

Abstract
To study the binding mode of the adipokine chemerin as well as the short peptide agonist chemerin‐9 (C9) to its two receptors chemokine‐like receptor 1 (CMKLR1) and G protein‐coupled receptor 1 (GPR1), we generated 5‐carboxytetramethylrhodamine (TAMRA) modified variants of both ligands. In addition, we labeled GPR1 and CMKLR1 with a nanoluciferase at the N‐terminus to perform NanoBRET binding assays. For GPR1, both ligands show high affinity and comparable binding. Significant differences were found for CMKLR1, whereby only full‐length chemerin binds with high affinity in saturation and displacement assays. For TAMRA‐C9 a biphasic binding consisting of two binding states has been found and no displacement studies could be performed. Thus, we conclude that CMKLR1 requires full‐length chemerin for stable binding in contrast to GPR1. This work demonstrates the NanoBRET binding assay as a new tool for binding studies at chemerin receptors and it enables deeper insights into the ligand binding parameters.
Keywords: chemerin, CMKLR1, GPR1, ligand binding, peptides, protein expression
The adipokine chemerin and the short peptide agonist chemerin‐9 activate two GPCRs: CMKLR1 and GPR1. Here, we established a nanoluciferase based binding assay to analyze differences in ligand binding. For GPR1, both ligands show high affinity and comparable binding. At CMKLR1, full‐length chemerin is required for stable binding, indicating a second interaction side for binding of the larger protein.

Introduction
Obesity is an increasing worldwide health issue that highly correlates with low‐grade inflammation and metabolic syndrome. [1] To gain deeper insight into the relationship between obesity and inflammation, it is necessary to investigate molecules being important for both such as the adipokine chemerin and its receptors. [2] Chemerin is a 16 kDa protein expressed in liver, skin, lung or white adipose tissue. [3] Secreted chemerin plays a major role in the immune response. Simultaneously, it was shown that obese patients have elevated chemerin levels. Thus, this protein links chronic inflammation of adipose tissue and its occurrence in obesity. [4]
Chemerin is expressed in a biological inactive form termed pre‐prochemerin with 163 amino acids. After cleavage of the N‐terminal signal peptide, prochemerin is secreted. [5] For activation of chemerin, C‐terminal procession by serine proteases is required. The most active chemerin forms are truncated at position S157 or F156 and thus are named chemerinS157 or chemerinF156 (ChemS157 or ChemF156) consisting of 137 or 136 amino acids respectively. [6] Derived from the C‐terminus of ChemS157, the short nonapeptide agonist chemerin‐9 (C9) was developed that shows the same activity as the native protein in several assays. [7]
Chemerin operates mainly by activating two G protein‐coupled receptors (GPCR), chemokine‐like receptor 1 (CMKLR1) and G protein‐coupled receptor 1 (GPR1). A third receptor, the C−C chemokine receptor‐like 2 (CCRL2), is described to bind chemerin but leads to no cellular response. Although CMKLR1 and GPR1 bind the active ChemS157 and share high sequence identity, they show differences in cellular signaling and function. The CMKLR1 is expressed by various leukocytes, e. g. natural killer cells, plasmacytoid dendritic cells and macrophages, as well as adipocytes.[ 3b , 5 , 8 ] Signaling by this receptor leads to the migration of immune cells towards sites of inflammation but it also regulates the differentiation of adipocytes.[ 3c , 5 ] CMKLR1 is a typical rhodopsin‐like GPCR that activates Gαi proteins, which inhibits the synthesis of cAMP by adenylyl cyclases. [9] Additionally, arrestin‐3 recruitment and subsequent internalization of the receptor was described.[ 9 , 10 ] Furthermore, downstream activation of MAP kinases like ERK1/2 or p38 has been shown. [11]
In contrast, GPR1 was first described as an orphan receptor in the human hippocampus in 1994. [12] In 2008, chemerin was discovered as its native ligand. [13] Until today, the biological role of GPR1 is still not fully understood. In knock‐out mice fed with high‐fat diet, an aggravated glucose intolerance was recognized as well as a decrease in serum testosterone and lower bone mineral density. [14] The GPR1 is classified as an atypical GPCR with no ligand‐induced G protein activation but arrestin recruitment and RhoA/ROCK‐mediated signaling.[ 13 , 15 ] Recently, we described ligand independent constitutive internalization and scavenging properties of this receptor. [16]
To enhance the knowledge about the similarities and differences of both receptors, we established a nanoluciferase (Nluc) based ligand‐binding assay. [17] We created N‐terminal Nluc‐receptor constructs and validated their activity. We synthesized the short peptide agonist C9 with a 5‐carboxytetramethylrhodamine (TAMRA) fluorophore label as well as a TAMRA‐labeled ChemS157 ([K141(TAMRA)]ChemS157) with a semi‐synthetic approach by ligating a TAMRA‐labeled peptide to the N‐terminal fragment of the ChemS157 protein by native chemical ligation. Surprisingly, in displacement binding assays, we found that CMKLR1 requires full‐length chemerin for stable binding in contrast to GPR1 where the short peptide agonist is sufficient.
Results and Discussion
Fluorescent labeling of ChemerinS157 and C9
To study the binding mode of GPR1 and CMKLR1, we set‐up a BRET‐based binding assay using Nluc‐fused chemerin receptors and TAMRA‐labeled ChemS157 protein and TAMRA‐labeled C9 peptide (Figure 1). Thus, we synthesized C9 and the corresponding TAMRA‐C9 as described previously. [18] Additionally, we expressed the full‐length ChemS157 by recombinant expression and developed a strategy to generate a TAMRA‐labeled ChemS157. This semi‐synthetic approach combines a TAMRA‐labeled peptide (part A) with a segment ChemS157 (part B) by using native chemical ligation (NCL) (Figure 2A).
Figure 1.
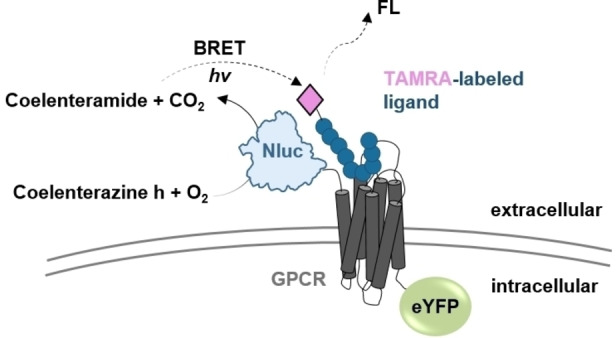
NanoBRET based approach for GPCRs. The binding behavior of TAMRA‐labeled ligands at GPCRs can be studied by using an N‐terminally Nluc‐tagged GPCR construct. The fused Nluc converts coelenterazine h to coelenteramide. The arising energy can excite the TAMRA fluorophore of the ligand only when it is bound in the GPCR binding pocket and thus in close proximity.
Figure 2.

Strategy and analysis of [K141(TAMRA)]ChemS157. (A) Schematic overview of the semi‐synthetic strategy. Part A was synthesized by solid phase peptide synthesis (SPPS) with an N‐terminal cysteine using Fmoc/tBu strategy to receive the peptide [K141(TAMRA)]ChemS157(135–157). Part B was expressed as a fusion protein with C‐terminal intein‐chitin‐binding domain (CBD) for intein‐mediated purification with an affinity chitin‐binding tag (IMPACT). Native chemical ligation (NCL) of the ChemS157(21–134)‐thioester and the peptide [K141(TAMRA)]ChemS157(135–157) fragment, forming the TAMRA‐labeled full‐length protein, was performed. (B) Reverse phase high pressure liquid chromatography (RP‐HPLC) of NCL over time. The upper panel shows the chromatogram with detection at 220 nm, the lower panel fluorescence at 572 nm after excitation at 553 nm. (C) RP‐HPLC chromatogram and (D) mass spectrum by matrix‐assisted laser desorption ionization ‐ time of flight (MALDI‐TOF) linear mode of the purified and refolded [K141(TAMRA)]ChemS157. The calculated molecular mass is 18885.4 Da
The C‐terminal fragment [K141(TAMRA)]ChemS157(135‐157) (part A) was synthesized with the attached TAMRA fluorophore by solid‐phase peptide synthesis. A purity of≥95 % was yielded for the peptide fragment and the identity was proven by MALDI‐TOF‐MS (Figure S1).
The N‐terminal fragment ChemS157(21‐134)‐intein‐CBD (part B) was recombinantly expressed to create a reactive thioester at the C‐terminus after using the intein‐mediated purification with affinity chitin‐binding tag (IMPACT) system.
The expression was monitored with SDS‐PAGE (Figure S2). IB were purified, re‐solubilized and the CBD domain was refolded by decreasing the urea concentration. The solution was loaded on chitin beads for the IMPACT purification. Samples were collected during purification to investigate the cleavage of the reactive MESNa‐thioester. After four cleavage steps, the samples were analyzed by SDS‐PAGE showing successful purification with a band at 15 kDa (calculated mass of thioester: 15.9 kDa, Figure S2).
In order to generate [K141(TAMRA)]ChemS157, the NCL was performed as described above (Figure 2A). The reaction was monitored by RP‐HPLC (Figure 2B). In the chromatogram for the 0 h sample, two peaks can be seen for educts part A and B. The first peak emerges also at the fluorescence channel and thus corresponds to the peptide part A. The second peak can be assigned to educt part B due to the missing fluorescence. This peak intensity decreased dramatically after 1 h, and a new peak arose. The signal occurs in the fluorescence channel as well indicating the potential ligation of both protein fragments. The product peak increased after 4 h (Figure 2B) and after 18 h the reaction was terminated. The product was purified, refolded by dialysis and afterwards concentrated. The purity of the protein was analyzed by RP‐HPLC (≥94 %) and the identity was confirmed by MALDI‐TOF‐MS (Figure 2C and D).
Proof of activity in signal transduction assay of TAMRA C9 and [K141(TAMRA)]ChemS157 at CMKLR1
To examine the functionality of [K141(TAMRA)]ChemS157, ChemS157, TAMRA‐C9 and C9, we analyzed G protein signaling with the IP‐One assay at CMKLR1 (Figure 3). TAMRA‐C9 showed a two‐fold increase in activity (EC50=171.5 nM,) compared to unlabeled C9 (EC50=387.6 nM, Figure 3A). [K141(TAMRA)]‐ChemS157 exhibits an EC50 value of 22.6 nM, which is 2.5‐fold higher than ChemS157 without the TAMRA label (EC50=9.0 nM, Figure 3B) and is not statistically significant (two‐tailed, unpaired t‐test, p=0.07). The C9 peptide has a lower potency than the full length chemerin at CMKLR1. However, labeling using TAMRA does not show any significant influence on protein and peptide.
Figure 3.
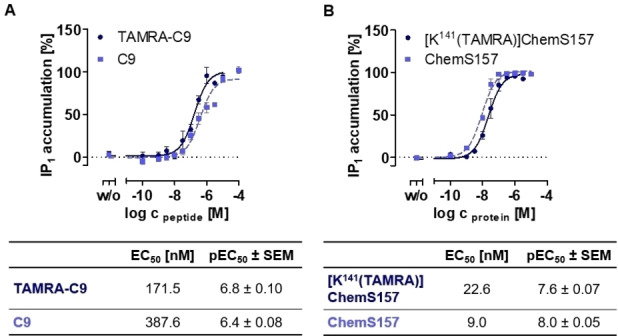
Activation of the CMKLR1 detected by IP‐accumulation after G protein activation. All assays were performed by using transiently transfected COS‐7 cells and were performed at least three times in triplicates. (A) Comparison of C9 and TAMRA‐C9 (B) Comparison of ChemS157 and [K141(TAMRA)]ChemS157.
Nluc‐fused receptor constructs
For the investigation of ligand binding at both chemerin receptor subtypes CMKLR1 and GPR1, Nluc was fused at the receptor N‐terminus with a Ser‐Gly4‐Ser linker (Tab. S1).
To evaluate the influence of the Nluc tag at receptor expression, we compared tagged and wild type receptor expression by fluorescence microscopy. Both, CMKLR1‐eYFP and Nluc‐CMKLR1‐eYFP are localized in the cell membrane exclusively (Figure 4A). In comparison, GPR1 and Nluc‐GPR1 are expressed in the cell membrane and in vesicles in the cytosol (Figure 4B). Thus, the Nluc addition to the N‐terminus has no influence on the receptor expression pattern.
Figure 4.
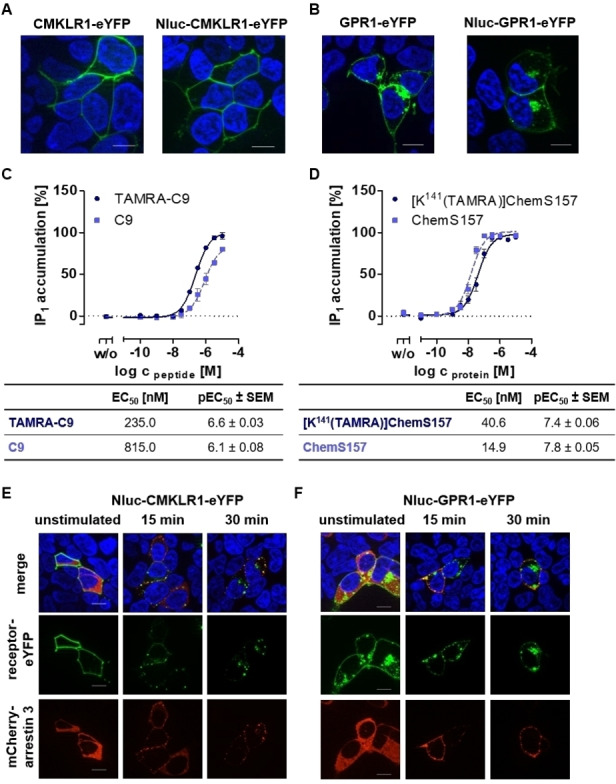
Characterization of Nluc‐fused receptor constructs. All assays were performed using transiently transfected HEK293 (A, B, E, F) or COS‐7 cells (C, D). Life cell fluorescence microscopy of CMKLR1‐eYFP and Nluc‐CMKLR1‐eYFP (A) and of GPR1‐eYFP and Nluc‐GPR1‐eYFP construct (B). Expression of receptors in the cell membrane for both is shown to be comparable for wildtype and Nluc‐fused receptors. Activation of Nluc‐CMKLR1 receptor was analyzed by IP‐One assay with peptide agonist C9 and TAMRA‐C9 (C) or with native ligand ChemS157 and TAMRA‐labeled variant [K141(TAMRA)]ChemS157 (D). Each assay was performed at least three times in triplicates. Arrestin recruitment to activated and phosphorylated receptors was studied after stimulation with 1 μM C9 for 15 min and 30 min for Nluc‐CMKLR1‐eYFP (E) and Nluc‐GPR1‐eYFP (F). Both receptors (green) recruit arrestin‐3 (red) and internalize upon ligand stimulation. For microscopy, cell nuclei were stained with Hoechst 33342 (blue); scale bar=10 μm.
For validation of receptor functionality with the Nluc‐tag, IP‐One assays were performed with Nluc‐CMKLR1‐eYFP using labeled and unlabeled peptide and protein ligand (Figure 4C, D). Both peptides are able to activate the Nluc‐CMKLR1 whereas TAMRA‐C9 shows a 4‐fold increased activity of 235.0 nM compared to C9 (EC50 815.0 nM, Figure 4C) and the protein ligands a 2.5 fold increased activity was found for the TAMRA labeled ligand (EC50 40.6 nM and EC50 14.9 nM, Figure 4D). Thus, the difference between CMKLR1 and at the Nluc‐CMKLR1 showed no significance after statistical evaluation with an unpaired two‐tailed t‐test.
Due to the lack of G protein signaling of GPR1, it is not possible to validate the receptor activity for Nluc‐GPR1‐eYFP with the IP‐One assay. Hence, we used microscopy to monitor recruitment of the intracellular adaptor molecule arrestin‐3, which binds activated and phosphorylated GPCRs (Figure 4E, F). Furthermore, similar results of internalization were obtained after stimulation of TAMRA‐C9 (Figure S3) in fluorescence microscopy studies. Thus, it was possible to show that both receptors with attached Nluc are fully active and can be activated similar to wild type receptor.
Saturation binding assay at CMKLR1 and GPR1
After validation of Nluc‐CMKLR1 and Nluc‐GPR1 construct functionality, NanoBRET assays were performed to compare binding of the short peptide C9 with the full‐length protein ChemS157. For saturation binding studies, cells were transfected with Nluc‐receptor‐eYFP and were stimulated with [K141(TAMRA)]ChemS157 (Figure 5A, B) or TAMRA‐C9 (Figure 5C, D) with increasing concentrations. Coelenterazine h was added and the BRET signal was detected directly. For the labeled full‐length protein, a sigmoidal concentration response curve with an EC50 of 75.9 nM was detected in saturation binding at CMKLR1 (Figure 5A). [K141(TAMRA)]ChemS157 protein binding to Nluc‐GPR1‐eYFP (Figure 5B) reached a comparable EC50 of 111.8 nM. Analyzing TAMRA‐C9 binding at Nluc‐CMKLR1, we observed that a biphasic model is necessary to fit the measured netBRET values (Figure 5C). Hence, the CMKLR1 exhibits two binding states: low ligand concentrations lead to a high affinity state at the receptor with an EC50 of 19.0 nM and to a low affinity state at higher ligand concentrations with EC50 of 1259.0 nM. At Nluc‐GPR1, TAMRA‐C9 leads to a fully saturated, sigmoidal binding curve with a low EC50 of 4.9 nM (Figure 5D).
Figure 5.
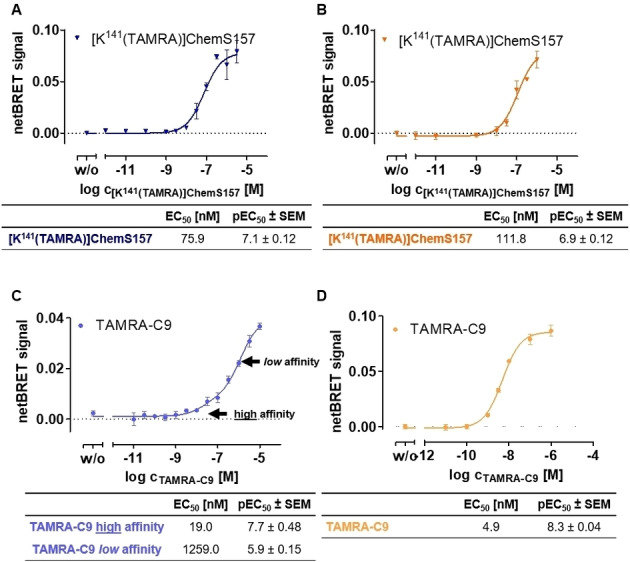
Saturation binding at Nluc‐CMKLR1 and Nluc‐GPR1 with TAMRA C9 and [K141(TAMRA)]ChemS157. All assays were performed using transiently transfected COS‐7 cells. BRET signal was detected directly after addition of coelenterazine h and TAMRA‐ligand. Each assay was performed at least three times in triplicates. A and C represent the saturation binding BRET assay results of [K141(TAMRA)]ChemS157 (A) and TAMRA‐C9 (C) at Nluc‐CMKLR1. Full‐length protein stimulation results in sigmoidal response curves at Nluc‐CMKLR1. Analysis of TAMRA‐C9 leads to biphasic curves at Nluc‐CMKLR1 with two BRET efficiencies for a high and a low affinity state. The saturation binding BRET assay of [K141(TAMRA)]ChemS157 (B) and TAMRA‐C9 (D) at Nluc‐GPR1 result in sigmoidal concentration‐response curves in both cases.
Displacement assays at CMKLR1 and GPR1
To increase the knowledge of binding parameters of unlabeled ligand, displacement assays were performed. For displacement BRET with ChemS157, the labeling was performed with 40 nM [K141(TAMRA)]ChemS157 and displaced with increasing amount of unlabeled ChemS157 (Figure 6A, B). To investigate the binding of the short peptide, 10 nM of TAMRA‐C9 were used to label the receptor and unlabeled C9 was added in increasing concentrations (Figure 6C, D). The full‐length TAMRA‐labeled ChemS157 can be displaced with the native protein ChemS157 at Nluc‐CMKLR1 with a Ki of 31.8 nM (Figure 6A). Displacement at Nluc‐GPR1 with full‐length chemerin leads to an even lower Ki value of 9.3 nM (Figure 6B). Surprisingly, in displacement studies of TAMRA‐C9 at Nluc‐CMKLR1 no displacement curves for TAMRA‐C9 with unlabeled C9 (Figure 6C) could be plotted, whereas at Nluc‐GPR1 displacement of TAMRA‐C9 was observed with a Ki value of 15 nM (Figure 6D).
Figure 6.

Displacement assay at Nluc‐CMKLR1 and Nluc‐GPR1 with TAMRA‐C9 and [K141(TAMRA)]ChemS157. All assays were performed using transiently transfected COS‐7 cells. TAMRA‐ligand was added with defined concentration and unlabeled ligand was used with increasing concentrations. Both were incubated for 1 h on ice to reach an equilibrium. BRET signal was detected directly after addition of coelenterazine h. Each assay was performed at least three times in triplicates. A and C represent the displacement binding BRET assay results of [K141(TAMRA)]ChemS157 (A) and TAMRA‐C9 (C) at Nluc‐CMKLR1. Full‐length protein results in sigmoidal displacement curves at Nluc‐CMKLR1. For TAMRA‐C9 no displacement curves at Nluc‐CMKLR1 can be plotted. The displacement BRET assay of [K141(TAMRA)]ChemS157 (B) and TAMRA‐C9 (D) with the corresponding unlabeled ligand at Nluc‐GPR1 result in sigmoidal displacement curves in both cases.
To verify the binding properties at CMKLR1, additional displacement assays were performed (Figure S4). For competition of TAMRA‐C9 with ChemS157, no stable signal for binding has been measured. Labeling by using [K141(TAMRA)]ChemS157 and displacement by C9 was also tested. Here, a BRET signal has been monitored, but no displacement was detected. This leads to the presumption that GPR1 binds TAMRA‐labeled peptide and protein in a similar manner and both variants can be displaced by the corresponding unlabeled ligand. For CMKLR1, the peptide TAMRA‐C9 shows binding in high and low affinity state and displacement of labeled ligands by C9 cannot be measured. Hence, the CMKLR1 requires full‐length chemerin protein for stable binding in contrast to GPR1.
Discussion
GPCRs are activated by a variety of ligands like peptides and proteins. They are responsible for many physiological processes and represent a diverse family of pharmacological targets, since 30 % of the current marketed drugs addressing this receptor family. [19] For the treatment of obesity, only few therapeutics are currently available, whereas Orlistat or Liraglutide are mostly prescribed. In 2021, the GPCR glucagon‐like peptide 1 receptor specific drug Semaglutide was approved and tested in clinic trials as the first orally available peptide drug. [20] This progress in obesity medication shows the great importance and potential of peptides and proteins as a base for the development of new therapeutics. To generate highly potent and selective drugs, an essential step is to understand the interaction of peptides and proteins to their cognate GPCRs, especially since the number of peptide based drug conjugates is constantly increasing. [21]
The chemotactic protein chemerin in its active form ChemS157 is known to bind the CMKLR1, GPR1 and the CCRL2 with high affinity,[ 13 , 22 ] but the functional relevance of the CCRL2 and the GPR1 is still uncertain. [23] GPR1 shows a sequence identity of 40 % compared to its homolog CMKLR1, but the mechanisms of signal transduction for both receptors are different. [9] Besides the natural ligand ChemS157, also the C‐terminally derived nonapeptide C9 is able to activate both receptors with similar effect.[ 7 , 13 , 14 , 24 ] In previous studies, we were already able to investigate the interaction of C9 with CMKLR1 and GPR1 by mutagenesis‐activity studies and computational modeling.[ 16 , 18 ] To directly study the binding modes of C9 and ChemS157 to both receptors, we established a NanoBRET binding assay. One major advantage of resonance energy transfer based assays is the investigation of binding and kinetic studies in a cellular based system, which may influence ligand binding to the receptors. [25] Additionally, this state of the art assay also enables high‐throughput testing without radioactive labeling, which facilitates the applicability, reduces health risks and avoids costs.
To investigate the binding of C9, we introduced the rhodamine‐based fluorophore TAMRA at the N‐terminus of the peptide. TAMRA is widely used as a fluorophore and energy acceptor in NanoBRET studies and can be attached easily during solid phase peptide synthesis to peptide ligands.
For analyzes of the endogenous protein ligand, ChemS157 was expressed and refolded as previously described. [6] Additionally, we generated a TAMRA‐labeled ChemS157, named [K141(TAMRA)]ChemS157 by a semisynthetic approach. A synthesized TAMRA‐labeled sequence of the protein is ligated to the recombinantly expressed major segment by NCL. This strategy was chosen to specifically modify the lysine side chain at position 141 of the protein by the fluorophore. Purification of the product was performed by RP‐HPLC since immobilized metal affinity chromatography was not possible due to the His‐tag on both, educt part B and the product [K141(TAMRA)]ChemS157.
The proteins and peptides were tested by an IP‐One assay at the CMKLR1 for their G protein activity. Differences in potency between C9 and ChemS157 are much higher compared to previously recorded calcium mobilization assays at the CMKLR1 receptor [7] and more in agreement with BRET based assays addressing the G protein signaling and arrestin recruitment. [9] We suggest that the differences are based on higher binding stability of the ChemS157, which might be visible in the IP‐One assay that is an equilibrium assay, instead of the calcium assay were the signal transduction is directly measured.
To use the chemerin receptors for binding studies, the Nluc is fused to the N‐terminus to ensure the energy transfer to the fluorophore after binding to the transmembrane domains and extracellular region of the GPCRs. [26] With respect to other energy donors like Rluc or Fluc, the Nluc construct has a small molecular weight (19 kDa), a higher stability, lower influence to the active receptor state and a 30‐fold higher brightness, representing a suitable donor for BRET assays.[ 17 , 27 ] We introduced the Nluc at the very N‐terminal part of the chemerin receptors, which is a promising approach and has already been described for other GPCRs like the β2‐adrenoceptors, Y1, Y2 and Y4 receptor or the CXCR4. [28] As expected, the Nluc constructs do not show any changes in respect to membrane localization compared to the receptor wild type.
To investigate the suitability of the Nluc‐receptor constructs IP‐One assays were performed for Nluc‐CMKLR1. Comparing wild type receptor with the Nluc‐fused CMKLR1 only a small 1.5 to 2‐fold shift can be seen for every peptide and protein variant, proving the applicability of the Nluc‐tagged receptor. Because of the missing G protein signaling of the GPR1, the activity and binding properties of the TAMRA C9 was investigated by internalization studies and arrestin‐3 recruitment by fluorescence microscopy.[ 9 , 29 ] These studies showed that the ligand is able to activate the GPR1 as well as its Nluc‐fused derivate.
In saturation binding assays for the labeled full‐length protein, comparable EC50 with a minor 1.5‐fold shift could be detected at Nluc‐CMKLR1 and Nluc‐GPR1, indicating the same affinity of [K141(TAMRA)]ChemS157 to both chemerin receptors. The saturation NanoBRET of the TAMRA‐C9 at Nluc‐GPR1 shows an EC50 value of 4.9 nM, which indicates high affinity to the GPR1 and an even higher affinity compared to the full‐length protein. Comparing the TAMRA‐C9 binding curve at CMKLR1, we observed a much lower signal intensity compared to the full‐length chemerin at both receptors, as well as the TAMRA‐labeled peptide at GPR1. This leads to the assumption, that the TAMRA label at the peptide shows a higher distance to the Nluc tag at the CMKLR1 N‐terminus and the orientation of the ligand is altered. In addition, the graph displays a biphasic shape at the CMKLR1 receptor, which points to the characterization of a low and a high affinity state of the C9. Similar biphasic binding curves are described for different GPCRs in binding assay such as Y1 or Y2 receptor. [28f] The shape of the curve indicates that the ligand first binds in a loose complex and second in a tightly bound state after conformational changes of the binding partner. [30] It can be speculated that the G protein might allosterically stabilize the high affinity state of GPCRs. [28f] This suggests that the binding mechanism at the CMKLR1 is more complex, compared to the GPR1.
Driven by the saturation binding experiments, we used constant amount of TAMRA‐labeled ligands in order to perform displacement experiments with the corresponding unlabeled ligands. Competition binding assays have the advantage that the physiological significance of the saturation binding assays can be determined and binding parameters for the native unlabeled agonist can be determined. The displacement of [K141(TAMRA)]ChemS157 with ChemS157 can be achieved at the CMKLR1 and at the GPR1 in the nanomolar range, which fits to the radio‐ligand binding and competition studies in literature. [9] The labeled TAMRA‐C9 was displaced at the GPR1 with a Ki value of 15 nM and shows a specific binding of the C9 to the receptor. It seems that the binding affinity of the wild type C9 to the GPR1 is slightly reduced in comparison to the TAMRA‐labeled variant (3‐fold shift). We suspect an interaction between the TAMRA label and the receptor may be responsible for the small difference in binding affinities. However, the GPR1 shows lower Ki values for ChemS157 and C9 through all the binding assays, indicating a higher affinity for chemerin compared to CMKLR1.
Furthermore, displacement experiments with TAMRA‐C9 and unlabeled C9 could not be performed successfully. We presume that binding of the small peptide ligand C9 is less stable compared to ChemS157 at CMKLR1 and therefore can not be measured after the time required to reach equilibrium in displacement assays. In general, our binding assays show that the difference in EC50 values between the nonapeptide and the protein is in agreement with the receptor activation studies at the CMKLR1.
Comparing the results of the NanoBRET, we hypothesize that both CMKLR1 and GPR1 differ in their binding mode for the full‐length protein. These data support the current hypothesis, that the C‐terminal part of the protein binds to the transmembrane area of the CMKLR1 and is stabilized by the N‐terminal part of the receptor.[ 5 , 7 , 20c ] In contrast, for the GPR1 the N‐terminal receptor part seems not to be essential to stabilize the ChemS157 binding. The chemerin ligand and CMKLR1 receptor show structural similarity to other chemokine‐receptor systems, like SDF‐1 and CXCR4. For these receptors, the concept of the “two‐site binding model” has been declared: first the “site‐one” docking domain binds at the receptor N‐terminus and afterwards “site‐two” interacts with the receptor binding pocket and operates as the cellular signal trigger. [31] This model might be applicable for chemerin and CMKLR1 as well.
Conclusion
Altogether, we successfully established the NanoBRET binding assay for the chemerin receptors CMKLR1 and GPR1 in order to investigate binding for the peptide agonist C9 as well as the native ligand chemerin. We developed a strategy to synthesize a specific TAMRA‐labeled ChemS157 by a semisynthetic approach. With our data, we gained new evidence suggesting that the CMKLR1 receptor requires a second interaction site with chemerin, indicated by the biphasic behavior of the C9 peptide and the data of displacement assays. In contrast, the short peptide agonist is sufficient for stable binding at the GPR1. These studies not only promote to clarify the binding mechanism of the ChemS157 to the CMKLR1 and GPR1, but provide a new state of the art tool to create and test potent peptide therapeutics and small molecule modulators for obesity treatment.
Experimental Section
Material
Peptide synthesis : Fmoc‐protected amino acids were purchased from ORPEGEN (Heidelberg, Germany). Peptide resins, 1‐hydroxy benzotriazole (HOBt), diiodomethane, ethanedithiol (EDT), diethyl ether, and trifluoracetic acid (TFA) were obtained from Merck (Darmstadt, Germany). N,N’‐diisopropyl carbodiimide (DIC) was purchased from Iris Biotech (Marktredwitz, Germany). Dimethylformamide (DMF) and dichloromethane (DCM) were purchased from Biosolve (Valkenswaard, Netherlands), acetonitrile (ACN) was obtained from VWR (Darmstadt, Germany). O‐(7‐Azabenzotriazol‐1‐yl)‐N,N,N,N‐tetramethyluronium hexafluorophosphate (HATU) and N,N‐diisopropylethylamine (DIPEA) were obtained from Sigma‐Aldrich (St. Louis, MO, USA). 6‐Carboxytetramethylrhodamine (TAMRA) was purchased from emp biotech (Berlin, Germany).
NCL : Buffer reagents ethylenediaminetetraacetic acid (EDTA), 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid (HEPES), sodium chloride (NaCl), L‐cysteine hydrochloride monohydrate, L‐arginine, and polyoxyethylene (20) sorbitan monolaurate (TWEEN‐20) were purchased from Sigma‐Aldrich (St. Louis, USA). Guanidine hydrochloride, magnesium chloride hexahydrate, urea, tris(hydroxymethyl) methylamine (Tris), Tris(2‐carboxyethyl)phosphine hydrochloride (TCEP) were obtained from CarlRoth (Kalrsruhe, Germany) and cystamine dihydrochloride from Fluka (Schwerte, Germany). Sodium hydroxide and hydrochloric acid were purchased from Grüssing (Filsum, Germany). For the expression and purification, ampicillin sodium salt and LB‐media was obtained from Sigma Aldrich (Taufkirchen, Germany). Isopropyl‐beta‐D‐thiogalactopyranoside (IPTG) was obtained from Thermo Fischer (Schwerte, Germany). The chemical competent E.coli stocks of BL21(DE3) and the chitin resin was purchased from New England Biolabs (Frankfurt am Main, Germany) and sodium 2‐mercaptoethanesulfonate (MESNa) as well as the Amicon® centrifugation filter units were received from Merck (Darmstadt, Deutschland).
Cell culture : Cell culture media (Dulbecco's Modified Eagle's Medium (DMEM), Ham's F12, MCDB131), as well as trypsin‐EDTA, Dulbecco's Phosphate‐Buffered Saline (DPBS) and Hank's Balanced Salt Solution (HBSS) were obtained from Lonza (Basel, Switzerland). Fetal bovine serum (FBS) was obtained from Biochrom GmbH (Berlin, Germany). Opti‐MEM was obtained from Life Technologies (Basel, Switzerland). LipofectamineTM 2000 was obtained from Invitrogen (Carlsbad, CA, USA). Metafectene® Pro was received from Biontex Laboratories GmbH (München, Germany). Coelenterazine H was purchased from DiscoverX (Fremont, CA, USA). Hoechst33342 nuclear stain was obtained from Sigma‐Aldrich (St. Louis, MO, USA). Bovine arrestin‐3 was fused to mCherry and cloned into the pcDNA3 vector for microscopy studies previously. [32] To perform G protein activation studies, IP‐One Gq kit from Cisbio Bioassays (Codolet, France) was used. The vector for the chimeric GαΔ6qi4myr protein was kindly provided by E. Kostenis (Rheinische Friedrich‐Wilhelms‐Universität, Bonn, Germany). [33]
Cloning : Primers for cloning were bought from Biomers (Ulm, Germany). NanoLuciferase was purchased by Promega (Madison, WO, USA). Receptor construct hCMKLR1b‐eYFP and hGPR1‐eYFP in pVitro2 were produced previously. [6]
Methods
Peptide synthesis : Peptide synthesis was performed using the fluorenylmethoxycarbonyl (Fmoc)/tertbutyl (tBu) protection group strategy and pre‐loaded Wang resin to obtain a C‐terminal serine. Automated synthesis was implemented on a SYRO I synthesis robot (Multisyntech/Biotage) using a scale of 15 μmol per peptide and double coupling procedures using 8 equiv. of amino acid, ethyl cyanohydroxyiminoacetate (Oxyma) and N,N‐diisopropylcarbodiimide (DIC) and a reaction time of 42 min per cycle. Fmoc cleavage was performed in two subsequent cycles by applying 40 % and 20 % piperidine in N,N‐dimethylformamide (DMF) for 3 min and 10 min, respectively. Labeling peptides with 6‐carboxytetramethylrhodamine (TAMRA) was performed manually by reaction with 2 equiv. TAMRA, 1.9 equiv. HATU, 2 equiv. DIPEA in DMF overnight, at room temperature (RT) under light protection. The TAMRA‐label was attached either directly to the N‐terminus to produce a labeled C9, or after Dde deprotection of the lysine side chain of [K141(TAMRA)]ChemS157(135–157) (part A, Figure 2) for ligation to ChemS157(21‐134)‐thioester. For Dde‐cleavage, the resin was incubated with 2 % (v/v) hydrazine in DMF for 10 min at RT, washed with DMF and the cleavage was performed ten times. All peptides were cleaved by incubation with 90 % TFA, 7 % thioanisole, 3 % ethanedithiol (v/v/v) for 3 h at RT. Precipitation of peptides was performed with cold diethyl ether at −20 °C for 3 h, followed by centrifugation and washing steps. Peptides were purified by preparative reversed‐phase high performance liquid chromatography (RP‐HPLC) on a Phenomenex Aeris Peptide 5 μm, 100 Å, XB−C18 column (Phenomenex, Torrence, CA, USA). Purity and identity were confirmed by RP‐HPLC on a Jupiter 4 μm Proteo, 90 Å, C12 and Phenomenex Aeris Peptide 3.6 μm, 100 Å, XB−C18 column (Phenomenex), matrix‐assisted laser desorption/ionization with a time‐of‐flight detector (MALDI‐TOF) MS on an Ultraflex II and electrospray ionization (ESI) MS on an HCT ESI (Bruker Daltonics, Billerica, MA, USA). RP‐HPLC was performed using linear gradients of eluent A (0.08 % TFA in ACN) in eluent B (0.1 % TFA in ACN).
Expression of ChemS157(21‐134)‐thioester : The synthesis of ChemS157(21‐134)‐thioester (part B, Figure 2) was prepared using the intein‐mediated purification with affinity chitin‐binding tag (IMPACT) system. Recombinant expression was started by heat shock transformation of pET16b plasmid DNA with the gene of interest [His]10‐SSGHIEGRH‐ChemS157(21‐134)‐intein‐CBD (ChemS157(21‐134)‐intein‐CBD) in E.coli BL21(DE3). The overnight culture was inoculated with the transformed BL21(DE3) cells and afterwards incubated at 37 °C in a shaking incubator for 16 h. The expression was carried out by inoculating 8 L of LB‐media containing 0.3 μM ampicillin with the overnight cultivated bacteria to an OD600 of 0.1. Cells were grown at 37 °C in a shaking incubator until an OD600 of 0.7 was reached. By adding a final concentration of 1 μM isopropyl β‐d‐thiogalactosid (IPTG), the ChemS157(21‐134)‐intein‐CBD expression was induced. After four hours, cells were harvested and the pellets were resuspended in 20 mM HEPES buffer with 500 mM NaCl with pH 8.0. Cells were mechanically lysed by FastPrep for 40 s and 6 m/s after adding silica beads. A final concentration of 10 mM MgCl2 was added for DNase I digest and subsequently incubated at RT at an orbital shaker for 30 min. The inclusion bodies (IB) were collected by centrifugation for one hour at 10 °C by 12000 x g and washed four times with washing buffer (20 mM HEPES, 500 mM NaCl, 1 mM EDTA, pH 8.0). The IB were resolubilized over night at 4 °C in washing buffer containing 8 M urea and ultra‐centrifuged for 1 h at 4 °C with 35000 rpm. Afterwards, the supernatant was diluted with a constant flow of≤1 ml/min with HEPES buffer at 4 °C to 3 M urea. The diluted protein was loaded to a column containing chitin‐beads with a flow of≤1 ml/min and washed with HEPES‐buffer containing 0.2 % TWEEN‐20. The cleavage of intein‐CBD was mediated by 0.2 M MESNa and 0.1 M DTT and the C‐terminal reactive thioester was collected and concentrated by centrifugation with amicon tubes with a molecular weight cut‐off (MWCO) of 3000 Da. The protein‐product was freeze dried and stored at −20 °C.
Synthesis of [K 141 (TAMRA)]ChemS157 by native chemical ligation : To generate the product [K141(TAMRA)]ChemS157, [K141(TAMRA)]ChemS157(135–157) (part A) and ChemS157(21–134)‐thioester (part B) were linked by a native chemical ligation (Figure 2). Both reaction partners were deployed with a molar ratio of 1 : 2 (part A : part B) with a final concentration of 1 mM part A. The ligation was performed in reaction buffer (6 M guanidinium hydrochloride, 100 mM dibasic sodium phosphate, pH 6.0) containing 50 mM MESNa and 25 mM TCEP. The pH was adjusted to 4.0 with 5 M HCl and reaction was incubated at RT for 30 min. Next, the pH‐value was increased to 7.6 by adding 10 M NaOH. Reaction was incubated at RT for 18 h and the progress was monitored by RP‐HPLC. To acidify the pH, 0.1 % TFA was added and the solution was purified by preparative RP‐HPLC. Product fractions were combined using 25 mM Tris buffer with 500 mM NaCl and 6 M urea (pH 7.6) and refolded by dialysis at 4 °C for 6 days, consequently reducing the urea concentration after every 24 h. The protein was concentrated using amicon centrifugation tubes with a MWCO of 3000 Da and characterized by analytical RP‐HPLC and MALDI‐TOF‐MS.
Cloning of Nluc‐receptor constructs : To create nanoluciferase(Nluc)‐receptor constructs for binding assays, Nluc was N‐terminally fused to the receptors with a Ser‐Gly4‐Ser linker using the overlap extension PCR technique. [34] Briefly, the overlap extension PCR consists of two consecutive PCR steps. The Nluc and the receptor were amplified with the Ser‐Gly4‐Ser linker at the C‐ or N‐terminus, respectively. This enables the overlap of both PCR products in the second PCR steps. Next, the Nluc‐CMKLR1‐eYFP and Nluc‐GPR1‐eYFP constructs were incorporated into pVitro2 vector using MluI and XbaI restriction sites (Tab. S 1).
Cell culture : COS‐7 and HEK293 cells were cultivated in DMEM supplemented with 10 % FBS or DMEM/Ham's F12 supplemented with 15 % FBS, respectively. All cells were maintained in 75 cm2 cell culture flasks at 37 °C, 95 % humidity, and 5 % CO2 (standard conditions).
IP‐One assay : To test ligand and receptor functionality, the activation of G proteins was analyzed. COS‐7 cells were cultured in 25 cm2 cell culture flasks and transfected at 70–80 % confluence. Cells were transiently transfected with 3000 ng receptor DNA and 1000 ng chimeric G Protein (GαΔ6qi4myr) using Metafectene® Pro according to the manufacturer's protocol. One day post transfection, cells were re‐seeded in white, solid 384‐well plates and grown overnight at standard conditions. The assay was performed using Cisbio Bioassays IP‐One Gq kit. Briefly, cells were stimulated with peptide or protein with final concentration of 10− 4 M to 10− 12 M at 37 °C for 3 h. The ligand dilution and assay was performed in HBSS with 20 mM LiCl to prevent IP1 degradation to myo‐inositol. Stimulation was stopped by adding lysis buffer containing d2‐labeled inositol monophosphate and cryptate‐labeled anti‐IP antibody. After incubation at RT for 1 h, the emission at 665/8 nm and 620/10 nm were measured using a Tecan Spark plate reader. The HTRF ratio was calculated [(Em665 nm/Em620 nm)×104] and nonlinear regression analysis was performed with GraphPad Prism 5.0. Each receptor construct was tested at least three times independently in technical triplicate.
Microscopy : To analyze functionality of Nluc‐receptor constructs, fluorescence microscopy was performed. HEK293 cells were seeded into Ibidi μ‐slides 8 well (120,000 cells in 200 μL/per well) coated with poly D‐lysine and grown overnight at standard conditions. To evaluate receptor expression, cells were transfected with 900 ng receptor‐eYFP or Nluc‐receptor‐eYFP. Transfection was achieved using Lipofectamine according to the manufacturer's protocol. One day post‐transfection, fluorescence microscopy experiments were performed on an AxioVision Observer.Z1 microscope equipped with an ApoTome imaging system. All microscopy experiments were carried out in Opti‐MEM. To document the unstimulated state, cells were starved in Opti‐MEM reduced serum medium containing 2.5 μg/mL Hoechst 33342 for 30 min. For observation of peptide uptake, cells were stimulated with 1 μM TAMRA‐C9 in Opti‐MEM, washed with acidic wash (50 mM glycine, 100 mM NaCl, pH 3.0) in HBSS after the indicated time, followed by two washing steps with Opti‐MEM. To analyze the ability to recruit intracellular adaptor molecules, 100 ng mCherry‐arrestin‐3 was co‐transfected with the receptor as described above. Receptor stimulation was performed with 1 μM C9 and monitored over 60 min.
NanoBRET ligand binding assay : Binding assays were performed with COS‐7 cells, transiently transfected using Metafectene® Pro with 1000 ng receptor plasmid per 75 cm2 cell culture flask. One day post transfection, cells were reseeded in solid black 96‐well microplates and incubated for 24 h. All binding assays were performed on ice to prevent receptor internalization. For saturation assays, cell medium was removed and replaced with 100 μL BRET‐buffer (HBSS, 25 mM HEPES, pH 7.3). 50 μL coelenterazine h with final concentration of 4.2 μM were added and 50 μL of the serially diluted TAMRA‐C9 or [K141(TAMRA)]ChemS157 were added to final concentrations between 10−5 M to 10−12 M. Measurement was performed immediately after ligand stimulation .In addition, displacement assays were performed. Medium was replaced with 150 μl BRET‐buffer and 20 μl of TAMRA‐ligand was added (final concentration of 10 nM for peptide and 40 nM for protein). Next, 20 μl of unlabeled ligand were added to a final concentration between 10−5 M to 10−12 M. Cells were incubated on ice on a tumbler for 1 h under light protection. Measurement was performed immediately after adding of 10 μL coelenterazine h to a final concentration of 4.2 μM. For all BRET experiments, the luminescence signal was detected between 430–470 nm and TAMRA‐fluorescence was detected between 550–700 nm using a plate reader (Tecan Spark). BRET was calculated as the ratio of fluorescence and luminescence and netBRET was obtained by subtraction of control wells without labeled ligand. Data analysis was performed with GraphPad Prism 5.0 plotting netBRET values against ligand concentrations. The assays were performed with at least three independent experiments as triplicates.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
The authors thank Ronny Müller, Kristin Löbner, Janet Schwesinger, Christina Dammann and Regina Reppich‐Sacher for expert technical assistance. K.K. is grateful to his student Marlene Fritsche for her contribution to this project. This research was funded by the German Research Foundation (DFG)‐Project number 209933838‐SFB1052‐C08. A.S.C. received funding from the “Studienstiftung des Deutschen Volkes”. Funding for A.S.C. and K.K. by the Graduate School “Leipzig School of Natural Sciences – Building with Molecules and Nanoobjects” (BuildMoNa) is gratefully acknowledged. Open Access funding enabled and organized by Projekt DEAL.
A. S. Czerniak, K. Kretschmer, T. Weiß, A. G. Beck-Sickinger, ChemMedChem 2022, 17, e202200413.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Kawai T., Autieri M. V., Scalia R., Am. J. Physiol. Cell Physiol. 2021, 320, C375-C391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bozaoglu K., Bolton K., McMillan J., Zimmet P., Jowett J., Collier G., Walder K., Segal D., Endocrinology 2007, 148, 4687–4694. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Banas M., Zegar A., Kwitniewski M., Zabieglo K., Marczynska J., Kapinska-Mrowiecka M., LaJevic M., Zabel B. A., Cichy J., PLoS One 2015, 10, e0117830; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Luangsay S., Wittamer V., Bondue B., De Henau O., Rouger L., Brait M., Franssen J. D., de Nadai P., Huaux F., Parmentier M., J. Immunol. 2009, 183, 6489–6499; [DOI] [PubMed] [Google Scholar]
- 3c. Goralski K. B., McCarthy T. C., Hanniman E. A., Zabel B. A., Butcher E. C., Parlee S. D., Muruganandan S., Sinal C. J., J. Biol. Chem. 2007, 282, 28175–28188. [DOI] [PubMed] [Google Scholar]
- 4. Ernst M. C., Sinal C. J., Trends Endocrinol. Metab. 2010, 21, 660–667. [DOI] [PubMed] [Google Scholar]
- 5. Wittamer V., Franssen J.-D., Vulcano M., Mirjolet J.-F., Le Poul E., Migeotte I., Brézillon S., Tyldesley R., Blanpain C., Detheux M., Mantovani A., Sozzani S., Vassart G., Parmentier M., Communi D., J. Exp. Med. 2003, 198, 977–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schultz S., Saalbach A., Heiker J. T., Meier R., Zellmann T., Simon J. C., Beck-Sickinger A. G., Biochem. J. 2013, 452, 271–280. [DOI] [PubMed] [Google Scholar]
- 7. Wittamer V., Grégoire F., Robberecht P., Vassart G., Communi D., Parmentier M., J. Biol. Chem. 2004, 279, 9956–9962. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Roh S. G., Song S. H., Choi K. C., Katoh K., Wittamer V., Parmentier M., Sasaki S., Biochem. Biophys. Res. Commun. 2007, 362, 1013–1018; [DOI] [PubMed] [Google Scholar]
- 8b. Parolini S., Santoro A., Marcenaro E., Luini W., Massardi L., Facchetti F., Communi D., Parmentier M., Majorana A., Sironi M., Tabellini G., Moretta A., Sozzani S., Blood 2007, 109, 3625–3632. [DOI] [PubMed] [Google Scholar]
- 9. De Henau O., Degroot G.-N., Imbault V., Robert V., De Poorter C., Mcheik S., Galés C., Parmentier M., Springael J.-Y., PLoS One 2016, 11, e0164179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhou J. X., Liao D., Zhang S., Cheng N., He H. Q., Ye R. D., Acta Pharmacol Sin 2014, 35, 653–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Berg V., Sveinbjornsson B., Bendiksen S., Brox J., Meknas K., Figenschau Y., Arthritis Res. Ther. 2010, 12, R228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marchese A., Docherty J. M., Nguyen T., Heiber M., Cheng R., Heng H. H., Tsui L. C., Shi X., George S. R., O′Dowd B. F., Genomics 1994, 23, 609–618. [DOI] [PubMed] [Google Scholar]
- 13. Barnea G., Strapps W., Herrada G., Berman Y., Ong J., Kloss B., Axel R., Lee K. J., Proc. Natl. Acad. Sci. USA 2008, 105, 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.
- 14a. Rourke J. L., Muruganandan S., Dranse H. J., McMullen N. M., Sinal C. J., J. Endocrinol. 2014, 222, 201–215; [DOI] [PubMed] [Google Scholar]
- 14b. Yang Y. L., Ren L. R., Sun L. F., Huang C., Xiao T. X., Wang B. B., Chen J., Zabel B. A., Ren P., Zhang J. V., J. Endocrinol. 2016, 230, 55–65; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14c. Li J., Xiang L., Jiang X., Teng B., Sun Y., Chen G., Chen J., Zhang J. V., Ren P. G., J Orthop Translat 2017, 10, 42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rourke J. L., Dranse H. J., Sinal C. J., Mol. Cell. Endocrinol. 2015, 417, 36–51. [DOI] [PubMed] [Google Scholar]
- 16. Fischer T. F., Czerniak A. S., Weiß T., Schoeder C. T., Wolf P., Seitz O., Meiler J., Beck-Sickinger A. G., Cell. Mol. Life Sci. 2021, 78, 6265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. England C. G., Ehlerding E. B., Cai W., Bioconjugate Chem. 2016, 27, 1175–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fischer T. F., Schoeder C. T., Zellmann T., Stichel J., Meiler J., Beck-Sickinger A. G., J. Med. Chem. 2021, 64, 3048–3058. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Hauser A. S., Attwood M. M., Rask-Andersen M., Schiöth H. B., Gloriam D. E., Nat. Rev. Drug Discovery 2017, 16, 829–842; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19b. Rask-Andersen M., Masuram S., Schiöth H. B., Annu. Rev. Pharmacol. Toxicol. 2014, 54, 9–26; [DOI] [PubMed] [Google Scholar]
- 19c. Santos R., Ursu O., Gaulton A., Bento A. P., Donadi R. S., Bologa C. G., Karlsson A., Al-Lazikani B., Hersey A., Oprea T. I., Overington J. P., Nat. Rev. Drug Discovery 2017, 16, 19–34; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19d. Sriram K., Insel P. A., Mol. Pharmacol. 2018, 93, 251–258; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19e. van Gastel J., Leysen H., Boddaert J., Vangenechten L., Luttrell L. M., Martin B., Maudsley S., Pharmacol. Ther. 2021, 223, 107793. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Davies M., Færch L., Jeppesen O. K., Pakseresht A., Pedersen S. D., Perreault L., Rosenstock J., Shimomura I., Viljoen A., Wadden T. A., Lingvay I., Group S. S., Lancet 2021, 397, 971–984; [DOI] [PubMed] [Google Scholar]
- 20b. Domenica Rubino M. D., Jama 2021, 325, 1414–1425; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20c. Müller T. D., Blüher M., Tschöp M. H., DiMarchi R. D., Nat. Rev. Drug Discovery 2022, 21, 201–223; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20d. Thomas P., Wadden A., Jama 2021, 325, 1403–1413; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20e. Wilding J. P. H., Batterham R. L., Calanna S., Davies M., Van Gaal L. F., Lingvay I., McGowan B. M., Rosenstock J., Tran M. T. D., Wadden T. A., Wharton S., Yokote K., Zeuthen N., Kushner R. F., N. Engl. J. Med. 2021, 384, 989–1002. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Henninot A., Collins J. C., Nuss J. M., J. Med. Chem. 2018, 61, 1382–1414; [DOI] [PubMed] [Google Scholar]
- 21b. Davenport A. P., Scully C. C. G., de Graaf C., Brown A. J. H., Maguire J. J., Nat. Rev. Drug Discovery 2020, 19, 389–413. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Zabel B. A., Nakae S., Zúñiga L., Kim J.-Y., Ohyama T., Alt C., Pan J., Suto H., Soler D., Allen S. J., Handel T. M., Song C. H., Galli S. J., Butcher E. C., J. Exp. Med. 2008, 205, 2207–2220; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22b. Otero K., Vecchi A., Hirsch E., Kearley J., Vermi W., Del Prete A., Gonzalvo-Feo S., Garlanda C., Azzolino O., Salogni L., Lloyd C. M., Facchetti F., Mantovani A., Sozzani S., Blood 2010, 116, 2942–2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bondue B., Wittamer V., Parmentier M., Cytokine Growth Factor Rev. 2022, 22, 331–338. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Southern C., Cook J. M., Neetoo-Isseljee Z., Taylor D. L., Kettleborough C. A., Merritt A., Bassoni D. L., Raab W. J., Quinn E., Wehrman T. S., Davenport A. P., Brown A. J., Green A., Wigglesworth M. J., Rees S., J. Biomol. Screening 2013, 18, 599–609; [DOI] [PubMed] [Google Scholar]
- 24b. Kennedy A. J., Yang P., Read C., Kuc R. E., Yang L., Taylor E. J. A., Taylor C. W., Maguire J. J., Davenport A. P., J Am Heart Assoc 2016, 5, e004421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Machleidt T., Woodroofe C. C., Schwinn M. K., Méndez J., Robers M. B., Zimmerman K., Otto P., Daniels D. L., Kirkland T. A., Wood K. V., ACS Chem. Biol. 2015, 10, 1797–1804. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Shonberg J., Kling R. C., Gmeiner P., Löber S., Bioorg. Med. Chem. 2015, 23, 3880–3906; [DOI] [PubMed] [Google Scholar]
- 26b. Vernall A. J., Stoddart L. A., Briddon S. J., Ng H. W., Laughton C. A., Doughty S. W., Hill S. J., Kellam B., Org. Biomol. Chem. 2013, 11, 5673–5682. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Sun S., Yang X., Wang Y., Shen X., Int. J. Mol. Sci. 2016, 17, 1704; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27b. Hall M. P., Unch J., Binkowski B. F., Valley M. P., Butler B. L., Wood M. G., Otto P., Zimmerman K., Vidugiris G., Machleidt T., Robers M. B., Benink H. A., Eggers C. T., Slater M. R., Meisenheimer P. L., Klaubert D. H., Fan F., Encell L. P., Wood K. V., ACS Chem. Biol. 2012, 7, 1848–1857; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27c. Dale N. C., Johnstone E. K. M., White C. W., Pfleger K. D. G., Front Bioeng Biotechnol 2019, 0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.
- 28a. Stoddart L. A., Johnstone E. K. M., Wheal A. J., Goulding J., Robers M. B., Machleidt T., Wood K. V., Hill Stephen J., Pfleger Kevin D. G., Nat. Methods 2015, 12, 661–663; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28b. White C. W., Johnstone E. K. M., See H. B., Pfleger K. D. G., Cell. Signalling 2019, 54, 27–34; [DOI] [PubMed] [Google Scholar]
- 28c. Boursier M. E., Levin S., Zimmerman K., Machleidt T., Hurst R., Butler B. L., Eggers C. T., Kirkland T. A., Wood K. V., Ohana R. F., J. Biol. Chem. 2020, 295, 5124–5135; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28d. White C. W., Caspar B., Vanyai H. K., Pfleger K. D. G., Hill S. J., Cell Chem. Biol. 2020, 27, 499–510.e497; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28e. Tang T., Hartig C., Chen Q., Zhao W., Kaiser A., Zhang X., Zhang H., Qu H., Yi C., Ma L., Han S., Zhao Q., Beck-Sickinger A. G., Wu B., Nat. Commun. 2021, 12, 737; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28f. Tang T., Tan Q., Han S., Diemar A., Lobner K., Wang H., Schuss C., Behr V., Morl K., Wang M., Chu X., Yi C., Keller M., Kofoed J., Reedtz-Runge S., Kaiser A., Beck-Sickinger A. G., Zhao Q., Wu B., Sci. Adv. 2022, 8, eabm1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Foster S. R., Hauser A. S., Vedel L., Strachan R. T., Huang X.-P., Gavin A. C., Shah S. D., Nayak A. P., Haugaard-Kedström L. M., Penn R. B., Roth B. L., Bräuner-Osborne H., Gloriam D. E., Cell 2019, 179, 895–908.e821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.
- 30a.S. R. J. Hoare, in Assay Guidance Manual [Internet], Eli Lilly & Company and the National Center for Advancing Translational Sciences, 2021; [PubMed]
- 30b. Strickland S., Palmer G., Massey V., J. Biol. Chem. 1975, 250, 4048–4052; [PubMed] [Google Scholar]
- 30c. Tummino P. J., Copeland R. A., Biochemistry 2008, 47, 5481–5492. [DOI] [PubMed] [Google Scholar]
- 31.
- 31a. Crump M. P., Gong J. H., Loetscher P., Rajarathnam K., Amara A., Arenzana-Seisdedos F., Virelizier J. L., Baggiolini M., Sykes B. D., Clark-Lewis I., EMBO J. 1997, 16, 6996–7007; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31b. Clark-Lewis I., Kim K. S., Rajarathnam K., Gong J. H., Dewald B., Moser B., Baggiolini M., Sykes B. D., J. Leukocyte Biol. 1995, 57, 703–711; [DOI] [PubMed] [Google Scholar]
- 31c. Wells T. N., Power C. A., Lusti-Narasimhan M., Hoogewerf A. J., Cooke R. M., Chung C. W., Peitsch M. C., Proudfoot A. E., J. Leukocyte Biol. 1996, 59, 53–60. [DOI] [PubMed] [Google Scholar]
- 32. Walther C., Nagel S., Gimenez L. E., Morl K., Gurevich V. V., Beck-Sickinger A. G., J. Biol. Chem. 2010, 285, 41578–41590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kostenis E., Degtyarev M. Y., Conklin B. R., Wess J., J. Biol. Chem. 1997, 272, 19107–19110. [DOI] [PubMed] [Google Scholar]
- 34. Heckman K. L., Pease L. R., Nat. Protoc. 2007, 2, 924–932. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.