

Abstract
Caenorhabditis elegans is an important genetic model for neuroscience studies, used for analyses of how genes control connectivity, neuronal function, and behavior. To date, however, most studies of neuronal function in C. elegans are incapable of obtaining microscopy imaging with subcellular resolution and behavior analysis in the same set of animals. This constraint stems from the immobilization requirement for high-resolution imaging that is incompatible with behavioral analysis using conventional immobilization techniques. Here, we present a novel microfluidic device that uses surface acoustic waves (SAW) as a non-contact method to temporarily immobilize worms for a short period (30 seconds). We optimize the SAW based protocol for rapid switching between free-swimming and immobilized states, facilitating non-invasive analysis of swimming behavior as well as high-resolution synaptic imaging in the same animal. We find that the coupling of heat and acoustic pressure play a key role in the immobilization process. We introduce a proof-of-concept longitudinal study, illustrating that the device enables repeated imaging of fluorescently tagged synaptic receptors in command interneurons and analysis of swimming behavior in the same animals for three days. This longitudinal approach provides the first correlative analysis of synaptic glutamatergic receptors and swimming behavior in aging animals. We anticipate that this device will enable further longitudinal analysis of animal motility and subcellular morphological changes during development and aging in C. elegans.
Introduction
The nematode Caenorhabditis elegans serves as a powerful tool to study a large range of biological processes relevant to human health, including responses to external stimuli, cellular and subcellular development, and the pathology of numerous diseases.1–8 Many of these studies require immobilization of the C. elegans for high resolution imaging and analysis. The most popular methods for immobilizing worms include administering chemical anesthetics9,10 or using polystyrene nanoparticles on agarose pads slowing animals by friction.11 These methods are effective at preventing worms from moving, but often lead to slow or no recovery of worm behavior, physical damage, or unwanted secondary signaling effects. Therefore, conducting longitudinal studies of behavior and cellular microscopy in the same animal have not been feasible. Common developmental and aging studies have applied correlative association of imaging and behavior or circuit activity analysis, involving different sets of animals for each respective assay.12–14 Despite groups of C. elegans being isogenic, the ability to perform behavior and microscopy within the same set of animals will improve our ability to correlate cellular function and behavior, distinguishing between individual variation in response to environmental manipulation and experimental variations. Currently, an immobilization technique that can facilitate this multiplex study design is lacking.
The size of C. elegans and their ability to live for several days in liquid environments15 have made them ideally suited for manipulation by lab-on-a-chip microfluidic devices. The transparent gas permeable and precise micrometer molding of the silicone polydimethylsiloxane (PDMS) allow the engineering of microfluidic devices that control the placement, environment, and behavior of C. elegans animals.16–19 Several devices have been previously described allowing for the controlled, reversible immobilization of C. elegans animals. Generally, they can be described as either passive or active techniques.
Passive immobilization techniques use specific tailoring of microfluidic channel geometry to trap animals temporarily. For example, Hulme et al. introduced a method which used an array of tapered channels that used the pressure difference between the inlet and outlet to physically immobilize worms in the chamber, which could then be released by reversing the pressure.20 Viri et al. described a similar tapered channel approach to study bacterial transit in C. elegans.21 Berger et al. used trapping channels to study programmed cell death in aging C. elegans.22 These techniques are useful when focusing on the high-resolution imaging aspect of long-term C. elegans studies. However, due to the constrained geometry that could potentially induce mechanical stress in animals,21 these groups either did not conduct concurrent behavior studies20,22 or performed them on separate groups of non-immobilized animals.21
Alternatively, active immobilization methods introduce an external force to temporarily cease animal movements. These methods allow for larger channel geometries where animals can swim freely when not actively immobilized. One such mechanical method consists of pressurizing a flexible PDMS membrane to compress the microfluidic channel, therefore trapping animals temporarily.23–25 Mechanical immobilization typically requires specific size constraints and must be adjusted depending on the age or average size of C. elegans animals used. Direct contact from the membrane can also potentially lead to physiological damage. In another study, Chokshi et al. introduced CO2 into a worm chamber that could reversibly immobilize single worms up to one hour.26 However, hypoxia27 has been shown to induce molecular stress signaling in neurons which could be a problem in studies of neurodegeneration and aging. Heating or cooling the animals’ environment has also been shown to temporarily immobilize animals. Heating a microfluidic chamber to a specific knockdown temperature range has been shown to introduce quiescence in animals.28 This approach required a complex integration of laser and electrical systems. Similarly, cooling a chamber could also reduce worm movement enough to allow high resolution imaging.29,30 Yet, this cooling process to reach a sufficient immobilization temperature can be time consuming. Therefore, a simple non-contact method for fast, reversible immobilization of animals is necessary to harness the potential of C. elegans in developmental and aging studies.
Surface acoustic wave (SAW) technology integrated with microfluidic devices has recently gained more traction for a variety of biological applications.31,32 SAW microfluidic devices have previously been used with C. elegans to precisely move free-swimming animals to specified locations in a microfluidic chamber.33 They have also been used to precisely rotate chemically anesthetized C. elegans such that animals could be fluorescently imaged at a required orientation.34 However, these previous applications of SAW have not shown the ability to fully immobilize animals directly.
Here, we introduce a novel SAW microfluidic device to reversibly immobilize a single C. elegans for up to 30 seconds. We optimize the immobilization protocol and demonstrate the ability to rapidly switch between high resolution fluorescent imaging up to the synaptic level and tracking of swimming behavior. Furthermore, we show that the device enables several rounds of immobilization followed by free swimming over a period of 3–4 days, facilitating the quantification of fluorescent neuron intensity and swimming behavior in the same animal as it ages. Compared to existing immobilization methods, the device uses a simple geometry, is easy to fabricate, and requires a limited environmental change inside the device chamber. The immobilization process is gentle and non-invasive, enabling rapid recovery immediately after the acoustic wave is turned off. Altogether, this technology could pave the way toward longitudinal and multiplexed analyses of C. elegans.
Experimental methods
Device design
The schematic of our SAW-integrated worm immobilization device is shown in Fig. 1A. The device consists of rectangular polydimethylsiloxane (PDMS) chamber filled with fluid bonded to a lithium niobate substrate (Fig. 1B). The chamber has a measured height of 80 μm and a width of 500 μm. 200 μm channels connect the chamber to the inlet and outlet. An interdigital transducer (IDT) is fabricated atop the substrate adjacent to the PDMS chamber. When a radio frequency (RF) signal (50 MHz) is input into the IDT, a traveling wave is generated and propagates along the top of the substrate towards the PDMS chamber (Fig. 1C). As the waves reach the fluid in the chamber, the difference in velocity of acoustic propagation along the fluid versus the substrate causes some of the wave energy to leak into the fluid. This leakage propagates as an acoustic radiation force that can be precisely controlled via the power of the RF signal input.28
Fig. 1.
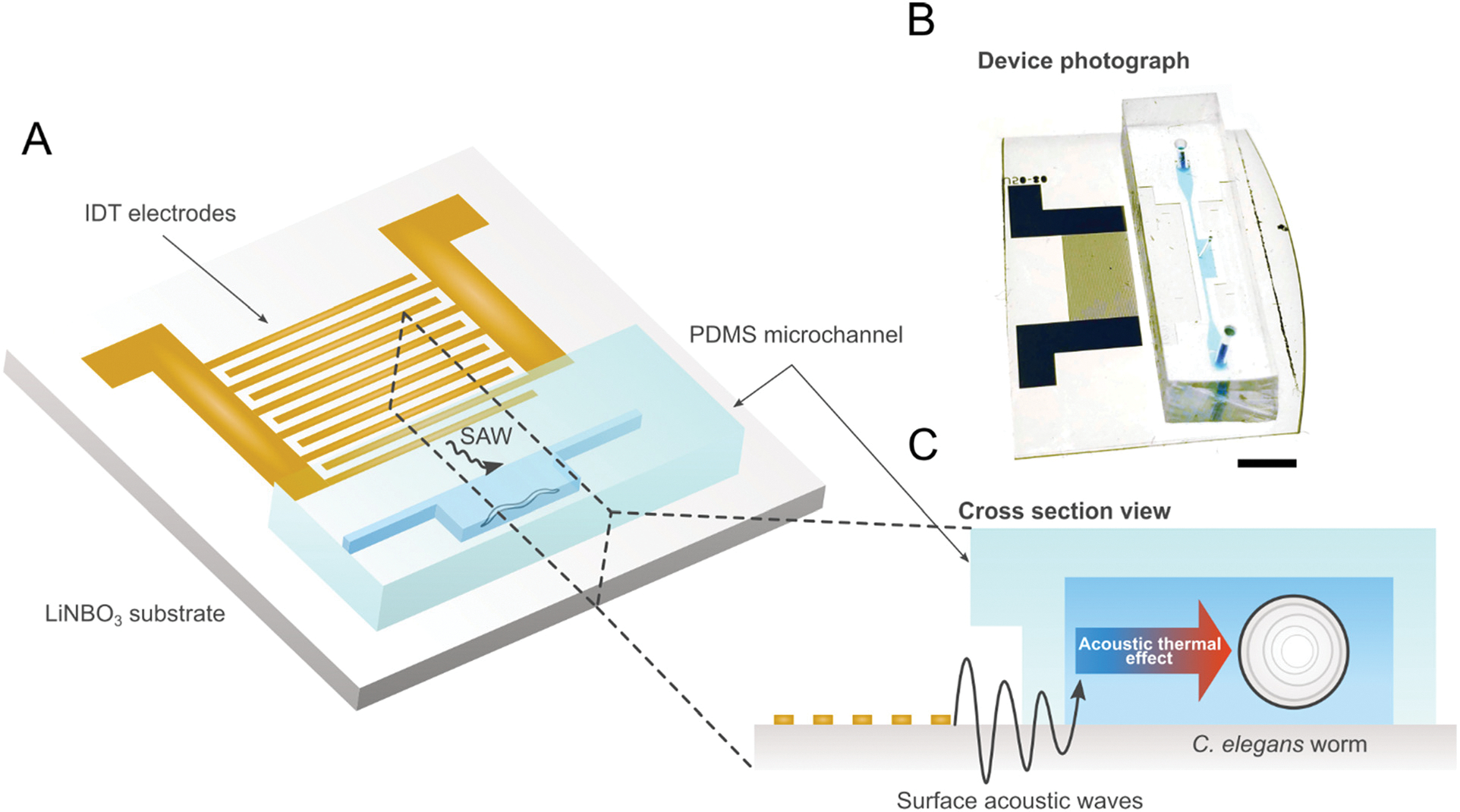
(A) Schematic and (B) photo of SAW device illustrating single IDT adjacent to PDMS worm chamber. Scale bar: 5 mm. (C) Cross-section view of device showing IDT producing traveling SAW acoustic force into chamber to immobilize worms.
Device fabrication
The immobilization device consists of the lithium niobate (LiNbO3) substrate patterned with a single IDT, and the PDMS chamber. The LiNbO3 substrate was patterned onto a 3 inch, Y + 36° X-propagation LiNbO3 wafer using standard photolithography techniques. First, a layer of photoresist (S1813, Dow, USA) was spin-coated onto the wafer. The IDT structures were then patterned onto the wafer using UV light exposure and then developed using a photoresist developer (MF319, Dow, USA). Next, layers of chrome and gold (Cr/Au, 10/100 nm) were deposited using e-beam evaporation. Excess photoresist was removed using a lift-off process (Remover PG, Kayaku, Japan). Finally, the wafer was diced into individual devices each containing a single IDT. Each individual IDT consists of 20 electrode pairs with a spacing of 20 μm and an aperture of 6 mm. The PDMS chamber was made using a negative replica from an SU8 master mold. To fabricate the SU8 mold, an 80 μm thick layer of SU8 2025 photoresist (MicroChem, USA) was spin-coated on a 3 inch silicon wafer. The wafer was then patterned using standard optical lithography. To create PDMS replicas, the master mold surface was first modified by placing it in a silane vapor for 30 minutes. Then, a mixture of PDMS base and crosslinker (Sylgard 184, Dow Corning, USA) with a ratio of 10 : 1 w/w was poured onto the master mold and cured at 65 °C for one hour. Inlet and outlet holes with a 0.75 mm diameter, as well as a 0.35 mm diameter hole in the center of the chamber to measure the temperature in control devices, were punched using a biopsy hole puncher. Finally, the PDMS chamber was bonded to the LiNbO3 substrate using oxygen plasma (PDC-001, Harrick Plasma, USA). To ensure a strong bond, the device was baked overnight at 65 °C.
Strains and maintenance
C. elegans strains were cultured and maintained at 20 °C under standard conditions.35 The initial characterization of the immobilization device and the longitudinal assays was completed using wild-type N2 Bristol strains. Animals used for fluorescent imaging of neurons and synapses either contained the integrated multicopy array nuIs25 (KP1148) or akIs141 (FJH18). nuIs25 and akIs141 both express the C. elegans ionotropic glutamate receptor homologue GLR-1 tagged with GFP either under its native glr-1p36 or AVA specific rig-3p promoters37 (Fig. S1†). Initially, animals used for fluorescence imaging contained the lite-1(ok530) mutation from the RB765 strain to prevent light-induced side effects. The ST66 or (ncIs17 [hsp16.2p::eGFP + pBluescript]) strain was used to measure the biological effect of SAW-induced heat.
Animals
The University of Colorado Boulder and Colorado State University do not require animal care (IACUC) protocols for C. elegans experimentation. Colorado State University invertebrate and transgenic animal care, fall under the review of the Institutional Biosafety Committee (IBC). This committee approves projects based on NIH and CDC/HHS Biosafety in Microbiological and Biomedical Laboratories guidelines (BMBL). As such the C. elegans animals used in this study were approved for use under the following IBC protocol number: 18-043B approved by the committee on January 28, 2022.
Device operation
An RF function generator (N5171B, Keysight, USA) and power amplifier (403LA, E&I, USA) were used to input the signal into the IDT. To identify the resonant frequency of the devices, we used a network analyzer (E5061B, Keysight, USA) in combination with visually tracking the velocity of polystyrene microparticles in the fluid that were pushed by the acoustic radiation force. Depending on the individual device, the resonant frequency was identified as approximately 46 MHz.
Initial characterization of the immobilization device was conducted on an inverted microscope (Eclipse Ti2, Nikon, Japan). Worms in the chamber were imaged with a digital CMOS camera (Orca-Flash 4.0, Hamamatsu, Japan) in combination with the included imaging software (HCImage Live, Hamamatsu, Japan). GFP-B fluorescent filter cubes (495 nm, ET 49002, Chroma, USA) were used to image fluorescent neurons in high resolution. Tracking the worm movement and further processing of images and videos were completed using ImageJ.38 For measuring the temperature inside the chamber, a digital thermocouple was used to track the change when SAW was applied.
All strains were initially synchronized by bleaching with 20% alkaline hypochlorite solution (3 mL bleach + 3.75 mL 1 M NaOH + 8.25 mL H2O). The resulting L1 larvae were transferred to OP50 bacteria seeded NGM plates. For all worm experiments, synchronized animals were allowed to grow until the adult day 1 stage in normal NGM plates. Worms were initially transferred from NGM plates to a microcentrifuge tube by washing with 300 mL M9 buffer. Concentration was then lowered by further diluting with M9 such that it was possible to pick out a single worm with a 10 μL micropipette. The single worm was then manually loaded into the immobilization device using the micropipette.
Immobilization characterization
The locomotion of C. elegans during SAW application was quantified by visually tracking the movement at the tip of the head of the animal inside the device. The Manual Tracking plugin39 in ImageJ was used to manually identify and click the tip of the head at each frame, measuring the distance the tip moved between frames. Tracking was performed on raw images with no post-processing. A worm was defined as immobilized when no noticeable head movement could be tracked over >5 frames.
For each test, 3 cycles of SAW were applied so that the animal was immobilized for 30 seconds. Between each cycle, there was a 3 minute recovery period, allowing the temperature in the chamber to decrease back to room-temperature, during which the worm was allowed to swim freely in the chamber. The head movement of the worm was tracked as the SAW was applied, as well as after the SAW was turned off to illustrate recovery. Various SAW powers and duty cycles were tested.
Quantification of longitudinal worm swimming
Experiments were conducted over a 72 hour period. SAW (2 W-50% duty cycle immobilization, 1 W-50% duty cycle hold) was applied at hour 0, 24, 48 to temporarily immobilize the worm for 30 seconds. SAW treatments were compared to control animals that were allowed to swim freely inside the chamber. Animal swimming was visually analyzed before SAW application at hour 0, 24, 48, and 72. Animals were scored as healthy if they exhibited a response to 10 seconds of blue light and were swimming at an average bending frequency greater than 1 Hz. One full bending cycle was defined as a full wave of dorsal–ventral bends through its body.40 To calculate average bending frequency, total dorsal–ventral bends were visually tracked over 10 seconds after blue light exposure and averaged per second. Every 12 hours, the liquid in the chamber, along with any offspring, was replaced with fresh liquid OP50 in an M9 medium. To prevent any evaporation between tests, devices were stored in a high humidity chamber at room temperature. The same individual animals were tracked over multiple days.
Quantification of longitudinal behavior tracking
Experiments were conducted over a 48 hour period. At hour 0, 24, and 48, worms were tracked for 30 seconds and the average bending frequency over the time frame was calculated in the same manner as described for the longitudinal worm swimming assay. SAW (2 W-50% duty cycle immobilization, 1 W-50% duty cycle hold) was then immediately applied at hour 0, 24, and 48 to temporarily immobilize the worm for 30 seconds for imaging. SAW animals were compared against control animals that were allowed to swim freely in the chamber for the same time frame. Feeding and storage methods were the same as previously described for the longitudinal worm swimming assay.
Quantification of worm deformation
For the single-cycle experiment, SAW (2 W-50% duty cycle immobilization, 1 W-50% duty cycle hold) was applied to temporarily immobilize the worm for 30 seconds. Length and width along the axes of the worm was manually measured immediately before, during, and after SAW immobilization using the measure tool on ImageJ. To calculate the percent change in length and width induced by SAW, the raw measured changes (length and width) during the immobilized state were divided by the initial measurements (length and width) before SAW application. To calculate recovery, the raw measured changes 10 seconds after SAW was turned off were divided by the measurements taken during the immobilized state. For the longitudinal study, the same previously described measurements were taken for a single animal when immobilizing with SAW at hour 0, 24, and 48.
Longitudinal fluorescent imaging of single neurons
Experiments were conducted over a 48 hour period. At hour 0, 24, and 48, SAW (2 W-50% duty cycle immobilization, 1 W-50% duty cycle hold) was applied to temporarily immobilize and hold the worm. Once the worm was visually deemed to be immobilized, imaging was switched from brightfield to wide field GFP fluorescence. Single 500 ms exposure wide field images of glutamatergic command interneurons (including AVA only) were taken using an inverted microscope (Eclipse Ti2, Nikon, Japan). Control images were taken of animals treated with sodium azide (50 mM). Since sodium azide treatment compromised animals’ health, different control animals were tracked as representative examples for fluorescent intensity for each day. For the SAW-treated animals, feeding and storage methods were the same as previously described for the longitudinal worm swimming assay. The fluorescence in the cell body of the command interneuron was quantified in ImageJ using the measure tool and ellipse tool to draw an ROI tightly constrained around the fluorescence of the cell body image at mid-range of the total intensity. The fluorescence within the ROI around the cell body was measured first and then the ROI was displaced in a region of the image without punctate fluorescence for a background measurement, which was subtracted from the cell body fluorescence for all measurements. Quantification of the command interneuron cell body fluorescence was done on all images except on those where movement of the animal could be detected in the images. Movement was defined as any noticeable change in >5 frames.
Temperature characterization
To measure SAW-induced temperature increase, a thermal probe was inserted into the device from above through the 0.35 mm central inlet. An initial standard room temperature of approximately 20 °C for the fluid was established. Then, temperature was tracked for various initial high power SAW applications, as well as switches to low power SAW for holding the immobilization. After the SAW was turned off, the cool-down temperature was tracked along 15–60 second intervals until room temperature was reached. For pure heating-only control tests, devices were placed upon a glass heating plate. The heating plate was turned on and the temperature was increased until the animal was immobilized. The heating plate was then immediately turned off and the fluid in the device was allowed to cool back to room temperature.
For heat stress quantification, SAW (2 W-50% duty cycle immobilization, 1 W-50% duty cycle hold) was applied to temporarily immobilize ST66 animals. Positive control animals were placed in a 32 °C hot bath for 1 hour. Negative control animals had no heat applied to them. Afterwards, all animals were allowed to swim in the chamber for one hour. Animals were then immobilized with sodium azide and single 500 ms exposure images were then taken of the worm bodies. For each image, the average intensity of the image was quantified in ImageJ by using the measure tool and the ellipse tool to draw an ROI around the entire worm body. The background of all images was initially normalized to an equivalent intensity range.
Statistical analysis
To estimate desired sample sizes to compare immobilization protocols and image analysis, we matched previously reported works.25,41 We then used standard statistical tests to determine significance. All statistical calculations were performed using Excel. For all tests of significance between data sets containing numerical means, a two-sided, unpaired Student’s t-test was performed. For tests of significance between categorical data sets, a chi-squared test of independence was performed. Significance was determined as follows: *P < 0.05, **P < 0.01, ***P < 0.001. Linear correlations were measured by calculating Pearson’s correlation coefficient.
Results
SAW induced rapid animal immobilization and instantaneous recovery
First, we visually confirmed the SAW device’s ability to immobilize worms by imaging single brightfield frames with a 100 ms exposure time. While a free-swimming worm produces a blurry image (Fig. 2A), we could see a clear outline of the animal when applying SAW (Fig. 2B). To evaluate the effectiveness and controllability of the device, we then tracked movement at the tip of the head of an individual worm over 3 cycles of immobilization and recovery (Fig. 2C and Video S1†). The immobilization process, initiated in this example with 2 W-50% duty cycle SAW, typically occurred in approximately 60 seconds. Interestingly, we saw that immediately after SAW was turned on, head movement briefly increased before subsequent immobilization for all 3 cycles. This is most likely caused by worms demonstrating avoidance behavior in response to mechanical wave stimuli, which has previously been reported.42,43 Moreover, we noticed a decrease in the movement response to SAW in cycle 2 and cycle 3. This could potentially be an example of worms demonstrating habituation upon repetitive stimulation.43 Switching the SAW to lower power (1 W-50%), the worm could be temporarily held immobilized for more than 30 seconds which is a sufficient timeframe for various high-resolution imaging needs. When the SAW was turned off, in only a few seconds, the worm started thrashing indicating that the worm can rapidly recover after the immobilization process.
Fig. 2.
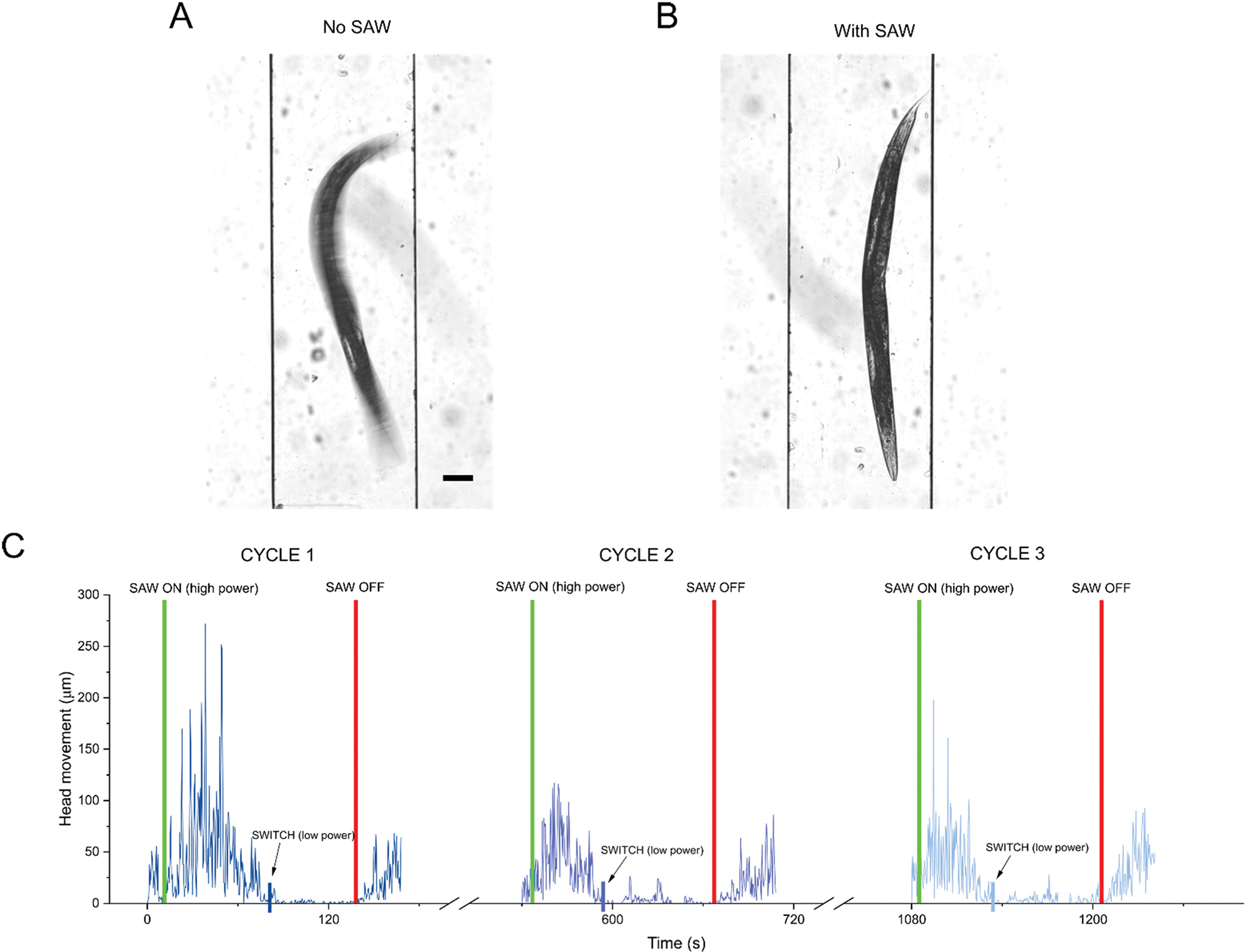
Imaging and characterization of worm immobilization process. Bright field images (100 ms exposure) of single C. elegans animal in chamber comparing (A) free worm swimming when no SAW is applied and (B) immobilization after SAW is applied. Scale bar: 100 μm. (C) Head movement of single representative C. elegans animal over 3 immobilization and recovery cycles (see methods). Initially, higher power SAW (2 W-50% duty cycle) was used to immobilize animals. Once animals stopped moving, SAW was switched to lower power (1 W-50% duty cycle) to hold animals in immobilized state for 30 seconds. Green vertical lines: high power SAW on. Blue vertical lines: switch SAW to lower power. Red vertical lines: SAW off.
We characterized various input powers along with comparing continuous and 50% duty cycles to find the most effective SAW protocol to fully immobilize an animal for a long enough time frame to focus on fluorescent structures of interest and capture images, while still allowing worms to recover after the 3 cycles (Table 1 and Fig. S2†). We found that the best way to extend the immobilization time without negatively affecting worm health was to apply SAW at two successive power levels. First, we applied higher power SAW to initially immobilize the animals. We then immediately switched to a 50% lower power SAW as a hold step to maintain the immobilized state. Keeping the high-power SAW on to hold immobilization reduced the worms’ ability to recover well. Meanwhile, simply turning SAW off without the hold step could only keep animals immobilized for approximately 10 seconds. We compared different combinations of high-power SAW (2 W continuous, 1 W continuous, 2 W-50% duty cycle) and low power SAW (0.5 W continuous, 1 W-50% duty cycle). We found that the best result was achieved using an approach combining both high-power and low-power steps. Thus, to achieve a sufficient immobilization time for imaging while still allowing healthy recovery, we used a 2 W-50% duty cycle step, immediately followed by a 1 W-50% duty cycle step which we designated as our standard protocol and used for all further experiments. Using this combinatorial approach, we were consistently able to hold worms immobilized for over 30 seconds for 3 cycles with 90% of animals swimming at the same rate as prior to SAW immobilization (Table 1). Applying high-power SAW until immobilization (between 30–70 seconds depending on the individual animal), then immediately switching to low-power SAW for 30 seconds using the standard protocol was selected as a safe immobilization length that allowed us enough time to fluorescently image animals under high resolution while still maintaining worm health over multiple cycles.
Table 1.
Swimming worms after 3 immobilization/recovery cycles for different SAW protocols
|
| ||||
|---|---|---|---|---|
| Power (high power/low power, duty cycle) | Worms tested | Swimming worms after 3 cycles | Percent swimming | Max immobilization hold – cycle 1 (s) |
|
| ||||
| 2 W/1 W, 50% duty cycle | 41 | a37*** | 90.2 | b39.13*** |
| 2 W/– – continuous | 14 | 10NS | 71.4 | 8.68 |
| 1 W/– – continuous | 12 | 10NS | 83.3 | 6.84 |
| 2 W/0.5 W continuous | 10 | 5 | 50.0 | 14.98NS |
| 1 W/0.5 W continuous | 10 | 4 | 40.0 | 26.17NS |
***P < 0.001, chi-square test of independence. NS = no significance.
***P < 0.001, two-sided, unpaired t-test. NS = no significance.
SAW immobilization did not lead to heat stress in animals
Generally, it is accepted that sustained exposure to temperatures greater than 25 °C has a negative effect on C. elegans health, reproduction, and lifespan.44–46 However, a previous study by Chuang et al. showed that short term exposure (seconds to minutes) to elevated temperatures in the so-called heat knockdown range of 31–37 °C will induce a temporary shutdown of neural function in worms.28 Once the temperature is lowered, regular neural functions resumed. This effect was shown to be repeatable for multiple cycles. Furthermore, there was no significant effect on worm lifespan and progeny.28
SAW produces a known heating effect along with the acoustic force. We characterized the SAW induced heating of a representative device and showed a temperature increase in the chamber from room temperature (21.7 °C) to 29.1 °C in 30 seconds, 33.1 °C in 60 seconds, and 36.2 °C in 90 seconds when applying high power 2 W-50% duty cycle SAW (Fig. S3†). After switching to the lower power hold step (1 W-50% duty cycle) the temperature stabilized at approximately 32 °C (Fig. S3†). We also measured the minimum temperature at which a worm was immobilized, and found it occurs at 28.1 °C. This is significantly lower than an immobilization temperature of 35 °C recorded when using a heating plate substrate to represent a pure heating effect (Video S2†). Moreover, this temperature is lower than the previously reported heat knockdown immobilization range of 31–37 °C.28 We believe the lower immobilization temperature indicates that both acoustic pressure and heat play a key role in the immobilization mechanism of our device, and therefore imparts less environmental stress on animals than just pure heating. We note that due to the SAW mechanism, it was difficult to decouple acoustic pressure and heating at the demonstrated power levels. However, we found that when we lowered the input power enough such there was a negligible temperature increase, immobilization with acoustic pressure alone was not possible.
To verify that heat stress was not having a significant effect during the immobilization process, we tested our protocol on the ST66 strain containing ncIs17 [hsp16.2p::eGFP] expressing a heat-shock inducible GFP marker. We compared up to 3 cycles of SAW immobilization with a positive control, where animals were placed in a 32 °C hot bath for 1 hour, and a negative control, where no heat was applied. One hour after heat or SAW was applied, we captured fluorescent images of animals immobilized with sodium azide (Fig. S4A†). For the different conditions, we compared the average fluorescent intensity in worm bodies (Fig. S4B†). The results showed that applying SAW for up to three cycles does not induce any significant heat shock response based on expression of GFP from the hsp16.2p::eGFP construct.
Worm immobilization via SAW facilitated high resolution imaging
We used the developed SAW immobilization protocol to capture high resolution fluorescent images of cell bodies of interest. Using a wide-field microscope, we were able to showcase the dynamic usability of the SAW device, which allowed for rapid switching between an immobilized and free-swimming state. Here, a single nuIs25 (KP1148) worm was immobilized with SAW and imaged for 3 cycles (Fig. 3A). In between each cycle, SAW was turned off and the animal was allowed to swim freely for 3 minutes. We proceeded to image single AVA neurons in akIs141 (Fig. 3B). At 40×magnification, we were able to image single synapses containing GFP tagged GLR-1 receptors at high resolution (Fig. 3B). Detailed illustrations of the imaged neurons for both strains can be found in Fig. S1.† The images confirmed that our device protocol was able to effectively immobilize animals for enough time to focus our camera on the cells of interest and use exposures up to 500 ms, which were required for clarity at higher magnifications.
Fig. 3.
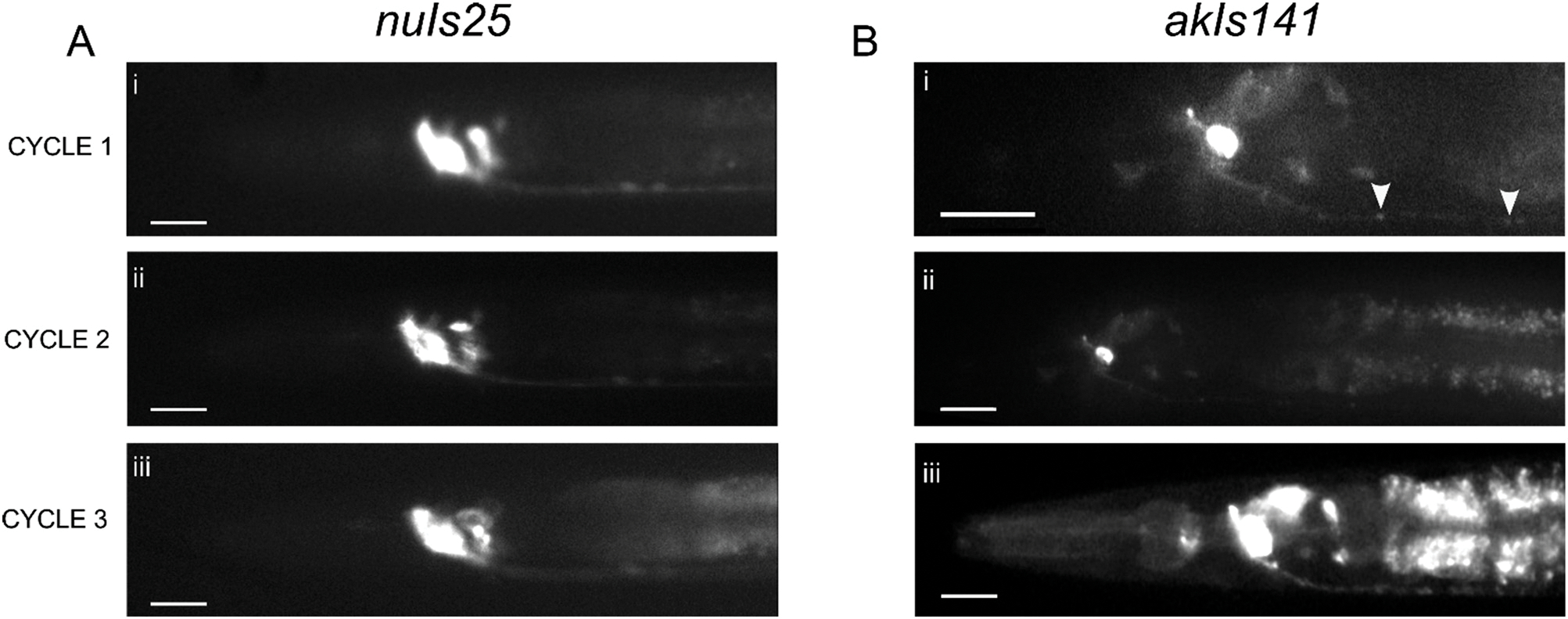
Imaging capability of SAW immobilization device. (A) Wide-field fluorescent images of GFP tagged GLR-1 in (i–iii) a single nuIs25 animal immobilized with SAW taken over 3 cycles, each 3 minutes apart taken with 20× objective. The animal was allowed to swim freely in chamber between each cycle. Scale bar: 20 μm. (B) Wide-field fluorescent images of GFP tagged GLR-1 in a single neuron (AVA) of akIs141 animals with (i) 40× objective and (ii and iii) 20× objective. Synaptic puncta are visible at 40× (arrowheads). Scale bar: 20 μm at 20×, 10 μm at 40×. All images were taken using 500 ms exposure.
SAW immobilization did not affect long term worm survival on-chip
We next tested how animal mobility and health would be affected by multiple immobilization recovery cycles during a span of 72 hours in the microfluidics device. The ratio of healthy worms immobilized with SAW once per day was compared against age matched control animals allowed to freely swim in the device for the same time frame (Fig. 4A). Worms that responded to 10 seconds of blue light stimulus and swimming at an average bending frequency >1 Hz were deemed as healthy.47,48 Over 80% of animals immobilized once a day with SAW survived in the chip over the 72 hour period. The results showed no significant difference in worm health between SAW immobilized and free-swimming control animals, suggesting that SAW application had a negligible impact on long-term worm health. In addition to swimming, a representative animal was imaged under bright field every 24 hours showing no superficial signs of worm stress such as internal organ damage or a clear body (Fig. 4B).
Fig. 4.

Longitudinal analysis of worm mobility and health. (A) Ratio of animals swimming in the chamber over 72 hours comparing animals that were immobilized with SAW once per day (at hour 0, 24, 48) versus control animals in chamber where no SAW was applied. (B) Representative bright field image of the same freely swimming animal in chamber imaged at 0, 24, 48, and 72 hours before SAW immobilization once per day. For A, at least 20 animals were tracked for each condition.
Animals recovered from SAW induced deformation over multiple days
As an additional parameter to quantify the health of animals and their ability to recover from mechanical stimulation, we measured the deformation in C. elegans length and width caused by SAW application. Deformation measurements are a way to provide insight into animals’ ability to contract and relax their muscles in response to external stimuli. Muscle activation has been previously characterized as a key regulator of C. elegans body mechanics.49 As acoustic waves increase pressure in the chamber, we expected to see some deformation in worms when applying SAW. To confirm the presence of SAW-induced deformation, we initially tracked the percent length change (Fig. S5A†) and width change (Fig. S5B†) after turning SAW on, as well as recovery after turning SAW off for one immobilization cycle. On average, worms contracted by 2.3% in length and expanded by 3.1% in width when transitioning to an immobilized state after turning SAW on. After turning SAW off, animals quickly (<10 seconds) showed a recovery trend towards their original state, expanding in length by 1.2% and contracting in width by 2.3%.
We then conducted a similar analysis on worms over multiple days to see if they would show comparable muscular dynamics as they aged. In this case, we tracked the length change (Fig. S5C†) and width change (Fig. S5D†) caused by SAW in the same animals at 0, 24 and 48 hours. At each of the three timepoints tested, we saw similar trends in contraction and expansion as our previous single cycle test. We found that on average across all longitudinal time points, animals recovered 85% percent of their original length and 90% of their original width after SAW treatment. This suggests that the muscular processes in C. elegans are minimally affected by multiple SAW immobilization cycles.
Multiplexed tracking of longitudinal swimming behavior and fluorescent imaging achieved using SAW immobilization
Finally, we demonstrated the potential for our SAW device to be used as a platform to track worm behavior and perform fluorescent microscopy imaging in the same animals over multiple days. For our model, we studied GLR-1 tagged with GFP expressed under its native promoter in command interneurons (see Fig. S1† for a list of neurons and illustration of their position in the head). The command interneurons are critical for the control of locomotory behavior in C. elegans.50,51 By correlating GLR-1 expression and concurrent animal movement, we could in the future gain insight into how aging affects C. elegans synaptic receptor content and neural circuits that control locomotory movements.
For the behavior assay, we analyzed animals at 0, 24, and 48 hours and compared the average bending frequency of single worms in the device that were immobilized with SAW and allowed to recover against control animals that could swim freely in the device (Fig. 5A). For all time points, there was no significant difference between the bending frequency of the SAW and control worms. Both the SAW and control animals exhibited similar trends in their swimming behavior as they aged. Although there was no significant difference in daily average bending frequency for either the SAW or control populations, individual animals for both the SAW and control tests displayed substantial differences in their swimming behavior each day. This considerable day-to-day change in swimming frequency has been previously reported.52 Moreover, for both SAW and control tests, comparing individual animals showed high variability in movement at each time point. For example, at 24 hours, SAW animals demonstrated bending frequencies between 0.49–1.86 Hz, while control animals frequencies ranged from 0.86–1.47 Hz. This substantial individual variability in different animals, despite genetic and environmental homogeneity, has also been previously reported.53
Fig. 5.

Multiplexed tracking of longitudinal behavior and fluorescence. (A) Bending frequency of animals at 0, 24, and 48 hours comparing animals immobilized with SAW once per day versus control animals freely swimming in chamber. (B) Representative wide-field fluorescent images (20×objective, 500 ms exposure) of (i and iii) a single nuIs25 animal immobilized with SAW taken once per day, and (iv–vi) different representative nuIs25 animals immobilized with sodium azide corresponding to the same age. Scale bar: 20 μm. (C) Normalized greyscale intensity in GLR-1::GFP in the cell body of the command interneurons of images taken once a day over 3 days. Note: For animals immobilized with SAW, the same individual animals were imaged over multiple days. For animals immobilized with sodium azide, representative images of different animals for the corresponding age were taken. (D) Correlation plot of normalized greyscale intensity and bending frequency for individual animals immobilized with SAW tracked daily. Data are mean ± s.e.m. For A, at least seven animals were tracked at 0, 24, and 48 hours. For C and D, at least four animals were imaged at 0, 24, and 48 hours for both SAW and sodium azide experiments.
For the fluorescent imaging assay, we used the fluorescent nuIs25 (KP1148) strain in a lite-1(lf) loss of function background (allele: ok530). The lite-1(lf), background was chosen to simplify the immobilization process by reducing the reactions of C. elegans animals to 488 nm light. Once each day, we captured fluorescent images of glutamatergic neuronal cell bodies in the head of immobilized animals (Fig. 5B). We compared the fluorescent intensity of GLR-1::GFP in the cell body of the command interneurons each day by normalizing to the overall average on day 1 (Fig. 5C). SAW immobilization facilitated the tracking of fluorescent intensity of the same individual worms over three days. As a comparison, we imaged control animals immobilized with sodium azide. Animals did not recover well after sodium azide exposure in the device, so we did not track single animals over multiple days. Instead, we imaged representative animals corresponding to the same age as their SAW immobilized counterparts (Fig. 5B). Comparing between SAW and control animals did not show a significant difference in the daily fluorescent intensity (Fig. 5C). Similar to the behavior assay results, different individual animals of the same age demonstrated a wide range of fluorescent intensities. By using SAW to track the same animal over 48 hours, we found that fluorescent intensity tended to decrease in single animals as they aged. However, the overall average for the entire sample did not show a significant difference between days, due to the high variability in fluorescence between different animals. Likewise, the population of age-matched representative control animals showed large variations between animals and no significant difference between days. Due to the lack of recoverability stemming from the sodium azide exposure, we could not track daily fluorescent trends in individual control animals. This limitation readily illustrates the value of SAW immobilization, which allows the tracking of relative fluorescence changes in single animals over time to gain a clearer picture of aging trends. On the other hand, simply analyzing an overall representative population can be complicated by large variations between different animals, even in organisms thought to be isogenic.
To test whether the fluorescent intensity and swimming behavior tracked using SAW showed a linear relationship, we plotted corresponding fluorescent intensity and bending frequencies of individual animals and performed a correlation analysis at each time point (Fig. 5D). At 0 and 24 hours, we found the correlation coefficient to be r = 0.971 and r = 0.768 respectively. At 48 hours, we calculated r = −0.604. Based on this preliminary analysis, we saw that fluorescence and bending were positively correlated on day 1 and day 2. However, by day 3 the positive linear relationship broke down. Previous studies suggest a direct relationship between neuron activity of GLR-1 receptors and locomotion.54,55 The loss of correlation we observed at 48 hours is a new finding that is not well-studied. There are recent reports that indicate aging in C. elegans cause a loss of signaling in neurons and functional decline.56 However, our preliminary results would need to be further repeated with larger datasets to confirm validity. Such a study could be directly enabled with our technology.
Conclusions
This microfluidic SAW device provides a novel non-contact method for repeated, reversible, and temporally limited fast immobilization of individual C. elegans animals. By applying SAW in a precise and controlled manner to a simple PDMS channel, we can use an acoustic approach to immobilize animals for a short period of time, allowing for high resolution fluorescent imaging. This enables the longitudinal analysis of single neurons of individual animals by fluorescence microscopy and their swimming behavior as they age in the same device. Here, we illustrate a proof-of-concept by imaging the glutamate receptor GLR-1 tagged with GFP in individual animals and tracking the fluorescent intensity change and bending frequency of animals over a 48 hour period. Our approach can accommodate longitudinal analysis of worms with improved precision by tracking single animals over multiple days. In comparison, performing similar analysis by using different sets of animals may result in inconsistent measurements (Fig. 5C).
Other microfluidic methods to immobilize C. elegans have previously been reported, such as tapered channels,20–22 flexible membranes23–25 CO2 introduction,26 cooling27 and heating.28 However, they suffer from drawbacks such as geometry constraints and significant environmental changes that may induce stress signaling in animals that could interfere with analysis. Our device facilitates an acoustic immobilization method with a standard channel geometry and a minimal environmental change, allowing for rapid switching between an immobilized and free-swimming state. The simple design of the chip lends itself to straightforward fabrication using standard photolithography techniques. Moreover, devices are indefinitely reusable (>50 cycles tested).
There are certain limitations to consider when using this immobilization method. Although SAW can immobilize worms quickly (~1 min), maintaining the immobilized state for multiple minutes tended to prevent full recoveries. Therefore, if longer-term immobilization is required, other methods may be more suitable. However, our device is an ideal candidate for rapid high-resolution imaging and dynamic behavior screening.
In the future, the device can be further improved by characterizing its compatibility with long working distance objectives and more powerful microscopes to achieve better clarity, particularly at synaptic resolutions. Additionally, an automatic worm loading system to ensure a single worm is quickly and precisely inserted into the immobilization channel can be integrated into the PDMS chamber. The conversion from high to low SAW for microscopy immobilization could be automated based on live tracking of swimming. These improvements could also easily be integrated with a new design incorporating multiple SAW channels allowing higher throughput. Environmental control channels could also be added to the system, allowing for precise inflows for food and chemical delivery. In addition, this system would be perfectly suited for optogenetic approaches controlling neuronal activity in combination with fluorescent imaging and aging. Overall, by allowing a combined analysis of microscopy-based cellular signaling and behavior, our device shows promise for use in future applications for multiplexed C. elegans aging studies.
Supplementary Material
Acknowledgements
The microfluidic devices were fabricated in the JILA clean room at University of Colorado Boulder. Wild-type C. elegans animals and worm culturing media were provided by the Ding Xue Lab at University of Colorado Boulder. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). This work was supported by startup fund, NIH MIRA Award 1R35GM142817, and W.M. Keck Foundation medical research grant to X. Ding, a CCTSI New Methods Development pilot grant shared by F. Hoerndli and X. Ding and part of the CCTSI NIH-NCATS CTSA Grant (UL1TR002535), and internal funding from Colorado State University College of Veterinary Medicine and Biomedical Sciences to F. Hoerndli.
Footnotes
Conflicts of interest
There are no conflicts of interest to declare.
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2lc00737a
Notes and references
- 1.Corsi AK, Wightman B and Chalfie M, WormBook, 2015, pp. 1–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nigon VM and Félix M-A, WormBook, 2017, pp. 1–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Apfeld J and Alper S, Methods Mol. Biol, 2018, 1706, 53–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liang JJH, McKinnon IA and Rankin CH, J. Neurogenet, 2020, 34, 527–548. [DOI] [PubMed] [Google Scholar]
- 5.Reddien PW and Horvitz HR, Annu. Rev. Cell Dev. Biol, 2004, 20, 193–221. [DOI] [PubMed] [Google Scholar]
- 6.Kenyon CJ, Nature, 2010, 464, 504–512. [DOI] [PubMed] [Google Scholar]
- 7.Chalasani SH, Chronis N, Tsunozaki M, Gray JM, Ramot D, Goodman MB and Bargmann CI, Nature, 2007, 450, 63–70. [DOI] [PubMed] [Google Scholar]
- 8.Goodman MB, Hall DH, Avery L and Lockery SR, Neuron, 1998, 20, 763–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sulston JE, Hodgkin J and Wood WB, The Nematode Caenorhabditis Elegans, Methods, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 1988, pp. 587–606. [Google Scholar]
- 10.Massie MR, Lapoczka EM, Boggs KD, Stine KE and White GE, Cell Stress Chaperones, 2003, 8, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim E, Sun L, Gabel CV and Fang-Yen C, PLoS One, 2013, 8, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Churgin MA, Jung S-K, Yu C-C, Chen X, Raizen DM and Fang-Yen C, eLife, 2017, 6, e26652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hess M, Gomariz A, Goksel O and Ewald CY, eNeuro, 2019, 6(4), 14–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Felker DP, Robbins CE and McCormick MA, Transl. Med. Aging, 2020, 4, 1–10. [PMC free article] [PubMed] [Google Scholar]
- 15.Stiernagle T, WormBook, 2006, pp. 51–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chronis N, Zimmer M and Bargmann CI, Nat. Methods, 2007, 4, 727–731. [DOI] [PubMed] [Google Scholar]
- 17.Ben-Yakar A, Chronis N and Lu H, Curr. Opin. Neurobiol, 2009, 19, 561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le KN, Zhan M, Cho Y, Wan J, Patel DS and Lu H, Commun. Biol, 2020, 3, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wan J, Sun G, Dicent J, Patel DS and Lu H, Lab Chip, 2020, 20, 266–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hulme SE, Shevkoplyas SS, Apfeld J, Fontana W and Whitesides GM, Lab Chip, 2007, 7, 1515–1523. [DOI] [PubMed] [Google Scholar]
- 21.Viri V, Cornaglia M, Atakan HB, Lehnert T and Gijs MAM, Lab Chip, 2020, 20, 2696–2708. [DOI] [PubMed] [Google Scholar]
- 22.Berger S, Lattmann E, Aegerter-Wilmsen T, Hengartner M, Hajnal A, deMello A and i Solvas XC, Lab Chip, 2018, 18, 1359–1368. [DOI] [PubMed] [Google Scholar]
- 23.Gilleland CL, Rohde CB, Zeng F and Yanik MF, Nat. Protoc, 2010, 5, 1888–1902. [DOI] [PubMed] [Google Scholar]
- 24.Shivers J, Uppaluri S and Brangwynne CP, Microfluid. Nanofluid, 2017, 21, 149. [Google Scholar]
- 25.Berger S, Spiri S, deMello A and Hajnal A, Development, 2021, 148, dev199674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chokshi TV, Ben-Yakar A and Chronis N, Lab Chip, 2009, 9, 151–157. [DOI] [PubMed] [Google Scholar]
- 27.Park EC, Ghose P, Shao Z, Ye Q, Kang L, Xu XZS, Powell-Coffman JA and Rongo C, EMBO J, 2012, 31, 1379–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chuang H-S, Chen H-Y, Chen C-S and Chiu W-T, Lab Chip, 2013, 13, 2980–2989. [DOI] [PubMed] [Google Scholar]
- 29.Chung K, Crane MM and Lu H, Nat. Methods, 2008, 5, 637–643. [DOI] [PubMed] [Google Scholar]
- 30.Rohde CB and Yanik MF, Nat. Commun, 2011, 2, 271. [DOI] [PubMed] [Google Scholar]
- 31.Ding Y, Ball KA, Webb KJ, Gao Y, D’Alessandro A, Old WM, Stowell MHB and Ding X, Small, 2020, 16, 2003506. [DOI] [PubMed] [Google Scholar]
- 32.Gao Y, Fajrial AK, Yang T and Ding X, Biomater. Sci, 2021, 9, 1574–1582. [DOI] [PubMed] [Google Scholar]
- 33.Ding X, Lin S-CS, Kiraly B, Yue H, Li S, Chiang I-K, Shi J, Benkovic SJ and Huang TJ, Proc. Natl. Acad. Sci. U. S. A, 2012, 109, 11105–11109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang J, Yang S, Chen C, Hartman JH, Huang P-H, Wang L, Tian Z, Zhang P, Faulkenberry D, Meyer JN and Huang TJ, Lab Chip, 2019, 19, 984–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brenner S, Genetics, 1974, 77, 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rongo C, Whitfield CW, Rodal A, Kim SK and Kaplan JM, Cell, 1998, 94, 751–759. [DOI] [PubMed] [Google Scholar]
- 37.Hoerndli FJ, Maxfield DA, Brockie PJ, Mellem JE, Jensen E, Wang R, Madsen DM and Maricq AV, Neuron, 2013, 80, 1421–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schneider CA, Rasband WSand Eliceiri KW, Nat. Methods, 2012, 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cordelires F, Manual Tracking, https://imagej.nih.gov/ij/plugins/track/track.html, (accessed May 11, 2021).
- 40.Pierce-Shimomura JT, Chen BL, Mun JJ, Ho R, Sarkis R and McIntire SL, Proc. Natl. Acad. Sci. U. S. A, 2008, 105, 20982–20987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang L, Zhao P, Wu J, Chuang H-S and Wang W, Sens. Actuators, B, 2018, 259, 703–708. [Google Scholar]
- 42.Kubanek J, Shukla P, Das A, Baccus SA and Goodman MB, J. Neurosci, 2018, 38, 3081–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou W, Wang J, Wang K, Huang B, Niu L, Li F, Cai F, Chen Y, Liu X, Zhang X, Cheng H, Kang L, Meng L and Zheng H, Lab Chip, 2017, 17, 1725–1731. [DOI] [PubMed] [Google Scholar]
- 44.Klass MR, Mech. Ageing Dev, 1977, 6, 413–429. [DOI] [PubMed] [Google Scholar]
- 45.Petrella LN, PLoS One, 2014, 9, e112377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stroustrup N, Anthony WE, Nash ZM, Gowda V, Gomez A, López-Moyado IF, Apfeld J and Fontana W, Nature, 2016, 530, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ward A, Liu J, Feng Z and Xu XZS, Nat. Neurosci, 2008, 11, 916–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee KH and Aschner M, Curr. Protoc. Toxicol, 2016, 67, 11.21.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Petzold BC, Park S-J, Ponce P, Roozeboom C, Powell C, Goodman MB and Pruitt BL, Biophys. J, 2011, 100, 1977–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hills T, Brockie PJ and Maricq AV, J. Neurosci, 2004, 24, 1217–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.de Bono M and Maricq AV, Annu. Rev. Neurosci, 2005, 28, 451–501. [DOI] [PubMed] [Google Scholar]
- 52.Hulme SE, Shevkoplyas SS, McGuigan AP, Apfeld J, Fontana W and Whitesides GM, Lab Chip, 2010, 10, 589–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Restif C, Ibáñez-Ventoso C, Vora MM, Guo S, Metaxas D and Driscoll M, PLoS Comput. Biol, 2014, 10, e1003702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zheng Y, Brockie PJ, Mellem JE, Madsen DM and Maricq AV, Neuron, 1999, 24, 347–361. [DOI] [PubMed] [Google Scholar]
- 55.Whittaker AJ and Sternberg PW, Curr. Opin. Neurobiol, 2004, 14, 450–456. [DOI] [PubMed] [Google Scholar]
- 56.Wirak GS, Florman J, Alkema MJ, Connor CW and Gabel CV, eLife, 2022, 11, e72135. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.