||||||
| Patient or population: patients with depression Settings: in‐ and out‐patients Intervention: paroxetine Comparison: venlafaxine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Venlafaxine | Paroxetine | |||||
| Failure to respond at endpoint (6‐12 weeks) | 433 per 1000 | 457 per 1000 (307 to 617) | OR 1.10 (0.58 to 2.11) | 747 (4 studies) | ⊕⊕⊝⊝ low1,2 | |
| Failure to respond at 1‐4 weeks | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| Failure to respond at 16‐24 weeks | See comment | See comment | Not estimable | 0 (0) | See comment | No trial reported this outcome. |
| Failure to remit at endpoint | 444 per 1000 | 557 per 1000 (464 to 647) | OR 1.57 (1.08 to 2.29) | 807 (4 studies) | ⊕⊕⊕⊝ moderate1 | |
| SMD at endpoint | The mean SMD at endpoint in the intervention groups was 0.07 standard deviations higher (0.13 lower to 0.26 higher) | 411 (2 studies) | ⊕⊕⊕⊝ moderate1 | This effect approaches zero. | ||
| Failure to complete ‐ any cause ‐ | 250 per 1000 | 265 per 1000 (215 to 324) | OR 1.08 (0.82 to 1.44) | 1079 (6 studies) | ⊕⊕⊕⊝ moderate1 | |
| Participants with at least some Side Effects | 500 per 1000 | 502 per 1000 (342 to 661) | OR 1.01 (0.52 to 1.95) | 200 (2 studies) | ⊕⊕⊕⊝ moderate1 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; SMD: standardized mean difference; OR: Odds ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Blinding stated but not tested. No information on randomisation procedures and allocation concealment. 2 I squared 76%
Background
Description of the condition
Major depression is a severe mental illness characterised by a persistent low mood and/or loss of interest and pleasure accompanied by a range of symptoms including appetite loss, insomnia, fatigue, loss of energy, poor concentration, psychomotor symptoms, inappropriate guilt and morbid thoughts of death (APA 1994). This condition is associated with marked personal, social and economic morbidity, loss of functioning and productivity, and creates significant demands on service providers in terms of workload (NICE 2010). It was the third leading cause of burden among all diseases in the year 2002, and it is expected to show a rising trend during the coming 20 years (WHO 2006).
Description of the intervention
Although pharmacological and psychological interventions are both effective for major depression, in primary and secondary care settings antidepressant (AD) drugs remain the mainstay of treatment (APA 2000; Ellis 2004; NICE 2010 ). Paroxetine hydrochloride is a component of the class of AD known as selective serotonin reuptake inhibitors (SSRIs). In vitro studies suggest that paroxetine is a potent and highly selective inhibitor of neuronal serotonin reuptake and has only very weak effects on norepinephrine and dopamine neuronal reuptake. (Germann 2013).
How the intervention might work
Paroxetine is the most potent inhibitor of the reuptake of serotonin (Ki = 65 pmol/L) of all SSRIs and shows an intermediate affinity profile between the other SSRIs and tricyclic antidepressants (TCAs) with regards to the norepinephrine transporter (Ki = 45 nmol/L). At higher concentrations paroxetine loses its serotonin transporter (SERT) selectivity and may therefore act as a dual serotonin/norepinephrine uptake inhibitor (SNRI), nevertheless it is necessary to administer high doses of paroxetine (40 mg/day and higher) to determine a sufficient plasma level (higher than 100 ng/mL). (Gibiino 2012; Germann 2013).
Why it is important to do this review
Amongst ADs many different agents are available for the treatment of depression, including TCAs, monoamine oxidase inhibitors (MAOIs), SSRIs, SNRIs, and other newer agents (such as agomelatine, mirtazapine, reboxetine, bupropion). During the last 20 years, AD consumption has risen dramatically worldwide, mainly because of the increasing consumption of SSRIs and newer ADs, which have progressively become the most commonly prescribed ADs (Ciuna 2004). SSRIs are generally better tolerated than TCAs and there is evidence of similar efficacy (Anderson 2000). However, head‐to‐head comparison has provided contrasting findings. Amitriptyline, for example, may have the edge over SSRIs in terms of efficacy, and individual SSRIs and SNRIs may differ in terms of efficacy and tolerability (Hansen 2005; Cipriani 2009). Starting from this consideration, and with the aim to shed light on the field of AD trials and treatment of major depression, a group of researchers agreed to join forces under the rubric of the Meta‐Analyses of New Generation Antidepressants Study Group (MANGA Study Group) to systematically review all available evidence for each specific newer AD to inform clinical practice and mental health policies. We have up to now completed some individual reviews (about fluoxetine (Magni 2013), duloxetine (Cipriani 2012a), citalopram (Cipriani 2012b), sertraline (Cipriani 2009a), escitalopram (Cipriani 2009b), mirtazapine (Watanabe 2011), fluvoxamine (Omori 2010), milnacipran (Nakagawa 2009), and a number of other reviews are now underway.
Objectives
To determine the efficacy of paroxetine in comparison with other anti‐depressive agents in alleviating the acute symptoms of Major Depressive Disorder.
To review acceptability of treatment with paroxetine in comparison with other anti‐depressive agents.
To investigate the adverse effects of paroxetine in comparison with other anti‐depressive agents.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials (RCTs) comparing paroxetine with all other active anti‐depressive agents as monotherapy in the acute‐phase treatment of major depression were included. Quasi‐randomised trials, such as those allocating by using alternate days of the week, were excluded. Cluster‐randomised trials were eligible for inclusion. For trials which have a cross‐over design only results from the first randomisation phase were considered.
Types of participants
The review included participants 18 years or older, of both sexes, with a primary diagnosis of unipolar major depression according to standardised criteria, DSM‐III, DSM‐III‐R, DSM‐IV (APA 2000), ICD‐10 (WHO 1992), Feighner criteria (Feighner 1972) or Research Diagnostic Criteria (Spitzer 1972). Studies using ICD‐9 were excluded, as it only lists disease names and does not have diagnostic criteria.
We included participants with the following subtypes of depression: chronic, with catatonic features, with melancholic features, with atypical features, with postpartum onset, and with seasonal pattern. We included studies in which up to 20% of participants presented depressive episodes in bipolar affective disorder. We also included participants with a concurrent secondary diagnosis of another psychiatric disorder.
We excluded participants with a concurrent primary diagnosis of Axis I or II disorders and participants with a serious concomitant medical illness.
Types of interventions
We examined paroxetine in comparison with conventional treatments for acute depression. We also examined paroxetine in comparison with the non‐conventional antidepressant hypericum.
We excluded trials in which paroxetine was compared with another type of psychopharmacological agent (i.e., anxiolytics, anticonvulsants, antipsychotics or mood‐stabilisers), and trials in which paroxetine was used as an augmentation strategy.
Eligible intervention
1. Paroxetine: any dose and pattern of administration
Eligible comparators
Conventional anti‐depressive agents: any dose and mode or pattern of administration:
1. Older ADs:
Tricyclics;
Heterocyclics;
MAOIs.
2. SSRIs
3. Newer or non‐conventional anti‐depressive agents, for example:
SNRIs;
Hypericum.
Types of outcome measures
Primary outcomes
1. Efficacy: response rate
(1) Number of patients who responded to treatment, showing a reduction of at least 50% on the Hamilton rating scale for depression (HDRS) (Hamilton 1960) or Montgomery and Asberg Depression Rating Scale (MADRS) (Montgomery 1979), or any other depression scale, or "much or very much improved" (score one or two) on Clinical Global Impression (CGI) ‐ Improvement. All response rates were calculated out of the total number of randomised patients. Where more than one criterion was provided, we preferred the former criterion for judging response. We used the first criterion whenever possible, even when we needed to impute standard deviations (SDs) or response rates according to the procedures described below. We applied intention‐to‐treat (ITT) analyses, whereby all the dropouts not included in the analyses were considered non‐responders.
When studies reported response rates at various time points of the trial, we decided a priori to subdivide the treatment indices as follows.
(a) Early response: between one and four weeks; the time point closest to two weeks was given preference. (b) Acute phase treatment response: between six and 12 weeks; the time point given in the original study as the study endpoint was given preference. (c) Follow‐up response: between four and six months; the time point closest to 24 weeks was given preference.
The acute phase treatment response (between six and 12 weeks) was our primary outcome of interest.
Secondary outcomes
2. Efficacy: remission rate and continuous outcomes
(1) Number of patients who achieved remission. The cut‐off point for remission was set a priori:
(a) at seven or less on the 17‐item HDRS and at eight or less for all the other longer versions of HDRS, or
(b) at 10 or less on the MADRS (Zimmerman 2004), or
(c) "not ill or borderline mentally ill" (score one or two) on CGI‐Severity (Guy 1970).
All remission rates were calculated out of the total number of randomised patients. Where two or more were provided, we preferred the first criteria for judging remission. We applied the ITT analyses, whereby all the dropouts not included in the analyses were considered non‐remitters. (2) Change scores from baseline to the time point in question (early response, acute phase response, or follow‐up response as defined above) on HDRS or MADRS, or any other depression scale. We applied a looser form of ITT analyses, whereby all the patients with at least one post‐baseline measurement were represented by their Last Observations Carried Forward (LOCF).
(3) Social adjustment, social functioning including the Global Assessment of Function (GAF) (Luborsky 1962) scores.
(4) Health‐related quality of life: we limited ourselves to SF‐12/SF‐36 (Ware 1993), HoNOS (Wing 1994) and WHO‐QOL (WHOQOL Group 1998).
(5) Costs to healthcare services.
3. Acceptability
(1) Number of patients who dropped out during the trial as a proportion of the total number of randomised patients ‐ Total dropout rate.
(2) Number of patients who dropped out due to inefficacy during the trial as a proportion of the total number of randomised patients ‐ Dropout rates due to inefficacy.
(3) Number of patients who dropped out due to side effects during the trial as a proportion of the total number of randomised patients ‐ Dropout rates due to side effects.
4. Tolerability
(1) Total number of patients experiencing at least some side effects.
(2) Total number of patients experiencing the following specific side effects.
(a) Sleepiness/drowsiness. (b) Insomnia. (c) Dry mouth. (d) Constipation. (e) Urination problem. (f) Hypotension. (g) Agitation/anxiety. (h) Suicide wishes/gestures/attempts. (i) Completed suicide. (j) Vomiting/nausea. (k) Diarrhoea.
In order not to miss any relatively rare or unexpected yet important side effects, in the data extraction phase, we collected all side effects data reported in the literature and discussed ways to summarise them post hoc.
Search methods for identification of studies
The Cochrane, Depression, Anxiety and Neurosis Review Group's Specialised Register (CCDANCTR)
The Cochrane Depression, Anxiety and Neurosis Group (CCDAN) registers were searched up to September, 2012. The CCDAN maintain two clinical trials registers at their editorial base in Bristol, UK, a references register and a studies based register. The CCDANCTR‐References Register contains over 31,500 reports of randomised controlled trials in depression, anxiety and neurosis. Approximately 65% of these references have been tagged to individual, coded trials. The coded trials are held in the CCDANCTR‐Studies Register and records are linked between the two registers through the use of unique Study ID tags. Coding of trials is based on the EU‐Psi coding manual, please contact the CCDAN Trials Search Coordinator for further details. Reports of trials for inclusion in the Group's registers are collated from routine (weekly), generic searches of MEDLINE (1950‐), EMBASE (1974‐) and PsycINFO (1967‐); quarterly searches of the Cochrane Central Register of Controlled Trials (CENTRAL) and review specific searches of additional databases. Reports of trials are also sourced from international trials registers c/o the World Health Organization’s trials portal (ICTRP), ClinicalTrials.gov, drug companies, the handsearching of key journals, conference proceedings and other (non‐Cochrane) systematic reviews and meta‐analyses.
Details of CCDAN's generic search strategies can be found on the Group's website.
Electronic searches
1. CCDANCTR
CDANCTR‐Studies was searched using the following search strategy: Diagnosis = Depress* or Dysthymi* or "Adjustment Disorder*" or "Mood Disorder*" or "Affective Disorder" or "Affective Symptoms" and Intervention = Paroxetine
CCDANCTR‐References was searched using a similar set of terms to find additional untagged/uncoded references: Keyword = Depress* or Dysthymi* or "Adjustment Disorder*" or "Mood Disorder*" or "Affective Disorder" or "Affective Symptoms" and Free‐Text = Paroxetine
There was no restriction on date, language or publication status applied to the search.
2. International Regulatory Authorities and Trial Registries
Websites of the following drug regulatory authorities were searched for additional unpublished data: The Food and Drug Administration (FDA) in the USA, the Medicines and Healthcare products Regulatory Agency (MHRA) in the UK, the European Medicines Agency (EMEA) in the EU, the Pharmaceuticals and Medical Devices Agency (PMDA) in Japan, the Therapeutic Goods Administration (TGA) in Australia). International trial registries were also searched for unpublished or ongoing research: Clinicaltrials.gov, ISRCTN, Nederlands Trial Register, EUDRACT, UMIN‐CTR and the Australian New Zealand Clinical Trials Registry.
Searching other resources
Personal communication
Pharmaceutical companies and experts in this field were contacted for additional information on studies meeting the inclusion criteria for this review.
Handsearching
Appropriate journals and conference proceedings relating to the treatment of depression with paroxetine have already been handsearched and incorporated into the CCDANCTR.
Reference lists of all included studies, previous systematic reviews and major textbooks of affective disorder written in English were checked for published reports and citations of unpublished research. A cited reference search was conducted (on the Web of Science) to identify new reports citing any of the included studies.
Data collection and analysis
Data were entered into RevMan 5.2 (RevMan 2012) software by two review authors (DP and CT) (double data entry).
Selection of studies
Studies which met the following rough inclusion criteria constituted the preliminary list and their full texts were retrieved. The rough inclusion criteria were: (a) randomised trial; (b) comparing paroxetine against any other antidepressant; (c) patients with major depression, regardless of the diagnostic criteria used.
Studies relating to paroxetine generated by the search strategies of the CCDANCTR‐References and the other complementary searches were checked by review authors CR and CT to see if they meet the rough inclusion criteria, firstly based on the title and abstracts. All the studies rated as possible candidates by either of the two review authors were added to the preliminary list and their full texts retrieved. All the full text articles in this preliminary list were then assessed by MP and DP to see if they met the strict inclusion criteria. If the raters disagreed the final rating was made by consensus with the involvement (if necessary) of CB. Considerable care was taken to exclude duplicate publications.
Data extraction and management
Two review authors, working independently and in duplicate (CR and CT) extracted data from the included studies. Data were extracted on: participant characteristics (age, sex, depression diagnosis, comorbidity, depression severity, antidepressant treatment history for the index episode, study setting); intervention details (dosage range, mean daily dosage actually prescribed, co‐intervention if any, paroxetine as investigational drug or as comparator drug, sponsorship); and outcome measures of interest from the included studies. The results were compared with those in the completed reviews of individual antidepressants in The Cochrane Library. If there were any discrepancies, a third review author (AC) intervened and the agreed‐upon results were used in the review as well as fed back to the authors of the completed reviews. If the trial was a three (or more) ‐armed trial involving a placebo arm, the data were extracted from the placebo arm as well.
Assessment of risk of bias in included studies
Two review authors (MP and CR) independently assessed trial risk of bias in accordance with the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). The Cochrane Collaboration’s tool for assessing risk of bias covers six domains of bias: selection bias (random sequence generation, allocation concealment), performance bias (blinding of participants and personnel), detection bias (blinding of outcome assessment), attrition bias (incomplete outcome data), reporting bias (selective reporting), and other bias not covered elsewhere. Particular attention was given to the adequacy of the random allocation concealment and double blinding. Where inadequate details of methodological characteristics of trials were provided, the authors were contacted in order to obtain further information. If the raters disagreed the final rating was made by consensus with the involvement (if necessary) of another member of the review group (CB). The ratings were also compared with those in the completed reviews of individual antidepressants in The Cochrane Library. If there were any discrepancies, they were fed back to the authors of the completed reviews.
Measures of treatment effect
All comparisons were performed between paroxetine and comparator ADs considered as individual ADs and as a class.
Dichotomous data
For dichotomous data, odds ratios (ORs) were calculated with a 95% confidence interval (CI). For statistically significant results, we calculated the number needed to treat to provide benefit (NNTb) and the number needed to treat to induce harm (NNTh).
Continuous data
For continuous data we calculated the standardized mean differences (SMDs) with a 95% CI.
Unit of analysis issues
Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (e.g. pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase, the participants can differ systematically from their initial state, despite a wash‐out phase. For the same reason, cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in major depression, we only used data from the first phase of the cross‐over studies.
Cluster‐randomised trials
No cluster‐randomised trials were identified for this version of the review. Should they be identified in a future update, we plan to use the generic inverse variance technique, if such trials have been appropriately analysed taking into account intra‐class correlation coefficients to adjust for cluster effects.
Multiple intervention groups
Studies that compared more than two intervention groups of the same drug (i.e. different dosages) were included in meta‐analysis by combining group arms of the study into a single group, for the intervention and for the control group respectively, as recommended in section 16.5 of the Cochrane Handbook (Higgins 2011).
Dealing with missing data
When dichotomous or continuous outcomes were not reported, we asked the trial authors to supply the data.
For dichotomous data, we applied intention‐to‐treat (ITT) analyses, whereby all the dropouts not included in the analyses were considered as non‐responders or non‐remitters (i.e. it was assumed they would have experienced the negative outcome by the end of the trial, e.g. failure to respond to treatment).
For continuous data, we applied a looser form of ITT analyses, whereby all patients with at least one post‐baseline measurement were represented by their Last Observations Carried Forward (LOCF). When only the standard error (SE) or t‐statistics or P values were reported, standard deviations (SDs) were calculated according to Altman (Altman 1996).
In the absence of supplemental data from the authors, the SDs of the HDRS (or any other depression scale) and response/remission rates were calculated according to the validated imputation methods (Furukawa 2005; Furukawa 2006). We examined the validity of these imputations in the sensitivity analyses.
Assessment of heterogeneity
Skewed data and non‐quantitative data were presented descriptively. An outcome whose minimum score is zero could be considered skewed when the mean was smaller than twice the SD. Heterogeneity between studies was investigated using the I2 statistic (Higgins 2003; Ioannidis 2008) and by visual inspection of the forest plots.
According to the Cochrane Handbook (Higgins 2011), the following thresholds for the interpretation of I2 were used: 0% to 40%: might not be important; 30% to 60%: may represent moderate heterogeneity; 50% to 90%: may represent substantial heterogeneity; 75% to 100%: considerable heterogeneity. Moreover, we considered the sample size, the magnitude and the direction of the treatment effects.
Assessment of reporting biases
Data from included studies were entered into a funnel plot (trial effect against trial variance) to investigate small‐study effects (Sterne 2000). We used the tests for funnel plot asymmetry only when there were at least 10 studies included in the meta‐analysis, and results were interpreted cautiously, with visual inspection of the funnel plots (Higgins 2011). When evidence of small‐study effects was identified, possible reasons for funnel plot asymmetry, including publication bias, were investigated.
Data synthesis
For dichotomous data, odds ratios (OR) were calculated with 95% confidence intervals. The primary analysis used a random‐effects model, which had the highest generalisability in our empirical examination of summary effect measures for meta‐analyses (Furukawa 2002a). The robustness of this summary measure was routinely examined by checking the results under a fixed‐effect model. Material differences between the models were reported.
Continuous data were analysed using standardized mean differences (SMD) (with 95% CIs) as different measurement scales were used. A random‐effects model was employed. Fixed‐effect analyses were used routinely for continuous outcomes to investigate the effect of the choice of method on the estimates. Material differences between the models were reported.
Subgroup analysis and investigation of heterogeneity
Subgroup analyses should be performed and interpreted with caution because multiple analyses will lead to false positive conclusions (Oxman 1992). However, we performed the following subgroup analyses for the following a priori reasons.
Paroxetine dosing (fixed low dosage, fixed standard dosage, fixed high dosage; flexible low dosage, flexible standard dosage, flexible high dosage), because there is evidence to suspect that low dosage antidepressant may be associated with better outcomes both in terms of effectiveness and side effects than standard or high dosage antidepressants (Bollini 1999; Furukawa 2002b) and also because fixed versus flexible dosing schedule may affect estimates of treatment effectiveness (Khan 2003).
Comparator dosing (low effective range, medium to high effective range), as it is easy to imagine that there are greater chances of completing the study on the experimental drug than on the comparator drug that is increased to the maximum dosage.
Depression severity (severe major depression, moderate/mild major depression).
Treatment settings (psychiatric inpatients, psychiatric outpatients, primary care).
Elderly patients (> 65 years of age), separately from other adult patients.
Sensitivity analysis
Funnel plot analysis was performed to check for existence of small‐study effects including publication bias. We performed the following sensitivity analyses. By limiting the studies to be included to those with higher quality, we examined if the results changed, and checked for the robustness of the observed findings.
Excluding trials with unclear concealment of random allocation and/or unclear double blinding.
Excluding trials whose dropout rate was greater than 20%.
Performing the worst‐case scenario ITT (all the patients in the experimental group experienced the negative outcome and all those allocated to the comparison group experienced the positive outcome) and the best‐case scenario ITT (all the patients in the experimental group experienced the positive outcome and all those allocated to the comparison group experienced the negative outcome).
Excluding trials for which the response rates had to be calculated based on the imputation method (Furukawa 2005) and those for which the SD had to be borrowed from other trials (Furukawa 2006).
Examination of "wish bias" by comparing paroxetine as investigational drug versus paroxetine as comparator, as there is evidence to suspect that a new antidepressant might perform worse when used as a comparator than when used as an experimental agent (Barbui 2004; Lundh 2012).
Excluding studies funded by the pharmaceutical company marketing paroxetine. This sensitivity analysis is particularly important in view of the recent repeated findings that funding strongly affects outcomes of research studies (Als‐Nielsen 2003; Bhandari 2004; Lexchin 2003; Montgomery 2004b; Perlis 2005; Procyshyn 2004) and because industry sponsorship and authorship of clinical trials are increasing over 20 years (Buchkowsky 2004).
We planned that if subgroups within any of the subgroup or sensitivity analyses turned out to be significantly different from one another, we would run meta‐regression for exploratory analyses of additive or multiplicative influences of the variables in question. Our routine application of random‐effects and fixed‐effect models as well as our secondary outcomes of remission rates and continuous severity measures may be considered additional forms of sensitivity analyses.
Summary of findings
The GRADE approach was employed to interpret findings (Langendam 2013) and the GRADE profiler (GRADEPRO) allowed us to import data from Review Manager 5.2 (Review Manager ) to create 'Summary of findings' tables. These tables provide outcome‐specific information concerning the overall quality of evidence from studies included in the comparison, the magnitude of effect of the interventions examined, and the sum of available data on the outcomes we considered.
The following outcomes were included in the 'Summary of findings' tables.
Failure to respond at endpoint (six to 12 weeks).
Failure to respond at one to four weeks.
Failure to respond at 16 to 24 weeks.
Failure to remit at endpoint.
SMD at endpoint.
Failure to complete ‐ any cause.
Participants with at least some SE.
Results
Description of studies
See Characteristics of included studies; Characteristics of excluded studies; Characteristics of studies awaiting classification
Results of the search
The original searches yielded 691 records: after reading abstracts, 187 papers were considered potentially relevant for this review. Of these, 13 were excluded because of the not relevant diagnostic status or other reasons, 58 studies were defined as awaiting assessment and one study was ongoing. The remaining 115 studies, retrieved for more detailed evaluation, met the inclusion criteria and were included in the review. A total of 94 studies contributed to the quantitative synthesis (meta‐analysis) (see Figure 1).
1.
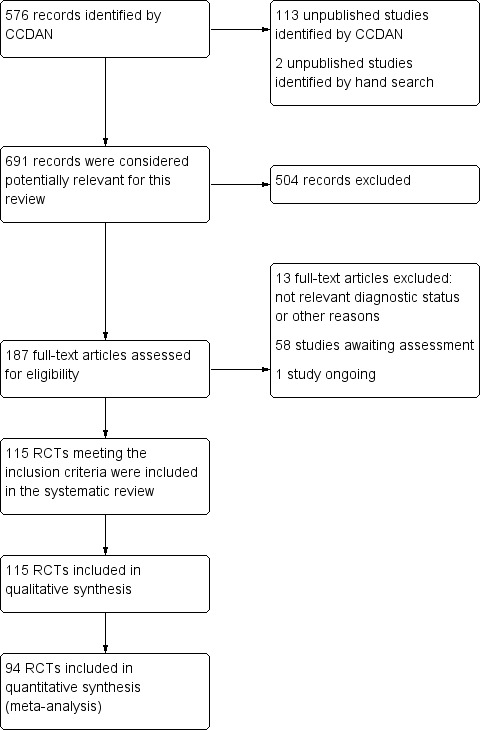
Flow diagram.
Included studies
See: Characteristics of included studies
Overall, a total of 115 studies (of which 25 were unpublished) were included in the present systematic review (26,134 participants). Unpublished data were obtained from the websites of pharmaceutical industries sponsoring the drugs under evaluation (23 trials), from the European Medicine Agency (one trial) and from conference proceedings (one trial).
Design
The great majority of included studies were reported to be double‐blind (99 out of 115 RCTs, that is 86%). The participants were followed up for six weeks (range: three to 52 weeks) in 57 trials.
Sample sizes
The mean number of participants per study was 226, with a minimum sample size of 10 (Javors 2000) and a maximum of 1057 (NCT00463242).
Setting
A total of 54 trials enrolled only outpatients, 14 trials enrolled only inpatients, and both inpatients and outpatients were enrolled in 18 trials. Twelve trials were conducted in general practice setting and for the remaining 17 trials the setting was unclear. Fifty‐two out of 115 trials took place in Europe; 34 trials took place in America (United States, Central America) and Canada; nine trials took place in Asia and two in Australia, For the remaining trials the country was unclear.
Participants
The majority of included trials (100 RCTs) enrolled patients with a diagnosis of major depression based on DSM‐III (30 studies), DSM‐III‐R (26 studies), DSM‐IV or ICD 10 criteria (44 studies). The remaining studies used other diagnostic criteria (15 studies). Forty‐one trials excluded patients over 65 years, while 15 trials included only elderly patients. We also included a minority of studies in which up to 20% of patients presented depressive episodes in bipolar disorder (six studies).
Intervention and Comparators
In 54 studies paroxetine was compared with older ADs (two RCTs versus dothiepin, two RCTs versus nortriptyline, 19 RCTs versus amitriptyline, 12 RCTs versus imipramine, three RCTs versus desipramine, three RCTs versus maprotiline, five RCTs versus mianserin, five RCTs versus clomipramine, two RCTs versus lofepramine, and one RCT versus doxepine). Twenty‐one RCTs compared paroxetine with other SSRIs (one RCT versus sertraline, two RCTs versus escitalopram, 14 RCTs versus fluoxetine and two RCTs versus fluvoxamine. There was one three‐armed study comparing paroxetine with fluoxetine and sertraline (Fava 2002), and one three‐armed study comparing paroxetine with citalopram and sertraline (Jefferson 2001 29060/785)). Forty studies compared paroxetine with newer or non‐conventional ADs (one RCT versus trazodone, three RCTs versus milnacipran, eight RCTs versus venlafaxine, two RCTs versus nefazodone, three RCTs versus reboxetine, two RCTs versus bupropion, one RCT versus hypericum, three RCTs versus tianeptine, four RCTs versus mirtazapine, six RCTs versus duloxetine, one RCT versus amisulpride, four RCTs versus agomelatine,one RCT versus aprepitant (MK‐869), and one RCT versus substance P).
Outcomes
At the end of the reviewing process, 94 RCTs were included in the meta‐analysis. For efficacy outcomes, 76 RCTs provided continuous data, and 81 dichotomous data. For acceptability outcomes, 94 RCTs provided data on total dropouts, 61 on dropouts due to inefficacy and 78 on dropouts due to side effects. In the majority of trials (93 out of 115, 81%) the HDRS scale was used for reporting outcomes.
Overall, 15,863 patients were included in the efficacy analysis, dichotomous outcome (7776 participants randomised to paroxetine and 8087 randomised to another antidepressant) and 14,637 were included in the efficacy analysis, continuous outcome (7326 participants randomised to paroxetine and 7311 randomised to another antidepressant). A total of 18,658 patients were included in the acceptability analysis (9037 partIcipants randomised to paroxetine and 9621 randomised to another antidepressant).
Excluded studies
See: Characteristics of excluded studies; Characteristics of studies awaiting classification
Thirteen articles initially selected did not meet our inclusion criteria and were excluded because of the wrong diagnosis or other reasons A total of 58 records were classified as "awaiting classification".
Ongoing studies
There is one ongoing study (Thomas 2008), This is a large multicentre trial including more than 500 participants with a primary diagnosis of major depression.
Studies awaiting classification
There were 58 studies for which sufficient information was not currently available to make a decision about inclusion or exclusion. The majority of studies in this section were written in Chinese (39 out of 58 studies). For the remaining studies, the number of participants or the outcomes were unclear.
Risk of bias in included studies
See:Characteristics of included studies.
Our judgment about the overall risk of bias in the individual studies is illustrated in Figure 2 and Figure 3. The Cochrane 'Risk of bias' tool highlighted poor reporting for a number of items in many of the included studies, although judging articles from some time ago by today’s standards can be problematic (Begg 1996; Turner 2012). Moreover, many articles failed to report methodologically relevant information on study procedure (in these cases the judgement was defined as "unclear"). In general, the reporting of studies was not good. This type of reporting has been associated with an overestimate of the estimate of effect (Schulz 1995) and this should be considered when interpreting the results.
2.

'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
3.
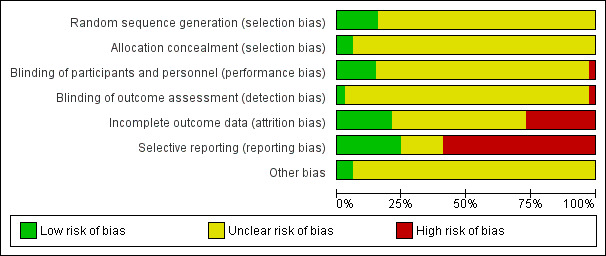
'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
Random sequence generation
The majority of studies (96 RCTs) did not report the methods of random sequence generation, while 18 studies (CL3‐023; 29060III/85/038; Aberg‐Wistedt 2000; Baldwin 2006; Boulenger 2006; Christiansen 1996; Freed 1996; Gallen 2001; Goldstein 2004 (HMAT B); Higuchi 2009; M/2020/0047; M/2020/0052; Owens 2008; Ravindran 1997; Shinkai 2004; Szegedi 2005; Wade 2003; Yoshimura 2007) specified this information, and they were classified as "low risk of bias".
Allocation concealment
Only a few trials (seven out of 115) (Baldwin 2006; Boulenger 2006; Gallen 2001; Higuchi 2009; M/2020/0047; M/2020/0052; Szegedi 2005) reported details on allocation concealment and were classified as "low risk".
Blinding
The great majority of included trials were undertaken under double‐blind conditions (99 out of 115 RCTs), however in many cases blinding was stated but not tested; two trials employed an open‐label design and for the remaining studies the design was unclear.
Incomplete outcome data
Twenty‐four trials were rated as "low" in terms of addressing incomplete outcome data, while 60 studies were classified as "unclear risk" and 31 as "high risk".
Selective reporting
The study protocol was not available for almost all studies so it is difficult to make a judgment on the possibility of outcome reporting bias. However, in 28 studies results were consistent with what was stated in the study protocols.
Other potential sources of bias
Most of the included studies were funded by the pharmaceutical industry or do not specify the source of funding. Seven studies were independent from commercial sponsorship (Javors 2000; Laghrissi‐Thode 1995; Laursen 1985; Montgomery 2004; Shinkai 2004; Steinmeyer 1992; Lepine 2001).
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8; Table 9; Table 10; Table 11; Table 12; Table 13; Table 14; Table 15; Table 16; Table 17; Table 18; Table 19
All the results of this systematic review need to be interpreted considering the characteristics and the risk of bias profile of each included study (Characteristics of included studies).
Comparison 1: Paroxetine versus older ADs
Pirmary outcome: Efficacy. Number of patients who responded to treatment
1.1 Responders at endpoint (six to 12 weeks)
We found no statistically significant difference in terms of response rate between paroxetine and older ADs as a class (odds ratio (OR): 1.04, 95% confidence interval (CI) 0.92 to 1.17, 34 RCTs, 4647 participants), and in head‐to‐head comparisons (see Analysis 1.1).
1.1. Analysis.
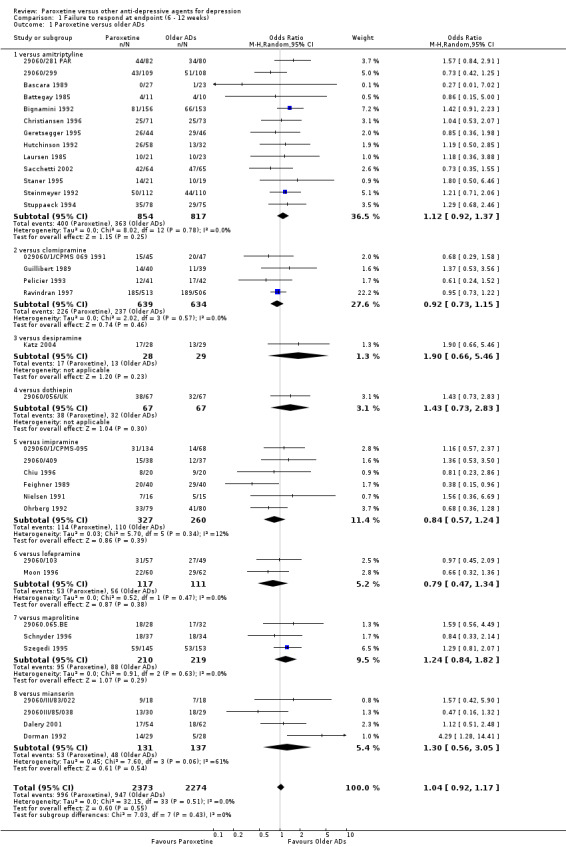
Comparison 1 Failure to respond at endpoint (6 ‐ 12 weeks), Outcome 1 Paroxetine versus older ADs.
1.2 Early response rate (one to four weeks)
We found no difference between paroxetine and older ADs for this outcome (see Analysis 2.1).
2.1. Analysis.
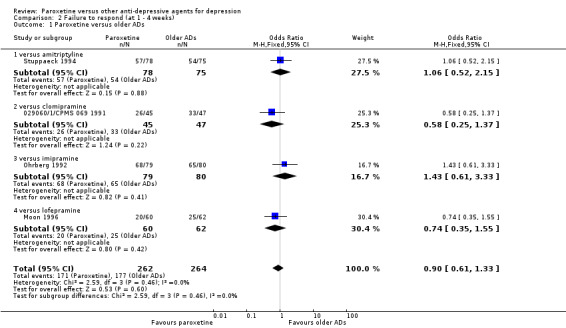
Comparison 2 Failure to respond (at 1 ‐ 4 weeks), Outcome 1 Paroxetine versus older ADs.
1.3 Follow‐up response rate (16 to 24 weeks)
No studies provided data for this outcome.
Secondary outcome: Efficacy. Number of patients who remitted
1.4 Remission at endpoint (six to 12 weeks)
There was no evidence that paroxetine was more effective than older ADs as a class in terms of remission rates at endpoint (see Analysis 4.1). In head‐to‐head comparisons we found a difference in favour of clomipramine over paroxetine (OR: 3.39, 95% CI 1.50 to 7.65, number needed to treat to provide benefit (NNTb) = 4, 95% CI 2 to 11, 1 RCT, 120 participants).
4.1. Analysis.
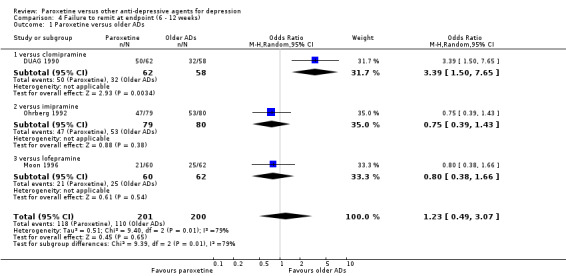
Comparison 4 Failure to remit at endpoint (6 ‐ 12 weeks), Outcome 1 Paroxetine versus older ADs.
1.5. Early remission rate (one to four weeks)
There was no evidence that paroxetine was more effective than older ADs in terms of early remissions (see Analysis 5.1).
5.1. Analysis.
Comparison 5 Failure to remit (at 1 ‐ 4 weeks), Outcome 1 Paroxetine versus older ADs.
1.6. Follow‐up remission rate (16 to 24 weeks)
No studies provided data for this outcome.
Secondary outcome: Efficacy. Standardized mean difference (SMD)
1.7 Standardized mean difference at endpoint (six to 12 weeks)
On this outcome, we found no statistically significant differences between paroxetine and older ADs (SMD: 0.01, 95% CI ‐0.08 to 0.10, 34 RCTs, 4712 participants), nor between paroxetine and individual ADs (see Analysis 7.1).
7.1. Analysis.
Comparison 7 Standardized mean difference at endpoint (6 ‐ 12 weeks), Outcome 1 Paroxetine versus older ADs.
1.8 Standardized mean difference at one to four weeks
We found a difference in favour of desipramine over paroxetine (SMD: 0.86, 95% CI 0.28 to 1.44, 1 RCT, 50 participants) (see Analysis 8.1).
8.1. Analysis.
Comparison 8 Standardized mean difference (at 1 ‐ 4 weeks), Outcome 1 Paroxetine versus older ADs.
1.9 Standardized mean difference at 16 to 24 weeks
No studies provided data for this outcome.
Secondary outcome: Acceptability
1.10 Failure to complete due to any cause
In terms of participants who dropped out due to any cause, we found a difference between paroxetine and older ADs as a class (OR: 0.84, 95% CI 0.73 to 0.96, number needed to treat to induce harm (NNTh) = 23, 95% CI 15 to 44, 43 RCTs, 6777 participants). In head‐to‐head comparisons paroxetine was better tolerated than clomipramine (OR: 0.67, 95% CI 0.52 to 0.87, NNTh = 14, 95% CI 8 to 39, 4 RCTs, 1273 participants), and than imipramine (OR: 0.65, 95% CI 0.50 to 0.85, NNTh= 8, 95% CI 6 to 14, 9 RCTs, 1268 participants) (see.Analysis 10.1)
10.1. Analysis.
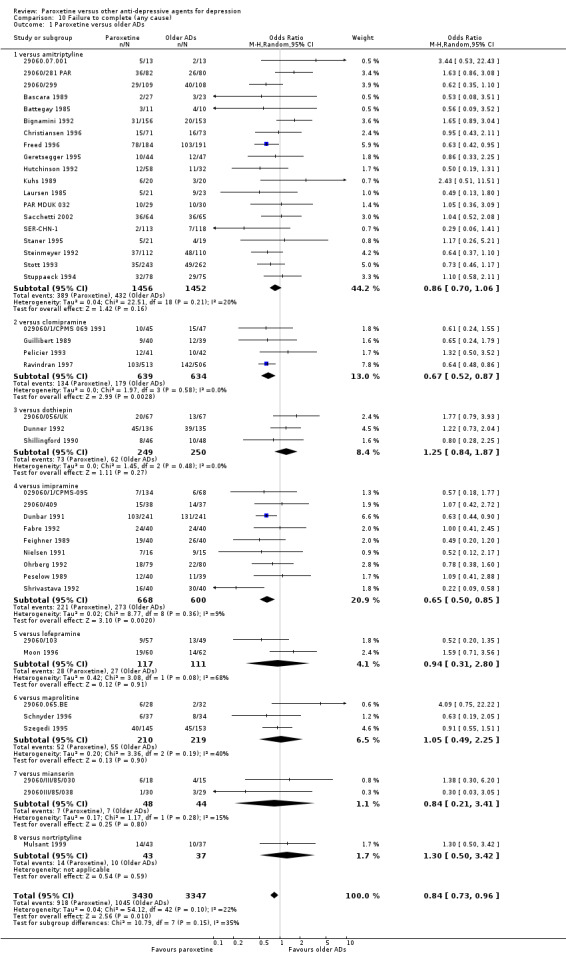
Comparison 10 Failure to complete (any cause), Outcome 1 Paroxetine versus older ADs.
1.11 Failure to complete due to inefficacy
We found no differences in terms of dropouts due to inefficacy between paroxetine and older ADs (OR: 1.22, 95% CI 0.93 to 1.61, 27 RCTs, 4436 participants) (see Analysis 11.1).
11.1. Analysis.
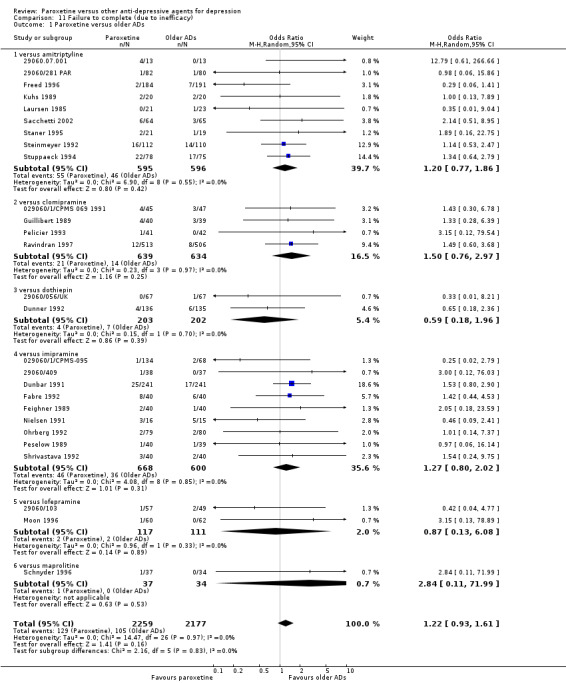
Comparison 11 Failure to complete (due to inefficacy), Outcome 1 Paroxetine versus older ADs.
1.12 Failure to complete due to side effects
The analysis of dropouts due to side effects revealed that amitriptyline (OR: 0.74, 95% CI 0.56 to 0.98, NNTh = 29, 95% CI 15 to 1318, 12 RCTs, 1698 participants), clomipramine (OR: 0.59, 95% CI 0.41 to 0.84, NNTh = 19, 95% CI 11 to 61, 4 RCTs, 1273 participants), imipramine (OR: 0.58, 95% CI 0.43 to 0.77, NNTh = 10, 95% CI 7 to 17, 9 RCTs, 1268 participants) and overall older ADs (OR: 0.76, 95% CI 0.63 to 0.92, NNTh = 25, 95% CI 17 to 49, 34 RCTs, 5175 participants) were significantly less well tolerated than paroxetine (see Analysis 12.1).
12.1. Analysis.
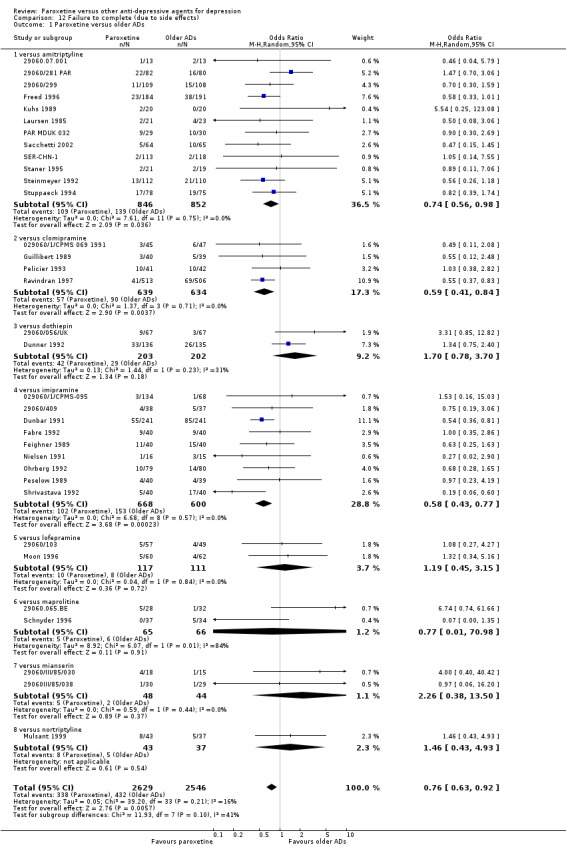
Comparison 12 Failure to complete (due to side effects), Outcome 1 Paroxetine versus older ADs.
Secondary outcome: Tolerability
1.13 Total number of patients experiencing at least some side effects
There was evidence that paroxetine was associated with a lower rate of adverse events than amitriptyline (OR: 0.53, 95% CI 0.39 to 0.72, NNTh = 7, 95% CI 6 to 10, 16 RCTs, 2492 participants), imipramine (OR: 0.62, 95% CI 0.42 to 0.94, NNTh = 10, 95% CI 7 to 19, 9 RCTs, 1189 participants) and than older ADs as a class (OR: 0.64, 95% CI 0.53 to 0.77, NNTh = 11, 95% CI 9 to 14, 41 RCTs, 6099 participants) (see Analysis 13.1).
13.1. Analysis.
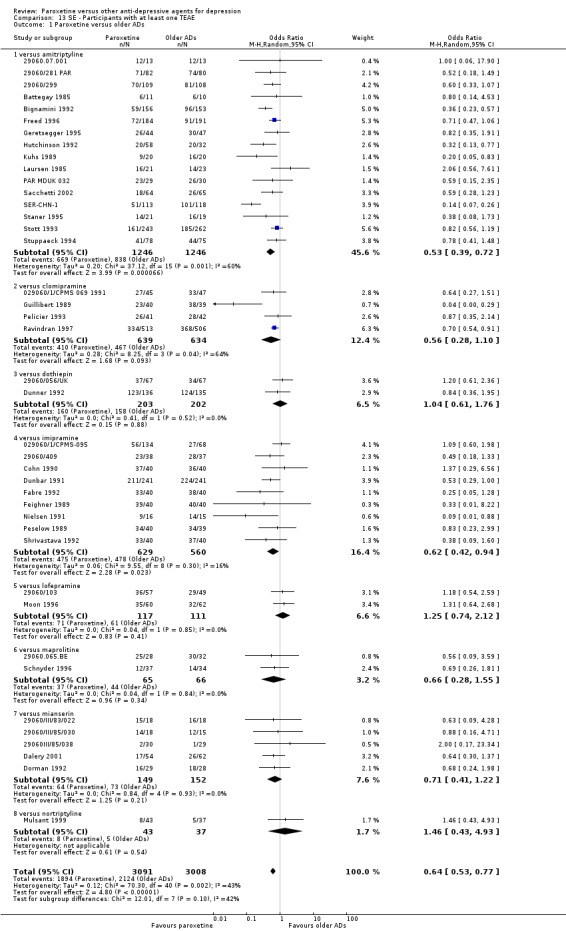
Comparison 13 SE ‐ Participants with at least one TEAE, Outcome 1 Paroxetine versus older ADs.
1.14 Number of patients experiencing specific side effects
(a) Sleepiness/drowsiness. We found a difference in favour of paroxetine over maprotiline (OR: 0.37, 95% CI 0.17 to 0.82, NNTh = 13, 95% CI 7 to 52, 2 RCTs, 358 participants) (see Analysis 79.1).
79.1. Analysis.
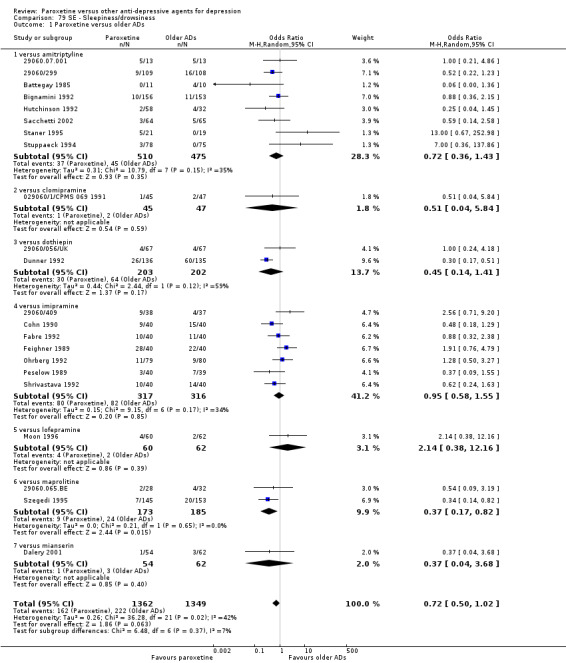
Comparison 79 SE ‐ Sleepiness/drowsiness, Outcome 1 Paroxetine versus older ADs.
(b) Insomnia. There was evidence that paroxetine was associated with a higher rate of insomnia than older ADs (OR: 2.17, 95% CI 1.51 to 3.12, NNTh = 17, 95% CI 12 to 29, 15 RCTs, 1986 participants). In head‐to‐head comparisons, paroxetine was associated with a higher rate of insomnia than amitriptyline (OR: 3.66, 95% CI 1.36 to 9.85, NNTh = 13, 95% CI 8 to 36, 4 RCTs, 352 participants), dothiepin (OR: 2.34, 95% CI 1.03 to 5.31, NNTh = 19, 95% CI 10 to 246, 2 RCTs, 405 participants) and than maprotiline (OR: 4.38, 95% CI 1.72 to 11.15, NNTh = 9, 95% CI 6 to 22, 1 RCTs, 298 participants) (see Analysis 54.1).
54.1. Analysis.
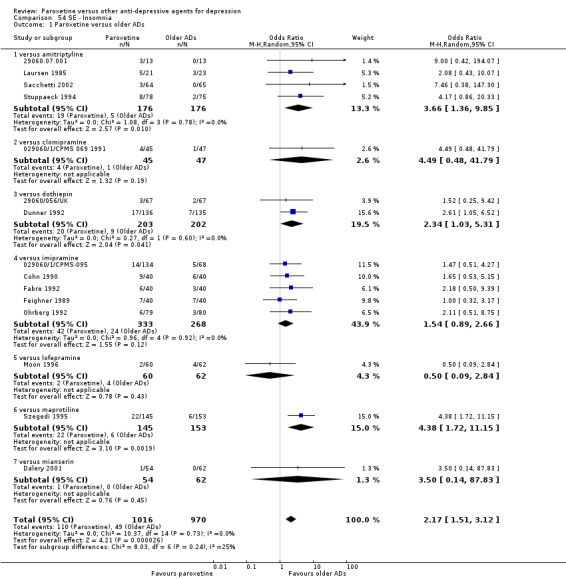
Comparison 54 SE ‐ Insomnia, Outcome 1 Paroxetine versus older ADs.
(c) Dry mouth. Paroxetine was associated with a lower rate of dry mouth than amitriptyline (OR: 0.27, 95% CI 0.17 to 0.43, NNTh = 4, 95% CI 4 to 5, 12 RCTs, 1576 participants), clomipramine (OR: 0.35, 95% CI 0.26 to 0.48, NNTh = 6, 95% CI 5 to 8, 2 RCTs, 1111 participants), dothiepin (OR: 0.22, 95% CI 0.10 to 0.50, NNTh = 3, 95% CI 3 to 5, 2 RCTs, 405 participants), imipramine (OR: 0.16, 95% CI 0.10 to 0.26, NNTh = 3, 95% CI 2 to 3, 8 RCTs, 835 participants), maprotiline (OR: 0.13, 95% CI 0.08 to 0.23, NNTh = 3, 95% CI 2 to 4, 3 RCTs, 429 participants), and older ADs as a class (OR: 0.23, 95% CI 0.18 to 0.30, NNTh = 4, 95% CI 3 to 5, 29 RCTs, 4578 participants) (see Analysis 38.1).
38.1. Analysis.
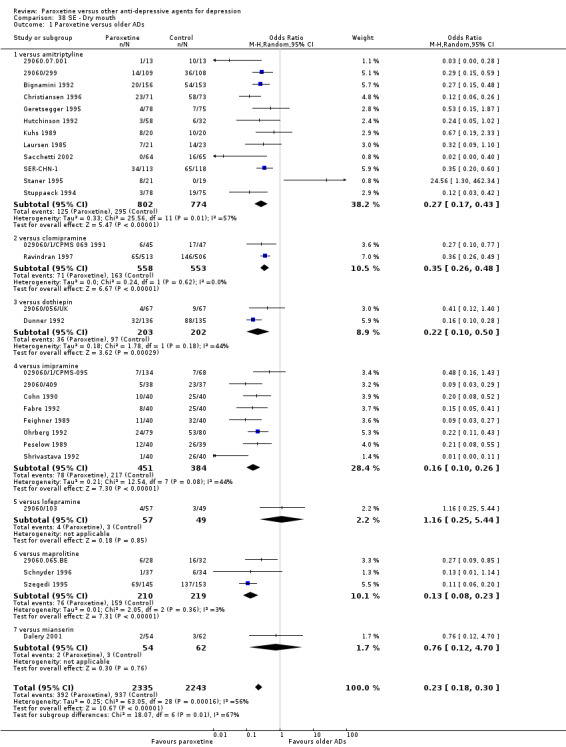
Comparison 38 SE ‐ Dry mouth, Outcome 1 Paroxetine versus older ADs.
(d) Constipation. Paroxetine was associated with a lower rate of constipation than older ADs as a class (OR: 0.49, 95% CI 0.40 to 0.60, NNTh = 12, 95% CI 9 to 16, 26 RCTs, 3934 participants). In head‐to‐head comparisons, paroxetine was associated with a lower rate of constipation than amitriptyline (OR: 0.61, 95% CI 0.37 to 0.99, NNTh = 18, 95% CI 11 to 65, 10 RCTs, 1146 participants), clomipramine (OR: 0.57, 95% CI 0.38 to 0.85, NNTh = 19, 95% CI 12 to 64, 2 RCTs, 1111 participants), dothiepin (OR: 0.57, 95% CI 0.32 to 0.99, NNTh = 10, 95% CI 5 to 378, 1 RCT, 271 participants), imipramine (OR: 0.40, 95% CI 0.25 to 0.63, NNTh = 6, 95% CI 5 to 11, 7 RCTs, 633 participants), and maprotiline (OR: 0.31, 95% CI 0.17 to 0.55, NNTh = 7, 95% CI 5 to 12, 3 RCTs, 429 participants) (see Analysis 31.1).
31.1. Analysis.
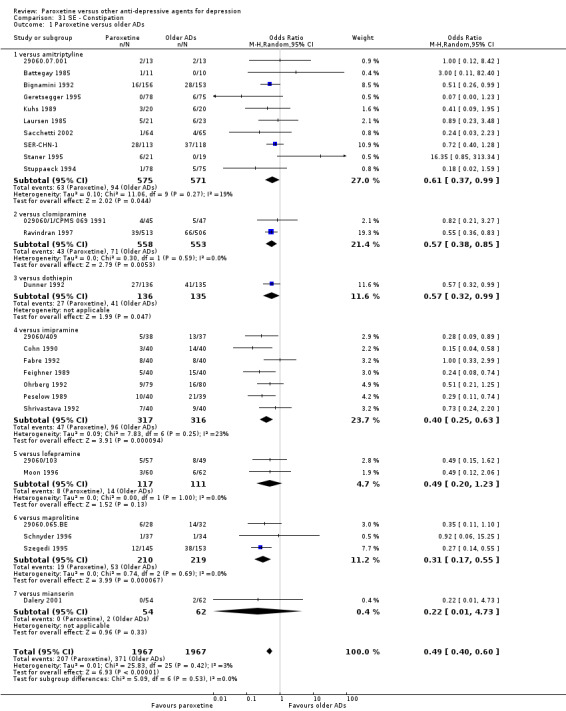
Comparison 31 SE ‐ Constipation, Outcome 1 Paroxetine versus older ADs.
(e) Urination/Urogenital problems. There was evidence in favour of paroxetine over imipramine for urinary retention (OR: 0.04, 95% CI 0.00 to 0.73, NNTh = 4, 95% CI 3 to 10, 1 RCT, 80 participants) (see Analysis 90.4) and for urogenital problems (Not Otherwise Specified) (OR: 0.10, 95% CI 0.01 to 0.82, NNTh = 10, 95% CI 6 to 38, 1 RCT, 159 participants) (see Analysis 90.5).
90.4. Analysis.
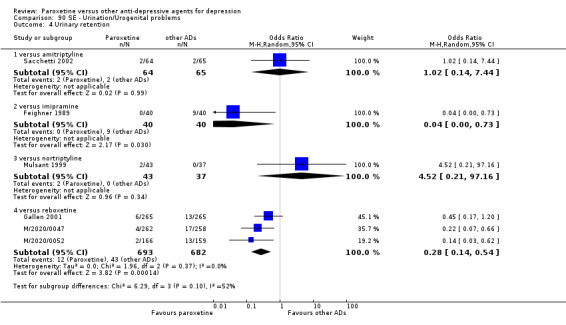
Comparison 90 SE ‐ Urination/Urogenital problems, Outcome 4 Urinary retention.
90.5. Analysis.
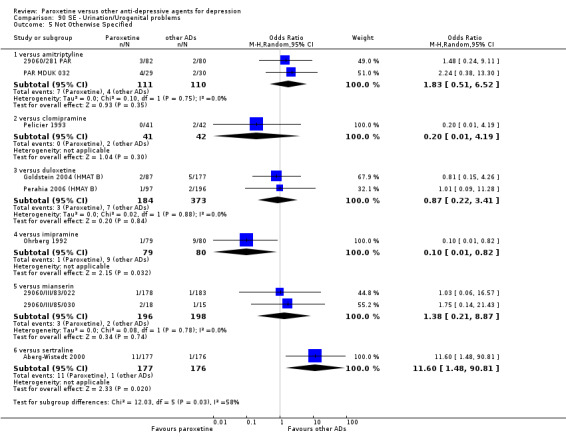
Comparison 90 SE ‐ Urination/Urogenital problems, Outcome 5 Not Otherwise Specified.
(f) Hypotension. We found no difference between paroxetine and older ADs (see Analysis 50.1).
50.1. Analysis.
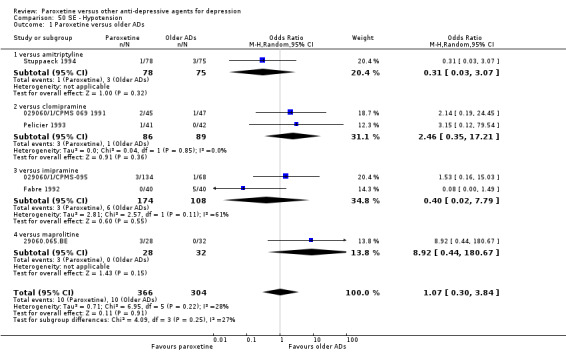
Comparison 50 SE ‐ Hypotension, Outcome 1 Paroxetine versus older ADs.
(g) Agitation/anxiety. We found no difference between paroxetine and older ADs (see Analysis 18.1).
18.1. Analysis.
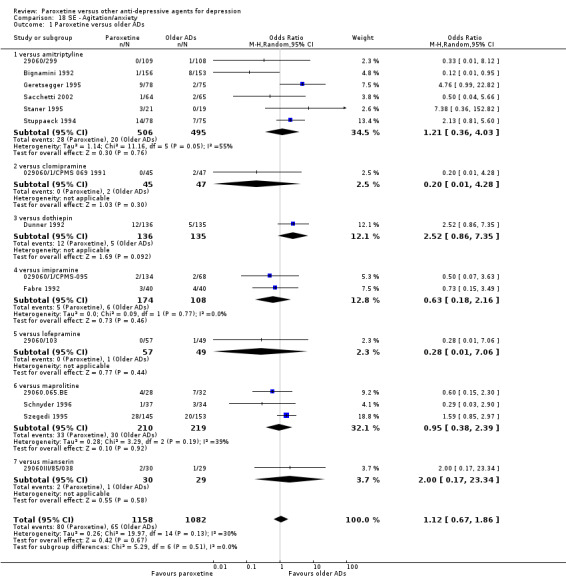
Comparison 18 SE ‐ Agitation/anxiety, Outcome 1 Paroxetine versus older ADs.
(h) Suicide wishes/gestures/attempts. We found no difference between paroxetine and older ADs (see Analysis 96.1; Analysis 96.3).
96.1. Analysis.
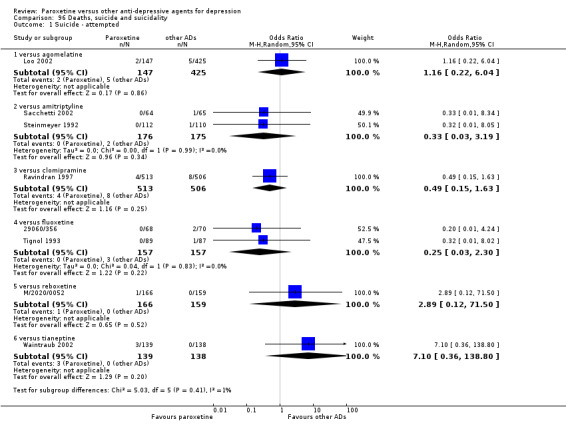
Comparison 96 Deaths, suicide and suicidality, Outcome 1 Suicide ‐ attempted.
96.3. Analysis.
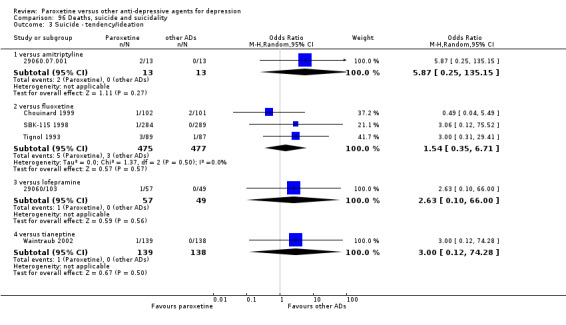
Comparison 96 Deaths, suicide and suicidality, Outcome 3 Suicide ‐ tendency/ideation.
(i) Completed suicide. We found no difference between paroxetine and older ADs (see Analysis 96.2).
96.2. Analysis.
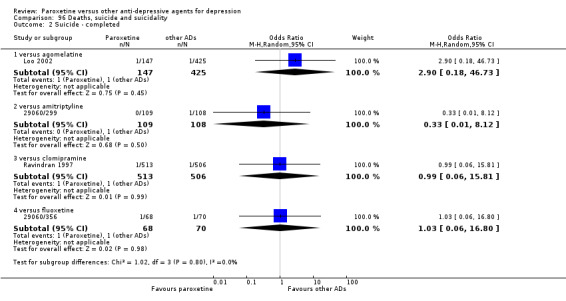
Comparison 96 Deaths, suicide and suicidality, Outcome 2 Suicide ‐ completed.
(j) Vomiting/nausea. Paroxetine was associated with a higher rate of vomiting/nausea than older ADs as a class (OR: 2.10, 95% CI 1.59 to 2.77, NNTh = 12, 95% CI 10 to 17, 30 RCTs, 4545 participants). In head‐to‐head comparisons we found evidence in favour of amitriptyline (OR: 2.17, 95% CI 1.43 to 3.29, NNTh = 15, 95% CI 10 to 29, 10 RCTs, 1282 participants), dothiepin (OR: 3.12, 95% CI 1.1 to 8.78, NNTh = 8, 95% CI 5 to 15, 2 RCTs, 405 participants), imipramine (OR: 2.05, 95% CI 1.23 to 3.42, NNTh = 11, 95% CI 7 to 26, 8 RCTs, 835 participants), and lofepramine (OR: 2.97, 95% CI 1.12 to 7.92, NNTh = 9, 95% CI 5 to 34, 2 RCTs, 228 participants) over paroxetine (see Analysis 59.1).
59.1. Analysis.
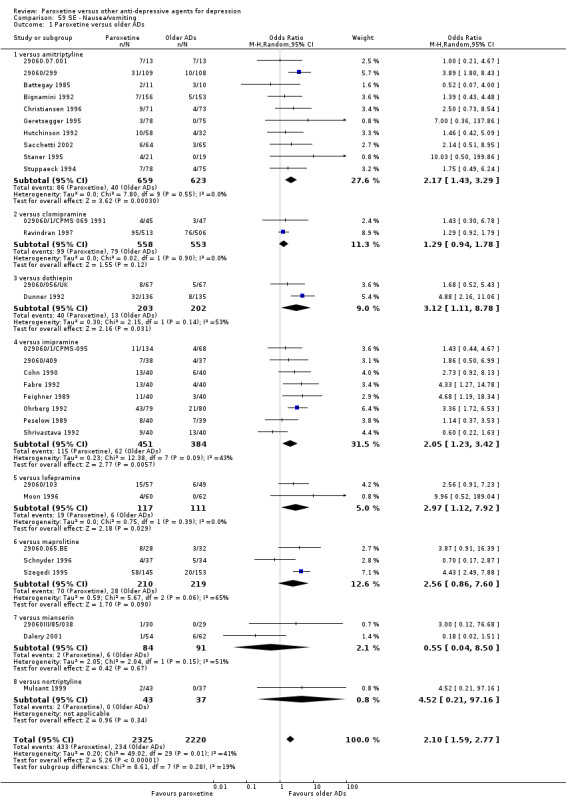
Comparison 59 SE ‐ Nausea/vomiting, Outcome 1 Paroxetine versus older ADs.
(k) Diarrhoea. We found that paroxetine was associated with a higher rate of diarrhoea than older ADs as a class (OR: 2.41, 95% CI 1.56 to 3.73, NNTh = 20, 95% CI 14 to 36, 13 RCTs, 1743 participants) and than dothiepin (OR: 3.47, 95% CI 1.23 to 9.75, NNTh = 12, 95% CI 7 to 56, 1 RCTs, 271 participants) and maprotiline (OR: 2.94, 95% CI 1.34 to 6.47, NNTh = 11, 95% CI 7 to 33, 2 RCTs, 358 participants) (see Analysis 36.1).
36.1. Analysis.
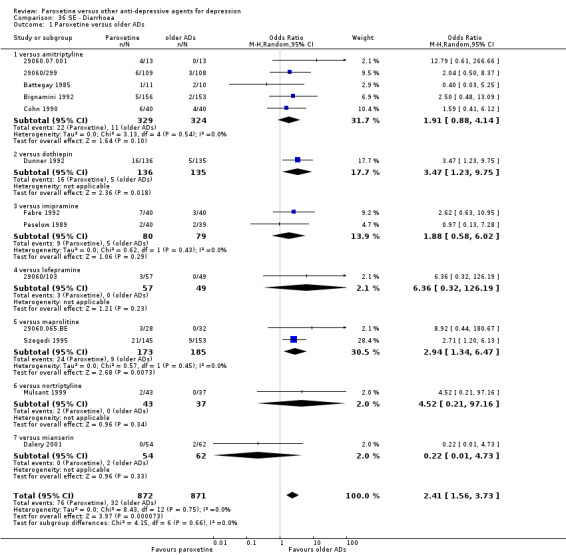
Comparison 36 SE ‐ Diarrhoea, Outcome 1 Paroxetine versus older ADs.
(l) Other side effects. Other statistically significant side effects are reported in Table 20.
1. Other significant tolerability outcomes not a priori listed in the protocol.
| Adverse event | Study | Paroxetine | Comparator | Odds Ratio, Random [95% CI] | NNTh [95% CI] | ||
| Events | Total | Events | Total | ||||
| Paroxetine versus older antidepressants | |||||||
| Paroxetine vs amitriptyline | |||||||
| Anticholinergic | Hutchinson 1992; Staner 1995 | 5 | 79 | 12 | 51 | 0.22 [0.07 to 0.76] | 6 [3 to 23] |
| Body as a whole | 29060/281 PAR; PAR MDUK 032 | 54 | 111 | 37 | 110 | 1.87 [1.08 to 3.23] | 7 [4 to 46] |
| Dizziness | 29060/299; 29060.07.001; Hutchinson 1992; Kuhs 1989; Laursen 1985; Sacchetti 2002; SER‐CHN‐1 | 31 | 398 | 57 | 379 | 0.42 [0.25 to 0.72] | 14 [9 to 36] |
| Behaviour (irritability) | Battegay 1985 | 7 | 11 | 1 | 10 | 15.75 [1.42 to 174.25] | 2 [1 to 5] |
| Palpitations | Battegay 1985; Bignamini 1992; Laursen 1985 | 4 | 188 | 13 | 186 | 0.42 [0.06 to 2.74] | 21 [11 to 152] |
| Tachycardia | Sacchetti 2002; SER‐CHN‐1 | 6 | 177 | 21 | 183 | 0.43 [0.04 to 4.44] | 12 [7 to 36] |
| Tremor | 29060/299; 29060.07.001; Battegay 1985; Geretsegger 1995; Laursen 1985; Sacchetti 2002; SER‐CHN‐1; Staner 1995; Stuppaeck 1994 | 25 | 508 | 53 | 506 | 0.44 [0.22 to 0.90] | 18 [11 to 44] |
| Paroxetine vs clomipramine | |||||||
| Anticholinergic | Guillibert 1989 | 7 | 40 | 16 | 39 | 0.30 [0.11 to 0.86] | 4 [2 to 24] |
| Paroxetine vs dothiepin | |||||||
| Headache | 29060/056/UK; Dunner 1992 | 39 | 203 | 18 | 202 | 2.46 [1.35 to 4.48] | 10 [6 to 28] |
| Paroxetine vs imipramine | |||||||
| Dyspnea | 29060/409 | 4 | 38 | 12 | 37 | 0.25 [0.07 to 0.85] | 5 [3 to 25] |
| Sweating | Cohn 1990; Feighner 1989; Peselow 1989; Shrivastava 1992;029060/1/CPMS‐095; Fabre 1992; Ohrberg 1992 | 61 | 373 | 97 | 308 | 0.43 [0.29 to 0.65] | 7 [5 to 11] |
| Weight gain | Ohrberg 1992 | 1 | 79 | 8 | 80 | 0.12 [0.01 to 0.95] | 11 [6 to 58] |
| Paroxetine vs maprotiline | |||||||
| Appetite decreased | Szegedi 1995 | 12 | 145 | 2 | 153 | 6.81 [1.50 to 30.99] | 14 [8 to 47] |
| Headache | 29060.065.BE; Schnyder 1996; Szegedi 1995 | 51 | 210 | 30 | 209 | 1.96 [1.13 to 3.39] | 10 [6 to 41] |
| Weight gain | 29060.065.BE; Schnyder 1996 | 0 | 65 | 8 | 66 | 0.10 [0.01 to 0.81] | 8 [5 to 24] |
| Paroxetine vs mianserin | |||||||
| Nervous system | 29060/III/85/030; Dorman 1992 | 15 | 47 | 25 | 43 | 0.25 [0.09 to 0.67] | 4 [2 to 16] |
| Gastrointestinal | 29060/III/83/022; 29060/III/85/030;Dorman 1992 | 32 | 65 | 13 | 61 | 3.56 [1.63 to 7.79] | 4 [2 to 8] |
| Paroxetine versus other SSRIs | |||||||
| Paroxetine vs fluoxetine | |||||||
| Dizziness | 29060/356; Gagiano 1993; Geretsegger 1994; MY‐1045/BRL‐029060/1; Ontiveros 1994 | 114 | 868 | 81 | 869 | 1.50 [1.11 to 2.04] | 26 [15 to 118] |
| Fatigue | 29060/356 | 2 | 68 | 9 | 70 | 0.21 [0.04 to 0.99] | 10 [5 to 90] |
| Nervous system | Cassano 2002; De Wilde 1993 | 24 | 173 | 47 | 169 | 0.41 [0.24 to 0.72] | 7 [4 to 18] |
| Sexual problems (ejaculation disorders) | MY‐1045/BRL‐029060/1 | 36 | 357 | 12 | 351 | 3.17 [1.62 to 6.20] | 15 [10 to 33] |
| Paroxetine vs fluvoxamine | |||||||
| Sweating | Kiev 1997 | 10 | 30 | 3 | 30 | 4.50 [1.09 to 18.50] | 4 [2 to 30] |
| Paroxetine vs sertraline | |||||||
| Fatigue | Aberg‐Wistedt 2000; Fava 2002 | 97 | 273 | 48 | 272 | 2.41 [1.21 to 4.77] | 6 [4 to 9] |
| Sexual problems (ejaculation disorders) | Aberg‐Wistedt 2000; Fava 2002 | 32 | 273 | 10 | 272 | 3.50 [1.68 to 7.28] | 12 [8 to 28] |
| Tremor | Aberg‐Wistedt 2000; Fava 2002 | 42 | 273 | 25 | 272 | 1.82 [1.07 to 3.09] | 16 [9 to 142] |
| Paroxetine versus newer or non‐conventional antidepressants and other agents | |||||||
| Paroxetine vs aprepitant | |||||||
| Sexual problems (ejaculation disorders) | Kramer 1998 | 14 | 72 | 2 | 71 | 8.33 [1.82 to 38.16] | 6 [4 to 15] |
| Paroxetine vs hypericum | |||||||
| Dizziness | Szegedi 2005 | 24 | 126 | 9 | 125 | 3.03 [1.35 to 6.82] | 8 [5 to 2]8 |
| Paroxetine vs mirtazapine | |||||||
| Fatigue | Benkert 1999; Schatzberg 2002; Wade 2003 | 33 | 360 | 54 | 366 | 0.47 [0.22 to 0.99] | 12 [7 to 43] |
| Flatulence | Schatzberg 2002 | 15 | 126 | 4 | 128 | 4.19 [1.35 to 13.00] | 11 [7 to 42] |
| Headache | Benkert 1999; Schatzberg 2002; Wade 2003 | 73 | 360 | 46 | 366 | 1.77 [1.12 to 2.79] | 11 [7 to 28] |
| Sexual problems (general) | Benkert 1999 | 18 | 136 | 4 | 139 | 5.15 [1.69 to 15.64] | 10 [6 to 25] |
| Sweating | Benkert 1999; Schatzberg 2002; Wade 2003 | 38 | 360 | 13 | 366 | 3.12 [1.61 to 6.03] | 14 [9 to 30] |
| Tremor | Benkert 1999; Schatzberg 2002 | 21 | 262 | 6 | 267 | 3.67 [1.43 to 9.42] | 17 [11 to 49] |
| Weight gain | Benkert 1999; Schatzberg 2002; Wade 2003 | 12 | 360 | 46 | 366 | 0.25 [0.13 to 0.47] | 11 [8 to 19] |
| Paroxetine vs nefazodone | |||||||
| Sweating | Hicks 2002 | 7 | 20 | 0 | 20 | 22.78 [1.20 to 432.58] | 3 [2 to 7] |
| Paroxetine vs reboxetine | |||||||
| Asthenia | Gallen 2001; M/2020/0047; M/2020/0052 | 91 | 693 | 48 | 682 | 2.01 [1.39 to 2.91] | 16 [11 to 34] |
| Chills | Gallen 2001; M/2020/0047; M/2020/0052 | 13 | 693 | 36 | 682 | 0.34 [0.18 to 0.65] | 29 [19 to 69] |
| Dyspnea | M/2020/0047; M/2020/0052 | 13 | 428 | 3 | 417 | 4.32 [1.22 to 15.31] | 43 [24 to 200] |
| Paraesthesia | Gallen 2001; M/2020/0047; M/2020/0052 | 10 | 693 | 22 | 682 | 0.44 [0.20 to 0.96] | 56 [30 to 535] |
| Sexual problems (anorgasmia) | Gallen 2001; M/2020/0047; M/2020/0052 | 24 | 693 | 1 | 682 | 9.84 [2.23 to 43.46] | 30 [21 to 52] |
| Sexual problems (libido decreased) | Gallen 2001; M/2020/0047; M/2020/0052 | 26 | 693 | 11 | 682 | 2.34 [1.14 to 4.82] | 47 [26 to 229] |
| Sleep disorders | Gallen 2001; M/2020/0047; M/2020/0052 | 16 | 693 | 4 | 682 | 3.67 [1.29 to 10.45] | 58 [34 to 215] |
| Sweating | Gallen 2001; M/2020/0047; M/2020/0052 | 55 | 693 | 104 | 682 | 0.48 [0.34 to 0.68] | 14 [9 to 25] |
| Tremor | Gallen 2001; M/2020/0047; M/2020/0052 | 31 | 693 | 11 | 682 | 2.75 [1.35 to 5.57] | 35 [21 to 95] |
| Weight gain | Gallen 2001; M/2020/0052 | 10 | 431 | 2 | 424 | 4.12 [1.02 to 16.62] | 54 [29 to 351] |
| Yawning | Gallen 2001;M/2020/0047 | 11 | 527 | 0 | 523 | 12.13 [1.57 to 93.64] | 48 [30 to 115] |
| Paroxetine vs tianeptine | |||||||
| Dizziness | Lepine 2001; Waintraub 2002 | 10 | 304 | 1 | 300 | 7.02 [1.25 to 39.32] | 34 [20 to 118] |
Comparison 2: Paroxetine versus SSRIs
Primary outcome: Efficacy. Number of patients who responded to treatment
2.1 Responders at endpoint (six to 12 weeks)
We found a difference in favour of citalopram over paroxetine (OR: 1.54, 95% CI 1.04 to 2.28, NNTb = 9, 95% CI 5 to 102, 1 RCT, 406 participants).
2.2 Early response rate (one to four weeks) and follow‐up response rate (16 to 24 weeks)
We found no differences between paroxetine and individual SSRIs (see Analysis 2.2; Analysis 3.1).
2.2. Analysis.
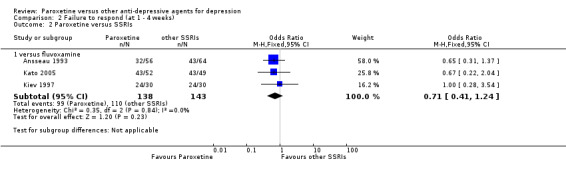
Comparison 2 Failure to respond (at 1 ‐ 4 weeks), Outcome 2 Paroxetine versus SSRIs.
3.1. Analysis.
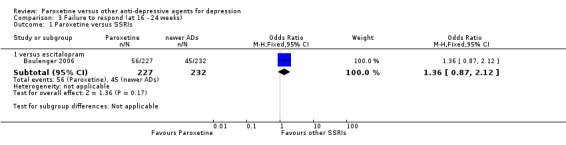
Comparison 3 Failure to respond (at 16 ‐ 24 weeks), Outcome 1 Paroxetine versus SSRIs.
Secondary outcome: Efficacy. Number of patients who remitted
2.3 Remission rate at endpoint (six to 12 weeks)
We found no differences between paroxetine and individual SSRIs (see Analysis 4.2).
4.2. Analysis.
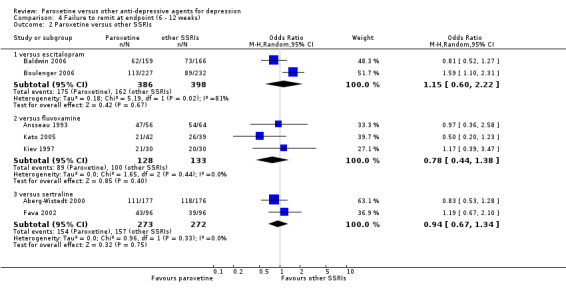
Comparison 4 Failure to remit at endpoint (6 ‐ 12 weeks), Outcome 2 Paroxetine versus other SSRIs.
2.4 Early remission rate (one to four weeks)
We found no difference between paroxetine and other SSRIs in terms of early remission rates (see Analysis 5.2).
5.2. Analysis.
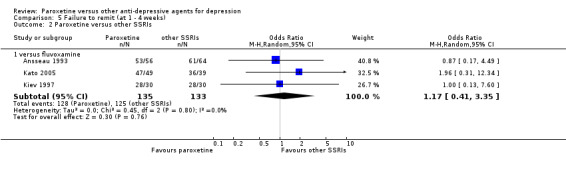
Comparison 5 Failure to remit (at 1 ‐ 4 weeks), Outcome 2 Paroxetine versus other SSRIs.
2.5. Follow‐up remission rate (16 to 24 weeks)
No studies provided data for this outcome.
Secondary outcome: Efficacy. Standardized mean difference
2.6 Standardized mean difference at endpoint (six to 12 weeks)
For this continuous outcome at endpoint, we found no difference between paroxetine and other SSRIs (see Analysis 7.2).
7.2. Analysis.
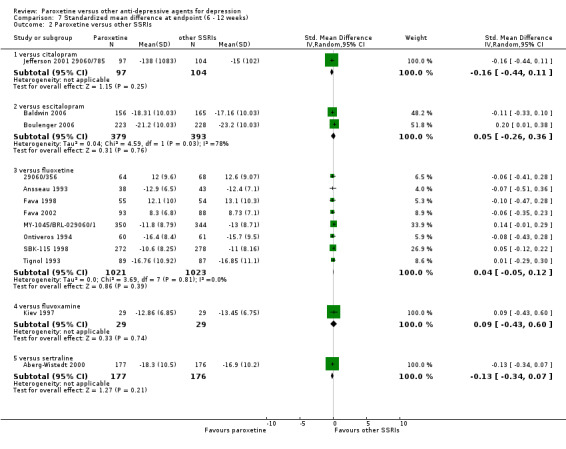
Comparison 7 Standardized mean difference at endpoint (6 ‐ 12 weeks), Outcome 2 Paroxetine versus other SSRIs.
2.7 Standardized mean difference at one to four weeks
For this outcome we found no difference between paroxetine and other SSRIs (see Analysis 8.2).
8.2. Analysis.
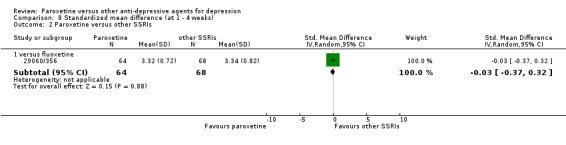
Comparison 8 Standardized mean difference (at 1 ‐ 4 weeks), Outcome 2 Paroxetine versus other SSRIs.
2.8 Standardized mean difference at 16 to 24 weeks
No difference was found for this outcome.
Secondary outcome: Acceptability
2.9 Failure to complete due to any cause
In terms of patients who dropped out during the trial for any reason, we found no difference between paroxetine and individual SSRIs (see Analysis 10.2).
10.2. Analysis.
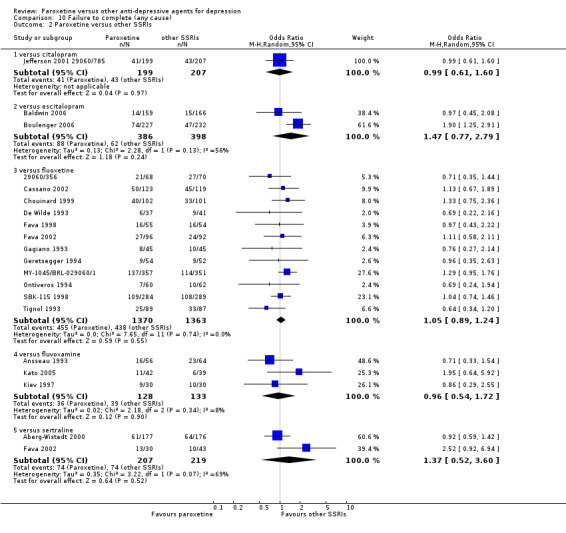
Comparison 10 Failure to complete (any cause), Outcome 2 Paroxetine versus other SSRIs.
2.10 Failure to complete due to inefficacy
We found no difference in terms of dropouts due to inefficacy (see Analysis 11.2).
11.2. Analysis.
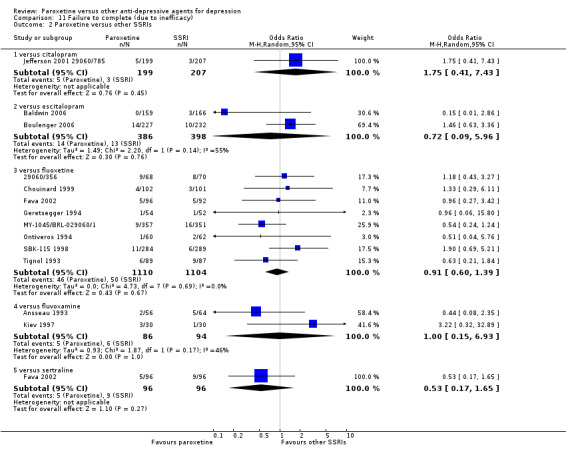
Comparison 11 Failure to complete (due to inefficacy), Outcome 2 Paroxetine versus other SSRIs.
2.11 Failure to complete due to side effects
In terms of dropouts due to side effects, we found that paroxetine was less well tolerated than fluoxetine (OR: 1.34, 95% CI 1.06 to 1.70, NNTh = 29, 95% CI 16 to 137, 11 RCTs, 2491 participants) (see Analysis 12.2).
12.2. Analysis.
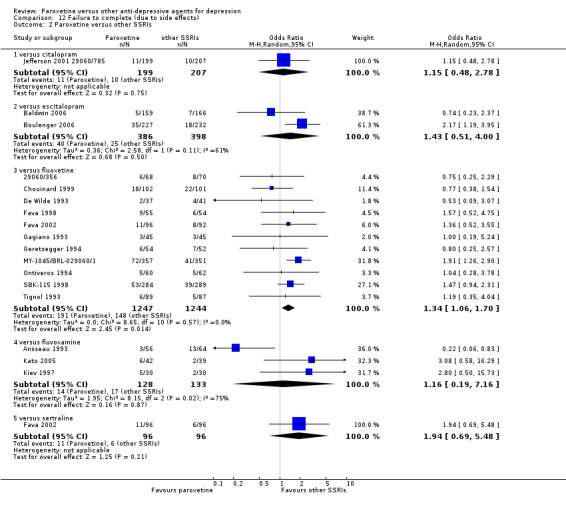
Comparison 12 Failure to complete (due to side effects), Outcome 2 Paroxetine versus other SSRIs.
Secondary outcome: Tolerability
2.13 Total number of patients experiencing at least some side effects
We found no difference between paroxetine and other SSRIs in terms of number of patients experiencing side effects (see Analysis 13.2).
13.2. Analysis.
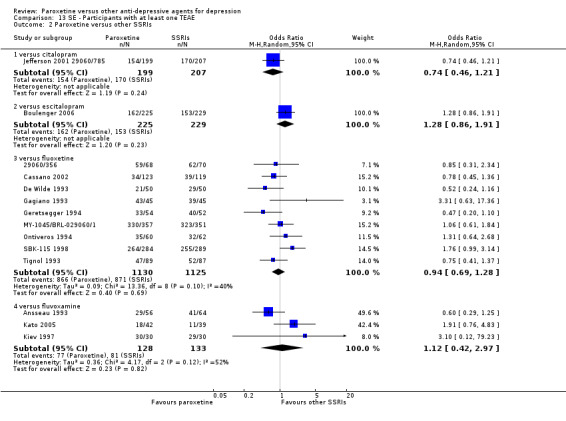
Comparison 13 SE ‐ Participants with at least one TEAE, Outcome 2 Paroxetine versus other SSRIs.
2.14 Number of patients experiencing specific side effects
(a) Sleepiness/drowsiness. We found a difference in favour of fluoxetine over paroxetine (OR: 1.48, 95% CI 1.16 to 1.88, NNTh = 20, 95% CI 12 to 53, 8 RCTs, 2116 participants) (see Analysis 79.2).
79.2. Analysis.
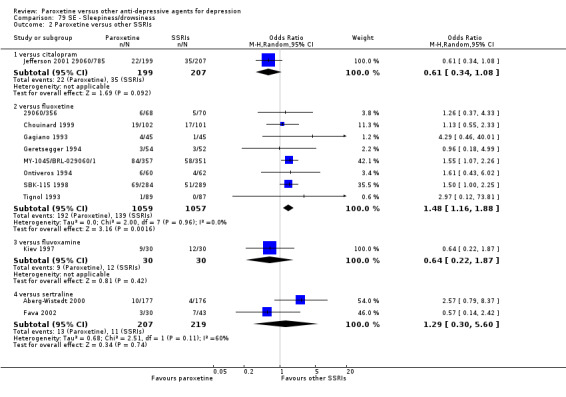
Comparison 79 SE ‐ Sleepiness/drowsiness, Outcome 2 Paroxetine versus other SSRIs.
(b) Insomnia. We found no difference between paroxetine and SSRIs (see Analysis 54.2).
54.2. Analysis.
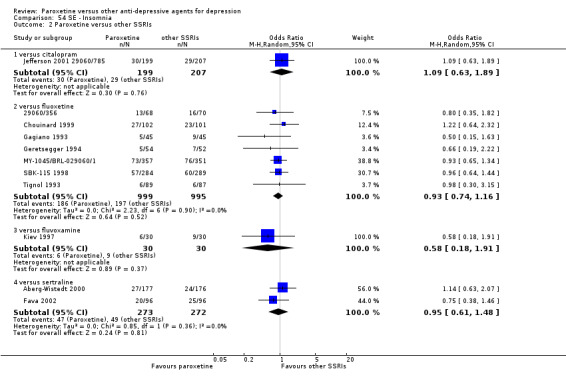
Comparison 54 SE ‐ Insomnia, Outcome 2 Paroxetine versus other SSRIs.
(c) Dry mouth. Paroxetine was associated with a higher rate of dry mouth than fluoxetine (OR: 1.67, 95% CI 1.17 to 2.38, NNTh = 18, 95% CI 12 to 38, 6 RCTs, 1920 participants) (see Analysis 38.2).
38.2. Analysis.
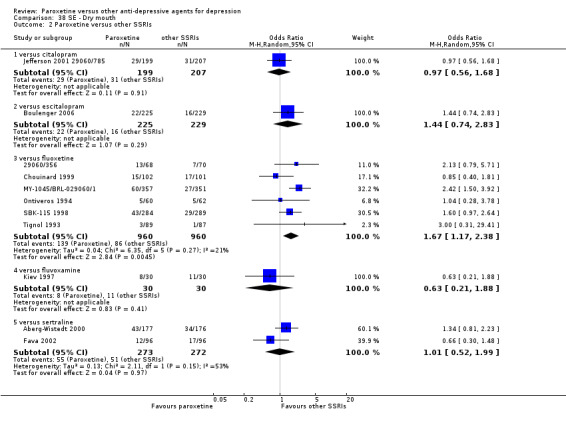
Comparison 38 SE ‐ Dry mouth, Outcome 2 Paroxetine versus other SSRIs.
(d) Constipation. Paroxetine was associated with a higher rate of constipation than fluoxetine (OR: 2.71, 95% CI 1.47 to 5.01, NNTh = 14, 95% CI 9 to 26, 3 RCTs, 1001 participants) and sertraline (OR: 3.26, 95% CI 1.73 to 6.14, NNTh = 10, 95% CI 7 to 20, 2 RCTs, 545 participants) (see Analysis 31.2).
31.2. Analysis.
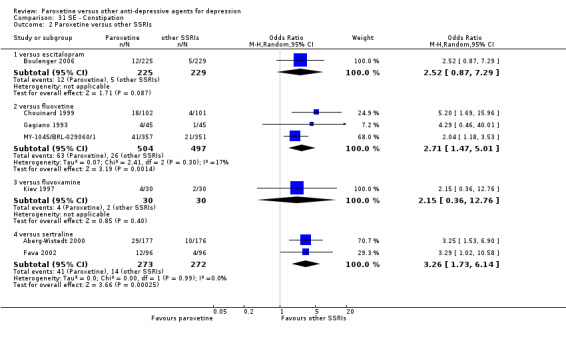
Comparison 31 SE ‐ Constipation, Outcome 2 Paroxetine versus other SSRIs.
(e) Urination/Urogenital problems. There was difference in favour of sertraline over paroxetine for this outcome (OR: 11.60, 95% CI 1.48 to 90.81, NNTh = 18, 95% CI 11 to 52, 1 RCT, 353 participants) (see Analysis 90.5).
(f) Hypotension. We found no difference between paroxetine and other SSRIs (see Analysis 50.2).
50.2. Analysis.
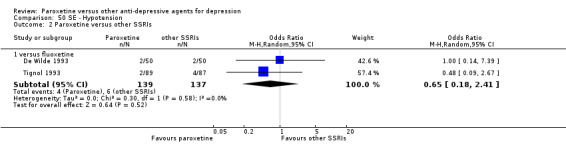
Comparison 50 SE ‐ Hypotension, Outcome 2 Paroxetine versus other SSRIs.
(g) Agitation/anxiety. We found no difference between paroxetine and other SSRIs (see Analysis 18.2).
18.2. Analysis.
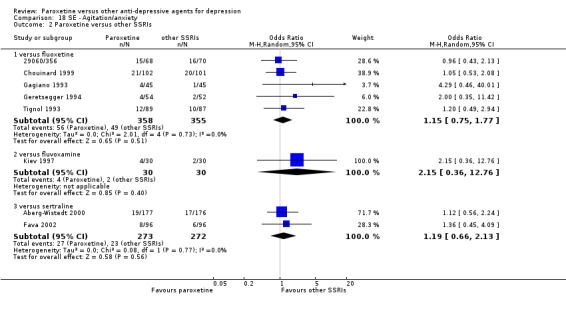
Comparison 18 SE ‐ Agitation/anxiety, Outcome 2 Paroxetine versus other SSRIs.
(h) Suicide wishes/gestures/attempts. We found no difference between paroxetine and other SSRIs (see Analysis 96.1; Analysis 96.3)..
(i) Completed suicide. We found no difference between paroxetine and other SSRIs (see Analysis 96.2).
(j) Vomiting/nausea. We found a difference in favour of fluoxetine over paroxetine (OR: 1.24, 95% CI 1.02 to 1.51, NNTh = 26, 95% CI 14 to 298, 10 RCTs, 2336 participants) (see Analysis 59.2).
59.2. Analysis.
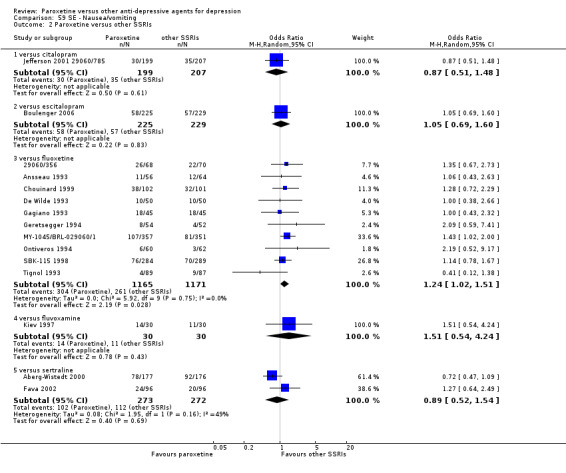
Comparison 59 SE ‐ Nausea/vomiting, Outcome 2 Paroxetine versus other SSRIs.
(k) Diarrhoea. We found a difference in favour of paroxetine over sertraline (OR: 0.40, 95% CI 0.26 to 0.60, NNTh = 6, 95% CI 4 to 11, 2 RCTs, 545 participants) (see Analysis 36.2).
36.2. Analysis.
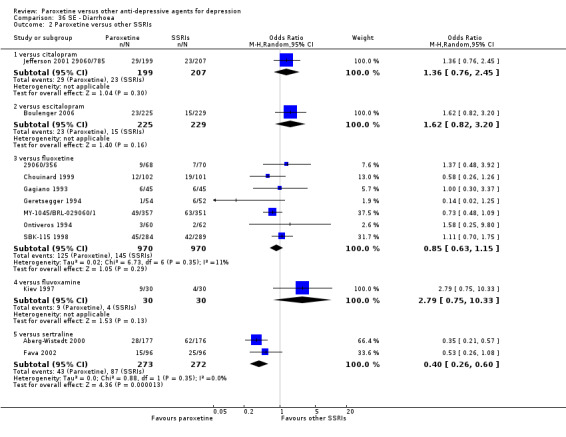
Comparison 36 SE ‐ Diarrhoea, Outcome 2 Paroxetine versus other SSRIs.
(l) Other side effects. Other statistically significant side effects are reported in Table 20.
Comparison 3: Paroxetine versus newer or non‐conventional ADs
Primary outcome: Efficacy. Number of patients who responded to treatment
3.1 Responders at endpoint (six to 12 weeks)
In terms of efficacy as number of patients who responded to treatment, there was a trend in favour of paroxetine over reboxetine (OR: 0.82, 95% CI 0.66 to 1.02, 3 RCTs, 1369 participants) (see Analysis 1.3).
1.3. Analysis.
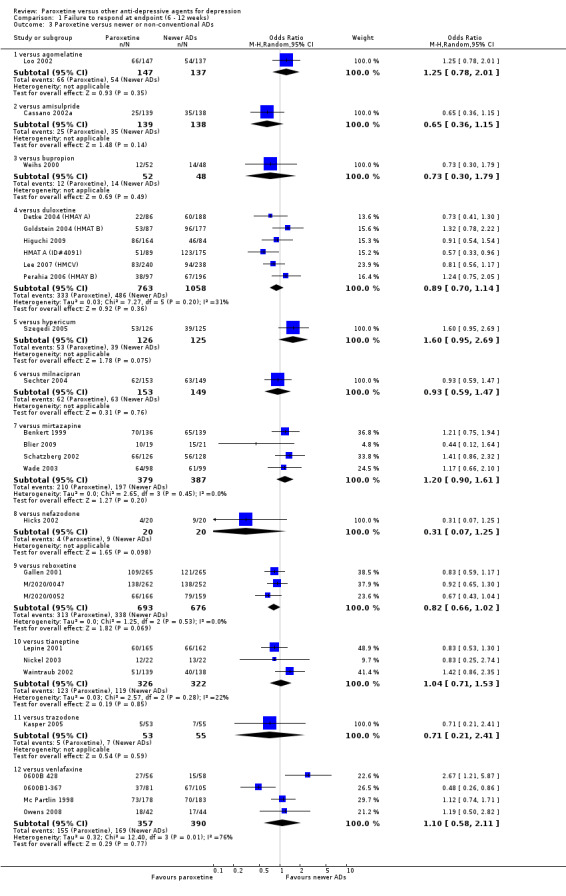
Comparison 1 Failure to respond at endpoint (6 ‐ 12 weeks), Outcome 3 Paroxetine versus newer or non‐conventional ADs.
3.2 Early response rate (one to four weeks)
In terms of efficacy as number of patients who responded to treatment at one to four weeks, we found a difference in favour of paroxetine over reboxetine (OR: 0.66, 95% CI 0.50 to 0.87, NNTb = 16, 95% CI 10 to 50, 3 RCTs, 1375 participants). By contrast, we found a difference in favour of mirtazapine over paroxetine for this outcome (OR: 2.39, 95% CI 1.42 to 4.02, NNTb = 8, 95% CI 5 to 14, 3 RCTs, 726 participants) (see Analysis 2.3).
2.3. Analysis.
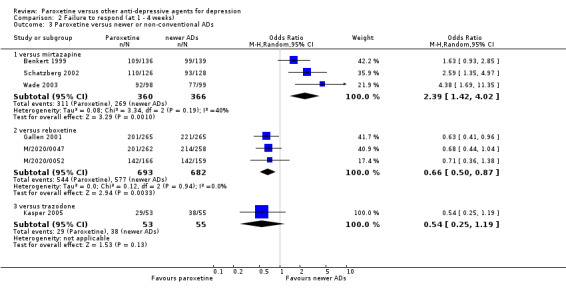
Comparison 2 Failure to respond (at 1 ‐ 4 weeks), Outcome 3 Paroxetine versus newer or non‐conventional ADs.
3.3 Follow‐up response rate (16 to 24 weeks)
In terms of efficacy as number of patients who responded to treatment at 16‐24 weeks, we found no difference between paroxetine and newer or non‐conventional ADs (see Analysis 3.2).
3.2. Analysis.
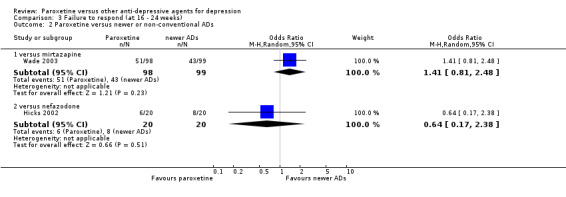
Comparison 3 Failure to respond (at 16 ‐ 24 weeks), Outcome 2 Paroxetine versus newer or non‐conventional ADs.
Secondary outcome: Efficacy. Number of patients who remitted
3.4 Remission rate at endpoint (six to 12 weeks)
In terms of efficacy as number of patients who remitted, we found a difference in favour of hypericum (OR: 1.84, 95% CI 1.11 to 3.06, NNTb = 7, 95% CI 4 to 38, 1 RCT, 251 participants), mirtazapine (OR: 1.52, 95% CI 1.13 to 2.06, NNTb = 11, 95% CI 6 to 37, 4 RCTs, 766 participants) and venlafaxine (OR: 1.57, 95% CI 1.08 to 2.29, NNTb = 11, 95% CI 6 to 54, 4 RCTs, 807 participants) over paroxetine (see Analysis 4.3).
4.3. Analysis.
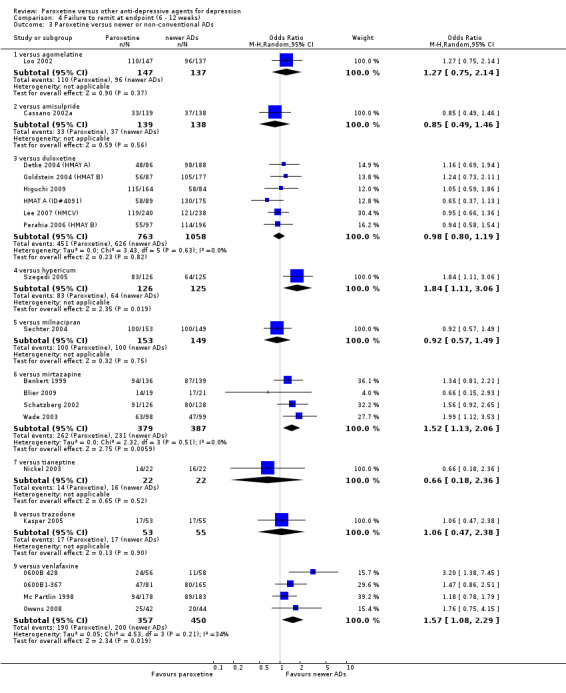
Comparison 4 Failure to remit at endpoint (6 ‐ 12 weeks), Outcome 3 Paroxetine versus newer or non‐conventional ADs.
3.5 Early remission rate (one to four weeks)
In terms of efficacy as number of patients who remitted at one to four weeks, we found a difference between paroxetine and mirtazapine, in favour of mirtazapine (OR: 2.31, 95% CI 1.04 to 5.11, NNTb = 18, 95% CI 11 to 54, 3 RCTs, 726 participants) (see Analysis 5.3).
5.3. Analysis.
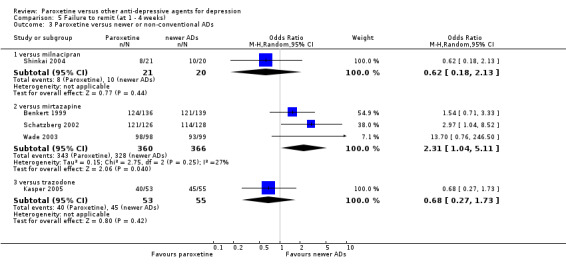
Comparison 5 Failure to remit (at 1 ‐ 4 weeks), Outcome 3 Paroxetine versus newer or non‐conventional ADs.
3.6. Follow‐up remission rate (16 to 24 weeks)
We found a difference in favour of mirtazapine over paroxetine for this outcome (OR: 1.89, 95% CI 1.01 to 3.54, NNTb = 8, 95% CI 4 to 265, 1 RCT, 197 participants) (see Analysis 6.1).
6.1. Analysis.
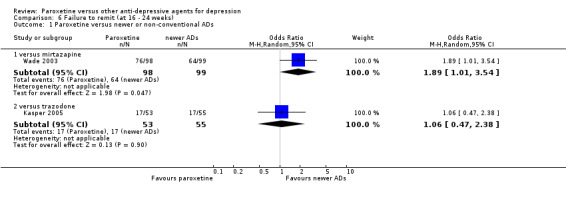
Comparison 6 Failure to remit (at 16 ‐ 24 weeks), Outcome 1 Paroxetine versus newer or non‐conventional ADs.
Secondary outcome: Efficacy. Standardized mean difference
3.7 Standardized mean difference at endpoint (six to 12 weeks)
In terms of continuous outcomes at 6‐12 weeks, we found a difference in favour of mirtazapine over paroxetine (SMD: 0.33, 95% CI 0.08 to 0.58, 1 RCTs, 246 participants). Moreover, there was a trend in favour of paroxetine over reboxetine (SMD: ‐0.10, 95% CI ‐0.21 to 0.00, 3 RCTs, 1291 participants). There was no evidence that paroxetine was different from other newer or non‐conventional ADs (see Analysis 7.3).
7.3. Analysis.
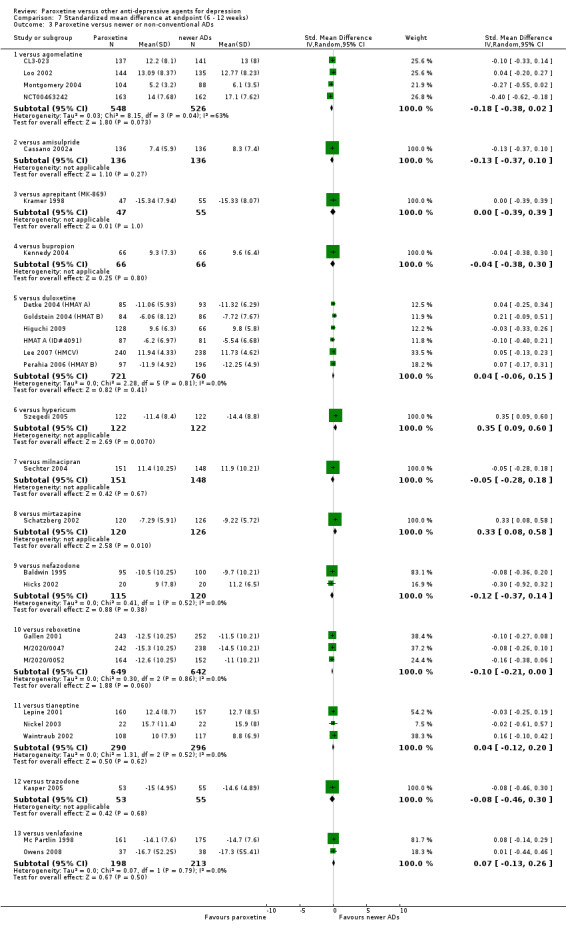
Comparison 7 Standardized mean difference at endpoint (6 ‐ 12 weeks), Outcome 3 Paroxetine versus newer or non‐conventional ADs.
3.8 Standardized mean difference at one to four weeks
For this outcome we found a difference in favour of paroxetine over reboxetine (SMD: ‐0.17, 95% CI ‐0.31 to ‐0.03, 2 RCTs, 805 participants) (see Analysis 8.3).
8.3. Analysis.
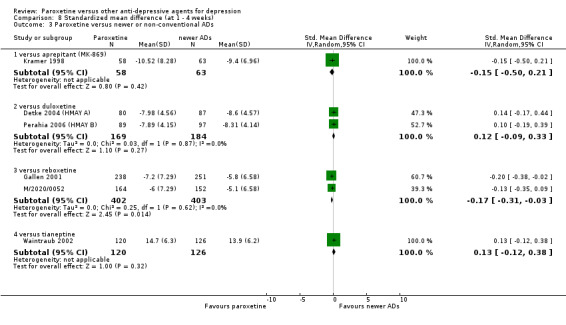
Comparison 8 Standardized mean difference (at 1 ‐ 4 weeks), Outcome 3 Paroxetine versus newer or non‐conventional ADs.
3.9 Standardized mean difference at 16 to 24 weeks
For this outcome, we found no difference between paroxetine and newer or non‐conventional ADs (see Analysis 9.2).
9.2. Analysis.
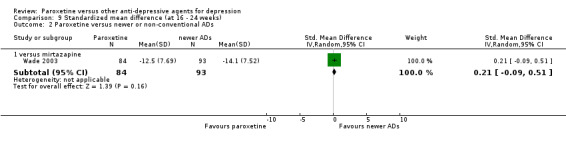
Comparison 9 Standardized mean difference (at 16 ‐ 24 weeks), Outcome 2 Paroxetine versus newer or non‐conventional ADs.
Secondary outcome: Acceptability
3.10 Failure to complete due to any cause
In terms of patients who dropped out for any reason, we found no difference between paroxetine and newer or non‐conventional ADs (see Analysis 10.3).
10.3. Analysis.
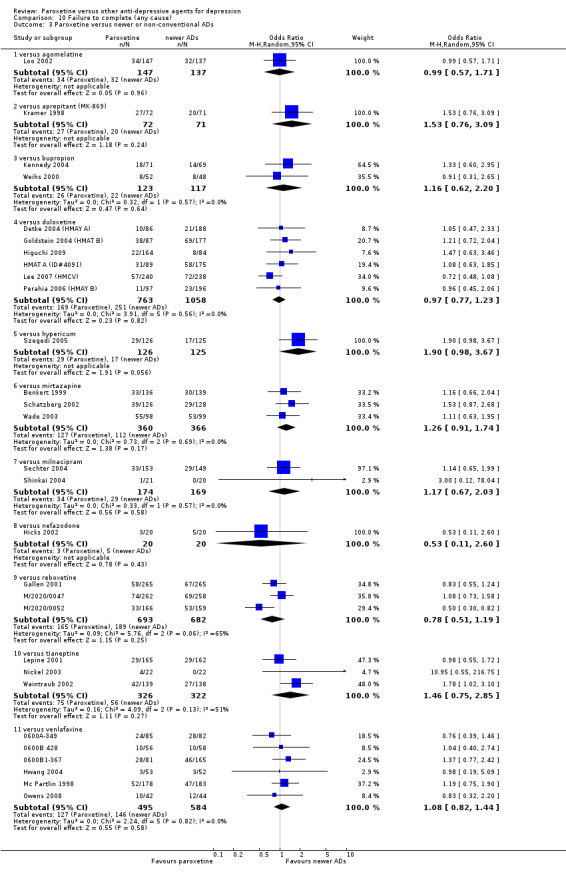
Comparison 10 Failure to complete (any cause), Outcome 3 Paroxetine versus newer or non‐conventional ADs.
3.11 Failure to complete due to inefficacy
No difference was found between paroxetine and newer or non‐conventional ADs in terms of discontinuation due to inefficacy (see Analysis 11.3).
11.3. Analysis.
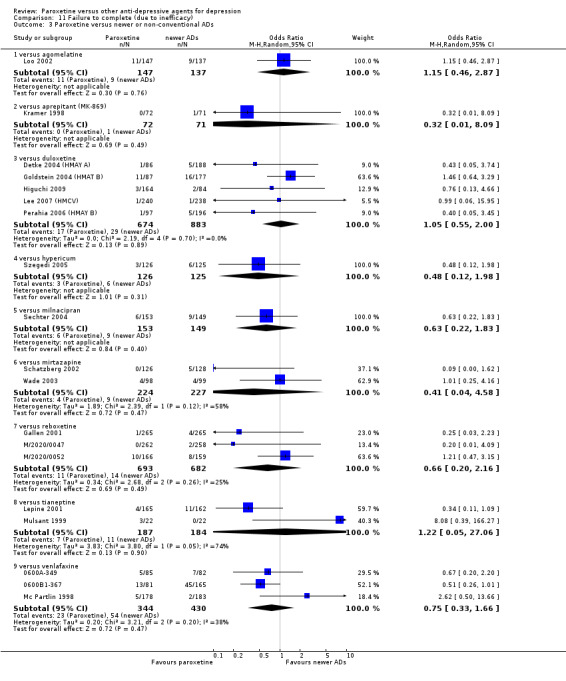
Comparison 11 Failure to complete (due to inefficacy), Outcome 3 Paroxetine versus newer or non‐conventional ADs.
3.12 Failure to complete due to side effects
In terms of patients who dropped out due to side effects, we found a difference between paroxetine and reboxetine (OR: 0.38, 95% CI 0.17 to 0.86, NNTh = 16, 95% CI 10 to 30, 3 RCTs, 1375 participants) in favour of paroxetine; and a difference between paroxetine and tianeptine (OR: 3.38, 95% CI 1.31 to 8.71, NNTh = 13, 95% CI 7 to 47, 1 RCT, 327 participants) in favour of tianeptine (see Analysis 12.3).
12.3. Analysis.
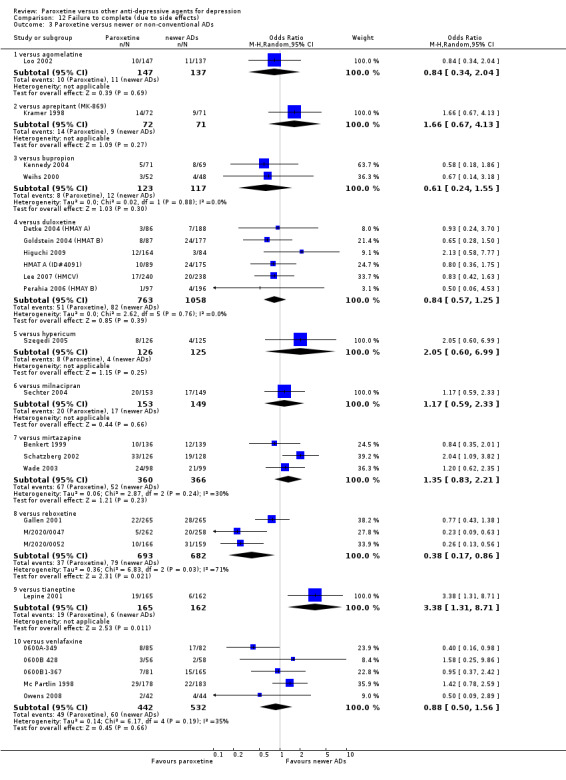
Comparison 12 Failure to complete (due to side effects), Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Secondary outcome: Tolerability
3.13 Total number of patients experiencing at least some side effects
There was evidence that paroxetine was less well tolerated than hypericum (OR: 2.60, 95% CI 1.51 to 4.46, NNTh = 5, 95% CI 3 to 10, 1 RCT, 251 participants) (see Analysis 13.3).
13.3. Analysis.
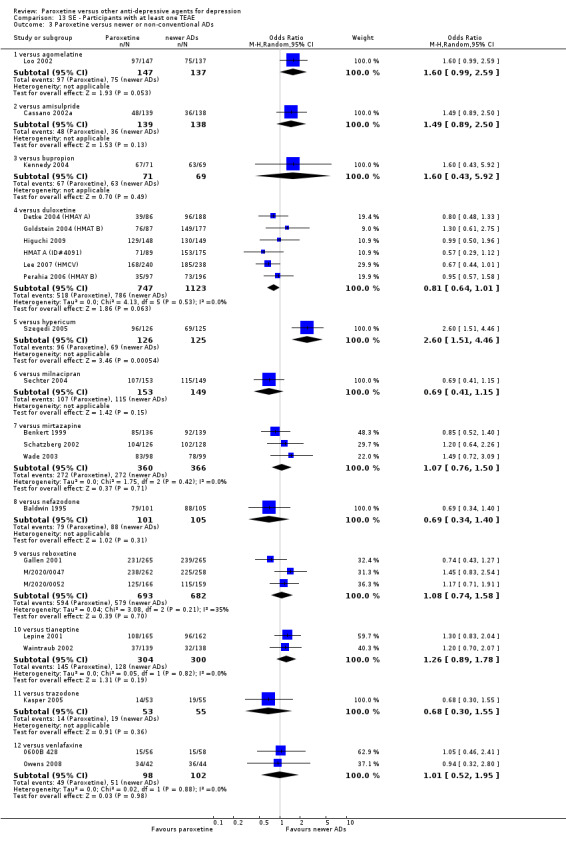
Comparison 13 SE ‐ Participants with at least one TEAE, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
3.14 Number of patients experiencing specific side effects
(a) Sleepiness/drowsiness. We found a difference in favour of bupropion (OR: 7.63, 95% CI 2.51 to 23.16, NNTh = 5, 95% CI 4 to 9, 2 RCTs, 240 participants) and in favour of reboxetine (OR: 2.66, 95% CI 1.45 to 4.89, NNTh = 13, 95% CI 9 to 22, 3 RCTs, 1375 participants) over paroxetine (see Analysis 79.3).
79.3. Analysis.
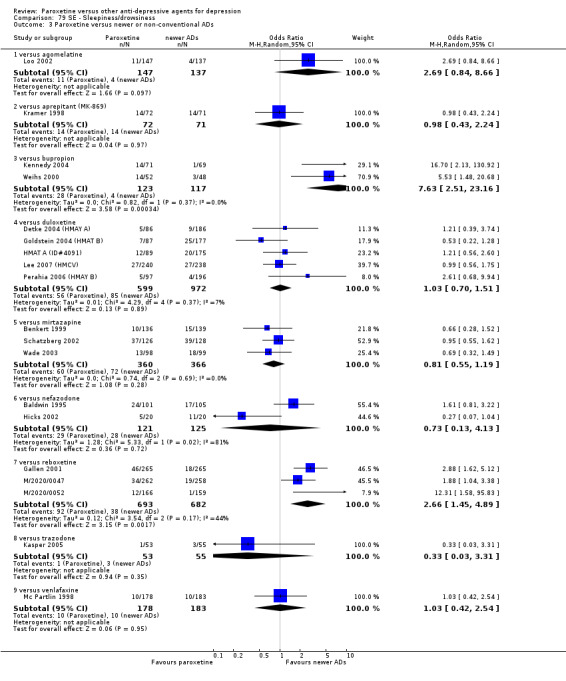
Comparison 79 SE ‐ Sleepiness/drowsiness, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
(b) Insomnia. There was evidence that paroxetine was associated with a lower rate of insomnia than reboxetine (OR: 0.48, 95% CI 0.31 to 0.74, NNTh = 8, 95% CI 6 to 11, 3 RCTs, 1375 participants) (see Analysis 54.3).
54.3. Analysis.
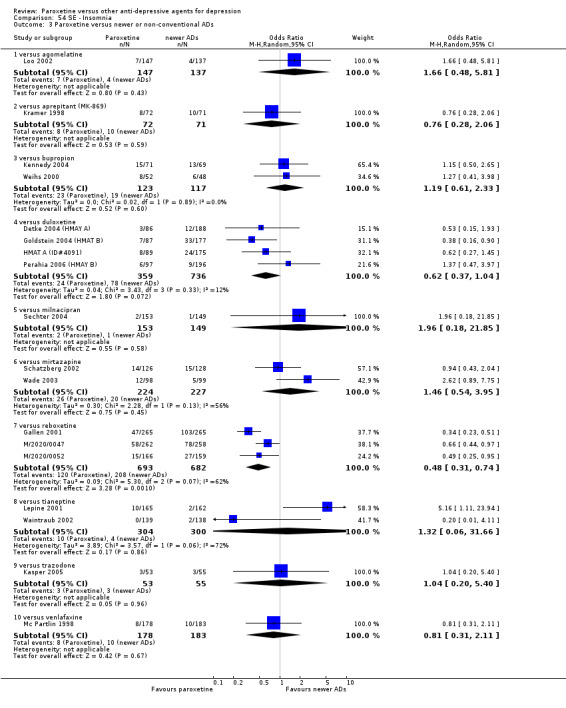
Comparison 54 SE ‐ Insomnia, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
(c) Dry mouth. Paroxetine was associated with a lower rate of dry mouth than reboxetine (OR: 0.35, 95% CI 0.27 to 0.45, NNTh = 5, 95% CI 4 to 6, 3 RCTs, 1375 participants). By contrast, we found a difference in favour of hypericum (OR: 2.62, 95% CI 1.36 to 5.04, NNTh = 7, 95% CI 4 to 19, 1 RCT, 251 participants) over paroxetine (see Analysis 38.3).
38.3. Analysis.
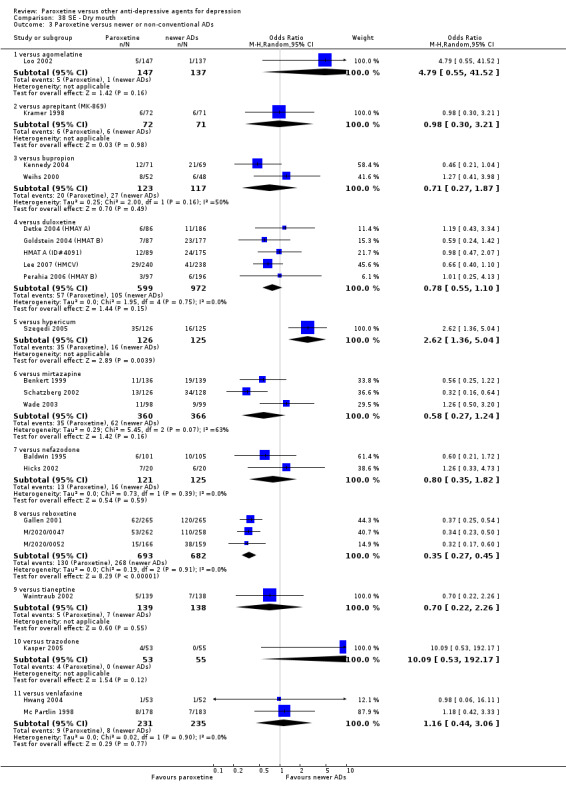
Comparison 38 SE ‐ Dry mouth, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
(d) Constipation. Paroxetine was associated with a lower rate of constipation than reboxetine (OR: 0.48, 95% CI 0.36 to 0.63, NNTh = 9, 95% CI 7 to 15, 3 RCTs, 1375 participants) (see Analysis 31.3).
31.3. Analysis.
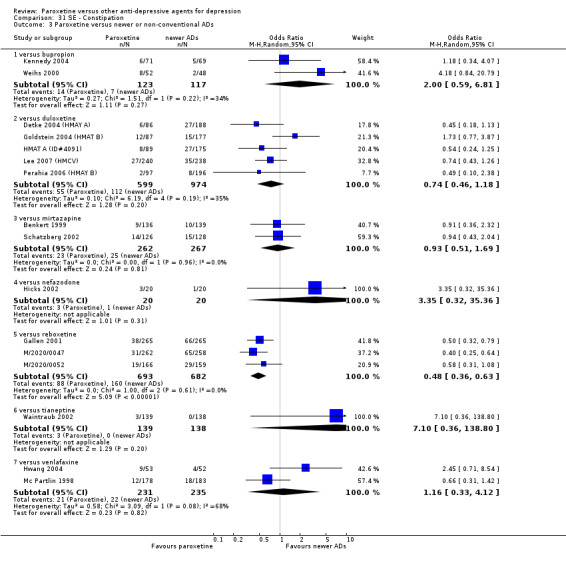
Comparison 31 SE ‐ Constipation, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
(e) Urination/Urogenital problems. There was evidence in favour of paroxetine over reboxetine, in particular for dysuria (OR: 0.16, 95% CI 0.04 to 0.75, NNTh = 42, 95% CI 25 to 151, 2 RCTs, 855 participants) (see Analysis 90.2) and urinary retention (OR: 0.28, 95% CI 0.14 to 0.54, NNTh = 22, 95% CI 15 to 40, 3 RCTs, 1375 participants) (see Analysis 90.4).
90.2. Analysis.
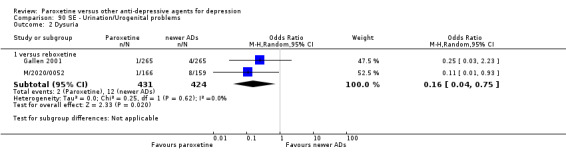
Comparison 90 SE ‐ Urination/Urogenital problems, Outcome 2 Dysuria.
(f) Hypotension. We found a difference in favour of paroxetine over reboxetine (OR: 0.37, 95% CI 0.19 to 0.75, NNTh = 19, 95% CI 13 to 36, 3 RCTs, 1375 participants) (see Analysis 50.3).
50.3. Analysis.
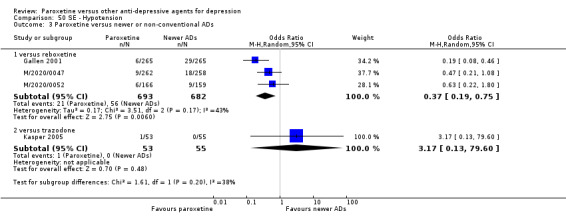
Comparison 50 SE ‐ Hypotension, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
(g) Agitation/anxiety. We found no difference between paroxetine and newer or non‐conventional ADs (see Analysis 18.3).
18.3. Analysis.
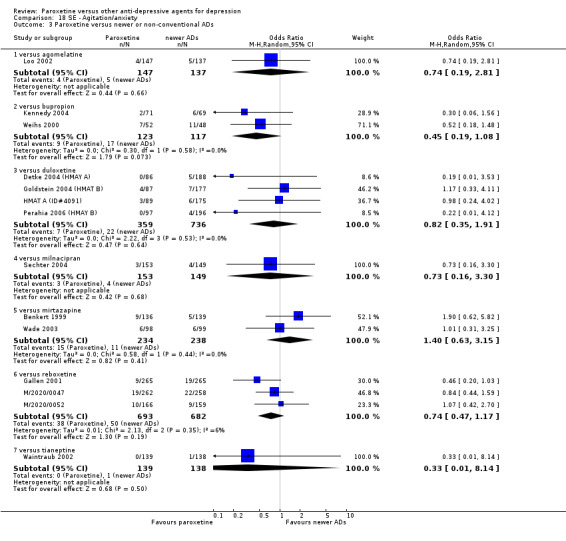
Comparison 18 SE ‐ Agitation/anxiety, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
(h) Suicide wishes/gestures/attempts. We found no difference between paroxetine and newer or non‐conventional ADs (see Analysis 96.1; Analysis 96.3).
(i) Completed suicide. We found no difference between paroxetine and newer or non‐conventional ADs (see Analysis 96.2) .
(j) Vomiting/nausea. Paroxetine was associated with a higher rate of vomiting/nausea than agomelatine (OR: 6.81, 95% CI 2.31 to 20.14, NNTh = 7, 95% CI 5 to 14, 1 RCT, 284 participants), amisulpride (OR: 7.13, 95% CI 2.06 to 24.68, NNTh = 9, 95% CI 6 to 19, 1 RCT, 277 participants), hypericum (OR: 2.58, 95% CI 1.13 to 5.88, NNTh = 11, 95% CI 6 to 65, 1 RCT, 251 participants), mirtazapine (OR: 3.03, 95% CI 1.91 to 4.82, NNTh = 8, 95% CI 6 to 14, 3 RCTs, 726 participants), reboxetine (OR: 2.07, 95% CI 1.60 to 2.69, NNTh = 8, 95% CI 6 to 12, 3 RCTs, 1375 participants) and tianeptine (OR: 2.54, 95% CI 1.38 to 4.67, NNTh = 14, 95% CI 9 to 38, 2 RCTs, 604 participants). By contrast, paroxetine was better than duloxetine (OR: 0.68, 95% CI 0.52 to 0.88, NNTh = 34, 95% CI 14 to 78, 5 RCTs, 1573 participants) (see Analysis 59.3).
59.3. Analysis.
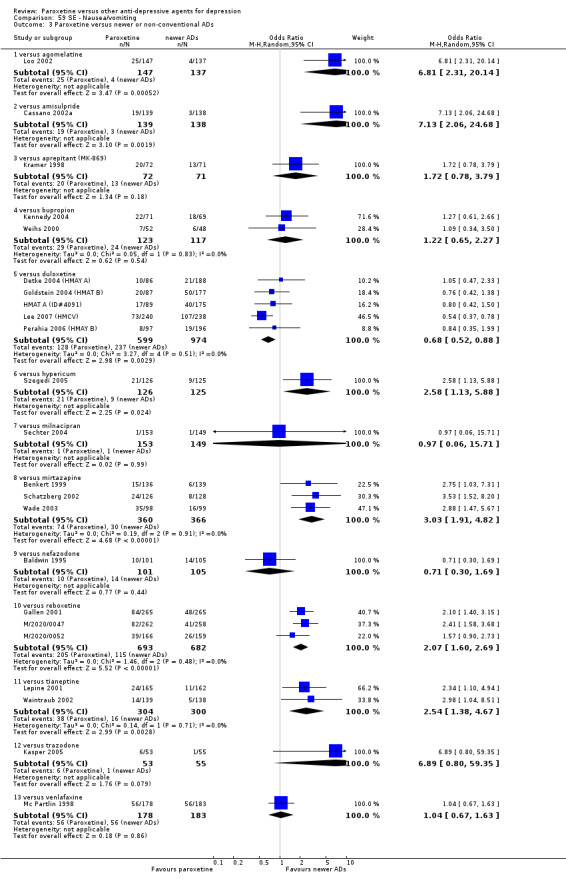
Comparison 59 SE ‐ Nausea/vomiting, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
(k) Diarrhoea. We found that paroxetine was associated with a higher rate of diarrhoea than bupropion (OR: 3.03, 95% CI 1.35 to 6.84, NNTh = 8, 95% CI 5 to 25, 2 RCTs, 240 participants) and reboxetine (OR: 3.45, 95% CI 2.31 to 5.15, NNTh = 9, 95% CI 7 to 13, 3 RCTs, 1375 participants). Moreover, a trend in favour of hypericum over paroxetine (OR: 2.10, 95% CI 1.00 to 4.44, 1 RCT, 251 participants) was found (see Analysis 36.3).
36.3. Analysis.
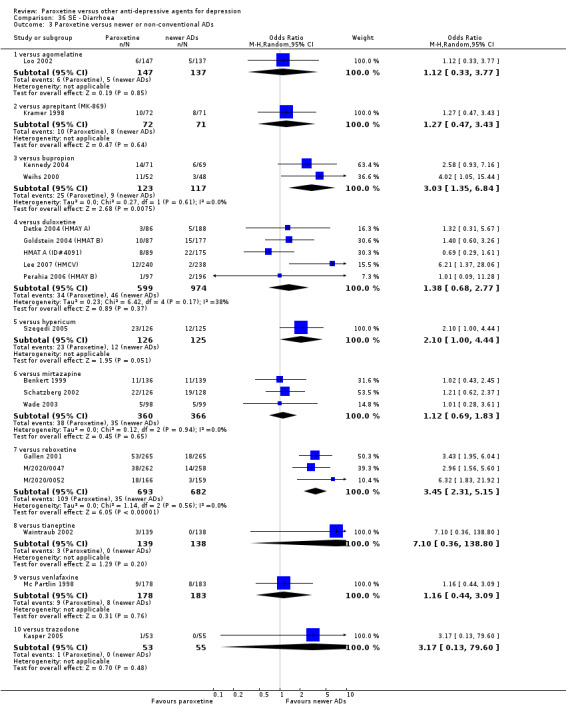
Comparison 36 SE ‐ Diarrhoea, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
(l) Other side effects. Other statistically significant side effects are reported in Table 20.
Subgroup analyses
1. Paroxetine dosing
The great majority of studies used paroxetine within the standard dose range of 20‐40 mg/day. Therefore, it was not meaningful to carry out this pre‐planned subgroup analysis.
2. Comparator dosing
All comparator doses were within the therapeutic range. Due to the small number of trials outside the therapeutic range, it was not considered meaningful to carry out this pre‐planned subgroup analysis.
3. Depression severity
The great majority of studies reported a mean baseline score corresponding to moderate to severe major depression. Therefore, it was not meaningful to carry out this pre‐planned subgroup analysis.
4. Treatment settings
Results from this subgroup analysis did not materially change the main findings (full details available on request from authors).
5. Elderly patients
Results from this subgroup analysis did not materially change the main findings (full details available on request from authors).
Sensitivity analysis
1. Excluding trials with unclear concealment of random allocation and/or unclear double blinding
Although technically possible to carry out these sensitivity analyses, they were not performed, because they would not have contributed useful information due to the small number of studies (only seven out of 115 trials) reporting clear details on concealment of random allocation (Baldwin 2006; Boulenger 2006; Gallen 2001; Higuchi 2009; M/2020/0047; M/2020/0052; Szegedi 2005).
2. Excluding trials whose dropout rate was greater than 20%
It was not meaningful to carry out this pre‐planned sensitivity analysis.
3. Performing the worst‐ and best‐case scenario analyses
Results from these sensitivity analyses did not materially change the main findings (full details available on request from authors).
4. Excluding trials for which the imputation methods were used
Excluding trials for which the SDs had to be borrowed from other trials, results for all comparisons did not materially change.
5. Examination of “wish bias” and exclusion of studies funded by the pharmaceutical company marketing paroxetine
These pre‐planned sensitivity analyses were not carried out because we found only a few studies per comparison.
6. Excluding studies funded by the pharmaceutical company marketing paroxetine
These pre‐planned sensitivity analyses were not carried out because we found only a few studies per comparison.
Assessment of heterogeneity
For primary outcomes (response rate at endpoint), we found I2 indicative of substantial heterogeneity level (I2 between 50 and 90%) in the comparison between paroxetine and mianserin (I2 = 61%), and between paroxetine and venlafaxine (I2 = 76%) . For the secondary outcome failure to remit at endpoint, substantial heterogeneity was found in the comparison between paroxetine and older ADs as a class (I2 = 79%) and between paroxetine and escitalopram (I2 = 81%). Moreover, for the secondary outcome standardized mean difference at endpoint, substantial heterogeneity was found in the comparison between paroxetine and clomipramine (I2 = 78%) and between paroxetine and escitalopram (I2 = 78%). For the outcome failure to complete (any cause), substantial heterogeneity was found in the comparisons between paroxetine and lofepramine (I2 = 68%), sertraline (I2 = 69%) and reboxetine (I2 = 65%). For the outcome failure to complete (due to side effects) heterogeneity was found in the comparison between paroxetine and maprotiline (I2 = 84%), fluvoxamine (I2 = 75%) and reboxetine (I2 = 71%) . For the secondary outcome failure to complete (due to inefficacy), substantial heterogeneity was found in the comparison between paroxetine and tianeptine (I2 = 74%).
Assessment of publication bias
Visual inspection of funnel plots did not reveal substantial asymmetry in any of the comparisons between paroxetine and other conventional and unconventional ADs.
Discussion
Summary of main results
The present systematic review included a total of 115 randomised controlled trials (RCTs), involving 26,134 participants. The included studies did not report all the outcomes that were pre‐specified in the protocol and for some comparisons only a small number of trials provided data. Overall, we detected differences between paroxetine and some comparator ADs in terms of efficacy, acceptability and tolerability. For the primary outcome, response rate, paroxetine was less effective than citalopram at endpoint. In head‐to‐head comparisons with newer or non‐conventional ADs, we found a difference in favour of paroxetine over reboxetine and in favour of mirtazapine over paroxetine in the early phase. For the secondary outcome remission rate, we found a difference in favour of clomipramine, hypericum, mirtazapine and venlafaxine over paroxetine. For the secondary outcome acceptability, paroxetine was better than reboxetine and amitriptyline (dropouts due to side effects) and better than clomipramine, imipramine and than older ADs as a class (dropouts due to side effects and dropouts due to any cause). By contrast, paroxetine was associated with a higher rate of dropouts due to side effects than fluoxetine and tianeptine. For the secondary outcome tolerability ‐ number of patients experiencing at least some side effects ‐ paroxetine was better than amitriptyline, imipramine and older ADs as a class, and worse than hypericum.
Overall completeness and applicability of evidence
All studies included in the present review recruited participants with a formal diagnosis of depression according to operationalised diagnostic criteria such as DSM‐III or DSM‐IV criteria, and therefore there was considerable homogeneity in the study populations. However, we have to consider that the majority of included studies were short in duration and some analyses were underpowered to demonstrate clinically meaningful differences between treatments. There was also considerable variation in the type of control medication used in the trials, and in some cases trials did not provide data for all the outcomes specified in the protocol, thus limiting the overall completeness of evidence. Another weakness of this analysis is that different subgroups of studies provided data for each efficacy and acceptability analyses, therefore raising the possibility of outcome reporting bias (Furukawa 2007).
The great majority of included trials were conducted in developed countries (Europe, United States, Canada) and this issue may be of limit in the application of these results in other countries of the world. Moreover, even though we collected a considerable amount of data on efficacy and acceptability, for some comparisons only a few trials were identified. In consequence, the reader is left with the decision of whether the findings from trials carried out in developed countries with standardized methods can be translated to routine clinical practice.
It has long been argued that placebo‐controlled trials are required to adequately demonstrate the efficacy of novel antidepressant drugs (Cipriani 2009), however, in the present review we focused only on the comparison between paroxetine and other active treatments. The background logic that guided the development of the present review was based on two considerations. First, the efficacy of paroxetine versus placebo has already been quantified in a systematic review of published and unpublished studies (Barbui 2008); second, the need to provide real‐world evidence for patients in need of pharmacological treatment. We therefore made the choice of including only studies that compared paroxetine with another active treatment, as we reasoned that clinicians need to know how paroxetine, a reference AD agent, compares with a selection of possible comparator ADs. Although the search was thorough, and we did our very best to retrieve as much data as possible (through asking pharmaceutical companies and study authors to supply all available information and searching the GSK website to retrieve unpublished data), it is still possible that there are unpublished studies that have not been identified. Of consequence, we can assume that data from some trials are still lacking, most of which are likely to be studies with negative findings. We are also aware of the possibility that a number of further RCTs comparing paroxetine with other antidepressant drugs are currently being conducted and will be included in future updates of this review.
Quality of the evidence
The 'Risk of bias' assessment is crucial in influencing the results' interpretation and deserves therefore due attention. All included studies were RCTs and were very similar in design and conduct. Using high‐quality research evidence is relevant to review results and to speed up the translation of research in a way that really responds to clinically relevant questions. However, the quality of RCTs is not easy to assess. Even though RCTs are the design of choice for evaluating the efficacy and acceptability of healthcare interventions (Jüni 2001; Purgato 2010), the evidence upon which the findings of this review are based is relatively poor as evaluated with the Cochrane 'Risk of bias' tool, and this is also reflected in our grading within the 'Summary of findings' tables. For example, allocation concealment and blinding of outcome assessment were "unclear" in the great majority of included studies and there is evidence that non‐blinded assessors in RCTs might generate biased results (Hróbjartsson 2013). The great majority of studies included in our review were described as "double‐blind", but information on the procedure followed to guarantee the blindness, and if blindness was successful was not reported in many cases. The investigators' and participants' belief about what treatment the participant is taking is a crucial issue that could influence the trIal's results. Also, the reporting of outcomes was often unclear or incomplete (for example, many RCTs did not report SDs) and the figures used to report or summarise the analyses were not immediately understandable. However, we have to consider that the scant information about allocation concealment may be a matter of reporting in the text than a real defect in the study design. The quality of evidence evaluated with the GRADE methodology (within‐study risk of bias, directness of evidence, heterogeneity, precision of effect estimates and risk of publication bias) was in general, from moderate to low.
Potential biases in the review process
Some possible limitations of this review should be noted and this means that the interpretation of results should remain tentative.
Although the search was thorough, it is possible that there are still unpublished studies which have not been identified. Moreover, the search date is September 2012 and there are a number of studies classified as "awaiting assessment", the eligibility of which has yet to be determined and the impact of which on the results of the review is uncertain.
It is important to bear in mind that the majority of the included studies were funded by the pharmaceutical industry;Hamilton 1960.
To assess efficacy, we used rating scales administered by clinicians or expert assessors. Even though both the Hamilton rating scale for depression (HDRS) and Montgomery and Asberg Depression Rating Scale (MADRS) are standardized tools commonly used in antidepressant trials, they are all potentially prone to observer bias. Self‐administered questionnaires were not used in the included studies, so this might represent a limitation of the methods used in the primary studies of the present review.
In this review we decided to consider the response rate as the primary outcome because it is one of the main goals for the treatment of major depressive disorder. The term “treatment response” describes a state of improvement in the patient’s condition of sufficient quality to result in the treating physician’s impression of at least a moderate degree of global improvement, conventionally defined as a reduction of at least 50% in depressive symptomatology. However, from a clinical point of view, the ultimate goal of the acute treatment phase of major depressive disorder may well be to achieve remission. Full remission from depression correlates with better longer‐term functional recovery, lower risk of relapse and higher level of patients satisfaction than partial response (without remission). Thus, one important limitation of the included trials (and consequently of the present review) is that only a few studies reported remission rates, underpowering the analysis and undermining the possibility to find significant differences between comparisons. Moreover, outcomes of clear relevance to patients and clinicians, in particular, patients’ and their carers’ attitudes to treatment, and the patient's ability to return to work and resume normal social functioning, were not reported in the included studies.
Agreements and disagreements with other studies or reviews
Even though it is a matter of ongoing discussion in the scientific literature (Gartlehner 2011), there is now robust evidence that there are statistically and clinically significant differences among antidepressants (Cipriani 2009). Results from this review suggest some differences in terms of efficacy, acceptability and tolerability between paroxetine and certain ADs, and might contribute to developing and keeping up to date an evidence‐based hierarchy of antidepressants to be used by clinicians (both specialists and general practitioners) when prescribing an antidepressant drug for moderate to severe acute major depression.
Authors' conclusions
Implications for practice.
The effects we have found in terms of efficacy, acceptability and tolerability of paroxetine compared with certain antidepressants (ADs), are of generally moderate quality. Data from the present review suggest some possible differences between paroxetine and other ADs, but the clinical meaning of these differences is uncertain, and no definitive implications for clinical practice can be drawn. Considering the methodological limitation of standard systematic reviews that rely only on evidence from direct comparisons and given the wide spectrum of available comparisons for the treatment of major depression, the use of the methodology of multiple treatments meta‐analysis (MTM) may provide a more informative and clinically useful summary of the results that can be used to guide treatment decisions.
Implications for research.
Results described in this systematic review come from evidence assessed as of low or moderate quality according to the GRADE methodology, a tool providing outcome‐specific information concerning the overall quality of evidence from each included study in the comparison and the magnitude of effect of the interventions examined. Moreover, in many cases studies were financially supported by pharmaceutical industries. Industry‐sponsored trials tend to follow a standard design which involves short‐term, double‐blind, parallel‐group studies of patients with acute episodes or exacerbations of chronic illness. Often, patients with medical and psychiatric comorbidity or taking concomitant medication are excluded. Moreover, it is known that economic support by drug manufacturers can strongly influence progress of research and its results. Consequently, there is a risk that these studies do not provide sufficient and adequate information to clinicians in real‐world settings. Studies should be conducted following high methodological standards and with the primary intent of providing clinicians with useful practical data regarding the comparative effectiveness of marketed medications, and consider rating scales but also pragmatic outcome measures (for example hospitalisations, return to work, social functioning and so on). If not, there is a risk that research will be guided only by economic interests instead of being based on clinical grounds.
Moreover, when dealing with summary statistics, the quality and the completeness of information is important. Meta‐analyses of poor quality studies may be seriously misleading (Savović 2012), because the bias associated with defects in the conduct of primary studies (randomised trials) can seriously affect the overall estimate of intervention. Systematic review authors (not only within The Cochrane Collaboration) should routinely assess the risk of bias in the results of trials, and should report meta‐analyses restricted to trials at low risk of bias (Wood 2008).
Feedback
June 2007
Summary
Feedback 1. The protocol does not refer to dosage change in the studies that will be included, or how data on the effects of dosage change and its timing will be analysed. A major problem with paroxetine are adverse effects early during treatment and withdrawal symptoms with reduction of dosage, omission of doses, and cessation of treatment. Many RCTs have not examined these aspects, and the review must state how far they have done so.
Feedback 2. Because RCTs are not the main source of information about harmful effects it is important to look at other types of study, including caseseries, in reviewing the evidence. The methods of reviewing adverse effects are discussed in detail in the Handbook for Reviews of Interventions, Appendix 6b (which will be updated and incorporated in theforthcoming 5th edition in the body of the Handbook).
Feedback 3. The evidence that treatment with paroxetine and other SSRIs or withdrawal from it can cause violent behaviour, albeit rarely, should also be considered in the protocol. It has been summarised by Healy, Herxheimer and Menkes. Antidepressants and violence: problems at the interface of medicine and law. PLoS Medicine 2006; 3(9):e372
Reply
Feedback 1. We agree with Dr. Herxheimer that all issues relating to dose change are really important clinical questions. Unfortunately, most RCTs have not reported anything about that. However, in order to be able to show all available information about safety/tolerability issues, in the protocol we stated that that "in order not to miss any relatively rare or unexpected yet important side effects, in the extraction phase, we will collect all side effects data reported in literature and will discuss ways to summarize them post hoc."
Feedback 2. RCTs are not the main source of information especially about side effects, because rare adverse events are unlikely to be observed in clinical trials. Furthermore, a through investigation should require the inclusion of observational evidence (cohort studies, case control and even case series). However, the main aim of the present review is to compare head‐to‐head paroxetine with other active treatments for depression. Thus, we decided to focus only on randomised evidence to reduce the risk of selection bias.
Feedback 3. This is an very interesting paper. We were aware of this issue (and other similar issues) related to tolerability profile of antidepressant drugs (and especially paroxetine). This is the reason why we reported in the protocol that we are going to collect and report in the full text review all available information "about rare or unexpected yet important side effects."
Contributors
Andrew Herxheimer Submitter agrees with default conflict of interest statement: I certify that I have no affiliations with or involvement in any organization or entity with a financial interest in the subject matter of my feedback.
What's new
| Date | Event | Description |
|---|---|---|
| 2 November 2008 | Amended | Converted to new review format. |
Notes
This review is one of a number of separate reviews examining head‐to‐head comparisons as part of the multiple Meta‐Analyses of New Generation Antidepressants (MANGA) Study . We have up to now completed individual reviews about sertraline (Cipriani 2009a), escitalopram (Cipriani 2009b), milnacipran (Nakagawa 2009), fluvoxamine (Omori 2010), mirtazapine (Watanabe 2011), duloxetine (Cipriani 2012a), citalopram (Cipriani 2012b), fluoxetine (Magni 2013) and a number of other reviews are now underway.
Acknowledgements
We would like to thank the CCDAN Editorial Team for their support, information and advice.
We also would like to thank all authors that provided additional data to be used in the present report and especially Drs. Jonathan Davidson, Stuart Montgomery and David Baldwin.
CRG Funding Acknowledgement The National Institute for Health Research (NIHR) is the largest single funder of the Cochrane Depression, Anxiety and Neurosis Group.
Disclaimer The views and opinions expressed therein are those of the review authors and do not necessarily reflect those of the NIHR, NHS or the Department of Health.
Data and analyses
Comparison 1. Failure to respond at endpoint (6 ‐ 12 weeks).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 34 | 4647 | Odds Ratio (M‐H, Random, 95% CI) | 1.04 [0.92, 1.17] |
| 1.1 versus amitriptyline | 13 | 1671 | Odds Ratio (M‐H, Random, 95% CI) | 1.12 [0.92, 1.37] |
| 1.2 versus clomipramine | 4 | 1273 | Odds Ratio (M‐H, Random, 95% CI) | 0.92 [0.73, 1.15] |
| 1.3 versus desipramine | 1 | 57 | Odds Ratio (M‐H, Random, 95% CI) | 1.90 [0.66, 5.46] |
| 1.4 versus dothiepin | 1 | 134 | Odds Ratio (M‐H, Random, 95% CI) | 1.43 [0.73, 2.83] |
| 1.5 versus imipramine | 6 | 587 | Odds Ratio (M‐H, Random, 95% CI) | 0.84 [0.57, 1.24] |
| 1.6 versus lofepramine | 2 | 228 | Odds Ratio (M‐H, Random, 95% CI) | 0.79 [0.47, 1.34] |
| 1.7 versus maprolitine | 3 | 429 | Odds Ratio (M‐H, Random, 95% CI) | 1.24 [0.84, 1.82] |
| 1.8 versus mianserin | 4 | 268 | Odds Ratio (M‐H, Random, 95% CI) | 1.30 [0.56, 3.05] |
| 2 Paroxetine versus other SSRIs | 17 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus citalopram | 1 | 406 | Odds Ratio (M‐H, Random, 95% CI) | 1.54 [1.04, 2.28] |
| 2.2 versus escitalopram | 2 | 784 | Odds Ratio (M‐H, Random, 95% CI) | 1.12 [0.76, 1.65] |
| 2.3 versus fluoxetine | 10 | 2353 | Odds Ratio (M‐H, Random, 95% CI) | 0.98 [0.77, 1.24] |
| 2.4 versus fluvoxamine | 3 | 261 | Odds Ratio (M‐H, Random, 95% CI) | 1.19 [0.72, 1.94] |
| 2.5 versus sertraline | 2 | 545 | Odds Ratio (M‐H, Random, 95% CI) | 1.21 [0.85, 1.73] |
| 3 Paroxetine versus newer or non‐conventional ADs | 27 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus agomelatine | 1 | 284 | Odds Ratio (M‐H, Random, 95% CI) | 1.25 [0.78, 2.01] |
| 3.2 versus amisulpride | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 0.65 [0.36, 1.15] |
| 3.3 versus bupropion | 1 | 100 | Odds Ratio (M‐H, Random, 95% CI) | 0.73 [0.30, 1.79] |
| 3.4 versus duloxetine | 6 | 1821 | Odds Ratio (M‐H, Random, 95% CI) | 0.89 [0.70, 1.14] |
| 3.5 versus hypericum | 1 | 251 | Odds Ratio (M‐H, Random, 95% CI) | 1.60 [0.95, 2.69] |
| 3.6 versus milnacipran | 1 | 302 | Odds Ratio (M‐H, Random, 95% CI) | 0.93 [0.59, 1.47] |
| 3.7 versus mirtazapine | 4 | 766 | Odds Ratio (M‐H, Random, 95% CI) | 1.20 [0.90, 1.61] |
| 3.8 versus nefazodone | 1 | 40 | Odds Ratio (M‐H, Random, 95% CI) | 0.31 [0.07, 1.25] |
| 3.9 versus reboxetine | 3 | 1369 | Odds Ratio (M‐H, Random, 95% CI) | 0.82 [0.66, 1.02] |
| 3.10 versus tianeptine | 3 | 648 | Odds Ratio (M‐H, Random, 95% CI) | 1.04 [0.71, 1.53] |
| 3.11 versus trazodone | 1 | 108 | Odds Ratio (M‐H, Random, 95% CI) | 0.71 [0.21, 2.41] |
| 3.12 versus venlafaxine | 4 | 747 | Odds Ratio (M‐H, Random, 95% CI) | 1.10 [0.58, 2.11] |
1.2. Analysis.
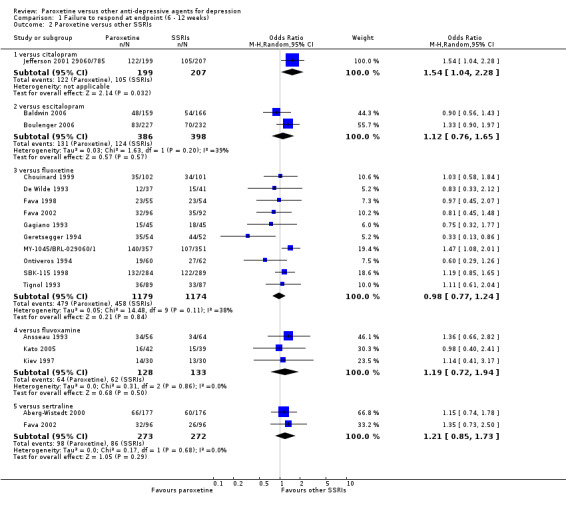
Comparison 1 Failure to respond at endpoint (6 ‐ 12 weeks), Outcome 2 Paroxetine versus other SSRIs.
Comparison 2. Failure to respond (at 1 ‐ 4 weeks).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 4 | 526 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.61, 1.33] |
| 1.1 versus amitriptyline | 1 | 153 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.52, 2.15] |
| 1.2 versus clomipramine | 1 | 92 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.58 [0.25, 1.37] |
| 1.3 versus imipramine | 1 | 159 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.43 [0.61, 3.33] |
| 1.4 versus lofepramine | 1 | 122 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.74 [0.35, 1.55] |
| 2 Paroxetine versus SSRIs | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 versus fluvoxamine | 3 | 281 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.71 [0.41, 1.24] |
| 3 Paroxetine versus newer or non‐conventional ADs | 7 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus mirtazapine | 3 | 726 | Odds Ratio (M‐H, Random, 95% CI) | 2.39 [1.42, 4.02] |
| 3.2 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 0.66 [0.50, 0.87] |
| 3.3 versus trazodone | 1 | 108 | Odds Ratio (M‐H, Random, 95% CI) | 0.54 [0.25, 1.19] |
Comparison 3. Failure to respond (at 16 ‐ 24 weeks).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus SSRIs | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 versus escitalopram | 1 | 459 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.36 [0.87, 2.12] |
| 2 Paroxetine versus newer or non‐conventional ADs | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus mirtazapine | 1 | 197 | Odds Ratio (M‐H, Random, 95% CI) | 1.41 [0.81, 2.48] |
| 2.2 versus nefazodone | 1 | 40 | Odds Ratio (M‐H, Random, 95% CI) | 0.64 [0.17, 2.38] |
Comparison 4. Failure to remit at endpoint (6 ‐ 12 weeks).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 3 | 401 | Odds Ratio (M‐H, Random, 95% CI) | 1.23 [0.49, 3.07] |
| 1.1 versus clomipramine | 1 | 120 | Odds Ratio (M‐H, Random, 95% CI) | 3.39 [1.50, 7.65] |
| 1.2 versus imipramine | 1 | 159 | Odds Ratio (M‐H, Random, 95% CI) | 0.75 [0.39, 1.43] |
| 1.3 versus lofepramine | 1 | 122 | Odds Ratio (M‐H, Random, 95% CI) | 0.80 [0.38, 1.66] |
| 2 Paroxetine versus other SSRIs | 7 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus escitalopram | 2 | 784 | Odds Ratio (M‐H, Random, 95% CI) | 1.15 [0.60, 2.22] |
| 2.2 versus fluvoxamine | 3 | 261 | Odds Ratio (M‐H, Random, 95% CI) | 0.78 [0.44, 1.38] |
| 2.3 versus sertraline | 2 | 545 | Odds Ratio (M‐H, Random, 95% CI) | 0.94 [0.67, 1.34] |
| 3 Paroxetine versus newer or non‐conventional ADs | 20 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus agomelatine | 1 | 284 | Odds Ratio (M‐H, Random, 95% CI) | 1.27 [0.75, 2.14] |
| 3.2 versus amisulpride | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 0.85 [0.49, 1.46] |
| 3.3 versus duloxetine | 6 | 1821 | Odds Ratio (M‐H, Random, 95% CI) | 0.98 [0.80, 1.19] |
| 3.4 versus hypericum | 1 | 251 | Odds Ratio (M‐H, Random, 95% CI) | 1.84 [1.11, 3.06] |
| 3.5 versus milnacipran | 1 | 302 | Odds Ratio (M‐H, Random, 95% CI) | 0.92 [0.57, 1.49] |
| 3.6 versus mirtazapine | 4 | 766 | Odds Ratio (M‐H, Random, 95% CI) | 1.52 [1.13, 2.06] |
| 3.7 versus tianeptine | 1 | 44 | Odds Ratio (M‐H, Random, 95% CI) | 0.66 [0.18, 2.36] |
| 3.8 versus trazodone | 1 | 108 | Odds Ratio (M‐H, Random, 95% CI) | 1.06 [0.47, 2.38] |
| 3.9 versus venlafaxine | 4 | 807 | Odds Ratio (M‐H, Random, 95% CI) | 1.57 [1.08, 2.29] |
Comparison 5. Failure to remit (at 1 ‐ 4 weeks).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 2 | 279 | Odds Ratio (M‐H, Random, 95% CI) | 2.38 [0.38, 14.85] |
| 1.1 versus clomipramine | 1 | 120 | Odds Ratio (M‐H, Random, 95% CI) | 10.32 [0.54, 196.06] |
| 1.2 versus imipramine | 1 | 159 | Odds Ratio (M‐H, Random, 95% CI) | 1.35 [0.45, 4.09] |
| 2 Paroxetine versus other SSRIs | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluvoxamine | 3 | 268 | Odds Ratio (M‐H, Random, 95% CI) | 1.17 [0.41, 3.35] |
| 3 Paroxetine versus newer or non‐conventional ADs | 5 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus milnacipran | 1 | 41 | Odds Ratio (M‐H, Random, 95% CI) | 0.62 [0.18, 2.13] |
| 3.2 versus mirtazapine | 3 | 726 | Odds Ratio (M‐H, Random, 95% CI) | 2.31 [1.04, 5.11] |
| 3.3 versus trazodone | 1 | 108 | Odds Ratio (M‐H, Random, 95% CI) | 0.68 [0.27, 1.73] |
Comparison 6. Failure to remit (at 16 ‐ 24 weeks).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus newer or non‐conventional ADs | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus mirtazapine | 1 | 197 | Odds Ratio (M‐H, Random, 95% CI) | 1.89 [1.01, 3.54] |
| 1.2 versus trazodone | 1 | 108 | Odds Ratio (M‐H, Random, 95% CI) | 1.06 [0.47, 2.38] |
Comparison 7. Standardized mean difference at endpoint (6 ‐ 12 weeks).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 34 | 4712 | Std. Mean Difference (IV, Random, 95% CI) | 0.01 [‐0.08, 0.10] |
| 1.1 versus amitriptyline | 13 | 1919 | Std. Mean Difference (IV, Random, 95% CI) | 0.07 [‐0.06, 0.20] |
| 1.2 versus clomipramine | 5 | 1242 | Std. Mean Difference (IV, Random, 95% CI) | 0.14 [‐0.21, 0.49] |
| 1.3 versus doxepin | 2 | 306 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.12 [‐0.35, 0.10] |
| 1.4 versus imipramine | 7 | 572 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.10 [‐0.37, 0.17] |
| 1.5 versus lofepramine | 1 | 92 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.06 [‐0.47, 0.35] |
| 1.6 versus maprolitine | 2 | 336 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.01 [‐0.23, 0.20] |
| 1.7 versus nortriptyline | 1 | 56 | Std. Mean Difference (IV, Random, 95% CI) | 0.20 [‐0.32, 0.73] |
| 1.8 versus mianserin | 3 | 189 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.21 [‐0.50, 0.07] |
| 2 Paroxetine versus other SSRIs | 13 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 versus citalopram | 1 | 201 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.16 [‐0.44, 0.11] |
| 2.2 versus escitalopram | 2 | 772 | Std. Mean Difference (IV, Random, 95% CI) | 0.05 [‐0.26, 0.36] |
| 2.3 versus fluoxetine | 8 | 2044 | Std. Mean Difference (IV, Random, 95% CI) | 0.04 [‐0.05, 0.12] |
| 2.4 versus fluvoxamine | 1 | 58 | Std. Mean Difference (IV, Random, 95% CI) | 0.09 [‐0.43, 0.60] |
| 2.5 versus sertraline | 1 | 353 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.13 [‐0.34, 0.07] |
| 3 Paroxetine versus newer or non‐conventional ADs | 27 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 versus agomelatine | 4 | 1074 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.18 [‐0.38, 0.02] |
| 3.2 versus amisulpride | 1 | 272 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.13 [‐0.37, 0.10] |
| 3.3 versus aprepitant (MK‐869) | 1 | 102 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.00 [‐0.39, 0.39] |
| 3.4 versus bupropion | 1 | 132 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.04 [‐0.38, 0.30] |
| 3.5 versus duloxetine | 6 | 1481 | Std. Mean Difference (IV, Random, 95% CI) | 0.04 [‐0.06, 0.15] |
| 3.6 versus hypericum | 1 | 244 | Std. Mean Difference (IV, Random, 95% CI) | 0.35 [0.09, 0.60] |
| 3.7 versus milnacipran | 1 | 299 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.05 [‐0.28, 0.18] |
| 3.8 versus mirtazapine | 1 | 246 | Std. Mean Difference (IV, Random, 95% CI) | 0.33 [0.08, 0.58] |
| 3.9 versus nefazodone | 2 | 235 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.12 [‐0.37, 0.14] |
| 3.10 versus reboxetine | 3 | 1291 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.10 [‐0.21, 0.00] |
| 3.11 versus tianeptine | 3 | 586 | Std. Mean Difference (IV, Random, 95% CI) | 0.04 [‐0.12, 0.20] |
| 3.12 versus trazodone | 1 | 108 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.08 [‐0.46, 0.30] |
| 3.13 versus venlafaxine | 2 | 411 | Std. Mean Difference (IV, Random, 95% CI) | 0.07 [‐0.13, 0.26] |
Comparison 8. Standardized mean difference (at 1 ‐ 4 weeks).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 7 | 723 | Std. Mean Difference (IV, Random, 95% CI) | 0.19 [‐0.02, 0.39] |
| 1.1 versus amitriptyline | 4 | 566 | Std. Mean Difference (IV, Random, 95% CI) | 0.14 [‐0.02, 0.31] |
| 1.2 versus clomipramine | 1 | 84 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.15 [‐0.58, 0.28] |
| 1.3 versus desipramine | 1 | 50 | Std. Mean Difference (IV, Random, 95% CI) | 0.86 [0.28, 1.44] |
| 1.4 versus imipramine | 1 | 23 | Std. Mean Difference (IV, Random, 95% CI) | 0.21 [‐0.61, 1.03] |
| 2 Paroxetine versus other SSRIs | 1 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluoxetine | 1 | 132 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.03 [‐0.37, 0.32] |
| 3 Paroxetine versus newer or non‐conventional ADs | 6 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 versus aprepitant (MK‐869) | 1 | 121 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.15 [‐0.50, 0.21] |
| 3.2 versus duloxetine | 2 | 353 | Std. Mean Difference (IV, Random, 95% CI) | 0.12 [‐0.09, 0.33] |
| 3.3 versus reboxetine | 2 | 805 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.17 [‐0.31, ‐0.03] |
| 3.4 versus tianeptine | 1 | 246 | Std. Mean Difference (IV, Random, 95% CI) | 0.13 [‐0.12, 0.38] |
Comparison 9. Standardized mean difference (at 16 ‐ 24 weeks).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus SSRIs | 1 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 versus sertraline | 1 | 353 | Std. Mean Difference (IV, Random, 95% CI) | 0.01 [‐0.20, 0.22] |
| 2 Paroxetine versus newer or non‐conventional ADs | 1 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 versus mirtazapine | 1 | 177 | Std. Mean Difference (IV, Random, 95% CI) | 0.21 [‐0.09, 0.51] |
9.1. Analysis.
Comparison 9 Standardized mean difference (at 16 ‐ 24 weeks), Outcome 1 Paroxetine versus SSRIs.
Comparison 10. Failure to complete (any cause).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 43 | 6777 | Odds Ratio (M‐H, Random, 95% CI) | 0.84 [0.73, 0.96] |
| 1.1 versus amitriptyline | 19 | 2908 | Odds Ratio (M‐H, Random, 95% CI) | 0.86 [0.70, 1.06] |
| 1.2 versus clomipramine | 4 | 1273 | Odds Ratio (M‐H, Random, 95% CI) | 0.67 [0.52, 0.87] |
| 1.3 versus dothiepin | 3 | 499 | Odds Ratio (M‐H, Random, 95% CI) | 1.25 [0.84, 1.87] |
| 1.4 versus imipramine | 9 | 1268 | Odds Ratio (M‐H, Random, 95% CI) | 0.65 [0.50, 0.85] |
| 1.5 versus lofepramine | 2 | 228 | Odds Ratio (M‐H, Random, 95% CI) | 0.94 [0.31, 2.80] |
| 1.6 versus maprolitine | 3 | 429 | Odds Ratio (M‐H, Random, 95% CI) | 1.05 [0.49, 2.25] |
| 1.7 versus mianserin | 2 | 92 | Odds Ratio (M‐H, Random, 95% CI) | 0.84 [0.21, 3.41] |
| 1.8 versus nortriptyline | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 1.30 [0.50, 3.42] |
| 2 Paroxetine versus other SSRIs | 19 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus citalopram | 1 | 406 | Odds Ratio (M‐H, Random, 95% CI) | 0.99 [0.61, 1.60] |
| 2.2 versus escitalopram | 2 | 784 | Odds Ratio (M‐H, Random, 95% CI) | 1.47 [0.77, 2.79] |
| 2.3 versus fluoxetine | 12 | 2733 | Odds Ratio (M‐H, Random, 95% CI) | 1.05 [0.89, 1.24] |
| 2.4 versus fluvoxamine | 3 | 261 | Odds Ratio (M‐H, Random, 95% CI) | 0.96 [0.54, 1.72] |
| 2.5 versus sertraline | 2 | 426 | Odds Ratio (M‐H, Random, 95% CI) | 1.37 [0.52, 3.60] |
| 3 Paroxetine versus newer or non‐conventional ADs | 29 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus agomelatine | 1 | 284 | Odds Ratio (M‐H, Random, 95% CI) | 0.99 [0.57, 1.71] |
| 3.2 versus aprepitant (MK‐869) | 1 | 143 | Odds Ratio (M‐H, Random, 95% CI) | 1.53 [0.76, 3.09] |
| 3.3 versus bupropion | 2 | 240 | Odds Ratio (M‐H, Random, 95% CI) | 1.16 [0.62, 2.20] |
| 3.4 versus duloxetine | 6 | 1821 | Odds Ratio (M‐H, Random, 95% CI) | 0.97 [0.77, 1.23] |
| 3.5 versus hypericum | 1 | 251 | Odds Ratio (M‐H, Random, 95% CI) | 1.90 [0.98, 3.67] |
| 3.6 versus mirtazapine | 3 | 726 | Odds Ratio (M‐H, Random, 95% CI) | 1.26 [0.91, 1.74] |
| 3.7 versus milnacipram | 2 | 343 | Odds Ratio (M‐H, Random, 95% CI) | 1.17 [0.67, 2.03] |
| 3.8 versus nefazodone | 1 | 40 | Odds Ratio (M‐H, Random, 95% CI) | 0.53 [0.11, 2.60] |
| 3.9 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 0.78 [0.51, 1.19] |
| 3.10 versus tianeptine | 3 | 648 | Odds Ratio (M‐H, Random, 95% CI) | 1.46 [0.75, 2.85] |
| 3.11 versus venlafaxine | 6 | 1079 | Odds Ratio (M‐H, Random, 95% CI) | 1.08 [0.82, 1.44] |
Comparison 11. Failure to complete (due to inefficacy).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 27 | 4436 | Odds Ratio (M‐H, Random, 95% CI) | 1.22 [0.93, 1.61] |
| 1.1 versus amitriptyline | 9 | 1191 | Odds Ratio (M‐H, Random, 95% CI) | 1.20 [0.77, 1.86] |
| 1.2 versus clomipramine | 4 | 1273 | Odds Ratio (M‐H, Random, 95% CI) | 1.50 [0.76, 2.97] |
| 1.3 versus dothiepin | 2 | 405 | Odds Ratio (M‐H, Random, 95% CI) | 0.59 [0.18, 1.96] |
| 1.4 versus imipramine | 9 | 1268 | Odds Ratio (M‐H, Random, 95% CI) | 1.27 [0.80, 2.02] |
| 1.5 versus lofepramine | 2 | 228 | Odds Ratio (M‐H, Random, 95% CI) | 0.87 [0.13, 6.08] |
| 1.6 versus maprolitine | 1 | 71 | Odds Ratio (M‐H, Random, 95% CI) | 2.84 [0.11, 71.99] |
| 2 Paroxetine versus other SSRIs | 13 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus citalopram | 1 | 406 | Odds Ratio (M‐H, Random, 95% CI) | 1.75 [0.41, 7.43] |
| 2.2 versus escitalopram | 2 | 784 | Odds Ratio (M‐H, Random, 95% CI) | 0.72 [0.09, 5.96] |
| 2.3 versus fluoxetine | 8 | 2214 | Odds Ratio (M‐H, Random, 95% CI) | 0.91 [0.60, 1.39] |
| 2.4 versus fluvoxamine | 2 | 180 | Odds Ratio (M‐H, Random, 95% CI) | 1.00 [0.15, 6.93] |
| 2.5 versus sertraline | 1 | 192 | Odds Ratio (M‐H, Random, 95% CI) | 0.53 [0.17, 1.65] |
| 3 Paroxetine versus newer or non‐conventional ADs | 19 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus agomelatine | 1 | 284 | Odds Ratio (M‐H, Random, 95% CI) | 1.15 [0.46, 2.87] |
| 3.2 versus aprepitant (MK‐869) | 1 | 143 | Odds Ratio (M‐H, Random, 95% CI) | 0.32 [0.01, 8.09] |
| 3.3 versus duloxetine | 5 | 1557 | Odds Ratio (M‐H, Random, 95% CI) | 1.05 [0.55, 2.00] |
| 3.4 versus hypericum | 1 | 251 | Odds Ratio (M‐H, Random, 95% CI) | 0.48 [0.12, 1.98] |
| 3.5 versus milnacipran | 1 | 302 | Odds Ratio (M‐H, Random, 95% CI) | 0.63 [0.22, 1.83] |
| 3.6 versus mirtazapine | 2 | 451 | Odds Ratio (M‐H, Random, 95% CI) | 0.41 [0.04, 4.58] |
| 3.7 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 0.66 [0.20, 2.16] |
| 3.8 versus tianeptine | 2 | 371 | Odds Ratio (M‐H, Random, 95% CI) | 1.22 [0.05, 27.06] |
| 3.9 versus venlafaxine | 3 | 774 | Odds Ratio (M‐H, Random, 95% CI) | 0.75 [0.33, 1.66] |
Comparison 12. Failure to complete (due to side effects).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 34 | 5175 | Odds Ratio (M‐H, Random, 95% CI) | 0.76 [0.63, 0.92] |
| 1.1 versus amitriptyline | 12 | 1698 | Odds Ratio (M‐H, Random, 95% CI) | 0.74 [0.56, 0.98] |
| 1.2 versus clomipramine | 4 | 1273 | Odds Ratio (M‐H, Random, 95% CI) | 0.59 [0.41, 0.84] |
| 1.3 versus dothiepin | 2 | 405 | Odds Ratio (M‐H, Random, 95% CI) | 1.70 [0.78, 3.70] |
| 1.4 versus imipramine | 9 | 1268 | Odds Ratio (M‐H, Random, 95% CI) | 0.58 [0.43, 0.77] |
| 1.5 versus lofepramine | 2 | 228 | Odds Ratio (M‐H, Random, 95% CI) | 1.19 [0.45, 3.15] |
| 1.6 versus maprolitine | 2 | 131 | Odds Ratio (M‐H, Random, 95% CI) | 0.77 [0.01, 70.98] |
| 1.7 versus mianserin | 2 | 92 | Odds Ratio (M‐H, Random, 95% CI) | 2.26 [0.38, 13.50] |
| 1.8 versus nortriptyline | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 1.46 [0.43, 4.93] |
| 2 Paroxetine versus other SSRIs | 17 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus citalopram | 1 | 406 | Odds Ratio (M‐H, Random, 95% CI) | 1.15 [0.48, 2.78] |
| 2.2 versus escitalopram | 2 | 784 | Odds Ratio (M‐H, Random, 95% CI) | 1.43 [0.51, 4.00] |
| 2.3 versus fluoxetine | 11 | 2491 | Odds Ratio (M‐H, Random, 95% CI) | 1.34 [1.06, 1.70] |
| 2.4 versus fluvoxamine | 3 | 261 | Odds Ratio (M‐H, Random, 95% CI) | 1.16 [0.19, 7.16] |
| 2.5 versus sertraline | 1 | 192 | Odds Ratio (M‐H, Random, 95% CI) | 1.94 [0.69, 5.48] |
| 3 Paroxetine versus newer or non‐conventional ADs | 24 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus agomelatine | 1 | 284 | Odds Ratio (M‐H, Random, 95% CI) | 0.84 [0.34, 2.04] |
| 3.2 versus aprepitant (MK‐869) | 1 | 143 | Odds Ratio (M‐H, Random, 95% CI) | 1.66 [0.67, 4.13] |
| 3.3 versus bupropion | 2 | 240 | Odds Ratio (M‐H, Random, 95% CI) | 0.61 [0.24, 1.55] |
| 3.4 versus duloxetine | 6 | 1821 | Odds Ratio (M‐H, Random, 95% CI) | 0.84 [0.57, 1.25] |
| 3.5 versus hypericum | 1 | 251 | Odds Ratio (M‐H, Random, 95% CI) | 2.05 [0.60, 6.99] |
| 3.6 versus milnacipran | 1 | 302 | Odds Ratio (M‐H, Random, 95% CI) | 1.17 [0.59, 2.33] |
| 3.7 versus mirtazapine | 3 | 726 | Odds Ratio (M‐H, Random, 95% CI) | 1.35 [0.83, 2.21] |
| 3.8 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 0.38 [0.17, 0.86] |
| 3.9 versus tianeptine | 1 | 327 | Odds Ratio (M‐H, Random, 95% CI) | 3.38 [1.31, 8.71] |
| 3.10 versus venlafaxine | 5 | 974 | Odds Ratio (M‐H, Random, 95% CI) | 0.88 [0.50, 1.56] |
Comparison 13. SE ‐ Participants with at least one TEAE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 41 | 6099 | Odds Ratio (M‐H, Random, 95% CI) | 0.64 [0.53, 0.77] |
| 1.1 versus amitriptyline | 16 | 2492 | Odds Ratio (M‐H, Random, 95% CI) | 0.53 [0.39, 0.72] |
| 1.2 versus clomipramine | 4 | 1273 | Odds Ratio (M‐H, Random, 95% CI) | 0.56 [0.28, 1.10] |
| 1.3 versus dothiepin | 2 | 405 | Odds Ratio (M‐H, Random, 95% CI) | 1.04 [0.61, 1.76] |
| 1.4 versus imipramine | 9 | 1189 | Odds Ratio (M‐H, Random, 95% CI) | 0.62 [0.42, 0.94] |
| 1.5 versus lofepramine | 2 | 228 | Odds Ratio (M‐H, Random, 95% CI) | 1.25 [0.74, 2.12] |
| 1.6 versus maprolitine | 2 | 131 | Odds Ratio (M‐H, Random, 95% CI) | 0.66 [0.28, 1.55] |
| 1.7 versus mianserin | 5 | 301 | Odds Ratio (M‐H, Random, 95% CI) | 0.71 [0.41, 1.22] |
| 1.8 versus nortriptyline | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 1.46 [0.43, 4.93] |
| 2 Paroxetine versus other SSRIs | 14 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus citalopram | 1 | 406 | Odds Ratio (M‐H, Random, 95% CI) | 0.74 [0.46, 1.21] |
| 2.2 versus escitalopram | 1 | 454 | Odds Ratio (M‐H, Random, 95% CI) | 1.28 [0.86, 1.91] |
| 2.3 versus fluoxetine | 9 | 2255 | Odds Ratio (M‐H, Random, 95% CI) | 0.94 [0.69, 1.28] |
| 2.4 versus fluvoxamine | 3 | 261 | Odds Ratio (M‐H, Random, 95% CI) | 1.12 [0.42, 2.97] |
| 3 Paroxetine versus newer or non‐conventional ADs | 23 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus agomelatine | 1 | 284 | Odds Ratio (M‐H, Random, 95% CI) | 1.60 [0.99, 2.59] |
| 3.2 versus amisulpride | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 1.49 [0.89, 2.50] |
| 3.3 versus bupropion | 1 | 140 | Odds Ratio (M‐H, Random, 95% CI) | 1.60 [0.43, 5.92] |
| 3.4 versus duloxetine | 6 | 1870 | Odds Ratio (M‐H, Random, 95% CI) | 0.81 [0.64, 1.01] |
| 3.5 versus hypericum | 1 | 251 | Odds Ratio (M‐H, Random, 95% CI) | 2.60 [1.51, 4.46] |
| 3.6 versus milnacipran | 1 | 302 | Odds Ratio (M‐H, Random, 95% CI) | 0.69 [0.41, 1.15] |
| 3.7 versus mirtazapine | 3 | 726 | Odds Ratio (M‐H, Random, 95% CI) | 1.07 [0.76, 1.50] |
| 3.8 versus nefazodone | 1 | 206 | Odds Ratio (M‐H, Random, 95% CI) | 0.69 [0.34, 1.40] |
| 3.9 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 1.08 [0.74, 1.58] |
| 3.10 versus tianeptine | 2 | 604 | Odds Ratio (M‐H, Random, 95% CI) | 1.26 [0.89, 1.78] |
| 3.11 versus trazodone | 1 | 108 | Odds Ratio (M‐H, Random, 95% CI) | 0.68 [0.30, 1.55] |
| 3.12 versus venlafaxine | 2 | 200 | Odds Ratio (M‐H, Random, 95% CI) | 1.01 [0.52, 1.95] |
Comparison 14. SE ‐ Abnormal dreams.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus maprolitine | 1 | 71 | Odds Ratio (M‐H, Random, 95% CI) | 2.84 [0.11, 71.99] |
| 2 Paroxetine versus other SSRIs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluvoxamine | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 13.16 [0.69, 249.48] |
| 3 Paroxetine versus newer or non‐conventional ADs | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus milnacipran | 1 | 302 | Odds Ratio (M‐H, Random, 95% CI) | 4.93 [0.23, 103.64] |
| 3.2 versus reboxetine | 2 | 1050 | Odds Ratio (M‐H, Random, 95% CI) | 0.99 [0.33, 2.99] |
14.1. Analysis.
Comparison 14 SE ‐ Abnormal dreams, Outcome 1 Paroxetine versus older ADs.
14.2. Analysis.
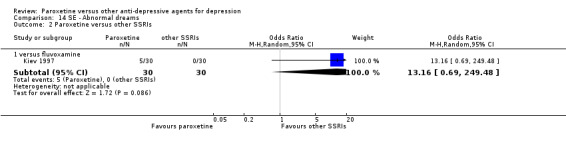
Comparison 14 SE ‐ Abnormal dreams, Outcome 2 Paroxetine versus other SSRIs.
14.3. Analysis.
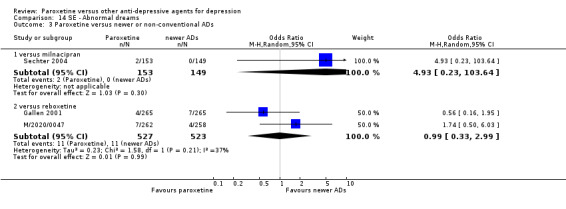
Comparison 14 SE ‐ Abnormal dreams, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 15. SE ‐ Abnormal laboratory values.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Abnormal white blood cells | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus fluoxetine | 1 | 138 | Odds Ratio (M‐H, Random, 95% CI) | 0.34 [0.01, 8.45] |
| 2 Leucocytosis | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus amitriptyline | 1 | 40 | Odds Ratio (M‐H, Random, 95% CI) | 1.0 [0.06, 17.18] |
| 3 ALT/AST increase | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus amitriptyline | 2 | 262 | Odds Ratio (M‐H, Random, 95% CI) | 2.13 [0.27, 16.88] |
| 4 NOS | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 versus clomipramine | 1 | 83 | Odds Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 8.42] |
15.1. Analysis.
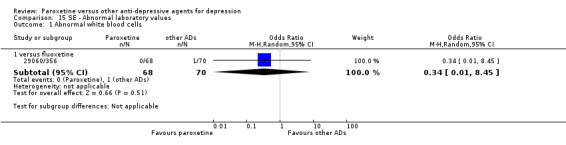
Comparison 15 SE ‐ Abnormal laboratory values, Outcome 1 Abnormal white blood cells.
15.2. Analysis.
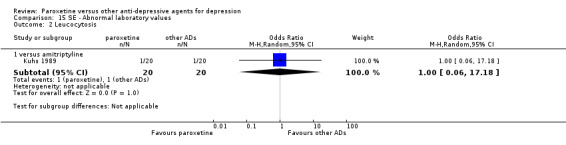
Comparison 15 SE ‐ Abnormal laboratory values, Outcome 2 Leucocytosis.
15.3. Analysis.
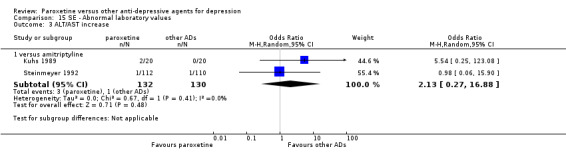
Comparison 15 SE ‐ Abnormal laboratory values, Outcome 3 ALT/AST increase.
15.4. Analysis.
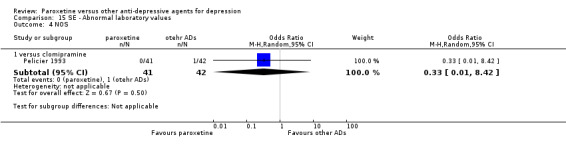
Comparison 15 SE ‐ Abnormal laboratory values, Outcome 4 NOS.
Comparison 16. SE ‐ Abnormal thinking.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus newer or non‐conventional ADs | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus duloxetine | 2 | 528 | Odds Ratio (M‐H, Random, 95% CI) | 0.93 [0.16, 5.46] |
| 1.2 versus reboxetine | 2 | 1050 | Odds Ratio (M‐H, Random, 95% CI) | 1.18 [0.06, 24.15] |
16.1. Analysis.
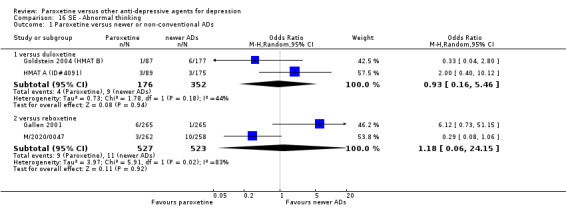
Comparison 16 SE ‐ Abnormal thinking, Outcome 1 Paroxetine versus newer or non‐conventional ADs.
Comparison 17. SE ‐ Accidental injury.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus other SSRIs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus fluoxetine | 1 | 573 | Odds Ratio (M‐H, Random, 95% CI) | 1.02 [0.49, 2.12] |
| 2 Paroxetine versus newer or non‐conventional ADs | 5 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 versus duloxetine | 2 | 528 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.37, 2.72] |
| 2.2 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.74 [0.88, 3.43] |
17.1. Analysis.
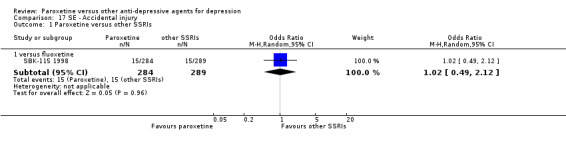
Comparison 17 SE ‐ Accidental injury, Outcome 1 Paroxetine versus other SSRIs.
17.2. Analysis.
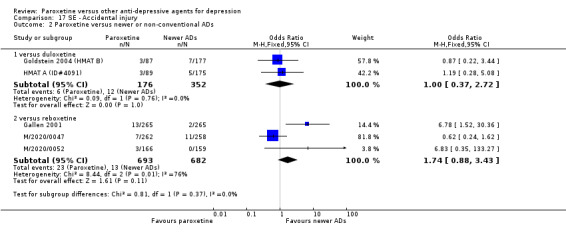
Comparison 17 SE ‐ Accidental injury, Outcome 2 Paroxetine versus newer or non‐conventional ADs.
Comparison 18. SE ‐ Agitation/anxiety.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 15 | 2240 | Odds Ratio (M‐H, Random, 95% CI) | 1.12 [0.67, 1.86] |
| 1.1 versus amitriptyline | 6 | 1001 | Odds Ratio (M‐H, Random, 95% CI) | 1.21 [0.36, 4.03] |
| 1.2 versus clomipramine | 1 | 92 | Odds Ratio (M‐H, Random, 95% CI) | 0.2 [0.01, 4.28] |
| 1.3 versus dothiepin | 1 | 271 | Odds Ratio (M‐H, Random, 95% CI) | 2.52 [0.86, 7.35] |
| 1.4 versus imipramine | 2 | 282 | Odds Ratio (M‐H, Random, 95% CI) | 0.63 [0.18, 2.16] |
| 1.5 versus lofepramine | 1 | 106 | Odds Ratio (M‐H, Random, 95% CI) | 0.28 [0.01, 7.06] |
| 1.6 versus maprolitine | 3 | 429 | Odds Ratio (M‐H, Random, 95% CI) | 0.95 [0.38, 2.39] |
| 1.7 versus mianserin | 1 | 59 | Odds Ratio (M‐H, Random, 95% CI) | 2.0 [0.17, 23.34] |
| 2 Paroxetine versus other SSRIs | 8 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluoxetine | 5 | 713 | Odds Ratio (M‐H, Random, 95% CI) | 1.15 [0.75, 1.77] |
| 2.2 versus fluvoxamine | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 2.15 [0.36, 12.76] |
| 2.3 versus sertraline | 2 | 545 | Odds Ratio (M‐H, Random, 95% CI) | 1.19 [0.66, 2.13] |
| 3 Paroxetine versus newer or non‐conventional ADs | 14 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus agomelatine | 1 | 284 | Odds Ratio (M‐H, Random, 95% CI) | 0.74 [0.19, 2.81] |
| 3.2 versus bupropion | 2 | 240 | Odds Ratio (M‐H, Random, 95% CI) | 0.45 [0.19, 1.08] |
| 3.3 versus duloxetine | 4 | 1095 | Odds Ratio (M‐H, Random, 95% CI) | 0.82 [0.35, 1.91] |
| 3.4 versus milnacipran | 1 | 302 | Odds Ratio (M‐H, Random, 95% CI) | 0.73 [0.16, 3.30] |
| 3.5 versus mirtazapine | 2 | 472 | Odds Ratio (M‐H, Random, 95% CI) | 1.40 [0.63, 3.15] |
| 3.6 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 0.74 [0.47, 1.17] |
| 3.7 versus tianeptine | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 8.14] |
Comparison 19. SE ‐ Akathisia.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 versus amitriptyline | 1 | 21 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.28 [0.01, 7.57] |
| 2 Paroxetine versus newer or non‐conventional ADs | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 versus reboxetine | 2 | 1050 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.37 [0.55, 3.44] |
19.1. Analysis.
Comparison 19 SE ‐ Akathisia, Outcome 1 Paroxetine versus older ADs.
19.2. Analysis.
Comparison 19 SE ‐ Akathisia, Outcome 2 Paroxetine versus newer or non‐conventional ADs.
Comparison 20. SE ‐ Anorexia.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus other SSRIs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus fluoxetine | 1 | 138 | Odds Ratio (M‐H, Random, 95% CI) | 3.90 [0.78, 19.50] |
| 2 Paroxetine versus newer or non‐conventional ADs | 10 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus aprepitant (MK‐869) | 1 | 143 | Odds Ratio (M‐H, Random, 95% CI) | 2.83 [0.72, 11.15] |
| 2.2 versus bupropion | 1 | 100 | Odds Ratio (M‐H, Random, 95% CI) | 6.13 [0.71, 52.93] |
| 2.3 versus duloxetine | 4 | 1299 | Odds Ratio (M‐H, Random, 95% CI) | 0.64 [0.39, 1.05] |
| 2.4 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 0.71 [0.47, 1.08] |
| 2.5 versus tianeptine | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 8.14] |
20.1. Analysis.
Comparison 20 SE ‐ Anorexia, Outcome 1 Paroxetine versus other SSRIs.
20.2. Analysis.
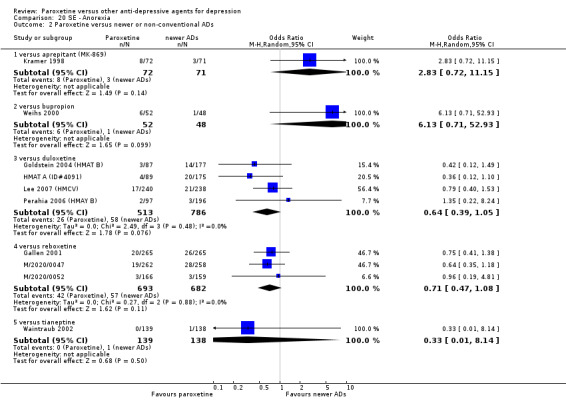
Comparison 20 SE ‐ Anorexia, Outcome 2 Paroxetine versus newer or non‐conventional ADs.
Comparison 21. SE ‐ Anticholinergic.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 paroxetine versus older ADs | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 versus amitriptyline | 2 | 130 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.22 [0.07, 0.76] |
| 1.2 versus clomipramine | 1 | 79 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.30 [0.11, 0.86] |
| 2 paroxerine versus other SSRIs | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 versus fluoxetine | 2 | 296 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.79 [0.31, 2.05] |
21.1. Analysis.
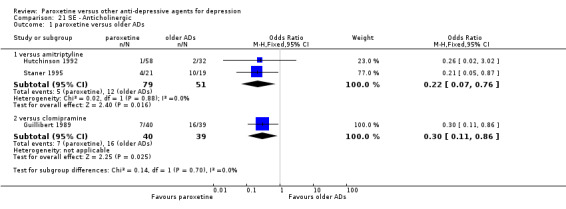
Comparison 21 SE ‐ Anticholinergic, Outcome 1 paroxetine versus older ADs.
21.2. Analysis.
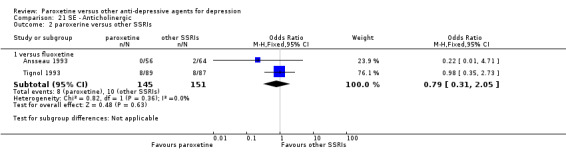
Comparison 21 SE ‐ Anticholinergic, Outcome 2 paroxerine versus other SSRIs.
Comparison 22. SE ‐ Appetite decreased.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 4 | 685 | Odds Ratio (M‐H, Random, 95% CI) | 4.00 [1.65, 9.71] |
| 1.1 versus amitriptyline | 1 | 26 | Odds Ratio (M‐H, Random, 95% CI) | 12.79 [0.61, 266.66] |
| 1.2 versus imipramine | 2 | 361 | Odds Ratio (M‐H, Random, 95% CI) | 2.45 [0.76, 7.90] |
| 1.3 versus maprotiline | 1 | 298 | Odds Ratio (M‐H, Random, 95% CI) | 6.81 [1.50, 30.99] |
| 2 Paroxetine versus other SSRIs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluoxetine | 1 | 122 | Odds Ratio (M‐H, Random, 95% CI) | 0.58 [0.20, 1.70] |
| 3 Paroxetine versus newer or non‐conventional ADs | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus duloxetine | 2 | 752 | Odds Ratio (M‐H, Random, 95% CI) | 1.09 [0.59, 2.01] |
| 3.2 versus reboxetine | 1 | 325 | Odds Ratio (M‐H, Random, 95% CI) | 0.76 [0.20, 2.88] |
22.1. Analysis.
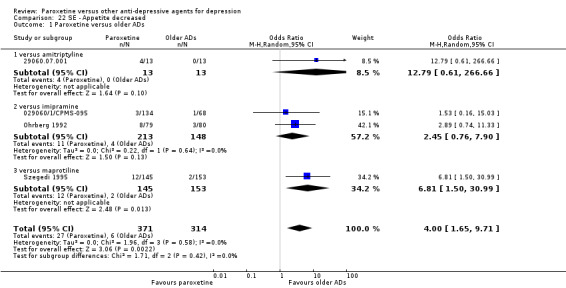
Comparison 22 SE ‐ Appetite decreased, Outcome 1 Paroxetine versus older ADs.
22.2. Analysis.
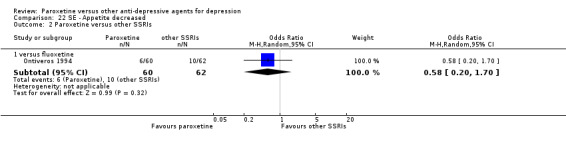
Comparison 22 SE ‐ Appetite decreased, Outcome 2 Paroxetine versus other SSRIs.
22.3. Analysis.
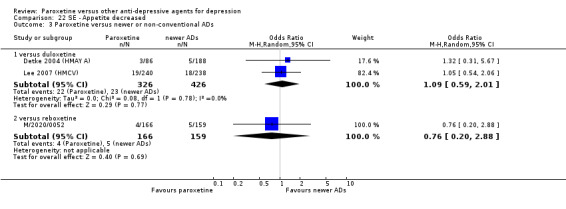
Comparison 22 SE ‐ Appetite decreased, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 23. SE ‐ Appetite increased.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus newer or non‐conventional ADs | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus bupropion | 1 | 100 | Odds Ratio (M‐H, Random, 95% CI) | 0.29 [0.03, 2.93] |
| 1.2 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 1.43 [0.22, 9.41] |
23.1. Analysis.
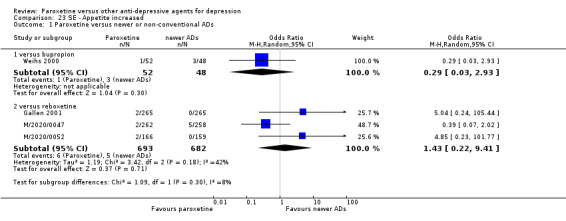
Comparison 23 SE ‐ Appetite increased, Outcome 1 Paroxetine versus newer or non‐conventional ADs.
Comparison 24. SE ‐ Asthenia.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 17 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus amitriptyline | 5 | 779 | Odds Ratio (M‐H, Random, 95% CI) | 0.44 [0.12, 1.55] |
| 1.2 versus dothiepin | 2 | 405 | Odds Ratio (M‐H, Random, 95% CI) | 1.58 [0.30, 8.47] |
| 1.3 versus imipramine | 6 | 680 | Odds Ratio (M‐H, Random, 95% CI) | 1.29 [0.73, 2.28] |
| 1.4 versus lofepramine | 1 | 122 | Odds Ratio (M‐H, Random, 95% CI) | 4.36 [0.47, 40.16] |
| 1.5 versus maprolitine | 3 | 429 | Odds Ratio (M‐H, Random, 95% CI) | 0.76 [0.49, 1.16] |
| 2 Paroxetine versus other SSRIs | 7 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus citalopram | 1 | 406 | Odds Ratio (M‐H, Random, 95% CI) | 0.59 [0.33, 1.04] |
| 2.2 versus fluoxetine | 5 | 1680 | Odds Ratio (M‐H, Random, 95% CI) | 1.27 [0.91, 1.77] |
| 2.3 versus fluvoxamine | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 0.51 [0.13, 1.95] |
| 3 Paroxetine versus newer ADs | 13 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus aprepitant (MK‐869) | 1 | 143 | Odds Ratio (M‐H, Random, 95% CI) | 1.47 [0.61, 3.58] |
| 3.2 versus bupropion | 1 | 140 | Odds Ratio (M‐H, Random, 95% CI) | 4.25 [0.87, 20.80] |
| 3.3 versus duloxetine | 3 | 1006 | Odds Ratio (M‐H, Random, 95% CI) | 0.79 [0.45, 1.38] |
| 3.4 versus nefazodone | 2 | 246 | Odds Ratio (M‐H, Random, 95% CI) | 1.15 [0.36, 3.62] |
| 3.5 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 2.07 [1.19, 3.60] |
| 3.6 versus tianeptine | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 0.99 [0.06, 16.03] |
| 3.7 versus venlafaxine | 1 | 361 | Odds Ratio (M‐H, Random, 95% CI) | 0.46 [0.17, 1.23] |
| 3.8 versus trazodone | 1 | 108 | Odds Ratio (M‐H, Random, 95% CI) | 3.17 [0.13, 79.60] |
24.1. Analysis.
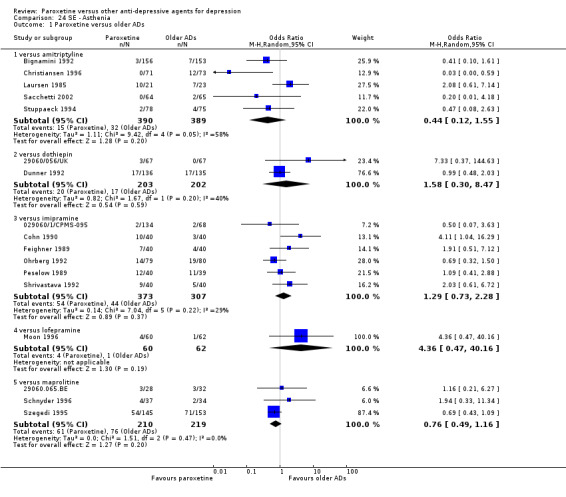
Comparison 24 SE ‐ Asthenia, Outcome 1 Paroxetine versus older ADs.
24.2. Analysis.
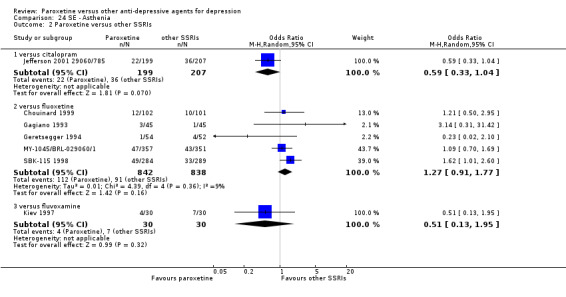
Comparison 24 SE ‐ Asthenia, Outcome 2 Paroxetine versus other SSRIs.
24.3. Analysis.
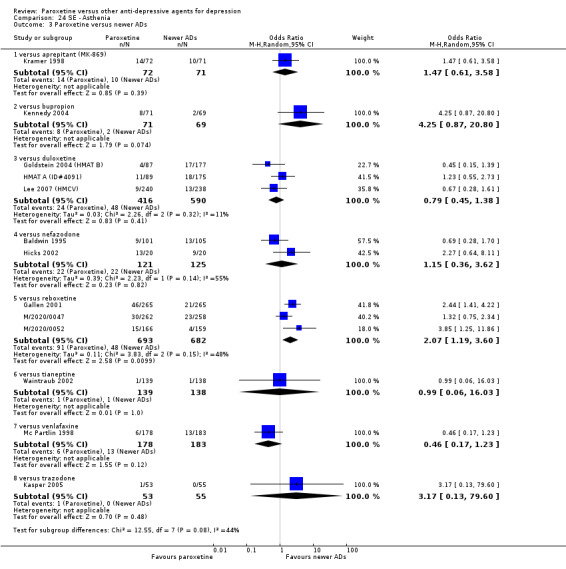
Comparison 24 SE ‐ Asthenia, Outcome 3 Paroxetine versus newer ADs.
Comparison 25. SE ‐ Behaviour.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Euphoria | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus reboxetine | 1 | 520 | Odds Ratio (M‐H, Random, 95% CI) | 0.14 [0.01, 2.71] |
| 2 Hostility | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus reboxetine | 1 | 325 | Odds Ratio (M‐H, Random, 95% CI) | 0.48 [0.04, 5.30] |
| 3 Impulsive | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus tianeptine | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 8.14] |
| 4 Irritability | 4 | 557 | Odds Ratio (M‐H, Random, 95% CI) | 1.34 [0.15, 12.23] |
| 4.1 versus amitriptyline | 1 | 21 | Odds Ratio (M‐H, Random, 95% CI) | 15.75 [1.42, 174.25] |
| 4.2 versus aprepitant (MK‐869) | 1 | 143 | Odds Ratio (M‐H, Random, 95% CI) | 0.19 [0.02, 1.63] |
| 4.3 versus mianserin | 1 | 116 | Odds Ratio (M‐H, Random, 95% CI) | 0.38 [0.02, 9.43] |
| 4.4 versus tianeptine | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 3.0 [0.12, 74.28] |
| 5 Paranoid reaction | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 versus maprolitine | 1 | 71 | Odds Ratio (M‐H, Random, 95% CI) | 4.86 [0.23, 104.92] |
| 6 Psychotic symptoms | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 versus fluvoxamine | 1 | 81 | Odds Ratio (M‐H, Random, 95% CI) | 9.23 [0.48, 177.35] |
25.1. Analysis.
Comparison 25 SE ‐ Behaviour, Outcome 1 Euphoria.
25.2. Analysis.
Comparison 25 SE ‐ Behaviour, Outcome 2 Hostility.
25.3. Analysis.
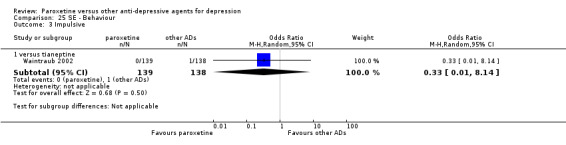
Comparison 25 SE ‐ Behaviour, Outcome 3 Impulsive.
25.4. Analysis.
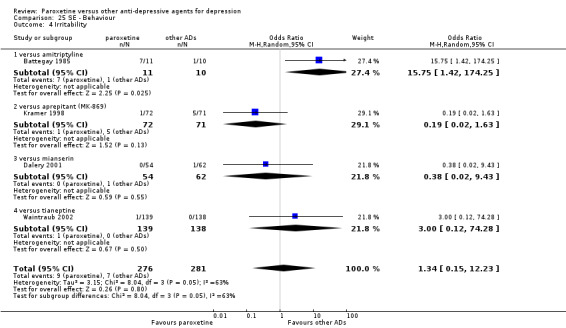
Comparison 25 SE ‐ Behaviour, Outcome 4 Irritability.
25.5. Analysis.
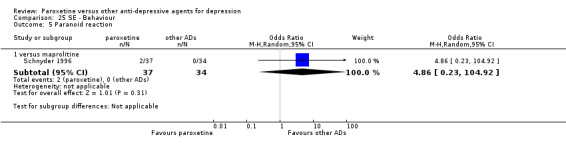
Comparison 25 SE ‐ Behaviour, Outcome 5 Paranoid reaction.
25.6. Analysis.
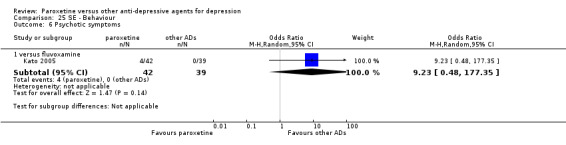
Comparison 25 SE ‐ Behaviour, Outcome 6 Psychotic symptoms.
Comparison 26. SE ‐ Body as a whole.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 6 | 463 | Odds Ratio (M‐H, Random, 95% CI) | 1.40 [0.83, 2.36] |
| 1.1 versus amitriptyline | 2 | 221 | Odds Ratio (M‐H, Random, 95% CI) | 1.87 [1.08, 3.23] |
| 1.2 versus clomipramine | 1 | 83 | Odds Ratio (M‐H, Random, 95% CI) | 0.40 [0.11, 1.41] |
| 1.3 versus mianserin | 1 | 33 | Odds Ratio (M‐H, Random, 95% CI) | 2.5 [0.41, 15.29] |
| 1.4 versus mianserin | 3 | 126 | Odds Ratio (M‐H, Random, 95% CI) | 1.29 [0.48, 3.52] |
| 2 Paroxetine versus other SSRIs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluoxetine | 1 | 242 | Odds Ratio (M‐H, Random, 95% CI) | 1.57 [0.65, 3.78] |
26.1. Analysis.
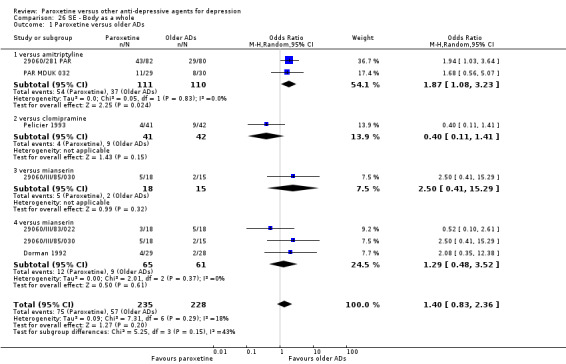
Comparison 26 SE ‐ Body as a whole, Outcome 1 Paroxetine versus older ADs.
26.2. Analysis.
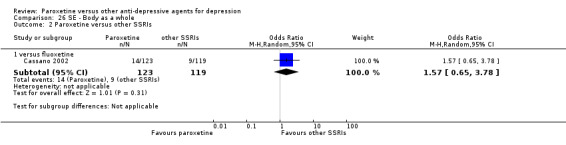
Comparison 26 SE ‐ Body as a whole, Outcome 2 Paroxetine versus other SSRIs.
Comparison 27. SE ‐ Bronchitis.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus other SSRIs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus fluoxetine | 1 | 138 | Odds Ratio (M‐H, Random, 95% CI) | 0.19 [0.02, 1.71] |
| 2 Paroxetine versus newer or non‐conventional ADs | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus reboxetine | 2 | 845 | Odds Ratio (M‐H, Random, 95% CI) | 0.42 [0.06, 2.85] |
27.1. Analysis.
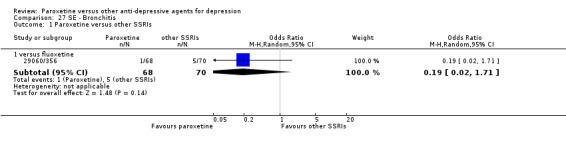
Comparison 27 SE ‐ Bronchitis, Outcome 1 Paroxetine versus other SSRIs.
27.2. Analysis.
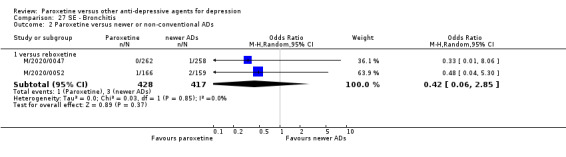
Comparison 27 SE ‐ Bronchitis, Outcome 2 Paroxetine versus newer or non‐conventional ADs.
Comparison 28. SE ‐ Cardiovascular system.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Syncope | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus maprolitine | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 1.16 [0.21, 6.27] |
| 2 NOS | 8 | 693 | Odds Ratio (M‐H, Random, 95% CI) | 0.73 [0.41, 1.31] |
| 2.1 versus amitriptyline | 3 | 242 | Odds Ratio (M‐H, Random, 95% CI) | 1.01 [0.24, 4.31] |
| 2.2 versus clomipramine | 1 | 83 | Odds Ratio (M‐H, Random, 95% CI) | 0.69 [0.20, 2.40] |
| 2.3 versus fluoxetine | 1 | 242 | Odds Ratio (M‐H, Random, 95% CI) | 0.85 [0.32, 2.28] |
| 2.4 versus mianserin | 3 | 126 | Odds Ratio (M‐H, Random, 95% CI) | 0.24 [0.05, 1.06] |
28.1. Analysis.
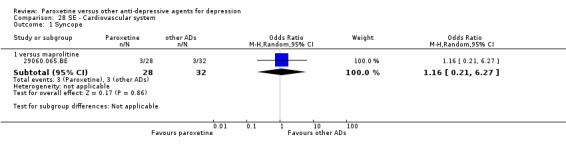
Comparison 28 SE ‐ Cardiovascular system, Outcome 1 Syncope.
28.2. Analysis.
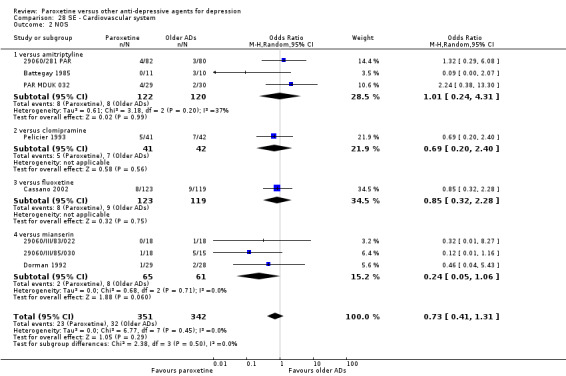
Comparison 28 SE ‐ Cardiovascular system, Outcome 2 NOS.
Comparison 29. SE ‐ Chills.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus newer or non‐conventional ADs | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 0.37 [0.18, 0.74] |
29.1. Analysis.
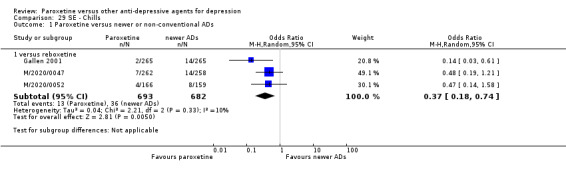
Comparison 29 SE ‐ Chills, Outcome 1 Paroxetine versus newer or non‐conventional ADs.
Comparison 30. SE ‐ Confusion.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 5 | 738 | Odds Ratio (M‐H, Random, 95% CI) | 0.48 [0.18, 1.30] |
| 1.1 versus amitriptyline | 1 | 153 | Odds Ratio (M‐H, Random, 95% CI) | 0.96 [0.13, 7.00] |
| 1.2 versus clomipramine | 1 | 92 | Odds Ratio (M‐H, Random, 95% CI) | 1.05 [0.14, 7.76] |
| 1.3 versus dothiepin | 1 | 271 | Odds Ratio (M‐H, Random, 95% CI) | 0.14 [0.02, 1.12] |
| 1.4 versus lofepramine | 1 | 106 | Odds Ratio (M‐H, Random, 95% CI) | 0.17 [0.01, 3.53] |
| 1.5 versus mianserin | 1 | 116 | Odds Ratio (M‐H, Random, 95% CI) | 0.57 [0.05, 6.42] |
| 2 Paroxetine versus newer or non‐conventional ADs | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus nefazodone | 1 | 40 | Odds Ratio (M‐H, Random, 95% CI) | 4.75 [0.48, 46.91] |
| 2.2 versus reboxetine | 2 | 1050 | Odds Ratio (M‐H, Random, 95% CI) | 1.58 [0.20, 12.39] |
30.1. Analysis.
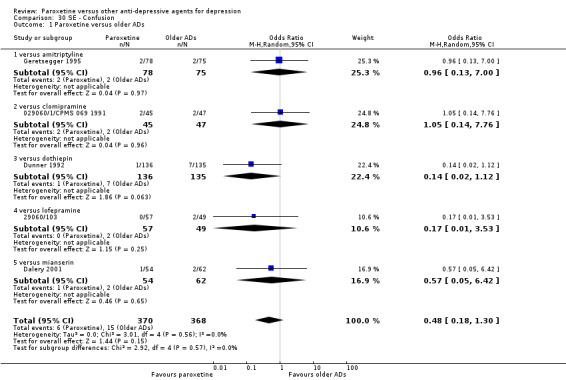
Comparison 30 SE ‐ Confusion, Outcome 1 Paroxetine versus older ADs.
30.2. Analysis.
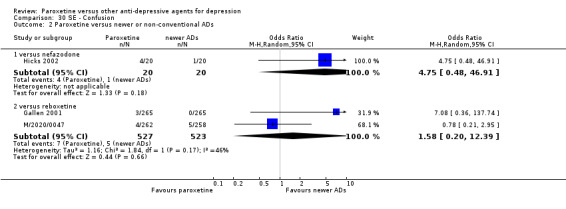
Comparison 30 SE ‐ Confusion, Outcome 2 Paroxetine versus newer or non‐conventional ADs.
Comparison 31. SE ‐ Constipation.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 26 | 3934 | Odds Ratio (M‐H, Random, 95% CI) | 0.49 [0.40, 0.60] |
| 1.1 versus amitriptyline | 10 | 1146 | Odds Ratio (M‐H, Random, 95% CI) | 0.61 [0.37, 0.99] |
| 1.2 versus clomipramine | 2 | 1111 | Odds Ratio (M‐H, Random, 95% CI) | 0.57 [0.38, 0.85] |
| 1.3 versus dothiepin | 1 | 271 | Odds Ratio (M‐H, Random, 95% CI) | 0.57 [0.32, 0.99] |
| 1.4 versus imipramine | 7 | 633 | Odds Ratio (M‐H, Random, 95% CI) | 0.40 [0.25, 0.63] |
| 1.5 versus lofepramine | 2 | 228 | Odds Ratio (M‐H, Random, 95% CI) | 0.49 [0.20, 1.23] |
| 1.6 versus maprolitine | 3 | 429 | Odds Ratio (M‐H, Random, 95% CI) | 0.31 [0.17, 0.55] |
| 1.7 versus mianserin | 1 | 116 | Odds Ratio (M‐H, Random, 95% CI) | 0.22 [0.01, 4.73] |
| 2 Paroxetine versus other SSRIs | 7 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus escitalopram | 1 | 454 | Odds Ratio (M‐H, Random, 95% CI) | 2.52 [0.87, 7.29] |
| 2.2 versus fluoxetine | 3 | 1001 | Odds Ratio (M‐H, Random, 95% CI) | 2.71 [1.47, 5.01] |
| 2.3 versus fluvoxamine | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 2.15 [0.36, 12.76] |
| 2.4 versus sertraline | 2 | 545 | Odds Ratio (M‐H, Random, 95% CI) | 3.26 [1.73, 6.14] |
| 3 Paroxetine versus newer or non‐conventional ADs | 16 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus bupropion | 2 | 240 | Odds Ratio (M‐H, Random, 95% CI) | 2.00 [0.59, 6.81] |
| 3.2 versus duloxetine | 5 | 1573 | Odds Ratio (M‐H, Random, 95% CI) | 0.74 [0.46, 1.18] |
| 3.3 versus mirtazapine | 2 | 529 | Odds Ratio (M‐H, Random, 95% CI) | 0.93 [0.51, 1.69] |
| 3.4 versus nefazodone | 1 | 40 | Odds Ratio (M‐H, Random, 95% CI) | 3.35 [0.32, 35.36] |
| 3.5 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 0.48 [0.36, 0.63] |
| 3.6 versus tianeptine | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 7.10 [0.36, 138.80] |
| 3.7 versus venlafaxine | 2 | 466 | Odds Ratio (M‐H, Random, 95% CI) | 1.16 [0.33, 4.12] |
Comparison 32. SE ‐ Cough.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus lofepramine | 1 | 106 | Odds Ratio (M‐H, Random, 95% CI) | 0.12 [0.01, 2.29] |
| 2 Paroxetine versus newer or non‐conventional ADs | 5 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus duloxetine | 2 | 528 | Odds Ratio (M‐H, Random, 95% CI) | 1.22 [0.43, 3.42] |
| 2.2 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 1.32 [0.50, 3.48] |
32.1. Analysis.
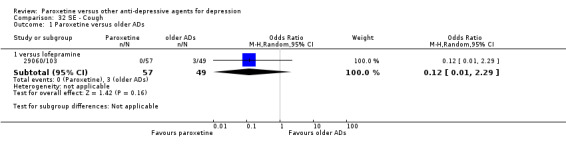
Comparison 32 SE ‐ Cough, Outcome 1 Paroxetine versus older ADs.
32.2. Analysis.
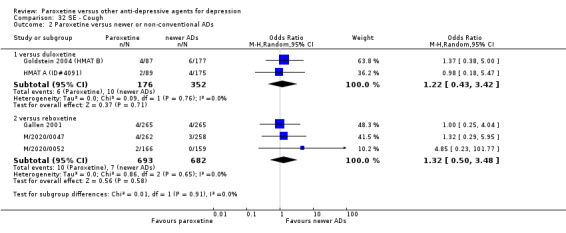
Comparison 32 SE ‐ Cough, Outcome 2 Paroxetine versus newer or non‐conventional ADs.
Comparison 33. SE ‐ Delirium.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus other SSRIs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus fluoxetine | 1 | 90 | Odds Ratio (M‐H, Random, 95% CI) | 3.07 [0.12, 77.32] |
| 2 Paroxetine versus older ADs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus clomipramine | 1 | 92 | Odds Ratio (M‐H, Random, 95% CI) | 0.2 [0.01, 4.28] |
33.1. Analysis.
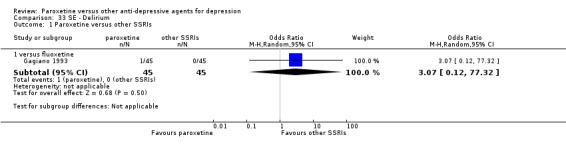
Comparison 33 SE ‐ Delirium, Outcome 1 Paroxetine versus other SSRIs.
33.2. Analysis.
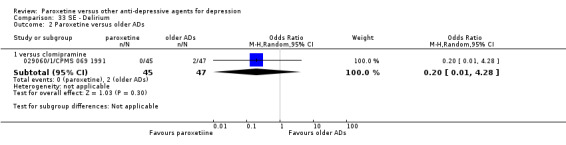
Comparison 33 SE ‐ Delirium, Outcome 2 Paroxetine versus older ADs.
Comparison 34. SE ‐ Depersonalization.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus amitriptyline | 1 | 21 | Odds Ratio (M‐H, Random, 95% CI) | 5.53 [0.23, 130.34] |
| 1.2 versus lofepramine | 1 | 106 | Odds Ratio (M‐H, Random, 95% CI) | 2.63 [0.10, 66.00] |
| 2 Paroxetine versus other SSRIs | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluoxetine | 1 | 176 | Odds Ratio (M‐H, Random, 95% CI) | 2.97 [0.12, 73.81] |
| 2.2 versus fluvoxamine | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 0.46 [0.08, 2.75] |
| 3 Paroxetine versus newer or non‐conventional ADs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus tianeptine | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 3.0 [0.12, 74.28] |
34.1. Analysis.
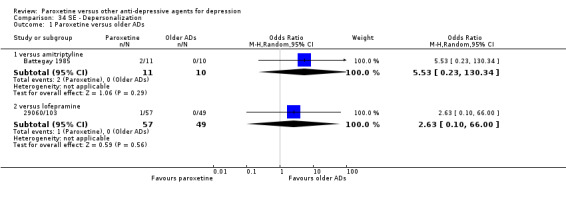
Comparison 34 SE ‐ Depersonalization, Outcome 1 Paroxetine versus older ADs.
34.2. Analysis.
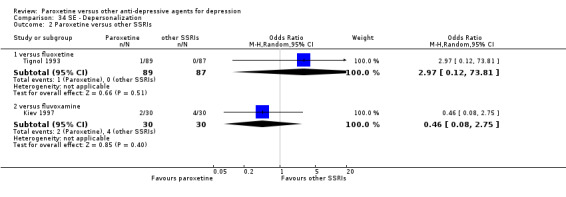
Comparison 34 SE ‐ Depersonalization, Outcome 2 Paroxetine versus other SSRIs.
34.3. Analysis.
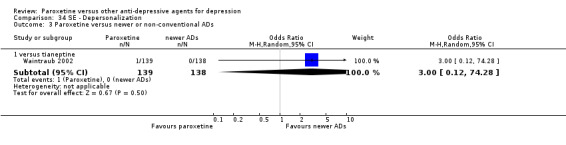
Comparison 34 SE ‐ Depersonalization, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 35. SE ‐ Depression.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 2 | 323 | Odds Ratio (M‐H, Random, 95% CI) | 0.92 [0.09, 9.38] |
| 1.1 versus amitriptyline | 1 | 217 | Odds Ratio (M‐H, Random, 95% CI) | 3.0 [0.12, 74.46] |
| 1.2 versus lofepramine | 1 | 106 | Odds Ratio (M‐H, Random, 95% CI) | 0.28 [0.01, 7.06] |
| 2 Paroxetine versus other SSRIs | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluoxetine | 4 | 1128 | Odds Ratio (M‐H, Random, 95% CI) | 0.97 [0.45, 2.09] |
| 3 Paroxetine versus newer or non‐conventional ADs | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus agomelatine | 1 | 284 | Odds Ratio (M‐H, Random, 95% CI) | 1.41 [0.39, 5.13] |
| 3.2 versus reboxetine | 2 | 1050 | Odds Ratio (M‐H, Random, 95% CI) | 0.76 [0.23, 2.46] |
35.1. Analysis.
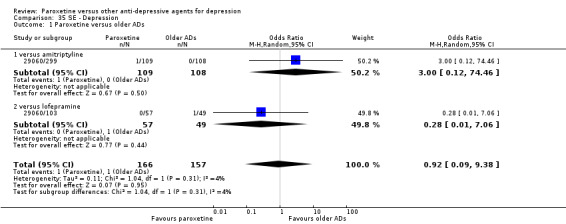
Comparison 35 SE ‐ Depression, Outcome 1 Paroxetine versus older ADs.
35.2. Analysis.
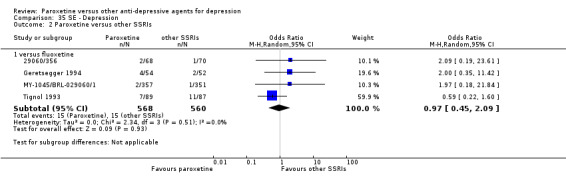
Comparison 35 SE ‐ Depression, Outcome 2 Paroxetine versus other SSRIs.
35.3. Analysis.
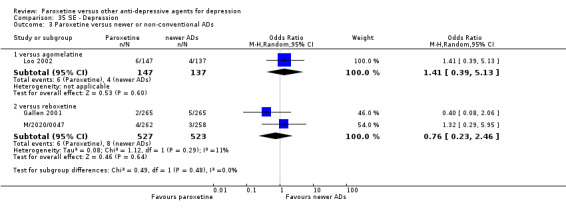
Comparison 35 SE ‐ Depression, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 36. SE ‐ Diarrhoea.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 13 | 1743 | Odds Ratio (M‐H, Random, 95% CI) | 2.41 [1.56, 3.73] |
| 1.1 versus amitriptyline | 5 | 653 | Odds Ratio (M‐H, Random, 95% CI) | 1.91 [0.88, 4.14] |
| 1.2 versus dothiepin | 1 | 271 | Odds Ratio (M‐H, Random, 95% CI) | 3.47 [1.23, 9.75] |
| 1.3 versus imipramine | 2 | 159 | Odds Ratio (M‐H, Random, 95% CI) | 1.88 [0.58, 6.02] |
| 1.4 versus lofepramine | 1 | 106 | Odds Ratio (M‐H, Random, 95% CI) | 6.36 [0.32, 126.19] |
| 1.5 versus maprolitine | 2 | 358 | Odds Ratio (M‐H, Random, 95% CI) | 2.94 [1.34, 6.47] |
| 1.6 versus nortriptyline | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 4.52 [0.21, 97.16] |
| 1.7 versus mianserin | 1 | 116 | Odds Ratio (M‐H, Random, 95% CI) | 0.22 [0.01, 4.73] |
| 2 Paroxetine versus other SSRIs | 12 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus citalopram | 1 | 406 | Odds Ratio (M‐H, Random, 95% CI) | 1.36 [0.76, 2.45] |
| 2.2 versus escitalopram | 1 | 454 | Odds Ratio (M‐H, Random, 95% CI) | 1.62 [0.82, 3.20] |
| 2.3 versus fluoxetine | 7 | 1940 | Odds Ratio (M‐H, Random, 95% CI) | 0.85 [0.63, 1.15] |
| 2.4 versus fluvoxamine | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 2.79 [0.75, 10.33] |
| 2.5 versus sertraline | 2 | 545 | Odds Ratio (M‐H, Random, 95% CI) | 0.40 [0.26, 0.60] |
| 3 Paroxetine versus newer or non‐conventional ADs | 19 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus agomelatine | 1 | 284 | Odds Ratio (M‐H, Random, 95% CI) | 1.12 [0.33, 3.77] |
| 3.2 versus aprepitant (MK‐869) | 1 | 143 | Odds Ratio (M‐H, Random, 95% CI) | 1.27 [0.47, 3.43] |
| 3.3 versus bupropion | 2 | 240 | Odds Ratio (M‐H, Random, 95% CI) | 3.03 [1.35, 6.84] |
| 3.4 versus duloxetine | 5 | 1573 | Odds Ratio (M‐H, Random, 95% CI) | 1.38 [0.68, 2.77] |
| 3.5 versus hypericum | 1 | 251 | Odds Ratio (M‐H, Random, 95% CI) | 2.10 [1.00, 4.44] |
| 3.6 versus mirtazapine | 3 | 726 | Odds Ratio (M‐H, Random, 95% CI) | 1.12 [0.69, 1.83] |
| 3.7 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 3.45 [2.31, 5.15] |
| 3.8 versus tianeptine | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 7.10 [0.36, 138.80] |
| 3.9 versus venlafaxine | 1 | 361 | Odds Ratio (M‐H, Random, 95% CI) | 1.16 [0.44, 3.09] |
| 3.10 versus trazodone | 1 | 108 | Odds Ratio (M‐H, Random, 95% CI) | 3.17 [0.13, 79.60] |
Comparison 37. SE ‐ Dizziness.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 21 | 2554 | Odds Ratio (M‐H, Random, 95% CI) | 0.80 [0.62, 1.03] |
| 1.1 versus amitriptyline | 7 | 777 | Odds Ratio (M‐H, Random, 95% CI) | 0.42 [0.25, 0.72] |
| 1.2 versus clomipramine | 1 | 92 | Odds Ratio (M‐H, Random, 95% CI) | 0.19 [0.02, 1.70] |
| 1.3 versus dothiepin | 2 | 405 | Odds Ratio (M‐H, Random, 95% CI) | 1.73 [0.94, 3.18] |
| 1.4 versus imipramine | 6 | 558 | Odds Ratio (M‐H, Random, 95% CI) | 0.80 [0.53, 1.22] |
| 1.5 versus lofepramine | 2 | 228 | Odds Ratio (M‐H, Random, 95% CI) | 0.76 [0.32, 1.81] |
| 1.6 versus maprotiline | 1 | 298 | Odds Ratio (M‐H, Random, 95% CI) | 1.08 [0.66, 1.78] |
| 1.7 versus mianserin | 1 | 116 | Odds Ratio (M‐H, Random, 95% CI) | 0.57 [0.05, 6.42] |
| 1.8 versus nortriptyline | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 0.28 [0.01, 7.08] |
| 2 Paroxetine versus other SSRIs | 10 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus escitalopram | 1 | 454 | Odds Ratio (M‐H, Random, 95% CI) | 0.97 [0.51, 1.84] |
| 2.2 versus fluoxetine | 6 | 1737 | Odds Ratio (M‐H, Random, 95% CI) | 1.50 [1.11, 2.04] |
| 2.3 versus fluvoxamine | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 1.45 [0.44, 4.86] |
| 2.4 versus sertraline | 2 | 545 | Odds Ratio (M‐H, Random, 95% CI) | 1.40 [0.61, 3.22] |
| 3 Paroxetine versus newer or non‐conventional ADs | 23 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus aprepitant (MK‐869) | 1 | 143 | Odds Ratio (M‐H, Random, 95% CI) | 1.2 [0.35, 4.13] |
| 3.2 versus bupropion | 2 | 240 | Odds Ratio (M‐H, Random, 95% CI) | 0.81 [0.37, 1.76] |
| 3.3 versus duloxetine | 5 | 1573 | Odds Ratio (M‐H, Random, 95% CI) | 0.83 [0.58, 1.18] |
| 3.4 versus hypericum | 1 | 251 | Odds Ratio (M‐H, Random, 95% CI) | 3.03 [1.35, 6.82] |
| 3.5 versus milnacipran | 1 | 302 | Odds Ratio (M‐H, Random, 95% CI) | 9.00 [0.48, 168.64] |
| 3.6 versus mirtazapine | 3 | 726 | Odds Ratio (M‐H, Random, 95% CI) | 1.19 [0.66, 2.11] |
| 3.7 versus nefazodone | 2 | 246 | Odds Ratio (M‐H, Random, 95% CI) | 0.48 [0.23, 1.03] |
| 3.8 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 0.84 [0.59, 1.21] |
| 3.9 versus tianeptine | 2 | 604 | Odds Ratio (M‐H, Random, 95% CI) | 7.02 [1.25, 39.32] |
| 3.10 versus trazodone | 1 | 108 | Odds Ratio (M‐H, Random, 95% CI) | 0.51 [0.04, 5.79] |
| 3.11 versus venlafaxine | 2 | 466 | Odds Ratio (M‐H, Random, 95% CI) | 1.39 [0.18, 10.62] |
37.1. Analysis.
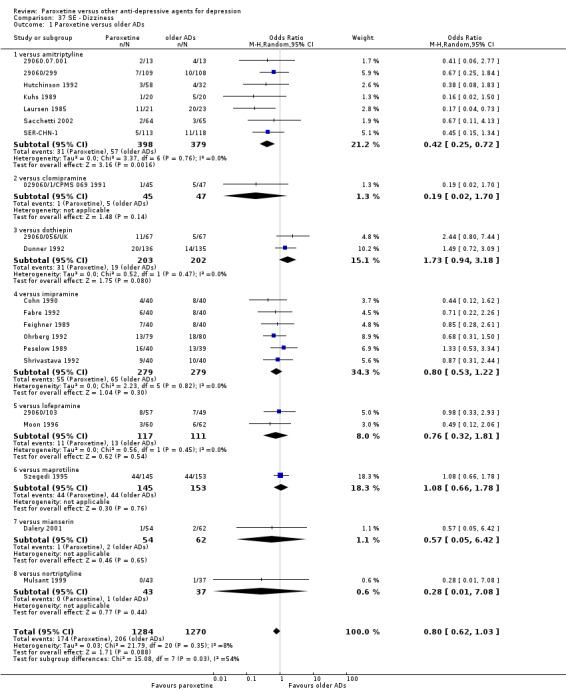
Comparison 37 SE ‐ Dizziness, Outcome 1 Paroxetine versus older ADs.
37.2. Analysis.
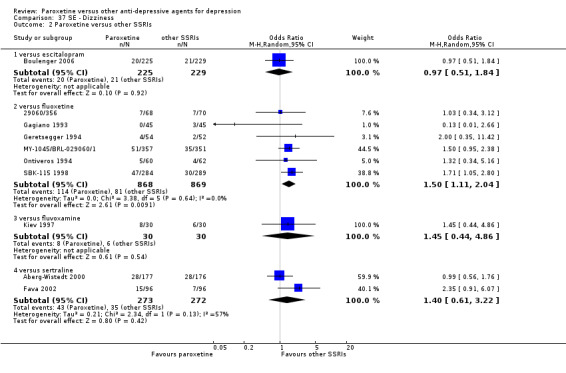
Comparison 37 SE ‐ Dizziness, Outcome 2 Paroxetine versus other SSRIs.
37.3. Analysis.
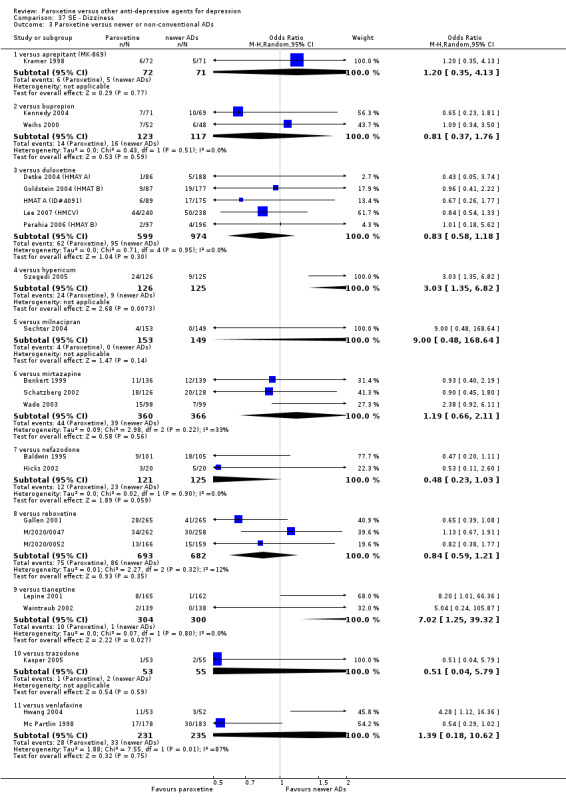
Comparison 37 SE ‐ Dizziness, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 38. SE ‐ Dry mouth.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 29 | 4578 | Odds Ratio (M‐H, Random, 95% CI) | 0.23 [0.18, 0.30] |
| 1.1 versus amitriptyline | 12 | 1576 | Odds Ratio (M‐H, Random, 95% CI) | 0.27 [0.17, 0.43] |
| 1.2 versus clomipramine | 2 | 1111 | Odds Ratio (M‐H, Random, 95% CI) | 0.35 [0.26, 0.48] |
| 1.3 versus dothiepin | 2 | 405 | Odds Ratio (M‐H, Random, 95% CI) | 0.22 [0.10, 0.50] |
| 1.4 versus imipramine | 8 | 835 | Odds Ratio (M‐H, Random, 95% CI) | 0.16 [0.10, 0.26] |
| 1.5 versus lofepramine | 1 | 106 | Odds Ratio (M‐H, Random, 95% CI) | 1.16 [0.25, 5.44] |
| 1.6 versus maprolitine | 3 | 429 | Odds Ratio (M‐H, Random, 95% CI) | 0.13 [0.08, 0.23] |
| 1.7 versus mianserin | 1 | 116 | Odds Ratio (M‐H, Random, 95% CI) | 0.76 [0.12, 4.70] |
| 2 Paroxetine versus other SSRIs | 11 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus citalopram | 1 | 406 | Odds Ratio (M‐H, Random, 95% CI) | 0.97 [0.56, 1.68] |
| 2.2 versus escitalopram | 1 | 454 | Odds Ratio (M‐H, Random, 95% CI) | 1.44 [0.74, 2.83] |
| 2.3 versus fluoxetine | 6 | 1920 | Odds Ratio (M‐H, Random, 95% CI) | 1.67 [1.17, 2.38] |
| 2.4 versus fluvoxamine | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 0.63 [0.21, 1.88] |
| 2.5 versus sertraline | 2 | 545 | Odds Ratio (M‐H, Random, 95% CI) | 1.01 [0.52, 1.99] |
| 3 Paroxetine versus newer or non‐conventional ADs | 22 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus agomelatine | 1 | 284 | Odds Ratio (M‐H, Random, 95% CI) | 4.79 [0.55, 41.52] |
| 3.2 versus aprepitant (MK‐869) | 1 | 143 | Odds Ratio (M‐H, Random, 95% CI) | 0.98 [0.30, 3.21] |
| 3.3 versus bupropion | 2 | 240 | Odds Ratio (M‐H, Random, 95% CI) | 0.71 [0.27, 1.87] |
| 3.4 versus duloxetine | 5 | 1571 | Odds Ratio (M‐H, Random, 95% CI) | 0.78 [0.55, 1.10] |
| 3.5 versus hypericum | 1 | 251 | Odds Ratio (M‐H, Random, 95% CI) | 2.62 [1.36, 5.04] |
| 3.6 versus mirtazapine | 3 | 726 | Odds Ratio (M‐H, Random, 95% CI) | 0.58 [0.27, 1.24] |
| 3.7 versus nefazodone | 2 | 246 | Odds Ratio (M‐H, Random, 95% CI) | 0.80 [0.35, 1.82] |
| 3.8 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 0.35 [0.27, 0.45] |
| 3.9 versus tianeptine | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 0.70 [0.22, 2.26] |
| 3.10 versus trazodone | 1 | 108 | Odds Ratio (M‐H, Random, 95% CI) | 10.09 [0.53, 192.17] |
| 3.11 versus venlafaxine | 2 | 466 | Odds Ratio (M‐H, Random, 95% CI) | 1.16 [0.44, 3.06] |
Comparison 39. SE ‐ Dyspepsia.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 5 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus amitriptyline | 3 | 552 | Odds Ratio (M‐H, Random, 95% CI) | 0.44 [0.05, 3.72] |
| 1.2 versus dothiepin | 1 | 271 | Odds Ratio (M‐H, Random, 95% CI) | 0.68 [0.25, 1.84] |
| 1.3 versus maprotiline | 1 | 298 | Odds Ratio (M‐H, Random, 95% CI) | 0.47 [0.20, 1.12] |
| 2 Paroxetine versus other SSRIs | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluoxetine | 3 | 1371 | Odds Ratio (M‐H, Random, 95% CI) | 0.77 [0.54, 1.09] |
| 2.2 versus fluvoxamine | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 0.51 [0.13, 1.95] |
| 3 Paroxetine versus newer or non‐conventional ADs | 6 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus duloxetine | 3 | 802 | Odds Ratio (M‐H, Random, 95% CI) | 0.67 [0.28, 1.61] |
| 3.2 versus mirtazapine | 1 | 197 | Odds Ratio (M‐H, Random, 95% CI) | 1.33 [0.47, 3.72] |
| 3.3 versus reboxetine | 2 | 1050 | Odds Ratio (M‐H, Random, 95% CI) | 1.12 [0.64, 1.96] |
39.1. Analysis.
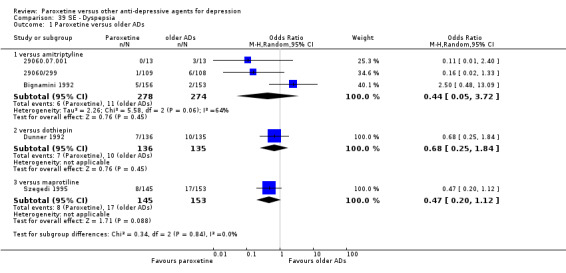
Comparison 39 SE ‐ Dyspepsia, Outcome 1 Paroxetine versus older ADs.
39.2. Analysis.
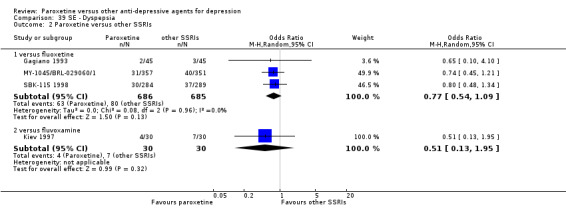
Comparison 39 SE ‐ Dyspepsia, Outcome 2 Paroxetine versus other SSRIs.
39.3. Analysis.
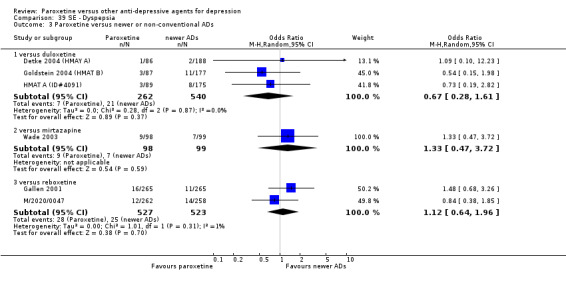
Comparison 39 SE ‐ Dyspepsia, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 40. SE ‐ Dyspnea.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus imipramine | 1 | 75 | Odds Ratio (M‐H, Random, 95% CI) | 0.25 [0.07, 0.85] |
| 2 Paroxetine versus other SSRIs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluoxetine | 1 | 203 | Odds Ratio (M‐H, Random, 95% CI) | 2.31 [0.84, 6.35] |
| 3 Paroxetine versus newer or non‐conventional ADs | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus duloxetine | 1 | 274 | Odds Ratio (M‐H, Random, 95% CI) | 0.43 [0.02, 9.08] |
| 3.2 versus reboxetine | 2 | 845 | Odds Ratio (M‐H, Random, 95% CI) | 4.30 [1.21, 15.26] |
40.1. Analysis.
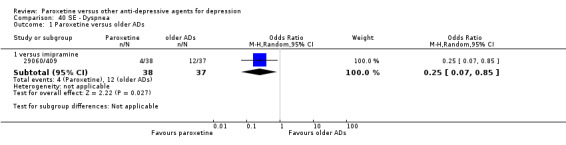
Comparison 40 SE ‐ Dyspnea, Outcome 1 Paroxetine versus older ADs.
40.2. Analysis.
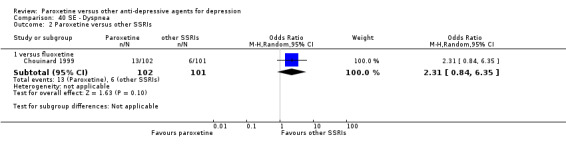
Comparison 40 SE ‐ Dyspnea, Outcome 2 Paroxetine versus other SSRIs.
40.3. Analysis.
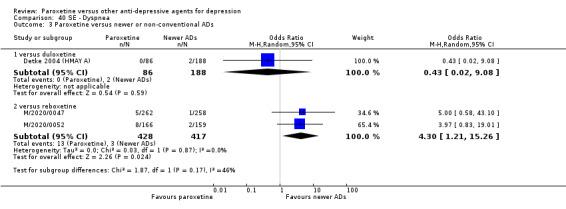
Comparison 40 SE ‐ Dyspnea, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 41. SE ‐ ECG abnormailities.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 3 | 382 | Odds Ratio (M‐H, Random, 95% CI) | 0.33 [0.08, 1.40] |
| 1.1 versus amitriptyline | 1 | 231 | Odds Ratio (M‐H, Random, 95% CI) | 0.25 [0.05, 1.19] |
| 1.2 versus maprolitine | 1 | 71 | Odds Ratio (M‐H, Random, 95% CI) | 2.84 [0.11, 71.99] |
| 1.3 versus nortriptyline | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 0.11 [0.01, 2.27] |
41.1. Analysis.
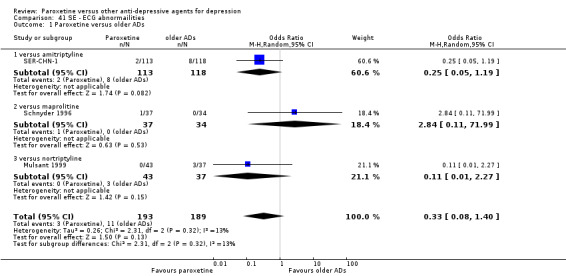
Comparison 41 SE ‐ ECG abnormailities, Outcome 1 Paroxetine versus older ADs.
Comparison 42. SE ‐ Emotional lability.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 7 | 953 | Odds Ratio (M‐H, Random, 95% CI) | 1.18 [0.54, 2.57] |
| 1.1 versus amitriptyline | 4 | 503 | Odds Ratio (M‐H, Random, 95% CI) | 1.66 [0.65, 4.20] |
| 1.2 versus clomipramine | 1 | 92 | Odds Ratio (M‐H, Random, 95% CI) | 0.34 [0.01, 8.58] |
| 1.3 versus maprolitine | 2 | 358 | Odds Ratio (M‐H, Random, 95% CI) | 0.60 [0.12, 2.91] |
| 2 Paroxetine versus other SSRIs | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluoxetine | 4 | 1139 | Odds Ratio (M‐H, Random, 95% CI) | 0.83 [0.31, 2.22] |
| 3 Paroxetine versus newer or non‐conventional ADs | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus reboxetine | 2 | 1050 | Odds Ratio (M‐H, Random, 95% CI) | 0.62 [0.08, 5.05] |
42.1. Analysis.
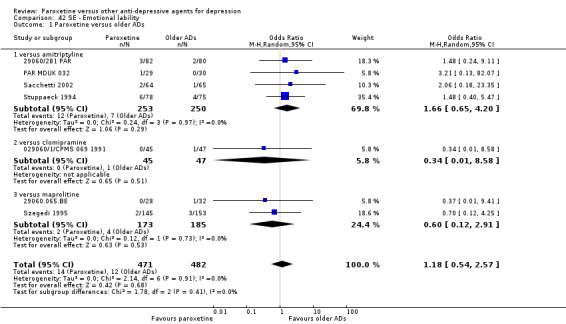
Comparison 42 SE ‐ Emotional lability, Outcome 1 Paroxetine versus older ADs.
42.2. Analysis.
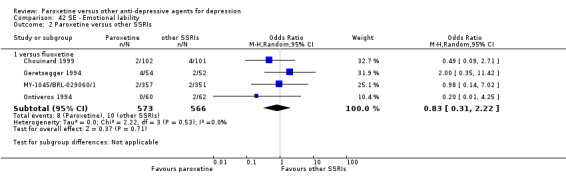
Comparison 42 SE ‐ Emotional lability, Outcome 2 Paroxetine versus other SSRIs.
42.3. Analysis.
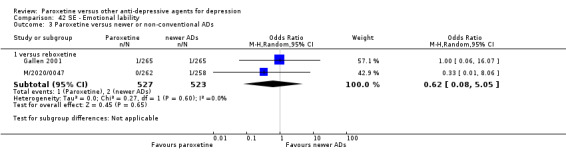
Comparison 42 SE ‐ Emotional lability, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 43. SE ‐ Fatigue.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus lofepramine | 1 | 106 | Odds Ratio (M‐H, Random, 95% CI) | 0.17 [0.01, 3.53] |
| 1.2 versus mianserin | 1 | 116 | Odds Ratio (M‐H, Random, 95% CI) | 3.50 [0.14, 87.83] |
| 2 Paroxetine versus other SSRIs | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluoxetine | 1 | 138 | Odds Ratio (M‐H, Random, 95% CI) | 0.21 [0.04, 0.99] |
| 2.2 versus sertraline | 2 | 545 | Odds Ratio (M‐H, Random, 95% CI) | 2.41 [1.21, 4.77] |
| 3 Paroxetine versus newer or non‐conventional ADs | 8 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus duloxetine | 3 | 1045 | Odds Ratio (M‐H, Random, 95% CI) | 1.19 [0.60, 2.39] |
| 3.2 versus hypericum | 1 | 251 | Odds Ratio (M‐H, Random, 95% CI) | 1.15 [0.54, 2.48] |
| 3.3 versus mirtazapine | 3 | 726 | Odds Ratio (M‐H, Random, 95% CI) | 0.58 [0.32, 1.06] |
| 3.4 versus reboxetine | 1 | 325 | Odds Ratio (M‐H, Random, 95% CI) | 4.91 [0.57, 42.47] |
43.1. Analysis.
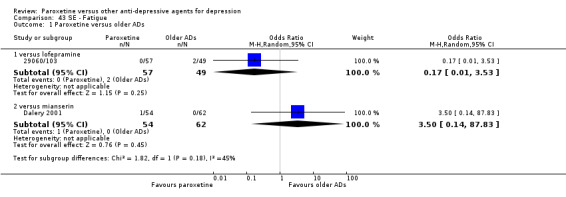
Comparison 43 SE ‐ Fatigue, Outcome 1 Paroxetine versus older ADs.
43.2. Analysis.
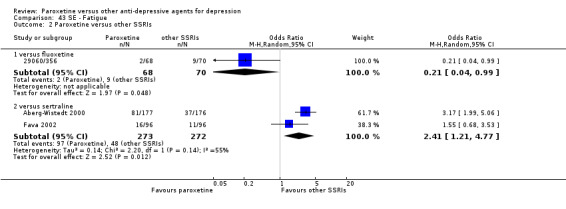
Comparison 43 SE ‐ Fatigue, Outcome 2 Paroxetine versus other SSRIs.
43.3. Analysis.
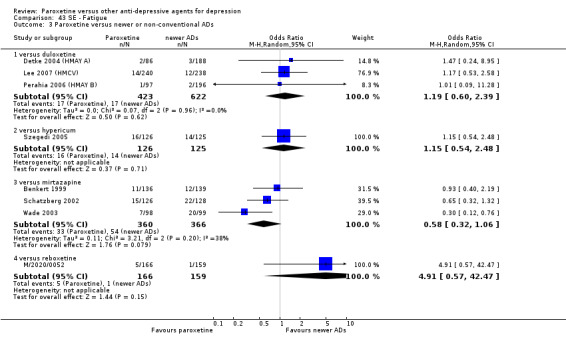
Comparison 43 SE ‐ Fatigue, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 44. SE ‐ Fever.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus imipramine | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 8.22] |
| 1.2 versus mianserin | 1 | 116 | Odds Ratio (M‐H, Random, 95% CI) | 3.50 [0.14, 87.83] |
| 2 Paroxetine versus newer or non‐conventional ADs | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus reboxetine | 2 | 1050 | Odds Ratio (M‐H, Random, 95% CI) | 0.99 [0.28, 3.54] |
44.1. Analysis.
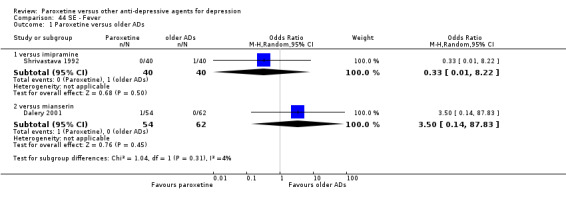
Comparison 44 SE ‐ Fever, Outcome 1 Paroxetine versus older ADs.
44.2. Analysis.
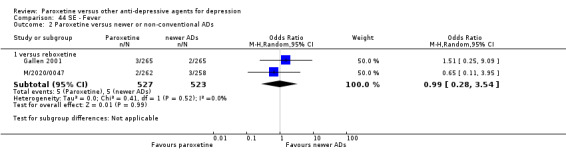
Comparison 44 SE ‐ Fever, Outcome 2 Paroxetine versus newer or non‐conventional ADs.
Comparison 45. SE ‐ Flatulence.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus amitriptyline | 1 | 21 | Odds Ratio (M‐H, Random, 95% CI) | 3.0 [0.11, 82.40] |
| 2 Paroxetine versus other SSRIs | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluoxetine | 1 | 573 | Odds Ratio (M‐H, Random, 95% CI) | 1.69 [0.87, 3.29] |
| 2.2 versus fluvoxamine | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 0.22 [0.02, 2.14] |
| 3 Paroxetine versus newer or non‐conventional ADs | 6 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus aprepitant (MK‐869) | 1 | 143 | Odds Ratio (M‐H, Random, 95% CI) | 0.38 [0.07, 2.01] |
| 3.2 versus mirtazapine | 1 | 254 | Odds Ratio (M‐H, Random, 95% CI) | 4.19 [1.35, 13.00] |
| 3.3 versus reboxetine | 2 | 1050 | Odds Ratio (M‐H, Random, 95% CI) | 1.84 [0.87, 3.87] |
| 3.4 versus duloxetine | 2 | 528 | Odds Ratio (M‐H, Random, 95% CI) | 2.34 [0.83, 6.59] |
45.1. Analysis.
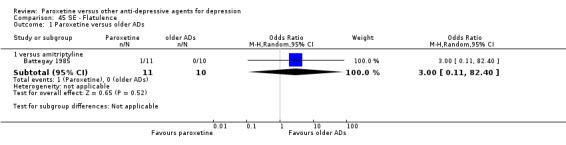
Comparison 45 SE ‐ Flatulence, Outcome 1 Paroxetine versus older ADs.
45.2. Analysis.
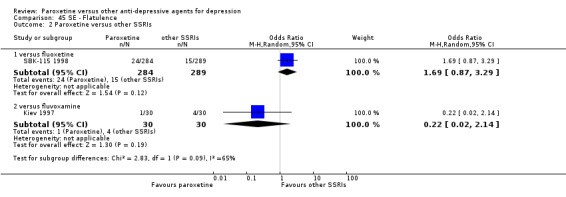
Comparison 45 SE ‐ Flatulence, Outcome 2 Paroxetine versus other SSRIs.
45.3. Analysis.
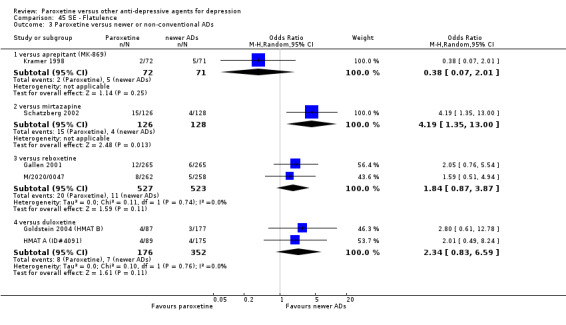
Comparison 45 SE ‐ Flatulence, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 46. SE ‐ Gastrointestinal disorder.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Appendicitis | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus fluoxetine | 1 | 138 | Odds Ratio (M‐H, Random, 95% CI) | 3.13 [0.13, 78.26] |
| 2 Cholelithiasis | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus imipramine | 1 | 202 | Odds Ratio (M‐H, Random, 95% CI) | 0.17 [0.01, 4.16] |
| 3 Colitis | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus fluoxetine | 1 | 122 | Odds Ratio (M‐H, Random, 95% CI) | 7.61 [0.38, 150.51] |
| 4 Eructation | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 versus reboxetine | 1 | 520 | Odds Ratio (M‐H, Random, 95% CI) | 0.14 [0.01, 2.71] |
| 5 Melena | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 versus maprolitine | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 3.55 [0.14, 90.59] |
| 6 Gastritis | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 versus imipramine | 1 | 202 | Odds Ratio (M‐H, Random, 95% CI) | 1.53 [0.16, 15.03] |
| 7 Gastroenteritis | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 versus reboxetine | 2 | 1050 | Odds Ratio (M‐H, Random, 95% CI) | 1.13 [0.42, 2.98] |
| 8 Intestinal obstruction | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 versus fluoxetine | 1 | 138 | Odds Ratio (M‐H, Random, 95% CI) | 3.13 [0.13, 78.26] |
| 9 Intestinal perforation | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 versus lofepramine | 1 | 106 | Odds Ratio (M‐H, Random, 95% CI) | 0.28 [0.01, 7.06] |
| 10 Peptic ulcer hemorrhage | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 versus fluoxetine | 1 | 573 | Odds Ratio (M‐H, Random, 95% CI) | 3.06 [0.12, 75.52] |
| 11 NOS | 18 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1 versus amisulpride | 2 | 298 | Odds Ratio (M‐H, Random, 95% CI) | 1.77 [0.14, 21.98] |
| 11.2 versus amitriptyline | 4 | 478 | Odds Ratio (M‐H, Random, 95% CI) | 0.68 [0.35, 1.33] |
| 11.3 versus clomipramine | 1 | 83 | Odds Ratio (M‐H, Random, 95% CI) | 0.56 [0.23, 1.38] |
| 11.4 versus imipramine | 1 | 159 | Odds Ratio (M‐H, Random, 95% CI) | 1.69 [0.53, 5.41] |
| 11.5 versus fluoxetine | 2 | 950 | Odds Ratio (M‐H, Random, 95% CI) | 0.75 [0.37, 1.49] |
| 11.6 versus fluvoxamine | 1 | 81 | Odds Ratio (M‐H, Random, 95% CI) | 0.92 [0.27, 3.12] |
| 11.7 versus maprolitine | 1 | 71 | Odds Ratio (M‐H, Random, 95% CI) | 0.54 [0.17, 1.71] |
| 11.8 versus mianserin | 3 | 126 | Odds Ratio (M‐H, Random, 95% CI) | 3.56 [1.62, 7.79] |
| 11.9 versus reboxetine | 2 | 855 | Odds Ratio (M‐H, Random, 95% CI) | 1.04 [0.08, 14.21] |
| 11.10 versus sertraline | 1 | 192 | Odds Ratio (M‐H, Random, 95% CI) | 1.28 [0.48, 3.39] |
46.1. Analysis.
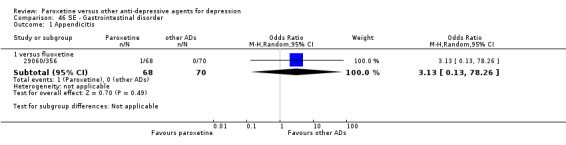
Comparison 46 SE ‐ Gastrointestinal disorder, Outcome 1 Appendicitis.
46.2. Analysis.
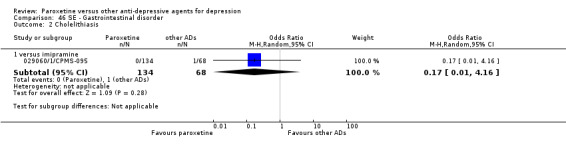
Comparison 46 SE ‐ Gastrointestinal disorder, Outcome 2 Cholelithiasis.
46.3. Analysis.
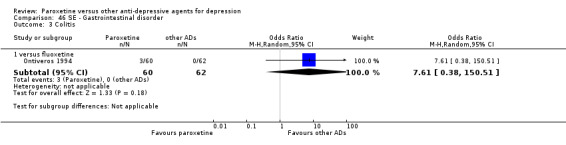
Comparison 46 SE ‐ Gastrointestinal disorder, Outcome 3 Colitis.
46.4. Analysis.
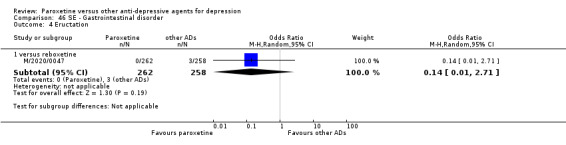
Comparison 46 SE ‐ Gastrointestinal disorder, Outcome 4 Eructation.
46.5. Analysis.
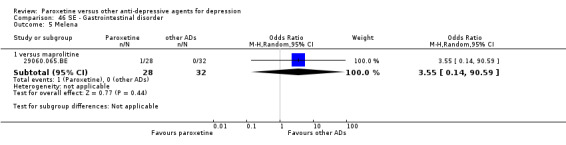
Comparison 46 SE ‐ Gastrointestinal disorder, Outcome 5 Melena.
46.6. Analysis.
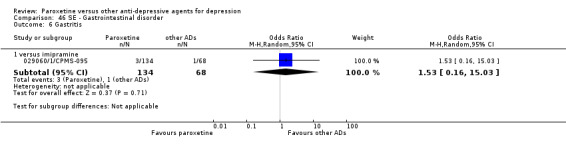
Comparison 46 SE ‐ Gastrointestinal disorder, Outcome 6 Gastritis.
46.7. Analysis.
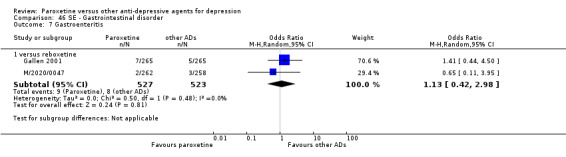
Comparison 46 SE ‐ Gastrointestinal disorder, Outcome 7 Gastroenteritis.
46.8. Analysis.
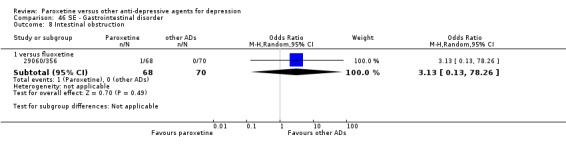
Comparison 46 SE ‐ Gastrointestinal disorder, Outcome 8 Intestinal obstruction.
46.9. Analysis.
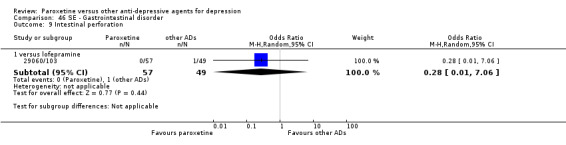
Comparison 46 SE ‐ Gastrointestinal disorder, Outcome 9 Intestinal perforation.
46.10. Analysis.
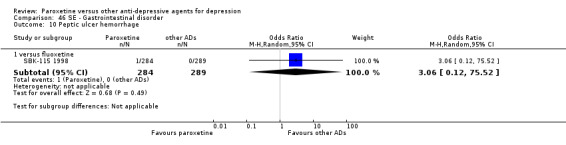
Comparison 46 SE ‐ Gastrointestinal disorder, Outcome 10 Peptic ulcer hemorrhage.
46.11. Analysis.
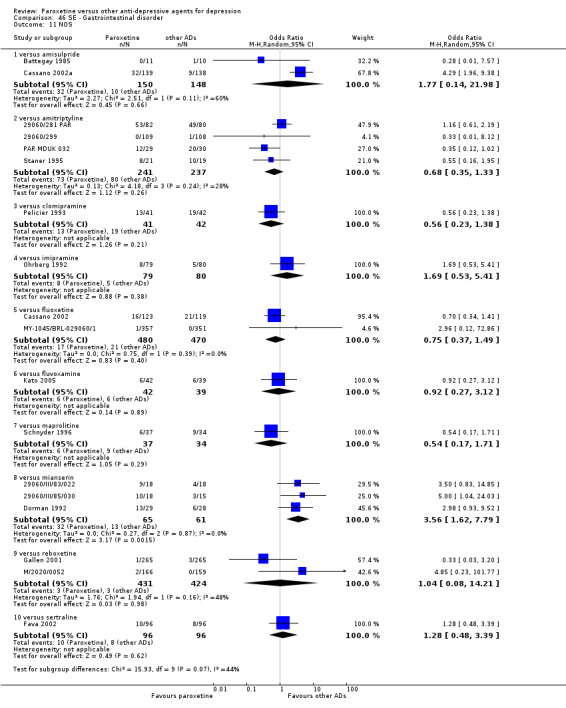
Comparison 46 SE ‐ Gastrointestinal disorder, Outcome 11 NOS.
Comparison 47. SE ‐ Gynecological.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Dysmenorrhea | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus fluvoxamine | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 0.13 [0.01, 2.61] |
| 1.2 versus reboxetine | 2 | 1050 | Odds Ratio (M‐H, Random, 95% CI) | 0.42 [0.07, 2.57] |
| 2 Mestrual disorder | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus reboxetine | 1 | 325 | Odds Ratio (M‐H, Random, 95% CI) | 4.85 [0.23, 101.77] |
| 3 Vaginal moniliasis | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus reboxetine | 2 | 1050 | Odds Ratio (M‐H, Random, 95% CI) | 0.80 [0.19, 3.36] |
| 4 Ectopic pregnancy | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 versus fluoxetine | 1 | 573 | Odds Ratio (M‐H, Random, 95% CI) | 0.20 [0.01, 4.23] |
| 5 Polycystic granuloma | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 versus fluoxetine | 1 | 573 | Odds Ratio (M‐H, Random, 95% CI) | 0.34 [0.01, 8.33] |
47.1. Analysis.
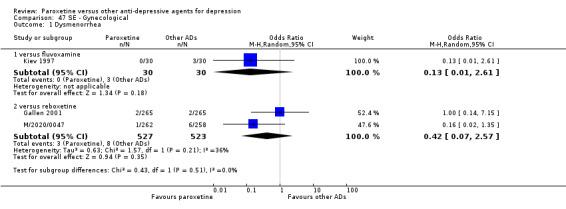
Comparison 47 SE ‐ Gynecological, Outcome 1 Dysmenorrhea.
47.2. Analysis.
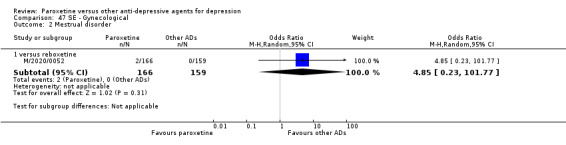
Comparison 47 SE ‐ Gynecological, Outcome 2 Mestrual disorder.
47.3. Analysis.
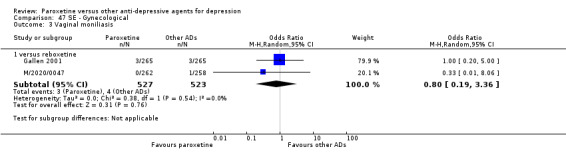
Comparison 47 SE ‐ Gynecological, Outcome 3 Vaginal moniliasis.
47.4. Analysis.
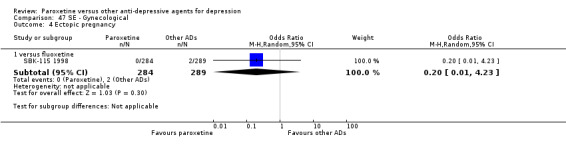
Comparison 47 SE ‐ Gynecological, Outcome 4 Ectopic pregnancy.
47.5. Analysis.
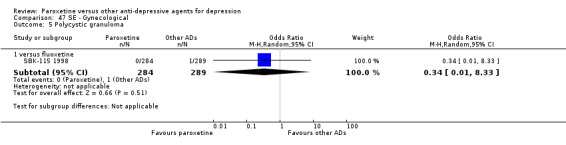
Comparison 47 SE ‐ Gynecological, Outcome 5 Polycystic granuloma.
Comparison 48. SE ‐ Headache.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 29 | 4496 | Odds Ratio (M‐H, Random, 95% CI) | 1.38 [1.13, 1.68] |
| 1.1 versus amitriptyline | 10 | 1323 | Odds Ratio (M‐H, Random, 95% CI) | 0.93 [0.59, 1.48] |
| 1.2 versus clomipramine | 2 | 1111 | Odds Ratio (M‐H, Random, 95% CI) | 1.23 [0.85, 1.78] |
| 1.3 versus dothiepin | 2 | 405 | Odds Ratio (M‐H, Random, 95% CI) | 2.46 [1.35, 4.48] |
| 1.4 versus imipramine | 8 | 835 | Odds Ratio (M‐H, Random, 95% CI) | 1.29 [0.87, 1.93] |
| 1.5 versus lofepramine | 2 | 228 | Odds Ratio (M‐H, Random, 95% CI) | 1.58 [0.55, 4.54] |
| 1.6 versus maprolitine | 3 | 419 | Odds Ratio (M‐H, Random, 95% CI) | 1.96 [1.13, 3.39] |
| 1.7 versus mianserin | 2 | 175 | Odds Ratio (M‐H, Random, 95% CI) | 1.14 [0.16, 7.93] |
| 2 Paroxetine versus other SSRIs | 14 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus citalopram | 1 | 406 | Odds Ratio (M‐H, Random, 95% CI) | 0.80 [0.51, 1.27] |
| 2.2 versus escitalopram | 1 | 454 | Odds Ratio (M‐H, Random, 95% CI) | 0.79 [0.51, 1.24] |
| 2.3 versus fluoxetine | 8 | 2116 | Odds Ratio (M‐H, Random, 95% CI) | 1.02 [0.84, 1.23] |
| 2.4 versus fluvoxamine | 2 | 141 | Odds Ratio (M‐H, Random, 95% CI) | 0.91 [0.10, 8.45] |
| 2.5 versus sertraline | 2 | 545 | Odds Ratio (M‐H, Random, 95% CI) | 0.80 [0.55, 1.14] |
| 3 Paroxetine versus newer or non‐conventional ADs | 23 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus agomelatine | 1 | 284 | Odds Ratio (M‐H, Random, 95% CI) | 1.43 [0.57, 3.62] |
| 3.2 versus aprepitant (MK‐869) | 1 | 143 | Odds Ratio (M‐H, Random, 95% CI) | 0.80 [0.39, 1.64] |
| 3.3 versus bupropion | 2 | 240 | Odds Ratio (M‐H, Random, 95% CI) | 0.92 [0.23, 3.75] |
| 3.4 versus duloxetine | 5 | 1573 | Odds Ratio (M‐H, Random, 95% CI) | 1.01 [0.73, 1.40] |
| 3.5 versus hypericum | 1 | 251 | Odds Ratio (M‐H, Random, 95% CI) | 1.08 [0.48, 2.39] |
| 3.6 versus mirtazapine | 3 | 726 | Odds Ratio (M‐H, Random, 95% CI) | 1.77 [1.12, 2.79] |
| 3.7 versus nefazodone | 2 | 246 | Odds Ratio (M‐H, Random, 95% CI) | 1.14 [0.65, 1.99] |
| 3.8 versus tianeptine | 2 | 604 | Odds Ratio (M‐H, Random, 95% CI) | 1.34 [0.67, 2.67] |
| 3.9 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 1.13 [0.84, 1.53] |
| 3.10 versus trazodone | 1 | 108 | Odds Ratio (M‐H, Random, 95% CI) | 0.11 [0.01, 2.04] |
| 3.11 versus venlafaxine | 2 | 466 | Odds Ratio (M‐H, Random, 95% CI) | 1.37 [0.70, 2.66] |
48.1. Analysis.
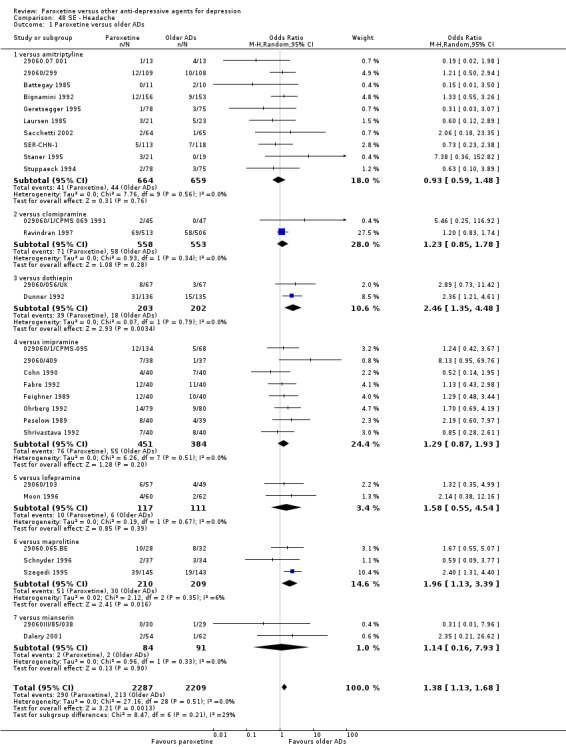
Comparison 48 SE ‐ Headache, Outcome 1 Paroxetine versus older ADs.
48.2. Analysis.
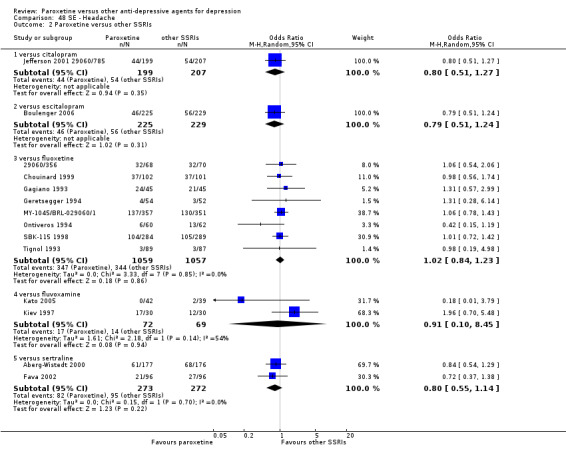
Comparison 48 SE ‐ Headache, Outcome 2 Paroxetine versus other SSRIs.
48.3. Analysis.
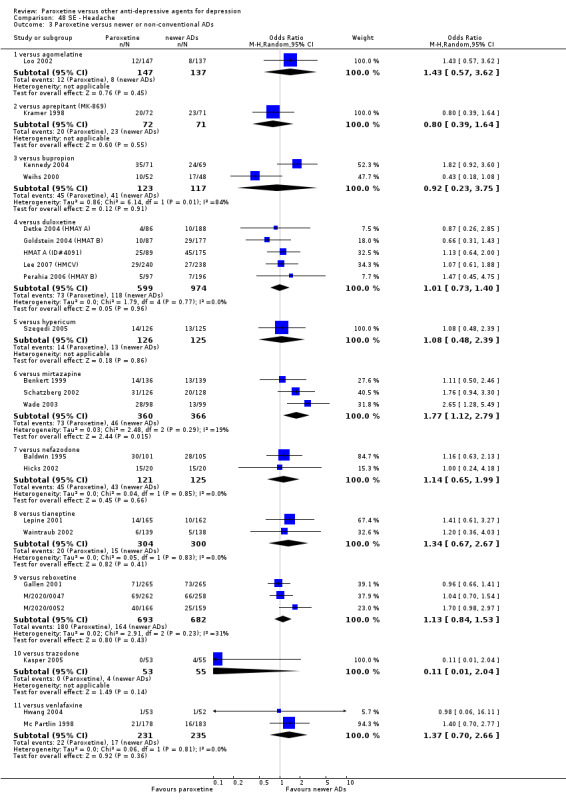
Comparison 48 SE ‐ Headache, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 49. SE ‐ Hypertension.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 3 | 202 | Odds Ratio (M‐H, Random, 95% CI) | 0.82 [0.22, 3.14] |
| 1.1 versus amitriptyline | 1 | 26 | Odds Ratio (M‐H, Random, 95% CI) | 0.17 [0.01, 3.92] |
| 1.2 versus maprolitine | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 1.8 [0.28, 11.64] |
| 1.3 versus mianserin | 1 | 116 | Odds Ratio (M‐H, Random, 95% CI) | 0.57 [0.05, 6.42] |
| 2 Paroxetine versus other SSRIs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluoxetine | 1 | 573 | Odds Ratio (M‐H, Random, 95% CI) | 3.06 [0.12, 75.52] |
| 3 Paroxetine versus newer or non‐conventional ADs | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus duloxetine | 1 | 274 | Odds Ratio (M‐H, Random, 95% CI) | 2.21 [0.31, 15.99] |
| 3.2 versus reboxetine | 2 | 855 | Odds Ratio (M‐H, Random, 95% CI) | 0.65 [0.15, 2.77] |
| 3.3 versus venlafaxine | 1 | 361 | Odds Ratio (M‐H, Random, 95% CI) | 3.12 [0.32, 30.28] |
49.1. Analysis.
Comparison 49 SE ‐ Hypertension, Outcome 1 Paroxetine versus older ADs.
49.2. Analysis.
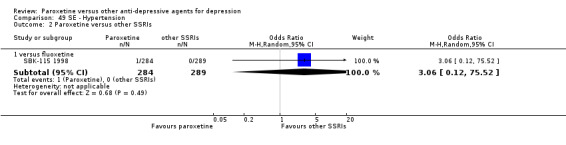
Comparison 49 SE ‐ Hypertension, Outcome 2 Paroxetine versus other SSRIs.
49.3. Analysis.
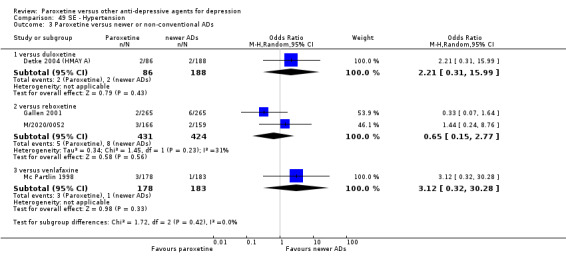
Comparison 49 SE ‐ Hypertension, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 50. SE ‐ Hypotension.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 6 | 670 | Odds Ratio (M‐H, Random, 95% CI) | 1.07 [0.30, 3.84] |
| 1.1 versus amitriptyline | 1 | 153 | Odds Ratio (M‐H, Random, 95% CI) | 0.31 [0.03, 3.07] |
| 1.2 versus clomipramine | 2 | 175 | Odds Ratio (M‐H, Random, 95% CI) | 2.46 [0.35, 17.21] |
| 1.3 versus imipramine | 2 | 282 | Odds Ratio (M‐H, Random, 95% CI) | 0.40 [0.02, 7.79] |
| 1.4 versus maprolitine | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 8.92 [0.44, 180.67] |
| 2 Paroxetine versus other SSRIs | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluoxetine | 2 | 276 | Odds Ratio (M‐H, Random, 95% CI) | 0.65 [0.18, 2.41] |
| 3 Paroxetine versus newer or non‐conventional ADs | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 0.37 [0.19, 0.75] |
| 3.2 versus trazodone | 1 | 108 | Odds Ratio (M‐H, Random, 95% CI) | 3.17 [0.13, 79.60] |
| 4 Postural hypotension | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 versus clomipramine | 1 | 92 | Odds Ratio (M‐H, Random, 95% CI) | 1.05 [0.14, 7.76] |
| 4.2 versus dothiepin | 1 | 134 | Odds Ratio (M‐H, Random, 95% CI) | 3.05 [0.12, 76.10] |
| 4.3 versus nortriptyline | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 2.65 [0.10, 66.96] |
| 4.4 versus reboxetine | 1 | 530 | Odds Ratio (M‐H, Random, 95% CI) | 0.25 [0.03, 2.23] |
50.4. Analysis.
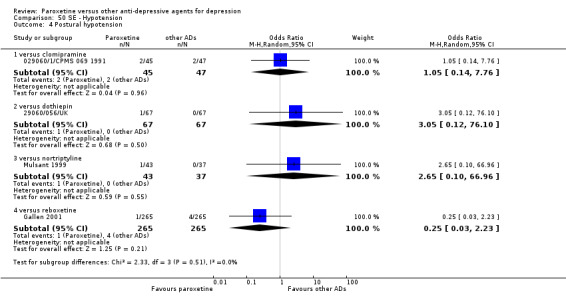
Comparison 50 SE ‐ Hypotension, Outcome 4 Postural hypotension.
Comparison 51. SE ‐ Hypertonia.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus newer or non‐conventional ADs | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.39, 1.84] |
51.1. Analysis.
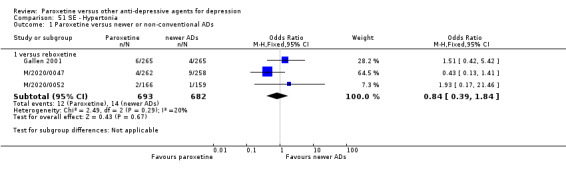
Comparison 51 SE ‐ Hypertonia, Outcome 1 Paroxetine versus newer or non‐conventional ADs.
Comparison 52. SE ‐ Infection.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Meningitis | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus amitriptyline | 1 | 162 | Odds Ratio (M‐H, Random, 95% CI) | 2.96 [0.12, 73.82] |
| 2 Otitis media | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus reboxetine | 1 | 530 | Odds Ratio (M‐H, Random, 95% CI) | 0.33 [0.03, 3.20] |
| 3 Respiratory upper | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus agomelatine | 1 | 572 | Odds Ratio (M‐H, Random, 95% CI) | 0.36 [0.04, 2.88] |
| 3.2 versus aprepitant (MK‐869) | 1 | 143 | Odds Ratio (M‐H, Random, 95% CI) | 1.52 [0.41, 5.64] |
| 3.3 versus lofepramine | 1 | 122 | Odds Ratio (M‐H, Random, 95% CI) | 3.21 [0.32, 31.76] |
| 3.4 versus tianeptine | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 8.14] |
| 4 Urinary | 9 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 versus amitriptyline | 1 | 153 | Odds Ratio (M‐H, Random, 95% CI) | 0.08 [0.00, 1.50] |
| 4.2 versus clomipramine | 1 | 92 | Odds Ratio (M‐H, Random, 95% CI) | 0.33 [0.03, 3.33] |
| 4.3 versus fluoxetine | 2 | 212 | Odds Ratio (M‐H, Random, 95% CI) | 4.39 [0.71, 27.09] |
| 4.4 versus lofepramine | 1 | 106 | Odds Ratio (M‐H, Random, 95% CI) | 6.36 [0.32, 126.19] |
| 4.5 versus mianserin | 1 | 116 | Odds Ratio (M‐H, Random, 95% CI) | 1.15 [0.07, 18.85] |
| 4.6 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 0.54 [0.19, 1.55] |
| 5 NOS | 6 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 versus amitriptyline | 1 | 217 | Odds Ratio (M‐H, Random, 95% CI) | 11.42 [0.62, 209.13] |
| 5.2 versus bupropion | 1 | 100 | Odds Ratio (M‐H, Random, 95% CI) | 0.29 [0.03, 2.93] |
| 5.3 versus mirtazapine | 1 | 197 | Odds Ratio (M‐H, Random, 95% CI) | 0.65 [0.22, 1.91] |
| 5.4 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 0.64 [0.38, 1.07] |
52.1. Analysis.
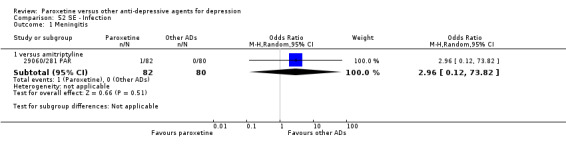
Comparison 52 SE ‐ Infection, Outcome 1 Meningitis.
52.2. Analysis.
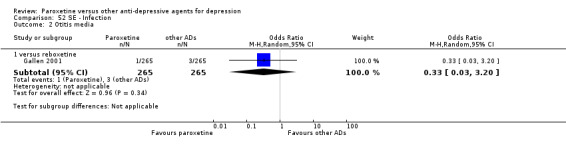
Comparison 52 SE ‐ Infection, Outcome 2 Otitis media.
52.3. Analysis.
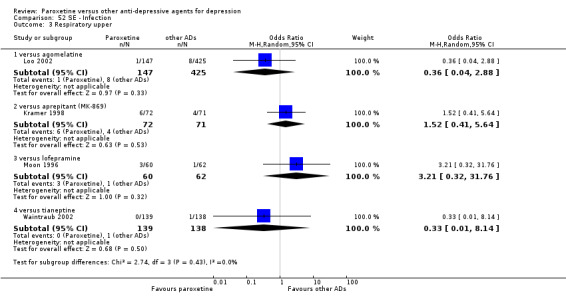
Comparison 52 SE ‐ Infection, Outcome 3 Respiratory upper.
52.4. Analysis.

Comparison 52 SE ‐ Infection, Outcome 4 Urinary.
52.5. Analysis.
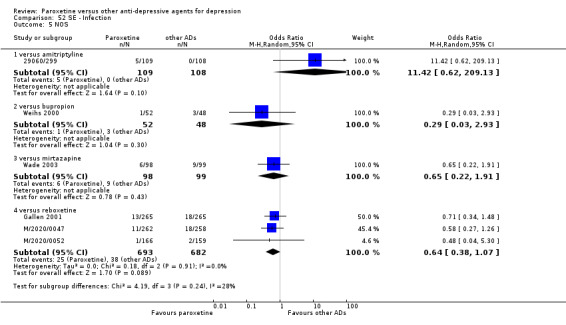
Comparison 52 SE ‐ Infection, Outcome 5 NOS.
Comparison 53. SE ‐ Influenza‐like symptoms.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus newer or non‐conventional ADs | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus nefazodone | 1 | 40 | Odds Ratio (M‐H, Random, 95% CI) | 2.25 [0.36, 13.97] |
| 1.2 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 2.01 [0.84, 4.80] |
53.1. Analysis.
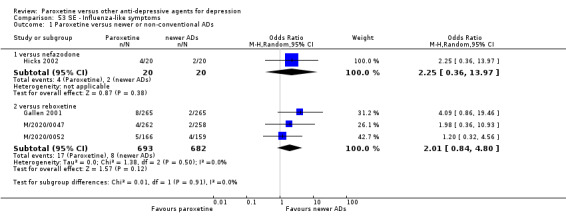
Comparison 53 SE ‐ Influenza‐like symptoms, Outcome 1 Paroxetine versus newer or non‐conventional ADs.
Comparison 54. SE ‐ Insomnia.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 15 | 1986 | Odds Ratio (M‐H, Random, 95% CI) | 2.17 [1.51, 3.12] |
| 1.1 versus amitriptyline | 4 | 352 | Odds Ratio (M‐H, Random, 95% CI) | 3.66 [1.36, 9.85] |
| 1.2 versus clomipramine | 1 | 92 | Odds Ratio (M‐H, Random, 95% CI) | 4.49 [0.48, 41.79] |
| 1.3 versus dothiepin | 2 | 405 | Odds Ratio (M‐H, Random, 95% CI) | 2.34 [1.03, 5.31] |
| 1.4 versus imipramine | 5 | 601 | Odds Ratio (M‐H, Random, 95% CI) | 1.54 [0.89, 2.66] |
| 1.5 versus lofepramine | 1 | 122 | Odds Ratio (M‐H, Random, 95% CI) | 0.5 [0.09, 2.84] |
| 1.6 versus maprotiline | 1 | 298 | Odds Ratio (M‐H, Random, 95% CI) | 4.38 [1.72, 11.15] |
| 1.7 versus mianserin | 1 | 116 | Odds Ratio (M‐H, Random, 95% CI) | 3.50 [0.14, 87.83] |
| 2 Paroxetine versus other SSRIs | 11 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus citalopram | 1 | 406 | Odds Ratio (M‐H, Random, 95% CI) | 1.09 [0.63, 1.89] |
| 2.2 versus fluoxetine | 7 | 1994 | Odds Ratio (M‐H, Random, 95% CI) | 0.93 [0.74, 1.16] |
| 2.3 versus fluvoxamine | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 0.58 [0.18, 1.91] |
| 2.4 versus sertraline | 2 | 545 | Odds Ratio (M‐H, Random, 95% CI) | 0.95 [0.61, 1.48] |
| 3 Paroxetine versus newer or non‐conventional ADs | 18 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus agomelatine | 1 | 284 | Odds Ratio (M‐H, Random, 95% CI) | 1.66 [0.48, 5.81] |
| 3.2 versus aprepitant (MK‐869) | 1 | 143 | Odds Ratio (M‐H, Random, 95% CI) | 0.76 [0.28, 2.06] |
| 3.3 versus bupropion | 2 | 240 | Odds Ratio (M‐H, Random, 95% CI) | 1.19 [0.61, 2.33] |
| 3.4 versus duloxetine | 4 | 1095 | Odds Ratio (M‐H, Random, 95% CI) | 0.62 [0.37, 1.04] |
| 3.5 versus milnacipran | 1 | 302 | Odds Ratio (M‐H, Random, 95% CI) | 1.96 [0.18, 21.85] |
| 3.6 versus mirtazapine | 2 | 451 | Odds Ratio (M‐H, Random, 95% CI) | 1.46 [0.54, 3.95] |
| 3.7 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 0.48 [0.31, 0.74] |
| 3.8 versus tianeptine | 2 | 604 | Odds Ratio (M‐H, Random, 95% CI) | 1.32 [0.06, 31.66] |
| 3.9 versus trazodone | 1 | 108 | Odds Ratio (M‐H, Random, 95% CI) | 1.04 [0.20, 5.40] |
| 3.10 versus venlafaxine | 1 | 361 | Odds Ratio (M‐H, Random, 95% CI) | 0.81 [0.31, 2.11] |
Comparison 55. SE ‐ Metabolic and nutritional system.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 5 | 441 | Odds Ratio (M‐H, Random, 95% CI) | 1.34 [0.50, 3.55] |
| 1.1 versus amitriptyline | 2 | 221 | Odds Ratio (M‐H, Random, 95% CI) | 0.98 [0.30, 3.19] |
| 1.2 versus clomipramine | 1 | 83 | Odds Ratio (M‐H, Random, 95% CI) | 1.03 [0.06, 16.95] |
| 1.3 versus nortriptyline | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 2.65 [0.10, 66.96] |
| 1.4 versus mianserin | 1 | 57 | Odds Ratio (M‐H, Random, 95% CI) | 7.53 [0.37, 152.73] |
| 2 Paroxetine versus other SSRIs | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluoxetine | 2 | 1281 | Odds Ratio (M‐H, Random, 95% CI) | 1.00 [0.10, 9.64] |
| 3 Paroxetine versus newer or non‐conventional ADs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus amisulpride | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 1.62 [0.52, 5.09] |
55.1. Analysis.
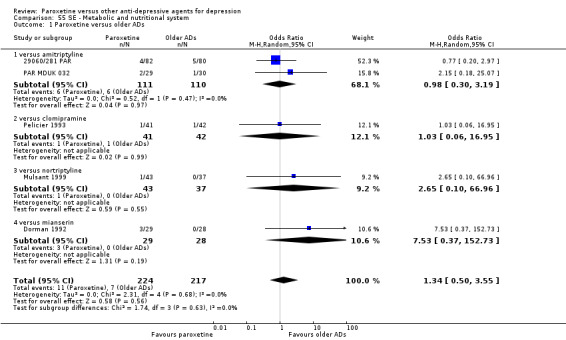
Comparison 55 SE ‐ Metabolic and nutritional system, Outcome 1 Paroxetine versus older ADs.
55.2. Analysis.
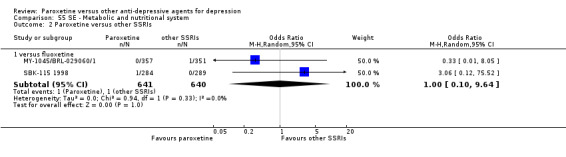
Comparison 55 SE ‐ Metabolic and nutritional system, Outcome 2 Paroxetine versus other SSRIs.
55.3. Analysis.
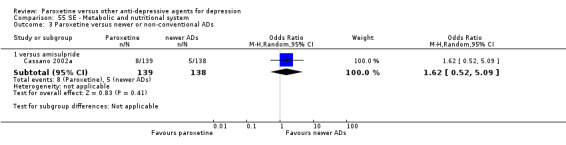
Comparison 55 SE ‐ Metabolic and nutritional system, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 56. SE ‐ Musculoskeletal and connective tissue disorders.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 3 | 566 | Odds Ratio (M‐H, Random, 95% CI) | 1.34 [0.26, 7.03] |
| 1.1 versus amitriptyline | 1 | 162 | Odds Ratio (M‐H, Random, 95% CI) | 1.98 [0.18, 22.22] |
| 1.2 versus lofepramine | 1 | 106 | Odds Ratio (M‐H, Random, 95% CI) | 0.28 [0.01, 7.06] |
| 1.3 versus maprotiline | 1 | 298 | Odds Ratio (M‐H, Random, 95% CI) | 3.19 [0.13, 78.86] |
| 2 Paroxetine versus newer or non‐conventional ADs | 5 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus duloxetine | 3 | 802 | Odds Ratio (M‐H, Random, 95% CI) | 0.89 [0.36, 2.20] |
| 2.2 versus mirtazapine | 1 | 197 | Odds Ratio (M‐H, Random, 95% CI) | 1.83 [0.52, 6.45] |
| 2.3 versus reboxetine | 1 | 530 | Odds Ratio (M‐H, Random, 95% CI) | 3.01 [0.12, 74.26] |
56.1. Analysis.
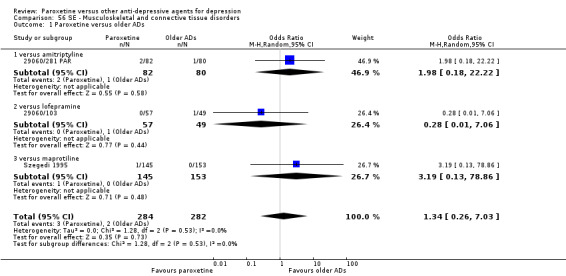
Comparison 56 SE ‐ Musculoskeletal and connective tissue disorders, Outcome 1 Paroxetine versus older ADs.
56.2. Analysis.
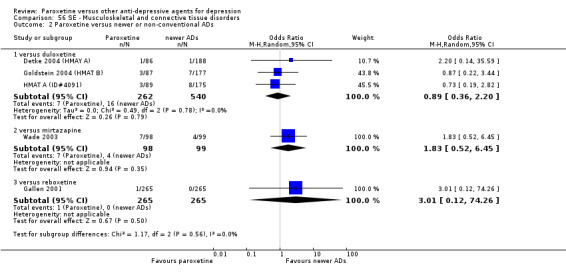
Comparison 56 SE ‐ Musculoskeletal and connective tissue disorders, Outcome 2 Paroxetine versus newer or non‐conventional ADs.
Comparison 57. SE ‐ Myalgia.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus other SSRIs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus fluoxetine | 1 | 90 | Odds Ratio (M‐H, Random, 95% CI) | 0.13 [0.01, 2.66] |
| 2 Paroxetine versus newer or non‐conventional ADs | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus reboxetine | 2 | 1050 | Odds Ratio (M‐H, Random, 95% CI) | 0.65 [0.16, 2.56] |
57.1. Analysis.
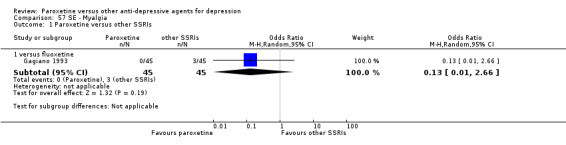
Comparison 57 SE ‐ Myalgia, Outcome 1 Paroxetine versus other SSRIs.
57.2. Analysis.
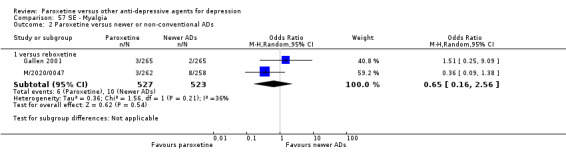
Comparison 57 SE ‐ Myalgia, Outcome 2 Paroxetine versus newer or non‐conventional ADs.
Comparison 58. SE ‐ Nervousness.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus imipramine | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 1.37 [0.29, 6.56] |
| 2 Paroxetine versus other SSRIs | 6 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluoxetine | 4 | 1660 | Odds Ratio (M‐H, Random, 95% CI) | 1.07 [0.75, 1.52] |
| 2.2 versus fluvoxamine | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 1.3 [0.31, 5.40] |
| 2.3 versus sertraline | 1 | 192 | Odds Ratio (M‐H, Random, 95% CI) | 0.93 [0.43, 2.00] |
| 3 Paroxetine versus newer or non‐conventional ADs | 7 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus aprepitant (MK‐869) | 1 | 143 | Odds Ratio (M‐H, Random, 95% CI) | 4.12 [0.45, 37.78] |
| 3.2 versus milnacipran | 1 | 302 | Odds Ratio (M‐H, Random, 95% CI) | 2.96 [0.30, 28.78] |
| 3.3 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 0.80 [0.48, 1.35] |
| 3.4 versus duloxetine | 2 | 528 | Odds Ratio (M‐H, Random, 95% CI) | 0.56 [0.22, 1.47] |
58.1. Analysis.
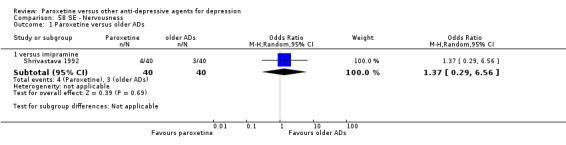
Comparison 58 SE ‐ Nervousness, Outcome 1 Paroxetine versus older ADs.
58.2. Analysis.
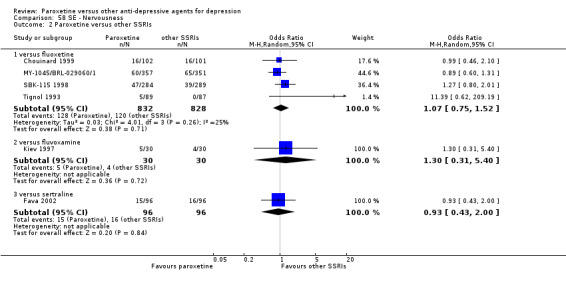
Comparison 58 SE ‐ Nervousness, Outcome 2 Paroxetine versus other SSRIs.
58.3. Analysis.
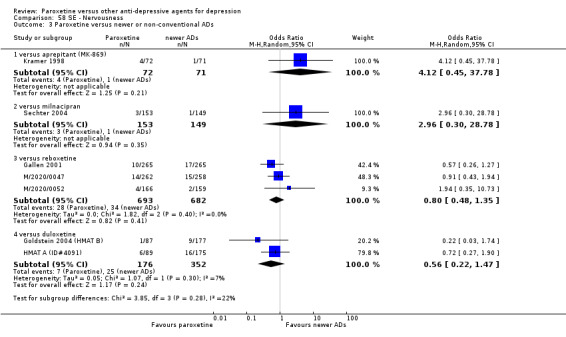
Comparison 58 SE ‐ Nervousness, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 59. SE ‐ Nausea/vomiting.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 30 | 4545 | Odds Ratio (M‐H, Random, 95% CI) | 2.10 [1.59, 2.77] |
| 1.1 versus amitriptyline | 10 | 1282 | Odds Ratio (M‐H, Random, 95% CI) | 2.17 [1.43, 3.29] |
| 1.2 versus clomipramine | 2 | 1111 | Odds Ratio (M‐H, Random, 95% CI) | 1.29 [0.94, 1.78] |
| 1.3 versus dothiepin | 2 | 405 | Odds Ratio (M‐H, Random, 95% CI) | 3.12 [1.11, 8.78] |
| 1.4 versus imipramine | 8 | 835 | Odds Ratio (M‐H, Random, 95% CI) | 2.05 [1.23, 3.42] |
| 1.5 versus lofepramine | 2 | 228 | Odds Ratio (M‐H, Random, 95% CI) | 2.97 [1.12, 7.92] |
| 1.6 versus maprolitine | 3 | 429 | Odds Ratio (M‐H, Random, 95% CI) | 2.56 [0.86, 7.60] |
| 1.7 versus mianserin | 2 | 175 | Odds Ratio (M‐H, Random, 95% CI) | 0.55 [0.04, 8.50] |
| 1.8 versus nortriptyline | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 4.52 [0.21, 97.16] |
| 2 Paroxetine versus other SSRIs | 15 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus citalopram | 1 | 406 | Odds Ratio (M‐H, Random, 95% CI) | 0.87 [0.51, 1.48] |
| 2.2 versus escitalopram | 1 | 454 | Odds Ratio (M‐H, Random, 95% CI) | 1.05 [0.69, 1.60] |
| 2.3 versus fluoxetine | 10 | 2336 | Odds Ratio (M‐H, Random, 95% CI) | 1.24 [1.02, 1.51] |
| 2.4 versus fluvoxamine | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 1.51 [0.54, 4.24] |
| 2.5 versus sertraline | 2 | 545 | Odds Ratio (M‐H, Random, 95% CI) | 0.89 [0.52, 1.54] |
| 3 Paroxetine versus newer or non‐conventional ADs | 23 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus agomelatine | 1 | 284 | Odds Ratio (M‐H, Random, 95% CI) | 6.81 [2.31, 20.14] |
| 3.2 versus amisulpride | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 7.13 [2.06, 24.68] |
| 3.3 versus aprepitant (MK‐869) | 1 | 143 | Odds Ratio (M‐H, Random, 95% CI) | 1.72 [0.78, 3.79] |
| 3.4 versus bupropion | 2 | 240 | Odds Ratio (M‐H, Random, 95% CI) | 1.22 [0.65, 2.27] |
| 3.5 versus duloxetine | 5 | 1573 | Odds Ratio (M‐H, Random, 95% CI) | 0.68 [0.52, 0.88] |
| 3.6 versus hypericum | 1 | 251 | Odds Ratio (M‐H, Random, 95% CI) | 2.58 [1.13, 5.88] |
| 3.7 versus milnacipran | 1 | 302 | Odds Ratio (M‐H, Random, 95% CI) | 0.97 [0.06, 15.71] |
| 3.8 versus mirtazapine | 3 | 726 | Odds Ratio (M‐H, Random, 95% CI) | 3.03 [1.91, 4.82] |
| 3.9 versus nefazodone | 1 | 206 | Odds Ratio (M‐H, Random, 95% CI) | 0.71 [0.30, 1.69] |
| 3.10 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 2.07 [1.60, 2.69] |
| 3.11 versus tianeptine | 2 | 604 | Odds Ratio (M‐H, Random, 95% CI) | 2.54 [1.38, 4.67] |
| 3.12 versus trazodone | 1 | 108 | Odds Ratio (M‐H, Random, 95% CI) | 6.89 [0.80, 59.35] |
| 3.13 versus venlafaxine | 1 | 361 | Odds Ratio (M‐H, Random, 95% CI) | 1.04 [0.67, 1.63] |
Comparison 60. SE ‐ Neoplasm.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Basal cell carcinoma | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus fluoxetine | 1 | 573 | Odds Ratio (M‐H, Random, 95% CI) | 0.20 [0.01, 4.23] |
| 2 Bone | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus amitriptyline | 1 | 217 | Odds Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 8.12] |
| 3 Meningioma | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus fluoxetine | 1 | 176 | Odds Ratio (M‐H, Random, 95% CI) | 0.32 [0.01, 8.02] |
| 4 Pulmonary | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 versus lofepramine | 1 | 106 | Odds Ratio (M‐H, Random, 95% CI) | 0.28 [0.01, 7.06] |
| 5 NOS | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 versus clomipramine | 1 | 92 | Odds Ratio (M‐H, Random, 95% CI) | 3.20 [0.13, 80.68] |
| 5.2 versus fluoxetine | 2 | 1281 | Odds Ratio (M‐H, Random, 95% CI) | 1.49 [0.24, 9.15] |
60.1. Analysis.
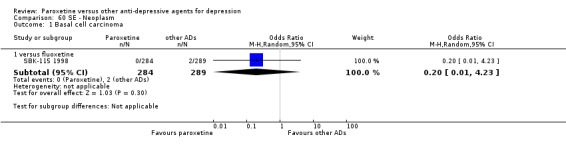
Comparison 60 SE ‐ Neoplasm, Outcome 1 Basal cell carcinoma.
60.2. Analysis.
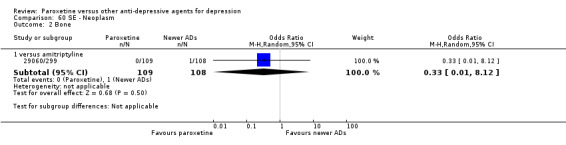
Comparison 60 SE ‐ Neoplasm, Outcome 2 Bone.
60.3. Analysis.
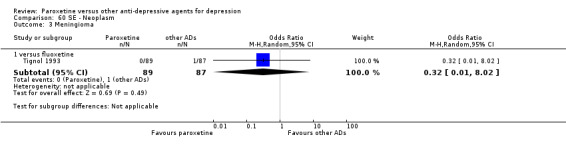
Comparison 60 SE ‐ Neoplasm, Outcome 3 Meningioma.
60.4. Analysis.
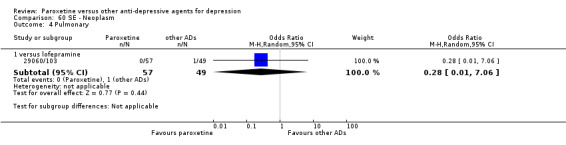
Comparison 60 SE ‐ Neoplasm, Outcome 4 Pulmonary.
60.5. Analysis.
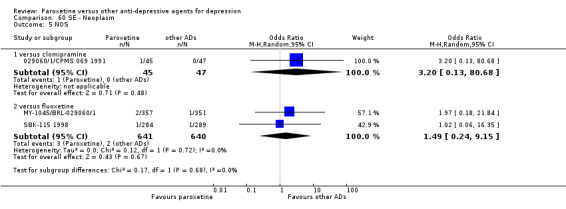
Comparison 60 SE ‐ Neoplasm, Outcome 5 NOS.
Comparison 61. SE ‐ Nervous system.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Amnesia | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus fluoxetine | 1 | 122 | Odds Ratio (M‐H, Random, 95% CI) | 1.58 [0.25, 9.80] |
| 1.2 versus reboxetine | 2 | 855 | Odds Ratio (M‐H, Random, 95% CI) | 4.87 [0.83, 28.50] |
| 2 Concentration impaired | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus reboxetine | 1 | 325 | Odds Ratio (M‐H, Random, 95% CI) | 4.85 [0.23, 101.77] |
| 3 Hyperesthesia | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus reboxetine | 2 | 1050 | Odds Ratio (M‐H, Random, 95% CI) | 0.75 [0.05, 10.74] |
| 4 Parkinsonism | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 versus nortriptyline | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 0.28 [0.01, 7.08] |
| 5 Seizure | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 versus nortriptyline | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 0.28 [0.01, 7.08] |
| 6 Stroke | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 versus lofepramine | 1 | 106 | Odds Ratio (M‐H, Random, 95% CI) | 0.28 [0.01, 7.06] |
| 7 NOS | 11 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 versus amitriptyline | 3 | 261 | Odds Ratio (M‐H, Random, 95% CI) | 0.76 [0.46, 1.26] |
| 7.2 versus clomipramine | 1 | 83 | Odds Ratio (M‐H, Random, 95% CI) | 0.49 [0.19, 1.23] |
| 7.3 versus fluoxetine | 2 | 342 | Odds Ratio (M‐H, Random, 95% CI) | 0.38 [0.14, 1.04] |
| 7.4 versus imipramine | 1 | 202 | Odds Ratio (M‐H, Random, 95% CI) | 0.24 [0.04, 1.36] |
| 7.5 versus maprolitine | 1 | 71 | Odds Ratio (M‐H, Random, 95% CI) | 1.5 [0.47, 4.78] |
| 7.6 versus mianserin | 2 | 90 | Odds Ratio (M‐H, Random, 95% CI) | 0.25 [0.09, 0.68] |
| 7.7 versus sertraline | 1 | 353 | Odds Ratio (M‐H, Random, 95% CI) | 0.62 [0.23, 1.63] |
61.1. Analysis.
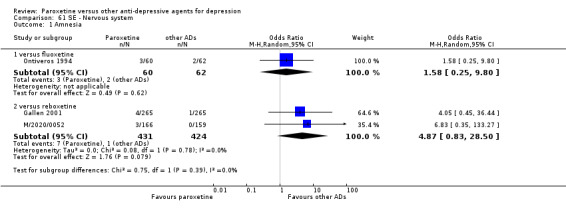
Comparison 61 SE ‐ Nervous system, Outcome 1 Amnesia.
61.2. Analysis.
Comparison 61 SE ‐ Nervous system, Outcome 2 Concentration impaired.
61.3. Analysis.
Comparison 61 SE ‐ Nervous system, Outcome 3 Hyperesthesia.
61.4. Analysis.
Comparison 61 SE ‐ Nervous system, Outcome 4 Parkinsonism.
61.5. Analysis.
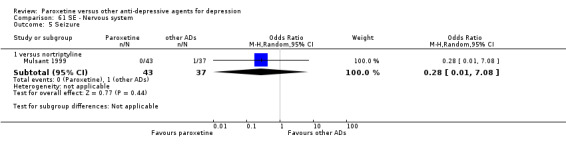
Comparison 61 SE ‐ Nervous system, Outcome 5 Seizure.
61.6. Analysis.
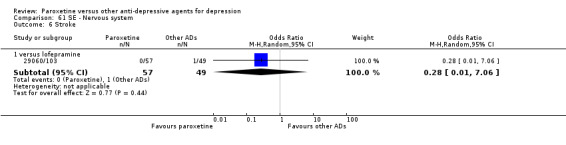
Comparison 61 SE ‐ Nervous system, Outcome 6 Stroke.
61.7. Analysis.
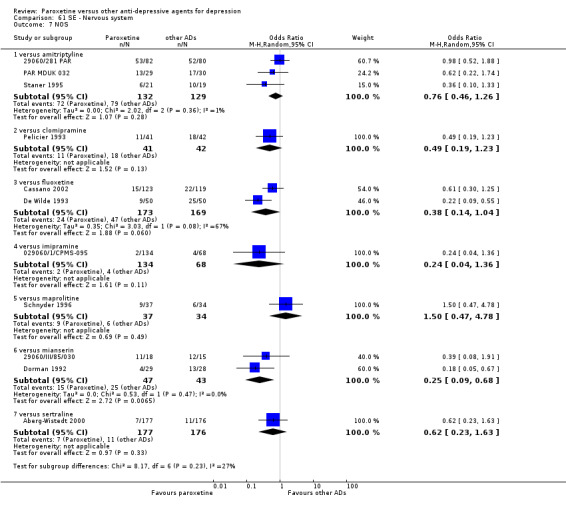
Comparison 61 SE ‐ Nervous system, Outcome 7 NOS.
Comparison 62. SE ‐ Pain (back).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus maprotiline | 1 | 298 | Odds Ratio (M‐H, Random, 95% CI) | 3.19 [0.13, 78.86] |
| 1.2 versus mianserin | 1 | 116 | Odds Ratio (M‐H, Random, 95% CI) | 3.50 [0.14, 87.83] |
| 2 Paroxetine versus other SSRIs | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluoxetine | 2 | 663 | Odds Ratio (M‐H, Random, 95% CI) | 0.63 [0.30, 1.31] |
| 3 Paroxetine versus newer or non‐conventional ADs | 8 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus duloxetine | 1 | 293 | Odds Ratio (M‐H, Random, 95% CI) | 1.35 [0.22, 8.24] |
| 3.2 versus duloxetine | 4 | 1095 | Odds Ratio (M‐H, Random, 95% CI) | 0.85 [0.37, 1.97] |
| 3.3 versus mirtazapine | 1 | 197 | Odds Ratio (M‐H, Random, 95% CI) | 1.24 [0.51, 3.02] |
| 3.4 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 1.36 [0.71, 2.62] |
62.1. Analysis.
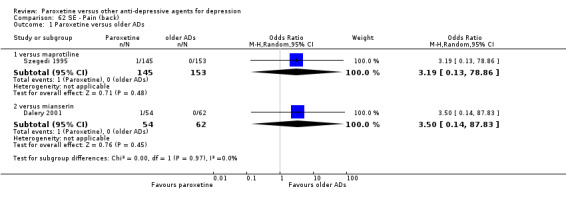
Comparison 62 SE ‐ Pain (back), Outcome 1 Paroxetine versus older ADs.
62.2. Analysis.
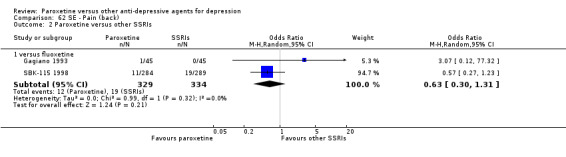
Comparison 62 SE ‐ Pain (back), Outcome 2 Paroxetine versus other SSRIs.
62.3. Analysis.
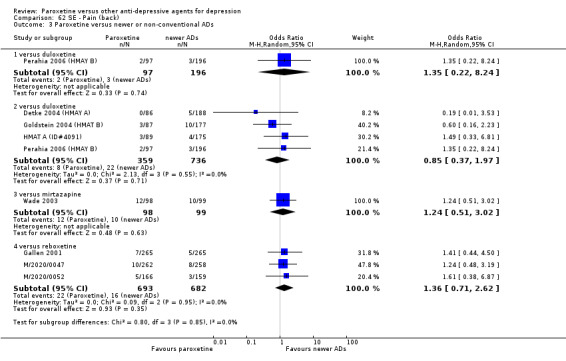
Comparison 62 SE ‐ Pain (back), Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 63. SE ‐ Pain (chest).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 3 | 235 | Odds Ratio (M‐H, Random, 95% CI) | 1.11 [0.17, 7.08] |
| 1.1 versus amitriptyline | 1 | 21 | Odds Ratio (M‐H, Random, 95% CI) | 3.0 [0.11, 82.40] |
| 1.2 versus dothiepin | 1 | 134 | Odds Ratio (M‐H, Random, 95% CI) | 3.05 [0.12, 76.10] |
| 1.3 versus imipramine | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 0.19 [0.01, 4.09] |
| 2 Paroxetine versus other SSRIs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluoxetine | 1 | 90 | Odds Ratio (M‐H, Random, 95% CI) | 7.49 [0.38, 149.40] |
| 3 Paroxetine versus newer or non‐conventional ADs | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus duloxetine | 1 | 264 | Odds Ratio (M‐H, Random, 95% CI) | 1.49 [0.33, 6.81] |
| 3.2 versus reboxetine | 2 | 1050 | Odds Ratio (M‐H, Random, 95% CI) | 0.86 [0.31, 2.40] |
63.1. Analysis.
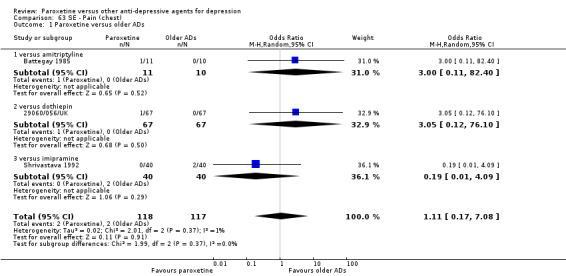
Comparison 63 SE ‐ Pain (chest), Outcome 1 Paroxetine versus older ADs.
63.2. Analysis.
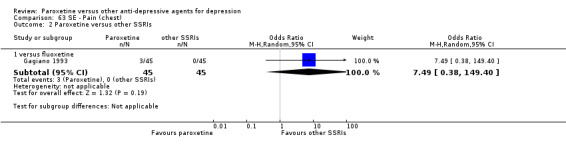
Comparison 63 SE ‐ Pain (chest), Outcome 2 Paroxetine versus other SSRIs.
63.3. Analysis.
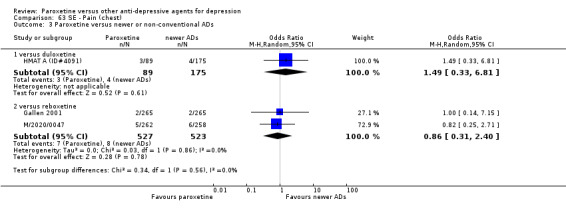
Comparison 63 SE ‐ Pain (chest), Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 64. SE ‐ Pain (abdominal).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 5 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus amitriptyline | 2 | 330 | Odds Ratio (M‐H, Random, 95% CI) | 0.61 [0.18, 2.07] |
| 1.2 versus imipramine | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 0.19 [0.01, 4.09] |
| 1.3 versus maprolitine | 2 | 369 | Odds Ratio (M‐H, Random, 95% CI) | 1.58 [0.76, 3.28] |
| 2 Paroxetine versus other SSRIs | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluoxetine | 3 | 1022 | Odds Ratio (M‐H, Random, 95% CI) | 0.55 [0.20, 1.53] |
| 3 Paroxetine versus newer or non‐conventional ADs | 13 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus agomelatine | 1 | 284 | Odds Ratio (M‐H, Random, 95% CI) | 0.93 [0.26, 3.28] |
| 3.2 versus aprepitant (MK‐869) | 1 | 143 | Odds Ratio (M‐H, Random, 95% CI) | 0.64 [0.17, 2.36] |
| 3.3 versus bupropion | 1 | 100 | Odds Ratio (M‐H, Random, 95% CI) | 0.92 [0.22, 3.89] |
| 3.4 versus duloxetine | 4 | 1095 | Odds Ratio (M‐H, Random, 95% CI) | 0.75 [0.33, 1.69] |
| 3.5 versus hypericum | 1 | 251 | Odds Ratio (M‐H, Random, 95% CI) | 0.72 [0.29, 1.79] |
| 3.6 versus mirtazapine | 1 | 197 | Odds Ratio (M‐H, Random, 95% CI) | 1.01 [0.34, 3.00] |
| 3.7 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 1.04 [0.59, 1.83] |
| 3.8 versus tianeptine | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 0.39 [0.07, 2.04] |
64.1. Analysis.
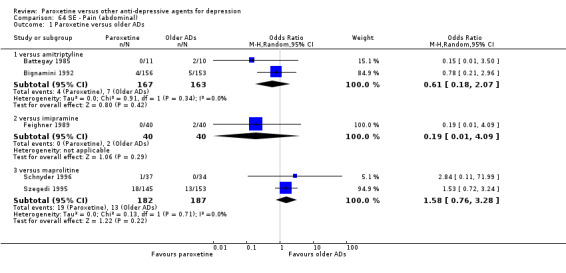
Comparison 64 SE ‐ Pain (abdominal), Outcome 1 Paroxetine versus older ADs.
64.2. Analysis.
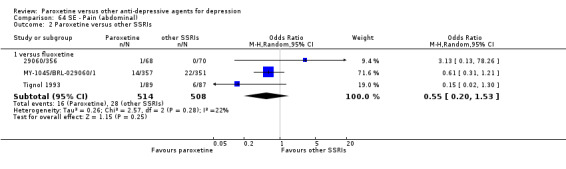
Comparison 64 SE ‐ Pain (abdominal), Outcome 2 Paroxetine versus other SSRIs.
64.3. Analysis.
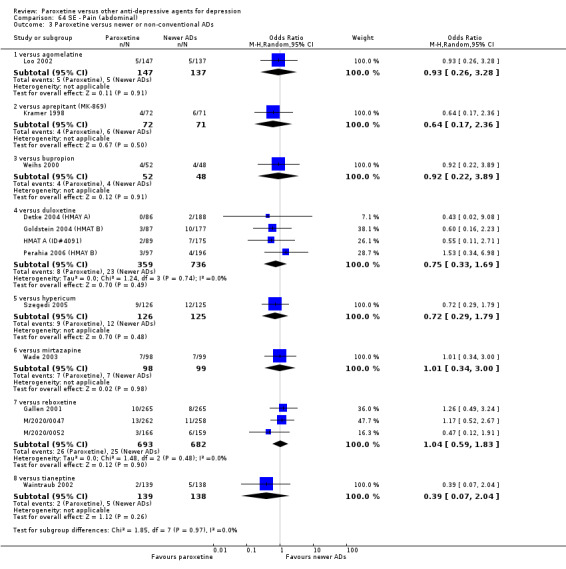
Comparison 64 SE ‐ Pain (abdominal), Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 65. SE ‐ Pain (general).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus dothiepin | 1 | 134 | Odds Ratio (M‐H, Random, 95% CI) | 9.57 [0.50, 181.29] |
| 1.2 versus mianserin | 1 | 116 | Odds Ratio (M‐H, Random, 95% CI) | 3.50 [0.14, 87.83] |
| 2 Paroxetine versus SSRIs | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluoxetine | 1 | 90 | Odds Ratio (M‐H, Random, 95% CI) | 7.49 [0.38, 149.40] |
| 2.2 versus sertraline | 2 | 545 | Odds Ratio (M‐H, Random, 95% CI) | 1.54 [0.26, 9.00] |
| 3 Paroxetine versus newer or non‐conventional ADs | 7 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus duloxetine | 2 | 530 | Odds Ratio (M‐H, Random, 95% CI) | 1.48 [0.74, 2.94] |
| 3.2 versus mirtazapine | 1 | 197 | Odds Ratio (M‐H, Random, 95% CI) | 0.56 [0.16, 1.97] |
| 3.3 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 0.93 [0.27, 3.25] |
| 3.4 versus tianeptine | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 8.14] |
65.1. Analysis.
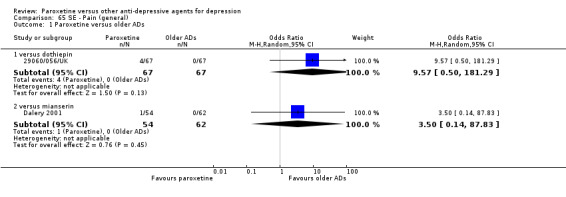
Comparison 65 SE ‐ Pain (general), Outcome 1 Paroxetine versus older ADs.
65.2. Analysis.
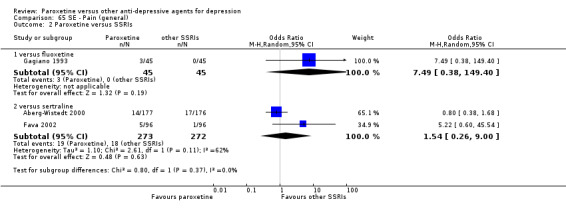
Comparison 65 SE ‐ Pain (general), Outcome 2 Paroxetine versus SSRIs.
65.3. Analysis.
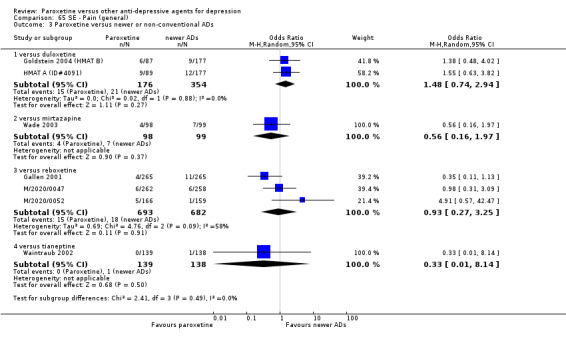
Comparison 65 SE ‐ Pain (general), Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 66. SE ‐ Pain (neck).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus newer or non‐conventional ADs | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus duloxetine | 1 | 264 | Odds Ratio (M‐H, Random, 95% CI) | 0.13 [0.01, 2.30] |
| 1.2 versus reboxetine | 1 | 325 | Odds Ratio (M‐H, Random, 95% CI) | 1.93 [0.17, 21.46] |
66.1. Analysis.
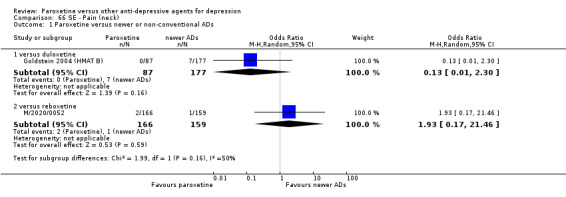
Comparison 66 SE ‐ Pain (neck), Outcome 1 Paroxetine versus newer or non‐conventional ADs.
Comparison 67. SE ‐ Palpitations.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 9 | 1171 | Odds Ratio (M‐H, Random, 95% CI) | 0.63 [0.27, 1.51] |
| 1.1 versus amitriptyline | 3 | 374 | Odds Ratio (M‐H, Random, 95% CI) | 0.42 [0.06, 2.74] |
| 1.2 versus dothiepin | 1 | 134 | Odds Ratio (M‐H, Random, 95% CI) | 7.33 [0.37, 144.63] |
| 1.3 versus imipramine | 3 | 441 | Odds Ratio (M‐H, Random, 95% CI) | 0.54 [0.18, 1.61] |
| 1.4 versus lofepramine | 1 | 106 | Odds Ratio (M‐H, Random, 95% CI) | 0.17 [0.01, 3.53] |
| 1.5 versus mianserin | 1 | 116 | Odds Ratio (M‐H, Random, 95% CI) | 3.50 [0.14, 87.83] |
| 2 Paroxetine versus other SSRIs | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluoxetine | 1 | 100 | Odds Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 8.21] |
| 2.2 versus fluvoxamine | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 0.22 [0.02, 2.14] |
| 3 Paroxetine versus newer or non‐conventional ADs | 7 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus duloxetine | 4 | 1280 | Odds Ratio (M‐H, Random, 95% CI) | 0.86 [0.34, 2.18] |
| 3.2 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 0.69 [0.36, 1.35] |
67.1. Analysis.
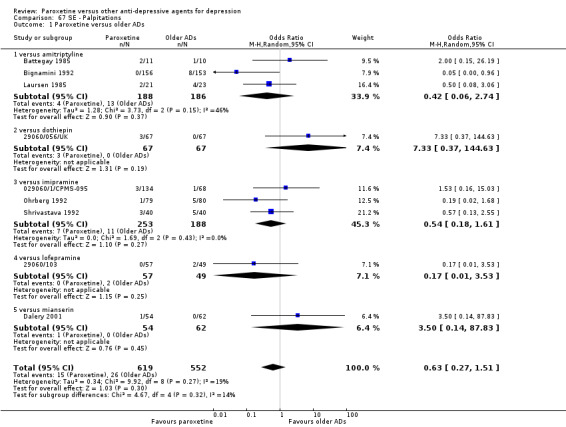
Comparison 67 SE ‐ Palpitations, Outcome 1 Paroxetine versus older ADs.
67.2. Analysis.
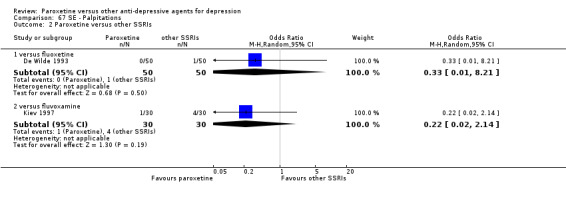
Comparison 67 SE ‐ Palpitations, Outcome 2 Paroxetine versus other SSRIs.
67.3. Analysis.
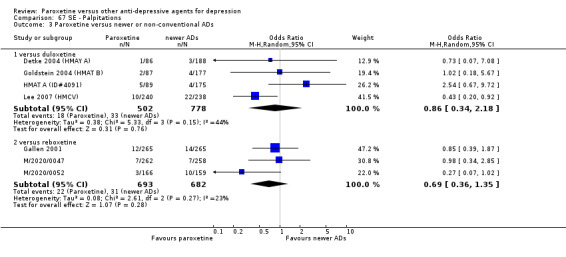
Comparison 67 SE ‐ Palpitations, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 68. SE ‐ Paraesthesia.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus amitriptyline | 1 | 21 | Odds Ratio (M‐H, Random, 95% CI) | 0.9 [0.05, 16.59] |
| 1.2 versus imipramine | 2 | 282 | Odds Ratio (M‐H, Random, 95% CI) | 0.77 [0.17, 3.43] |
| 1.3 versus maprotiline | 1 | 298 | Odds Ratio (M‐H, Random, 95% CI) | 3.19 [0.13, 78.86] |
| 2 Paroxetine versus SSRIs | 2 | 528 | Odds Ratio (M‐H, Random, 95% CI) | 1.21 [0.17, 8.41] |
| 2.1 versus duloxetine | 2 | 528 | Odds Ratio (M‐H, Random, 95% CI) | 1.21 [0.17, 8.41] |
| 3 Paroxetine versus newer or non‐conventional ADs | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 0.44 [0.20, 0.96] |
68.1. Analysis.
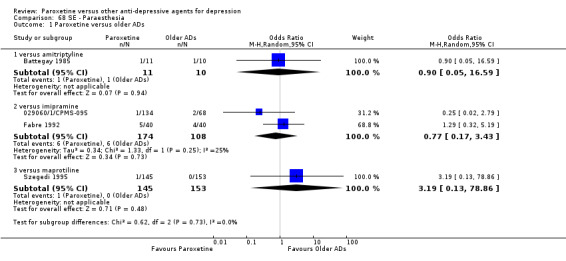
Comparison 68 SE ‐ Paraesthesia, Outcome 1 Paroxetine versus older ADs.
68.2. Analysis.
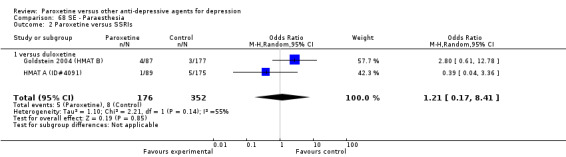
Comparison 68 SE ‐ Paraesthesia, Outcome 2 Paroxetine versus SSRIs.
68.3. Analysis.
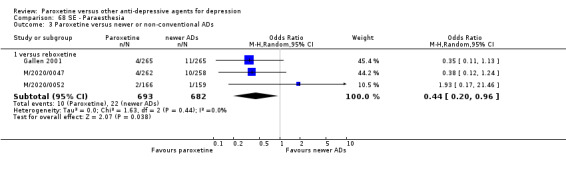
Comparison 68 SE ‐ Paraesthesia, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 69. SE ‐ Pharyngitis.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus amitriptyline | 1 | 217 | Odds Ratio (M‐H, Random, 95% CI) | 3.09 [0.61, 15.65] |
| 1.2 versus imipramine | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 3.16 [0.31, 31.78] |
| 2 Paroxetine versus SSRIs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluoxetine | 1 | 203 | Odds Ratio (M‐H, Random, 95% CI) | 3.0 [0.12, 74.52] |
| 3 Paroxetine versus newer or non‐conventional ADs | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus duloxetine | 1 | 264 | Odds Ratio (M‐H, Random, 95% CI) | 2.06 [0.29, 14.87] |
| 3.2 versus reboxetine | 2 | 1050 | Odds Ratio (M‐H, Random, 95% CI) | 0.74 [0.37, 1.48] |
69.1. Analysis.
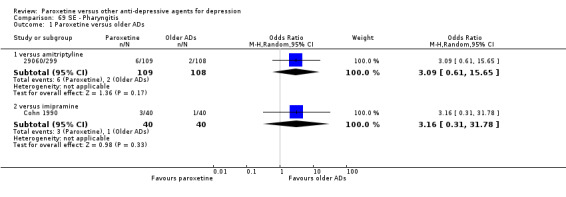
Comparison 69 SE ‐ Pharyngitis, Outcome 1 Paroxetine versus older ADs.
69.2. Analysis.
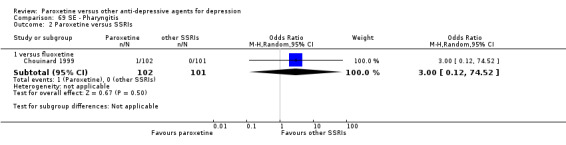
Comparison 69 SE ‐ Pharyngitis, Outcome 2 Paroxetine versus SSRIs.
69.3. Analysis.
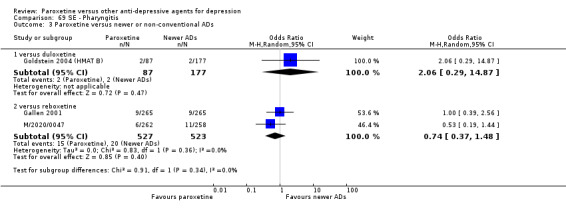
Comparison 69 SE ‐ Pharyngitis, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 70. SE ‐ Pruritus.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus other SSRIs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus fluoxetine | 1 | 176 | Odds Ratio (M‐H, Random, 95% CI) | 0.32 [0.03, 3.12] |
| 2 Paroxetine versus newer or non‐conventional ADs | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 0.81 [0.19, 3.43] |
| 2.2 versus duloxetine | 1 | 264 | Odds Ratio (M‐H, Random, 95% CI) | 1.36 [0.22, 8.32] |
70.1. Analysis.
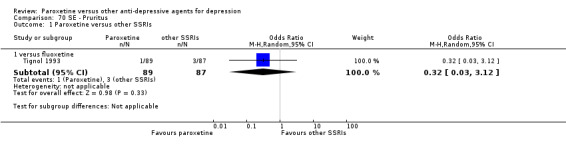
Comparison 70 SE ‐ Pruritus, Outcome 1 Paroxetine versus other SSRIs.
70.2. Analysis.
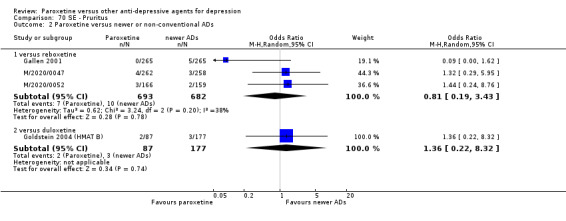
Comparison 70 SE ‐ Pruritus, Outcome 2 Paroxetine versus newer or non‐conventional ADs.
Comparison 71. SE ‐ RARE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Abdomen enlarged | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus reboxetine | 1 | 520 | Odds Ratio (M‐H, Random, 95% CI) | 0.98 [0.24, 3.98] |
| 2 Acute pyelonephritis | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluoxetine | 1 | 573 | Odds Ratio (M‐H, Random, 95% CI) | 0.20 [0.01, 4.23] |
| 3 Alcohol abuse | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus fluoxetine | 1 | 573 | Odds Ratio (M‐H, Random, 95% CI) | 3.06 [0.12, 75.52] |
| 3.2 versus maprotiline | 1 | 298 | Odds Ratio (M‐H, Random, 95% CI) | 0.35 [0.01, 8.65] |
| 3.3 versus tianeptine | 1 | 278 | Odds Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 8.19] |
| 4 Angina attack | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 versus dothiepin | 1 | 134 | Odds Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 8.21] |
| 5 Coronary artery disorder | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 versus fluoxetine | 1 | 708 | Odds Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 8.05] |
| 6 Deep thrombophlebitis | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 versus fluoxetine | 3 | 1419 | Odds Ratio (M‐H, Random, 95% CI) | 0.33 [0.05, 2.13] |
| 7 Edema generalized | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 versus reboxetine | 2 | 1050 | Odds Ratio (M‐H, Random, 95% CI) | 0.99 [0.10, 9.57] |
| 8 Balance difficulty | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 versus dothiepin | 1 | 134 | Odds Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 8.21] |
| 9 Enucleation of eyeball | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 versus dothiepin | 1 | 134 | Odds Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 8.21] |
| 10 Epistaxis | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 versus fluoxetine | 2 | 911 | Odds Ratio (M‐H, Random, 95% CI) | 0.99 [0.10, 9.53] |
| 11 Falls | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1 versus dothiepin | 1 | 134 | Odds Ratio (M‐H, Random, 95% CI) | 3.05 [0.12, 76.10] |
| 11.2 versus mianserin | 1 | 116 | Odds Ratio (M‐H, Random, 95% CI) | 3.50 [0.14, 87.83] |
| 12 Hiccup | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.1 versus tianeptine | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 3.0 [0.12, 74.28] |
| 13 Hot flushes | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.1 versus tianeptine | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 0.99 [0.06, 16.03] |
| 14 Hyperkinesia | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 14.1 versus reboxetine | 2 | 1050 | Odds Ratio (M‐H, Random, 95% CI) | 1.49 [0.42, 5.33] |
| 15 Incoordination | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 15.1 versus reboxetine | 1 | 530 | Odds Ratio (M‐H, Random, 95% CI) | 3.02 [0.31, 29.25] |
| 16 Irregular pulse | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 16.1 versus dothiepin | 1 | 134 | Odds Ratio (M‐H, Random, 95% CI) | 5.15 [0.24, 109.38] |
| 17 Light headedness | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 17.1 versus dothiepin | 1 | 134 | Odds Ratio (M‐H, Random, 95% CI) | 3.05 [0.12, 76.10] |
| 17.2 versus tianeptine | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 8.14] |
| 18 Malaise | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 18.1 versus tianeptine | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 3.0 [0.12, 74.28] |
| 19 Overdose | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 19.1 versus mianserin | 1 | 36 | Odds Ratio (M‐H, Random, 95% CI) | 0.32 [0.01, 8.27] |
| 19.2 versus fluoxetine | 1 | 138 | Odds Ratio (M‐H, Random, 95% CI) | 0.68 [0.11, 4.18] |
| 20 Peripheal vascular disorder | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 20.1 versus reboxetine | 1 | 530 | Odds Ratio (M‐H, Random, 95% CI) | 0.33 [0.03, 3.20] |
| 21 Pregnancy | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 21.1 versus amitriptyline | 1 | 217 | Odds Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 8.12] |
| 22 Psychosomatic disorders | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 22.1 versus fluoxetine | 1 | 176 | Odds Ratio (M‐H, Random, 95% CI) | 0.32 [0.01, 8.02] |
| 23 Renal failure | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 23.1 versus fluoxetine | 1 | 138 | Odds Ratio (M‐H, Random, 95% CI) | 3.13 [0.13, 78.26] |
| 24 Stage 1 coma | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 24.1 versus fluoxetine | 1 | 176 | Odds Ratio (M‐H, Random, 95% CI) | 2.97 [0.12, 73.81] |
| 25 Tooth disorder | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 25.1 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 0.90 [0.12, 6.97] |
| 26 Voice alteration | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 26.1 versus reboxetine | 1 | 325 | Odds Ratio (M‐H, Random, 95% CI) | 4.85 [0.23, 101.77] |
71.1. Analysis.
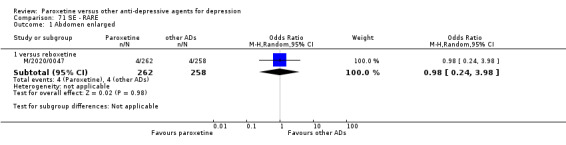
Comparison 71 SE ‐ RARE, Outcome 1 Abdomen enlarged.
71.2. Analysis.
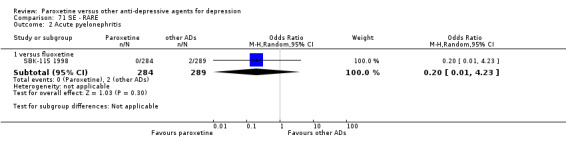
Comparison 71 SE ‐ RARE, Outcome 2 Acute pyelonephritis.
71.3. Analysis.
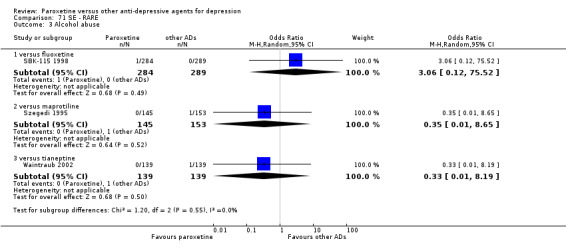
Comparison 71 SE ‐ RARE, Outcome 3 Alcohol abuse.
71.4. Analysis.
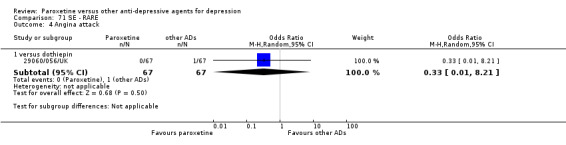
Comparison 71 SE ‐ RARE, Outcome 4 Angina attack.
71.5. Analysis.
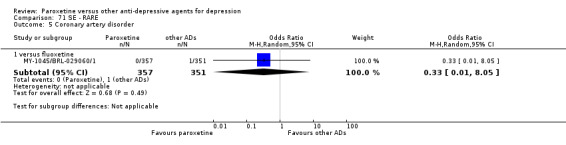
Comparison 71 SE ‐ RARE, Outcome 5 Coronary artery disorder.
71.6. Analysis.
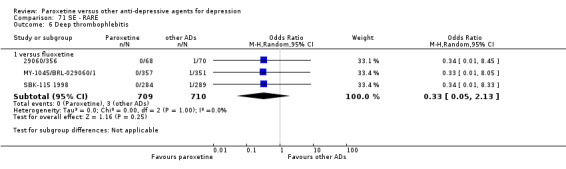
Comparison 71 SE ‐ RARE, Outcome 6 Deep thrombophlebitis.
71.7. Analysis.
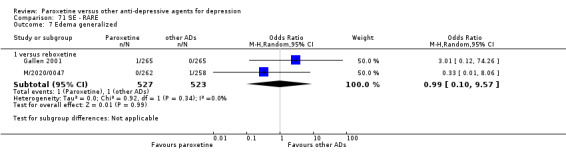
Comparison 71 SE ‐ RARE, Outcome 7 Edema generalized.
71.8. Analysis.
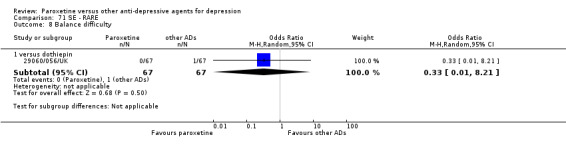
Comparison 71 SE ‐ RARE, Outcome 8 Balance difficulty.
71.9. Analysis.
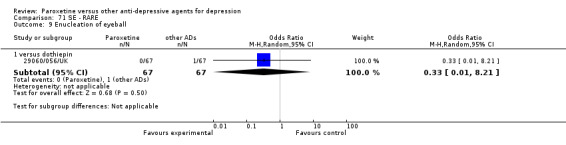
Comparison 71 SE ‐ RARE, Outcome 9 Enucleation of eyeball.
71.10. Analysis.
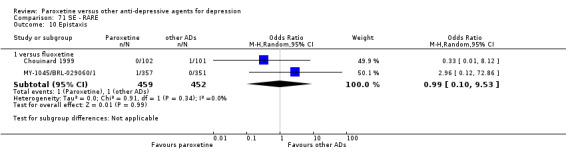
Comparison 71 SE ‐ RARE, Outcome 10 Epistaxis.
71.11. Analysis.
Comparison 71 SE ‐ RARE, Outcome 11 Falls.
71.12. Analysis.
Comparison 71 SE ‐ RARE, Outcome 12 Hiccup.
71.13. Analysis.
Comparison 71 SE ‐ RARE, Outcome 13 Hot flushes.
71.14. Analysis.
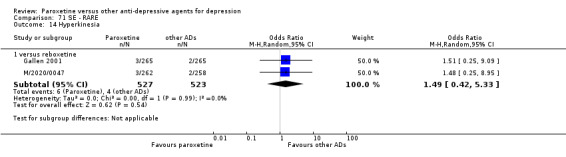
Comparison 71 SE ‐ RARE, Outcome 14 Hyperkinesia.
71.15. Analysis.
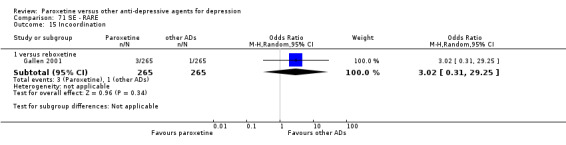
Comparison 71 SE ‐ RARE, Outcome 15 Incoordination.
71.16. Analysis.
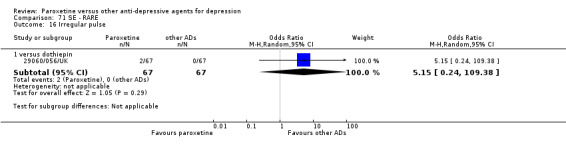
Comparison 71 SE ‐ RARE, Outcome 16 Irregular pulse.
71.17. Analysis.
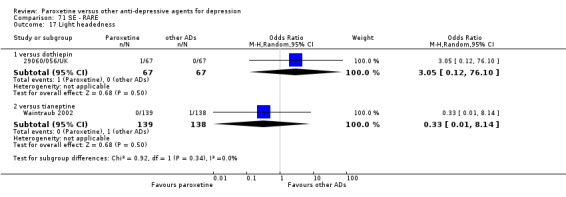
Comparison 71 SE ‐ RARE, Outcome 17 Light headedness.
71.18. Analysis.
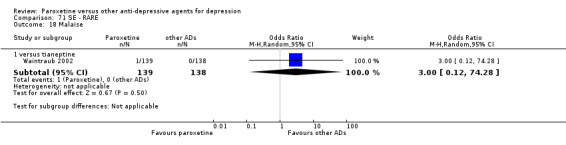
Comparison 71 SE ‐ RARE, Outcome 18 Malaise.
71.19. Analysis.
Comparison 71 SE ‐ RARE, Outcome 19 Overdose.
71.20. Analysis.
Comparison 71 SE ‐ RARE, Outcome 20 Peripheal vascular disorder.
71.21. Analysis.
Comparison 71 SE ‐ RARE, Outcome 21 Pregnancy.
71.22. Analysis.
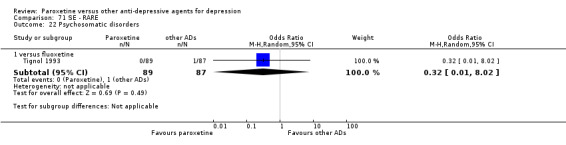
Comparison 71 SE ‐ RARE, Outcome 22 Psychosomatic disorders.
71.23. Analysis.
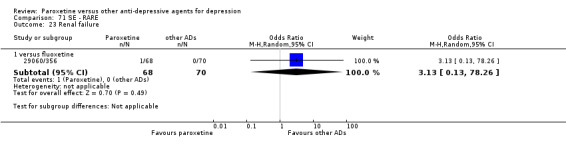
Comparison 71 SE ‐ RARE, Outcome 23 Renal failure.
71.24. Analysis.
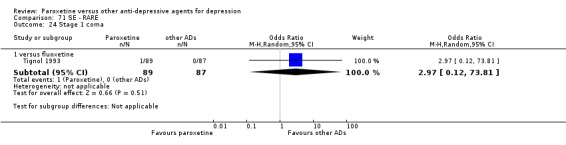
Comparison 71 SE ‐ RARE, Outcome 24 Stage 1 coma.
71.25. Analysis.
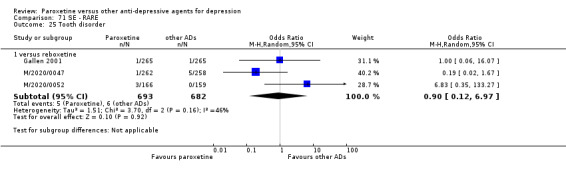
Comparison 71 SE ‐ RARE, Outcome 25 Tooth disorder.
71.26. Analysis.
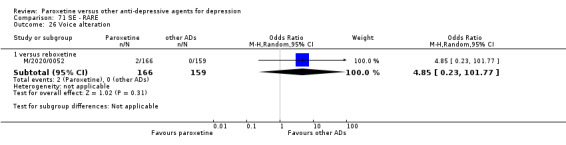
Comparison 71 SE ‐ RARE, Outcome 26 Voice alteration.
Comparison 72. SE ‐ Rash, itching, allergic reactions.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine verus older ADs | 3 | 444 | Odds Ratio (M‐H, Random, 95% CI) | 0.47 [0.08, 2.87] |
| 1.1 versus imipramine | 1 | 75 | Odds Ratio (M‐H, Random, 95% CI) | 0.13 [0.01, 2.57] |
| 1.2 versus maprolitine | 2 | 369 | Odds Ratio (M‐H, Random, 95% CI) | 0.99 [0.10, 9.63] |
| 2 Paroxetine versus other SSRIs | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus duloxetine | 1 | 264 | Odds Ratio (M‐H, Random, 95% CI) | 0.18 [0.01, 3.28] |
| 2.2 versus fluoxetine | 1 | 138 | Odds Ratio (M‐H, Random, 95% CI) | 0.34 [0.01, 8.45] |
| 3 Paroxetine versus newer or non‐conventional ADs | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 0.62 [0.36, 1.05] |
| 3.2 versus tianeptine | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 3.0 [0.12, 74.28] |
72.1. Analysis.
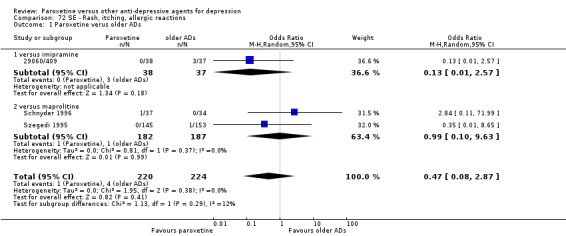
Comparison 72 SE ‐ Rash, itching, allergic reactions, Outcome 1 Paroxetine verus older ADs.
72.2. Analysis.
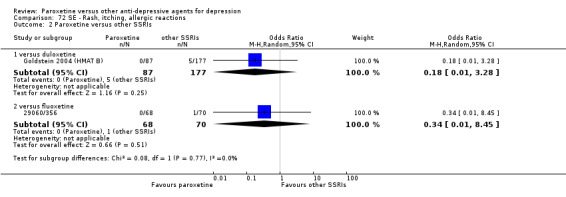
Comparison 72 SE ‐ Rash, itching, allergic reactions, Outcome 2 Paroxetine versus other SSRIs.
72.3. Analysis.
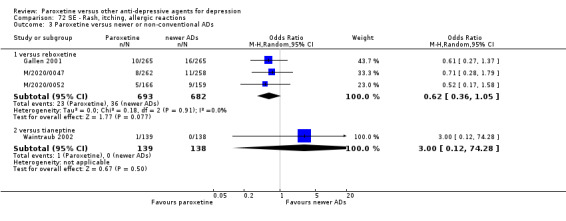
Comparison 72 SE ‐ Rash, itching, allergic reactions, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 73. SE ‐ Respiratory disorder.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 11 | 1102 | Odds Ratio (M‐H, Random, 95% CI) | 1.08 [0.54, 2.18] |
| 1.1 versus amitriptyline | 2 | 221 | Odds Ratio (M‐H, Random, 95% CI) | 0.47 [0.15, 1.42] |
| 1.2 versus dothiepin | 1 | 271 | Odds Ratio (M‐H, Random, 95% CI) | 1.35 [0.55, 3.33] |
| 1.3 vesus imipramine | 4 | 319 | Odds Ratio (M‐H, Random, 95% CI) | 2.62 [0.90, 7.64] |
| 1.4 versus lofepramine | 1 | 106 | Odds Ratio (M‐H, Random, 95% CI) | 6.36 [0.32, 126.19] |
| 1.5 versus mianserin | 3 | 185 | Odds Ratio (M‐H, Random, 95% CI) | 0.17 [0.03, 1.02] |
| 2 Paroxetine versus other SSRIs | 5 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluoxetine | 5 | 1674 | Odds Ratio (M‐H, Random, 95% CI) | 0.95 [0.66, 1.35] |
73.1. Analysis.
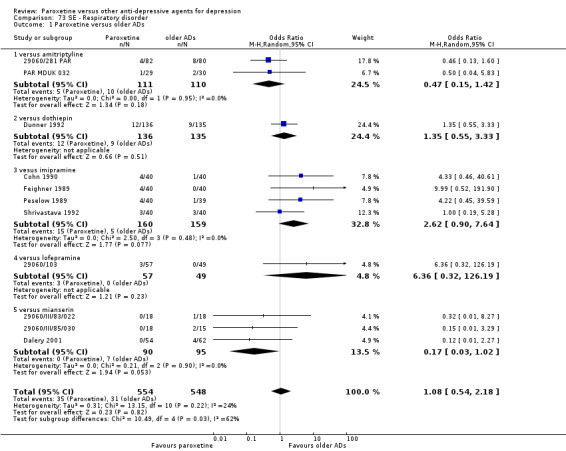
Comparison 73 SE ‐ Respiratory disorder, Outcome 1 Paroxetine versus older ADs.
73.2. Analysis.
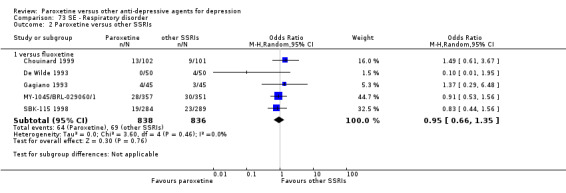
Comparison 73 SE ‐ Respiratory disorder, Outcome 2 Paroxetine versus other SSRIs.
Comparison 74. SE ‐ Restlessness.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus SSRIs | 1 | 274 | Odds Ratio (M‐H, Random, 95% CI) | 6.61 [0.27, 164.02] |
| 1.1 versus duloxetine | 1 | 274 | Odds Ratio (M‐H, Random, 95% CI) | 6.61 [0.27, 164.02] |
| 2 Paroxetine versus newer or non‐conventional ADs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus reboxetine | 1 | 325 | Odds Ratio (M‐H, Random, 95% CI) | 4.85 [0.23, 101.77] |
74.1. Analysis.
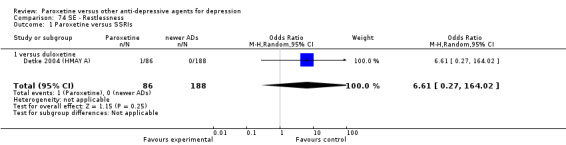
Comparison 74 SE ‐ Restlessness, Outcome 1 Paroxetine versus SSRIs.
74.2. Analysis.
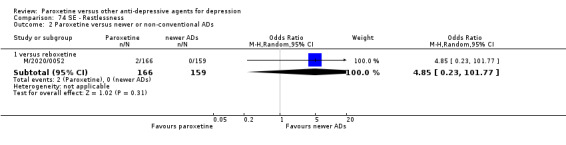
Comparison 74 SE ‐ Restlessness, Outcome 2 Paroxetine versus newer or non‐conventional ADs.
Comparison 75. SE ‐ Rhinitis.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus mianserin | 1 | 116 | Odds Ratio (M‐H, Random, 95% CI) | 0.38 [0.02, 9.43] |
| 2 Paroxetine versus SSRIs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus sertraline | 1 | 192 | Odds Ratio (M‐H, Random, 95% CI) | 0.94 [0.46, 1.92] |
| 3 Paroxetine versus newer or non‐conventional ADs | 7 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus agomelatine | 1 | 284 | Odds Ratio (M‐H, Random, 95% CI) | 0.31 [0.03, 2.98] |
| 3.2 versus mirtazapine | 1 | 197 | Odds Ratio (M‐H, Random, 95% CI) | 1.38 [0.46, 4.13] |
| 3.3 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 1.39 [0.32, 6.05] |
| 3.4 versus duloxetine | 2 | 528 | Odds Ratio (M‐H, Random, 95% CI) | 1.10 [0.59, 2.06] |
75.1. Analysis.
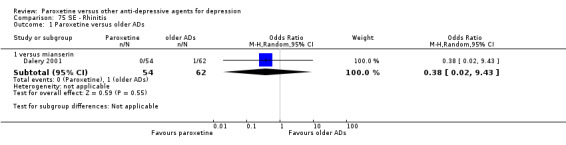
Comparison 75 SE ‐ Rhinitis, Outcome 1 Paroxetine versus older ADs.
75.2. Analysis.
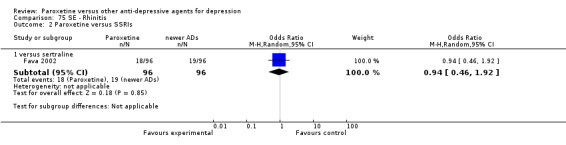
Comparison 75 SE ‐ Rhinitis, Outcome 2 Paroxetine versus SSRIs.
75.3. Analysis.
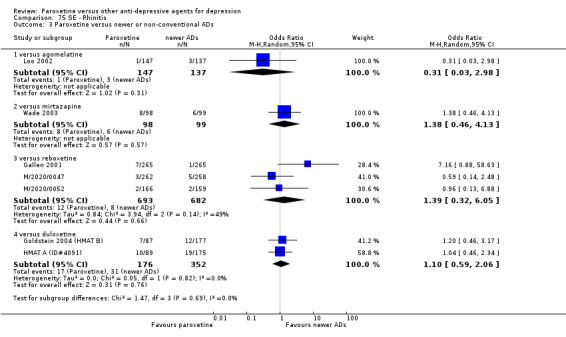
Comparison 75 SE ‐ Rhinitis, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 76. SE ‐ Sexual problems.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Anorgasmia | 7 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus duloxetine | 2 | 538 | Odds Ratio (M‐H, Random, 95% CI) | 0.69 [0.22, 2.18] |
| 1.2 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 9.84 [2.23, 43.46] |
| 1.3 versus sertraline | 2 | 545 | Odds Ratio (M‐H, Random, 95% CI) | 2.22 [0.43, 11.36] |
| 2 Ejaculation disorder | 11 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus aprepitant (MK‐869) | 1 | 143 | Odds Ratio (M‐H, Random, 95% CI) | 8.33 [1.82, 38.16] |
| 2.2 versus fluoxetine | 1 | 708 | Odds Ratio (M‐H, Random, 95% CI) | 3.17 [1.62, 6.20] |
| 2.3 versus fluvoxamine | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 3.22 [0.32, 32.89] |
| 2.4 versus imipramine | 1 | 79 | Odds Ratio (M‐H, Random, 95% CI) | 12.24 [0.65, 229.29] |
| 2.5 versus maprolitine | 1 | 71 | Odds Ratio (M‐H, Random, 95% CI) | 2.84 [0.11, 71.99] |
| 2.6 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 0.73 [0.37, 1.46] |
| 2.7 versus sertraline | 2 | 545 | Odds Ratio (M‐H, Random, 95% CI) | 3.49 [1.67, 7.26] |
| 2.8 versus tianeptine | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 3.0 [0.12, 74.28] |
| 3 Impotence | 6 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus aprepitant (MK‐869) | 1 | 143 | Odds Ratio (M‐H, Random, 95% CI) | 3.72 [0.74, 18.54] |
| 3.2 versus duloxetine | 1 | 264 | Odds Ratio (M‐H, Random, 95% CI) | 1.02 [0.25, 4.17] |
| 3.3 versus fluvoxamine | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 1.56 [0.24, 10.05] |
| 3.4 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 0.60 [0.30, 1.17] |
| 4 Libido decreased | 11 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 versus aprepitant (MK‐869) | 1 | 143 | Odds Ratio (M‐H, Random, 95% CI) | 9.39 [0.50, 177.78] |
| 4.2 versus bupropion | 1 | 140 | Odds Ratio (M‐H, Random, 95% CI) | 1.93 [0.67, 5.53] |
| 4.3 versus duloxetine | 3 | 802 | Odds Ratio (M‐H, Random, 95% CI) | 1.36 [0.36, 5.14] |
| 4.4 versus fluvoxamine | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 1.3 [0.31, 5.40] |
| 4.5 versus maprotiline | 1 | 298 | Odds Ratio (M‐H, Random, 95% CI) | 5.35 [0.25, 112.36] |
| 4.6 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 2.34 [1.14, 4.82] |
| 4.7 versus tianeptine | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 0.99 [0.06, 16.03] |
| 5 Penis disorder | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 versus reboxetine | 1 | 530 | Odds Ratio (M‐H, Random, 95% CI) | 0.14 [0.01, 2.75] |
| 6 NOS | 14 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 versus amitriptyline | 1 | 21 | Odds Ratio (M‐H, Random, 95% CI) | 2.0 [0.15, 26.19] |
| 6.2 versus aprepitant (MK‐869) | 1 | 143 | Odds Ratio (M‐H, Random, 95% CI) | 11.65 [0.63, 214.77] |
| 6.3 versus bupropion | 1 | 140 | Odds Ratio (M‐H, Random, 95% CI) | 2.35 [0.77, 7.15] |
| 6.4 versus citalopram | 1 | 406 | Odds Ratio (M‐H, Random, 95% CI) | 0.87 [0.38, 2.00] |
| 6.5 versus clomipramine | 1 | 1019 | Odds Ratio (M‐H, Random, 95% CI) | 1.60 [0.72, 3.55] |
| 6.6 versus duloxetine | 2 | 528 | Odds Ratio (M‐H, Random, 95% CI) | 1.25 [0.40, 3.90] |
| 6.7 versus fluvoxamine | 1 | 81 | Odds Ratio (M‐H, Random, 95% CI) | 1.36 [0.39, 4.70] |
| 6.8 versus mirtazapine | 1 | 275 | Odds Ratio (M‐H, Random, 95% CI) | 5.15 [1.69, 15.64] |
| 6.9 versus nefazodone | 1 | 40 | Odds Ratio (M‐H, Random, 95% CI) | 14.55 [0.75, 283.37] |
| 6.10 versus nortriptyline | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 2.65 [0.10, 66.96] |
| 6.11 versus reboxetine | 2 | 1050 | Odds Ratio (M‐H, Random, 95% CI) | 1.95 [0.48, 7.98] |
| 6.12 versus venlafaxine | 1 | 361 | Odds Ratio (M‐H, Random, 95% CI) | 1.29 [0.34, 4.90] |
76.1. Analysis.
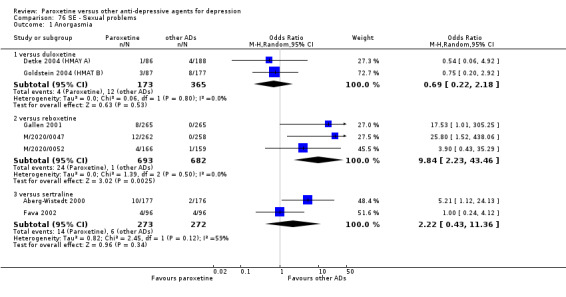
Comparison 76 SE ‐ Sexual problems, Outcome 1 Anorgasmia.
76.2. Analysis.
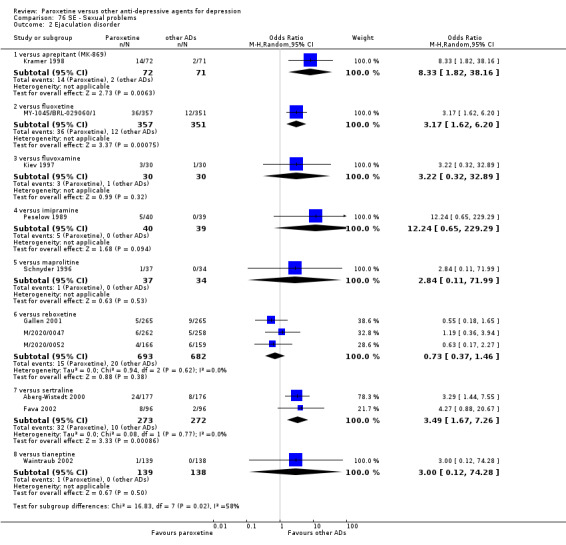
Comparison 76 SE ‐ Sexual problems, Outcome 2 Ejaculation disorder.
76.3. Analysis.
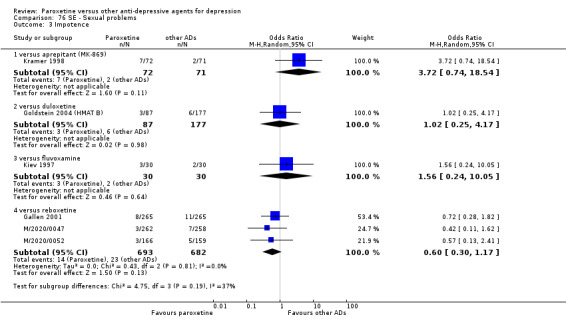
Comparison 76 SE ‐ Sexual problems, Outcome 3 Impotence.
76.4. Analysis.
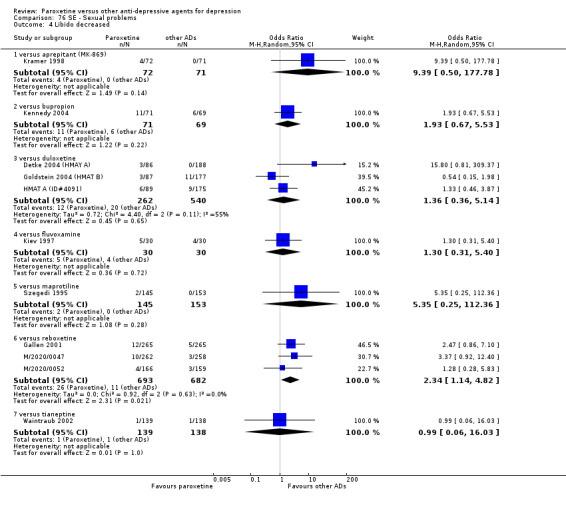
Comparison 76 SE ‐ Sexual problems, Outcome 4 Libido decreased.
76.5. Analysis.
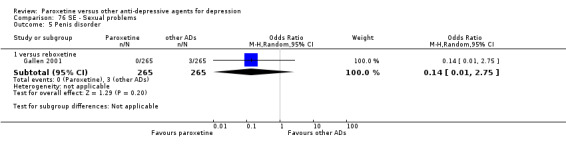
Comparison 76 SE ‐ Sexual problems, Outcome 5 Penis disorder.
76.6. Analysis.
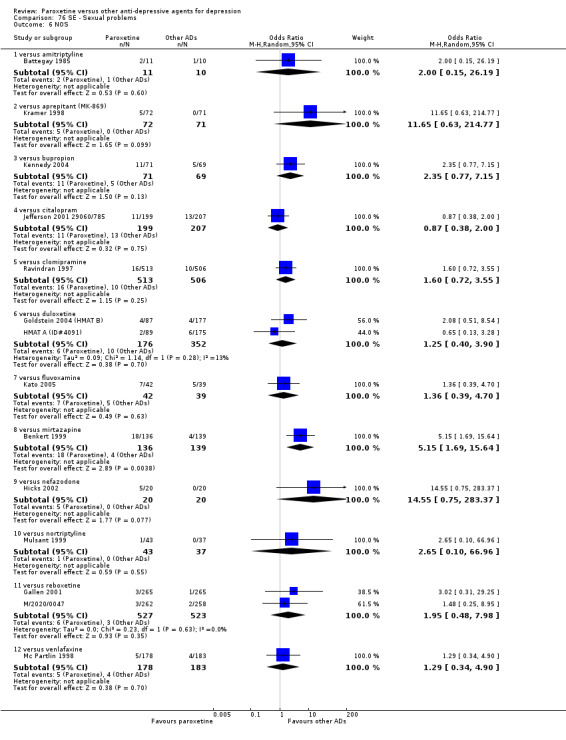
Comparison 76 SE ‐ Sexual problems, Outcome 6 NOS.
Comparison 77. SE ‐ Skin and appendages.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 5 | 659 | Odds Ratio (M‐H, Random, 95% CI) | 1.26 [0.63, 2.52] |
| 1.1 versus amitriptyline | 2 | 221 | Odds Ratio (M‐H, Random, 95% CI) | 1.36 [0.61, 3.05] |
| 1.2 versus clomipramine | 1 | 83 | Odds Ratio (M‐H, Random, 95% CI) | 1.03 [0.19, 5.41] |
| 1.3 versus maprotiline | 1 | 298 | Odds Ratio (M‐H, Random, 95% CI) | 3.19 [0.13, 78.86] |
| 1.4 versus mianserin | 1 | 57 | Odds Ratio (M‐H, Random, 95% CI) | 0.31 [0.01, 7.95] |
| 2 Paroxetine versus other SSRIs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluoxetine | 1 | 100 | Odds Ratio (M‐H, Random, 95% CI) | 0.13 [0.01, 1.06] |
| 3 Paroxetine versus newer or non‐conventional ADs | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus reboxetine | 2 | 1050 | Odds Ratio (M‐H, Random, 95% CI) | 0.35 [0.10, 1.20] |
77.1. Analysis.
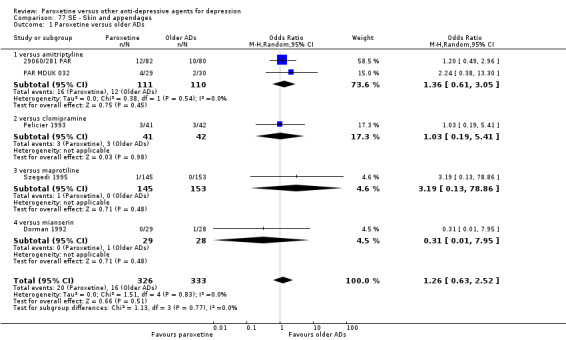
Comparison 77 SE ‐ Skin and appendages, Outcome 1 Paroxetine versus older ADs.
77.2. Analysis.
Comparison 77 SE ‐ Skin and appendages, Outcome 2 Paroxetine versus other SSRIs.
77.3. Analysis.
Comparison 77 SE ‐ Skin and appendages, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 78. SE ‐ Sleep disorders.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus imipramine | 1 | 159 | Odds Ratio (M‐H, Random, 95% CI) | 0.49 [0.12, 2.02] |
| 1.2 versus mianserin | 1 | 116 | Odds Ratio (M‐H, Random, 95% CI) | 0.38 [0.02, 9.43] |
| 2 Paroxetine versus newer or non‐conventional ADs | 9 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus duloxetine | 2 | 538 | Odds Ratio (M‐H, Random, 95% CI) | 2.92 [0.70, 12.22] |
| 2.2 versus hypericum | 1 | 251 | Odds Ratio (M‐H, Random, 95% CI) | 2.07 [0.69, 6.24] |
| 2.3 versus mirtazapine | 2 | 451 | Odds Ratio (M‐H, Random, 95% CI) | 1.46 [0.54, 3.95] |
| 2.4 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 3.13 [1.04, 9.43] |
| 2.5 versus trazodone | 1 | 108 | Odds Ratio (M‐H, Random, 95% CI) | 0.11 [0.01, 2.04] |
78.1. Analysis.
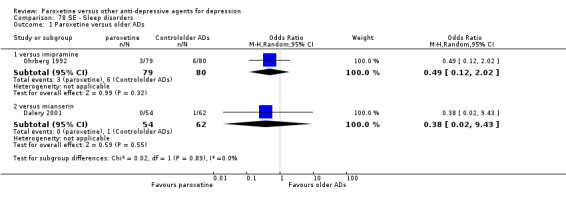
Comparison 78 SE ‐ Sleep disorders, Outcome 1 Paroxetine versus older ADs.
78.2. Analysis.
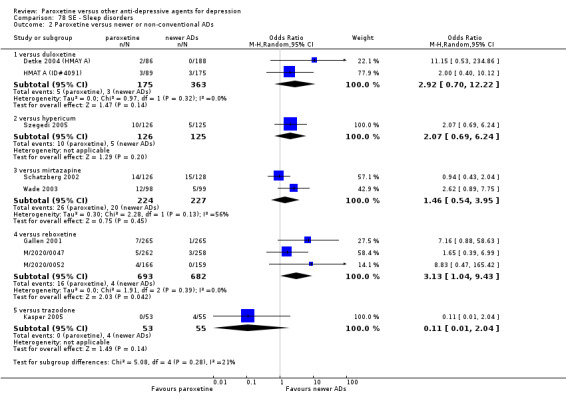
Comparison 78 SE ‐ Sleep disorders, Outcome 2 Paroxetine versus newer or non‐conventional ADs.
Comparison 79. SE ‐ Sleepiness/drowsiness.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 22 | 2711 | Odds Ratio (M‐H, Random, 95% CI) | 0.72 [0.50, 1.02] |
| 1.1 versus amitriptyline | 8 | 985 | Odds Ratio (M‐H, Random, 95% CI) | 0.72 [0.36, 1.43] |
| 1.2 versus clomipramine | 1 | 92 | Odds Ratio (M‐H, Random, 95% CI) | 0.51 [0.04, 5.84] |
| 1.3 versus dothiepin | 2 | 405 | Odds Ratio (M‐H, Random, 95% CI) | 0.45 [0.14, 1.41] |
| 1.4 versus imipramine | 7 | 633 | Odds Ratio (M‐H, Random, 95% CI) | 0.95 [0.58, 1.55] |
| 1.5 versus lofepramine | 1 | 122 | Odds Ratio (M‐H, Random, 95% CI) | 2.14 [0.38, 12.16] |
| 1.6 versus maprolitine | 2 | 358 | Odds Ratio (M‐H, Random, 95% CI) | 0.37 [0.17, 0.82] |
| 1.7 versus mianserin | 1 | 116 | Odds Ratio (M‐H, Random, 95% CI) | 0.37 [0.04, 3.68] |
| 2 Paroxetine versus other SSRIs | 12 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus citalopram | 1 | 406 | Odds Ratio (M‐H, Random, 95% CI) | 0.61 [0.34, 1.08] |
| 2.2 versus fluoxetine | 8 | 2116 | Odds Ratio (M‐H, Random, 95% CI) | 1.48 [1.16, 1.88] |
| 2.3 versus fluvoxamine | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 0.64 [0.22, 1.87] |
| 2.4 versus sertraline | 2 | 426 | Odds Ratio (M‐H, Random, 95% CI) | 1.29 [0.30, 5.60] |
| 3 Paroxetine versus newer or non‐conventional ADs | 19 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus agomelatine | 1 | 284 | Odds Ratio (M‐H, Random, 95% CI) | 2.69 [0.84, 8.66] |
| 3.2 versus aprepitant (MK‐869) | 1 | 143 | Odds Ratio (M‐H, Random, 95% CI) | 0.98 [0.43, 2.24] |
| 3.3 versus bupropion | 2 | 240 | Odds Ratio (M‐H, Random, 95% CI) | 7.63 [2.51, 23.16] |
| 3.4 versus duloxetine | 5 | 1571 | Odds Ratio (M‐H, Random, 95% CI) | 1.03 [0.70, 1.51] |
| 3.5 versus mirtazapine | 3 | 726 | Odds Ratio (M‐H, Random, 95% CI) | 0.81 [0.55, 1.19] |
| 3.6 versus nefazodone | 2 | 246 | Odds Ratio (M‐H, Random, 95% CI) | 0.73 [0.13, 4.13] |
| 3.7 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 2.66 [1.45, 4.89] |
| 3.8 versus trazodone | 1 | 108 | Odds Ratio (M‐H, Random, 95% CI) | 0.33 [0.03, 3.31] |
| 3.9 versus venlafaxine | 1 | 361 | Odds Ratio (M‐H, Random, 95% CI) | 1.03 [0.42, 2.54] |
Comparison 80. SE ‐ Sinusitis.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus newer or non‐conventional ADs | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 1.31 [0.60, 2.86] |
80.1. Analysis.
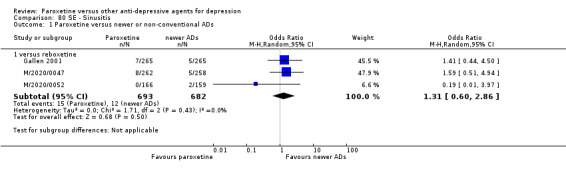
Comparison 80 SE ‐ Sinusitis, Outcome 1 Paroxetine versus newer or non‐conventional ADs.
Comparison 81. SE ‐ Special senses.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 NOS | 6 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus amitriptyline | 2 | 221 | Odds Ratio (M‐H, Random, 95% CI) | 1.22 [0.59, 2.53] |
| 1.2 versus clomipramine | 1 | 83 | Odds Ratio (M‐H, Random, 95% CI) | 0.75 [0.16, 3.58] |
| 1.3 versus mianserin | 3 | 126 | Odds Ratio (M‐H, Random, 95% CI) | 1.23 [0.32, 4.73] |
| 2 Taste perversion | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus amitriptyline | 2 | 150 | Odds Ratio (M‐H, Random, 95% CI) | 0.71 [0.05, 10.26] |
| 2.2 versus reboxetine | 1 | 530 | Odds Ratio (M‐H, Random, 95% CI) | 1.25 [0.33, 4.73] |
| 3 Auditory disorders | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus reboxetine | 1 | 325 | Odds Ratio (M‐H, Random, 95% CI) | 0.19 [0.01, 3.97] |
81.1. Analysis.
Comparison 81 SE ‐ Special senses, Outcome 1 NOS.
81.2. Analysis.
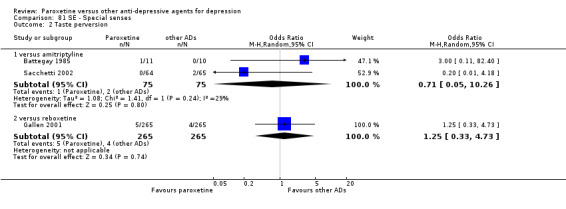
Comparison 81 SE ‐ Special senses, Outcome 2 Taste perversion.
81.3. Analysis.
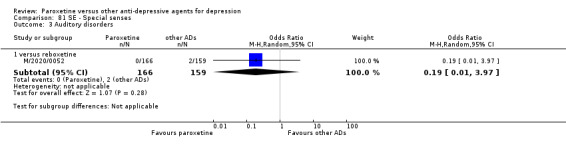
Comparison 81 SE ‐ Special senses, Outcome 3 Auditory disorders.
Comparison 82. SE ‐ Surgical procedure.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus dothiepin | 1 | 134 | Odds Ratio (M‐H, Random, 95% CI) | 3.05 [0.12, 76.10] |
| 2 Paroxetine versus other SSRIs | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluoxetine | 2 | 663 | Odds Ratio (M‐H, Random, 95% CI) | 4.87 [0.55, 43.27] |
82.1. Analysis.
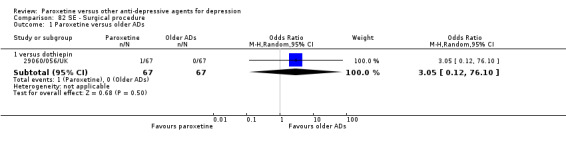
Comparison 82 SE ‐ Surgical procedure, Outcome 1 Paroxetine versus older ADs.
82.2. Analysis.
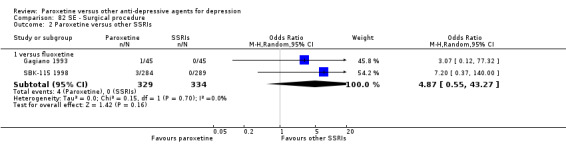
Comparison 82 SE ‐ Surgical procedure, Outcome 2 Paroxetine versus other SSRIs.
Comparison 83. SE ‐ Sweating.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 15 | 2970 | Odds Ratio (M‐H, Random, 95% CI) | 0.75 [0.50, 1.12] |
| 1.1 versus amitriptyline | 5 | 774 | Odds Ratio (M‐H, Random, 95% CI) | 1.72 [0.60, 4.90] |
| 1.2 versus clomipramine | 2 | 1111 | Odds Ratio (M‐H, Random, 95% CI) | 0.73 [0.48, 1.10] |
| 1.3 versus imipramine | 6 | 681 | Odds Ratio (M‐H, Random, 95% CI) | 0.43 [0.29, 0.65] |
| 1.4 versus lofepramine | 1 | 106 | Odds Ratio (M‐H, Random, 95% CI) | 0.13 [0.01, 1.10] |
| 1.5 versus maprotiline | 1 | 298 | Odds Ratio (M‐H, Random, 95% CI) | 1.51 [0.77, 2.96] |
| 2 Paroxetine versus other SSRIs | 8 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus escitalopram | 1 | 454 | Odds Ratio (M‐H, Random, 95% CI) | 1.49 [0.81, 2.72] |
| 2.2 versus fluoxetine | 6 | 1431 | Odds Ratio (M‐H, Random, 95% CI) | 0.95 [0.43, 2.12] |
| 2.3 versus fluvoxamine | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 4.5 [1.09, 18.50] |
| 3 Paroxetine versus newer or non‐conventional ADs | 18 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus aprepitant (MK‐869) | 1 | 143 | Odds Ratio (M‐H, Random, 95% CI) | 4.31 [0.88, 21.07] |
| 3.2 versus bupropion | 1 | 140 | Odds Ratio (M‐H, Random, 95% CI) | 1.36 [0.53, 3.46] |
| 3.3 versus duloxetine | 5 | 1573 | Odds Ratio (M‐H, Random, 95% CI) | 0.80 [0.50, 1.28] |
| 3.4 versus hypericum | 1 | 251 | Odds Ratio (M‐H, Random, 95% CI) | 1.48 [0.61, 3.61] |
| 3.5 versus mirtazapine | 3 | 726 | Odds Ratio (M‐H, Random, 95% CI) | 3.12 [1.61, 6.03] |
| 3.6 versus nefazodone | 1 | 40 | Odds Ratio (M‐H, Random, 95% CI) | 22.78 [1.20, 432.58] |
| 3.7 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 0.48 [0.34, 0.68] |
| 3.8 versus tianeptine | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 13.49 [0.75, 241.76] |
| 3.9 versus trazodone | 1 | 108 | Odds Ratio (M‐H, Random, 95% CI) | 3.17 [0.13, 79.60] |
| 3.10 versus venlafaxine | 1 | 361 | Odds Ratio (M‐H, Random, 95% CI) | 2.95 [0.92, 9.44] |
83.1. Analysis.
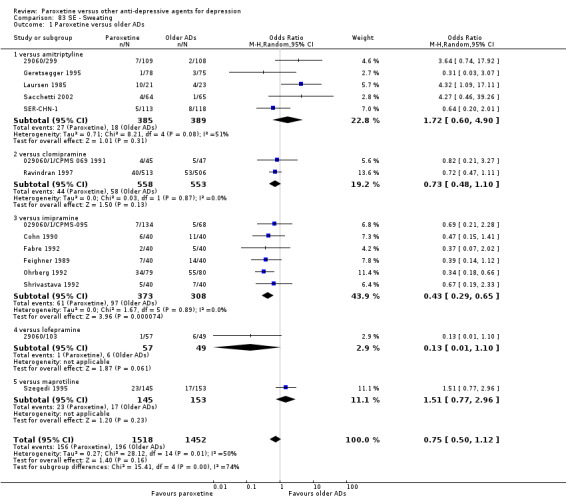
Comparison 83 SE ‐ Sweating, Outcome 1 Paroxetine versus older ADs.
83.2. Analysis.
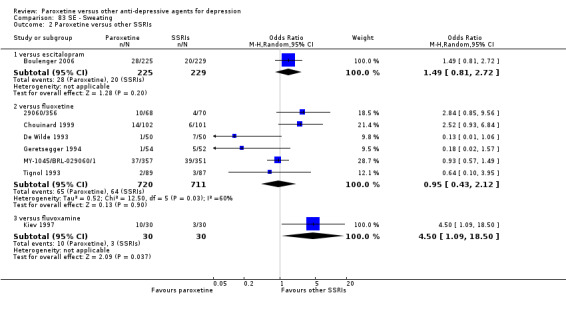
Comparison 83 SE ‐ Sweating, Outcome 2 Paroxetine versus other SSRIs.
83.3. Analysis.
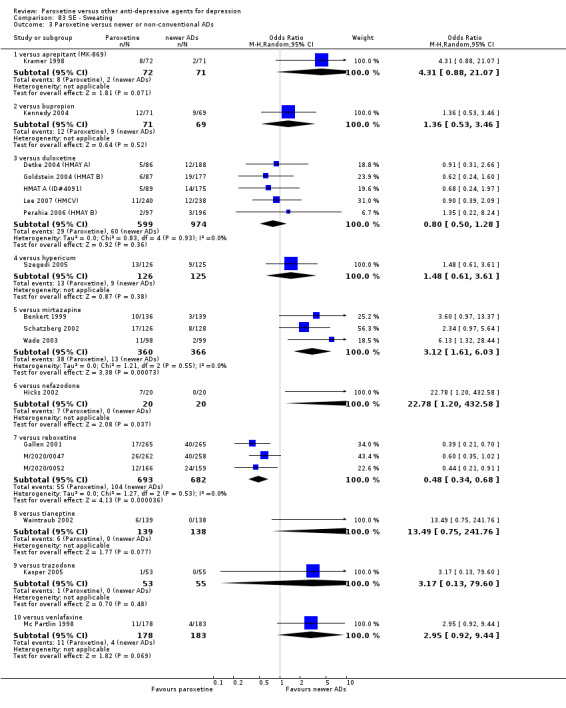
Comparison 83 SE ‐ Sweating, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 84. SE ‐ Tachycardia.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 5 | 717 | Odds Ratio (M‐H, Random, 95% CI) | 0.37 [0.12, 1.13] |
| 1.1 versus amitriptyline | 2 | 360 | Odds Ratio (M‐H, Random, 95% CI) | 0.43 [0.04, 4.44] |
| 1.2 versus imipramine | 3 | 357 | Odds Ratio (M‐H, Random, 95% CI) | 0.36 [0.09, 1.47] |
| 2 Paroxetine versus SSRIs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus sertraline | 1 | 353 | Odds Ratio (M‐H, Random, 95% CI) | 1.29 [0.47, 3.55] |
| 3 Paroxetine versus newer or non‐conventional ADs | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus duloxetine | 1 | 293 | Odds Ratio (M‐H, Random, 95% CI) | 0.67 [0.07, 6.53] |
| 3.2 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 0.39 [0.15, 1.01] |
84.1. Analysis.
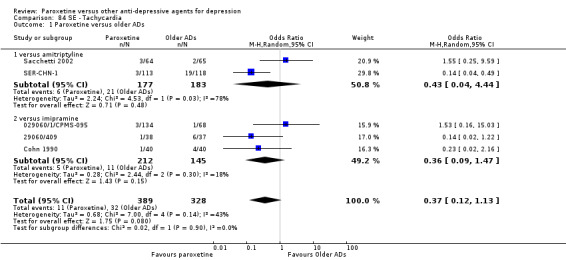
Comparison 84 SE ‐ Tachycardia, Outcome 1 Paroxetine versus older ADs.
84.2. Analysis.
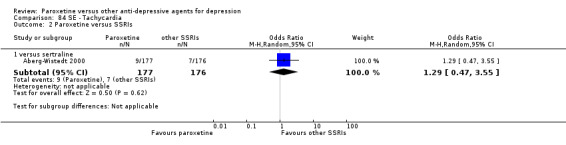
Comparison 84 SE ‐ Tachycardia, Outcome 2 Paroxetine versus SSRIs.
84.3. Analysis.
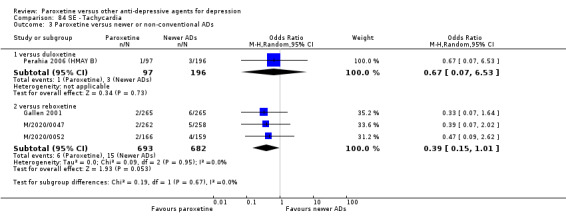
Comparison 84 SE ‐ Tachycardia, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 85. SE ‐ Tension.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus newer or non‐conventional ADs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus duloxetine | 1 | 293 | Odds Ratio (M‐H, Random, 95% CI) | 0.50 [0.06, 4.53] |
85.1. Analysis.
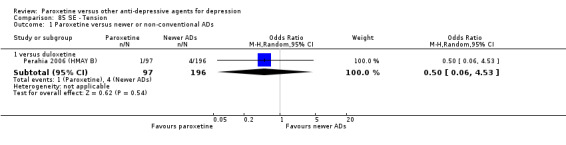
Comparison 85 SE ‐ Tension, Outcome 1 Paroxetine versus newer or non‐conventional ADs.
Comparison 86. SE ‐ Thirst.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus newer or non‐conventional ADs | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus reboxetine | 2 | 1050 | Odds Ratio (M‐H, Random, 95% CI) | 1.74 [0.51, 6.01] |
86.1. Analysis.
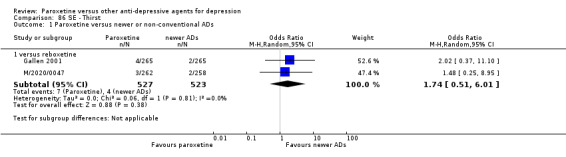
Comparison 86 SE ‐ Thirst, Outcome 1 Paroxetine versus newer or non‐conventional ADs.
Comparison 87. SE ‐ Tinnitus.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus mianserin | 1 | 116 | Odds Ratio (M‐H, Random, 95% CI) | 0.38 [0.02, 9.43] |
| 2 Paroxetine versus newer or non‐conventional ADs | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus duloxetine | 2 | 538 | Odds Ratio (M‐H, Random, 95% CI) | 0.56 [0.11, 2.77] |
| 2.2 versus tianeptine | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 3.0 [0.12, 74.28] |
87.1. Analysis.
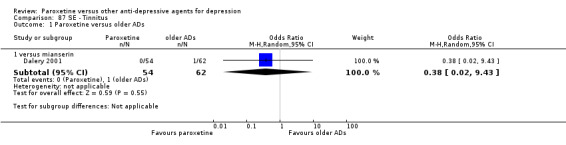
Comparison 87 SE ‐ Tinnitus, Outcome 1 Paroxetine versus older ADs.
87.2. Analysis.
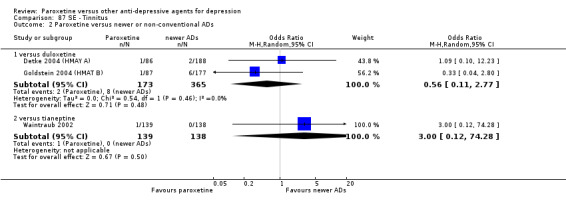
Comparison 87 SE ‐ Tinnitus, Outcome 2 Paroxetine versus newer or non‐conventional ADs.
Comparison 88. SE ‐ Tremor.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 23 | 3313 | Odds Ratio (M‐H, Random, 95% CI) | 0.69 [0.50, 0.94] |
| 1.1 versus amitriptyline | 9 | 1014 | Odds Ratio (M‐H, Random, 95% CI) | 0.44 [0.22, 0.90] |
| 1.2 versus clomipramine | 2 | 1111 | Odds Ratio (M‐H, Random, 95% CI) | 0.76 [0.52, 1.12] |
| 1.3 versus imipramine | 8 | 835 | Odds Ratio (M‐H, Random, 95% CI) | 0.85 [0.51, 1.42] |
| 1.4 versus lofepramine | 1 | 106 | Odds Ratio (M‐H, Random, 95% CI) | 0.85 [0.12, 6.30] |
| 1.5 versus maprolitine | 2 | 131 | Odds Ratio (M‐H, Random, 95% CI) | 0.84 [0.03, 25.65] |
| 1.6 versus mianserin | 1 | 116 | Odds Ratio (M‐H, Random, 95% CI) | 3.50 [0.14, 87.83] |
| 2 Paroxetine versus other SSRIs | 10 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluoxetine | 7 | 1994 | Odds Ratio (M‐H, Random, 95% CI) | 0.98 [0.67, 1.44] |
| 2.2 versus fluvoxamine | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 1.38 [0.28, 6.80] |
| 2.3 versus sertraline | 2 | 545 | Odds Ratio (M‐H, Random, 95% CI) | 1.82 [1.07, 3.09] |
| 3 Paroxetine versus newer or non‐conventional ADs | 13 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus bupropion | 1 | 100 | Odds Ratio (M‐H, Random, 95% CI) | 0.91 [0.25, 3.38] |
| 3.2 versus duloxetine | 4 | 1095 | Odds Ratio (M‐H, Random, 95% CI) | 1.45 [0.64, 3.29] |
| 3.3 versus mirtazapine | 2 | 529 | Odds Ratio (M‐H, Random, 95% CI) | 3.67 [1.43, 9.42] |
| 3.4 versus nefazodone | 1 | 40 | Odds Ratio (M‐H, Random, 95% CI) | 11.18 [0.56, 222.98] |
| 3.5 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 2.75 [1.35, 5.57] |
| 3.6 versus tianeptine | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 2.0 [0.18, 22.31] |
| 3.7 versus trazodone | 1 | 108 | Odds Ratio (M‐H, Random, 95% CI) | 1.04 [0.14, 7.66] |
88.1. Analysis.
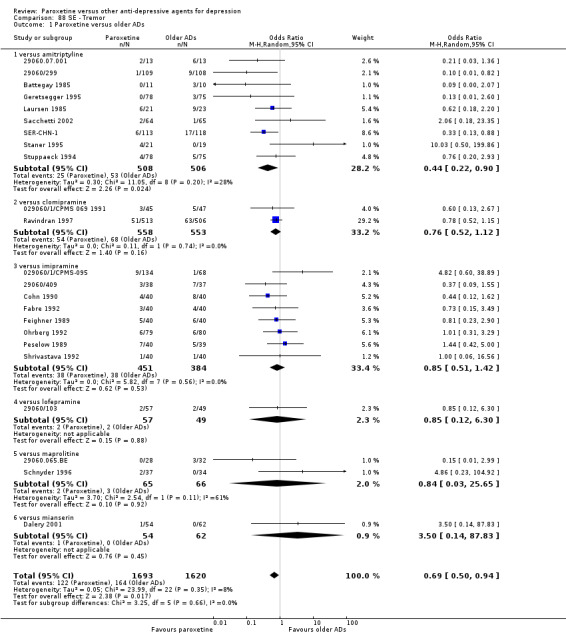
Comparison 88 SE ‐ Tremor, Outcome 1 Paroxetine versus older ADs.
88.2. Analysis.
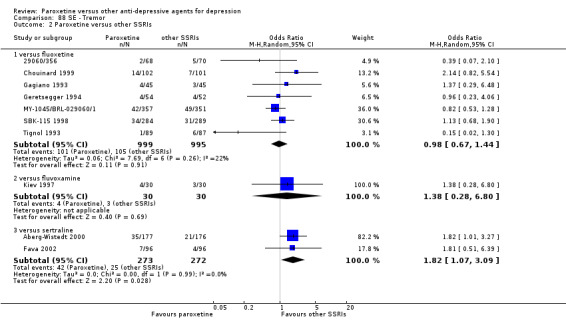
Comparison 88 SE ‐ Tremor, Outcome 2 Paroxetine versus other SSRIs.
88.3. Analysis.
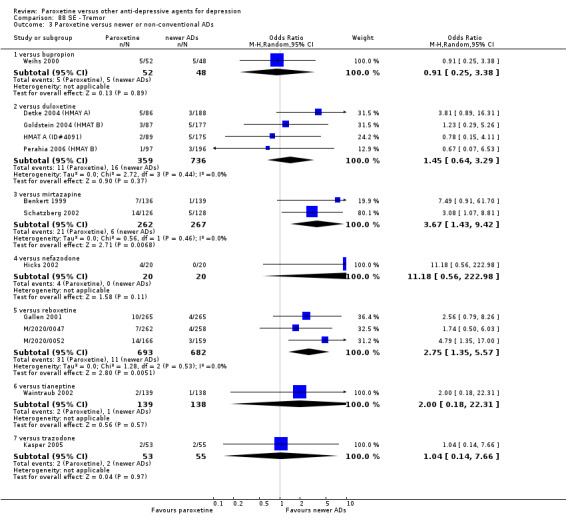
Comparison 88 SE ‐ Tremor, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 89. SE ‐ Upper respiratory tract infection.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus newer or non‐conventional ADs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus reboxetine | 1 | 325 | Odds Ratio (M‐H, Random, 95% CI) | 2.63 [0.69, 10.11] |
89.1. Analysis.
Comparison 89 SE ‐ Upper respiratory tract infection, Outcome 1 Paroxetine versus newer or non‐conventional ADs.
Comparison 90. SE ‐ Urination/Urogenital problems.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Difficulty micturating | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus amitriptyline | 2 | 65 | Odds Ratio (M‐H, Random, 95% CI) | 0.85 [0.23, 3.19] |
| 2 Dysuria | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus reboxetine | 2 | 855 | Odds Ratio (M‐H, Random, 95% CI) | 0.16 [0.04, 0.75] |
| 3 Urination frequency | 5 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus bupropion | 1 | 100 | Odds Ratio (M‐H, Random, 95% CI) | 0.12 [0.01, 2.46] |
| 3.2 versus duloxetine | 1 | 293 | Odds Ratio (M‐H, Random, 95% CI) | 1.01 [0.09, 11.28] |
| 3.3 versus imipramine | 1 | 79 | Odds Ratio (M‐H, Random, 95% CI) | 0.08 [0.00, 1.45] |
| 3.4 versus reboxetine | 2 | 1050 | Odds Ratio (M‐H, Random, 95% CI) | 1.30 [0.56, 2.99] |
| 4 Urinary retention | 6 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 versus amitriptyline | 1 | 129 | Odds Ratio (M‐H, Random, 95% CI) | 1.02 [0.14, 7.44] |
| 4.2 versus imipramine | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 0.04 [0.00, 0.73] |
| 4.3 versus nortriptyline | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 4.52 [0.21, 97.16] |
| 4.4 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 0.28 [0.14, 0.54] |
| 5 Not Otherwise Specified | 9 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 versus amitriptyline | 2 | 221 | Odds Ratio (M‐H, Random, 95% CI) | 1.83 [0.51, 6.52] |
| 5.2 versus clomipramine | 1 | 83 | Odds Ratio (M‐H, Random, 95% CI) | 0.20 [0.01, 4.19] |
| 5.3 versus duloxetine | 2 | 557 | Odds Ratio (M‐H, Random, 95% CI) | 0.87 [0.22, 3.41] |
| 5.4 versus imipramine | 1 | 159 | Odds Ratio (M‐H, Random, 95% CI) | 0.10 [0.01, 0.82] |
| 5.5 versus mianserin | 2 | 394 | Odds Ratio (M‐H, Random, 95% CI) | 1.38 [0.21, 8.87] |
| 5.6 versus sertraline | 1 | 353 | Odds Ratio (M‐H, Random, 95% CI) | 11.60 [1.48, 90.81] |
90.1. Analysis.
Comparison 90 SE ‐ Urination/Urogenital problems, Outcome 1 Difficulty micturating.
90.3. Analysis.
Comparison 90 SE ‐ Urination/Urogenital problems, Outcome 3 Urination frequency.
Comparison 91. SE ‐ Visual problems (accommodation disorders, amblyopia, blepharoptosis, blurred vision, detached retina, dry eye, eye strain, mydriasis).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 8 | 830 | Odds Ratio (M‐H, Random, 95% CI) | 1.21 [0.74, 1.98] |
| 1.1 versus amitriptyline | 3 | 278 | Odds Ratio (M‐H, Random, 95% CI) | 0.76 [0.37, 1.58] |
| 1.2 versus clomipramine | 1 | 92 | Odds Ratio (M‐H, Random, 95% CI) | 2.20 [0.38, 12.62] |
| 1.3 versus imipramine | 2 | 238 | Odds Ratio (M‐H, Random, 95% CI) | 1.60 [0.65, 3.92] |
| 1.4 versus lofepramine | 1 | 106 | Odds Ratio (M‐H, Random, 95% CI) | 6.36 [0.32, 126.19] |
| 1.5 versus mianserin | 1 | 116 | Odds Ratio (M‐H, Random, 95% CI) | 1.15 [0.07, 18.85] |
| 2 Paroxetine versus SSRIs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus sertraline | 1 | 353 | Odds Ratio (M‐H, Random, 95% CI) | 1.21 [0.51, 2.87] |
| 3 Paroxetine versus newer or non‐conventional ADs | 10 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus duloxetine | 4 | 1299 | Odds Ratio (M‐H, Random, 95% CI) | 1.06 [0.61, 1.82] |
| 3.2 versus nefazodone | 1 | 206 | Odds Ratio (M‐H, Random, 95% CI) | 0.6 [0.21, 1.72] |
| 3.3 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 0.72 [0.30, 1.71] |
| 3.4 versus tianeptine | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 3.0 [0.12, 74.28] |
| 3.5 versus venlafaxine | 1 | 105 | Odds Ratio (M‐H, Random, 95% CI) | 2.0 [0.18, 22.75] |
91.1. Analysis.
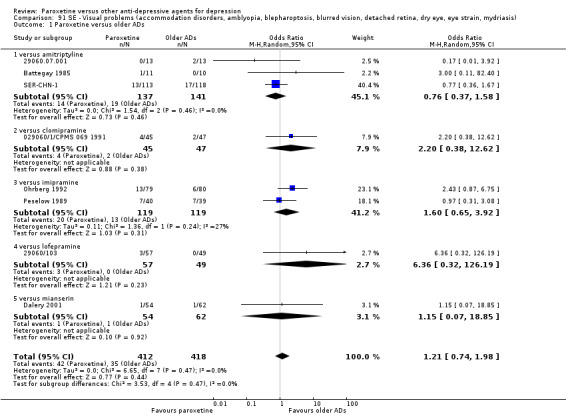
Comparison 91 SE ‐ Visual problems (accommodation disorders, amblyopia, blepharoptosis, blurred vision, detached retina, dry eye, eye strain, mydriasis), Outcome 1 Paroxetine versus older ADs.
91.2. Analysis.
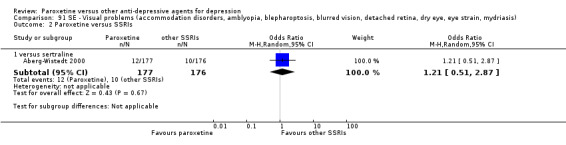
Comparison 91 SE ‐ Visual problems (accommodation disorders, amblyopia, blepharoptosis, blurred vision, detached retina, dry eye, eye strain, mydriasis), Outcome 2 Paroxetine versus SSRIs.
91.3. Analysis.
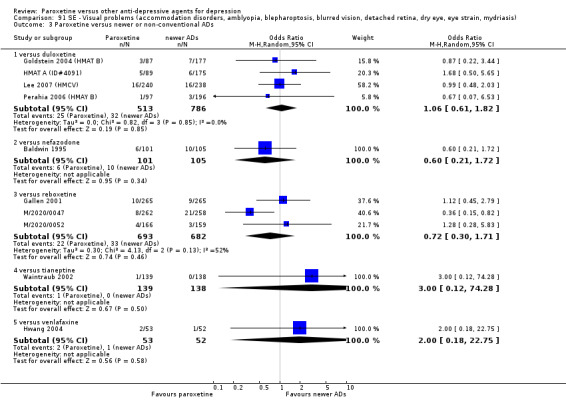
Comparison 91 SE ‐ Visual problems (accommodation disorders, amblyopia, blepharoptosis, blurred vision, detached retina, dry eye, eye strain, mydriasis), Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 92. SE ‐ Vertigo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 6 | 767 | Odds Ratio (M‐H, Random, 95% CI) | 0.70 [0.32, 1.50] |
| 1.1 versus amitriptyline | 4 | 636 | Odds Ratio (M‐H, Random, 95% CI) | 0.57 [0.17, 1.91] |
| 1.2 versus maprolitine | 2 | 131 | Odds Ratio (M‐H, Random, 95% CI) | 0.91 [0.27, 3.03] |
| 2 Paroxetine versus other SSRIs | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluoxetine | 1 | 106 | Odds Ratio (M‐H, Random, 95% CI) | 0.96 [0.23, 4.06] |
| 2.2 versus fluvoxamine | 1 | 81 | Odds Ratio (M‐H, Random, 95% CI) | 1.9 [0.17, 21.82] |
| 3 Paroxetine versus newer or non‐conventional ADs | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus duloxetine | 1 | 293 | Odds Ratio (M‐H, Random, 95% CI) | 1.01 [0.09, 11.28] |
| 3.2 versus reboxetine | 1 | 325 | Odds Ratio (M‐H, Random, 95% CI) | 0.48 [0.04, 5.30] |
| 3.3 versus trazodone | 1 | 108 | Odds Ratio (M‐H, Random, 95% CI) | 0.34 [0.01, 8.52] |
92.1. Analysis.
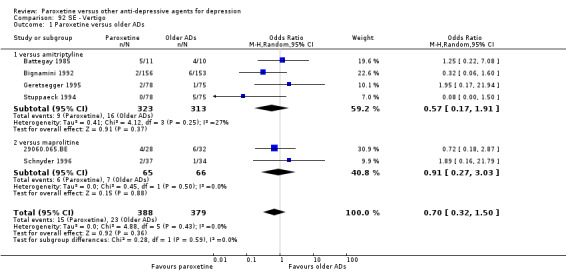
Comparison 92 SE ‐ Vertigo, Outcome 1 Paroxetine versus older ADs.
92.2. Analysis.
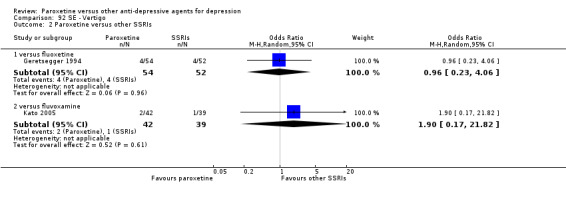
Comparison 92 SE ‐ Vertigo, Outcome 2 Paroxetine versus other SSRIs.
92.3. Analysis.
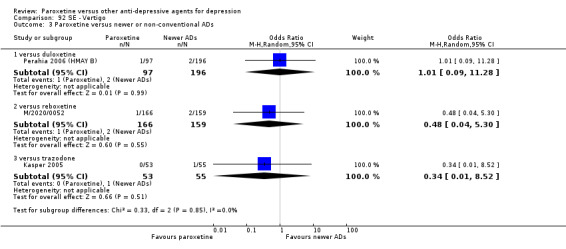
Comparison 92 SE ‐ Vertigo, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 93. SE ‐ Weight gain.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 6 | 729 | Odds Ratio (M‐H, Random, 95% CI) | 0.52 [0.14, 1.98] |
| 1.1 versus amitryptline | 1 | 231 | Odds Ratio (M‐H, Random, 95% CI) | 1.05 [0.36, 3.09] |
| 1.2 versus clomipramine | 1 | 92 | Odds Ratio (M‐H, Random, 95% CI) | 5.46 [0.25, 116.92] |
| 1.3 versus imipramine | 1 | 159 | Odds Ratio (M‐H, Random, 95% CI) | 0.12 [0.01, 0.95] |
| 1.4 versus maprolitine | 2 | 131 | Odds Ratio (M‐H, Random, 95% CI) | 0.10 [0.01, 0.81] |
| 1.5 versus mianserin | 1 | 116 | Odds Ratio (M‐H, Random, 95% CI) | 3.50 [0.14, 87.83] |
| 2 Paroxetine versus other SSRIs | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluoxetine | 2 | 276 | Odds Ratio (M‐H, Random, 95% CI) | 0.99 [0.27, 3.59] |
| 3 Paroxetine versus newer or non‐conventional ADs | 9 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus duloxetine | 2 | 567 | Odds Ratio (M‐H, Random, 95% CI) | 0.59 [0.09, 3.63] |
| 3.2 versus mirtazapine | 3 | 726 | Odds Ratio (M‐H, Random, 95% CI) | 0.26 [0.08, 0.84] |
| 3.3 versus reboxetine | 2 | 855 | Odds Ratio (M‐H, Random, 95% CI) | 4.12 [1.02, 16.62] |
| 3.4 versus tianeptine | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 3.0 [0.12, 74.28] |
| 3.5 versus venlafaxine | 1 | 105 | Odds Ratio (M‐H, Random, 95% CI) | 2.09 [0.49, 8.82] |
93.1. Analysis.
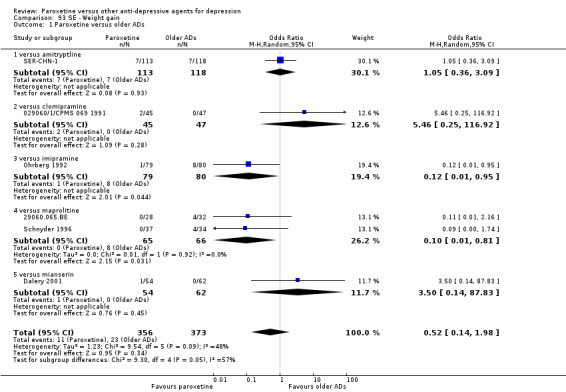
Comparison 93 SE ‐ Weight gain, Outcome 1 Paroxetine versus older ADs.
93.2. Analysis.
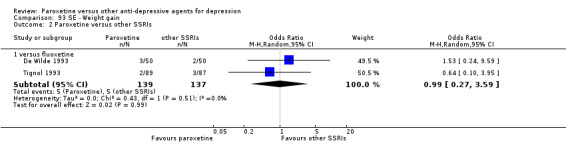
Comparison 93 SE ‐ Weight gain, Outcome 2 Paroxetine versus other SSRIs.
93.3. Analysis.
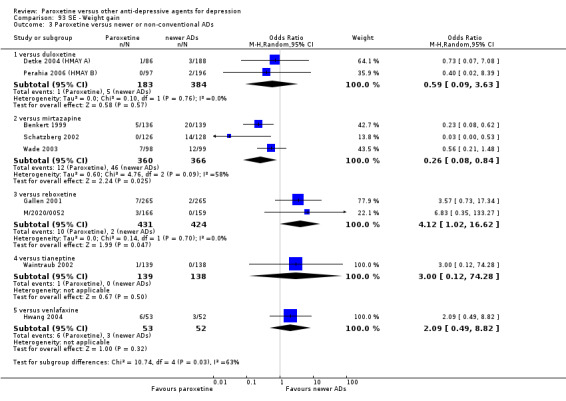
Comparison 93 SE ‐ Weight gain, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 94. SE ‐ Weight loss.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus maprolitine | 1 | 71 | Odds Ratio (M‐H, Random, 95% CI) | 2.84 [0.11, 71.99] |
| 2 Paroxetine versus other SSRIs | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus fluoxetine | 3 | 398 | Odds Ratio (M‐H, Random, 95% CI) | 0.42 [0.11, 1.55] |
| 3 Paroxetine versus newer or non‐conventional ADs | 6 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus duloxetine | 2 | 567 | Odds Ratio (M‐H, Random, 95% CI) | 0.37 [0.06, 2.18] |
| 3.2 versus mirtazapine | 1 | 275 | Odds Ratio (M‐H, Random, 95% CI) | 7.31 [0.37, 142.96] |
| 3.3 versus reboxetine | 3 | 1375 | Odds Ratio (M‐H, Random, 95% CI) | 0.88 [0.37, 2.07] |
94.1. Analysis.
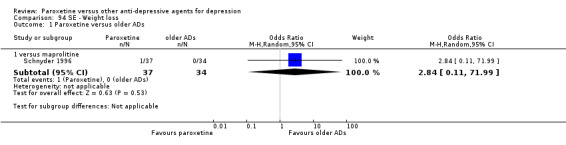
Comparison 94 SE ‐ Weight loss, Outcome 1 Paroxetine versus older ADs.
94.2. Analysis.
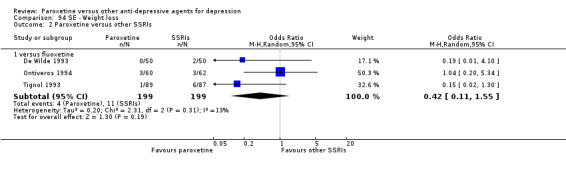
Comparison 94 SE ‐ Weight loss, Outcome 2 Paroxetine versus other SSRIs.
94.3. Analysis.
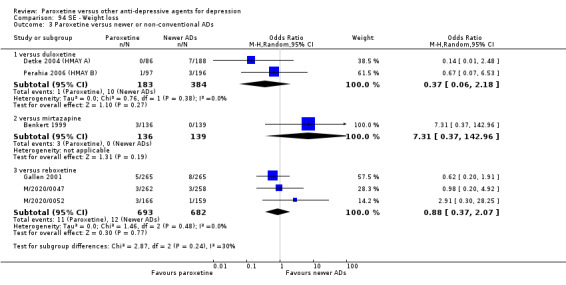
Comparison 94 SE ‐ Weight loss, Outcome 3 Paroxetine versus newer or non‐conventional ADs.
Comparison 95. SE ‐ Yawning.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Paroxetine versus older ADs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus imipramine | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 5.57 [0.62, 50.03] |
| 2 Paroxetine versus newer or non‐conventional ADs | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus reboxetine | 2 | 1050 | Odds Ratio (M‐H, Random, 95% CI) | 12.13 [1.57, 93.64] |
| 2.2 versus tianeptine | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 3.0 [0.12, 74.28] |
95.1. Analysis.
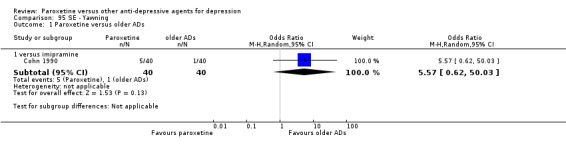
Comparison 95 SE ‐ Yawning, Outcome 1 Paroxetine versus older ADs.
95.2. Analysis.
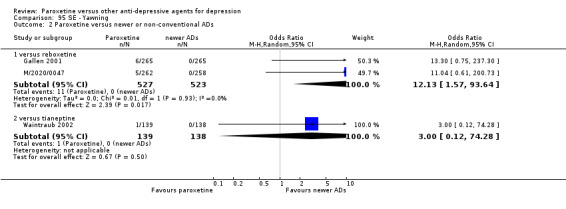
Comparison 95 SE ‐ Yawning, Outcome 2 Paroxetine versus newer or non‐conventional ADs.
Comparison 96. Deaths, suicide and suicidality.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Suicide ‐ attempted | 8 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 versus agomelatine | 1 | 572 | Odds Ratio (M‐H, Random, 95% CI) | 1.16 [0.22, 6.04] |
| 1.2 versus amitriptyline | 2 | 351 | Odds Ratio (M‐H, Random, 95% CI) | 0.33 [0.03, 3.19] |
| 1.3 versus clomipramine | 1 | 1019 | Odds Ratio (M‐H, Random, 95% CI) | 0.49 [0.15, 1.63] |
| 1.4 versus fluoxetine | 2 | 314 | Odds Ratio (M‐H, Random, 95% CI) | 0.25 [0.03, 2.30] |
| 1.5 versus reboxetine | 1 | 325 | Odds Ratio (M‐H, Random, 95% CI) | 2.89 [0.12, 71.50] |
| 1.6 versus tianeptine | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 7.10 [0.36, 138.80] |
| 2 Suicide ‐ completed | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 versus agomelatine | 1 | 572 | Odds Ratio (M‐H, Random, 95% CI) | 2.90 [0.18, 46.73] |
| 2.2 versus amitriptyline | 1 | 217 | Odds Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 8.12] |
| 2.3 versus clomipramine | 1 | 1019 | Odds Ratio (M‐H, Random, 95% CI) | 0.99 [0.06, 15.81] |
| 2.4 versus fluoxetine | 1 | 138 | Odds Ratio (M‐H, Random, 95% CI) | 1.03 [0.06, 16.80] |
| 3 Suicide ‐ tendency/ideation | 6 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 versus amitriptyline | 1 | 26 | Odds Ratio (M‐H, Random, 95% CI) | 5.87 [0.25, 135.15] |
| 3.2 versus fluoxetine | 3 | 952 | Odds Ratio (M‐H, Random, 95% CI) | 1.54 [0.35, 6.71] |
| 3.3 versus lofepramine | 1 | 106 | Odds Ratio (M‐H, Random, 95% CI) | 2.63 [0.10, 66.00] |
| 3.4 versus tianeptine | 1 | 277 | Odds Ratio (M‐H, Random, 95% CI) | 3.0 [0.12, 74.28] |
| 4 Deaths (any cause) | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 versus fluoxetine | 2 | 711 | Odds Ratio (M‐H, Random, 95% CI) | 0.64 [0.08, 5.23] |
| 4.2 versus lofepramine | 1 | 106 | Odds Ratio (M‐H, Random, 95% CI) | 0.86 [0.05, 14.07] |
96.4. Analysis.
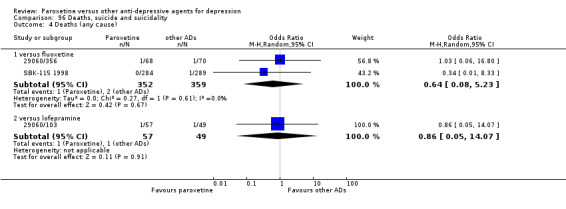
Comparison 96 Deaths, suicide and suicidality, Outcome 4 Deaths (any cause).
Characteristics of studies
Characteristics of included studies [ordered by study ID]
029060/1/CPMS 069 1991.
| Methods | Nine‐week, randomised, double‐blind, double‐dummy, parallel group, multicentre study. | |
| Participants | Participants of either sex, over the age of 60 years, with a diagnosis of a major episode of depression according to the DSM III, scoring 18 or more on the Hamilton rating scale for depression (HDRS‐21), and being a hospital inpatient who gave written informed consent. | |
| Interventions | Paroxetine: 45 participants. Clomipramine: 47 participants. Paroxetine dose range: 20‐50 mg/day. Clomipramine dose range: 150‐150 mg/day. |
|
| Outcomes | HDRS‐21, Montgomery and Asberg Depression Rating Scale (MADRS), Clinical Global Impression (CGI), the Widlocher rating scale, the Wang anxiety rating scale. Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | None. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly assigned", no further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind, double dummy". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "the analyses were based on the Intention‐to‐treat (ITT). This consisted of the all subjects who had received at least one dose of study medication and for whom any data were available after the start of treatment (...) the safety analysis was based on the ITT population". |
| Selective reporting (reporting bias) | Low risk | Outcomes data were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
029060/1/CPMS‐095.
| Methods | Randomised, double‐blind, double‐dummy, parallel group, multicentre study. | |
| Participants | In‐ or out‐patients of either sex in the age range of 18‐65 with a major depressive episode (DSM III‐R) and who had a score of at least 18 in the Hamilton rating scale for depression (HDRS‐17) total were included in the trial. A total of 210 participants were enrolled in the study and 202 participants proceeded to the active treatment phase; 68 were randomised to low‐dose paroxetine, 66 to high‐dose paroxetine and 68 to imipramine. All these participants were included in the intent‐to‐treat analysis. | |
| Interventions | Paroxetine: 134 participants. Imipramine: 68 participants. Paroxetine dose range: 20‐40 mg/day. Imipramine dose: 150 mg/day. |
|
| Outcomes | HDRS‐17, HDRS factor sub‐scales (anxiety, melancholia, retardation and sleep disturbance), Clinical Global Impression (CGI), Hamilton rating scale for anxiety (HAM‐A). Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Funding: paroxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "double blind (...) a double dummy technique was used. Paroxetine was given as white, oval, film‐coated tablets containing 20 mg paroxetine. Placebo tablets of identical appearance were supplied. Subjects took two tablets in the morning. (...) Imipramine was given as brown capsules containing 50 mg imipramine. Placebo capsules of identical appearance were supplied". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "the Intention‐to‐treat (ITT) population for efficacy was defined as subjects who were randomized and received treatment and if at least one efficacy assessment was made. The ITT population for safety and tolerability was defined as those subjects who were randomized and received treatment. The withdrawal effects population was defined as those included in the last 10 days of the run‐out period". Withdrawn: paroxetine 7/134 participants; imipramine 6/68 participants. |
| Selective reporting (reporting bias) | Low risk | Outcome data were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
0600A‐349.
| Methods | Data from eight randomised, double‐blind, controlled studies were pooled to compare efficacy in depressed patients receiving venlafaxine/venlafaxine extended release (XR), SSRIs or placebo for < 8 weeks. | |
| Participants | Patients could be enrolled in the studies if they were at least 18 and met the criteria of the DSM‐III‐R or DSM‐IV for major depression or major depressive disorder, respectively, for at least 1 month before enrolment. | |
| Interventions | Paroxetine: 85 participants. Venlafaxine XR: 82 participants. Paroxetine dose range: 20‐40 mg/day. Venlafaxine dose range: 75‐150 mg/day. |
|
| Outcomes | Hamilton rating scale for depression (HDRS‐21), Montgomery and Asberg Depression Rating Scale (MADRS), Clinical Global Impression (CGI) Improvement, Severity. Total dropout, dropout due to side effects, dropout due to inefficacy. | |
| Notes | Funding: venlafaxine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "analyses were performed on data from Intention‐to‐treat (ITT) patients; this ITT population included all patients who began the double blind treatment phase, received at least one dose of study medication, and had at least one HDRS evaluation during therapy or within 3 days of the last treatment day. The Last Observation Carried Forward (LOCF) approach was used for missing observations and to include results from the 6‐week studies in the week 8 efficacy assessment". |
| Selective reporting (reporting bias) | Unclear risk | Outcomes data not clearly reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
0600B 428.
| Methods | A multicentre, double‐blind, randomised study. | |
| Participants | Patients who had symptoms of major depression meeting DSM‐IV criteria for at least 2 weeks. | |
| Interventions | Paroxetine: 56 participants. Venlafaxine XR: 58 participants. Paroxetine dose: 20 mg/day. Venlafaxine dose: 75 mg/day. |
|
| Outcomes | Montgomery and Asberg Depression Rating Scale (MADRS), Hamilton rating scale for depression (HDRS), Clinical Global Impression (CGI). Total dropout, dropout due to side effects. | |
| Notes | Funding: venlafaxine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information on data analysis and patients missed during the follow‐up. |
| Selective reporting (reporting bias) | Unclear risk | Baseline characteristics and outcomes data not clearly reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
0600B1‐367.
| Methods | Pooled analysis of data from 5 double‐blind, parallel group trials conducted in depressed patients in the USA, Canada and Europe. | |
| Participants | Participants were 18 years of age who met the criteria for moderate to severe depression. Patients were eligible if they reported symptoms of depression for at least 1 month before the beginning of the study and scored a minimum of 20 on the Hamilton rating scale for depression (HDRS‐21 or 26 items) on the Montgomery and Asberg Depression Rating Scale (MADRS). | |
| Interventions | Paroxetine: 81 participants. Venlafaxine XR: 165 participants. Paroxetine dose: 20 mg/day. Venlafaxine dose range: 75‐150 mg/day. |
|
| Outcomes | HDRS‐21, MADRS, Clinical Global Impression (CGI) Severity. Total dropout, dropout due to side effects, dropout due to inefficacy. | |
| Notes | Exclusion criteria included significant cardiovascular, renal, hepatic or seizure disorders. In addition, patients were excluded if they had significant abnormalities on baseline physical examination, laboratory tests, ECG or a history of alcohol or drug abuse. Funding: venlafaxine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized (...) patients were randomly assigned". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "all study medications, including placebo, were given with food in the morning and were provided in identical capsules". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "scores based on the HDRS‐17 were generated for all Intention‐to‐treat (ITT) patients and were calculated on a Last Observation Carried Forward (LOCF) basis, such that the last observation for a patient who withdrew prematurely from the study was carried forward into all subsequent periods, up to 8 weeks". |
| Selective reporting (reporting bias) | Unclear risk | Outcomes data not clearly reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
29060.065.BE.
| Methods | Six‐week, randomised, double‐blind, double‐dummy, active‐controlled, parallel group study. | |
| Participants | Hospital in‐patients of either sex in the age range 18‐65 years with a major depressive episode according to the DSM‐III and a Hamilton rating scale for depression (HDRS‐21) score of 18 or more in the 21‐item total after a 1‐week placebo wash‐out. | |
| Interventions | Paroxetine: 28 participants. Maprolitine: 32 participants. Paroxetine dose range: 30‐40 mg/day. Maprolitine dose range: 100‐150 mg/day. |
|
| Outcomes | HDRS‐21, Clinical Global Impression (CGI) Severity, Montgomery and Asberg Depression Rating Scale (MADRS), the Hopkins' Symtom Checklist (HSCL‐58). Total dropout, dropout due to side effects. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Funding: paroxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind, double dummy". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind, double dummy". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "analyses were based on the Intention‐to‐treat (ITT) population. The ITT population consists of subjects who received at least one dose of active trial medication and a subsequent assessment. primary efficacy results were presented for the ITT population using the visit‐wise data set which consists of each person assessment at each visit". More than 20% of participants in the paroxetine group abandoned the study prematurely. |
| Selective reporting (reporting bias) | Low risk | Outcomes data were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
29060.07.001.
| Methods | Six‐week, multicentre, double‐blind, randomised, parallel group controlled trial. | |
| Participants | Inpatients, aged 18 or older, diagnosed with moderately severe to severe major depressive disorder with melancholia (DSM‐III), characterized by disorder of mood with symptoms such as depressed mood, sadness, hopelessness and worthlessness were eligible for the study. | |
| Interventions | Paroxetine: 13 participants. Amitriptyline: 13 participants. Paroxetine dose range: 10‐60 mg/day. Amitriptyline dose: 100 mg/day. |
|
| Outcomes | Hamilton rating scale for depression (HDRS), Symptom checklist (SCL), Clinical Global Impression (CGI), Montgomery and Asberg Depression Rating Scale (MADRS). Endpoint was identified at week 6. Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Funding: paroxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind (...) subjects received two bottles of screen medication (a morning and evening bottle) and were instructed to take two capsules from each bottle morning and evening". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "the primary analysis of the study is based on the all efficacy subject population using the extender data set and analysis of variance (...) The Intention to treat (ITT) population consisted of subjects who were evaluable for safety analysis. These subjects must have entered the active treatment phase of the study, taken double blind medication, and at least had an opportunity to report the presence or absence of an adverse event". |
| Selective reporting (reporting bias) | Low risk | Outcomes data were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
29060/056/UK.
| Methods | A double‐blind, multicentre, parallel group study. | |
| Participants | Inclusion criteria: male and female participants, aged > 65 years; major depressive illness according to the DSM‐III‐R; total score >18 on the Montgomery and Asberg Depression Rating Scale (MADRS). Exclusion criteria: participants with other psychiatric disorders; participants with suicidal tendencies. |
|
| Interventions | Paroxetine: 67 participants. Dotiepine: 67 participants. Paroxetine dose: 20 mg/day. Dotiepine dose: 75 mg/day. |
|
| Outcomes | MADRS, Clinical Global Impression (CGI), Leeds Sleep Evaluation Questionnaire (LSEQ). Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Funding: paroxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "the Intention‐to‐treat (ITT) population included all subjects who received at least one dose of study medication and had at least one assessment. All subjects who took active medication were analyzed for safety. MADRS, CGI and LSEQ data were subject to a Last Observation Carried Forward (LOCF) procedure before analysis". Withdrawn: paroxetine 12/67 participants; dothiepin 8/67 participants. |
| Selective reporting (reporting bias) | High risk | Standard deviations for rating scales scores not reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
29060/103.
| Methods | A double‐blind, multicentre, randomised, comparative study. | |
| Participants | Male or female hospital inpatients or outpatients aged 65 to 85 years suffering a major depressive episode defined according to DSM‐III‐R were eligible for inclusion. Participants scored > 20 on the Montgomery and Asberg Depression Rating Scale (MADRS) and 23 on the Folstein Mini Mental State Examination (FMMSE) at day ‐7 were included as well. | |
| Interventions | Paroxetine: 57 participants. Lofepramine: 49 participants. Paroxetine dose range: 20‐30 mg/day. Lofepramine dose range: 70‐210 mg/day. |
|
| Outcomes | MADRS, Global Deterioration Scale (GDS), Clinical Global Impression (CGI). Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Participants with severe disease, dementia, mania, bipolar affective disorder and schizophrenia were excluded from the study. Funding: paroxetine manufacturer. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "all subjects randomized to active treatment and not excluded by failure to take > or = 1 dose of study medication and who had data captured were included in the Intention‐to‐treat (ITT), clinical, safety, and tolerability analyses. Subjects who also had > or = 1 valid efficacy evaluation after the start of treatment were eligible for the ITT efficacy analysis. Subjects included in the ITT analysis were also included in the Per Protocol (PP) analysis". More than 20% of participants in the lofepramine group abandoned the study prematurely. |
| Selective reporting (reporting bias) | Low risk | Outcome data were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
29060/281 PAR.
| Methods | Seven‐week, double‐blind, double‐dummy, parallel group study. | |
| Participants | Participants aged 18 to 70 years; suffering from unipolar (endogenous or reactive) depression; fulfilling Feighner's criteria for depression; had a minimum Hamilton rating scale for depression (HDRS‐21) score of 18. | |
| Interventions | Paroxetine: 82 participants.
Amitriptyline: 80 participants. Paroxetine dose: 30 mg/day. Amitriptyline dose range: 75‐150 mg/day. |
|
| Outcomes | HDRS‐21, Clinical Global Impression (CGI), Self‐Rating Scale. Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Funding: paroxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "subjects were randomly allocated". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind, double dummy (...) all medication was administered orally once daily in the evening with matching placebo tablets or capsules according to a double dummy design". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "the main analyses were carried out on the Intention‐to‐treat (ITT) population ‐ patients who were randomized and had on‐treatment data available. An "extender" dataset was created using a Last Observation Carried Forward (LOCF) technique to impute missing values using the previous on‐treatment data for that subject". |
| Selective reporting (reporting bias) | High risk | Outcomes data reported without standard deviations. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
29060/299.
| Methods | A double‐blind, multicentre, randomised, flexible dose, parallel group study. | |
| Participants | Male or non‐pregnant, non‐lactating female patients using adequate contraception were eligible if they were aged 18 to 65 years; were hospitalised; had a diagnosis of severe, unipolar depression (without psychotic symptoms), defined according to ICD‐10 criteria; and had a score of at least 24 points on the Hamilton rating scale for depression (HDRS‐17) at screening and at the baseline assessment. | |
| Interventions | Paroxetine: 109 participants.
Amitriptyline: 108 participants. Paroxetine dose range: 20‐50 mg/day. Amitriptyline dose range: 100‐250 mg/day. |
|
| Outcomes | Clinical Global Impression (CGI), Hospital Anxiety and Depression scale (HAD). Total dropout, dropout due to side effects. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Funding: paroxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were randomized (1:1) to either paroxetine or amitriptyline". |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "the primary analysis was on the Intention‐to‐treat (ITT) data set at endpoint. No efficacy data was available for 15 of the randomized patients. The ITT analysis was therefore performed on 202 subjects of whom 102 were randomized to paroxetine and 100 to amitriptyline. The safety population included all randomized subjects". |
| Selective reporting (reporting bias) | Low risk | Outcomes data were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
29060/356.
| Methods | Eight‐week, double‐blind, multicentre, randomised, comparative study. | |
| Participants | Moderate to moderately severe depression with a diagnosis of Major Depressive Disorder (MDD) (DSM‐III‐R), male or female at least 18 years old, inpatients or outpatients with a Hamilton rating scale for depression (HDRS‐17) total score 18 or more, HAMD‐10 of 1 or more. | |
| Interventions | Fluoxetine: 68 participants. Paroxetine: 70 participants. Fluoxetine dose: 20 mg/day. Paroxetine dose: 20 mg/day. | |
| Outcomes | HDRS, Hamilton rating scale for anxiety (HAM‐A), Clinical Global Impression (CGI). Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Funding: paroxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "Intention‐to‐treat (ITT) population consisted of all subjects randomized to active treatment and for whom one valid post‐randomization evaluation was available. Per Protocol (PP) population consisted of all subjects who had no major protocol violation with regard to inclusion/exclusion criteria, duration of treatment was at least 42 days, and no major protocol violation occurred during the first 2 weeks of active treatment. Safety population consisted of all subjects who were randomized to treatment. Primary efficacy analysis was based on both ITT and PP population". More than 20% of participants abandoned the study prematurely. |
| Selective reporting (reporting bias) | Low risk | Outcomes data were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
29060/409.
| Methods | Controlled,double‐blind, double‐dummy study. | |
| Participants | Male and female participants with a diagnosis of MDD (DSM‐III), Hamilton rating scale for depression (HDRS‐17) total score >/= 22. Age range: 18‐65 years old. |
|
| Interventions | Paroxetine: 38 participants. Imipramine: 37 participants. Paroxetine dose: 20 mg/day. Imipramine dose range: 150‐300 mg/day. |
|
| Outcomes | HDRS‐25, HDRS‐17, Hamilton rating scale for anxiety (HAM‐A), Clinical Global Impression (CGI).Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Funding: paroxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind, double dummy". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind, double dummy". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "the subject population used for the efficacy analysis was the Intent‐to‐treat (ITT) Last Observation Carried Forward (LOCF)." More than 20% of participants abandoned the study prematurely. |
| Selective reporting (reporting bias) | Low risk | Outcome data were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
29060/III/83/022.
| Methods | Double‐blind, randomised, double‐dummy, parallel group study. | |
| Participants | Male or female participants with reactive or endogenous unipolar depression, and who were considered suitable for treatment with tricyclic‐like anti‐depressant, could be eligible for inclusion in the study. Participants also had to meet Spizer's criteria for depression and score a minimum of 15 on the Hamilton rating scale for depression (HDRS‐17) at baseline. Age range: 18‐70 years. |
|
| Interventions | Paroxetine: 18 participants. Mianserin: 18 participants. Paroxetine dose range: 15‐45 mg/day. Mianserine dose range: 30‐90 mg/day. |
|
| Outcomes | HDRS‐17, Clinical Global Impression (CGI). Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | None. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "subject were randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind, double dummy". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind, double dummy". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | No details on the Intention‐to‐treat (ITT) population. Number of randomized, and number of subjects withdrawn not reported for the paroxetine arm. |
| Selective reporting (reporting bias) | High risk | Outcomes data not clearly reported. Standard deviations on rating scales scores not reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
29060/III/85/030.
| Methods | Randomised, double‐blind, double‐dummy parallel group study. | |
| Participants | Participants suffering from reactive or endogenous depression (fulfilling the DSM criteria for major depressive illness) and having a minimum score of 17 on the Hamilton rating scale for depression (HDRS‐17) at baseline were eligible for inclusion in the study. Age range: 18‐65 years. |
|
| Interventions | Paroxetine: 18 participants. Mianserin: 15 participants. Paroxetine dose range: 15‐30 mg/day. Mianserine dose range: 30‐90 mg/day. |
|
| Outcomes | HDRS‐17. Total dropout, dropout due to side effects. Number of patients experiencing at least one side effect. | |
| Notes | No specific exclusion criteria were defined. Funding: paroxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "subject were randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "the population of interest was the Intent‐to‐treat (ITT) population and the timepoint of interest was the 6 week extender endpoint". More than 20% of participants in both groups abandoned the study prematurely. |
| Selective reporting (reporting bias) | High risk | Outcomes data not clearly reported. Standard deviations for rating scales scores not reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
29060III/85/038.
| Methods | A double‐bind, parallel group, active controlled, double‐dummy, randomised study. | |
| Participants | Participants suffering from either reactive or endogenous unipolar depression considered suitable for treatment with "tricyclic‐like" antidepressants were eligible for the study. Participants had to score al least 17 on the first 17‐itemof the Hamilton rating scale for depression (HDRS‐21) and fulfil the DSM‐III criteria for the diagnosis of depression. Subjects were excluded in they were suffering from severe co‐existing diseases, intolerant to "tricyclic‐like" drugs, scored 4 on the suicide item of the HDRS‐21, had received ECT in the previous 3 months required hospitalisation for ECT treatment, had taken IMAO with 2 weeks of the active phase of the study and female participants who were pregnant or lactating. Age range: 18‐70 years old. |
|
| Interventions | Paroxetine: 30 participants. Mianserin: 29 participants. Paroxetine dose range: 15‐30 mg/day Mianserine dose range: 30‐90 mg/day. |
|
| Outcomes | HDRS‐21, Clinical Global Impression (CGI). Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | None. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "subjects were randomized (...) allocation of treatment by random code". |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind, using a double dummy technique". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind, using a double dummy technique". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "results are presented for the Intention‐to‐treat (ITT) population, extender data set". Less than 20% of participants in each study arm abandoned the study prematurely. |
| Selective reporting (reporting bias) | Low risk | Outcomes data were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Aberg‐Wistedt 2000.
| Methods | Twenty‐four week, double‐blind, randomised study. | |
| Participants | Outpatients meeting DSM‐III‐R criteria for major depression (no patients with bipolar disorder). Age range: over 18 years. | |
| Interventions | Paroxetine: 177 participants Sertraline: 176 participants. Paroxetine dose: 20‐40 mg/day. Sertraline dose: 50‐150 mg/day. The association of short half‐time benzodiazepines was allowed for insomnia in those patients who already been receiving concomitant treatment before the study began. |
|
| Outcomes | Montgomery‐Asberg Depression Rating Scale (MADRS), Clinical Global Impression (CGI). Total dropout, side‐effect profile. |
|
| Notes | Funding: sertraline manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomly assigned". Probably done, as a similar trial by these investigators included the same phrase and used a proper method of allocation. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double‐blind"; no further information. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double‐blind"; no further information. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Three efficacy analyses were performed: an ITT analysis performed on all randomly assigned patients who were available at each study visit; an Intention‐to‐treat‐Last Observation Carried Forward (ITT‐LOCF) analysis and an analysis performed on all patients who completed study treatment with no major protocol deviation. Study endpoint: 61/177 missing from paroxetine group; 64/176 missing from control group. |
| Selective reporting (reporting bias) | Low risk | The study protocol is not available but it is clear that the published reports include all expected outcomes, including those that were pre‐specified. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Ansseau 1993.
| Methods | Six‐week, randomised, double‐blind, multicentre study. | |
| Participants | Psychiatric in‐ and outpatients meeting DSM‐III‐R for major depressive episode, with a minimum baseline score of 18 on the Hamilton rating scale for depression (HRSD‐21). Age range: 18‐65 years. | |
| Interventions | Fluvoxamine: 64 participants.
Paroxetine: 56 participants.
Fluvoxamine dose range: 50‐200 mg/day.
Paroxetine dose range: 20‐30 mg/day. For patients who had received benzodiazepines for at least two weeks prior to continue these agents, providing the dose remained unchanged throughout the study period. In addition, low dose lormethazepam or chloral hydrate were permitted in case of severe insomnia. |
|
| Outcomes | HRSD‐21, Clinical Global Impression‐ Severity (CGI‐S), Hamilton rating scale for anxiety (HAM‐A). Total dropout, dropout due to inefficacy, dropout due to side effects, number of patients experiencing at least one side effect, side‐effect profile. |
|
| Notes | Funding: paroxetine manufacturer. Patients with major depressive episode (DSM‐III‐R) were included, so there might be some bipolar depression, but correct number was not reported. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly assigned", no further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "the trial used a double‐blind design", no further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "the trial used a double‐blind design", no further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The Intention‐to‐treat (ITT) population was analysed using an endpoint dataset as well as a visit‐wise dataset. Study endpoint: 16/56 missing from paroxetine group; 23/64 missing from control group. |
| Selective reporting (reporting bias) | High risk | Standard deviations of change score for depression were not reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Aoba 2004.
| Methods | Six‐week, double‐blind, randomised study. | |
| Participants | Inpatients and outpatients meeting DSM‐IV criteria for major depression (no patients with bipolar disorder). Age range: 18‐65 years. | |
| Interventions | Paroxetine: 106 participants. Imipramine: 104 participants. Paroxetine dose: 20‐40 mg/day. Imipramine dose: 50‐150 mg/day. |
|
| Outcomes | Hamilton rating scale for depression (HDRS‐17), Clinical Global Impression (CGI). Total dropout, dropout due to inefficacy, dropout due to side effects, number of patients experiencing at least one side effect, side‐effect profile. |
|
| Notes | Funding: paroxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Study endpoint: 22/106 missing from paroxetine group; 31/104 missing from control group. All safety analyses were performed on safety population. Efficacy analyses were performed on FAS population and the Last Observation Carried Forward (LOCF) dataset was used. The FAS population consisted in all subjects who have entered the treatment phase with the exception of those who did not meet the major eligibility criteria, those who did not take any study medication during the treatment phase and those with no valid post baseline assessment. |
| Selective reporting (reporting bias) | High risk | Mean change from baseline only reported as "rate of decrease from baseline at endpoint". |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Bakish 1997.
| Methods | Double‐blind, active‐controlled study comparing the efficacy of paroxetine and fluoxetine. | |
| Participants | All patients fulfilled the DSM III‐R criteria for Major Affective Disorder, unipolar (either first episode or recurrent) and had met the diagnostic criteria for major depressive episode for at least one month prior to entering the study. To be included in the study, patients had to score 18 or more on the Hamilton rating scale for depression (HDRS‐17), with a score of two or more on item one. Patients with a concurrent DSM III‐R Axis I diagnosis or an unstable medical condition were excluded from the study. |
|
| Interventions | Paroxetine: 12 participants. Fluoxetine: 9 participants. Paroxetine dose: 20‐50 mg/day. Fluoxetine dose: 20‐80 mg/day. No concomitant psychotropic medications were allowed with the exception of chloral hydrate at bedtime on an "as needed" basis to a maximum dose of 1000 mg/day. |
|
| Outcomes | HDRS‐17, Clinical Global Impression (CGI). | |
| Notes | This study was partly supported by SmithKline Beecham. Outcomes data were not available. The study was primarily designed to investigate the effect of antidepressants on platelet serotonin parameters. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly assigned". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No clear information on incomplete outcome data management. |
| Selective reporting (reporting bias) | Unclear risk | Outcomes data not available. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Baldwin 1995.
| Methods | Eight‐week, double‐blind, randomised study. | |
| Participants | Outpatients meeting DSM‐III‐R criteria for major depression (there are some bipolars but less than 20% of the total randomised sample). Age range: over 18 years. | |
| Interventions | Paroxetine: 101 participants. Nefazodone: 105 participants. Paroxetine dose: 20‐40 mg/day. Nefazodone dose: 200‐600 mg/day. The association of short half‐time benzodiazepines was allowed for insomnia in those patients who already been receiving concomitant treatment before the study began. |
|
| Outcomes | Montgomery and Asberg Depression Rating Scale (MADRS), Hamilton rating scale for depression (HDRS‐17), Clinical Global Impression (CGI). Total dropout, dropout due to inefficacy, dropout due to side effects, number of patients experiencing at least one side effect, side‐effect profile. |
|
| Notes | Funding: nefazodone manufacturer. Adverse events reported only if occurring in >= 10% of any treatment group. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly assigned", no further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "a double dummy technique was used to maintain the blind". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "a double dummy technique was used to maintain the blind". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Safety analyses comprised the data from all patients who were randomly assigned to treatment and received the study medication. Efficacy analyses were performed on all patients who had received study medication and who had undergone at least one efficacy evaluation during treatment. Study endpoint: 28/101 missing from paroxetine group; 28/105 missing from control group. |
| Selective reporting (reporting bias) | High risk | Standard deviations of change score for depression were not reported. Adverse events were reported only if occurring in >= 10% of any treatment group. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Baldwin 2006.
| Methods | Twenty‐seven‐week double‐blind, randomised, multicentre study. For efficacy data we consider only the first eight weeks. | |
| Participants | Setting unclear, patients meeting DSM‐IV for major depressive episode, with a minimum baseline score of 22 on the Montgomery and Asberg Depression Rating Scale (MADRS). Age: older than 18 years. | |
| Interventions | Paroxetine: 101 participants.
Escitalopram: 105 participants. Paroxetine dose range: 20‐40 mg/day. Escitalopram dose range: 10‐20 mg/day. For patients who had received benzodiazepines for at least 6 months prior to continue these agents, providing the dose remained unchanged throughout the study period. |
|
| Outcomes | MADRS, Arizona sexual experience scale (ASEX). Total dropout, dropout due to inefficacy, dropout due to side effects, number of patients experiencing at least one side effect, side‐effect profile. |
|
| Notes | Funding: escitalopram manufacturer. Adverse events reported only if occurring in >= 5% of any treatment group. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were assigned to escitalopram or paroxetine treatment according to a computer‐generated randomization list". |
| Allocation concealment (selection bias) | Low risk | Quote: "the details of the randomization series were unknown to any of the investigators and were unknown to any of the investigators and were contained in a set of opaque envelops". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "all study personnel and participants were blinded to treatment assignment for the duration of the study". "Treatment was in the form of tablets of identical appearance, taste and smell". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "all study personnel and participants were blinded to treatment assignment for the duration of the study". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Efficacy analyses were conducted for the modified Intention‐to‐treat (ITT) population which included all randomized patients who took at least one dose of double blind study medication and who had least one valid post baseline MADRS assessment. |
| Selective reporting (reporting bias) | High risk | Standard deviations of change score for depression were not reported. Adverse events were reported only if occurring in >= 5% of any treatment group. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out |
Bascara 1989.
| Methods | Six‐week, double‐blind, randomised study. | |
| Participants | Setting unclear, patients meeting DSM‐III criteria for major depression with a minimum score of 18 on the Hamilton rating scale for depression (HDRS‐21). Age: over 18 years. | |
| Interventions | Paroxetine: 27 participants.
Amitriptyline: 23 participants. Paroxetine dose: 30 mg/day. Amitriptyline dose: 75 mg/day. |
|
| Outcomes | HDRS‐21. Dropout due to side effects. | |
| Notes | None. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were randomly allocated". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: " in a double blind fashion (...) material was prepared using a double dummy technique. all study medication was given in the morning". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information on incomplete outcome data management. |
| Selective reporting (reporting bias) | High risk | Standard deviations of change scores for depression were not reported. Efficay and safety analyses reported only as rate of patients who "improved" and for whom treatment was "well tolerated". |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Battegay 1985.
| Methods | Seven‐week, double‐blind, randomised study. | |
| Participants | Outpatients meeting DSM III endogenous and reactive depression, with a minimum score of 20 on the Hamilton rating scale for depression (HDRS‐17). Age range: 18‐60 years. | |
| Interventions | Paroxetine: 11 participants. Amitriptyline: 10 participants. Paroxetine dose range: 10‐30 mg/day. Amitriptyline dose range: 50‐100 mg/day. In case of severe insomnia tranquillisers with a short half life were permitted. |
|
| Outcomes | HDRS‐17, Montgomery and Asberg Depression Rating Scale (MADRS), Clinical Global Impression (CGI). Dropout, number of patients experiencing at least one side effect, side‐effect profile. |
|
| Notes | Small sample; more than 50% of patients did not complete the trial. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "the patients received (...) in a randomized procedure (...)". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No informations provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "the patients received in a double blind procedure identically looking white tablets containing either 10 mg paroxetine ore 50 mg amitriptyline". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Study endpoint: 3/11 missing from paroxetine group; 8/10 missing from control group. |
| Selective reporting (reporting bias) | High risk | Standard deviations of change scores for depression were not reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Benkert 1999.
| Methods | Six‐week, randomised, double‐blind, multicentre study. | |
| Participants | Outpatients meeting DSM‐IV criteria for major depression (no patients with bipolar disorder). Age range: 18‐70 years. | |
| Interventions | Paroxetine: 136 participants. Mirtazapine: 139 participants. Paroxetine dose range: 20‐40 mg/day. Mirtazapine dose range:15‐45 mg/day. In case of severe insomnia chloral hydrate was permitted. |
|
| Outcomes | The measure used for response and remission in the review: Hamilton rating scale for depression (HDRS‐17). Other measures: Hamilton rating scale for anxiety (HAM‐A), Beck Depression Inventory (BDI), Welzil‐Kohnen Color Scales (WKFS), Short Form‐36, Clinical Global Impression (CGI) improvement, CGI‐severity. |
|
| Notes | Funding: mirtazapine manufacturer. Adverse events reported only if occurring in >= 5% of any treatment group. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double‐blind", "both drugs were given once daily, mirtazapine in the evening and paroxetine in the morning using a double dummy technique. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double‐blind". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | More than 20% of the allocated patients to both of the intervention arms dropped out during the study. Efficacy analyses were based on the Intention‐to‐treat (ITT) patients sample, including all randomly assigned patients who received at least one dose of study medication and had at least one post‐baseline efficacy assessment. A Last Observation Carried Forward (LOCF) analysis was performed for the endpoint assessment. |
| Selective reporting (reporting bias) | High risk | Both the response and the remission outcomes at end of the acute‐phase treatment are reported with the proportion of the patients who achieved these. Standard deviations of change scores were not reported. Adverse events were reported only if occurring in >= 5% of any treatment group. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Bignamini 1992.
| Methods | Six‐week, randomised, double‐blind, multicentre study. | |
| Participants | Outpatients meeting DSM‐III diagnostic criteria for depression. Age range: 18‐70 years. |
|
| Interventions | Paroxetine: 156 participants.
Amitriptyline: 153 participants. Paroxetine dose range: 20‐30 mg/day. Amitriptyline dose range: 75‐150 mg/day. |
|
| Outcomes | Hamilton rating scale for depression (HDRS‐21). Total dropout, dropout due to inefficacy, dropout due to side effects, number of patients experiencing at least one side effect, side‐effect profile. |
|
| Notes | Funding: paroxetine manufacturer. Adverse events reported only if occurring in >=5% and only on graphs. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind"; "double dummy". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind"; "double dummy". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Study endpoint: 31/156 missing from Paroxetine group; 20/153 missing from control group. The population on which efficacy and safety analyses have been conducted was not well specified. |
| Selective reporting (reporting bias) | High risk | Two rating scales for depression Clinical Global Impression (CGI) and HDRS listed in methods but only one is reported (HDRS). Adverse events were reported only if occurring in >=5% and only on graphs. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Blier 2009.
| Methods | Eight‐week, randomised, double‐blind study. We consider only the first six weeks of treatment because in the subsequent two weeks the possibility of combining the two treatment is introduced and randomisation is lost. | |
| Participants | In and outpatients meeting DSM IV diagnostic criteria for major depression (no bipolar patients included in the study). Age range:18‐65 years. |
|
| Interventions | Paroxetine: 19 participants. Mirtazapine: 21 participants. Paroxetine dose range: 20‐30 mg/day. Mirtazapine dose range: 30‐45 mg/day. |
|
| Outcomes | Montgomery and Asberg Depression Rating Scale (MADRS), Hamilton rating scale for depression (HDRS‐17), Clinical Global Impression (CGI). | |
| Notes | Funding: mirtazapine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "patients received either two mirtazapine tablets at bedtime with two placebo in the morning or two paroxetine tablets in the morning with placebo in the evening". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number of total dropout not specified. |
| Selective reporting (reporting bias) | High risk | Three rating scales in method, but only one reported in results (MADRS). |
| Other bias | Unclear risk | Quote: "this investigator‐initiated study was fully funded by Organon Pharmaceuticals. The sponsor had no role in the study design, in the collection and interpretation of the data, in the preparation of this report and in the decision to publish this manuscript. |
Boulenger 2006.
| Methods | Twenty‐four‐week, randomised, double‐blind, multicentre study. | |
| Participants | Outpatients meeting DSM‐IV criteria for major depression (no patients with bipolar disorder) with a minimum score of 30 on the Montgomery and Asberg Depression Rating Scale (MADRS) rating scale (severe depression). Age range: 18‐75 years. | |
| Interventions | Paroxetine: 227 participants. Escitalopram: 232 participants. Paroxetine dose: 40 mg/day. Escitalopram dose: 20 mg/day. |
|
| Outcomes | MADRS (primary outcome), Hamilton rating scale for depression (HDRS‐24, HDRS‐17), Clinical Global Impression (CGI). Total dropout, dropout due to inefficacy, dropout due to side effects, number of patients experiencing at least one side effect, side‐effect profile. |
|
| Notes | Funding: escitalopram manufacturer. Adverse events reported only if occurring in >=5%. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients (...) were assigned in a 1:1 ratio of escitalopram or paroxetine according to a computer generated randomization list. (...) At each study centre, sequentially enrolled patients were assigned to the lowest randomization number in blocks of four". |
| Allocation concealment (selection bias) | Low risk | Quote: "the details of the randomization series were unknown to any of the investigators and were contained in a set of sealed opaque envelopes". |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "all study personnel and participants were blinded to treatment assignment for the duration of the entire study". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "all study personnel and participants were blinded to treatment assignment for the duration of the entire study". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Study endpoint (24 week) : 72/227 missing from paroxetine group; 44/232 missing from control group. Efficacy analyses were based on the Intention‐to‐treat (ITT) set and using Last Observation Carried Forward (LOCF). The ITT population comprised 451 patients (paroxetine: 223, escitalopram: 228). |
| Selective reporting (reporting bias) | High risk | Proportion of remitters and responders reported only for endpoint, data for other weeks reported only as a proportion on graphs. Standard deviations of change scores for depression were not reported. Adverse events were reported only if occurring in >=5%. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Cassano 2002.
| Methods | Fifty‐two‐week double‐blind, randomised, multicentre study. | |
| Participants | Outpatients fulfilling ICD‐10 criteria for major depression, with a Mini Mental State Examination (MMSE) score of at least 22, Hamilton rating scale for depression (HDRS) score of at least 18. Age range: over 65 years. | |
| Interventions | Paroxetine: 123 participants. Fluoxetine: 119 participants. Paroxetine dose range: 20‐40 mg/day. Fluoxetine dose range: 20‐60 mg/day. Stabilised treatment for concomitant systemic disease, temazepam for occasional treatment of insomnia and short or intermediate half‐life benzodiazepine for anxiety were permitted. |
|
| Outcomes | HDRS‐21, Clinical Anxiety Scale (CAS), Buschke Selective Reminding Test (BSRT), Blessed Information and Memory Test (BIMT), Clifton Assessment Schedule (CLAS), Cancellation Task Test (CTT), Wechsler Paired Word Test (WPW), MMSE, Clinical Global Impression (CGI). Total dropout, number of patients experiencing at least one side effect, side‐effect profile. |
|
| Notes | Funding: paroxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Study endpoint: 50/123 missing from paroxetine group; 45/119 missing from control group. Statistical evaluation was performed on the basis of an endpoint analysis and all the data were analysed on an observed‐case basis. |
| Selective reporting (reporting bias) | High risk | Standard deviations of change scores for depression were not reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Cassano 2002a.
| Methods | Eight‐week, double‐blind, randomised, multicentre study. | |
| Participants | Outpatients meeting DSM‐IV criteria for major depression (no patients with bipolar disorder) with a minimum score of 18 on the Hamilton rating scale for depression (HDRS). Age range: 18‐75 years. |
|
| Interventions | Paroxetine: 139 participants. Amisulpride: 138 participants. Paroxetine dose range: 20 mg/day. Amisulpride dose range: 50 mg/day. Benzodiazepine or hypnotic given at a stable dose were permitted. |
|
| Outcomes | HDRS, Montgomery and Asberg Depression Rating Scale (MADRS), Clinical Global Impression (CGI). Total dropout, dropout due to inefficacy, dropout due to side effects, number of patients experiencing at least one side effect, side‐effect profile. |
|
| Notes | Funding: amisulpride manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Less than 20% of the allocated patients to both of the intervention arms dropped out during the study. The primary efficacy analysis was performed on the Per Protocol (PP) population that included all randomized patients fully compliant with the protocol during the active treatment period and having at least one evaluation of the primary endpoint after baseline. Consistency of results was assessed in the Intention‐to‐treat (ITT) population. For patients not completing the trial the Last Observation Carried Forward (LOCF) was analysed. |
| Selective reporting (reporting bias) | High risk | Side effects reported only if statistically significant. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Chiu 1996.
| Methods | Six‐week, double‐blind, randomised study. | |
| Participants | Inpatients and Outpatients meeting DSM‐III‐R criteria for major depression with a minimum score of 18 at the Hamilton rating scale for depression (HDRS) rating scale. Age range: 18‐70 years. | |
| Interventions | Paroxetine: 20 participants. Imipramine: 20 participants. Paroxetine dose: 20‐30 mg/day. Imipramine dose: 75‐150 mg/day. No other psychoactive drugs were used except for patients who had initiated benzodiazepines within 14 days before the screening visit. Patients requiring hypnotics were allowed only one benzodiazepine on a p.n.r basis at night. |
|
| Outcomes | HDRS, Clinical Global Impression (CGI), Treatment Emergent Symptom Scale (TESS). Total dropout. Dropout due inefficacy. Side‐effect profile. | |
| Notes | Funding: paroxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "the blindness of the study was maintained by using a double dummy technique. Paroxetine Imipramine and placebo were all prepared in physically identical capsules. To maintain a double blind condition blister packs containing either active drugs or placebo capsules and the blisters used daily were the same in each group". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Study endpoint: 5/20 missing from paroxetine group; 5/20 missing from control group. 35 patients who did not violate the trial protocol were included in the analysis of adverse effects. 30 patients who had completed six weeks of active treatment according to the trail protocol were included for calculation of mean reduction of HDRS score. |
| Selective reporting (reporting bias) | High risk | HDRS scores for each week of treatment reported only on graphs. Side effects reported only on graphs. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Chouinard 1999.
| Methods | Twelve‐week, double‐blind, randomised, multicentre study. | |
| Participants | Patients fulfilling DSM‐III criteria for major depressive disorder, with a score of at least 20 on Hamilton rating scale for depression (HDRS‐21). Age range: not stated. |
|
| Interventions | Paroxetine: 102 participants. Fluoxetine: 101 participants. Paroxetine dose range: 20‐50 mg/day. Fluoxetine dose range: 20‐80 mg/day. Chloral hydrate was allowed just during the first two weeks of the study. |
|
| Outcomes | HDRS‐21, Clinical Global Impression(CGI), Hamilton rating scale for anxiety (HAM‐A). Total dropout, dropout due to inefficiency, dropout due to side effects, side‐effect profile. |
|
| Notes | Two participants abandoned prematurely the trial (1 in the fluoxetine and 1 in the paroxetine group) due to attempted suicide.
Funding: paroxetine manufacturer. Adverse events reported only if occurring in >= 5% of any treatment group. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | A total of 203 patients were randomized and 130 (64.04%) completed the study. A total of 100 patients in the paroxetine group and 98 in the fluoxetine one were evaluable for the Last Observation Carried Forward (LOCF) endpoint analysis. |
| Selective reporting (reporting bias) | High risk | Adverse events were reported only if occurring in >= 5% of any treatment group. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Christiansen 1996.
| Methods | Eight‐week, double‐blind, randomised, multicentre study. | |
| Participants | General practise patients with depression (unclear diagnostic criteria used) with a minimum score of 15 on the Hamilton rating scale for depression (HDRS). Age range: 18‐65 years. |
|
| Interventions | Paroxetine: 71 participants. Amitriptyline: 73 participants. Paroxetine dose range: 20‐40 mg/day. Amitriptyline dose range: 100‐150 mg/day. In case of severe insomnia tranquillisers with a short half life were permitted. |
|
| Outcomes | HDRS, Clinical Global Impression (CGI), Visual Analogue Scale (VAS). Total dropout, dropout due to side effect, dropout due to lack of efficacy. Tolerability profile. | |
| Notes | Funding: paroxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote:"patient identification numbers were randomly generated to indicate treatment options. These numbers were randomized and packed in blocks of four". |
| Allocation concealment (selection bias) | Unclear risk | Patients who fulfilled the entrance criteria were allocated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "a double dummy technique was employed using placebo tablets identical in appearance to the active drugs. The number of tablets taken daily was four at low dose and eight at high dose". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Study endpoint: 15/71 missing from paroxetine group, 16/73 from control group. Two different set of data were analysed for efficacy: the actual data produced at each visit from day 7 to day 55 and a Intention‐to‐treat (ITT) analysis of every randomised patient participating in the study and including for withdrawals last values carried forward. |
| Selective reporting (reporting bias) | High risk | Side effects reported only if statistically significant and only as a percentage. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
CL3‐023.
| Methods | Double‐blind, randomised study. | |
| Participants | Male and female adults, 18‐70 years of age, with a diagnosis of Major Depressive Disorder according to DSM‐IV criteria. Hamilton Depression Rating Scale (HDRS) total score > or = 22 at screening and baseline. | |
| Interventions | Paroxetine: 20 mg/day. Agomelatine: 25 mg/day. |
|
| Outcomes | Change from baseline to endpoint at HDRS, remission, sexual functions. | |
| Notes | Funding: by industry. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The randomisation of treatments (agomelatine, placebo, fluoxetine or paroxetine) was non‐adaptive, non‐centralised, and balanced with a 1:1:1 ratio. There was no stratification and permutation blocks were of fixed size = 6". |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double blind. Quote: "Agomelatine, placebo and fluoxetine (or paroxetine) were disguised in tablets or capsules (or tablets) of identical appearance and taste". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double blind. Quote: "Agomelatine, placebo and fluoxetine (or paroxetine) were disguised in tablets or capsules (or tablets) of identical appearance and taste". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient information. |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Cohn 1990.
| Methods | Six‐week, double‐blind, randomised, single‐centre, placebo‐controlled. | |
| Participants | Outpatients meeting DSM‐III criteria for major depression with a minimum score of 18 on Hamilton rating scale for depression (HDRS‐17). Age: older than 18 years. | |
| Interventions | Paroxetine: 40 participants Imipramine: 40 participants Placebo:40 participants. Paroxetine dose range: 10‐50 mg/day (mean dose: 30.9 mg/day). Imipramine dose range: 65‐275 mg/day (mean dose: 144.9 mg/day). Chloral hydrate (500 mg) was allowed for a maximum of 4 consecutive nights during the first two weeks of the study. |
|
| Outcomes | HDRS‐21, Clinical Global Impression (CGI) Improvement, Severity, Patient Global Experience (PGE). Total dropout, dropout due to side effect, dropout due to lack of efficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Funding: unclear. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Study endpoint: 21/40 missing from paroxetine group, 22/40 missing from control group. |
| Selective reporting (reporting bias) | High risk | Dichotomous outcome not reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Dalery 2001.
| Methods | Six‐week, double‐blind, randomised, multicentre study. | |
| Participants | Inpatients and outpatients meeting DSM‐III‐R criteria for major depression with a minimum score of 25 on the Montgomery and Asberg Depression Rating Scale (MADRS) rating scale. Age: older than 60 years. | |
| Interventions | Paroxetine: 54 participants. Mianserin: 62 participants. Paroxetine dose range: 20 mg/day. Mianserin dose range: 30 mg/day. No other psychoactive drugs were used except for patients who had initiated benzodiazepines within 14 days before the screening visit. |
|
| Outcomes | MADRS, Mini Mental State Examination (MMSE), Aubin Jouvent Rating Scale (AJRS). Total dropout, dropout due to side effect, dropout due to lack of efficacy. Tolerability profile. |
|
| Notes | None. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Study endpoint: 11/54 missing from Paroxetine group; 9/62 missing from control group. Efficacy analyses were based on the Intention‐to‐treat (ITT) patients sample, including all randomly assigned patients who received at least one dose of study medication and had at least one post‐baseline efficacy assessment. |
| Selective reporting (reporting bias) | High risk | Side‐effect profile incomplete. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Davidson 2005.
| Methods | Three‐week, double‐blind, randomised, study. | |
| Participants | Outpatients fulfilling DSM‐IV criteria for major depressive disorder. (No patients with bipolar disorder). Age range: 20‐60 years. |
|
| Interventions | Paroxetine: 25 participants. Venlafaxine XR: 24 participants. Paroxetine dose range: 20‐40 mg/day. Venlafaxine dose range: 150‐225 mg/day. |
|
| Outcomes | Heart rate variability. Total dropout. Responders at endpoint (3 weeks). | |
| Notes | This study has been conducted in order to evaluate cardiological effects of both paroxetine and venlafaxine in the treatment of depression. For this reason most efficacy and safety data are not available. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "medication assignment was based on a predetermined randomized allocation to treatment". |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Efficacy analyses were based on the Intention‐to‐treat (ITT) patients sample, including all randomly assigned patients who received at least one dose of study medication and had at least one post‐baseline efficacy assessment. Study endpoint: 11/54 missing from Paroxetine group; 9/62 missing from control group. |
| Selective reporting (reporting bias) | Unclear risk | Protocol not available. Reported cardiological parameters only. |
| Other bias | Unclear risk | Sponsorship bias cannot ruled out. |
De Wilde 1993.
| Methods | Six‐week, double‐blind, randomised, study. | |
| Participants | Patients fulfilling DSM‐III criteria for major depression, with a score of at least 18 on the Hamilton rating scale for depression (HDRS‐21). Age range: 18‐65 years. | |
| Interventions | Paroxetine: 50 participants. Fluoxetine: 50 participants. Paroxetine dose range: 20‐40 mg/day. Fluoxetine dose range: 20‐60 mg/day. Temazepam or other short‐acting benzodiazepines were permitted as hypnotic. |
|
| Outcomes | HDRS‐21, Montgomery and Asberg Depression Rating Scale (MADRS), Hopkins Symptoms Check List (HSCL), Clinical Global Impression (CGI). Total dropout, dropout due to side effects, number of patients experiencing at least one side effect, side‐effect profile. |
|
| Notes | Funding: paroxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | For the evaluation of efficacy data, the extender Last Observation Carried Forward (LOCF) dataset was used. Study endpoint: 6/37 missing from paroxetine group; 9/41 missing from control group. |
| Selective reporting (reporting bias) | High risk | Total number of randomised patients not reported. Only reported Intention‐to‐treat (ITT) population. Data per week reported only when statistically significant. Side effects were reported only by body. |
| Other bias | Unclear risk | Sponsorhip bias cannot be ruled out. |
Demyttenaere 2002.
| Methods | Double‐blind, randomised, multicentre study. | |
| Participants | Patients with DSM‐IV diagnosis of major depressive disorder. | |
| Interventions | Paroxetine: 43 participants. Fluoxetine: 42 participants. Paroxetine dose: 20 mg/day. Fluoxetine dose: 20 mg/day. |
|
| Outcomes | Hamilton rating scale for depression (HDRS‐17). | |
| Notes | None. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No clear information on incomplete data management. Number and reasons for dropouts not clear. |
| Selective reporting (reporting bias) | Unclear risk | The type of adverse events was not reported. |
| Other bias | Unclear risk | Sponsorhip bias cannot be ruled out. |
Detke 2004 (HMAY A).
| Methods | Eight‐week, double‐blind, randomised, multicentre, placebo‐ and active comparator‐controlled study comparing paroxetine with placebo and duloxetine. The protocol consisted of two identical studies conducted in parallel. Investigators were divided into the two separate study groups prior to beginning the studies. | |
| Participants | Outpatients of at least 18 years of age with a primary diagnosis of major depression as defined by the DSM‐IV. Patients were required to have a Hamilton rating scale for depression (HDRS‐17) total score ≥15 and a CGI‐Severity total score ≥4 at both Visits 1 and 2. | |
| Interventions | Paroxetine: 86 participants. Duloxetine 80 mg: 95 participants. Duloxetine 120 mg: 93 participants. Placebo: 93 participants. Paroxetine dose range: 20 mg once daily. Duloxetine 80 mg dose range: 40 mg twice daily. Duloxetine 120 mg dose range: 60 mg twice daily. |
|
| Outcomes | HDRS‐17, Montgomery and Asberg Depression Rating Scale (MADRS), Clinical Global Impression (CGI), Visual Analogue Scale (VAS), Patient Global Impression (PGI), Self‐rating depression scale (SDS), Scale for Suicidal Ideation (SSI). Total dropout, dropout due to side effects, side‐effect profile. | |
| Notes | Funding: duloxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were randomly assigned in a 1:1:1:1 ratio to placebo, duloxetine 80 mg/day, duloxetine 120 mg/day or paroxetine 20 mg/day". |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Treatments were administered in a double‐blind fashion via the use of a double‐dummy study drug design. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "All analyses were conducted on an Intention‐to‐treat (ITT) basis. (...) The change from baseline to endpoint in the HDRS‐ 17 item total score and other continuous efficacy variables were also calculated using the Last Observation Carried Forward (LOCF). Study endpoint: 10/86 missing from Paroxetine group, 12/95 from Duloxetine 80 mg, 9/93 missing from Duloxetine 120 mg, 18/93 missing from placebo group. |
| Selective reporting (reporting bias) | High risk | In the duloxetine 120 mg/day arm only treatment emergent side effects experienced by > or = 5% of patients are reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Dichter 2005.
| Methods | Twelve‐week, randomised trial. | |
| Participants | Participants were adult outpatients who met DSM‐IV criteria for major depression as determined by the Structured Clinical Interview for DSM‐IV patient version. Participants: (1) had scores on the Hamilton rating scale for depression (HDRS‐17) that were greater than 17; (2) were free of benzodiazepines for at least 2 weeks prior to their baseline assessment, antidepressant medication for at least 3 weeks prior to their baseline assessment, and fluoxetine, antipsychotics, lithium, carbamazepine, or valproate for at least 5 weeks prior to their baseline assessment; and (3) did not have: (a) any clinically significant physical illness that would limit treatment with either study drug; (b) a history of bipolar affective disorder; (c) any history of a psychotic Axis I disorder, including major depression with psychotic features; (d) current predominant nonpsychotic Axis I disorder, antisocial, borderline, or schizotypal Axis II personality disorders; (e) subnormal intellectual potential; (f) a history of substance abuse in the past 6 months or substance dependence in the past 12 months; (g) a known hypersensitivity to either study drug; or (h) any history of a seizure disorder. |
|
| Interventions | 20 participants were randomly assigned to receive either venlafaxine XR (n = 10; age
range: 27.3–54.5 years, mean (S.D.)=42.6 (9.2), 6 women) or paroxetine (n = 10; age range: 22.2–50.2 years, mean (S.D.) = 37.6 (9.0), 7 women). Paroxetine dose range:10‐30 mg/day. Venlafaxine dose range: 75‐225 mg/day. |
|
| Outcomes | HDRS‐17, Clinical Global Impression (CGI), Hamilton rating scale for anxiety (HAM‐A), Beck Depression Inventory (BDI), Beck Anxiety Scale (BAI). | |
| Notes | None. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "participants were randomly assigned". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information on incomplete outcome data management. |
| Selective reporting (reporting bias) | Unclear risk | Outcomes data not clearly reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Dorman 1992.
| Methods | Six‐week double‐blind, randomised, study. | |
| Participants | Outpatients fulfilling DSM‐III criteria for major depression, with a score of at least 17 on the Hamilton rating scale for depression (HDRS‐17). Age: older than 65 years. | |
| Interventions | Paroxetine: 29 participants. Mianserin: 28 participants. Paroxetine dose range: 15‐30 mg/day. Mianserin dose range: 30‐60 mg/day. With the exception of short‐ acting benzodiazepines for hypnotic purposes, no other concomitant psychotropic medication was permitted during the study. |
|
| Outcomes | HDRS‐17 Total dropout, dropout due to side effects, number of patients experience at least once side effect, side‐effect profile. |
|
| Notes | This study has been conducted in order to evaluate the effects on sleeping of both paroxetine and mianserin in the treatment of depression. For this reason most efficacy and safety data were not available. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Double‐blind, double‐dummy". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "Double‐blind, double‐dummy". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Study endpoint: 5/29 missing from paroxetine group, 3/28 missing from control group. |
| Selective reporting (reporting bias) | High risk | Side effects reported only per body system. Mean difference reported on graphs only. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
DUAG 1990.
| Methods | Six‐week double‐blind, randomised, multicentre study. | |
| Participants | Inpatients fulfilling DSM III criteria for major depressive disorder with a minimum score of 18 on the Hamilton rating scale for depression (HDRS‐17) rating scale. Patients remained hospitalised throughout the trial period, except that patients who responded were permitted to be discharged after two weeks of active treatment but only if the trial program could be followed strictly. Age range: 19‐67 years. |
|
| Interventions | Paroxetine: 62 participants. Clomipramine: 58 participants. Paroxetine dose range: 30 mg/day. Clomipramine dose range: 150 mg/day. No other psychotropic medication was allowed except occasional use of oxazepam as a sedative/hypnotic. |
|
| Outcomes | HDRS, HDRS subscales, Bech‐Rafaelsen Melancholia Scale (BRMS), UKU side effect scale. Total dropout. Reasons for dropout. | |
| Notes | We considered dropout at 4 weeks instead of 6 because patients who were rated non‐responders after 4 weeks of active treatment "were terminated" in the study. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly assigned". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Patients who were rated non‐responders after 4 weeks of active treatment "were terminated" in the study. 27/62 completed 6 weeks of treatment in the paroxetine group (23 were considered non‐responders, 12 were dropout); 33/58 completed 6 weeks treatment in the clomipramine group (4 were considered non‐responders, 19 were dropout). |
| Selective reporting (reporting bias) | High risk | Number of patients experiencing each side effect not reported. Mean change on HAMD reported only on graph. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Dunbar 1991.
| Methods | Six‐week double‐blind, randomised, parallel group, single‐centre, placebo‐controlled. | |
| Participants | Outpatients meeting DSM‐III criteria for major depression with a minimum score of 18 on Hamilton rating scale for depression (HDRS‐17). Age range:18‐65 years. | |
| Interventions | Paroxetine: 41 participants. Imipramine: 42 participants. Placebo: 42 participants. Paroxetine dose range: 10‐50 mg/day. Imipramine dose range: 65‐275 mg/day. |
|
| Outcomes | HDRS‐21, Clinical Global Impression (CGI) Improvement, Severity, Montgomery and Asberg Depression Rating Scale (MADRS), Patient Global Experience (PGE). Total dropout, dropout due to side effect, dropout due to lack of efficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Funding: paroxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Study endpoint: 21/41 missing from paroxetine group, 25/42 missing from control group. |
| Selective reporting (reporting bias) | High risk | Dichotomous outcome not reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Dunner 1992.
| Methods | Six‐week, double‐blind, randomised, multicentre study. | |
| Participants | Outpatients fulfilling DSM‐III diagnostic criteria for major depression (moderate to severe). Age: older than 60 years. |
|
| Interventions | Paroxetine: 136 participants. Doxepin: 135 participants. Paroxetine dose‐range: 10‐40 mg/day (mean dose: 23.4 mg/day). Doxepin dose‐range: max 200 mg/day (mean dose: 105.2 mg/day). Occasional use of chloral hydrate 500 mg as a hypnotic, for a maximum of 4 consecutive nights during the first 2 weeks of the study. |
|
| Outcomes | Hamilton rating scale for depression (HDRS), Clinical Global Impression (CGI), Montgomery and Asberg Depression Rating Scale (MADRS), Hopkins Symptom Checklist (SCL). Total dropout, dropout due to inefficiency, dropout due to side effects. |
|
| Notes | Adverse events reported only on graphs. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Standard deviations for mean change not reported (borrowed from The Cochrane Library); side effects reported incompletely. Number of patients experiencing each side effect not reported. |
| Selective reporting (reporting bias) | High risk | Standard deviation for mean change not reported (borrowed from The Cochrane Library); side effects reported incompletely. Number of patients experiencing each side effect not reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Fabre 1992.
| Methods | Six‐week double‐blind, randomised, parallel group, single‐centre, placebo‐controlled study. | |
| Participants | Outpatients meeting DSM‐III criteria for major depression with a minimum score of 18 on Hamilton rating scale for depression (HDRS‐17). Age: older than 18 years. | |
| Interventions | Paroxetine: 40 participants. Imipramine: 40 participants. Placebo: 40 participants. Paroxetine dose range: 10‐50 mg/day. Imipramine dose range: 65‐275 mg/day. Chloral hydrate (500 mg) was allowed for a maximum of 4 consecutive nights during the first two weeks of the study. |
|
| Outcomes | HDRS‐21, Clinical Global Impression (CGI) Improvement, Severity, Montgomery and Asberg Depression Rating Scale (MADRS), Patient Global Experience (PGE). Total dropout, dropout due to side effect, dropout due to lack of efficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Funding: paroxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Study endpoint: 24/40 missing from paroxetine group, 24/40 missing from control group. |
| Selective reporting (reporting bias) | High risk | Dichotomous outcome not reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Fava 1998.
| Methods | Twelve‐week randomised, double‐blind, multicentre study. | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for moderate to moderately severe major depression without a history of mania or hypomania, with a score of at least 18 on the Hamilton Rating Scale for Depression‐21 item (HDRS‐21), of at least 8 on the Raskin Depression Scale (and grater than Covi Anxiety Scale [CAS] score).Mean age: 41.3 years.Exclusion criteria: schizophrenia, adjustment disorder, bipolar disorder, panic disorder, social phobia, obsessive compulsive disorder, psychotic depression, atypical depression, serious concomitant medical illness, significant abnormal laboratory values, history of seizure disorder, high suicidal risk, recent history of alcohol or drug abuse, use other psychotropic drug within 14 days of baseline, ECT within 3 months of baseline, any investigational drug within 30 days of baseline, previous treatment with paroxetine, pregnancy, childbearing potential without contraceptive. | |
| Interventions | Paroxetine: 55 participants. Fluoxetine: 54 participants. Placebo: 19 participants. Paroxetine dose range: 20‐50 mg/day. Fluoxetine dose range: 20‐80 mg/day. |
|
| Outcomes | HDRS‐21, Covy Anxiety Scale (CAS), Raskin Depression Scale. | |
| Notes | Response: decrease of at least 50% in the HDRS‐21 total. score. Funding: by industry. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind, no further information. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double‐blind, no further information. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "we chose to conduct all analyses with an ITT approach". Number and reasons of dropout reported. |
| Selective reporting (reporting bias) | Unclear risk | Adverse events reported. Primary and secondary endpoint reported with standard deviations. |
| Other bias | Unclear risk | Sponsorship bias can not be ruled out. |
Fava 2002.
| Methods | Ten‐week randomised, double‐blind, multicentre study. | |
| Participants | Outpatients fulfilling DSM‐IV criteria for major depression or atypical major depression, with a baseline score of at least 16 on the first 17 items of the Hamilton rating scale for depression (HDRS‐28). No patients with bipolar disorder. | |
| Interventions | Paroxetine: 96 participants. Fluoxetine: 92 participants. Sertraline: 96 participants. Paroxetine dose range: 20‐60 mg/day. Fluoxetine dose range: 20‐60 mg/day. Sertraline dose range: 50‐200 mg/day. |
|
| Outcomes | HDRS‐17 , Clinical Global Impression (CGI) Severity and HDRSsleep disturbance, A/S, R, Cognitive Disturbance (COG) factors. Total dropout, dropout due to side effects, side‐effect profile. |
|
| Notes | Funding: fluoxetine manufacturer. Adverse events reported only if occurring in >=10%. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Study endpoint: 27/96 missing from paroxetine group; 24/92 missing from fluoxetine one, 26/96 from sertraline one. All enrolled patients who had at least one post‐baseline visit at which appropriate assessment was taken were included in the analyses of efficacy. All randomly assigned patients were included in the analyses of safety. |
| Selective reporting (reporting bias) | High risk | Adverse events reported only if occurring in >=10%. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Feighner 1989.
| Methods | Six‐week double‐blind, randomised, parallel group, single‐centre, placebo‐controlled study. | |
| Participants | Outpatients meeting DSM‐III criteria for major depression with a minimum score of 18 on Hamilton rating scale for depression (HDRS‐17). Age: older than 18 years. | |
| Interventions | Paroxetine: 40 participants. Imipramine: 40 participants. Placebo: 40 participants. Paroxetine dose range: 10‐50 mg/day. Imipramine dose range: 65‐275 mg/day. |
|
| Outcomes | HDRS‐21, Clinical Global Impression (CGI) Improvement, Severity, Montgomery and Asberg Depression Rating Scale (MADRS), Patient Global Experience (PGE). Total dropout, dropout due to side effect, dropout due to lack of efficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Funding: paroxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Study endpoint: 19/40 missing from paroxetine group, 26/40 missing from control group. |
| Selective reporting (reporting bias) | High risk | Dichotomous outcome not reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Freed 1996.
| Methods | Nine‐week, randomised, double‐blind study. | |
| Participants | Inclusion criteria: patients with Montgomery and Asberg Depression Rating Scale (MADRS) 20+. Age range: 19‐85 years. Country: Australia. Setting: family practice. | |
| Interventions | Paroxetine versus amitriptyline | |
| Outcomes | Montgomery and Asberg Depression Rating Scale (MADRS), Clinical Global Impression (CGI), dropouts. | |
| Notes | None. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were randomly assigned (...) using a computer generated randomization list". |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "double blind (...) medication was blinded by over‐encapsulation of marketed forms of paroxetine and amitriptyline in identical capsules. To maintain the blinding, placebo was given at night to patients receiving paroxetine and in the morning to patients receiving amitriptyline". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "all patients randomized to active treatment and fro whom one valid post‐randomization evaluation was available within 4 days of intake of medication, were included in the Intention‐to‐treat (ITT) analysis. Three hundred and six patients were eligible for the ITT analysis and all efficacy variables were analysed using this population. All patients (381) were included in the safety analysis; however, six patients were not evaluable by treatment due to inconsistent treatment allocation". |
| Selective reporting (reporting bias) | Low risk | Outcomes data were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Gagiano 1993.
| Methods | Six‐week randomised, double‐blind study. | |
| Participants | Outpatients fulfilling DSM‐III‐R criteria for major depressive episode, with a score of at least 18 on the Hamilton rating scale for depression (HDRS‐21). Age range: 18‐65 years. | |
| Interventions | Paroxetine: 45 participants. Fluoxetine: 45 participants. Paroxetine dose range: 20‐40 mg/day. Fluoxetine dose range: 20‐60 mg/day. Short acting benzodiazepines were permitted for use as hypnotics during the study period. |
|
| Outcomes | HDRS, Montgomery and Asberg Depression Rating Scale (MADRS), Hamilton rating scale for anxiety (HAM‐A), Clinical Global Impression (CGI). Total dropout, dropout due to inefficiency, dropout due to side effects, side‐effect profile. |
|
| Notes | Funding: paroxetine manufacturer. Adverse events reported only if occurring in >=10%. Unpublished data were retrieved. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "The double dummy technique using matching placebos was employed to maintain the double blind". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Study endpoint: 8/45 missing from paroxetine group; 10/45 missing from control group. All data from the Intention‐to‐treat (ITT) population were included in the analyses up to the time of withdrawal; thereafter the last observed value of a variable was carried forward for all subsequent missing data points. |
| Selective reporting (reporting bias) | Low risk | The study protocol was not available but the unpublished reports retrieved include all expected outcomes. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Gallen 2001.
| Methods | Multicentre, randomised, double‐blind, placebo‐ and active‐controlled, parallel‐group study. | |
| Participants | Inclusion Criteria: To be included in the study, patients must have met all of the following criteria: diagnosis of Major Depressive Disorder (MDD) without psychotic features, as defined by DSM‐IV; male or female, of any race, between the ages of 18 and 65 years. If female, must have been postmenopausal or must have met all of the following criteria: agreed to avoid pregnancy during the study; had a negative serum pregnancy test at screen; used an accepted means of birth control (as determined by the investigator), such as abstinence, oral contraceptive, implantable or injectable contraceptive, intrauterine device, or barrier method, or have been surgically sterilized; total score of ≥20 on the Hamilton rating scale for depression (HDRS‐17), which was administered via the IVRS prior to the screening visit; general good health, as confirmed by routine clinical laboratory safety findings. Voluntary consent to participate in the study, documented in a written Patient Informed Consent Form that was signed prior to the start of any study procedures at the screening visit. Exclusion Criteria: Patients were excluded from the study for any of the following reasons: DSM‐IV diagnosis of the following concomitant psychiatric disorders: MDD with psychotic features, cyclothymic disorder, bipolar I or bipolar II disorders, substance‐related disorders (within the preceding 12 months), schizophrenia, or other psychotic disorders; resistance to antidepressive treatment, defined as a lack of response to at least 2 previous courses of antidepressant medications administered at full doses for more than 1 month; participation in a previous clinical trial of reboxetine or lack of response to previous treatment with paroxetine, administered at a dose of ≥20 mg/day for more than 1 month; use of antidepressant medication for the treatment of depression in the 2 months preceding the start of the study; history of MDD associated with endocrine disorders: hypo‐ or hyper‐thyroidism tested by thyroid‐stimulating hormone and thyroxine, adrenal insufficiency, or Cushing’s syndrome; positive pregnancy test for females of childbearing potential; breast‐feeding by female patients; refusal by female patients of childbearing age to use an effective contraceptive method during the study; participation in any clinical study with an investigational compound in the 4 weeks preceding the study; history or presence of gastrointestinal, liver, or kidney disease or other conditions known to interfere with the absorption, distribution, metabolism, and excretion of drugs; history of seizures or brain injury; current evidence of clinically important hematopoietic, respiratory, or cardiovascular diseases; current evidence of urinary retention or glaucoma. Clinically significant illness in the 4 weeks preceding the study that might have interfered with the conduct of the trial. Clinically relevant abnormal findings in the physical examination, laboratory tests, or ECG at admission. Positive urine drug screen for amphetamines, barbiturates, marijuana metabolites, cocaine metabolites, methadone, methaqualone, opiates, phencyclidine, or propoxyphene. A positive urine drug screen for benzodiazepines did not exclude the patient. Treatment with electroconvulsive therapy in the 6 months preceding the study. Major risk of suicide as assessed by the investigator, a score of ≥3 on Item 3 of the HDRS at screen or baseline, or a history of suicide attempt during the current depressive episode. History of hypersensitivity to reboxetine or paroxetine. Use of the following medications, which are potent inhibitors of the drug‐metabolizing enzyme cytochrome p450‐3A4: azole antifungals, macrolide antibiotics, or fluvoxamine. Use of the following medications, which are known to be substrates or inhibitors of the drug‐metabolizing enzyme cytochrome p450‐2D6: Type 1C antiarrhythmics, quinidine, or cimetidine. Use of oral anticoagulants that are known to inhibit vitamin K‐dependent coagulation factors. Use of concomitant psychotropic medications other than the protocol‐specified sedatives/hypnotics, which could be taken on an as‐needed basis for sleep. Inability of the patient to comply with the conditions of the study, based on the investigator’s assessment. |
|
| Interventions | Paroxetine: 265 participants.
Reboxetine: 265 participants. Placebo: 257 participants. |
|
| Outcomes | Montgomery Asberg Depression Rating Scale (MADRS) total score at day 56 in the intent‐to‐treat (ITT) patient population.Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Funding: reboxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Pharmacia & Upjohn (P&U) prepared a randomization list for assignment of the patients to 1 of the 3 treatment groups. Study medication for each treatment group was prepared on this basis by P&U and was labeled with the corresponding patient number". |
| Allocation concealment (selection bias) | Low risk | Quote: "at the baseline visit, the investigator assigned each patient to a treatment group based on the patient's temporal entry into the study (i.e. by assigning the lowest patient number available). A list of patient numbers and medication assignments was provided only after the data for the study had been analyzed". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "double blind (...) study medications were provided as identically appearing capsules (...) Study medications for the randomized treatments consisted of identically appearing capsules that contained , paroxetine or placebo. The capsules were provided in clinical supply packages that were labeled with the protocol number, patient number, treatment period, dosing, directions and storage conditions. Investigators were given sealed drug‐disclosure sheets that contained information about each patient's treatment". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "the Intention‐to‐treat (ITT) population includes all patients who were randomized into the trial and who received at least one dose of study medication. All analyses were based on the ITT population. Efficacy analyses were based on the population of ITT patients who had at least one post‐baseline evaluation for the specified efficacy measure". |
| Selective reporting (reporting bias) | Low risk | Outcomes data were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Geretsegger 1994.
| Methods | Six‐week, double‐blind, randomised, multicentre study. | |
| Participants | Patients fulfilling DSM‐III criteria for major depression, with a score of at least 18 on the Hamilton rating scale for depression (HDRS‐21). Age range: 61‐85 years. | |
| Interventions | Paroxetine: 54 participants. Fluoxetine: 52 participants. Paroxetine dose range: 20‐40 mg/day. Fluoxetine dose range: 20‐60 mg/day. |
|
| Outcomes | HDRS‐21 item, Clinical Global Impression (CGI), Montgomery and Asberg Depression Rating Scale (MADRS). Total dropout, dropout due to side effects, number of patients experiencing at least one side effect, side‐effect profile. |
|
| Notes | Funding: paroxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Less than 20% of patients abandoned the study prematurely. Patients with at least one valid evaluation during treatment were included in the Intention‐to‐treat (ITT) analysis of efficacy (visit‐wise data). All randomised patients who received active treatment were included in ITT analysis of clinical tolerability and safety. |
| Selective reporting (reporting bias) | Low risk | The study protocol was not available but the unpublished reports we retrieved include all expected outcomes. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Geretsegger 1995.
| Methods | Six‐week, double‐blind, randomised, multicentre study. | |
| Participants | Inpatients for the first 3 weeks fulfilling DSM‐III criteria for major depression, with a score of at least 18 on the Hamilton rating scale for depression (HDRS‐17). Age: older than 65 years. | |
| Interventions | Paroxetine: 78 participants. Amitriptyline: 75 participants. Paroxetine dose range: 20‐30 mg/day. Amitriptyline dose range: 100‐150 mg/day. No other psychotropic medication was allowed except temazepam or oxazepam or chloral hydrate as hypnotics. |
|
| Outcomes | HDRS, Clinical Global Impression (CGI), Montgomery and Asberg Depression Rating Scale (MADRS). Total dropout, dropout due to side effects, number of patients experiencing at least one side effect, side‐effect profile. |
|
| Notes | Funding: paroxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly allocated", no further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double‐blind, double dummy technique". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double‐blind, double dummy technique". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | More than 20% of patients abandoned the study prematurely. Study endpoint: 32/78 missing from paroxetine group; 30/75 missing from control group. |
| Selective reporting (reporting bias) | Unclear risk | Mean change on HDRS and MADRS reported only on graph. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Gilmor 2002.
| Methods | Seven‐week randomised, open‐label, parallel‐group, forced‐titration multicentre study. | |
| Participants | Outpatients fulfilling DSM‐IV criteria for major depressive disorder, with a score of at least > or = 20 on the Montgomery and Asberg Depression Rating Scale (MADRS). Age range: 18‐57 years. | |
| Interventions | Paroxetine: 38 participants. Desipramine: 14 participants. Paroxetine dose range: 10‐60 mg/day. Desipramine dose range: 50‐300 mg/day. |
|
| Outcomes | MADRS, Clinical Global Impression (CGI). Total dropout. |
|
| Notes | Funding: paroxetine manufacturer. This study was conducted in order to evaluate inhibition of norepinephrine uptake in patient with major depression, so it is not a study of efficacy or tolerability. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly assigned to paroxetine or desipramine in a 3‐to‐1 ratio respectively". |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "open‐label". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "open‐label". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Dichotomous outcome not reported. Adverse events were not reported. Incomplete reporting of dropout due to side effects, dropout due to inefficacy. |
| Selective reporting (reporting bias) | High risk | Dichotomous outcome not reported. Adverse events were not reported. Incomplete reporting of dropout due to side effects, dropout due to inefficacy. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Goldstein 2004 (HMAT B).
| Methods | Multicentre, parallel, double‐blind, randomised, placebo‐ and active comparator‐controlled study with blinded placebo lead‐in and placebo lead‐out. The protocol consisted of two identical studies conducted in parallel and reported separately (Study Group A and Study Group B). The study consisted of two study periods. Study Period I was the 1‐week screening phase of the study, and Study Period II was an 11‐week acute therapy phase in which patients were assessed weekly from Visit 2 (Week 0) to Visit 5 (Week 3) and every other week from Visit 5 (Week 3) to Visit 9 (Week 11). This study design employed double‐blind, variable‐duration placebo lead‐in and lead‐out periods to blind patients and investigators at the start and end of active therapy. | |
| Participants | Planned: 356 patients (89 per treatment group) Randomized: 87 Paroxetine 20 mg once daily; 86 Duloxetine 20 mg BID (twice daily); 91 Duloxetine 40 mg BID ; 89 Placebo. Completed:49 Paroxetine 20 mg once daily; 55 Duloxetine 20 mg BID; 53 Duloxetine 40 mg BID; 52 Placebo. Criteria for Inclusion: Male and female outpatients of at least 18 years of age with a primary diagnosis of MDD as defined by the DSM‐IV, and confirmed by use of the Mini International Neuropsychiatric Interview. Patients were required to have a HAMD17 total score ≥15 and a CGI‐Severity total score ≥4 at both Visit 1 and Visit 2. |
|
| Interventions | Paroxetine 20 mg once daily (20 mg/day) Duloxetine 20 mg twice daily (40 mg/day) Duloxetine 40 mg twice daily (80 mg/day) Placebo |
|
| Outcomes | Efficacy: the primary efficacy measure was the Hamilton rating scale for depression (HDRS‐17) total score. Secondary efficacy measures included HDRS‐17 response rates (50% reduction from baseline to endpoint), HDRS‐17 remission rates (endpoint score ≤7), time to sustained response, and time to sustained remission. Other secondary measures included the HDRS‐17 subfactors and individual items, Montgomery and Asberg Depression Rating Scale (MADRS), Clinical Global Impression (CGI) Severity, Patient Global Impression (PGI) Improvement, Hamilton rating scale for anxiety (HAM‐A), Somatic Symptom Inventory 26‐ and 28‐item scale (SSI), and Visual Analog Scales for pain (VAS). Safety: safety was evaluated through the collection and reporting of discontinuation rates, treatment‐emergent adverse events, discontinuation‐emergent adverse events, laboratory analyses, vital signs, ECGs, and the Arizona Sexual Experiences (ASEX). Health Outcomes: Health outcomes were evaluated using the Quality of Life in Depression (QLDS) scale and Health Resource Utilization scales (HRUS). |
|
| Notes | Funding: duloxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were allocated within each study site by a computer‐generated random table and had an equal probability of receiving placebo, duloxetine 40 mg/day, administered as 20 mg twice daily, duloxetine 80 mg/day, administered as 40 mg twice daily, or paroxetine 20 mg/day administered once daily during the 8 week acute therapy phase. No stratification was employed". |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Number of patients experiencing each side effect not reported. Study endpoint: 38/87 missing from paroxetine group; 69/177 missing from control group. |
| Selective reporting (reporting bias) | High risk | Quote: "Treatment emergent adverse events experience by at least 5% of patients treated with duloxetine 80 mg/day and statistically significantly different from placebo are reported". |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Guillibert 1989.
| Methods | Six‐week double‐blind, randomised, multicentre study. | |
| Participants | Outpatients fulfilling DSM‐III diagnostic criteria for major depressive disorder with a minimum score at baseline of 20 on the Hamilton rating scale for depression (HDRS‐21) rating scale. Age: over 60 years. |
|
| Interventions | Paroxetine: 40 randomised. Clomipramine: 39 randomised. Paroxetine dose range: 20‐30 mg/day. Clomipramine dose range: 25‐75 mg/day. No other psychotropic medication was permitted except short half‐life benzodiazepine of the oxazepam type for the control of agitation, anxiety or insomnia. |
|
| Outcomes | HDRS, Wang Anxiety and Widlocher scales (WAW), Clinical global Impression (CGI). Total dropout, dropout due to inefficiency, dropout due to side effects, side‐effect profile. |
|
| Notes | None. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Study endpoint: 9/40 missing from Paroxetine group, 12/39 missing from Clomipramine one. All 79 patients were evaluable for tolerability and for an ITT evaluation with the respect of the HAMD scale. |
| Selective reporting (reporting bias) | High risk | SD for mean change not reported (borrowed from The Cochrane Library); side effects reported incompletely. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Hicks 2002.
| Methods | Eight‐week, double‐blind, randomised study. | |
| Participants | Outpatients fulfilling DSM‐IV diagnostic criteria for major depression with a minimum score of 18 on the Hamilton rating scale for depression (HDRS). Age range: 18‐65 years. |
|
| Interventions | Paroxetine: 20 randomised. Nefazodone: 20 randomised. Paroxetine dose range: 20‐40 mg/day (mean daily dose: 29.5 [SD 8.9]). Nefazodone dose range: 400‐600 mg/day (mean daily dose: 495 [SD 82.6]). No addition psychoactive medication was allowed during the study period. |
|
| Outcomes | HDRS, Clinical Global Impression (CGI), Montgomery and Asberg Depression Rating Scale (MADRS), sleeping analysis, dropout, side‐effect profile. | |
| Notes | Funding: nefazodone manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Study endpoint: 3/20 missing from paroxetine group, 5/20 missing from nefazodone group. |
| Selective reporting (reporting bias) | High risk | Three rating scales reported in the methods but just one reported in the results; side effect reported only if occurring in more than 5% of participants. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Higuchi 2009.
| Methods | Multicentre, parallel, double‐blind, randomised, placebo‐ and active comparator‐controlled study comparing duloxetine with placebo and paroxetine in the acute treatment of patients with DSM‐IV MDD. | |
| Participants | Male and female patients with a primary diagnosis of MDD as defined by the DSM‐IV, and confirmed by use of the MINI. Patients were required to have a HAM‐D17 total score ≥ 15 and a CGI‐S total score ≥ 4 at both Visit 1 and Visit 2. Country: Japan Setting: inpatients and outpatients Age: 18 to 65 years inclusive |
|
| Interventions | Paroxetine 20‐40 mg/day, once in the evening. Duloxetine 40 mg/day, once in the morning. Duloxetine 60 mg/day, once in the morning. Placebo. Duration: 6 weeks active treatment with 1‐week placebo lead‐in and 2‐week lead‐out periods Cointervention: No psychotropic drug except for one short or ultra‐short acting benzodiazepine sleeping pill. |
|
| Outcomes | Efficacy: Primary efficacy measure was the HAM‐D17 total score. Secondary efficacy measures included HAM‐D17 response rates (a 50% reduction from baseline to endpoint) and HDRS 17 remission rates (endpoint score <7), VAS for pain, CGI‐I. | |
| Notes | Randomised: 495 patients, however the first 50 patients were randomised according to a wrong randomisation scheme and were therefore omitted from the main analyses. The correct ITT sample would then be: 75 for duloxetine 40 mg, 76 for duloxetine 60 mg, 148 for paroxetine and 146 for placebo. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Probably done. Quote: "The sequence was generated by an independent investigator." |
| Allocation concealment (selection bias) | Low risk | Probably done. Quote: "The allocation was handled by an independent investigator." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Both the clinician and the patient were blinded. The capsules have been tested to be indistinguishable. The clinician was the assessor. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Both the clinician and the patient were blinded. The capsules have been tested to be indistinguishable. The clinician was the assessor. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Of the 445 patients appropriately randomised, 5 patients did not contribute at all to the analysis and about 10% of the patients had to have their outcomes LOCFed (the exact numbers cannot be known due to the way the authors summarised the data.) |
| Selective reporting (reporting bias) | Unclear risk | The protocol has not been published and we do not know if all the intended outcomes are reported. |
| Other bias | Unclear risk | Five‐armed placebo‐ and active drug‐controlled trial appears more trustworthy than straightforward 2‐arm trial. |
HMAT A (ID#4091).
| Methods | Multicentre, parallel, double‐blind, randomised, placebo‐ and active comparator‐controlled study. | |
| Participants | Planned: 356 patients (89 per treatment arm). Randomized: Duloxetine 20 mg BID 91; Duloxetine 40 mg BID 84; Placebo 90; Paroxetine 89. Completed: Duloxetine 20 mg BID 61; Duloxetine 40 mg BID 56; Placebo 61; Paroxetine 58. Criteria for Inclusion: Male and female outpatients of at least 18 years of age with a primary diagnosis of MDD as defined by the DSM‐IV, and confirmed by use of the Mini International Neuropsychiatric Interview. Patients were required to have a HAMD17 total score of ≥15 and a CGI‐Severity total score of ≥4 at both Visit 1 and Visit 2. |
|
| Interventions | Duloxetine 20 mg BID (40 mg/day). Duloxetine 40 mg BID (80 mg/day). Paroxetine 20 mg once daily (20 mg/day). Placebo. |
|
| Outcomes | Efficacy: the primary efficacy measure was the Hamilton rating scale for depression (HDRS‐17) total score. Secondary efficacy measures included HAMD17 response rates (a 50% reduction from baseline to endpoint), HDRS‐17 remission rates (endpoint score of ≤7), time to sustained response, and time to sustained remission. Other secondary measures included the HDRS‐17 subfactors and individual items, Montgomery and Asberg Depression Rating Scale (MADRS), Clinical Global Impression (CGI) Severity, Patient Global Impression (PGI) Improvement, Hamilton rating scale for anxiety (HAM‐A), Somatic Symptom Inventory (SSI), and Visual Analog Scales for pain (VAS). Safety: safety was evaluated through the collection and reporting of discontinuation rates, Treatment Emergent Adverse Effects, discontinuation‐emergent adverse events, laboratory analyses, vital signs, ECGs, and the Arizona Sexual Experiences (ASEX). Health Outcomes: Health outcomes were evaluated using the Quality of Life in Depression scale and Health Resource Utilization scales. | |
| Notes | Date first patient enrolled: 10 March 2000.
Date last patient completed: 10 April 2001. For all total scores calculated from individual items, if any of the individual items was missing, the corresponding total score was considered missing. Sites with fewer than 8 randomly assigned patients with baseline and at least one post baseline (Visit 4 to Visit 8) HAMD17 total score were pooled. If this resulted in a pooled site with fewer than 8 patients, these patients were pooled with the next smallest site. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "an Intention‐to‐treat (ITT) principle was applied in all efficacy and safety analyses". |
| Selective reporting (reporting bias) | High risk | Only adverse effects with total incidence > or = 2% were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Hutchinson 1992.
| Methods | Six‐week, double‐blind, randomised study. | |
| Participants | General practice patients fulfilling DSM‐III diagnostic criteria for major depression with a minimum score of 18 on the Hamilton rating scale for depression (HDRS). Age:older than 65 years. |
|
| Interventions | Paroxetine: 58 randomised. Amitriptyline: 32 randomised. Paroxetine dose range: 20‐30 mg/day. Amitriptyline dose range: 50‐100 mg/day. No other psychotropic medication was allowed during the study period except temazepam as hypnotic. |
|
| Outcomes | HDRS, Clinical Global Impression (CGI), Leeds Sleep Evaluation Questionnaire (LSEQ) Total dropout, dropout due to inefficiency, dropout due to side effects, number of patients experiencing at least one side effect, side‐effect profile. |
|
| Notes | Funding: paroxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "a biased randomization of treatment was adopted to ensure that two‐thirds of the patients received paroxetine and one‐third amitriptyline". |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind, double dummy". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind, double dummy". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Study endpoint: 12/58 missing from paroxetine group, 11/32 missing from Amitriptyline one. |
| Selective reporting (reporting bias) | High risk | Only "the most commonly" adverse events were reported. Mean change on HDRS reported only on graph. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Hwang 2004.
| Methods | Four‐week, open‐label, randomised study. | |
| Participants | Inpatients fulfilling DSM‐IV diagnostic criteria for major depression. Age:older than 65 years. |
|
| Interventions | Paroxetine: 53 randomised. Venlafaxine: 52 randomised. Paroxetine dose range: 20‐40 mg/day (mean daily dose: 37.4 mg/day [SD 20.9]). Venlfaxine dose range: 75‐150 mg/day (mean daily dose: 131.6 mg/day [SD 24.4]). Benzodiazepines were given to patients with anxiety or insomnia. |
|
| Outcomes | Hamilton rating scale for depression (HDRS), Clinical Global Impression (CGI). Total dropouts. | |
| Notes | None. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "open label trial". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "open label trial". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Study endpoint: 3/53 missing from paroxetine group; 3/52 missing from venlafaxine one. |
| Selective reporting (reporting bias) | High risk | Incomplete reporting of dropout due to side effects, dropout due to inefficiency. Incomplete reporting of number of patients experiencing at least one side effect. |
| Other bias | Unclear risk | No sufficient information in order to establish if other bias are present. |
Javors 2000.
| Methods | Six‐week, randomised study. | |
| Participants | Inpatients fulfilling DSM‐IV diagnostic criteria for major depression. Age range: unclear. |
|
| Interventions | Paroxetine: 3 participants. Desipramine: 5 participants.. Paroxetine dose range: 30‐60 mg/day. Desipramine dose range: 50‐250 mg/day. |
|
| Outcomes | Hamilton rating scale for depression (HDRS); platelet serotonin content. | |
| Notes | The study was primarily designed to assess the reduction of platelet serotonin content in depressed patients treated with paroxetine or desipramine. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly assigned". |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No clear information on incomplete outcome data management. |
| Selective reporting (reporting bias) | High risk | Adverse events, dropouts and side effects were not reported. As dichotomous outcome, only endpoint response rate are reported. (The study was primarily designed to assess the reduction of platelet serotonin content in depressed patients treated with paroxetine or desipramine). |
| Other bias | Low risk | This study was independent from pharmaceutical industry. |
Jefferson 2001 29060/785.
| Methods | Six‐week, double‐blind, placebo‐controlled, multicentre, parallel‐group, randomised study. | |
| Participants | Patients with major depressive disorder (DSM‐IV criteria), with a Montgomery and Asberg Depression Rating Scale (MADRS) score of at least 17 (both at the screening and baseline visits). Exclusion criteria: patient who have taken other psychotropic drugs, had a history of schizophrenia or schizoaffective disorder, had current (or within 6 months prior to screening) Axis I anxiety disorder or Axis I affective disorder other than major depressive disorder. Patient who, in the investigator's judgement, posed a current homicidal or suicidal risk. Woman who had a positive pregnancy test or who were lactating, women of child‐bearing potential who were not practicing a clinically accepted method of contraception. Subject with a serious medical disorder or condition that, in the investigator's opinion, precluded the administration of paroxetine controlled release (CR) or citalopram. Patient undergoing any form of psychotherapy. Age range: 18‐65 years. |
|
| Interventions | Citalopram 20 mg/day: 107 participants. Citalopram 40 mg/day: 100 participants. Paroxetine CR 12.5 mg/day: 96 participants. Paroxetine CR 25 mg/day: 103 participants. Placebo: 105 participants. |
|
| Outcomes | MADRS, Clinical Global Impression (CGI), Hamilton rating scale for anxiety (HAM‐A), Hospital Anxiety and Depression (HAD), Self‐rating depression scale (SDS). Total dropout, dropout due to inefficacy, dropout due to side effects, number of patients experiencing at least one side effect, side‐effect profile. |
|
| Notes | Funding: paroxetine manufacturer. One suicide in placebo group. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "subject were randomized (1:1:1:1:1) to either paroxetine CR 12.5 mg, paroxetine CR 25 mg, citalopram 20 mg, citalopram 40 mg, or placebo". |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "paroxetine CR and citalopram were provided as over‐encapsulated tablets (...) placebo capsules were identical in appearance to the active study medication capsules". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Study endpoint: "23/103 missing from paroxetine 25 mg group, 18/96 from paroxetine 12.5 one, 17/107 from citalopram 20 mg one, 26/100 from citalopram 40 mg one. Quote: "all subjects who were randomized to double‐blind medication and had at least one valid post baseline efficacy assessment comprised the Intention‐to‐treat (ITT) efficacy population. The Last Observation Carried Forward (LOCF) data at week 6 were the primary dataset of interest". |
| Selective reporting (reporting bias) | High risk | Only most frequent adverse events reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Kasper 2005.
| Methods | Six‐week, double‐blind,multicentre, parallel‐group, randomised study. | |
| Participants | Outpatients fulfilling DSM‐IV diagnostic criteria for major depression with a minimum score of 18 on the Hamilton rating scale for depression (HDRS‐17). Age range: 18‐65 years. |
|
| Interventions | Paroxetine: 53 randomised Trazodone: 55 randomised Paroxetine dose range: 20‐40 mg/day. Trazodone dose range: 300‐450 mg/day. No other psychotropic medication was allowed during the study period, with the exception of patients stabilised on BDZ. |
|
| Outcomes | HDRS, Clinical Global Impression (CGI), Montgomery and Asberg Depression Rating Scale (MADRS). Total dropout, dropout due to inefficiency, dropout due to side effects, number of patients experiencing at least one side effect and side‐effect profile. | |
| Notes | Funding: paroxetine and trazodone manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly assigned". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Double‐blind, double‐dummy". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "Double‐blind, double‐dummy". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | More than 20% of patients abandoned the study prematurely. Study endpoint: 14/53 missing from paroxetine group; 19/55 missing from trazodone one. |
| Selective reporting (reporting bias) | Low risk | CGI‐Improvement and CGI‐Severity reported only on graphs. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Kato 2005.
| Methods | Open‐label study with two parallel groups of patients randomly assigned to either paroxetine or fluvoxamine. | |
| Participants | Eighty‐one patients meeting DSM‐IV diagnosis of major depressive disorder (excluding bipolar disorder) were included in the study. Exclusion criteria were additional diagnoses on Axis 1 and Axis 2, pregnancy and major medical and neurological disorders. |
|
| Interventions | Fluvoxamine: 39 participants.
Paroxetine: 42 participants.
Fluvoxamine dose range: 50‐150 mg/day.
Paroxetine dose range: 20‐40 mg/day. Patients who had been receiving benzodiazepines for at least 10 days before entering the study were permitted to continue these agents, providing that the dose remained unchanged throughout the study period. A low‐dose sleep‐inducing hypnotic agent, either brotizolam or triazolam, was permitted for severe insomnia as an additional medication. |
|
| Outcomes | Hamilton rating scale for depression (HDRS). Total dropout, dropout due to side effects. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | In this study "lack of response" was defined as a 40% of less reduction of the HDRS. This study was partly sponsored by Glaxo Smith Kline and Meiji Seika Kaisha Ltd. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients randomly assigned". |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "open label". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "open label". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | No information regarding incomplete outcome data management. This study was primarily designed to assess the influence of the serotonin type 2A, 3A and 3B receptor genes in addition to a polymorphism in the promoter region of the serotonin transporter. |
| Selective reporting (reporting bias) | Unclear risk | Outcomes data not clearly reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Katz 2004.
| Methods | Six‐week double‐blind, parallel‐group, randomised study. | |
| Participants | Inpatients fulfilling DSM‐III‐R diagnostic criteria for major depression with a minimum score of 18 on the Hamilton rating scale for depression (HDRS). Age range: 20‐69 years. |
|
| Interventions | Paroxetine: 28 participants. Desipramine: 29 participants. Placebo: 25 participants. Paroxetine dose range: 20‐60 mg/day. Desipramine dose range: 50‐350 mg/day. |
|
| Outcomes | HDRS, Clinical Global Impression (CGI), Global Assessment Scale (GAS), Schedule for Affective Disorders and Schizophrenia (SADS), VIBES, Symptom checklist (SCL‐90), National Institute of Mental Health Mood Scale (NIMH) . | |
| Notes | None. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly assigned", no further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information provided about number of patients missing from paroxetine group and desipramine group at study endpoint. |
| Selective reporting (reporting bias) | High risk | Adverse events, dropouts and side effects were not reported. As dichotomous outcome, only endpoint response rate were reported. |
| Other bias | Unclear risk | No sufficient information in order to establish if other biases are present. |
Kennedy 2004.
| Methods | Eight‐week, double‐blind, double‐dummy, parallel‐group, randomised study | |
| Participants | Males and non‐pregnant females using adequate contraception were eligible if they were between 18 and 65. Participants had to have a diagnosis of moderate to severe major depressive disorder without psychotic features. Participants were excluded if they had a predisposition to seizures, serious suicidal risk, poor previous therapeutic response to antidepressant medication, significant DSM‐IV Axis II diagnosis, history of or current diagnosis of anorexia nervosa or bulimia, recent history of drug dependence or abuse, including alcohol. Age range: 18‐65 years. |
|
| Interventions | Paroxetine: 71 randomised. Bupropion: 69 randomised. Paroxetine dose: 20‐40 mg/day. Bupropion dose: 150‐300 mg/day. |
|
| Outcomes | Hamilton rating scale for depression (HDRS‐17), Clinical Global Impression (CGI), Hamilton rating scale for anxiety (HAM‐A), Quality of Life in Depression (QLDS). Total dropout, dropout due to side effects. Number of patients experiencing at least one side effect, side ‐ profile. | |
| Notes | Poor therapeutic response is defined as at least two failed responses to antidepressant medications to from two different antidepressant classes, at adequate dose and duration. Funding: bupropion manufacturer. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were randomly assigned". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind, double dummy". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind, double dummy". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "we performed statistical analysis using the Last Observation Carried Forward (LOCF) method on all subjects who took at least one dose of medication and completed at least a second visit". |
| Selective reporting (reporting bias) | Unclear risk | Outcomes data not clearly reported (the study was primarily designed to evaluate sexual functioning during paroxetine or bupropion treatment). |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Kiev 1997.
| Methods | Seven‐week, double‐blind, multicentre, randomised study. | |
| Participants | Psychiatric outpatients meeting DSM‐III‐R for recurrent major depressive disorder, with a minimum baseline score of 20 on the Hamilton rating scale for depression (HDRS‐21). Age range: 18‐65 years. | |
| Interventions | Paroxetine: 30 participants. Fluvoxamine: 30 participants. Paroxetine dose range: 20‐50 mg/day. Fluvoxamine dose range: 50‐150 mg/day. Concomitant use of any psychotropic medication was prohibited. While medications to treat gastrointestinal disturbances (antacids, laxatives), and headache (acetaminophen, aspirin, ibuprofen) and to provide nighttime sedation (chloral hydrate only) were permitted, all other medication use was prohibited unless approved by the study physician. |
|
| Outcomes | HRSD‐21, Clinical Global Impression (CGI), Hamilton rating scale for anxiety (HAM‐A), Symptom checklist (SCL‐56). Total dropout, dropout due to inefficiency, dropout due to side effects, number of patients experiencing at least one side effect, side‐effect profile. |
|
| Notes | Funding: fluvoxamine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly assigned", no further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Study endpoint: 11/30 missing from paroxetine group (1 due to lack of efficacy, 2 due to adverse effects); 9/30 missing from control group (3 due to lack of efficacy, 5 due to adverse effects). |
| Selective reporting (reporting bias) | High risk | Standard deviations of endpoint score for depression were not reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Kramer 1998.
| Methods | Six‐week, randomised, double‐blind, placebo‐controlled study. | |
| Participants | Out‐patients with a DSM‐IV diagnosis of major depression disorder (single or recurrent), a current episode of depression lasting at least 4 weeks (but less than 2 years) a score ≥22 (moderately depressed) on the Hamilton rating scale for depression (HDRS‐17), a score ≥15 (moderately high anxiety) on the Hamilton rating scale for anxiety (HAM‐A) and a score ≥4 (moderately ill) on the Clinical Global Impression (CGI) Severity. Patients considered at risk for suicide or violence were excluded. | |
| Interventions | Paroxetine:72 participants. Aprepitant (MK‐869): 71 participants. Paroxetine dose: 20 mg/day. Aprepitant (MK‐869): 300 mg/day. |
|
| Outcomes | HDRS‐21, HAM‐A, CGI‐S. | |
| Notes | Funding: aprepitant manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | More than 20% of participants in each arm abandoned the study prematurely. |
| Selective reporting (reporting bias) | High risk | Outcomes data not clearly reported. Only side effects with an incidence of > or = 5% were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Kramer 2001.
| Methods | Eight‐week, randomised, multicentre study. | |
| Participants | Outpatients with Major Depressive Disorder (MDD). | |
| Interventions | Paroxetine: 20 mg/day. Compound A: low dose (not specified). Compound A: high dose (not specified). Placebo. |
|
| Outcomes | Hamilton rating scale for depression (HDRS), Hamilton rating scale for anxiety (HAM‐A), adverse effects. | |
| Notes | None. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information on incomplete outcome data management. |
| Selective reporting (reporting bias) | High risk | Efficacy and tolerability data not clearly reported. |
| Other bias | Unclear risk | Insufficient information to establish the presence of other biases. |
Kuhs 1989.
| Methods | Six‐week, double‐blind, randomised, double‐dummy, parallel‐group, comparative study. | |
| Participants | Inclusion criteria: DSM III major depression, Hamilton rating scale for depression (HDRS‐21) 18+. Age: 18‐65 years. Country: Germany. Setting: inpatients. | |
| Interventions | Paroxetine: 20 participants.
Amitriptyline: 20 participants. Paroxetine dose: 30 mg/day. Amitriptyline dose: 150 mg/day. During the study 13 patients were treated with oxazepam (<20mg/d) as a co‐medication in cases of psychomotor agitation (paroxetine n = 5; amitriptyline n = 8). Short‐acting benzodiazepines (triazolam, lorazepam) were given in case of persistent insomnia (paroxetine n = 3; amitriptyline n = 1). Because of postural hypotension, 3 patients in the paroxetine group and 7 in the amitriptyline group were treated with dihydroergotamine (<5 mg/d). |
|
| Outcomes | HDRS‐21. Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | None. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "all subjects entered were eligible for the Intention‐to‐treat (ITT) population. Safety assessment were performed on the ITT population". No further details. More than 20% of participants in both arms abandoned the study prematurely. |
| Selective reporting (reporting bias) | High risk | Outcomes data not clearly reported. Continuous data not reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Laghrissi‐Thode 1995.
| Methods | Six‐week, randomised, double‐blind study. | |
| Participants | Patients suffering from a major depressive episode according to DSM III criteria using the Structured Clinical Interview for DSM III R. The current episode must have lasted at least 1 month and the Hamilton rating scale for depression (HDRS) score must have been 15 or higher for patient inclusion. Patients were excluded from the study if they suffered from an instable medical illness, had taken psychotropic medications within 2 weeks (or within 6 weeks for fluoxetine) or had contraindications to nortriptyline treatment. | |
| Interventions | Paroxetine: 20 mg/day. Nortriptyline: 50/120 ng/mL. |
|
| Outcomes | Postural stability in older depressed patients. | |
| Notes | This study was independent from pharmaceutical industry. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No clear information on incomplete outcome data management. |
| Selective reporting (reporting bias) | Unclear risk | No efficacy and tolerability data. This study was primarily designed to investigate the effect of antidepressants on postural stability in older depressed patients. |
| Other bias | Low risk | This study was independent from pharmaceutical industry. |
Laursen 1985.
| Methods | Six‐week, randomised, double‐blind,parallel‐group. | |
| Participants | Inclusion criteria: ICD 8 Manic‐depressive psychosis, Hamilton rating scale for depression (HDRS‐17) 15+; three of the four following criteria had to be present: phasic course of illness, changes in psychomotor activity, diurnal variation in symptom and unfounded lost of self‐esteem. Age: 35‐81 years. Country: Denmark. Setting: inpatients with outpatient follow‐up. | |
| Interventions | Paroxetine: 21 participants.
Amitriptyline: 23 participants. Paroxetine dose range: 30‐60 mg/day. Amitriptyline dose range: 150‐200 mg/day. In the event of need for an anxiolytic or hypnotic drug, benzodiazepines and chlorhydrate could be used. |
|
| Outcomes | HDRS‐17 and 6‐item subscale ratings.Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile | |
| Notes | Funding: independent from industry. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "random allocation to treatment (...) randomized". |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The statistical analyses were based on results from completers. No further details. |
| Selective reporting (reporting bias) | High risk | Outcomes data not clearly reported. Rating scales scores reported only in figures. |
| Other bias | Low risk | This study was independent from pharmaceutical industry. |
Lee 2007 (HMCV).
| Methods | Randomised, double‐blind, double‐dummy, parallel‐group study. | |
| Participants | Planned: Total 480, duloxetine 240, paroxetine 240.
Randomised: Total 478, duloxetine 238, paroxetine 240.
Completed: Total 349, duloxetine 166, paroxetine 183. Criteria for Inclusion: Patients of either sex and at least 18 years of age who met the DSM‐IV criteria for nonpsychotic Major Depressive Disorder (MDD), and had a Hamilton rating scale for depression (HDRS‐17) total score ≥15 and Clinical Global Impression (CGI) Severity score ≥4 were included. |
|
| Interventions | Duloxetine: 60 mg/day (given orally once a day as two 30 mg capsules). Paroxetine: 20 mg/day, given orally once a day as two 10 mg capsules. | |
| Outcomes | Efficacy: The primary measure of efficacy was to assess the severity of depression and its improvement during the course of therapy, using the HDRS‐17. Secondary measures of efficacy included: presence and severity of anxiety (HAM‐A), severity of illness (CGI‐Severity), degree of improvement (PGI‐Improvement), degree to which physical complaints were bothersome to patient (SSI), and experience of overall pain (VAS). Safety: Assessment of adverse events, including serious adverse events, routine laboratory tests, vital signs, and ECGs. | |
| Notes | For the primary efficacy analysis, a one‐sided 97.5% confidence interval of the adjusted mean HDRS‐17 total score was used to determine whether duloxetine was noninferior to paroxetine. Non‐inferiority was declared if the upper bound of the one‐sided 97.5% confidence interval was less than 2.2. Date of first patient enrolled: 17 February 2004. Date of last patient completed: 12 June 2005. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients meeting the entry criteria were randomized in a ratio of 1:1 to either duloxetine 60 mg/day or paroxetine 20 mg/day". |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind, double dummy". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind, double dummy". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "the primary analysis was based on the Per Protocol (PP) population. The analysis of treatment emergent adverse effects was based on all randomized patients with at least one post‐baseline observation and taking at least one dose of their randomzed medication (safety set). All other results are based on all randomized patients with at least one post‐baseline observation (Intention‐to‐treat population)". |
| Selective reporting (reporting bias) | High risk | Only adverse events occurring with frequency > or = 5% were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Lepine 2001.
| Methods | Multicentre, randomised, double‐blind study. | |
| Participants | Male of female, in‐ or outpatients fulfilling DSM‐IV criteria for Major Depressive Disorder (MDD) single episode or recurrent, severity moderate to severe, without psychotic features, with or without melancholic features and, for recurrent disorders, with or without seasonal pattern and inter‐episode recovery), were eligible for the study. To be selected, patients had to present at D7 a minimum severity score of 25 on the Montgomery and Asberg Depression Rating Scale (MADRS) and of 9 on the Hamilton rating scale for depression (HDRS) (items 1,2,7,8,10 and 13). These severity criteria also had to be present at inclusion, and those patients with a decrease of 30% or more on MADRS or HDRS global scores were excluded. Patients fulfilling DSM‐IV criteria for other Major Depressive Disorders: Bipilar I or II Disorders, dysthymic disorder, cyclothymic disorder, mixed anxio‐depressive disorder, recurrent brief depressive disorder, schizophrenia or any other acute or chronic psychosis and patients whose present disorder was due to a general medical condition or to a substance were not eligible for the study. |
|
| Interventions | Paroxetine: 165 participants.
Tianeptine: 162 participants. Paroxetine dose: 20 mg/day. Tianeptine dose: 37,5 mg/day. Treatment with a benzodiazepine during the last 2 weeks, or for more then 1 month during the last 6 months, or at daily dose equal to or greater the an equivalent of 15mg of diazepam on a lifetime period, were also non‐inclusion criteria. |
|
| Outcomes | MADRS, CPRS, HDRS, Hamilton rating scale for anxiety (HAM‐A), Clinical Global Impression (CGI), Severity of Depression Questionnaire (QSD), Hospital Anxiety and Depression (HAD), Mood and Anxiety Symptom Questionnaire (MASQ), Visual Analogue Scale (VAS). Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | This study was independent from pharmaceutical industry. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "analysis of the efficacy data was performed both on the Intention‐to‐treat (ITT) population (ITT: all patients with at least one evaluation under treatment) and on the Per Protocol (PP) population (PP: all completed patients without major deviation, according to the study protocol). The analysis was done at the endpoint for the ITT population and visit by visit for the PP population. The analysis of safety used all patients who had taken at least one dose of randomized treatment". Less than 20% of participants in both arms withdrew from the study prematurely. |
| Selective reporting (reporting bias) | Low risk | Outcomes data were reported. |
| Other bias | Low risk | This study was independent from pharmaceutical industry. |
Loo 2002.
| Methods | Eight‐week, double‐blind, randomised multicentre study. | |
| Participants | Seven hundred and eleven patients (238 males, 33.5%; 473 females, 66.5%; mean age 42.3 years) in 102 centres located in Belgium, France and the UK were included in this study. There was no significant difference between groups at inclusion for demo‐graphic characteristics or disease factors: 67.1% of patients met DSM‐IV criteria for a recurrent major depressive disorder, 33.5% of patients had an episode of severe intensity. | |
| Interventions | Patients were to take orally one capsule twice daily: one in the morning, one in the evening. Following a placebo run‐in period of 1 week, patients were randomised in double‐blind conditions to receive fixed doses of agomelatine (1mg, 5mg or 25mg in the evening capsule), paroxetine 20 mg (in the morning capsule) or placebo. Concomitant treatment with psychotropic drugs was not allowed with the exception of benzodiazepines at restricted doses. High potency benzodiazepines, such as alprazolam and triazolam, were not permitted. Drugs which were thought to be able to influence study evaluations by acting on patient’s mood or circadian rhythms, such as b‐blockers, central a‐blockers,non‐steroidal anti‐inflammatory drugs and exogenous melatonin,were not allowed. |
|
| Outcomes | The Hamilton rating scale for depression (HDRS) and the Clinical Global Impression (CGI) were assessed at baseline, and at weeks 1, 2, 4, 6and 8.The Montgomery and Asberg Depression Rating Scale (MADRS) and the Hamilton rating scale for anxiety (HAM‐A) were assessed at baseline, and at weeks 4 and 8. Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | In this Cochrane Review data referring to dichotomic outcomes were add up together, data referring to continuous outcomes were matched together as suggested by the Cochrane Handbook (Table 7.7.a: Formulae for combining groups). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind conditions". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind conditions". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Primary measure of efficacy was based on the Intention‐to‐treat (ITT) population, defined as "patients who has received at least one post‐randomization capsule and had a baseline measurement and at least one post‐baseline measurement". |
| Selective reporting (reporting bias) | Low risk | Outcomes data were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
M/2020/0047.
| Methods | Multicentre, randomised, double‐blind, placebo‐ and active‐controlled, parallel‐group study. | |
| Participants | Inclusion Criteria. To be included in the study, patients must have met all of the following criteria: diagnosis of Major Depressive Disorder (MDD) without psychotic features, as defined by DSM‐IV. Male or female, of any race, between the ages of 18 and 65 years. If female, must have been postmenopausal or must have met all of the following criteria: agreed to avoid pregnancy during the study; had a negative serum pregnancy test at screen; used an accepted means of birth control (as determined by the investigator), such as abstinence, oral contraceptive, implantable or injectable contraceptive, intrauterine device, or barrier method, or have been surgically sterilized. Total score of ≥20 on the Hamilton rating scale for depression (HDRS‐17), which was administered via the IVRS prior to the screening visit. General good health, as confirmed by routine clinical laboratory safety findings. Voluntary consent to participate in the study, documented in a written Patient Informed Consent Form that was signed prior to the start of any study procedures at the screening visit. Exclusion Criteria. Patients were excluded from the study for any of the following reasons: DSM‐IV diagnosis of the following concomitant psychiatric disorders: MDD with psychotic features, cyclothymic disorder, bipolar I or bipolar II disorders, substance‐related disorders (within the preceding 12 months), schizophrenia, or other psychotic disorders. Resistance to antidepressive treatment, defined as a lack of response to at least 2 previous courses of antidepressant medications administered at full doses for more than 1 month. Participation in a previous clinical trial of reboxetine or lack of response to previous treatment with paroxetine, administered at a dose of ≥20 mg/day for more than 1 month. Use of antidepressant medication for the treatment of depression in the 2 months preceding the start of the study. History of MDD associated with endocrine disorders: hypo‐ or hyper‐thyroidism tested by thyroid‐stimulating hormone and thyroxine, adrenal insufficiency, or Cushing’s syndrome. Positive pregnancy test for females of childbearing potential. Breast‐feeding by female patients. Refusal by female patients of childbearing age to use an effective contraceptive method during the study. Participation in any clinical study with an investigational compound in the 4 weeks preceding the study. History or presence of gastrointestinal, liver, or kidney disease or other conditions known to interfere with the absorption, distribution, metabolism, and excretion of drugs. History of seizures or brain injury; current evidence of clinically important hematopoietic, respiratory, or cardiovascular diseases; current evidence of urinary retention or glaucoma. Clinically significant illness in the 4 weeks preceding the study that might have interfered with the conduct of the trial. Clinically relevant abnormal findings in the physical examination, laboratory tests, or ECG at admission. Positive urine drug screen for amphetamines, barbiturates, marijuana metabolites, cocaine metabolites, methadone, methaqualone, opiates, phencyclidine, or propoxyphene. A positive urine drug screen for benzodiazepines did not exclude the patient. Treatment with electroconvulsive therapy in the 6 months preceding the study. Major risk of suicide as assessed by the investigator, a score of ≥3 on Item 3 of the HDRS at screen or baseline, or a history of suicide attempt during the current depressive episode. History of hypersensitivity to reboxetine or paroxetine. Use of the following medications, which are potent inhibitors of the drug‐metabolizing enzyme cytochrome p450‐3A4: azole antifungals, macrolide antibiotics , or fluvoxamine. Use of the following medications, which are known to be substrates or inhibitors of the drug‐metabolizing enzyme cytochrome p450‐2D6: Type 1C antiarrhythmics, quinidine, or cimetidine. Use of oral anticoagulants that are known to inhibit vitamin K‐dependent coagulation factors. Use of concomitant psychotropic medications other than the protocol‐specified sedatives/hypnotics, which could be taken on an as‐needed basis for sleep. Inability of the patient to comply with the conditions of the study, based on the investigator’s assessment. |
|
| Interventions | Paroxetine: 262 participants.
Reboxetine: 258 participants. Placebo: 254 participants. |
|
| Outcomes | Montgomery Asberg Depression Rating Scale (MADRS) total score at day 56 in the Intent‐to‐treat (ITT) patient population.Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Funding: reboxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Pharmacia & Upjohn (P&U) prepared a randomization list for assignment of the patients to 1 of the 3 treatment groups. Study medication for each treatment group was prepared on this basis by P&U and was labeled with the corresponding patient number". |
| Allocation concealment (selection bias) | Low risk | Quote: "at the baseline visit, the investigator assigned each patient to a treatment group based on the patient's temporal entry into the study (i.e. by assigning the lowest patient number available). A list of patient numbers and medication assignments was provided only after the data for the study had been analyzed". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "double blind (...) study medications were provided as identically appearing capsules (...) The randomized medications consisted of identically appearing capsules containing reboxetine, paroxetine or placebo. The capsules were provided in clinical supply packages that were labeled with the protocol number, patient number, treatment period, dosing, directions and storage conditions. Investigators were given sealed drug‐disclosure sheets that contained information about each patient's treatment". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "the Intention‐to‐treat (ITT) dataset, which includes all patients who were randomized into the trial and who received at least one dose of study medication, was used for all the analyses. Two types of analyses were performed for all efficacy variables: Last Observation Carried Forward (LOCF) and Observed Cases (OC). The LOCF analyses used the last valid assessment as an estimate for all subsequent missing values. The OC analysis did not replace missing data. The LOCF analyses were the primary analyses and the OC analyses were the secondary analyses". |
| Selective reporting (reporting bias) | Low risk | Outcomes data were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
M/2020/0052.
| Methods | Eight‐week, multicentre, randomised, double‐blind, parallel‐group study. | |
| Participants | Inclusion Criteria. To be included in the study, patients must have met all of the following criteria: diagnosis of Major depressive Disorder (MDD) without psychotic features, as defined by DSM‐IV. Male or female, of any race, between the ages of 18 and 65 years. If female, must have been postmenopausal or must have met all of the following criteria: agreed to avoid pregnancy during the study; had a negative serum pregnancy test at screen; used an accepted means of birth control (as determined by the investigator), such as oral contraceptive, implantable or injectable contraceptive, intrauterine device, or barrier method, or have been surgically sterilized. Total score of ≥22 and ≤35 on the 21‐Item Hamilton rating scale for depression (HDRS) at screen and confirmed at baseline. Voluntary consent to participate in the study documented in a written Patient Informed Consent Form that was signed prior to the start of any study procedures at the screening visit. Exclusion Criteria. Patients were excluded from the study for any of the following reasons: DSM‐IV diagnosis of the following concomitant psychiatric disorders: MDD with psychotic features, dysthymic or cyclothymic disorder, bipolar I or bipolar II disorders, substance‐related disorders, schizophrenia, or other psychotic disorders; a lack of response to a previous course of either reboxetine or paroxetine; history of MDD associated with endocrine disorders: hypo‐ or hyper‐thyroidism tested by thyroid‐stimulating hormone and thyroxine, adrenal insufficiency, or Cushing’s syndrome, etc.; positive serum pregnancy test for females of childbearing potential. Breast‐feeding female patients. Participation in a clinical study with an investigational compound in the 4 weeks preceding the study. Presence of gastrointestinal, liver, or kidney disease or other conditions known to interfere with the absorption, distribution, metabolism, and excretion of drugs. History of seizures or brain injury; current evidence of clinically important hematopoietic, respiratory, or cardiovascular diseases; current evidence of urinary retention or glaucoma. Clinically significant illness in the 4 weeks preceding the study that might have interfered with the conduct of the trial. Clinically relevant abnormal findings in the physical examination, laboratory tests, or ECG at admission. Treatment with electroconvulsive therapy in the 6 months preceding the study. Major risk of suicide as assessed by the investigator, a score of ≥3 on Item 3 of the HDRS at screen or baseline, or a history of suicide attempt during the current depressive episode. History of hypersensitivity to reboxetine or paroxetine. Use of the following medications, which are known to inhibit major drug‐metabolizing enzymes other than cytochrome p450‐2D6: azole antifungals, macrolide antibiotics, or fluvoxamine. Use of oral anticoagulants that are known to inhibit vitamin K‐dependent coagulation factors. Use of concomitant psychotropic medications other than the protocol‐specified sedatives/hypnotics, which could be taken on an as‐needed basis for sleep. Inability of the patient to comply with the conditions of the study based on the investigator’s assessment. |
|
| Interventions | Paroxetine: 166 participants. Reboxetine: 159 participants. | |
| Outcomes | To assess efficacy and tolerability of reboxetine in comparison with paroxetine in patients suffering from MDD as determined by the HDRS scale. Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Funding: reboxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Pharmacia & Upjohn (P&U) prepared a randomization list for assignment of the patients to 1 of the 2 treatment groups. Study medication for each treatment group was prepared on this basis by P&U and was labled with the corresponding patient number". |
| Allocation concealment (selection bias) | Low risk | Quote: "at the baseline visit, the investigator assigned each patient to a treatment group based on the patient's temporal entry into the study (i.e. by assigning the lowest patient number available). A list of patient numbers and medication assignments was provided only after the data for the study had been analyzed". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "double blind (...) study medications were provided in product packages that were labeled with the protocol number, the patient number, the study week, and the dose level. The randomized medications consisted of identically appearing capsules containing reboxetine, paroxetine with placebo. The capsules were provided in clinical supply packages that were labeled with the protocol number, patient number, treatment period, dosing, directions and storage conditions. Investigators were given sealed drug‐disclosure sheets that contained information about each patient's treatment assignment". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "the Intention‐to‐treat (ITT) dataset, which includes all patients randomized into the trial and who received at least one treatment dose with at least one post‐baseline efficacy follow up evaluation, was to be used for the analysis. Two types of analyses were performed for all efficacy variables: Last Observation Carried Forward (LOCF) and Observed Cases (OC). The LOCF analyses used the last valid assessment as an estimate for all subsequent missing values. The OC analysis did not replace missing data. The ITT dataset using the LOCF technique was to be the primary analysis and the OC analysis was to be included as a secondary analysis". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Outcomes data were reported. |
| Selective reporting (reporting bias) | Low risk | Outcomes data were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Mc Partlin 1998.
| Methods | Twelve‐week, parallel‐group, randomised, double‐blind trial. | |
| Participants | Outpatients at least 18 years of age were eligible if they satisfied DSM‐IV criteria for major depression, had symptoms of depression for at least 14 days, and had a minimum baseline score of 19 on the Montgomery and Asberg Depression Rating Scale (MADRS). | |
| Interventions | Paroxetine: 178 participants. Venlafaxine XR: 183 participants. Paroxetine dose: 20 mg/day. Venlafaxine dose: 75 mg/day. |
|
| Outcomes | MADRS, Hamilton rating scale for depression (HDRS), Clinical Global Impression (CGI). Total dropout, dropout due to side effects, dropout due to inefficacy. Side‐effect profile. | |
| Notes | Funding: venlafaxine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "data from patients who took at least one dose of study medication and who had at least one efficacy evaluation during the treatment period comprised the Intention‐to‐treat (ITT) population". |
| Selective reporting (reporting bias) | High risk | Only most common adverse effects were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Montgomery 2004.
| Methods | Twelve‐week, randomised, double‐blind study. | |
| Participants | Depressed outpatients, aged 18 years or above, who fulfilled the diagnostic criteria for Major Depressive Disorder (MDD) of DSM IV, single or recurrent episode. The diagnosis of MDD and any comorbid psychiatric disorders was documented using the Mini International Neuropsychiatric Interview (MINI). No other Axis I or II disorders could be included. Patients having any concomitant severe and/or unstable medical illnesses likely to interfere with the conduct of the study were also excluded. | |
| Interventions | Paroxetine:168 participants. Agomelatine: 167 participants. Paroxetine dose: 20 mg/day. Agomelatine dose: 25 mg/day. |
|
| Outcomes | Montgomery and Asberg Depression Rating Scale (MADRS), Hamilton rating scale for anxiety (HAM‐A), side effects. | |
| Notes | This study was independent from pharmaceutical industry. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind conditions". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind conditions". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information on incomplete outcome data management. |
| Selective reporting (reporting bias) | Unclear risk | Some data were not reported. |
| Other bias | Low risk | This study was independent from pharmaceutical industry. |
Moon 1996.
| Methods | Six‐week, multicentre, randomised, double‐blind, comparative study. | |
| Participants | Inclusion criteria: male and female patients; aged 18‐65 years; patients who met DSM‐III criteria for a major depressive episode; subjects with Montgomery and Asberg Depression Rating Scale (MADRS) score of 18+. Exclusion criteria: patients with significant suicidal tendencies; psychosis or rapid cyclic manic‐depressive disorder; patients who had received other psychotropic agents, such as IMAO within 2 weeks of entering the study, or concurrent treatment with clonidine or anticoagulants. |
|
| Interventions | Paroxetine: 60 participants. Lofepramine: 62 participants. Paroxetine dose range: 20‐30 mg/day. Lofepramine dose range: 140‐210 mg/day. |
|
| Outcomes | MADRS, Clinical Global Impression (CGI). Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Only side effects occurring in >5% of patients. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were randomly allocated". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "the intent‐to‐treat (ITT) population comprised all subjects who received at least 1 dose of trial medication and were assessed at least once during the active phase of the study". Less than 20% of participants in both arms withdrew from the study prematurely. |
| Selective reporting (reporting bias) | High risk | Only side effects occurring in >5% of patients were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Mulsant 1999.
| Methods | Six‐week, randomised, double‐blind study. | |
| Participants | For inclusion in the analyses, patients had to meet the following criteria: age of 60 years or older, DSM‐IV major depressive episode without psychotic features or history of bipolar or schizoaffective disorder, baseline Hamilton rating scale for depression (HDRS) score of 15 or above, Mini Mental State Examination (MMSE) score of 18 or above, no history of alcohol or other substance abuse or dependency during at least the past year, and no specific medical condition contraindicating treatment with either nortriptyline or paroxetine (e.g., QRS longer than 120 ms or bradycardia with heart rate below 50 beats per minute). | |
| Interventions | Paroxetine: 43 participants. Nortriptyilne: 37 participants. Paroxetine dose range: 20‐30 mg/day. Nortriptyline dose range: 50 mg/day. Patients complaining of severe anxiety and/or insomnia were prescribed lorazepam on a regular basis (e.g., twice a day or at bedtime); the lowest possible doses were used and, as much as clinically possible, doses were kept constant. |
|
| Outcomes | Study participants were also evaluated with the Structured Clinical Interview for DSM‐IV Axis I Disorders (SCID‐IV) and several rating scales, including a semi‐structured version of the HDRS‐17, the Cumulative Illness Rating Scale adapted for Geriatrics (CIRS‐G), the UKU side effect rating scale (UKU), and a standardised version of MMSE. | |
| Notes | Sponsor unclear. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Subjects were randomly assigned". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind conditions". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind conditions". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | An Intention‐to‐treat (ITT) Last Observation Carried Forward (LOCF) analysis was performed. |
| Selective reporting (reporting bias) | Low risk | Outcomes data were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
MY‐1045/BRL‐029060/1.
| Methods | Multicentre, randomised, double‐blind, placebo‐controlled study. | |
| Participants | Outpatients >18 years old with a primary diagnosis meeting DSM‐III‐R criteria for Major depressive Disorder (MDD) (single or recurrent episode) with a total score of >18 on the first 17 items of the Hamilton rating scale for depression (HDRS‐21). Total score could not have decreased by 25% between the screen and baseline visits. Exclusion criteria: other diagnosis than MDD, serious suicidal or homicidal risk, substance abuse/dependence, prior ECT (within 3 months of the study), serious concomitant medical conditions and patients with a history of hypersensitivity to fluoxetine or who had previously taken paroxetine. |
|
| Interventions | Paroxetine:357 participants. Fluoxetine: 251 participants. Paroxetine dose range: 20‐50 mg/day. Fluoxetine dose range: 20‐80 mg/day. |
|
| Outcomes | HDRS, Clinical Global Impression (CGI), Raskin Depression Score (RDS), Global Assessment of Functioning (GAF), Symptoms Checklist (SCL‐90). Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Funding: paroxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "the Intention‐to‐treat (ITT) analysis included all randomized subjects. All randomized subjects who had an on‐therapy efficacy evaluation were included in the efficacy analysis; all randomized subjects were included in the safety analysis. The main analysis was based on the extender dataset which modified the observed data so that missing data for a given week were estimated by bringing forward (extending) the data from the previous week. If week 1 data were missing, no estimate was made". |
| Selective reporting (reporting bias) | Low risk | Outcomes data were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
NCT00463242.
| Methods | Double‐blind, randomised study. | |
| Participants | Male and female adults, 18‐70 years of age, with a diagnosis of Major Depressive Disorder according to DSM‐IV criteria. Hamilton Depression Rating Scale (HDRS) total score > or = 22 at screening and baseline. | |
| Interventions | Paroxetine: 20‐40 mg/day. Agomelatine: 25‐50 mg/day. |
|
| Outcomes | Change from baseline to endpoint at HDRS, remission, sexual functions. | |
| Notes | Funding: by industry. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no further information. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind. No further information. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double‐blind. No further information. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient information. |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Nickel 2003.
| Methods | Double‐blind, randomised, controlled study of depressed inpatients. | |
| Participants | Hospitalised patients of either sex (25–65 years of age) fulfilling the DSM‐IV criteria for a major depressive episode or bipolar I or II disorder (single‐episode or recurrent depression, without psychotic features) and having a total score of 18 on the Hamilton rating scale for depression (HDRS‐21) were eligible for enrolment in the study. The difference in the total HDRS score between selection and inclusion on day 1 had to be 6 points in order to minimise confounding by spontaneous remission. Patients with other affective disorders, i.e., dysthymia, double depression, recurrent brief depressed disorder, anxiety disorder, or acute or chronic psychotic disorder, were not enrolled. Other exclusion criteria were pregnancy and absences of effective contraception in women of childbearing potential; suicidality; history of drug or alcohol abuse or dependence; other severe diseases such as uncontrolled cardiovascular, neurological, or metabolic disorders. | |
| Interventions | Paroxetine: 22 participants.
Tianeptine: 22 participants. Paroxetine dose range: 20‐40 mg/day. Tianeptine dose range: 37,5‐ 75 mg/day. During the study, no other psychotropic drugs was allowed except chloralhydrate in a dosage of up to 2 g/d on an as‐required basis. The prescription of medications that would not interfere with the antidepressant treatment was acceptable. |
|
| Outcomes | HDRS, Montgomery and Asberg Depression Rating Scale (MADRS), Clinical Global Impression (CGI), Beck Depression Inventory (BDI). Total dropout, dropout due to inefficacy. | |
| Notes | Funding: tianeptine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were enrolled and randomly assigned". No further details". |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "the analysis of changes in the efficacy measures was performed on the Intention‐to‐treat (ITT) population with use of the Last Observation Carried Forward (LOCF) method". |
| Selective reporting (reporting bias) | High risk | Side effects not reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Nielsen 1991.
| Methods | Double‐blind, randomised, multicentre study. | |
| Participants | Inclusion criteria were depression (unipolar or bipolar) calling for pharmacotherapeutic treatment, Hamilton rating scale for depression (HDRS) 18+, age 18‐70 years and all patients met the DSM‐III criteria for major depressive episode. Exclusion criteria were concurrent somatic conditions contraindicating treatment with antidepressants, i.e. cardiovascular conditions, severe liver or renal disease, severe hypertension and unstable endocrinological disease. |
|
| Interventions | Paroxetine: 16 participants. Imipramine: 15 participants. Paroxetine dose: 30 mg/day. Imipramine dose: 150 mg/day. Concomitant medications was restricted to occasional doses of oxazepam (or similar benzodiazepines) as sedative and paracetamol as analgesic, when necessary. Other psychotropic medication, including lithium, was not allowed. |
|
| Outcomes | HDRS‐17, Melancholia Scale (MES), UKU scale. Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect. | |
| Notes | Funding unclear. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "the patients were randomly allocated". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "to reduce bias caused by patient withdrawals, the last value was carried forward in the analyses". More than 20% of participants in both arms withdrew from the study prematurely. |
| Selective reporting (reporting bias) | High risk | Side effects were not reported. Continuous outcomes only in graph. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Ohrberg 1992.
| Methods | Multicentre, randomised, double‐blind, parallel‐group, active‐controlled study. | |
| Participants | Male and female participants, aged 18 to 70 years inclusive, with depression requiring drug treatment, with a diagnosis confirmed by observation of signs and symptoms, and a score >15 on the Hamilton rating scale for depression (HDRS) scale. Key exclusion criteria: other mental disorder, serious suicidal risk (score of >3 on HDRS item 3), treatment with a IMAO within the 14 days preceding entry to the study. | |
| Interventions | Paroxetine: 79 participants. Imipramine:80 participants. Paroxetine dose range: 10‐50 mg/day. Imipramine dose range: 50‐250 mg/day. With the exception of oxazepam or lorazepam as sedative/hypnotics, no other psychopharmacological drugs were allowed. |
|
| Outcomes | HDRS, Bech‐Rafaelsen Melancholia Scale (BRMS). Total dropout, dropout due to side effects, dropout due to inefficacy. Side‐effect profile. | |
| Notes | Only side effects occurring in >5% of patients. Funding unclear. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "subjects were randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "the Intention‐to‐treat (ITT) dataset included all participants who were randomised and received study medication. The efficacy dataset was the subset of the ITT population who completed at least two weeks of treatment. To reduce bias due to subjects who withdrew prematurely, the last value recorded was carried forward". More than 20% of participants in the imipramine arm withdrew from the study prematurely. |
| Selective reporting (reporting bias) | High risk | Only side effects occurring in >5% of patients were reported. |
| Other bias | Unclear risk | Insufficient information to establish the presence of other biases. |
Ontiveros 1994.
| Methods | Randomised, double‐blind, parallel‐group study. | |
| Participants | Male and female participants, aged 18 to 75 years inclusive, who were suffering from a major depressive episode according to the DSM‐III criteria, with a diagnosis confirmed by observation of signs and symptoms, and a score >18 on the Hamilton rating scale for depression (HDRS) scale at the pretreatment and baseline assessment. Participants with severe co‐existing disease, who had received ECT therapy in the previous 3 months, IMAO in the previous 2 weeks, lithium in the previous 3 months, oral neuroleptics in the previous 2 weeks, depot neuroleptics in the previous 4 weeks, benzodiazepines in the previous 3 days or coadministration of any drug with know psychotropic effects, participants at severe risk of suicide were excluded from the study. | |
| Interventions | Paroxetine: 60 participants. Fluoxetine: 62 participants. Paroxetine dose: 20 mg/day. Fluoxetine dose: 20 mg/day. |
|
| Outcomes | HDRS, Clinical Global Impression (CGI). Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Funding: paroxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind (...) in order to maintain the blind, all medication was concealed in coloured capsules". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "the primary analyses were performed on the Intention‐to‐treat (ITT) population, which included all subjects who were randomized to active treatment and had at least one valid efficacy evaluation after the start of treatment". The primary dataset was the ITT endpoint assessment (defined as the patient's last available assessment between weeks 4 and 6). Less than 20% of participants withdrew from the study prematurely. |
| Selective reporting (reporting bias) | High risk | Only adverse events by more than 5% of participants were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Owens 2008.
| Methods | Eight‐week, prospective, multicentre, randomised, double‐blind, parallel‐group study. | |
| Participants | Male and female patients (18–65 years) meeting diagnostic criteria for Major Depressive Disorder (MDD) were eligible (American Psychiatric Association, 1994). The diagnosis of MDD was made by the principal investigator using the Mini International Neuropsychiatric Interview (MINI) ‐ a structured diagnostic interview for DSM‐IV. A total score of >20 on the Montgomery and Asberg Depression Rating Scale (MADRS) was required at screening and baseline. Patients were excluded if they had a clinically predominant axis I disorder other than MDD. Other key exclusion criteria were: history of unresponsiveness to either paroxetine or venlafaxine or exhibited prior hypersensitivity/intolerance to either paroxetine CR or venlafaxine XR, substance abuse/dependence, prior non‐response to SSRIs, suicidal/homicidal risk, concurrent psychotherapy or psychotropic pharmacotherapy, or any serious medical condition or clinically significant finding in the screening or baseline evaluation that would preclude the administration of paroxetine CR or venlafaxine XR. Patients were excluded if they required concomitant therapy with psychoactive medication or patients who have taken other psychoactive medication within the time frames specified below prior to the screening visit: antidepressants other than MAOIs or fluoxetine (e.g., tricyclic antidepressants, SSRIs, and NSRIs), lithium and oral antipsychotics‐14 days; hypnotics, benzodiazepines, and all other sedatives (including chlorpheniramine and other sedating antihistamines)‐14 days; fluoxetine, MAOIs‐4 weeks; depot neuroleptics‐12 weeks; any CNS‐active herbal preparations/supplement (e.g., St John’s wort, kava kava, etc.)F‐14 days. | |
| Interventions | Paroxetine: 42 participants. Venlafaxine XR: 44 participants. Paroxetine dose range: 12.5‐75 mg/day. Venlafaxine dose range: 75‐375 mg/day. |
|
| Outcomes | MADRS, Clinical Global Impression (CGI). Total dropout, dropout due to side‐effects. Number of patients experiencing at least one side effect. | |
| Notes | Funding: paroxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomized (...) patients meeting eligibility criteria at baseline were randomized (1:1) to receive over‐encapsulated paroxetine or venlafaxine tables using a computer‐generated randomization list". |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind (...) over‐encapsulated paroxetine or venlafaxine tables". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "all patients who were randomized, received at least one dose of study medication and had at least one post‐baseline efficacy assessment, were included in the modified intent‐to‐treat (ITT) efficacy analyses (...) Patients withdrawing before week 2 without MADRS assessments are not included in the analyses of the MADRS. The analyses on observed data were repeated using the Last Observation Carried Forward (LOCF) approach to impute missing values (...). Because five of the randomized patients did not have a post‐baseline efficacy assessment, the evaluable population (modified ITT) consisted of 81 patients". More than 20% of participants withdrew from the study prematurely. |
| Selective reporting (reporting bias) | Low risk | Outcomes data were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
PAR MDUK 032.
| Methods | Six‐week, randomised, double‐blind, double‐dummy, parallel‐group comparative study. | |
| Participants | Male and female patients, aged 18 to 75 years, suffering from reactive or endogenous unipolar depression according to the DSM‐III criteria, considered suitable for treatment with anti‐depressant drugs, and also having a minimum score of 17 on the first 17 items of the Hamilton rating scale for depression (HDRS‐21), were eligible for the inclusion in the study. | |
| Interventions | Paroxetine: 29 participants.
Amitriptyline: 30 participants. Paroxetine dose range: 20‐30 mg/day. Amitriptyline dose range: 100‐150 mg/day. |
|
| Outcomes | HDRS‐21, Physician's Global Assessment of Severity of Illness (PGAS). Total dropout, dropout due to side‐effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Funding: paroxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "the main analyses were carried out on the Inten‐to‐treat (ITT) population using the extender dataset ‐ a dataset that has missing values imputed by Last Observation Carried Forward (LOCF)". |
| Selective reporting (reporting bias) | High risk | Continuous outcomes data reported without Standard deviations. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Pelicier 1993.
| Methods | Five‐week, multicentre, randomised, double‐blind, double‐dummy, parallel group, comparative study. | |
| Participants | Male and female patients, aged 60 or more, with moderate reactive depression who could be treated as out‐patients were eligible for the study. Participants also had to have moderate reactive depression by Feighner's Criteria and the Zung self‐rating depressions scale. | |
| Interventions | Paroxetine: 51 participants. Clomipramine: 42 participants. Paroxetine dose: 20 mg/day. Clomipramine dose: 60 mg/day. |
|
| Outcomes | Montgomery and Asberg Depression Rating Scale (MADRS), Zung Self‐rating Depressions Scale (SDS), the Visual Analogue Scale (VAS), Global Assessment Scale (GAS). Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Funding: unclear. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "subjects were randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind, double dummy (...) placebo tablets were used to maintain blinding". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No clear information regarding the management of incomplete outcomes data. |
| Selective reporting (reporting bias) | Unclear risk | Outcomes data not clearly reported. Only most frequent adverse effects were reported. |
| Other bias | Unclear risk | Insufficient information to establish the presence of other biases. |
Perahia 2006 (HMAY B).
| Methods | Eight‐week, double‐blind, randomised multicentre study, followed by a 26‐week continuation phase for responders. | |
| Participants | Outpatients of at least 18 years of age with a primary diagnosis of major depression as defined by the DSM‐IV. Patients were required to have a Hamilton rating scale for depression (HDRS) total score ≥15 and a Clinical Global Impression (CGI) Severity total score ≥4 at both Visits 1 and 2. No patients with bipolar disorder were included. |
|
| Interventions | Duloxetine 40 mg twice daily (80 mg/day): 93 participants. Duloxetine 60 mg twice daily (120 mg/day): 103 participants. Paroxetine 20 mg once daily (20 mg/day): 97 participants. Placebo: 99 participants. |
|
| Outcomes | Efficacy: HDRS‐17 total score; HDRS‐17 response rates (a 50% reduction from baseline to endpoint), and HAMD17 remission rates (endpoint score ≤7); Montgomery and Asberg Depression Rating Scale (MADRS), CGI‐Severity, Patient Global Impression (PGI) Improvement, Hamilton rating scale for anxiety (HAM‐A), Scale for Suicidal Ideation (SSI), and Visual Analogue Scale (VAS) for pain. Total dropout, dropout due to inefficiency, dropout due to side effects, number of patients experiencing at least one side effect, side‐effect profile. |
|
| Notes | Funding: duloxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: Patients were randomly assigned in a 1:1:1:1 ratio to placebo, duloxetine 80 mg/day, duloxetine 120 mg/day or paroxetine 20 mg/day". |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The study utilized a double blind, variable duration placebo lead‐in at the beginning of the acute phase and a placebo lead out at the end of the continuation phase to minimize possible bias in the ratings of efficacy and tolerability associated with patients and investigators knowledge of the onset and conclusion of active drug therapy". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "The study utilized a double blind, variable duration placebo lead‐in at the beginning of the acute phase and a placebo lead out at the end of the continuation phase to minimize possible bias in the ratings of efficacy and tolerability associated with patients and investigators knowledge of the onset and conclusion of active drug therapy". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Study endpoint: 11/97 missing from paroxetine group; 23/196 missing from control group. |
| Selective reporting (reporting bias) | High risk | In duloxetine 120 mg/day group only adverse events with an incidence > or = 2.5% were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Peselow 1989.
| Methods | Six‐week, single‐centre, double‐blind, randomised, parallel‐group study. | |
| Participants | Only moderate to moderately severe depressed patients with a DSM‐III diagnosis of major depressive disorder without mania, characterised by disorder of mood with symptoms such as depressed mood, sadness, hopelessness, and worthlessness‐were to be admitted to the study. In addition to the DSM‐III diagnostic criteria, the participant had to: (1) be al least 18 years old and (2) have Hamilton rating scale for depression (HDRS‐21) score at the screen and baseline visits of at least 18 on the first 17 items of the 21‐item scale, and the HDRS‐21 total could not decrease by 20% or more between the screen and baseline visits, and (3) have a Raskin Depression Scale (RDS) score at baseline of at least 8, and the RDS score had to be higher than the Covi Anxiety Scale score (CAS). | |
| Interventions | Paroxetine: 40 participants. Imipramine: 39 participants. Paroxetine dose: 20‐50 mg/day. Imipramine dose: 65‐275 mg/day. |
|
| Outcomes | HDRS‐21, RDS, Clinical Global Impression (CGI) Severity, Improvement, Patients Global Evaluation (PGE). Total dropout, dropout due to side‐effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Funding: unclear. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind (...) the subject received two medication bottles and was instructed to take two capsules from the bottle labeled "morning" and one capsule from the bottle labeled "evening". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "the primary analysis of the study is based on the wee 6 evaluation from the All Efficacy Population using a Last Observation Carried Forward (LOCF) approach". More than 20% of participants in the paroxetine arm withdrew from the study prematurely. |
| Selective reporting (reporting bias) | High risk | Only most frequent adverse effects were reported. |
| Other bias | Unclear risk | Insufficient information to establish the presence of other biases. |
Ravindran 1997.
| Methods | Twelve‐week, randomised, double‐blind, multicentre, parallel‐group study. | |
| Participants | Patients with a diagnosis of depression with associated anxiety, suitable for treatment with antidepressant, a Montgomery and Asberg Depression Rating Scale (MADRS) score of at least 20 and a Covi Anxiety Score (CAS) score of at least 11 could qualify for study entry. | |
| Interventions | Paroxetine: 513 participants. Clomipramine: 503 participants. Paroxetine dose range: 20‐40 mg/day. Clomipramine dose range: 75‐150 mg/day. The only concurrent psychotropic medication permitted during active treatment was temazepam, up to 20 mg at night as a hypnotic, on an as‐needed basis. |
|
| Outcomes | MADRS, CAS, Clinical Global Impression (CGI). Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Only side effects occurring in >10% of patients. Funding: paroxetine manufacturer. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were randomly assigned on the basis of a computer‐generated schedule in which treatments were balanced within blocks of consecutive patients to receive double blind treatment with either paroxetine or clomipramine for 12 weeks". |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "endpoint data were generated from the last available on‐treatment assessment for each patient. The endpoint for each analysis was taken as the visit at which at least 70% of the Intent‐to‐treat (ITT) population (randomized patient with at least one valid on‐treatment efficacy assessment) remained". More than 20% of participants in both arms withdrew from the study prematurely. |
| Selective reporting (reporting bias) | Low risk | Outcomes data were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Sacchetti 2002.
| Methods | Twelve‐week, randomised double‐blind, double‐dummy, parallel‐group study. | |
| Participants | Adult male and female patients aged between 18 and 70 years were eligible for inclusion if they met DSM‐III‐R criteria for recurrent major depression, had a total score of 518 on the Hamilton Rating Scale for Depression (HDRS‐21) 16 and had scores of one or more for each of the HDRS core symptoms of depressed mood, suicide, insomnia, and retardation. In addition, patients had to have experienced at least one of the following: one or more episodes of affective illness per year for the previous 3 years; three or more episodes of affective illness in the previous 2 years; two or more episodes of affective illness in the previous year. Patients were excluded if they had a schizoaffective disorder; had a high risk of suicide, serious concurrent physical disease or abnormal laboratory findings; were pregnant or lactating or likely to become pregnant; were taking certain prescribed medications including oral anticoagulants, non‐steroidal anti‐inflammatory drugs, diuretics, Type 1C anti‐arrhythmics or any investigational drugs; were known to be abusing drugs or alcohol; or had a history of allergic drug reactions. Patients were excluded who had recently received other psychoactive therapy including: electroconvulsive therapy (ECT) in the previous 3 months; depot neuroleptics in the past 4 weeks; oral neuroleptics in the past 2 weeks; monoamine oxidase inhibitors in the past 2 weeks; other prophylactic antidepressant or antiepileptic drugs in the previous 2 weeks. In addition, patients stabilised on lithium therapy in the previous 3 months were excluded. |
|
| Interventions | Paroxetine: 64 participants.
Amitriptyline: 65 participants. Paroxetine dose range: 20‐50 mg/day. Amitriptyline dose range: 100‐250 mg/day. The only psychotropic medication permitted was temazepam, which could be used as a hypnotic if required. |
|
| Outcomes | HDRS, Clinical Global Impression (CGI). Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Patients taking essential non‐psychotropic medication (unless specifically excluded) which had been started at least one month before study entry were permitted to continue with it throughout,preferably at unchanged dosage. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind, double dummy". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind, double dummy". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: the Intention‐to‐treat (ITT) study endpoint assessment was assessed at the 70% Last Observation Carried Forward (LOCF) endpoint, the latest visit during the acute phase at which at least 70% of patients provided data. For the remaining patients, data were carried forward from the last visit where the relevant measure was recorded. All patients who were randomly allocated to treatment, received their treatment, and for whom at least one assessment was available after taking active treatment, were eligible for inclusion in the ITT population (...) week 5 was the last visit in the acute phase where at least 70% of subjects had efficacy data". More than 20% of participants in both arms withdrew from the study prematurely. |
| Selective reporting (reporting bias) | Low risk | Outcomes data were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
SBK‐115 1998.
| Methods | Twelve‐week, randomised, double‐blind, comparative, placebo‐controlled study. | |
| Participants | Outpatients >18 years old with moderate to moderately severe depression (DSM: single episode or recurrent). At both the screen visit and baseline the Hamilton rating scale for depression (HDRS‐21) had to be at least 18 for the HDRS‐21 first 17 items; the could not decrease by more than 25% between screen and baseline visit. The Raskin depression score had to be at least 8 at baseline and must have exceeded the Covi Anxiety Score (CAS). Key exclusion criteria were patients with a primary psychiatric diagnosis other than depression, or those with serious concomitant diseases. Patients were excluded who had a serious suicidal threat, recent ECT or with substance abuse. |
|
| Interventions | Paroxetine:287 participants. Fluoxetine: 289 participants. Paroxetine dose range: 20‐50 mg/day. Fluoxetine dose range: 20‐80 mg/day. |
|
| Outcomes | HDRS, Clinical Global Impression (CGI), the Raskin Depression score (RDS), CAS. Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Funding: paroxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "subjects were randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "analyses were performed on the Intention‐to‐treat (ITT) population, which included all subjects who were randomized to study medication. For the safety analysis, this included the total patient population. The efficacy ITT population (depression, anxiety) only included all randomized patients with at least one on‐therapy efficacy evaluation". More than 20% of participants in both arms abandoned the study prematurely. |
| Selective reporting (reporting bias) | Low risk | Outcomes data were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Schatzberg 2002.
| Methods | Eight‐week, double‐blind, randomised, comparative trial. | |
| Participants | Patients at least 65 years of age were eligible. Participant in the study were outpatients recruited through advertising, private practices, and routine intake at clinics and other healthcare facilities. At the screening visit, patients were required to satisfy DSM‐IV criteria for a single or recurrent major depressive episode, have a Mini‐Mental State Exam (MMSE) score above the 25th percentile for age and educational level, and score at least 18 on the Hamilton rating scale for depression (HDRS‐17). Patients were excluded if their HDRS score decreased by 20% or more between the screening and baseline visits. Patients with concomitant medical illnesses were eligible for the study if their condition was stable for at least 3 months and they had been receiving standard therapy for the condition for at least 1 month. | |
| Interventions | Paroxetine: 126 participants. Mirtazapine: 128 participants. Paroxetine dose range: 10‐40 mg/day (mean daily dose: 26.5 [SD 5.5]). Mirtazapine dose range: 15‐45 mg/day (mean daily dose: 25.7 [SD 6.7]). Patients were allowed to take chloral hydrate (500mg‐1000mg) or zolpidem (5mg‐10mg) as needed for sleep induction. |
|
| Outcomes | HDRS, Clinical Global Impression (CGI). Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Only side effects occurring in >10% of patients. Funding: paroxetine manufacturer. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "study drugs were supplied as identical‐appearing capsules of mirtazapine 15 mg and paroxetine 10 mg. Because active treatment would involve fewer capsules of mirtazapine than paroxetine, matching placebo capsules were supplied for use with mirtazapine, to maintain the blind". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: the primary analysis was conducted on the Intent‐to‐treat (ITT) population, which included all randomized patients who received at least one dose of study medication and at least one post treatment assessment of efficacy. we used a Last Observation Carried Forward (LOCF) analysis (i.e., missing data were replaced by the last recorded value)". |
| Selective reporting (reporting bias) | High risk | Outcomes data not clearly reported (continuous data not reported, only side effects occurring in at least 10% of participants reported). |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Schnyder 1996.
| Methods | Phase III multicentre, double‐blind, randomised, parallel‐group study. | |
| Participants | Patients were in‐ or out‐patients of either sex, aged 18 to 65 years, had been diagnosed with major depression according to the DSM‐III‐R criteria, and had a score of at least 18 on the Hamilton rating scale for depression (HDRS‐21) on the day before the start of treatment. Exclusion criteria were pregnancy or lactation period, severe co‐existing physical diseases, MAOI or neuroleptics shortly prior to the study, chronic lithium therapy, intolerance to tricyclic antidepressants, schizophrenia, dementia, or a history of ECT treatment, manic episodes or bipolar disorder. Before entering the study, all patients had to undergo a general medical examination. |
|
| Interventions | Paroxetine: 37 participants. Maprolitine: 34 participants. Paroxetine dose range: 20‐40 mg/day. Maprolitine dose range: 50‐150 mg/day. In case of sleep disturbances, the following concurrent medications were permitted: lormetazepam, dipotassium chloracepate, chloral hydrate, diazepam, flurazepam, oxazepam, bromazepam, flunitrazepam and zolpidem. |
|
| Outcomes | HDRS, Montgomery and Asberg Depression Rating Scale (MADRS), Clinical Global Impression (CGI). Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Reasons for withdrawal were reported. Funding: paroxetine manufacturer. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were randomly assigned to treatment". |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double‐blind study". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double‐blind study". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No clear information about data analysis. |
| Selective reporting (reporting bias) | High risk | Rating scales scores at follow‐up assessments were not clearly reported. The denominators (for continuous outcomes) were unclear. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Sechter 2004.
| Methods | Double‐blind, randomised controlled study conducted in 42 European centres. | |
| Participants | Outpatients meeting DSM‐IV criteria for unipolar major depression, without psychotic features, with Montgomery and Asberg Depression Rating Scale (MADRS) total score >20, aged 18–70 years, were included in the study. Patients with a significant suicide risk, a lack of response to two adequate antidepressant treatments, a history of psychotic disorder, a major personality disorder, a current primary diagnosis of panic disorder, agoraphobia, social phobia, or obsessive compulsive disorder, current alcohol or drug abuse or dependence, were excluded. | |
| Interventions | Paroxetine: 153 participants. Milnacipran: 149 participants. Patients were randomized to receive milnacipran 100 mg/day (50 mg bid) or paroxetine 20 mg/day (20 mg od) for 6 weeks. |
|
| Outcomes | Patients were evaluated at inclusion and on Day 7; 14; 28; 42; with a post‐treatment assessment 7 days after treatment discontinuation. The Hamilton rating scale for depression (HDRS‐17) total score, the MADRS total score, the Clinical Global Impression (CGI) for severity of illness and global improvement from Day 0 were determined at each visit. Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. |
|
| Notes | Only events reported more than once were included. Funding: milnacipran manufacturer. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized (...) patients were randomized". |
| Allocation concealment (selection bias) | Unclear risk | no information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "data were analyzed on an Intention‐to‐treat (ITT) basis with Last Observation Carried Forward (LOCF)". |
| Selective reporting (reporting bias) | High risk | Continuous outcomes data were reported without Standard deviations. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
SER‐CHN‐1.
| Methods | Double‐blind randomised comparative control study. | |
| Participants | Inclusion criteria: after 1 week placebo single blind wash out, only patients with Hamilton rating scale for depression (HDRS) score of 18 or more, or total score decreased less than 20% at baseline were enrolled. Male and female patients aged 18‐65 years who met the diagnostic criteria for depression on DSM‐III‐R and Chinese Classification of Mental Disease. Exlusion criteria: Patients with severe renal disease, severe cardiac disease, active thyroid disease, severe gastrointestinal or hepatic disease, unstable diabetes, Addison's disease, severe neurological diseases including Parkinsonism and organic brain pathology, rapid and cycling type of depression, epilepsy, schizophrenia or psychosis. Patients who have received ECT in the previous 12 weeks, lithium in the previous 4 weeks, oral neuroleptics in the previous 2 weeks or depot neuroleptics in the previous 4 weeks, MAOIs in the previous 2 weeks, investigational drugs in the previous 12 weeks, SSRIs in the previous 4‐5 weeks. |
|
| Interventions | Paroxetine: 113 participants.
Amitriptyline: 118 participants. Paroxetine dose range: 20‐30 mg/day. Amitriptyline dose: 150 mg/day. |
|
| Outcomes | HDRS, Hamilton rating scale for anxiety (HAM‐A), Clinical Global Impression (CGI). Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Funding: paroxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized study". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Intention‐to‐treat (ITT) population consisted of all subjects who received treatment and had one post treatment evaluation". Less than 20% of participants in both arms withdrew from the study prematurely. |
| Selective reporting (reporting bias) | High risk | Outcomes data not clearly reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Shillingford 1990.
| Methods | Six‐week, randomised, double‐blind study. | |
| Participants | Patients with major depressive episode according to DSM III criteria, in general practice setting. | |
| Interventions | Paroxetine versus dothiepin. | |
| Outcomes | Efficacy and tolerability. | |
| Notes | None. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information on incomplete outcome data management. |
| Selective reporting (reporting bias) | Unclear risk | No information provided. |
| Other bias | Unclear risk | Insufficient information to establish the presence of other biases. |
Shinkai 2004.
| Methods | Four‐week, randomised, double‐blind study. | |
| Participants | Inpatients with DSM‐IV major depressive disorder without psychotic features with a minimum score of 15 on the Hamilton rating scale for depression (HDRS). Age range: 20‐78 years. | |
| Interventions | Paroxetine: 21 participants. Milnacipram: 20 participants. Paroxetine mean dose: 34.28 mg. Milnacipran mean dose: 80.25 mg. No other drugs were administered throughout the study with the exception of a few patients who received benzodiazepines for insomnia. |
|
| Outcomes | HDRS‐17. | |
| Notes | Funding: independent from industry. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomly divided into either milnacipran or the paroxetine group using StatView, a computerized statistical package" |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Study endpoint: 1/21 missing from paroxetine group, 0/20 missing from control group. |
| Selective reporting (reporting bias) | High risk | Adverse events were not reported so that could not be entered in to the meta‐analysis. |
| Other bias | Low risk | This study was independent from pharmaceutical industry. |
Shrivastava 1992.
| Methods | Six‐week, randomised, double‐blind study. | |
| Participants | Outpatients of at least 18 years of age with a primary diagnosis of major depression as defined by the DSM‐III. Patients were required to have a Hamilton rating scale for depression (HDRS‐17) total score ≥18. | |
| Interventions | Paroxetine: 40 participants. Imipramine: 40 participants. Placebo: 40 participants. Paroxetine dose range: 10‐50 mg/day. Imipramine dose range: 65‐275 mg/day. 500‐mg dose of chloral hydrate was permitted for treatment of insomnia. |
|
| Outcomes | HDRS‐21. Total dropout, dropout due to inefficiency, dropout due to side effects, number of patients experiencing at least one side effect, side‐effect profile. |
|
| Notes | Funding: paroxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "After randomization to double blind treatment at a baseline visit, the subject received two medication bottles and was instructed to take two capsules from the bottle labeled "morning" (each capsule 10 mg paroxetine, 15 mg imipramine or placebo) and one from the bottle labeled "evening" (each capsule 50 mg imipramine or placebo)". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Study endpoint: "16/40 missing from Paroxetine group, 30/40 missing from Imipramine group". |
| Selective reporting (reporting bias) | High risk | Response and remission rates not reported. On the published paper adverse events reported only if occurring with frequency > or = 20%. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Staner 1995.
| Methods | Four‐week, double‐blind, parallel‐group trial. | |
| Participants | Inclusion criteria: RDC major depression, Hamilton rating scale for depression (HDRS‐21) of 18+.
Exclusion criteria: pregnant or lactating women; patients with severe concomitant disease; schizophrenics, known abusers of alcohol or illicit drugs; those who had received ECT in the previous 3 months, MAOIs or oral neuroleptics in the previous 14 days and/or depot neuroleptics in the previous 4 weeks. Age: 18‐65 years. Country: Belgium. Setting: inpatients. |
|
| Interventions | Paroxetine: 21 participants.
Amitriptyline: 19 participants. Paroxetine dose range: 20‐30 mg/day. Amitriptyline dose range: 100‐150 mg/day. |
|
| Outcomes | HDRS. Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Only the most common side effects were reported. Funding: paroxetine manufacturer. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "double blind". The evaluation of sleep (sleep EEG changes during antidepressant treatment was the primary outcome) consisted in sleep recording. Records were scored blind to group membership (paroxetine and amitriptyline). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "for the evaluation of the antidepressant effect, the Intention‐to‐treat (ITT) population extended or Last Observation Carried Forward (LOCF) dataset was used". |
| Selective reporting (reporting bias) | Low risk | Outcomes data were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Steinmeyer 1992.
| Methods | Six‐week, double‐blind, randomised study. | |
| Participants | The inclusion criteria were: a diagnosis of a major depressive disorder according to DSM‐III, a score of 18 or more on the Hamilton rating scale for depression (HDRS‐21) and the status of inpatients. All patients with an organic illness, a psychosis, psychosomatic disorder, addiction, or severe suicidal tendencies had to be excluded. |
|
| Interventions | Paroxetine: 112 participants Amitriptyline: 110 participants Paroxetine dose: 30 mg/day Amitriptyline dose: 150 mg/day Additional psychopharmacological treatment was not allowed, apart from temazepam in case of sleep disturbance. |
|
| Outcomes | The main efficacy measures were: HDRS, Clinical Global Impression (CGI). In addition, the subfactors of the HDRS were taken into consideration. Safety and tolerance were assessed from spontaneous adverse‐event reports, a standardised symptom checklist, laboratory data, physical examination, and vital signs". Total dropout, dropout due to side effects, dropout due to inefficacy. | |
| Notes | Funding: independent from industry. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were randomly allocated". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No clear information on incomplete outcome data management. |
| Selective reporting (reporting bias) | High risk | Only side effects experienced by more than 10% of participants reported (only in figure). |
| Other bias | Low risk | This study was independent from pharmaceutical industry. |
Stott 1993.
| Methods | Eight‐week, double‐blind study. | |
| Participants | Inclusion criteria: patients with depression and anxiety, Montgomery and Asberg Depression Rating Scale (MADRS) 16+, Covi Anxiety Score (CAS) 11+. Age range: 18‐65 years. Country: UK. Setting: family practice. | |
| Interventions | Paroxetine: 243 participants. Amitriptyline: 262 participants. Paroxetine: 20 mg/day. Amitriptyline: 25‐75 mg/day. |
|
| Outcomes | MADRS, CAS, adverse events. | |
| Notes | Outcomes data not available. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote "patients (...) were evaluated on an Intention‐to‐treat basis". |
| Selective reporting (reporting bias) | High risk | Outcomes data not available. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Stuppaeck 1994.
| Methods | Six‐week, randomised, double‐blind study. | |
| Participants | Inclusion criteria: DSM III major depression, melancholic subtype, Hamilton rating scale for depression (HDRS) 18+ Exclusion criteria: senile dementia (DSM‐III) alcohol or drug addiction, patients with a high risk for suicide, or ECT during the last 3 months, as well as relevant somatic diseases. Patients were also excluded if they received long‐term treatment with lithium. Age: 18‐65 years. Country: Austria, Germany. Setting: inpatients. |
|
| Interventions | Paroxetine: 78 participants.
Amitriptyline: 75 participants. Paroxetine dose range: 20‐50 mg/day (mean daily dose: 33.3). Amitriptyline dose range: 50‐250 mg/day (mean daily dose: 166). |
|
| Outcomes | Montgomery and Asberg Depression Rating Scale (MADRS), Clinical Global Impression (CGI). Total dropout. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Funding: paroxetine manufacturer. quality rating: 21 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: patients were randomly allocated". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "double blind (...) paroxetine was administered as a single daily dose in the morning, whereas amitriptyline was given each morning, noon, and evening. Patients in the paroxetine group received matching placebo tablets at noon and in the evening". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "analyses were performed on the Intention‐to‐treat (ITT) population with the Last Observation Carried Forward (LOCF) and visit wise datasets". More than 20% of participants in both arms abandoned the study prematurely. |
| Selective reporting (reporting bias) | High risk | Only most frequent adverse effects reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Szegedi 1995.
| Methods | Randomised, multicentre, parallel‐group, double‐ blind clinical trial in depressed out‐patients. | |
| Participants | Age range: 18–71 years. Exclusion criteria were requirement of hospitalisation, presence of psychotic symptoms, acute risk of suicidality or severe existing physical disease, pregnancy or lactation, or the need of additional benzodiazepine treatment. |
|
| Interventions | Paroxetine: 145 participants. Maprolitine: 153 participants. Paroxetine dose range: 20‐40 mg/day. Maprolitine dose range: 100‐150 mg/day. |
|
| Outcomes | Hamilton rating scale for depression (HDRS), Clinical Global Impression (CGI), Montgomery and Asberg Depression Rating Scale (MADRS), Hamilton rating scale for anxiety (HAM‐A), Bech‐Rafaelsen Mania Scale (BRMS), Raskin Depression score (RDS), Covi Anxiety Score (CAS). | |
| Notes | Funding: unclear. The study population includes subjects with diagnoses of major and minor depression. According to the protocol of this review, only data referring to patients with major depression were considered. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized". No further details. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind (...) a double dummy design was used". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind (...) a double dummy design was used". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "for all patients receiving active medication an Intention‐to‐treat (ITT) analysis was performed using the Last Observation Carried Forward (LOCF) method". |
| Selective reporting (reporting bias) | High risk | Only most often experienced adverse effects reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Szegedi 2005.
| Methods | Double‐blind, double‐dummy, randomised, phase III trial. | |
| Participants | All participants were 18‐70 years old and had single or recurrent moderate or severe episodes of unipolar major depression without psychotic features (DSM‐IV) persisting for two weeks to a year. At screening and baseline all participants had to have a total score ≥ 22 points on the Hamilton rating scale for depression (HDRS‐17) and ≥ 2 points for the item “depressive mood.” The diagnosis of depression was based on the mini‐international neuropsychiatric interview. There were no restrictions regarding ethnic group. We excluded anyone with a decrease in total depression score of ≥ 25% during the run‐in, or with a diagnosis of schizophrenia, acute anxiety disorder, adjustment disorder, depressive disorder of any type not stated above, bipolar disorder, organic mental disorder, acute post‐traumatic stress disorder, or substance abuse disorder. We also excluded patients with increased risk of suicide (defined by a score ≥ 4 for item 10 of the Montgomery and Asberg Depression Rating Scale (MADRS), who had previously attempted suicide, or who had not responded to more than one adequate treatment (equivalent to 150 mg/day amitriptyline for ≥ 6 weeks) in the present episode. | |
| Interventions | Paroxetine: 126 participants. Hypericum: 125 participants. Paroxetine dose range: 20‐40 mg/day. Hypericum dose range: 15‐45 mg/day. |
|
| Outcomes | HDRS, MADRS, Clinical Global Impression (CGI). Total dropout, dropout due to side‐effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Funding: hypericum manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomized (...) randomization was performed in blocks stratified by trial centre. A biometrician otherwise not involved in the trial generated the code using a validated computer program. The study drugs were dispensed to the centres in numbered containers". |
| Allocation concealment (selection bias) | Low risk | Quote: "On inclusion of a patient into randomized treatment the local investigator allocated each participants the lowest available number. The block size was withheld from the investigators". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "double blind, double dummy (...) coated tables; high and low dose tablets were indistinguishable in all aspects of their outward appearance. For each drug an identically matched placebo was available (the success of blinding was evaluated by examining the drugs before distribution). During the six weeks randomized treatment patients allocated to hypericum always took three coated tablets of hypericum/day plus one paroxetine placebo capsule in the morning whereas those in the paroxetine group took one capsule of paroxetine in the morning and three coated tables of hypericum placebo/day". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind, double dummy". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "the primary analysis was based on the Intention‐to‐treat (ITT) analysis to mirror clinical practice. we also performed as per protocol (PP) analysis to demonstrate robustness of the trial result to the choice of the analysis set". More than 20% of participants in the paroxetine arm abandoned the study prematurely. |
| Selective reporting (reporting bias) | High risk | Only adverse effects that occurred in at least 10 patients in one group reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Tignol 1993.
| Methods | Multicentre, randomised, double‐blind, double‐dummy, parallel group, comparative study | |
| Participants | Inclusion criteria: patients meeting criteria for Major depression (DSM‐III); patients with Montgomery and Asberg Depression Rating Scale (MADRS) score of >24. Age range 18‐65 years. Exclusion criteria: subjects with severe risk of suicide; patients who were schizophrenic or psychotic; patients who had received 1 or more specified psychiatric drugs prior to the study. |
|
| Interventions | Paroxetine: 89 participants. Fluoxetine: 87 participants. Paroxetine dose: 20 mg/day. Fluoxetine dose: 20 mg/day. |
|
| Outcomes | MADRS, Clinical Global Impression (CGI), Hamilton rating scale for anxiety (HAM‐A), Hospital Anxiety and Depression (HAD).Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Funding: paroxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "eligible patients were randomized". |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "the Intent‐to‐treat (ITT) population comprised all subjects who received at least one dose of randomized active treatment and who had at least one post‐treatment assessment". More than 20% of participants in both arms abandoned the study prematurely. |
| Selective reporting (reporting bias) | Unclear risk | Only endpoint outcomes data were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Wade 2003.
| Methods | Twenty‐four‐week, prospective, multicentre, randomised, double‐blind study. | |
| Participants | Male and female patients who were at least 18 years old, fulfilling DSM‐IV criteria for a single or recurrent depressive episode according to the DSM‐IV checklist, with a total baseline score > 18 on the Hamilton rating scale for depression (HDRS‐17) and who provided their written informed consent were eligible for participation in the study. Patients were excluded if they were suffering from schizophrenia or bipolar affective disorder, were suicidal, or were diagnosed with current illicit drugs abuse or alcohol dependence. Treatment with any other psychotropic drug, including any anxiolytic or hypnotic within 1 week before entry to the study, with mirtazapine or paroxetine during the present episode or hypersensitivity to either compound, with fluoxetine within 5 weeks, or with any other antidepressant within 2 weeks of entry, prohibited inclusion. In addition, patients of both sexes were not eligible if they had any clinically meaningful renal, hepatic, respiratory, cardiovascular or cerebrovascular disease. Pregnant or lactating women and those of childbearing age not employing any adequate contraception were also excluded. | |
| Interventions | Paroxetine: 98 participants. Mirtazapine: 99 participants. Paroxetine dose range: 20‐30 mg/day (mean daily dose: 23.9 [SD 3.96]). Mirtazapine dose range: 30‐45 mg/day (mean daily dose: 34.6 [SD 5.7]). Use of any psychotropic concomitant drug, including anxiolytics (benzodiazepines) and hypnotics was prohibited during the study. |
|
| Outcomes | HDRS‐17, Clinical Global Impression (CGI).Total dropout, dropout due to side effects, dropout due to inefficacy. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Only side effects occurring in >5% of patients in either groups. Funding: mirtazapine manufacturer. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomization was performed according to centrally prepared randomization lists". |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "double blind oral treatment with either drug, prepared as indistinguishable‐looking capsules containing 15 mg mirtazapine or 10 mg paroxetine". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "efficacy analyses were based on the Intention‐to‐treat (ITT) sample, thus including all randomly assigned subjects who received at least one dose of blinded study medication, and who had at least one post‐dose efficacy assessment. The primary inferences concerning efficacy were made using the Last Observation Carried Forward (LOCF) method at endpoint. Additional Observed Case (OC) analysis was used at the week 24 assessment to ensure robustness of the LOCF results and at other visits (for the primary efficacy variable only) to determine effect of treatment. Tolerability assessments were performed on All Subjects Treated (AST) group, consisting of all randomized patients who took at least one dose of blinded medication". More than 20% of participants in both arms abandoned the study prematurely. |
| Selective reporting (reporting bias) | High risk | Outcomes data not clearly reported. Only side effects experienced by > or = 5% of participants were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Waintraub 2002.
| Methods | Three‐month controlled, randomised, double‐blind, clinical trial. | |
| Participants | Male and female patients aged 18 to 60 years who met, according to the investigator, the DSM‐IV criteria for a major depressive episode of moderate or severe intensity were included. Patients had to have a total Montgomery and Asberg Depression Rating Scale (MADRS) score ≥20, a total Hamilton rating scale for depression (HDRS) score ≥18, a MADRS item 10 score (suicidal thoughts) ≤2, and a HDRS item 3 score (suicide) ≤1. Patients with significant physical illness or epilepsy, or who were pregnant or drug abusers were excluded, as were patients who were currently being treated with barbiturates, thymoregulators, antipsychotics, electroconvulsive therapy, or other antidepressant treatments during 7 days prior to the start of the trial. Other exclusion criteria were anxiety disorders, acute or chronic psychoses, or neurodegenerative diseases. | |
| Interventions | Paroxetine: 139 participants.
Tianeptine: 138 participants. Paroxetine dose range: 20‐40 mg/day. Tianeptine dose range: 37,5‐75 mg/day. If clinically required, bromazepam (maximal dosage 9 mg/day) and zopiclone (maximal dosage 7.5 mg/day) could be prescribed. |
|
| Outcomes | MADRS, HDRS, Clinical Global Impression (CGI). Total dropout. Number of patients experiencing at least one side effect, side‐effect profile. | |
| Notes | Funding: tianeptine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were randomly allocated". |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind conditions". No further details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind conditions". No further details. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "the efficacy analysis was performed both on the full analysis set (FAS), which is a 'complete as possible' and a 'close as possible' to the Intention‐to‐treat (ITT) ideal of including all randomized subjects, and on the per protocol (PP) populations. The safety analysis was performed on all patients who received at least one dose of the study drug (...) Seven patients with no data inclusion visit were excluded from the randomized population". Dropout not clearly reported. |
| Selective reporting (reporting bias) | High risk | Dropout not clearly reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Weihs 2000.
| Methods | Six‐week randomised, double‐blind, double‐dummy, parallel‐group, active‐treatment phase. | |
| Participants | Men and women 60 years of age and older with a minimum baseline score of 18 on theHamilton rating scale for depression (HDRS‐21) who presented with a recurrent episode of nonpsychotic major depressive disorder (DSM‐IV) with a duration of at least 8 weeks, but not more than 24 months, and who were considered clinically appropriate for treatment with either bupropion SR or paroxetine were eligible for the study. Patients who had a known predisposition to seizures were excluded from the study. Patients taking medications or treatments that lower the seizure threshold were also excluded. Patients were excluded if they were actively suicidal, had a history or current diagnosis of anorexia nervosa or bulimia nervosa, had an unstable medical disorder, or had a history of non‐responsiveness to pharmacotherapy for depression. Patients were also excluded if they had a history of alcohol or substance abuse within 1 year prior to the study or myocardial infarction, uncontrolled hypertension, or unstable heart disease within 6 months prior to the study. | |
| Interventions | Paroxetine: 52 participants.
Bupropion: 48 participants. Paroxetine dose range: 10‐40 mg/day (mean daily dose: 22 [SD 7]). Bupropion: dose range: 100‐300 mg/day.(mean daily dose: 197 [SD 53]). |
|
| Outcomes | HDRS, Clinical Global Impression (CGI), Hamilton rating scale for anxiety (HAM‐A). Total dropout, dropout due to side effects. Side‐effect profile. | |
| Notes | Funding: paroxetine manufacturer. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Of the 100 patients enrolled in the study, 48 were randomly assigned to treatment with bupropion SR and 52 were randomly assigned to treatment with paroxetine." |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind, double dummy". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "double blind, double dummy". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "all patients who received at least one dose of study medication and completed one or more treatment‐phase efficacy assessments beyond baseline were included in the efficacy analyses (Intent‐to‐treat ITT population). Both Observed and Last Observation Carried Forward (LOCF) scores were analyzed". Less than 20% of participants in both arms abandoned the study prematurely. |
| Selective reporting (reporting bias) | Low risk | Outcomes data were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Yoshimura 2007.
| Methods | Eight‐week, randomised study. | |
| Participants | In‐ or out‐patients who met the DSM‐IV‐TR criteria for major depressive disorder (MDD) without psychotic features and who scored at least 16 on the Hamilton Rating Scale for Depression (HDRS). Fifteen patients were male and 27 were female. The age of the participants ranged from 28 to 74 years old. | |
| Interventions | Paroxetine: 21 participants. Milnacipran: 21 participants. Paroxetine dose range: 10‐40 mg/day (mean: 31; SD: 13). Milnacipran dose range: 25‐150 mg/day (mean 83; SD: 31). |
|
| Outcomes | Serum brain‐derived neurotrophic factor (BDNF); HDRS. | |
| Notes | Sponsorship bias cannot be ruled out. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "the patients were randomly divided into either a paroxetine group or a milnacipran group using StatView, a computerized statistical package". |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information provided. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information on incomplete outcome data management. |
| Selective reporting (reporting bias) | Low risk | Outcomes data were reported. |
| Other bias | Unclear risk | Sponsorship bias cannot be ruled out. |
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Arminen 1992 | More than 20% of bipolars included in this study. |
| Benkert 1997 | Not a relevant diagnostic status and study design. |
| Bird 2000 | Not a relevant diagnostic status (comorbidity with other disorders). |
| Cocchi 1997 | Not a relevant diagnostic status (comorbidity with alcohol dependence). |
| Ferrando 1997 | Not a relevant diagnostic status (comorbidity with other disorders). |
| Gulseren 2005 | Not a relevant diagnostic status (comorbidity with other disorders). |
| Hazleman 1997 | Not a relevant diagnostic status (comorbidity with other disorders). |
| Katona 1998 | Not a relevant diagnostic status (comorbidity with dementia). |
| Poirier 1997 | Patients with treatment resistant depression. |
| Rapaport 2003 | Wrong design (no active comparator). |
| Sacchetti 1997 | Patients with treatment resistant depression. |
| SAD‐PD 2004 | Not a relevant diagnostic status (comorbidity with other disorders). |
| Zanardi 1996 | More than 20% of people with bipolar included in this study. |
Characteristics of studies awaiting assessment [ordered by study ID]
29060/III/83/12.
| Methods | Six‐week double‐blind, double‐dummy, randomised study. |
| Participants | Paroxetine: 19 participants. Mianserin: 16 participants. Placebo: 10 participants. |
| Interventions | Paroxetine: 30 mg/day. Mianserin: 30‐90 mg/day. Placebo. |
| Outcomes | Hamilton rating scale for depression (HDRS), Global Assessment and Final Assessment of efficacy and tolerance. |
| Notes | Funding: probably by industry. |
Ballus 1997.
| Methods | Twenty‐four‐week double‐blind, randomised study. |
| Participants | Paroxetine: 43 participants. Venlafaxine: 41 participants. |
| Interventions | Paroxetine: 20‐40 mg/day. Venlafaxine: 75‐150 mg/day. |
| Outcomes | Hamilton rating scale for depression (HDRS), Clinical Global Impression (CGI), Montgomery and Asberg Depression Rating Scale (MADRS). |
| Notes | Unclear diagnosis. |
Benattia 2004.
| Methods | Randomised, double‐blind, multicentre study. |
| Participants | Paroxetine: 39 participants. Mianserin: 34 participants. |
| Interventions | Paroxetine: 30 mg/day. Mianserin: 60 mg/day. |
| Outcomes | Hamilton rating scale for depression (HDRS), dropout. |
| Notes | The number of bipolar participants was unclear. |
Blackwell 1967.
| Methods | Unclear. |
| Participants | Unclear. |
| Interventions | Psychotropic drugs. |
| Outcomes | Unclear. |
| Notes | No abstract available. |
Chen 2001.
| Methods | Controlled study. Unclear design. |
| Participants | Patients with depression. |
| Interventions | Venlafaxine and paroxetine. |
| Outcomes | Unclear. |
| Notes | Unclear study design; waiting for translation from Chinese to English. |
Deuschle 2003.
| Methods | Double‐blind study. |
| Participants | A total of 126 adult inpatients were included in this study. |
| Interventions | Paroxetine: 40 mg/day. Amitriptyline: 150 mg/day. |
| Outcomes | Saliva cortisol concentrations, Hamilton rating scale for depression (HDRS). |
| Notes | Unclear study design. |
Feng 2005.
| Methods | Comparative study. Unclear design. |
| Participants | Patients with depression. |
| Interventions | Paroxetine and clomipramine. |
| Outcomes | Unclear outcomes. |
| Notes | Waiting for translation from Chinese to English. |
Geiger 1998.
| Methods | Randomised, open‐label, multicentre trial. |
| Participants | Paroxetine: 176 participants. Fluoxetine: 322 participants. Nortriptyline: 79 participants. |
| Interventions | Paroxetine: 20‐30 mg/day. Fluoxetine: unclear. Nortriptyline: unclear. |
| Outcomes | Hamilton rating scale for depression (HDRS), Clinical Global Impression (CGI). |
| Notes | Funding: unclear. Waiting for data from study authors. |
Gonul 1999.
| Methods | Randomised, single blind study. |
| Participants | Fluoxetine: number of randomised unclear. Paroxetine: number of randomised unclear. Setraline: number of randomised unclear. Flovoxamine: number of randomised unclear. |
| Interventions | Fluoxetine: 20 mg/day. Paroxetine: 20 mg/day. Setraline: 50 mg/day. Flovoxamine: 150 mg/day. |
| Outcomes | Hamilton rating scale for depression (HDRS), dropout. |
| Notes | Funding: unclear. |
Gou 2002.
| Methods | A clinical control study. Unclear design. |
| Participants | Patients with depression. |
| Interventions | Paroxetine and venlafaxine. |
| Outcomes | Unclear. |
| Notes | Waiting for translation from Chinese to English. |
Guan 2001.
| Methods | Unclear study design. |
| Participants | Patients with depression. |
| Interventions | Sertraline, fluoxetine and paroxetine. |
| Outcomes | Quality of life and cost of treatment. |
| Notes | Waiting for translation from Chinese to English. |
Gurovich 1997.
| Methods | Six‐week, parallel group study. |
| Participants | Toatal number of participants: 150 outpatients. |
| Interventions | Amitriptyline; Fluoxetine; Paroxetine; Sertraline; Tianeptine. |
| Outcomes | Hamilton rating scale for depression (HDRS). |
| Notes | Unclear study design. |
Han 2004.
| Methods | A controlled study. Unclear design. |
| Participants | Patients with depression. |
| Interventions | Trazodone and paroxetine. |
| Outcomes | Unclear. |
| Notes | Waiting for translation from Chinese to English. |
Hang 2005.
| Methods | A comparative study. |
| Participants | Patients with depression. |
| Interventions | Mirtazapine and paroxetine. |
| Outcomes | Unclear. |
| Notes | Waiting for translation from Chinese to English. |
Henning 2002.
| Methods | Parallel group study. |
| Participants | Paroxetine: 23 participants. Amitriptyline: 17 participants. |
| Interventions | Paroxetine: 40 mg/day. Amitriptyline: 150 mg/day. |
| Outcomes | Peripheral Nervous System functions. |
| Notes | Unclear study design. |
Huang 2006.
| Methods | Control study. Unclear design. |
| Participants | Patients with depression. |
| Interventions | Paroxetine and imipramine. |
| Outcomes | Unclear. |
| Notes | Waiting for translation from Chinese to English. |
Jakitowicz 2000.
| Methods | Parallel group study. |
| Participants | Paroxetine: 14 participants. Tianeptine: 16 participants. |
| Interventions | Paroxetine: 20‐50 mg/day. Tianeptine: 37.5 mg/day. |
| Outcomes | EEG profile, Clinical Global Impression (CGI). |
| Notes | Funding: unclear. |
Lemoine 1998.
| Methods | Eight‐week, randomised, double‐blind study. |
| Participants | Toatal number of participants: 173 outpatients. |
| Interventions | Nefazodone, paroxetine. |
| Outcomes | Hamilton rating scale for depression (HDRS), Clinical Global Impression (CGI) ‐ Improvement. |
| Notes | Funding: unclear. Waiting for data from study authors. |
Li 2001.
| Methods | A contrast study. Unclear design. |
| Participants | Patients with depression. |
| Interventions | Paroxetine and amitriptyline. |
| Outcomes | Unclear. |
| Notes | Waiting for translation from Chinese to English. |
Li 2003.
| Methods | A comparative study. Unclear design. |
| Participants | Patients with depression. |
| Interventions | Paroxetine and amitriptyline. |
| Outcomes | Unclear. |
| Notes | Waiting for translation from Chinese to English. |
Li 2004.
| Methods | Eight‐weeks, randomised study. |
| Participants | Patients with depression. |
| Interventions | Paroxetine and venlafaxine. |
| Outcomes | Efficacy (HDRS) and side effects. |
| Notes | Waiting for translation from Chinese to English. |
Li 2006.
| Methods | A comparative study. Unclear design. |
| Participants | Patients with depression. |
| Interventions | Citalopram and paroxetine. |
| Outcomes | Unclear. |
| Notes | Waiting for translation from Chinese to English. |
Liao 2002.
| Methods | Unclear design. |
| Participants | Patients with depression. |
| Interventions | Fluoxetine, paroxetine, and venlafaxine. |
| Outcomes | Pharmcy‐economic evaluation. |
| Notes | Waiting for translation from Chinese to English. |
Liu 2002.
| Methods | A comparative study. Unclear design. |
| Participants | Patients with senile melancholia. |
| Interventions | Paroxetine and amitriptyline. |
| Outcomes | Unclear. |
| Notes | Waiting for translation from Chinese to English. |
Liu 2005.
| Methods | A comparative study. Unclear design. |
| Participants | Patients with depression. |
| Interventions | Venlafaxine and paroxetine. |
| Outcomes | Unclear. |
| Notes | Waiting for translation from Chinese to English. |
Ma 2000.
| Methods | Clinical comparative study. Unclear design. |
| Participants | Patients with depression. |
| Interventions | Study on paroxetine (8 comparators unclear). |
| Outcomes | Unclear. |
| Notes | Waiting for translation from Chinese to English. |
Mertens 1988.
| Methods | Randomised, open‐label study. |
| Participants | Outpatients with major depression. |
| Interventions | Paroxetine, fluoxetine, venlafaxine, citalopram, sertraline. |
| Outcomes | Hamilton rating scale for depression (HDRS). |
| Notes | Pooled data. |
Montoya 1998.
| Methods | Six‐week, randomised, double‐blind study. |
| Participants | Toatal number of participants: 38 outpatients. |
| Interventions | Imipramine, paroxetine. |
| Outcomes | Sexual functioning, dropout. |
| Notes | Funding: unclear. Waiting for data from study authors. |
Peng 2004a.
| Methods | A comparative study. Unclear design. |
| Participants | Depressed patients. |
| Interventions | Mirtazapine and paroxetine. |
| Outcomes | Unclear. |
| Notes | Waiting for translation from Chinese to English. |
Peng 2004b.
| Methods | A comparative study. Unclear design. |
| Participants | Depressed patients. |
| Interventions | Tianeptine and paroxetine. |
| Outcomes | Unclear. |
| Notes | Waiting for translation from Chinese to English. |
Qiao 2005.
| Methods | A comparative study. Unclear design. |
| Participants | Depressed patients. |
| Interventions | Citalopram and paroxetine. |
| Outcomes | Unclear. |
| Notes | Waiting for translation from Chinese to English. |
R228060 2004.
| Methods | Six‐week, randomised, double‐blind study. |
| Participants | Toatal number of participants: 688 outpatients. |
| Interventions | R228060, paroxetine, placebo. |
| Outcomes | Unclear. |
| Notes | Funding: by industry. |
Ren 2004.
| Methods | A comparative study. Unclear design. |
| Participants | Depressed patients. |
| Interventions | Mirtazapine and paroxetine. |
| Outcomes | Unclear. |
| Notes | Waiting for translation from Chinese to English. |
Salzman 1993.
| Methods | Thirteen‐week, parallel group study. |
| Participants | Outpatients with major depression. |
| Interventions | Fluoxetine, paroxetine, placebo. |
| Outcomes | Prolactin levels, tryptophan levels, neutral amino acids levels. |
| Notes | Unclear study design. |
Serretti 2001.
| Methods | Six‐week, double‐blind study. |
| Participants | Patients with major and bipolar depression. |
| Interventions | Paroxetine, fluvoxamine and either placebo or pindolol. |
| Outcomes | Hamilton rating scale for depression (HDRS). |
| Notes | The number of bipolar patients included in this study was unclear. |
Shu 2004.
| Methods | Control study. Unclear design. |
| Participants | Elderly patients with depression. |
| Interventions | Paroxetine and mianserin. |
| Outcomes | Unclear. |
| Notes | Waiting for translation from Chinese to English. |
Song 2004.
| Methods | A comparative study. Unclear design. |
| Participants | Elderly patients with depression. |
| Interventions | Paroxetine and amitriptyline. |
| Outcomes | Unclear. |
| Notes | Waiting for translation from Chinese to English. |
Starmer 1996.
| Methods | Four‐week, randomised, double‐blind study. |
| Participants | Patients with depression in general practice. |
| Interventions | Paroxetine, amitriptyline. |
| Outcomes | To assess the effect of paroxetine and amitriptyline on driving and psychomotor performance. |
| Notes | Funding: unclear. |
Su 2005.
| Methods | Comparative study. Unclear design. |
| Participants | Patients with depression. |
| Interventions | Mirtazapine and paroxetine. |
| Outcomes | Unclear. |
| Notes | Waiting for translation from Chinese to English. |
Sun 2001.
| Methods | Comparative study. Unclear design. |
| Participants | Patients with depression. |
| Interventions | Paroxetine and amitriptyline. |
| Outcomes | Unclear. |
| Notes | Waiting for translation from Chinese to English. |
Tao 2005.
| Methods | Comparative study. Unclear design. |
| Participants | Patients with depression. |
| Interventions | Citalopram and paroxetine. |
| Outcomes | Unclear. |
| Notes | Waiting for translation from Chinese to English. |
Tao 2006.
| Methods | Comparative study. Unclear design. |
| Participants | Patients with depression. |
| Interventions | Tianeptine and paroxetine. |
| Outcomes | Unclear. |
| Notes | Waiting for translation from Chinese to English. |
Wang 2003.
| Methods | Unclear. |
| Participants | Patients with depressive disorder. |
| Interventions | Fluoxetine, paroxetine and venlafaxine. |
| Outcomes | Unclear. |
| Notes | Waiting for translation from Chinese to English. |
Wang 2004.
| Methods | Unclear. |
| Participants | Patients with depressive disorder. |
| Interventions | Paroxetine and tricyclic antidepressants. |
| Outcomes | Unclear. |
| Notes | Waiting for translation from Chinese to English. |
Wang 2005.
| Methods | Unclear. |
| Participants | Patients with depressive disorder. |
| Interventions | Paroxetine (unclear comparators). |
| Outcomes | Unclear. |
| Notes | Waiting for translation from Chinese to English. |
Wang 2007.
| Methods | Unclear. |
| Participants | Patients with depressive disorder. |
| Interventions | Participants assigned to fluoxetine or paroxetine. |
| Outcomes | Quality of life. |
| Notes | Waiting for translation from Chinese to English. |
Wieck 2001.
| Methods | Randomised, double‐blind study. |
| Participants | Patients with major depression. |
| Interventions | Paroxetine, reboxetine. |
| Outcomes | Hamilton rating scale for depression (HDRS). |
| Notes | Funding: unclear. |
Wu 2000.
| Methods | Double‐blind, randomised study. |
| Participants | Patients with major depressive disorder. |
| Interventions | Paroxetine and doxepine. |
| Outcomes | Unclear. |
| Notes | Waiting for translation from Chinese to English. |
Wu 2007.
| Methods | Twenty‐four‐week, open‐label study. |
| Participants | Patients with major depressive disorder. |
| Interventions | Venlafaxine and paroxetine. |
| Outcomes | HDRS scores, remission rate at endpoint. |
| Notes | Waiting for translation from Chinese to English. |
Xie 1999.
| Methods | A randomised double‐blind ‐controlled study. |
| Participants | Patients with major depressive disorder. |
| Interventions | Paroxetine and amitriptyline. |
| Outcomes | Unclear. |
| Notes | Waiting for translation from Chinese to English. |
Yang 1998.
| Methods | Double‐blind, randomised study. |
| Participants | Toatal number of participants: 30 outpatients. |
| Interventions | Paroxetine, amitriptyline, fluoxetine. |
| Outcomes | Hamilton rating scale for depression (HDRS). |
| Notes | Funding: unclear. |
Yang 2004.
| Methods | Unclear design. |
| Participants | Patients with depression. |
| Interventions | Paroxetine and amitriptyline. |
| Outcomes | Adverse reactions, Hamilton rating scale for depression (HDRS). |
| Notes | Waiting for translation from Chinese to English. |
Yang 2006.
| Methods | Comparative study. Unclear design. |
| Participants | Patients with depression. |
| Interventions | Reboxetine and paroxetine. |
| Outcomes | Unclear. |
| Notes | Waiting for translation from Chinese to English. |
Ye 2002.
| Methods | A clinical control study. Unclear design. |
| Participants | Outpatients with depression. |
| Interventions | Paroxetine and venlafaxine. |
| Outcomes | Unclear. |
| Notes | Waiting for translation from Chinese to English. |
Zhang 2003.
| Methods | Comparative study. Unclear design. |
| Participants | Patients with depression. |
| Interventions | Venlafaxine extended release versus paroxetine. |
| Outcomes | Unclear. |
| Notes | Waiting for translation from Chinese to English. |
Zhou 2005.
| Methods | Comparative study. Unclear design. |
| Participants | Patients with depression. |
| Interventions | Paroxetine and imipramine. |
| Outcomes | Unclear. |
| Notes | Waiting for translation from Chinese to English. |
Zimbroff 2004.
| Methods | Two‐week, open‐label, randomised study. |
| Participants | Non responders outpatients. |
| Interventions | Paroxetine, citalopram, fluoxetine, sertraline. |
| Outcomes | Montgomery‐Asberg Depression Rating Scale (MADRS). |
| Notes | Funding: pharmaceutical industry. |
Zou 2006.
| Methods | Comparative study. Unclear design. |
| Participants | Patients with depression. |
| Interventions | Mirtazapine and paroxetine. |
| Outcomes | Unclear. |
| Notes | Waiting for translation from Chinese to English. |
Characteristics of ongoing studies [ordered by study ID]
Thomas 2008.
| Trial name or title | Thomas 2008 (GENPOD study). |
| Methods | Multicentre randomised controlled trial. |
| Participants | Patients with a primary diagnosis of depression according to ICD10 criteria and a Beck Depression Inventory (BDI) score > 14. |
| Interventions | Antidepressant treatments. |
| Outcomes | BDI score at 6 weeks. |
| Starting date | 2008 |
| Contact information | Thomas L. |
| Notes |
ICD: International Classification of Disease
Differences between protocol and review
In the present review we added the 'Summary of findings' tables using the GRADE methodology.
Contributions of authors
MP, CG, DP, LRM, CT, CR, TAF, NW, CB, AC collected the data; MP, AC, CB ran the analyses; MP, AC, CB drafted and critically revised the manuscript.
TAF and NW critically revised the manuscript.
Sources of support
Internal sources
Department of Public Health and Community Medicine, Section of Psychiatry, University of Verona, Italy.
External sources
No sources of support supplied
Declarations of interest
MP, CG, DP, CT, LRM, CR, CB, AC: none.
TAF has received honoraria for speaking at CME meetings sponsored by Asahi Kasei, Eli Lilly, GlaxoSmithKline, Mochida, MSD, Otsuka, Pfizer, Shionogi and Tanabe‐Mitsubishi. He is diplomate of the Academy of Cognitive Therapy. He has received royalties from Igaku‐Shoin, Seiwa‐Shoten and Nihon Bunka Kagaku‐sha. He is on advisory board for Sekisui Chemicals and Takeda Science Foundation. The Japanese Ministry of Education, Science, and Technology, the Japanese Ministry of Health, Labor and Welfare, and the Japan Foundation for Neuroscience and Mental Health have funded his research projects.
NW has received research grants from the Japanese Ministry of Education, Science, Sports and Culture; and from the Japanese Ministry of the Health, Labour and Welfare. He has also received speaking fees and research funds from Asahi Kasei, Dai‐Nippon Sumitomo, Eli Lilly, GlaxoSmithKline, Janssen, MSD, Otsuka, Pfizer and Schering‐Plough.
New
References
References to studies included in this review
029060/1/CPMS 069 1991 {unpublished data only}
- GlaxomithKline. A multicentre, parallel group study to compare the efficacy and tolerability of paroxetine with clomipramine in elderly patients. www.gsk‐clinicalstudyregister.com 1989.
- Rouillon F, et al. A double‐blind, multicentre study comparing increasing doses of paroxetine (20‐50 mg) and clomipramine (50‐150mg) in elderly patients with major depression. World congress of biological psychiatry (poster presentation). 1991.
029060/1/CPMS‐095 {unpublished data only}
- GlaxoSmithKline. A double‐blind comparative study of withdrawal effects following abrupt discontinuation of treatment with paroxetine in low or high dose or imipramine. www.gsk‐clinicalstudyregister.com 1992.
0600A‐349 {unpublished data only}
- Stahl SM, Entsuah R, Rudolph RL. Comparative efficacy between venlafaxine and SSRIs: A pooled analysis of patients with depression. Biological Psychiatry 2002;52:1166‐74. [DOI] [PubMed] [Google Scholar]
0600B1‐367 {published data only}
- Entsuah R, Shaffer M, Zhang J. A critical examination of the sensitivity of underdimensional subscales derived from the Hamilton Depression Rating Scale to antidepressant drug effects. Journal of Psychiatric Research 2002;36:437‐48. [DOI] [PubMed] [Google Scholar]
- Salinas, Ipr the Venlalaxlne XR 367 Study Group. Wyeth‐AyerstResearch, Paris, France. Once‐dally extended release (XR) venlafaxine versus paroxetlne in outpatients with major depression. Biological psychiatry 1997;42(1):244S. [Google Scholar]
- Shelton C, Entsuah R, Padmanabhan SK, Vinall PE. Venlafaxine XR demonstrates higher rates of sustained remission compared to fluoxetine, paroxetine or placebo. Internal Clinical Psychopharmacology 2005;20(4):233‐8. [DOI] [PubMed] [Google Scholar]
0600B 428 {unpublished data only}
- Casabona GM, Silenzi V, Guazzelli M. A randomized, double blind, comparison of venlafaxine ER and paroxetine in outpatients with moderate to severe major depression. European Neuropsychopharmacology 2002;12(Suppl 3):208. [Google Scholar]
29060/056/UK {unpublished data only}
- GlaxoSmithKline. A double‐blind, between patient, multicentre study in general practice comparing the efficacy and tolerability of paroxetine with those of dothiepin in the treatment of elderly depressed patients. www.gsk‐clinicalstudyregister.com.
29060/103 {unpublished data only}
- Moon CAL, et al. A double blind, multicentre study comparing the efficacy, tolerability and effects on cognitive function of paroxetine with those of lofepramine in elderly depressed hospital in‐ or out‐patients. British Journal of Clinical Practice 1996.
29060/281 PAR {unpublished data only}
- GlaxoSmithKline. A trial to assess the effectiveness and tolerance of paroxetine by double‐blind comparison with amitriptyline in the treatment of depressed patient in General Practice. www.gsk‐clinicalstudyregister.com.
29060/299 {unpublished data only}
- GlaxoSmithKline. Double‐blind Study comparing the efficacy and tolerability of paroxetine and amitriptyline in patients with severe depression. www.gsk‐clinicalstudyregister.com.
29060/356 {unpublished data only}
- GlaxoSmithKline. A double‐blind, multicentre study to compare paroxetine and fluoxetine in the treatment of patients with major depressive disorder with regard to antidepressant efficacy, effects on associated anxiety and tolerability. www.gsk‐clinicalstudyregister.com 1994.
29060/409 {unpublished data only}
- GlaxoSmithKline. A comparative, controlled double blind, double dummy study about the efficacy and safety of paroxetine vs imipramine in ambulatory patients with major depression. www.gsk‐clinicalstudyregister.com.
29060/III/83/022 {unpublished data only}
- GlaxoSmithKline. A study to assess the efficacy and tolerance of paroxetine in patients with depression by double‐blind comparison with mianserin. www.gsk‐clinicalstudyregister.com.
29060/III/85/030 {unpublished data only}
- GlaxoSmithKline. An assessment of the efficacy and tolerability of paroxetine by double blind comparison with mianserin in patients referred to a psychiatric clinic. www.gsk‐clinicalstudyregister.com.
29060.065.BE {unpublished data only}
- Laurelle M, et al. A multicenter double‐blind comparative study between paroxetine and maprolitine in major depression. European Neuropsychopharmacology 1:439 (poster presentation). 1991.
29060.07.001 {unpublished data only}
- GlaxoSmithKline. A double‐blind comparison of paroxetine, amitriptyline and placebo in inpatients with major depressive disorder with melancholia. www.gsk‐clinicalstudyregister.com.
29060III/85/038 {unpublished data only}
- GlaxoSmithKline. To assess the effectiveness and tolerance of paroxetine in depressed patients by double‐blind comparison with mianserin. www.gsk‐clinicalstudyregister.com.
Aberg‐Wistedt 2000 {published data only}
- Aberg‐Wistedt A, Agren H, Ekselius L, Bengtson F, Akerblad AC. Sertraline versus paroxetine in major depression: clinical outcome after six months of continuous therapy. Journal of Clinical Psychopharmacology 2000;20(6):645‐52. [DOI] [PubMed] [Google Scholar]
- Khoury A, Aberg Wistedt A, Stain Malmgren R. Effect of sertraline and paroxetine treatment on depression scores and peripheral indices of serotonergic function in major depression. 11th European College of Neuropsychopharmacology Congress. Paris, France. 31st October 4th November 1998. Utrecht: ECNP, 1998.
- Stain‐Malmgren R, Khoury AE, Aberg‐Wistedt A, Tham A. Serotonergic function in major depression and effect of sertraline and paroxetine treatment. International Clinical Psychopharmacology 2001;16(2):93‐101. [DOI] [PubMed] [Google Scholar]
Ansseau 1993 {published data only}
- Ansseau M, Gabriels A, Loyens J, Bartholome F, Evrard J, Nayer A, et al. A double‐blind comparison of paroxetine and fluvoxamine in major depression. European Neuropsychopharmacology 1993;3:323‐4. [Google Scholar]
- Ansseau M, Gabriels A, Loyens J, Bartholome F, Evrard JL, Nayer A, et al. Controlled comparison of paroxetine and fluvoxamine in major depression. Human Psychopharmacology 1994;9:329‐36. [Google Scholar]
Aoba 2004 {published and unpublished data}
- Aoba A, Tanaka Y, Kageyama S, Matsutani S, Mori N, Narita H, et al. A post‐marketing clinical study of Paroxetine hydrochloride hydrate in patients with depression or depressive episodes. A double blind Imipramine hydrochloride controlled comparative study. Japanese Journal of Clinical Psychopharmacology 2004;7(5):831‐53. [Google Scholar]
Bakish 1997 {published data only}
- Bakish D, Cavazzoni P, Chudzik J, Ravindran A, Hrdina PD. Effects of selective serotonin reuptake inhibitors on platelet serotonin parameters in major depressive disorder. Biological Psychiatry 1997;41(2):184‐90. [DOI] [PubMed] [Google Scholar]
Baldwin 1995 {published data only}
- Baldwin D, Hawley CJ, Abed RT, Maragakis BP, Cox J, Buckingham SA, et al. A multicenter double blind comparison of Nefazodone and Paroxetine in the treatment of outpatients with moderate to severe depression. Journal of Clinical Psychiatry 1996;57(Suppl 2):46‐52. [PubMed] [Google Scholar]
Baldwin 2006 {published data only}
- Baldwin DS, James AC, Huusom AKT, Hindmarch I. A double blind, randomized, parallel‐group, flexible‐dose study to evaluate the tolerability and effects of treatment discontinuation with escitalopram and paroxetine in patients with major depressive disorder. International Clinical Psychopharmacology 2006;21:159‐69. [DOI] [PubMed] [Google Scholar]
Bascara 1989 {published data only}
- Bascara B. A double blind study to compare the effectiveness and tolerability of paroxetine and amitriptyline in depressed patients. Acta Psychiatrica Scandinavica, Supplementum 1989;350:141‐2. [DOI] [PubMed] [Google Scholar]
Battegay 1985 {published data only}
- Battegay R, Hager M, Rauchfleisch U. Double‐blind comparative study of paroxetine and amitriptyline in depressed patients of a university psychiatric outpatient clinic. Neuropsychobiology 1985;13:31‐7. [DOI] [PubMed] [Google Scholar]
Benkert 1999 {published data only}
- Benkert O, Szegedi A, Kohnen R. Mirtazapine compared with paroxetine in major depression. Journal of Clinical Psychiatry 2000;61(9):656‐63. [DOI] [PubMed] [Google Scholar]
Bignamini 1992 {published and unpublished data}
- Bignamini A, Rapisarda V. A double‐blind multicentre study of paroxetine and amitriptyline in depressed outpatients. International Clinical Psychopharmacology 1992;6 Suppl 4:37‐41. [DOI] [PubMed] [Google Scholar]
Blier 2009 {published data only}
- Blier P, Gobbi G, Turcotte JE, Montigny C, Boucher N, Hebert C, et al. Mirtazapine and paroxetine in major depression: a comparison of monotherapy versus their combination for treatment initiation. European Neuropsychopharmacology 2009;19:457‐65. [DOI] [PubMed] [Google Scholar]
Boulenger 2006 {published data only}
- Boulenger JP, Huusom AKT, Florea I, Baekdal T, Sarchiapone M. A comparative study of the efficacy of long‐term treatment with escitalopram and paroxetine in severely depressed patients. Current Medical Research and Opinion 2006;22(7):1331‐41. [DOI] [PubMed] [Google Scholar]
Cassano 2002 {published data only}
- Cassano GB, Puca F, Scapicchio PL, Trabucchi M. Paroxetine and fluoxetine effects on mood and cognitive functions in depressed nondemented elderly patients. Journal of Clinical Psychiatry 2002;63(5):396‐402. [PubMed] [Google Scholar]
Cassano 2002a {published data only}
- Cassano GB, Jori MC. Efficay and safety of amisulpride 50 mg versus paroxetine 20 mg in major depression: a randomized, double‐blind, parallel group study. Internetional Clinical Psychopharmacology 2002;17:27‐32. [DOI] [PubMed] [Google Scholar]
Chiu 1996 {published data only}
- Chiu HJ, Hong CJ, Chan CH. Paroxetine in the treatment of chinese patients with depressive episode: a double blind randomized comparison with Imipramine. Chinese Medical Journal 1996;57:418‐23. [PubMed] [Google Scholar]
Chouinard 1999 {published and unpublished data}
- Chouinard G, Saxena B, Belanger MC, Ravindran A, Bakish D, Beauclair L, et al. A Canadian multicenter, double‐blind study of paroxetine and fluoxetine in major depressive disorder. Journal of Affective Disorders 1999;54(1‐2):39‐48. [DOI] [PubMed] [Google Scholar]
Christiansen 1996 {published data only}
- Christiansen PE, Behnke K, Black CH, Ohrstrom JK, Bork‐Rasmussen H, Nilsson J. Paroxetine and amitriptyline in the treatment of depression in general practice. Acta Psychiatrica Scandinavica 1996;93:158‐63. [DOI] [PubMed] [Google Scholar]
CL3‐023 {published and unpublished data}
- European Medicines Agency. Evaluation of Medicines for Human Use: CHMP assessment report for Valdoxan‐Agomelatine. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/000915/WC500046226.pdf, Procedure No. EMEA/H/C/000915 2008.
- Hickie IB, Rogers NL. Novel melatonin‐based therapies: potential advances in the treatment of major depression. Lancet 2011;378:621‐31. [DOI] [PubMed] [Google Scholar]
Cohn 1990 {published and unpublished data}
- Cohn JB, Crowder JE, Wilcox CS, Ryan PJ. A placebo‐ and imipramine‐controlled study of paroxetine. Psychopharmacology Bulletin 1990;26(2):185‐9. [PubMed] [Google Scholar]
- Cohn JB, Wilcox CS. Paroxetine in major depression: a double blind trial with imipramine and placebo. Journal of Clinical Psychiatry 1992;53(2):52‐6. [PubMed] [Google Scholar]
Dalery 2001 {published data only}
- Dalery J, Aubin V. Comparative study of Paroxetine and Mianserin in depression of the elderly: efficacy, tolerance, serotonine dependance. L'Encephale 2001;27:71‐81. [PubMed] [Google Scholar]
Davidson 2005 {published data only}
- Davidson J, Watkins L, Owens M, Krulewicz S, Connor K, Carpenter D, et al. Effects of Paroxetine and Venlafaxine XR on heart rate variability in depression. Journal of Clinical Psychopharmacology 2005;25(5):480‐4. [DOI] [PubMed] [Google Scholar]
Demyttenaere 2002 {published data only}
- Demyttenaere K, Albert A, Mesters P, Dewe W, Bruyckere K, Sangeleer M. What happens with adverse events during 6 months of treatment with selective serotonin reuptake inhibitors?. Journal of Clinical Psychiatry 2005;66(7):859‐63. [DOI] [PubMed] [Google Scholar]
- Demyttenaere K, Bruffaerts R, Albert A, Mesters P, Dewe W, Debruyckere K, Sangeleer M. Development of an antidepressant compliance questionnaire. Acta Psychiatrica Scandinavica 2004;110(3):201‐7. [DOI] [PubMed] [Google Scholar]
- Demyttenaere K, Mesters P, Dewe W, Boulanger B, Bruyckere K, Sangeleer M, et al. Six month compliance with fluoxetine or paroxetine treatment in depressed outpatients. European Neuropsychopharmacology 2002;12(Suppl 3):208. [Google Scholar]
Detke 2004 (HMAY A) {published and unpublished data}
- Detke MJ, Wiltse CG, Mallinckrodt CH, McNamara RK, Demitrack MA, Bitter I. Duloxetine in the acute and long‐term treatment of major depressive disorder: a placebo‐ and paroxetine‐controlled trial. European Neuropsychopharmacology 2004;14(6):457‐70. [DOI] [PubMed] [Google Scholar]
De Wilde 1993 {published data only}
- Wilde J, Spiers R, Mertens C, Bartholome F, Schotte G, Leyman S. A double‐blind, comparative, multicentre study comparing paroxetine with fluoxetine in depressed patients. Acta Psychiatrica Scandinavica 1993;87(2):141‐5. [DOI] [PubMed] [Google Scholar]
Dichter 2005 {published data only}
- Dichter GS, Tomarken AJ, Freid CM, Addington S, Shelton RC. Do venlafaxine XR and paroxetine equally influence negative and positive affect?. Journal of Affective Disorders 2005;85(3):333‐9. [DOI] [PubMed] [Google Scholar]
Dorman 1992 {published data only}
- Dorman T. Sleep and paroxetine: a comparison with mianserin in elderly depressed patients. International Clinical Psychopharmacology 1992;6(4):53‐8. [PubMed] [Google Scholar]
DUAG 1990 {published data only}
- Danish University Antidepressant Group. Paroxetine: A selective serotonine reuptake inhibitor showing better tolerance, but weaker antidepressant effect than clomipramine in a controlled multicenter study. Journal of Affective Disorder 1990;18:289‐99. [DOI] [PubMed] [Google Scholar]
Dunbar 1991 {published and unpublished data}
- Dunbar GC, Cohn JB, Fabre LF, Feighner JP, Fieve RR, Mendels, et al. A comparison of paroxetine, imipramine and placebo in depressed outpatients. British Journal of Psychiatry 1991;159:394‐8. [DOI] [PubMed] [Google Scholar]
- Lapierre Y, Bentkover J, Schainbaum S, Manners S. Direct cost of depression: analysis of treatment costs of paroxetine versus Imipramine in Canada. Canadian Journal of Psychiatry 1995;40(7):370‐7. [DOI] [PubMed] [Google Scholar]
Dunner 1992 {published and unpublished data}
- Dunner DL, Cohn J, Walshe T, Cohn CK, Feighner JP, Fieve RR, et al. Two combined, multicenter double‐blind studies of Paroxetine and Doxepin in geriatric patients with major depression. Journal of Clinical Psychiatry 1992;53(2):57‐60. [PubMed] [Google Scholar]
Fabre 1992 {published and unpublished data}
- Fabre LF. A 6‐week, double‐blind trial of paroxetine, imipramine and placebo in depressed outpatients. Clinical Journal of Psychiatry 1992;53(2):40‐3. [PubMed] [Google Scholar]
Fava 1998 {published data only}
- Fava M, Amsterdam JD, Deltito JA, Salzman C, Schwaller M, Dunner DL. A double‐blind study of paroxetine, fluoxetine, and placebo in outpatients with major depression. Annals of Clinical Psychiatry 1998;10(4):145‐50. [DOI] [PubMed] [Google Scholar]
Fava 2002 {published data only}
- Fava M, Hoog SL, Judge RA, Kopp JB, Nilsson ME, Gonzales JS. Acute efficacy of fluoxetine versus sertraline and paroxetine in major depressive disorder including effects of baseline insomnia. Journal of Clinical Psychopharmacology 2002;22(2):137‐47. [DOI] [PubMed] [Google Scholar]
- Fava M, Judge R, Hoog SL, Nilsson ME, Koke SC. Fluoxetine versus sertraline and paroxetine in major depressive disorder: changes in weight with long‐term treatment. Journal of Clinical Psychiatry 2000;61(11):863‐7. [DOI] [PubMed] [Google Scholar]
- Fava M, Rosenbaum JF, Hoog SL, Tepner RG, Kopp JB, Nilsson ME. Fluoxetine versus sertraline and paroxetine in major depression: tolerability and efficacy in anxious depression. Journal of Affective Disorders 2000;59(2):119‐26. [DOI] [PubMed] [Google Scholar]
Feighner 1989 {published and unpublished data}
- Feighner JP, Boyer WF. Paroxetine in the treatment of depression: a comparison with imipramne and placebo. Acta Psychiatrica Scandinavica 1989;80(350):125‐9. [DOI] [PubMed] [Google Scholar]
Freed 1996 {published and unpublished data}
- Freed E, George T, Goldney R, Lambert T, Tiller J. A double‐blind multicentre comparison of paroxetine and amitriptyline in Australian general‐practice. XXth Collegium Internationale Neuro psychopharmacologicum, Melbourne, Australia. 1996.
- Freed E, Goldney R, Lambert T, Tiller J, Johnston R. A double‐blind, multicentre study to assess the tolerability and efficacy of paroxetine compared with amitriptyline in the treatment of depressed patients in Australian general practice. Australian and New Zealand Journal of Psychiatry 1999;33:416‐21. [DOI] [PubMed] [Google Scholar]
Gagiano 1993 {published and unpublished data}
- Gagiano CA. A double blind comparison of paroxetine and fluoxetine in patients with major depression. British Journal of Clinical Research 1993;4:145‐52. [Google Scholar]
- Gagiano CA, Judge R. Paroxetine vs. fluoxetine. Clinical Neuropharmacology 1992;15(1):334. [Google Scholar]
Gallen 2001 {published data only}
- Brown MT, Brinkman SA, Reisner JK, Rowland CR. Reboxetine, placebo, and paroxetine comparison in patients with major depressive disorder. Clinical Research. Pharmacia & Upjoh 2001.
- Ferguson J, Wesnes K, Barker K, Schwartz G. Reboxetine effects on cognitive functioning in depressed patients. European Neuropsychopharmacology 2002;12(Suppl 3):206. [Google Scholar]
- Ferguson JM, Wesnes KA, Schwartz GE. Reboxetine effects on cognitive functioning in depressed patients. 155th Annual Meeting of the American Psychiatric Association, Philadelphia, PA. 2002.
- Ferguson JM, Wesnes KA, Schwartz GE. Reboxetine versus paroxetine versus placebo: effects on cognitive functioning in depressed patients. International Clinical Psychopharmacology 2003;18(1):9‐14. [DOI] [PubMed] [Google Scholar]
- Gallen CC, Brown MT. Reboxetine versus paroxetine versus placebo in patients with major depressive disorder. European Neuropsycopharmacology. 2001; Vol. 11, issue suppl3:S216.
Geretsegger 1994 {published and unpublished data}
- Geretsegger C, Bohmer F, Ludwig M. Paroxetine in the elderly depressed patient: randomized comparison with fluoxetine of efficacy, cognitive and behavioural. International Clinical Psychopharmacology 1994;9:25‐9. [PubMed] [Google Scholar]
- Schone W, Ludwig M. A double‐blind study of paroxetine compared with fluoxetine in geriatric patients with major depression. Journal of Clinical Psychopharmacology 1993;13:34‐9. [DOI] [PubMed] [Google Scholar]
- Schone W, Ludwig M. Paroxetine in the treatment of depression in geriatric patientsa double‐blind comparative study with fluoxetine. Fortschritte der Neurologie Psychiatrie 1994;62(Suppl1):16‐8. [PubMed] [Google Scholar]
Geretsegger 1995 {published and unpublished data}
- Geretsegger C, Stuppaeck CH, Mair M, Platz T, Fartacek R, Heim M. Multicentre double‐blind study of paroxetine and amitriptyline in elderly depressed inpatients. Psychopharmacology 1995;119:277‐81. [DOI] [PubMed] [Google Scholar]
Gilmor 2002 {published data only}
- Gilmor ML, Owens MJ, Nemeroff CB. Inhibition of Norepinephrine uptake in patients with major depression treated with Paroxetine. American Journal of Psychiatry 2002;159(10):1702‐10. [DOI] [PubMed] [Google Scholar]
Goldstein 2004 (HMAT B) {published and unpublished data}
- Goldstein DJ, Lu Y, Dekte MJ, Wiltse CG, Mallinckrodt C, Demitrack MA. Duloxetine versus Paroxetine in the treatment of depression. 155th Annual Meeting of American Psychiatric Association. 2002 May 18‐23; Philadelphia.
- Goldstein DJ, Lu Y, Detke M, Wiltse C, Mallinckrodt C, Demitrack MA. Duloxetine in the treatment of depression: a double blind placebo controlled comparison with Paroxetine. European Neuropsychopharmacology 2002;12:43‐4. [DOI] [PubMed] [Google Scholar]
- Goldstein DJ, Lu Y, Detke MJ, Wiltse C, Mallinckrodt C, Demitrack MA. Duloxetine in the treatment of depression: a double‐blind placebo‐controlled comparison with paroxetine. Journal of Clinical Psychopharmacology 2004;24(4):389‐99. [DOI] [PubMed] [Google Scholar]
- Mallinckrodt C, Goldstein DJ, Lu Y, Detke M, Wiltse C, Demitrack MA. Duloxetine in the treatment of depression: a double blind placebo controlled comparison with Paroxetine. European Neuropsychopharmacology 2002;12(Suppl 3):S214. [DOI] [PubMed] [Google Scholar]
Guillibert 1989 {published data only}
- Guillibert E, Pelicier Y, Archambault JC, Chabannes JP, Clerc G, Desvilles M, et al. A double blind, multicentre study of Paroxetine versus Clomipramine in depressed elderly patients. Acta Psychiatrica Scandinavica 1989;80(Suppl 350):132‐4. [DOI] [PubMed] [Google Scholar]
Hicks 2002 {published data only}
- Argyropuolos SV, Hicks JA, Nash JR, Bell CJ, Rich AS, Nutt DJ, et al. Correlation of subjective and objective sleep measurements at different statges of the treatment of depression. Psychiatry Research 2003;120:179‐90. [DOI] [PubMed] [Google Scholar]
- Hicks JA, Argyropoulos SB, Rich AS, Nash JR, Bell CJ, Edwards C, et al. Randomized controlled study of sleep after Nefazodone or Paroxetine treatment in outpatients with depression. British Journal of Psychiatry 2002;180:528‐35. [DOI] [PubMed] [Google Scholar]
Higuchi 2009 {published data only}
- Higuchi T, Murasaki M, Kamijima K. Clinical evaluation of duloxetine in the treatment of major depressive disorder placebo and paroxetine controlled double‐blind comparative study. Japanese Journal of Clinical Psychopharmacology 2009;12:1613‐34. [Google Scholar]
HMAT A (ID#4091) {unpublished data only}
- Eli Lilly. Duloxetine versus placebo and paroxetine in the acue treatment of major depression, study group a. www.lillytrials.com www.clinicalstudyresults.org.
- Eli Lilly and company (www.clinicalstudyresults.org). Duloxetine versus placebo and paroxetine in the acute treatment of major depression, Study group A. www.clinicalstudyresults.org 2004.
Hutchinson 1992 {published data only}
- Hutchinson DR, Tong S, Moon CA, Vince M, Clarke A. Paroxetine in the treatment of elderly depressed patients in general practice: a double‐blind comparison with amitriptyline. International Clinical Psychopharmacolology 1992;6(4):43‐51. [DOI] [PubMed] [Google Scholar]
- Hutchinson DR, Tong S, Moon CAL, Vince M, Clarke A. A double blind study in general practice to compare the efficacy and tolerability of paroxetine and amitriptyline in depressed elderly patients. British Journal of Clinical Research 1991;2:43‐57. [Google Scholar]
Hwang 2004 {published data only}
- Hwang J‐P, Yang C‐H, Tsai S‐J. Comparison study of venlafaxine and paroxetine for the treatment of depression in elderly Chinese inpatients. Internationa Journal of Geriatric Psychiatry 2004;19:189‐90. [DOI] [PubMed] [Google Scholar]
Javors 2000 {published data only}
- Javors MA, Houston JP, Tekell JL, Brannan SK, Frazen A. Reduction of platelet serotonin content in depressed patients treated with either paroxetine or desipramine. International Journal of Neuropsychopharmacology 2000;3:229‐35. [DOI] [PubMed] [Google Scholar]
Jefferson 2001 29060/785 {unpublished data only}
- Jefferson JW, Griest J. A double blind comparison of citalopram and paroxetine in the treatment of patients with depression and anxiety. 39th Annual Meeting of the American College of Neuropsychopharmacology. 2000, Dec 10‐14; San Juan; Puerto Rico.
- Jefferson JW, Griest J. Citalopram compared with paroxetine in depressed patients with associated anxiety. 154th Annual Meeting of the American Psychiatric Association; New Orleans; LA. 2001 May 5‐10.
Kasper 2005 {published data only}
- Kasper S, Olivieri L, loreto G, Dionisio P. A comparative, randomised, double‐blind study of trazodone prolonged‐release and paroxetine in the treatment of patients with major depressive disorder. Current Medical Research and Opinion 2005;21(8):1139‐46. [DOI] [PubMed] [Google Scholar]
Kato 2005 {published data only}
- Kato M, Ikenaga Y, Wakeno M, Okugawa G, Nobuhara K, Fukuda T, et al. Controlled clinical comparison of paroxetine and fluvoxamine considering the serotonin transporter promoter polymorphism. International Clinical Psychopharmacology 2005;20:151‐56. [DOI] [PubMed] [Google Scholar]
- Kato M, Ikenaga Y, Wakeno M, Okugawa G, Nobuhara K, Fukuda T, et al. Effects of the serotonin type 2A, 3A and 3B receptor and the serotonintransporter genes on paroxetine and fluvoxamine efficacy and adverse drug reactions in depressed japanese patients. Neuropsychobiology 2006;53:186‐95. [DOI] [PubMed] [Google Scholar]
- Kato M, Wakeno M, Okugawa G, Nagata M, Nobuhara K, Ochi T, et al. Clinical comparison of paroxetine and fluvoxamine considering of the serotonin transporter promoter polymorphism in patients with affective disorder. International Clinical Psychopharmacology 2004;19(3):175. [DOI] [PubMed] [Google Scholar]
Katz 2004 {published data only}
- Katz MM, Tekell JL, Bowden CL, Brannan S, Houston JP, Berman N, et al. Onset and early behavioral effects of pharmacologically different antidepressants and placebo in depression. Neuropsychopharmacology 2004;29:566‐79. [DOI] [PubMed] [Google Scholar]
Kennedy 2004 {published and unpublished data}
- GlaxoSmithKline. An eight‐week doubleblind study comparing the effects of 20 mg of paroxetine to 150 mg Wellbutrin SR in patients with Major Depressive Disorder. www.gsk‐clinicalstudyregistrer.com.
- Kennedy SH. Differences in sexual disfunction between men and women with depression: a comparison before and after treatment with bupropion and paroxetine. Neuropsycophamacology 2004;29(Suppl1):S148‐9. [Google Scholar]
- Kennedy SH, Fulton KA, Bagby M, Greene A, Cohen NL, Shahryar RT. Sexual function during bupropion or paroxetine treatment of major depressive disorder. Canadian Journal of Psychiatry 2006;51(4):234‐42. [DOI] [PubMed] [Google Scholar]
Kiev 1997 {published and unpublished data}
- Kiev A, Feiger A. A double‐blind comparison of fluvoxamine and paroxetine in the treatment of depressed outpatients. Journal of Clinical Psychiatry 1997;58(4):146‐52. [DOI] [PubMed] [Google Scholar]
- Kiev A, Feiger A. Comparison of fluvoxamine and paroxetine in depression. Xth World Congress of Psychiatry. Madrid, Spain, 1996.
- Kiev A, Feiger AD. A double‐blind comparison of fluvoxamine and paroxetine in major depressive disorder. 149th Annual Meeting of the American Psychiatric Association. New York, NY, 1996.
Kramer 1998 {published data only}
- Feighner JP. Substance P. 11th European College of Neuropsychopharmacology Congress. Paris, France. 1998.
- Kramer MS, Cutler N, Feighner J, Shrivastava R, Carman J, Sramek JJ, et al. Distinct mechanism for antidepressant activity by blockade of central substance P receptors. Science 1998;281:1640‐5. [DOI] [PubMed] [Google Scholar]
Kramer 2001 {published data only}
- Kramer MS. Substance P (NKI) receptor antagonists (SPAs) in patients with major depression. 40th Annual Meeting, American College of Neuropsychopharmacology, Waikoloa, HI. 2001.
- Krishnan RR. Clinical experience with substance p receptor (nk1) antagonists in depression. Journal of Clinical Psychiatry 2002;63(Suppl 11):25‐9. [PubMed] [Google Scholar]
Kuhs 1989 {published and unpublished data}
- Khus H, et al. A double‐blind study of paroxetine versus amitriptyline, with particular reference to cardiovascular effects. Proceedings of the World Congress of Biological Psychiatry. 1991:22‐23 (poster presentation).
- Kuhs H, Rudolf GA. A double‐blind study of the comparative antidepressant effect of paroxetine and amitriptyline. Acta Psychiatrica Scandinavica 1989;80(350):145‐6. [DOI] [PubMed] [Google Scholar]
- Kuhs H, Rudolf GA. Cardiovascular effects of paroxetine. Psychopharmacology 1990;102(3):379‐82. [DOI] [PubMed] [Google Scholar]
- Kuhs H, Rudolf GA, Schlake HP, Rolf LH. Relationship between parameters of serotonin transport and antidepressant plasma levels or therapeutic response in depressive patients treated with paroxetine and amitriptyline. Acta Psychiatrica Scandinavica 1992;85(5):364‐9. [DOI] [PubMed] [Google Scholar]
Laghrissi‐Thode 1995 {published data only}
- Laghrissi‐Thode F, Pollock BG, Miller MC, Mulsant BH, Altieri L, Finkel MS. Double‐blind comparison of paroxetine and nortriptyline on the postural stability of late‐life depressed patients. Psychopharmacology Bulletin 1995;31(4):659‐63. [PubMed] [Google Scholar]
Laursen 1985 {published and unpublished data}
- Laursen AL, Mikkelsen PL, Fievre Honore P. Paroxetine in the treatment of depression ‐ a randomised comparison with amitriptyline. Acta Psychiatrica Scandinavica 1985;71:249‐55. [DOI] [PubMed] [Google Scholar]
Lee 2007 (HMCV) {published data only}
- Lee P, Shu L, Xu X, Wang CY, Lee MS, Liu CY, et al. Once‐daily duloxetine 60 mg in the treatment of major depressive disorder: multicenter, double‐blind, randomized, paroxetine‐controlled, non‐inferiority trial in China, Korea, Taiwan and Brazil. Psychiatry and Clinical Neurosciences 2007;61(3):295‐307. [DOI] [PubMed] [Google Scholar]
Lepine 2001 {published data only}
- Lepine JP, Altamura C, Ansseau M, Ayuso Gutierrez JL, Bitter I, Lader M, et al. Tienapine and paroxetine in major depressive disorder, with a special focus on the anxious component in depression: an international, 6 week double‐blind study. Human Psychopharmacology: Clinical and Experimental 2001;16(3):219‐27. [DOI] [PubMed] [Google Scholar]
Loo 2002 {published data only}
- Loo H, Hale A, D'haenen H. Determination of the dose of agomelatine, a melatoninergic agonist and selective 5‐HT antagonist, in the treatment of major depressive disorder: a placebo‐controlled dose range study. International Clinical Psychopharmacology 2002;17:239‐47. [DOI] [PubMed] [Google Scholar]
M/2020/0047 {unpublished data only}
- Brown MT, Brinkman SA, Reisner JK, Rowland CR. Reboxetine, placebo, and paroxetine comparison in patients with major depressive disorder. Clinical Research. Pharmacia & Upjohn; 6/2/2001.
M/2020/0052 {unpublished data only}
- Baldwin D, Bridgman K, Buis C. Resolution of sexual dysfunction during double‐blind treatment of major depression with reboxetine or paroxetine. Journal of Psychopharmacology 2006;20(1):91‐6. [DOI] [PubMed] [Google Scholar]
- Linde AS, Bartlett C, Math M, Richard KA. Reboxetine (PNU‐1555950E) vs Paroxetine in a double‐blind, multinational study of treatment in major depressive disorder. Clinical Research. Pharmacia & Upjohn 9/9/2004.
Mc Partlin 1998 {published data only}
- McPartlin GM, Reynolds A, Anderson C, Casoy J. A comparison of once‐daily venlafaxine XR and paroxetine in depressed out patients treated in general practice. Primary Care Psychiatry 1998;4:127‐32. [Google Scholar]
Montgomery 2004 {published data only}
- Montgomery SA, Kennedy SH, Burrows GD, Lejoyeux M, Hindmarch I. Absence of discontinuation symptoms with agomelatine and occurrence of discontinuation symptoms with paroxetine: a randomized, double‐blind, placebo‐controlled discontinuation study. International Clinical Psychopharmacology 2004;19(5):271‐80. [DOI] [PubMed] [Google Scholar]
Moon 1996 {published and unpublished data}
- Moon C, Vince M. Treatment of major depression in general practice: a double‐blind comparison of paroxetine and lofepramine. British Journal of Clinical Practice August 1996;50(5):240‐4. [PubMed] [Google Scholar]
Mulsant 1999 {published data only}
- Fabian TJ, Dew MA, Pollock BG, Reynolds CF, Mulsant BH, Butters MA, et al. Endogenous concentrations of DHEA and DHEA‐S. Decrease with remission of depression in older adults. Biological Psychiatry 2001;50(10):767‐74. [DOI] [PubMed] [Google Scholar]
- Fabian TJ, Kroboth PD, Butters MA, et al. Are DHEA and/or DHEA‐S concentrations associated with remission of depression or improvements in mental status scores in older adults. 12th Annual Meeting of the American Association for Geriatric Psychiatry, New Orleans LA. 1999.
- Mulsant BH, Pollock BG, Nebes RD, Miller MD, Little JT, Stack J, et al. A double‐blind randomized comparison of nortriptyline and paroxetine in the treatment of late‐life depression: 6‐week outcome. Journal of Clinical Psychiatry 1999;60(Suppl 20):16‐20. [PubMed] [Google Scholar]
MY‐1045/BRL‐029060/1 {unpublished data only}
- GlaxoSmithKline. A multicenter, randomized, double‐blind, placebo‐controlled comparison of paroxetine and fluoxetine in the treatment of major depressive disorder. www.gsk‐clinicalstudyregister.com.
NCT00463242 {published and unpublished data}
- European Medicines Agency. Evaluation of Medicines for Human Use: CHMP assessment report for Valdoxan‐Agomelatine. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/000915/WC500046226.pdf, Procedure No. EMEA/H/C/000915 2008.
- Hickie IB, Rogers NL. Novel melatonin‐based therapies: potential advances in the treatment of major depression. Lancet 2011;378:621‐31. [DOI] [PubMed] [Google Scholar]
Nickel 2003 {published data only}
- Nickel T, Sonntag A, Schill J, Zobel AW, Nibal A, Brunnauer A, et al. Clinical and neurobiological effects of tianeptine and paroxetine in major depression. Journal of Clinical Psychopharmacology 2003;23:155‐168. [DOI] [PubMed] [Google Scholar]
Nielsen 1991 {published data only}
- Nielsen OA, Morsing I, Petersen JS, Larsen T, Moller SE, Manniche PM, et al. Paroxetine and imipramine treatment of depressive patients, in a controlled multicentre study with plasma amino acid measurements. Acta Psychiatrica Scandinavica 1991;84:233‐41. [DOI] [PubMed] [Google Scholar]
- Skausig OB, Nielsen OA, Morsing I, Petersen JS, Larsen T, Moller SE, et al. Paroxetine and imipramine treatment of depressed patients in a controlled, multicentre study with plasma amino acid measurements. Nordic Journal of Psychiatry 1992;46(Suppl 27):23‐6. [Google Scholar]
Ohrberg 1992 {published and unpublished data}
- Chiristainsen PE. Paroxetine and imipramine in the treatment of depressed patients in psychiatric specialist practice. Nordic Journal of Psychiatry 1992;27:33‐9. [Google Scholar]
- Ohrberg S, Christainsen PE, Severin B, Calberg H, Nilakantan B, Borup A, et al. Paroxetine and imipramine in the treatment of depressive patients in psychiatric practice. Acta Psychiatrica Scandinavica 1992;86:437‐44. [DOI] [PubMed] [Google Scholar]
Ontiveros 1994 {published and unpublished data}
- Garcia Barriga, et al. A double‐blind study with paroxetine vs fluoxetine in depressive patients. Xth World Congress of Psychiatry, Madrid, Spain. 1996.
- Ontiveros A, et al. A double blind study with paroxetine vs fluoxetine in depressive patients. Biological Psychiatry 1994;35:677. [Google Scholar]
Owens 2008 {published data only}
- Owens MJ, Krulewicz S, Simon JS, Sheehan DV, Thase ME, Carpenter DJ, et al. Estimates of serotonin and norepinephrine transporter inhibition in depressed patients treated with paroxetine or venlafaxine. Neuropsychopharmacology 2008;33:3201‐12. [DOI] [PubMed] [Google Scholar]
PAR MDUK 032 {unpublished data only}
- GlaxoSmithKline. A double ‐blind study to compare the efficacy and tolerability of paroxetine and amitriptyline in a multicentre general practice study in depressed patients. www.gsk‐clinicalstudyregister.com.
Pelicier 1993 {published and unpublished data}
- Pelicier Y, Schaeffer P. A multicentre, double‐blind study to compare the efficacy and tolerability of paroxetine and clomipramine in elderly patients with reactive depression. Acta Psychiatrica Scandinavica 1989;80:132. [DOI] [PubMed] [Google Scholar]
- Pelicier Y, Schaeffer P. A multicentre, double‐blind study to compare the efficacy and tolerability of paroxetine and clomipramine in elderly patients with reactive depression [Etude multicentrique en double aveugle comparant l'efficacité et la tolérance de la paroxétine et de la clomipramine dans la dépression réactionnelle du suject agé]. L'Encephale 1993;19:257‐61. [PubMed] [Google Scholar]
Perahia 2006 (HMAY B) {published and unpublished data}
- Eli Lilly, company. Clinical study summary ID#4298. www.lilly.com 2006.
- Perahia DG, Wang F, Mallinckrodt CH, Walker DJ, Detke MJ. Duloxetine in the treatment of major depressive disorder: a placebo‐ and paroxetine‐controlled trial. European Psychiatry 2006;21(6):367‐78. [DOI] [PubMed] [Google Scholar]
Peselow 1989 {published and unpublished data}
- Dunbar GC, Cohn JB, Fabre LF, Feighner JP, Fieve RR, Mendels J, et al. A comparison of paroxetine, imipramine and placebo in depressed out‐patients. British Journal of Psychiatry 1991;159:394‐8. [DOI] [PubMed] [Google Scholar]
- Feighner JP, Cohn JB, Fabre LF Jr, Fieve RR, Mendels J, Shrivastava RK, et al. A study comparing Paroxetine placebo and imipramine in depressed patients. Journal of Affective Disorders 1993;28(2):71‐9. [DOI] [PubMed] [Google Scholar]
- Peselow ED, Filippi AM, Goodnick P, Barouche F, Fieve RR. A double‐blind, Imipramine‐ and placebo‐controlled study of paroxetine in depressed outpatients. Psychopharmacology Bulletin 1989;25(2):272‐6. [PubMed] [Google Scholar]
Ravindran 1997 {published and unpublished data}
- Hunter B. A double‐blind multicentre study in primary care comparing paroxetine and clomipramine in patients with depression with associated anxiety. European College of Neuropsychopharmacology Congress (ECNP), Venice. 1995. [DOI] [PubMed]
- Ravindran AV, et al. A double‐blind, multicenter study in primary care comparing paroxetine and clomipramine in subjects with depression and associated anxiety. Journal of Clinical Psychiatry 1997;58:112‐8. [DOI] [PubMed] [Google Scholar]
Sacchetti 2002 {published and unpublished data}
- Sacchetti E, Cassano GB, Penati G, Pavan L, Nardini M, Casacchia M, et al. Paroxetine versus amitriptyline in patients with recurrent major depression: a double‐blind trial. International Journal of Psychiatry in Clinical Practice 2002;6:23‐9. [DOI] [PubMed] [Google Scholar]
SBK‐115 1998 {unpublished data only}
- GlaxoSmithKline. A multicenter, randomized, double‐blind, placebo‐controlled comparison of paroxetine and fluoxetine in the treatment of major depressive disorder. www.gsk‐clinicalstudyregister.com.
Schatzberg 2002 {published data only}
- Schatzberg AF, Kremer C, Rodrigues HE, Murphy GM Jr, Mirtazapine vs. Paroxetine Study Group. Double‐blind randomized comparison of mirtazapine and paroxetine in elderly depressed patients. American Journal of Geriatric Psychiatry September‐October 2002;10(5):541‐50. [PubMed] [Google Scholar]
Schnyder 1996 {published data only}
- Schnyder U. Sudden recovery from depression under treatment with paroxetine. European Journal of Psychiatry 1996;10(3):184‐7. [Google Scholar]
- Schnyder U, Koller‐Leiser A. A double‐blind, multicentre study of paroxetine and maprolitine in major depression. Canadian Journal of Psychiatry 1996;41:239‐44. [DOI] [PubMed] [Google Scholar]
Sechter 2004 {published data only}
- Sechter D, Vandel P, Weiller E, Pezous N, Cabanac F, Tournoux. A comparative study of milnacipran and paroxetine in outpatients with major depression. Journal of Affective Disorders 2004;83:233‐6. [DOI] [PubMed] [Google Scholar]
- Vandel P, Sechter D, Weiller E, Pezous N, Cabanac F, Tournoux A, et al. Post‐treatment emergent adverse events in depressed patients following treatment with milnacipran and paroxetine. Human Psychopharmacololy 2004;19:585‐6. [DOI] [PubMed] [Google Scholar]
SER‐CHN‐1 {unpublished data only}
- GlaxoSmithKline. Antidepressant efficacy and safety of paroxetine; a double blind amitriptyline controlled multicenter comparison study in depressive patients. www.gsk‐clinicalstudyregister.com 1994.
Shillingford 1990 {unpublished data only}
- Shillingford JS, Shrivastava SH, Overweg N, Blumhardt CL. A double‐blind comparison of paroxetine and dothiepin on efficacy and tolerability in depressed community patients. Collegium Internationale Neuropsychopharmacologium Congress, Kyoto, Japan 1990.
Shinkai 2004 {published data only}
- Shinkai K, Yoshimura R, Ueda N, Okamoto K, Nakamura J. Associations between baseline plasma MHPG (3‐methoxy‐4‐hydroxyphenylglycol) levels and clinical responses with respect to milnacipran versus paroxetine treatment. Journal of Clinical Psychopharmacology 2004;24(1):11‐7. [DOI] [PubMed] [Google Scholar]
Shrivastava 1992 {published and unpublished data}
- Shrivastava RK, Shrivastava P, Overweg N, Blumhardt C. A double blind comparison of Paroxetine, Imipramine and placebo in Major Depression. Journal of Clinical Psychiatry 1992;53(2 (Suppl)):48‐51. [PubMed] [Google Scholar]
Staner 1995 {published data only}
- Staner L, Kerkhofs M, Detroux D, Leyman S, Linkowski P, Mendlewicz J. Acute, subchronic and withdrawal sleep EEG changes during treatment with paroxetine and amitriptyline: a double‐blind randomized trial in major depression. Sleep 1995;18:470‐7. [PubMed] [Google Scholar]
Steinmeyer 1992 {published data only}
- Moller HJ, Berzewski H, Eckmann F, Gonzalves N, Kissling W, Knorr W, et al. Double‐blind multicenter study of paroxetine and amitriptyline in depressed patients. Pharmacopsychiatry 1993;26:75‐8. [DOI] [PubMed] [Google Scholar]
- Moller HJ, Steinmeyer EM. Are serotonergic reuptake inhibitors more potent in reducing suicidality? An empirical study on paroxetine. Eurpean Neuropsychopharmacology 1994;4:55‐9. [DOI] [PubMed] [Google Scholar]
Stott 1993 {published data only}
- Stott PC, Blagden MD, Aitken CA. Depression and associated anxiety in primary care: a double‐blind comparison of paroxetine and amitriptyline. European Neuropsychopharmacology 1993;3:324‐5. [Google Scholar]
Stuppaeck 1994 {published data only}
- Stuppaeck CH, Geretsegger C, Whitworth AB, Schubert H, Platz T, Konig P, et al. A multicentre double‐blind trial of paroxetine versus amitriptyline in depressed inpatients. Journal Clinical Psychopharmacology 1994;14:241‐6. [PubMed] [Google Scholar]
Szegedi 1995 {published and unpublished data}
- Szegedi A, Wetzel H, Angersbach D, Philipp M, Benkert O. Response to treatment in minor and major depression: results of a double‐blind comparative study with paroxetine and maprotiline. Journal of Affective Disorders 1997;45:167‐78. [DOI] [PubMed] [Google Scholar]
- Szegedi A, Wetzel H, Angersbach D, Philipp M, Benkert O, Dunbar GC, et al. A double‐blind study comparing paroxetine and maprotiline in depressed outpatients. Pharmacopsychiatry 1997;30:97‐105. [DOI] [PubMed] [Google Scholar]
Szegedi 2005 {published data only}
- Szegedi A, Kohnen R, Dienel A, Kieser M. Acute treatment of moderate to severe depression with hypericum extract WS 5570 (ST John's wort): randomised controlled double‐blind non‐inferiority trial versus paroxetine. BMJ 2005;330:503. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tignol 1993 {published and unpublished data}
- Tignol J. A double‐blind, randomised, multicentre study comparing paroxetine 20mg daily versus fluoxetine 20mg daily in the treatment of adults with major depression. Clinical Neuropharmacology 1992;15:177. [Google Scholar]
- Tignol J. Double‐blind, randomized, fluoxetine‐controlled multicenter study of paroxetine in the treatment of depression. Journal of Clinical Psychopharmacology 1993;6(2):18S‐22S. [DOI] [PubMed] [Google Scholar]
- Tignol J, et al. A double blind, randomized, multicentre study comparing paroxetine 20 mg daily in te treatment of adults with major depression with regard to antidepressant efficacy, tolerance and anxiolytic effect [Etude multicentrique randomisee, en duoble aveugle comparant la paroxetine 20mg a fluoxetine 20mg chez des patients depressif hopitalises]. Acta med Int Psychiatr 1994;12:2558‐62. [Google Scholar]
Wade 2003 {published data only}
- Wade A, Crawford GM, Angus M, Wilson R Hamilton L. A randomized, double‐blind, 24‐week study comparing the efficacy and tolerability of mirtazapine and paroxetine in depressed patients in primary care. International Clinical Psychopharmacology 2003;18:133‐41. [DOI] [PubMed] [Google Scholar]
Waintraub 2002 {published data only}
- Waintraub L, Septien L, Azoulay P. Efficacy and safety of tianeptine in major depression. CNS Drugs 2002;16(1):65‐75. [DOI] [PubMed] [Google Scholar]
Weihs 2000 {published and unpublished data}
- Weihs KL, Settle EC Jr, Batley SR, Houser TL, Donahue RM, Ascher JA. Bupropion sustained release versus paroxetine for the treatment of depression in the elderly. Journal of Clinical Psychiatry 2000;61:196‐202. [DOI] [PubMed] [Google Scholar]
Yoshimura 2007 {published data only}
- Yoshimura R, Mitoma M, Sugita A, Hori H, Okamoto T, Umene W, et al. Effects of paroxetine or milnacipran on serum brain‐derived neurotrophic factor in depressed patients. Progress in Neuro‐Psychopharmacology and Biological Psychiatry 2007;31(5):1034‐7. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Arminen 1992 {published data only}
- Arminem S‐L, Ikonen U, Pulkkinen M, Leinonen E, Mahlanen A, Koponen H, et al. Paroxetine and imipramine:a 12‐week, double blind, multicentre study in hospitalized depressed patient. Nordic Journal of Psichiatry 1992;46, Suppl. 27:27‐31. [Google Scholar]
- Arminen SL, Ikonen U, Pulkkinen P, Leinonen E, Mahlanen A, Koponen H, et al. A 12‐week double‐blind multi‐centre study of paroxetine and imipramine in hospitalized depressed patients. Acta Psychiatrica Scandinavica 1994;89(6):382‐9. [DOI] [PubMed] [Google Scholar]
Benkert 1997 {published data only}
- Benkert O, Szegedi A, Wetzel H, Staab HJ, Meister W, Philipp M. Dose escalation vs continued doses of paroxetine and maprotiline: a prospective study in depressed out‐patients with inadequate treatment response. Acta Psychiatrica Scandinavica 1997;95(4):288‐96. [DOI] [PubMed] [Google Scholar]
Bird 2000 {published data only}
- Bird H, Broggini M. Paroxetine versus amitriptyline for treatment of depression associated with rheumatoid arthritis: A randomized, double blind, parallel group study. Journal of Rheumatology 2000;27(12):2791‐7. [PubMed] [Google Scholar]
Cocchi 1997 {published data only}
- Cocchi R. Paroxetine vs amitryptiline in depressed alcoholics. 10th European College of Neuropsychopharmacology Congress. Vienna, Austria. 1997.
Ferrando 1997 {published data only}
- Ferrando SJ, Goldman JD, Charness WE. Selective serotonin reuptake inhibitor treatment of depression in symptomatic HIV infection and AIDS: Improvements in affective and somatic symptoms. General Hospital Psychiatry 1997;19(2):89‐97. [DOI] [PubMed] [Google Scholar]
Gulseren 2005 {published data only}
- Gulseren L, Gulseren S, Hekimsoy Z, Mete L. Comparison of fluoxetine and paroxetine in type II diabetes mellitus patients. Archives of Medical Research 2005;36(2):159‐65. [DOI] [PubMed] [Google Scholar]
Hazleman 1997 {published data only}
- Hazleman BL. A double blind, multicentre study to assess the efficacy and tolerability of paroxetine and amitriptyline in patients with a mild, moderate or severe depressive episode associated with rheumatoid arthritis. National Research Register 1997.
Katona 1998 {published data only}
- Katona C, Hunter B, Judge R. A double‐blind comparison of the efficacy and safety of paroxetine and imipramine in the treatment of depression with dementia. Xth World Congress of Psychiatry, Madrid, Spain. 1996. [DOI] [PubMed]
- Katona CL, Hunter BN, Bray J. A double‐blind comparison of the efficacy and safely of paroxetine and imipramine in the treatment of depression with dementia. International Journal of Geriatric Psychiatry 1998;13(2):100‐8. [DOI] [PubMed] [Google Scholar]
Poirier 1997 {published data only}
- Poirier MF. The concept of resistant depression and therapeutic strategies, particularly with venlafaxine. L'Encephale 1999;25(2):55‐7. [PubMed] [Google Scholar]
- Poirier MF. Venlafaxine versus paroxetine in the treatment of resistant depression. Sixth World Congress of Biological Psychiatry, Nice, France. 1997.
- Poirier MF, Boyer P. Venlafaxine and paroxetine in treatment‐resistant depression. Double‐ blind, randomised comparison. British Journal of Psychiatry 1999;174:12‐6. [DOI] [PubMed] [Google Scholar]
- Poirier MF, Boyer P. Venlafaxine versus paroxetine for treatment resistant depression. XXIst Collegium Internationale Neuro psychopharmacologicum, Glasgow, Scotland. 12th 16th July, 1998.
- Schaeffer P, Poirier FM, Boyer P. Double‐blind trial of venlafaxine and paroxetine for treatment‐resistant depression. 11th European College of Neuropsychopharmacology Congress. Paris, France. 1998.
Rapaport 2003 {published data only}
- Rapaport MH, Schneider LS, Dunner DL, Davies JT, Pitts CD. Efficacy of controlled‐release paroxetine in the treatment of late‐life depression. Journal of Clinical Psychiatry 2003;64(9):1065‐74. [DOI] [PubMed] [Google Scholar]
Sacchetti 1997 {published data only}
- Sacchetti E, Conte G, Guarneri L, Calzeroni A, Bertini M, Panariello A. Effectiveness of fluvoxamine and paroxetine in major depressives with psychotic features. Human Psychopharmacology 1997;12:277‐8. [Google Scholar]
SAD‐PD 2004 {published data only}
- Richard I, McDonald W, Pearson N, LaDonna K, Guerrette B, Hui J, et al. Study of antidepressants in Parkinson’s Disease (SAD‐PD). ClinicalTrials.gov 2004.
Zanardi 1996 {published data only}
- Zanardi R, Franchini L, Gasperini M, Perez J, Smeraldi E. Double‐blind controlled trial of sertraline versus paroxetine in the treatment of delusional depression. American Journal of Psychiatry 1996;153(12):1631‐3. [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
29060/III/83/12 {unpublished data only}
- Glaxo SmithKline. A study to assess the effectiveness and tolerance of paroxetine by double blind comparison with placebo and mianserin. www.gsk‐clinicalstudyregister.com Accessed 26/10/2012.
Ballus 1997 {published data only}
- Ball s C, Quiros G, Flores T, Torre J, Palao D, Rojo L, et al. The efficacy and tolerability of venlafaxine and paroxetine in outpatients with mild to moderate depression or dysthimia. 10th European College of Neuropsychopharmacology Congress. Vienna, Austria. 1997.
- Ballus C, Quiros G, Flores T, Torre J, Palao D, Rojo L, et al. The efficacy and tolerability of venlafaxine and paroxetine in outpatients with mild to moderate depression or dysthimia. 11th European College of Neuropsychopharmacology Congress. Paris, France. 1998.
Benattia 2004 {published data only}
- Benattia I, Musgnung J, Graepel J. Treatment algorithms in depression: Remission with venlafaxine extended release versus SSRIs. 157th Annual Meeting of the American Psychiatric Association, New York, NY. 2004.
Blackwell 1967 {published data only}
- Blackwell B, Shepherd M. Early evaluation of psychotropic drugs in man. A trial that failed. Lancet 1967;2(520):819‐22. [DOI] [PubMed] [Google Scholar]
Chen 2001 {published data only}
- Chen H, Chen S, Li X, Lin J. A controlled study of the efficacy of venlafaxine capsules and paroxetine tablets in the treatment of depression. Herald of Medicine 2001;20(8):488‐9. [Google Scholar]
Deuschle 2003 {published data only}
- Deuschle M, Hamann B, Meichel C, Krumm B, Lederbogen F, Kniest A, et al. Antidepressive treatment with amitriptyline and paroxetine: effects on saliva cortisol concentrations. Journal of Clinical Psychopharmacology 2003;23(2):201‐5. [DOI] [PubMed] [Google Scholar]
- Deuschle M, Kniest A, Niemann H, Erb‐Bies M, Colla N, Hamann B, Heuser I. Impaired declarative memory in depressed patients is slow to recover: clinical experience. Pharmacopsychiatry 2004;37(4):147‐51. [DOI] [PubMed] [Google Scholar]
- Deuschle M, Krumm B, Bindeballe N, Colla M, Hamann B, Lederbogen F, et al. Open‐label non‐randomized versus double‐blind randomized antidepressive treatment: what are the advantages of clinical decision over randomization?. Pharmacopsychiatry 2004;37(6):299‐302. [DOI] [PubMed] [Google Scholar]
- Deuschle M, Luppa P, Gilles M, Hamann B, Heuser I. Antidepressant treatment and dehydroepiandrosterone sulfate: Different effects of amitriptyline and paroxetine. Neuropsychobiology 2004;50(3):252‐6. [DOI] [PubMed] [Google Scholar]
- Deuschle M, Luppa P, Hamann B, Nonell A, Heuser I. Increased concentration of corticosteroid‐binding globulin due to antidepressant treatment with amitritpyline, but not paroxetine. Journal of Psychiatric Research 2003;37(1):85‐7. [DOI] [PubMed] [Google Scholar]
- Kopf D, Westphal S, Luley CW, Ritter S, Gilles M, Weber‐Hamann B, et al. Lipid metabolism and insulin resistance in depressed patients: significance of weight, hypercortisolism, and antidepressant treatment. Journal of Clinical Psychopharmacology 2004;24(5):527‐31. [DOI] [PubMed] [Google Scholar]
Feng 2005 {published data only}
- Feng G, Xie W. A comparative study of paroxetine and clomipramine in the treatment of depression. Chinese Journal of Health Psychology 2005;13(1):35‐6. [Google Scholar]
Geiger 1998 {unpublished data only}
- Geiger ME, McCafferty J, Gergel I. Paroxetine: the preferred choice of patients in the treatment of depression.. 11th European College of Neuropsychopharmacology Congress. Paris, France. 1998.
- McCafferty JP, Oakes R, Gergel IP. Patient preference for paroxetine antidepressant medication. XXIst Collegium Internationale Neuro psychopharmacologicum, Glasgow, Scotland. 1998.
Gonul 1999 {unpublished data only}
- Gonul AS, Yabanoglu I, Reyhancan M, Oguz A. Selective serotonin reuptake inhibitors: discontinuation rates due to side effects. European Neuropsychopharmacology 1999;9(Suppl 5):215. [Google Scholar]
Gou 2002 {published data only}
- Gou Q, Feng E, Ren H. A clinical control study of venlafaxine and paroxetine in the treatment of depression. Journal of Chinese Civil Administration Medicine 2002;14(6):354‐5. [Google Scholar]
Guan 2001 {published data only}
- Guan N, Zhang J, Han Z. A study on quality of life and cost of treatment for depression with sertraline, fluoxetine and paroxetine. Chinese Journal of Behavioral Medical Science 2002;11(6):637‐8. [Google Scholar]
Gurovich 1997 {published data only}
- Gurovich IY, Shmukler AB, Storozhakova YA. Psycho‐social evaluation of antidepressive medication efficacy (quality‐of‐life approach). WPA Thematic Conf Jerusalem. 1997.
Han 2004 {published data only}
- Han Y, Ma Z, Wan Y. A controlled study of trazodone and paroxetine in the treatment of depression. Health Psychology Journal 2004;12(3):174‐5. [Google Scholar]
Hang 2005 {published data only}
- Hang R, Xu P, Wang R. A comparative study of mirtazapine and paroxetine in the treatment of depression. Shandong Archives of Psychiatry 2005;18(4):225‐6. [Google Scholar]
Henning 2002 {published data only}
- Henning O, Niedermaier N, Kniest A, Heuser I, Deuschle M. Amitriptyline and paroxetine: effects upon peripheral nervous system (PNS). Journal of Clinical Psychopharmacology 2002;22(2):229‐30. [DOI] [PubMed] [Google Scholar]
Huang 2006 {published data only}
- Huang K, Luo J, Ran J. Control study of paroxetine and imipramine in the treatment of depression. Medical Journal of Chinese People Health 2006;18(5):343‐55. [Google Scholar]
Jakitowicz 2000 {unpublished data only}
- Jakitowicz J, Nowicki Z, Pankewicz P, Wisniewski G. Pharmaco‐EEG profile of tianeptine (Coaxil) and paroxetine (Seroxat) in depressive patients. European Neuropsychopharmacology 2000;10(Suppl 2):51. [Google Scholar]
Lemoine 1998 {published data only}
- Lemoine P, Smeraldi E, Palomo T, Cosson J‐P, Zibellini M, Rico‐Villademoros F, Marcus RN. Nefazodone versus paroxetine in depressed outpatients. 151st Annual Meeting of the American Psychiatric Association. Toronto, Ontario, Canada. 1998.
Li 2001 {published data only}
- Li Z, Ma Y. A contrast study of paroxetine and amitriptyline in the treatment of depression. Journal of Henan University 2001;20(2):21‐2. [Google Scholar]
Li 2003 {published data only}
- Li W, Li Z, Lu J. A comparative study between paroxetine and amitriptyline in the treatment of depression. Shandong Archives of Psychiatry 2003;16(1):17‐8. [Google Scholar]
Li 2004 {published data only}
- Li Y, Zhang XN, Wu ZM. Comparative study on the effects of paroxetine and venlafaxine in treating depression. Chinese Journal of Clinical Rehabilitation 2004;8(21):4174‐5. [Google Scholar]
Li 2006 {published data only}
- Li D, Wang C, Yu J. A comparative study of citalopram and paroxetine in the treatment of depression. Medical Journal of Chinese People Health 2006;18(6):460‐1. [Google Scholar]
Liao 2002 {published data only}
- Liao R, Wen Y. Pharmcy‐economic evaluation of fluoxetine, paroxetine, and venlafaxine in treatment of major depression. Strait Pharmaceutical Journal 2002;14(6):80‐2. [Google Scholar]
Liu 2002 {published data only}
- Liu YH, Xu MX. A comparative study of paroxetine and amitriptyline in the treatment of senile melancholia. Journal of Linyi Medical College 2002;24(5):321‐3. [Google Scholar]
Liu 2005 {published data only}
- Liu W, Gao H. A study of venlafaxine and paroxetine in the treatment of depression. Medical Journal of Chinese People Health 2005;17(12):723‐4, 727. [Google Scholar]
Ma 2000 {published data only}
- Ma J, Wei H, Niu Y. Clinical comparative study on paroxetine in treatment of depression. Herald of Medicine 2000;19(2):144‐5. [Google Scholar]
Mertens 1988 {published data only}
Montoya 1998 {published data only}
- Montoya F, Ontiveros A, Valdes M, Costilla A. Sexual dysfunction on imipramine and paroxetine. 151st Annual Meeting of the American Psychiatric Association. Toronto, Ontario, Canada. 1998.
- Ontiveros A, Lugoleos J. A double‐blind study with paroxetine and imipramine. 152nd Annual Meeting of the American Psychiatric Association, Washington, DC. 1999.
Peng 2004a {published data only}
- Peng J, Xu Y, Piao S. A comparative study of mirtazapine and paroxetine for depressed patients. Medical Journal of Chinese People Health 2004;16(11):664‐5. [Google Scholar]
Peng 2004b {published data only}
- Peng J, Xu Y, Piao S. A comparative study of tianeptine and paroxetine for depressive patients. Shandong Archives of Psychiatry 2004;14(4):195‐6, 199. [Google Scholar]
Qiao 2005 {published data only}
- Qiao J, Yu J, Hao Z. Comparative study of citalopram and paroxetine in treatment of depression. Journal of Clinical Psychological Medicine 2005;15(5):281‐2. [Google Scholar]
R228060 2004 {published data only}
- Anon. A 6‐ week, randomized, double‐blind, parallel‐group, active‐and placebo‐controlled trial to assess the efficacy of R228060 in adult subjects with major depressive disorder (MDD). ClinicalTrials.gov 2004.
Ren 2004 {published data only}
- Ren H, Guo Q, Cheng M. A study of mirtazapine and paroxetine in the treatment of depression. Journal of Clinical Psychological Medicine 2004;14(2):88‐9. [Google Scholar]
Salzman 1993 {unpublished data only}
- Salzman C, Jimerson D, Vasile R, et al. Predictors of serotonergic antidepressant response. Clinical Neuropharmacology 1992;15:437. [Google Scholar]
Serretti 2001 {published data only}
- Serretti A, Zanardi R, Galbusera E, Cusin C, Rossini D, Lattuada E, et al. Genetics of the response to antidepressants in mood disorders. Giornale Italiano di Psicopatologia 2001;7(3):244‐58. [Google Scholar]
Shu 2004 {published data only}
- Shu D, Zhang K, He H. Control study of paroxetine and amitriptyline in treatment of aged depression. Modern Medicine and Health 2011;20(5):311. [Google Scholar]
Song 2004 {published data only}
- Song R, Liu Y. A comparative study of paroxetine and mianserin in the treatment of senile depresison. Shandong Archives of Psychiatry 2004;17(3):144‐6. [Google Scholar]
Starmer 1996 {unpublished data only}
- Starmer G, Mascord D. Comparison of effects of paroxetine and amitriptyline on driving and psychomotor performance in depression. XXth Collegium Internationale Neuro psychopharmacologicum, Melbourne, Australia. 1996.
Su 2005 {published data only}
- Su H, Lei D, Yu H. Comparative study of mirtazapine and paroxetine in the treatment of depression. Modern Medicine and Health 2005;21(12):1480‐1. [Google Scholar]
Sun 2001 {published data only}
- Sun Z, Yu X. A comparative study of paroxetine and amitriptyline in the treatment of depression. Shandong Archives of Psychiatry 2001;14(4):226‐8. [Google Scholar]
Tao 2005 {published data only}
- Tao W, Lv C, Li W. A comparative study between citalopram and paroxetine in the treatment of depression. Medical Journal of Chinese People Health 2005;17(9):491‐2. [Google Scholar]
Tao 2006 {published data only}
- Tao L. Comparative study between tianeptine and paroxetine in treatment of depression. Journal of Clinical Psychological Medicine 2006;16(4):219‐20. [Google Scholar]
Wang 2003 {published data only}
- Wang Q, Zhou Z. A comparative study of fluoxetine, paroxetine and venlafaxine in the treatment of depression. Health Psychology Journal 2003;11(4):255‐6, 259. [Google Scholar]
Wang 2004 {published data only}
- Wang X, Ma A, Sun H. A comparative study of paroxetine versus tricyclic antidepressants for depression. Journal of Clinical Psychological Medicine 2004;14(1):14‐5. [Google Scholar]
Wang 2005 {published data only}
- Wang X, Zhou D. A comparative study of domestic and imported products of paroxetine on efficacy and safety in patients with major depression. Shanghai Archives of Psychiatry 2005;17(6):334‐6. [Google Scholar]
Wang 2007 {published data only}
- Wang D, Wang X, Li X. Comparison between the effects of fluoxetine and paroxetine on the life quality of the depressive disorder. Nervous Diseases and Mental Health 2007;7:23‐5. [Google Scholar]
Wieck 2001 {published data only}
- Wieck A. Reboxetine versus Paroxetine in treatment of major depressive disorder. National Research Register 2001.
Wu 2000 {published data only}
- Wu XQ, Yang LQ, Lu C, Xiong YY, Liu GX, Ma WT, et al. Comparson of paroxetine and doxepin in depressed patients: a double‐blind randomized study. Pharmaceutical Journal of Chinese People's Liberation Army 2000;16(1):20‐2. [Google Scholar]
Wu 2007 {published data only}
- Wu YS, Chen YC, Lu RB. Venlafaxine vs. paroxetine in the acute phase of treatment for major depressive disorder among Han Chinese population in Taiwan. Journal of Clinical Pharmacy and Therapeutics 2007;32(4):353‐63. [DOI] [PubMed] [Google Scholar]
Xie 1999 {published data only}
- Xie GR, Huang MS, Xue MT, Fan CH. A randomized double‐blind amitriptyline‐controlled study of paroxetine treatment for major depression. Chinese Journal Clinical Pharmacology 1999;15(1):18‐21. [Google Scholar]
Yang 1998 {published data only}
- Yang Q, Li J. Amitriptyline enhanced the effect of the selective serotonin reuptake inhibitors: A pilot trial. XXIst Collegium Internationale Neuro psychopharmacologicum, Glasgow, Scotland. 1998.
Yang 2004 {published data only}
- Yang JZ, Ma HB, Yang SE. Paroxetine vs amitriptyline in treating depression. Chinese Journal of New Drugs and Clinical Remedies 2004;23(5):293‐6. [Google Scholar]
Yang 2006 {published data only}
- Yang X, Wang H, Shao G. Comparative study of reboxetine and paroxetine in the treatment of depression. Journal of Clinical Psychosomatic Diseases 2006;12(6):413‐5. [Google Scholar]
Ye 2002 {published data only}
- Ye Q, Gu XY, Liu JF, Zhao YL. A clinical control study of venlafaxine and paroxetine in the treatment of depression in outpatients. Journal of Clinical Psychological Medicine 2002;12(2):84‐5. [Google Scholar]
Zhang 2003 {published data only}
- Zhang Y, Zhang J, Liang W. A comparative study of venlafaxine extended release vs. paroxetine in treatment of depression. Shanghai Archives of Psychiatry 2003;15(6):341‐3, 327. [Google Scholar]
Zhou 2005 {published data only}
- Zhou J, Xu X, Liu Y. A comparative study of paroxetine and imipramine in the treatment of depression. Medical Journal of Chinese People Health 2005;17(12):731‐2. [Google Scholar]
Zimbroff 2004 {published data only}
- Zimbroff DL, Bose A, Li D. Escitalopram treatment of SSRI nonresponders can lead to remission in patients who fail initial SSRI therapy. 157th Annual Meeting of the American Psychiatric Association, New York, NY. 2004.
Zou 2006 {published data only}
- Zou J, Jing Y, Song K, Hu Y. Comparative study on mirtazapine and paroxetine in the treatment of depression. Journal of Clinical Psychological Medicine 2006;16(4):235‐6. [Google Scholar]
References to ongoing studies
Thomas 2008 {published data only}
- Thomas L, Mulligan J, Mason V, Tallon D, Wiles N, Cowen P, et al. GENetic and clinical Predictors Of treatment response in Depression: the GenPod randomised trial protocol. Trials 2008;9:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Additional references
Als‐Nielsen 2003
- Als‐Nielsen B, Chen W, Gluud C, Kjaergard LL. Association of funding and conclusions in randomized drug trials: a reflection of treatment effect or adverse events?. JAMA 2003;290(7):921‐8. [DOI] [PubMed] [Google Scholar]
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313(7066):1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Anderson 2000
- Anderson IM, Nutt DJ, Deakin JF. Evidence‐based guidelines for treating depressive disorders with antidepressants: a revision of the 1993 British Association for Psychopharmacology guidelines. Journal of Psychopharmacology 2000;14(1):3‐20. [DOI] [PubMed] [Google Scholar]
APA 1994
- American Psychiatric Association. .. Diagnostic and Statistical Manual of Mental Disorders (DSM‐IV). 4th Edition. Washington, DC: American Psychiatric Association, 1994. [Google Scholar]
APA 2000
- American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder (revision). American Journal of Psychiatry 2000;157(4 Suppl):1‐45. [PubMed] [Google Scholar]
Barbui 2004
- Barbui C, Cipriani A, Brambilla P, Hotopf M. "Wish bias" in antidepressant drug trials?. Journal of Clinical Psychopharmacology 2004;24(2):126‐30. [DOI] [PubMed] [Google Scholar]
Barbui 2008
- Barbui C, Furukawa TA, Cipriani A. Effectiveness of paroxetine in the treatment of acute major depression in adults: a systematic re‐examination of published and unpublished data from randomized trials. Canadian Medical Association Journal 2008;178(3):296‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
Begg 1996
- Begg C, Cho M, Eastwood S, Horton R, Moher D, Olkin I, et al. Improving the quality of reporting of randomized controlled trials. The CONSORT statement. JAMA 1996;276(8):637‐9. [DOI] [PubMed] [Google Scholar]
Bhandari 2004
- Bhandari M, Busse JW, Jackowski D, Montori VM, Schunemann H, Sprague S, et al. Association between industry funding and statistically significant pro‐industry findings in medical and surgical randomized trials. Canadian Medical Association Journal 2004;170(4):477‐80. [PMC free article] [PubMed] [Google Scholar]
Bollini 1999
- Bollini P, Pampallona S, Tibaldi G, Kupelnick B, Munizza C. Effectiveness of antidepressants. Meta‐analysis of dose‐effect relationships in randomised clinical trials. British Journal of Psychiatry 1999;174:297‐303. [DOI] [PubMed] [Google Scholar]
Buchkowsky 2004
- Buchkowsky SS, Jewesson PJ. Industry sponsorship and authorship of clinical trials over 20 years. Annals of Pharmacotherapy 2004;38(4):579‐85. [DOI] [PubMed] [Google Scholar]
Cipriani 2009
- Cipriani A, Furukawa TA, Salanti G, Geddes JR, Higgins JP, Churchill R, et al. Comparative efficacy and acceptability of 12 new‐generation antidepressants: a multiple‐treatments meta‐analysis. Lancet 2009;373:746‐58. [DOI] [PubMed] [Google Scholar]
Cipriani 2009a
- Cipriani A, Ferla T, Furukawa TA, Signoretti A, Nakagawa A, Churchill R, et al. Sertraline versus other antidepressive agents for depression. Cochrane Database of Systematic Reviews 2009, Issue 2. [DOI: 10.1002/14651858.CD006117.pub4] [DOI] [PubMed] [Google Scholar]
Cipriani 2009b
- Cipriani A, Santilli C, Furukawa TA, Signoretti A, Nakagawa A, McGuire H, et al. Escitalopram versus other antidepressive agents for depression. Cochrane Database of Systematic Reviews 2009, Issue 2. [DOI: 10.1002/14651858.CD006532.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cipriani 2012a
- Cipriani A, Koesters M, Furukawa TA, Nosè M, Purgato M, Omori IM, et al. Duloxetine versus other anti‐depressive agents for depression. Cochrane Database of Systematic Reviews 2012, Issue 10. [DOI: 10.1002/14651858.CD006533.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cipriani 2012b
- Cipriani A, Purgato M, Furukawa TA, Trespidi C, Imperadore G, Signoretti A, et al. Citalopram versus other anti‐depressive agents for depression. Cochrane Database of Systematic Reviews 2012, Issue 7. [DOI: 10.1002/14651858.CD006534.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ciuna 2004
- Ciuna A, Andretta M, Corbari L, Levi D, Mirandola M, Sorio A, et al. Are we going to increase the use of antidepressants up to that of benzodiazepines?. European Journal of Clinical Pharmacology 2004;60(9):629‐34. [DOI] [PubMed] [Google Scholar]
Cohen 1992
- Cohen J. A power primer. Psychological Bulletin 1992;112(1):155‐9. [DOI] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne DR, Altman DG, Higgins JP, Curtin F, Worthington HV, Vail A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Ellis 2004
- Ellis P. Australian and New Zealand clinical practice guidelines for the treatment of depression. Australian and New Zealand Journal of Psychiatry 2004;38(6):389‐407. [DOI] [PubMed] [Google Scholar]
Feighner 1972
- Feighner JP, Robins E, Guze SB, Woodruff RA, Winokur G, Munoz R. Diagnostic criteria for use in psychiatric research. Archives of General Psychiatry 1972;26:57‐63. [DOI] [PubMed] [Google Scholar]
Furukawa 2002a
- Furukawa TA, Guyatt GH, Griffith LE. Can we individualize the 'number needed to treat'? An empirical study of summary effect measures in meta‐analyses. International Journal of Epidemiology 2002;31(1):72‐6. [DOI] [PubMed] [Google Scholar]
Furukawa 2002b
- Furukawa TA, McGuire H, Barbui C. Meta‐analysis of effects and side effects of low dosage tricyclic antidepressants in depression: systematic review. BMJ 2002;325(7371):991‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Furukawa 2005
- Furukawa TA, Cipriani A, Barbui C, Brambilla P, Watanabe N. Imputing response rates from means and standard deviations in meta‐analysis. International Clinical Psychopharmacology 2005;20(1):49‐52. [DOI] [PubMed] [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59(1):7‐10. [DOI] [PubMed] [Google Scholar]
Furukawa 2007
- Furukawa TA, Watanabe N, Omori IM, Montori VM, Guyatt GH. Association between unreported outcomes and effect size estimates in Cochrane meta‐analyses. JAMA 2007;297(5):468‐70. [DOI] [PubMed] [Google Scholar]
Gartlehner 2011
- Gartlehner G, Hansen RA, Reichenpfader U, Kaminski A, Kien C, Strobelberger M, et al. Drug Class Review: Second‐Generation Antidepressants: Final Update 5 Report. Drug Class Reviews 2011. [PubMed]
Germann 2013
- Germann D, Ma G, Han F, Tikhomirova A. Paroxetine hydrochloride. Profiles of Drug Substances, Excipients, and Related Methodology 2013;38:367‐406. [DOI] [PubMed] [Google Scholar]
Gibiino 2012
- Gibiino S, Serretti A. Paroxetine for the treatment of depression: a critical update. Expert Opinion on Pharmacotherapy 2012;13(3):421‐31. [DOI] [PubMed] [Google Scholar]
Guy 1970
- Guy W, Bonato RR. Manual for the ECDEU Assessment Battery.2. Chevy Chase, MD: National Institute of Mental Health, 1970. [Google Scholar]
Hamilton 1960
- Hamilton M. A rating scale for depression. Journal of Neurology, Neurosurgery and Psychiatry 1960;23:56‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hansen 2005
- Hansen RA, Gartlehner G, Lohr KN, Gaynes BN, Carey TS. Efficacy and safety of second‐generation antidepressants in the treatment of major depressive disorder. Annals of Internal Medicine 2005;143(6):415‐26. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Green S, editors. Cochrane Handbook for Systematic Reviews of Interventions. Chichester: John Wiley & Sons, Ltd, 2011. [Google Scholar]
Hróbjartsson 2013
- Hróbjartsson A, Thomsen AS, Emanuelsson F, Tendal B, Hilden J, Boutron I, et al. Observer bias in randomized clinical trials with measurement scale outcomes: a systematic review of trials with both blinded and nonblinded assessors. Canadian Medical Association Journal 2013;ahead of print:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ioannidis 2008
- Ioannidis JP. Interpretation of tests of heterogeneity and bias in meta‐analysis. Journal of Evaluation in Clinical Practice 2008;14(5):951‐7. [DOI] [PubMed] [Google Scholar]
Jüni 2001
- Jüni P, Altman DG, Egger M. Systematic reviews in health care: assessing the quality of controlled clinical trials. BMJ 2001;323:42‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Khan 2003
- Khan A, Khan SR, Walens G, Kolts R, Giller EL. Frequency of positive studies among fixed and flexible dose antidepressant clinical trials: an analysis of the food and drug administration summary basis of approval reports. Neuropsychopharmacology 2003;28(3):552‐7. [DOI] [PubMed] [Google Scholar]
Langendam 2013
- Langendam MW, Akl EA, Dahm P, Glasziou P, Guyatt G, Schünemann HJ. Assessing and presenting summaries of evidence in Cochrane Reviews. Systematic Reviews 2013;2(1):81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lexchin 2003
- Lexchin J, Bero LA, Djulbegovic B, Clark O. Pharmaceutical industry sponsorship and research outcome and quality: systematic review. BMJ 2003;326(7400):1167‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Luborsky 1962
- Luborsky L. Clinician's judgments of mental health. Archives of General Psychiatry 1962;7:407‐17. [DOI] [PubMed] [Google Scholar]
Lundh 2012
- Lundh A, Sismondo S, Lexchin J, Busuioc OA, Bero L. Industry sponsorship and research outcome. Cochrane Database of Systematic Reviews 2012, Issue 12. [DOI: 10.1002/14651858.MR000033.pub2] [DOI] [PubMed] [Google Scholar]
Magni 2013
- Magni LR, Purgato M, Gastaldon C, Papola D, Furukawa TA, Cipriani A, et al. Fluoxetine versus other types of pharmacotherapy for depression. Cochrane Database of Systematic Reviews 2013, Issue 7. [DOI: 10.1002/14651858.CD004185.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Montgomery 1979
- Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. British Journal of Psychiatry 1979;134:382‐9. [DOI] [PubMed] [Google Scholar]
Montgomery 2004b
- Montgomery JH, Byerly M, Carmody T, Li B, Miller DR, Varghese F, et al. An analysis of the effect of funding source in randomized clinical trials of second generation antipsychotics for the treatment of schizophrenia. Controlled Clinical Trials 2004;25(6):598‐612. [DOI] [PubMed] [Google Scholar]
Nakagawa 2009
- Nakagawa A, Watanabe N, Omori IM, Barbui C, Cipriani A, McGuire H, et al. Milnacipran versus other antidepressive agents for depression. Cochrane Database of Systematic Reviews 2009, Issue 3. [DOI: 10.1002/14651858.CD006529.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
NICE 2010
- National Institute for Clinical Excellence. Depression: management of depression in primary and secondary care ‐ NICE guidance. National Institute for Clinical Excellence. National Institute for Clinical Excellence, 2010. [Google Scholar]
Omori 2010
- Omori IM, Watanabe N, Nakagawa A, Cipriani A, Barbui C, McGuire H, et al. Fluvoxamine versus other anti‐depressive agents for depression. Cochrane Database of Systematic Reviews 2010, Issue 3. [DOI: 10.1002/14651858.CD006114.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Oxman 1992
- Oxman AD, Guyatt GH. A consumer's guide to subgroup analyses. Annals of Internal Medicine 1992;116(1):78‐84. [DOI] [PubMed] [Google Scholar]
Perlis 2005
- Perlis RH, Perlis CS, Wu Y, Hwang C, Joseph M, Nierenberg AA. Industry sponsorship and financial conflict of interest in the reporting of clinical trials in psychiatry. American Journal of Psychiatry 2005;162(10):1957‐60. [DOI] [PubMed] [Google Scholar]
Procyshyn 2004
- Procyshyn RM, Chau A, Fortin P, Jenkins W. Prevalence and outcomes of pharmaceutical industry‐sponsored clinical trials involving clozapine, risperidone, or olanzapine. Canadian Journal of Psychiatry ‐ Revue Canadienne De Psychiatrie 2004;49(9):601‐6. [DOI] [PubMed] [Google Scholar]
Purgato 2010
- Purgato M, Barbui C, Cipriani A. Assessing risk of bias in randomized controlled trials. Epidemiologia e Psichiatria Sociale 2010;19(4):296‐7. [PubMed] [Google Scholar]
RevMan 2012 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version Version 5.2. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2012.
Savović 2012
- Savović J, Jones HE, Altman DG, Harris RJ, Jüni P, Pildal J, et al. Influence of reported study design characteristics on intervention effect estimates from randomized, controlled trials. Annals of Internal Medicine 2012;157(6):429‐38. [DOI] [PubMed] [Google Scholar]
Schulz 1995
- Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias. Dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 1995;273(5):408‐12. [DOI] [PubMed] [Google Scholar]
Spitzer 1972
- Spitzer RL, Endicott J, Robins E. Research diagnostic criteria: rationale and reliability. Archives General Psychiatry 1978;35(6):773‐82. [DOI] [PubMed] [Google Scholar]
Sterne 2000
- Sterne JA, Gavaghan D, Egger M. Publication and related bias in meta‐analysis: power of statistical tests and prevalence in the literature. Journal of Clinical Epidemiology 2000;53(11):1119‐29. [DOI] [PubMed] [Google Scholar]
Turner 2012
- Turner L, Shamseer L, Altman DG, Weeks L, Peters J, Kober T, et al. Consolidated standards of reporting trials (CONSORT) and the completeness of reporting of randomised controlled trials (RCTs) published in medical journals. Cochrane Database of Systematic Reviews 2012, Issue 11. [DOI: 10.1002/14651858.MR000030.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ware 1993
- Ware JE, Snow KK, Kosinski M, & Gandek B. SF‐36 Health Survey Manual and Interpretation Guide. Boston, MA: New England Medical Centre, 1993. [Google Scholar]
Watanabe 2011
- Watanabe N, Omori IM, Nakagawa A, Cipriani A, Barbui C, Churchill R, et al. Mirtazapine versus other antidepressive agents for depression. Cochrane Database of Systematic Reviews 2011, Issue 12. [DOI: 10.1002/14651858.CD006528.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
WHO 1992
- World Health Organization. The Tenth Revision of the International Classification of Diseases and Related Health Problems (ICD‐10). Geneva: World Health Organization, 1992. [Google Scholar]
WHO 2006
- World Health Organization. WHO Collaborative Centre for Drug Statistics Methodology. http://www.whocc.no/atcddd/.
WHOQOL Group 1998
- WHOQOL Group. The World Health Organization quality of life assessment (WHOQOL): Development and general psychometric properties. Social Science and Medicine 1998;46(12):1569‐85. [DOI] [PubMed] [Google Scholar]
Wing 1994
- Wing J. Measuring mental health outcomes: a perspective from the Royal College of Psychiatrists. Outcomes into Clinical Practice. London: British Medical Journal Publishing, 1994. [Google Scholar]
Wood 2008
- Wood L, Egger M, Gluud LL, Schulz KF, Jüni P, Altman DG, et al. Empirical evidence of bias in treatment effect estimates in controlled trials with different interventions and outcomes: meta‐epidemiological study. BMJ 2008;336(7644):601‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zimmerman 2004
- Zimmerman M, Posternak MA, Chelminski I. Derivation of a definition of remission on the Montgomery‐Asberg depression rating scale corresponding to the definition of remission on the Hamilton rating scale for depression. Journal of Psychiatric Research 2004;38(6):577‐82. [DOI] [PubMed] [Google Scholar]