

Abstract
Melanin is a free‐radical scavenger, antioxidant, and broadband absorber of ultraviolet (UV) radiation which protects the skin from environmental carcinogenesis. However, melanin synthesis and UV‐induced reactive melanin species are also implicated in melanocyte genotoxicity. Here, we attempted to reconcile these disparate functions of melanin using a UVB‐sensitive, NRAS‐mutant mouse model, TpN. We crossed TpN mice heterozygous for an inactivating mutation in Tyrosinase to produce albino and black littermates on a C57BL/6J background. These animals were then exposed to a single UVB dose on postnatal day three when keratinocytes in the skin have yet to be melanized. Approximately one‐third (35%) of black mice were protected from UVB‐accelerated tumor formation. However, melanoma growth rates, tumor mutational burdens, and gene expression profiles were similar in melanomas from black and albino mice. Skin from albino mice contained more cyclobutane pyrimidine dimer (CPD) positive cells than black mice 1‐h post‐irradiation. However, this trend gradually reversed over time with CPDs becoming more prominent in black than albino melanocytes at 48 h. These results show that in the absence of epidermal pigmentation, melanocytic melanin limits the tumorigenic effects of acute UV exposure but fails to protect melanocytes from UVB‐induced mutagenesis.
Keywords: carcinogenesis, dark CPD, melanin, melanocyte, mouse model, UV radiation
Significance.
Melanoma incidence is lower in people of color even though melanin synthesis and reactive species are genotoxic. Here, we show that without epidermal pigmentation, melanocytic melanin provides little to no cell‐intrinsic protection from UVB mutagenesis yet limits tumor formation in a mouse model genetically predisposed to melanoma. These observations support the idea that melanocytic melanin limits non‐mutagenic, UVB‐mediated events that promote melanoma progression. While ethnic differences in keratinocyte melanosomes largely determine skin pigmentation and UV sensitivity, our findings suggest that melanocytic melanin production mitigates melanoma risk through mechanisms other than resistance to UVB‐mediated DNA damage.
1. INTRODUCTION
Individuals with fair skin are at an increased risk of developing skin cancers, including melanoma (Brenner & Hearing, 2008; Rees, 2004). This risk is often attributed to differences in the composition of melanin, which protects the skin against DNA damage by absorbing ultraviolet (UV) radiation and scavenging free radicals (Brenner & Hearing, 2008). However, the ability of melanin to prevent melanoma is at odds with evidence that melanin production, and reactive species are genotoxic (Premi, 2020; Premi et al., 2015, 2019; Swope & Abdel‐Malek, 2018). Furthermore, even the darkest human skin types (Fitzpatrick Phototypes V and VI) have a sun protection factor (SPF) of less than 10 (Brenner & Hearing, 2008; Cripps, 1981; Mohania et al., 2017). Epidemiological studies have found that African albinos have a higher risk of developing keratinocyte carcinomas than pigmented individuals within the same population (Kiprono et al., 2014; Yakubu & Mabogunje, 1993). However, cases of melanoma are rare in African albinos, which could be attributed to decreased melanoma susceptibility, challenges in detecting amelanotic tumors, or the fact that these individuals rarely live past 30 years of age (Yakubu & Mabogunje, 1993). Taken together, such observations highlight uncertainties regarding the role of melanocytic melanin in protecting these cells from UV‐mediated mutagenesis and transformation.
Genetically engineered mouse models (GEMMs) provide a way to dissect the individual roles of melanin and UV in melanoma progression. Unfortunately, results from these studies have yet to establish a consensus in the field. Mitra et al. showed that red/yellow pheomelanin production results in oxidative stress, which is sufficient to promote UV‐independent melanoma formation in a melanoma‐prone, BRAF‐mutant GEMM (Mitra et al., 2012). Additional studies in a mouse model of melanocytic hyperproliferation found that melanin was required for UVA‐ but not UVB‐induced melanoma formation (Noonan et al., 2012). Differences in UVA and UVB carcinogenesis may explain this result. Thymine‐thymine (T–T) dimers are the predominant DNA lesions induced by UVA in the absence of melanin (Cadet et al., 2005). These lesions are often correctly replicated by DNA polymerase and are thus non‐carcinogenic (Johnson et al., 2000). However, the chemiexcitation of melanin in UVA‐exposed, pigmented animals is reported to cause cytosine‐containing dark cyclobutane pyrimidine dimers (CPDs), like those that occur after UVB exposure (Premi et al., 2015, 2019). Such lesions are more likely to generate thymine (C>T) transition mutations and promote subsequent tumor formation. These observations highlight a need to clarify how melanin production may impact UV mutagenesis and melanoma onset.
Few studies have examined the consequences of melanin in a single exposure model of UV‐accelerated tumorigenesis. However, as mentioned above, reactive melanin species, formed as a result of UV‐mediated chemiexcitation, are proposed to damage DNA through the generation of dark CPDs (Premi et al., 2015; Premi et al., 2019). The idea that such melanin induced‐DNA lesions may contribute to melanoma formation is supported by evidence that melanomas from individuals with germline MC1R variants, who produce more pheomelanin, have higher levels of intrinsic ROS, and develop melanomas with a greater number of C>T transitions (Mitra et al., 2012; Robles‐Espinoza et al., 2016). While this may be due to the role of MC1R in enhancing DNA repair (Wolf Horrell et al., 2016), it is also possible that pigment‐dependent mutagenesis is specific to certain bands of the UV spectrum or types of melanin metabolism (Wakamatsu et al., 2021). For example, UVB equally accelerated melanoma onset in black and albino HGF‐transgenic mice, while UVA enhanced melanoma formation only in the black transgenic mice (Noonan et al., 2012). Recent studies from our laboratory and others contradict this idea, showing that UVA has negligible effects on melanoma onset and mutational burden in pigmented GEMMs (Bowman et al., 2021; Viros et al., 2014). These observations highlight a need to better understand the ultimate impact of melanocyte‐intrinsic melanin on the mutational landscape and development of melanoma.
Here, we explored the role of melanocytic melanin in melanoma progression and UV‐mutagenesis using a UVB‐sensitive, NRAS‐mutant, p16 INK4a ‐deficient, mouse model (TpN; Burd et al., 2014). The TpN model mimics the expression of oncogenic NRAS in human melanocytic nevi and melanomas, and models p16INK4a loss of function, which occurs in ~70% of these tumors (Bauer et al., 2007; Cancer Genome Atlas, 2015; Roh et al., 2015). UVB irradiation on postnatal Day 3 catches a portion of melanocytes in a transitory, non‐follicular position and accelerates the formation of melanomas with mutational patterns akin to those observed in the human disease (Bowman et al., 2021; Hennessey et al., 2017; Mukhopadhyay et al., 2016; Trucco et al., 2019; Viros et al., 2014). We crossed TpN mice to albino mice carrying an inactivating mutation in Tyrosinase (Tyr c ) to produce litters containing black (Tyr WT/WT ) and albino (Tyr c/c ) animals on the same genetic background (C57BL/6J). These mice were exposed to a single, neonatal dose of UVB irradiation prior to epidermal melanization and monitored for tumor formation. Melanomas from black and albino mice were subjected to RNA‐ and whole exome‐sequencing (WES) to identify UVB‐induced, melanin‐dependent mutational patterns. Our findings suggest that in the absence of epidermal pigmentation, melanocytic melanin does not prevent UVB mutagenesis, but provides protection from UVB‐driven, non‐mutagenic processes that promote melanoma formation.
2. MATERIALS AND METHODS
2.1. Animal models and UV irradiation
Animal research protocols were reviewed and approved by The Ohio State University's Institutional Animal Care and Use Committee (Protocol #2012A00000134). TpN mice were backcrossed 7 generations to C57BL/6J (Hennessey et al., 2017). These animals were then bred to albino C57BL/6J mice containing a spontaneous, inactivating Tyrosinase mutation (Tyr c‐2j , Jackson Labs #000058) to establish a colony of albino TpN mice (Figure 1a).
FIGURE 1.
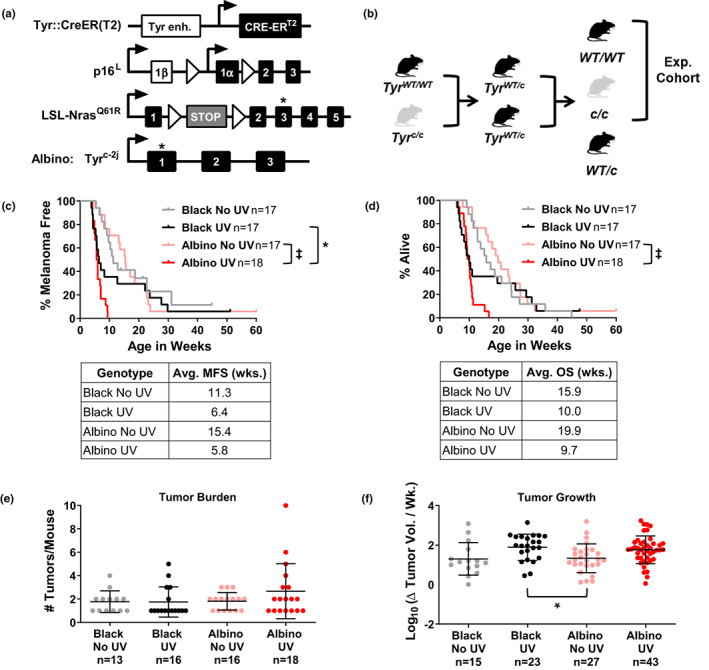
Melanoma onset and growth are independent of pigmentation in TpN mice. (a) Diagram of homozygous alleles in the TpN model. Alleles include a melanocyte‐specific, tamoxifen‐inducible CRE transgene (Tyr::CreERT2), a conditional p16 INK4a knockout allele (p16 L ), and an inducible, oncogenic Nras61R allele (LSL‐Nras 61R ). Albino TpN mice are also homozygous for an inactivating tyrosinase gene mutation (G291T; Tyr c‐2j ). (b) Breeding scheme used to generate experimental cohorts of black and albino mice. Note that mice heterozygous for the Tyr c‐2j allele (abbreviated Tyr c in the figure) were not analyzed. (c, d) Kaplan–Meier curves showing the melanoma free (“c”) and overall (“d”) survival of black and albino TpN mice mock irradiated (no UV) or treated with a single dose of 4.5 kJ/m2 UVB. Survival was compared among cohorts using log‐rank tests. Average melanoma free (MFS) and overall (OS) survivals are shown in the table below each graph. (e) Average number of tumors per mouse at the time of euthanasia. Each dot represents a single mouse with the mean ± SD indicated. No groups were statistically different as determined by a Kruskal–Wallis test with multiple comparisons. (f) Average tumor growth rates for black and albino TpN mice. Each dot represents a single tumor with the mean ± SD indicated. Statistical significance was determined by an ordinary one‐way ANOVA with multiple comparisons. *p < .05, ‡ p < .0001.
Albino and black TpN mice, homozygous for the Tyr::CreER(T2), p16 L , and LSL‐Nras Q61R alleles, were crossed as shown in Figure 1b to produce the experimental cohort. After birth, pups were treated topically with 20 mM 4‐hydroxytamoxifen (4‐OHT) on Days 1 and 2. On Day 3, unfurred albino and pigmented pups, lacking epidermal pigmentation, could be easily differentiated (Figure S1) and were randomly assigned to receive 4.5 kJ/m2 of ultraviolet B irradiation or ambient light. UVB was delivered to the dorsal side of each mouse using a solar simulator with a 300 W Xenon Short Arc lamp equipped with a UVB bandpass filter (Solar Light, Model 16S‐300‐009; Figure S1). This exposure models approximately 3.9 minimal erythema doses in an individual with phototype II skin (Hennessey et al., 2017). Treated pups were genotyped and only homozygous black (Tyr WT/WT ) and albino (Tyr c/c ) animals were monitored for tumor formation. Once discovered, tumors were measured with calipers three times per week (width x length) until the mouse met exclusion criteria. Upon euthanasia, tumor and skin samples were harvested and divided in half. One portion was fixed in 10% neutral buffered formalin and the other was flash‐frozen and stored at −80°C for future processing.
2.2. DNA and RNA isolation
Flash‐frozen tumor and skin samples were ground into a fine powder under liquid nitrogen using a mortar and pestle. RNA and DNA were isolated from the resulting powder. Tumor RNA was isolated using TRIzol reagent (Invitrogen, #15596026). The pelleted RNA was resuspended and run through the RNA Clean & Concentrator kit (Zymo Research, #R1018) for DNAse treatment and purification. Eluted RNA was quantified by Qubit (Invitrogen, #Q32866) and stored at −80°C. Tumor DNA was isolated using the Quick‐DNA Miniprep Kit protocol for Solid Tissues (Zymo Research, #D3024). DNA was quantified by Qubit and stored at −20°C.
2.3. RNA sequencing
Libraries were generated from 100 ng of DNase‐treated RNA using the NEBNext Ultra II Directional RNA Library Prep Kit and NEBNext rRNA Depletion Kit (New England BioLabs, #E6310S and #E7760S). The libraries were then subjected to paired‐end 150 bp sequencing on an Illumina HiSeq4000. The average read depth was 60X (range 46‐79X). Sequences were aligned to mm10 using STAR Alignment (Dobin et al., 2013; version 2.7.3a) and duplicates removed using Picard (a set of tools in Java for working with next‐generation sequencing data in the BAM format; version 2.17.11). After alignment, the resulting sequences were processed using the SplitNTrim, Indel Realigner, and Base Calibration tools from the Genome Analysis ToolKit (GATK, version 3.6) (McKenna et al., 2010). Differential gene expression was identified using DESeq2 (Love et al., 2014) and a volcano plot was created using the EnhancedVolcano function from Bioconductor with R. Raw RNA sequencing data were deposited in the NCBI Sequence Read Archive (SRA) under BioProject PRJNA591179.
2.4. Whole exome sequencing (WES) and variant analysis
Indexed WES libraries were generated from tumors and pooled normal controls using the Kapa Hyper Prep (Kapa Biosystems, #KK8500) and Agilent SureSelectXT Mouse All Exon (Agilent Technologies, #G7550A) target enrichment systems. Normal controls consisted of toe clip DNAs derived from 10 progenitors of the pigmented TpN colony. Indexed libraries were subjected to paired‐end 150 bp sequencing on an Illumina HiSeq4000. Sequences were aligned to mm10 using Burrows–Wheeler Aligner (BWA, version 0.7.15) (Li & Durbin, 2009) and duplicates were removed using Picard (version 2.17.11). After alignment, the resulting sequences were processed using the Indel Realigner and Base Calibration tools from GATK (versions 2.1 and 4.1) (McKenna et al., 2010). The average target coverage was 103X (range 55–145X). Raw WES data are publicly available in the NCBI SRA under BioProject PRJNA591179.
Variants (SNVs and indels) were identified from WES using VarScan2 (GATK version 3.6, VarScan2 version 2.4; Koboldt et al., 2012), Mutect2 (GATK version 4.1), and Strelka2 (version 2.9; Kim et al., 2018) to call differences between tumor and pooled normal DNA. Identified variants were annotated using the Ensembl Variant Effect Predictor (VEP; McLaren et al., 2016). Existing variants (dbSNP build 153) and duplicates were removed and only those mutations called by two or more algorithms were included in subsequent analyses. An OncoPrint was generated for deleterious mutations (annotated as “High” or “Moderate” by VEP) using ComplexHeatmap (version 2.0.0; Gu et al., 2016).
2.5. Lego and linear mutation plots
SNVs from each tumor were classified into one of 96 possible trinucleotide contexts. The average number of alterations within each trinucleotide context was calculated for each tumor genotype. This average was then normalized to the frequency of each trinucleotide sequence in the mm10 exome. Lego and linear plots were generated from these normalized data using MATLAB.
2.6. CPD dot blots
Dot blots were performed on whole skin lysates from black or albino mice subjected to ambient light (mock) or UVB irradiation (4.5 kJ/m2) on postnatal day 3 and harvested 1, 6, 24, or 48 h later. Isolated DNA (200 or 150 ng) was diluted in 150 μl of 0.4 M NaOH, boiled for 10 min, and then neutralized on ice with 150 μl of 2 M ammonium acetate, pH 7.0. Samples were applied to a nitrocellulose membrane using a dot blot apparatus and immobilized by drying overnight or baking at 80°C for 15 min in a vacuum oven. The membrane was blocked in 5% non‐fat dry milk before probing with anti‐thymine dimer (Sigma T1192; 1:1000) or anti‐8‐oxo‐2′‐deoxyguanosine (8‐oxoG; Abcam ab48508; 1:1000) antibodies followed by incubation in IRDye 800CW goat anti‐mouse secondary antibody (LI‐COR; 1:15,000). Thymine dimer and 8‐oxoG signals were visualized on an Odyssey CLx at 800 nm. Blots were quantified using LI‐COR ImageStudio software and signals of baked membranes normalized to methylene blue staining intensity (total DNA).
For methylene blue staining, the membrane was placed in mild stripping buffer (200 mM glycine, 3.5 mM sodium dodecyl sulfate, and 1% Tween 20, pH 2.3) for 5 min at room temperature twice, followed by two 10‐min washes in PBS and one 5‐min wash with TBST. The membrane was then incubated in methylene blue solution (0.3 M Sodium Acetate pH 5.0, 0.1% methylene blue) for 3 min at room temperature and excess stain was removed by washing in distilled water. Methylene blue stain was visualized on an Odyssey Fc and quantified using ImageStudio software.
2.7. Immunohistochemistry
Tumor sections were fixed overnight in 10% neutral buffered formalin and embedded in paraffin. Tumor sections (5 μm) were deparaffinized and rehydrated. Antigen retrieval was performed in a steamer with Dako Antigen Retrieval Solution (#S1699) for 30 min. The tumor sections were then blocked with Avidin and Biotin and incubated overnight in primary antibody directed against Ki67 (1:300, Abcam, #ab16667) or CD45 (1:500, BD Biosciences, #550539). Slides were treated with biotinylated secondary antibody (1:200, Vectra #BA9400, Ki67; 1:200, Vectra #BA1000, CD45) and Vectastain ABC‐HRP Reagent (Vector Labs #ZE1001). Lastly, DAB chromogen was added to each section until light brown staining was observed under a microscope. The slides were counterstained with hematoxylin. Six images of each slide were taken on a Vectra Automated Multispectral Imaging System (PerkinElmer). The average DAB positivity for each slide was determined using InForm Advanced Image Analysis Software (PerkinElmer).
Harvested skin was fixed overnight in 10% neutral buffered formalin, embedded in paraffin, and 5 μm tissue sections prepared. Melan‐A and CPD or 8‐oxoG staining was conducted as previously described (Rahman et al., 2021). Skin sections from unirradiated, postnatal day 3 mice were Fontana–Masson stained and imaged.
3. RESULTS
3.1. Melanocytic melanin provides protection from UVB‐accelerated melanomagenesis
We adapted the TpN mouse model to study the effects of melanin on melanoma progression. In this model, melanocyte‐specific Nras Q61R expression and p16 INK4a loss are initiated via the 4‐hydroxytamoxifen (4‐OHT)‐dependent activation of CreER(T2) (Figure 1a) (Bosenberg et al., 2006; Burd et al., 2014; Monahan et al., 2010). These genetic changes mimic the expression of NRAS oncogenes in benign human nevi (Bauer et al., 2007; Roh et al., 2015), the increased melanoma susceptibility of individuals with germline p16 INK4a loss, and the functional inactivation of p16INK4a in >70% of NRAS‐mutant melanomas (Cancer Genome Atlas, 2015; Foulkes et al., 1997). The TpN model was originally created on a pigmented, C57BL/6J background (Burd et al., 2014; Hennessey et al., 2017). Here, we established a complementary albino TpN model by crossing pigmented TpN mice to a C57BL/6J strain that contains an inactivating mutation in Tyrosinase (Tyr c‐2j , abbr. Tyr c ). The resulting mice have unpigmented skin, white fur, and red eyes (Townsend et al., 1981).
Albino and pigmented TpN mice were bred to produce founders heterozygous for Tyr c . These Tyr WT/c animals were then intercrossed to produce the experimental cohort (Figure 1b). Experimental pups were treated with 4‐OHT on postnatal days one and two to activate Cre and induce recombination of the LSL‐Nras Q61R and p16 L alleles. On day three, albino and black pups were divided into groups for either UVB irradiation or ambient light exposure (see Section 2). Fontana–Masson staining confirmed that the epidermis was not melanized at this developmental time point (Figure S2). Only homozygous black (Tyr WT/WT ) and albino (Tyr c/c ) mice were followed for tumor onset and growth.
A single UV exposure accelerated melanoma formation and decreased the overall survival (OS) of most TpN mice in the black and albino cohorts (Figure 1c,d). However, more animals in the black cohort escaped the tumorigenic effects of UV, yielding bi‐phasic melanoma‐free (MFS) and OS curves. Comparison of the black and albino MFS and OS curves using a log‐rank test showed a modest increase in the MFS of irradiated black versus albino mice (p = .047; Figure 1c). We also examined the influence of melanin on tumor burden and growth rates. Black and albino mice developed the same number of melanomas regardless of UV status (Figure 1e). Tumor growth was also equivalent among UV‐irradiated and unirradiated black and albino mice (Figure 1f).
Immunohistochemistry was performed on tumors from both black and albino UV‐irradiated mice to assess proliferation (Ki67) and total immune infiltration (CD45). Quantification of DAB‐positive cells showed there was no difference in cell proliferation or immune infiltration between the black and albino TpN tumors (Figure S3). These data show that in the absence of epidermal pigmentation, melanocytic melanin provides some protection from UV‐accelerated melanoma formation but does not alter the proliferative features of established TpN tumors.
3.2. Black and albino TpN melanomas have a similar genomic landscape
We used next generation sequencing to compare the gene expression profiles and mutation types found in melanomas from UV‐irradiated black and albino TpN mice. RNA sequencing of black and albino TpN melanomas identified only four genes that differed in expression between the groups with a greater than 50%‐fold change and adjusted p‐value < .05 (Figure 2a). These four genes, Ddx3y, Uty, Kdm5a and Elf2s3y, are all located on the Y chromosome and likely reflect differences in the sex distribution of our sequenced tumors (5 male and 1 female black tumor versus 6 female albino tumors). Thus, the overall gene expression patterns of black and albino TpN tumors were comparable.
FIGURE 2.
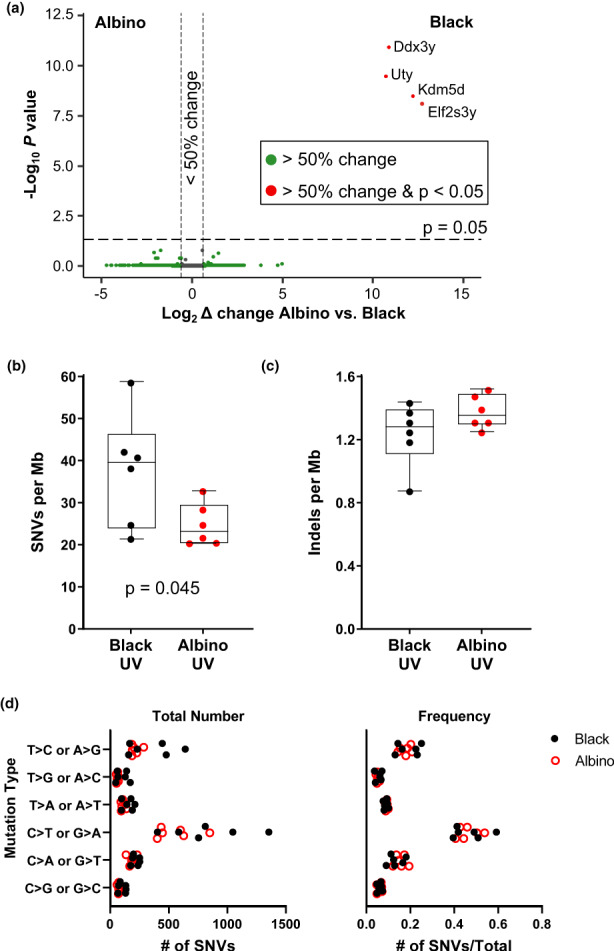
Gene expression and mutational burden are similar in melanomas from UV‐irradiated black and albino mice. (a) Volcano plot of gene expression changes identified by RNA‐seq in melanomas from UV‐irradiated black and albino mice. Genes exhibiting significant expression changes (p‐value ≤ .05 with 5% FDR) are located above the horizontal line. Genes that differ ≥50% in expression between black and albino tumors fall outside of the vertical lines. Transcripts with higher expression in melanomas from black mice appear on the right‐hand side of the plot. (b, c) Total number of single nucleotide variants (SNVs, “a”) and indels (“b”) found in whole exome sequencing of melanomas from UV‐irradiated black and albino mice. Box plots show the mean with 5th and 95th percentiles indicated. Dots represent individual tumors. Statistical significance was determined by one‐tailed, unpaired t‐tests with Welch's correction. (d) Total number (left) and overall frequency (right) of the designated alterations in melanomas from UV‐irradiated black and albino mice. Each dot represents an individual tumor. Statistical significance was determined by multiple t‐tests with a two‐stage, 5% FDR approach. ‡ p < .001
Whole exome sequencing (WES) was also performed on tumors from black and albino TpN mice to assess the types of genetic alterations present in each group. First, the number of single nucleotide variants (SNVs) and insertions and deletions (indels) were compared between pigmentation groups. On average, SNVs were more frequent in the black TpN tumors (p = .045 Figure 2b). However, there was no difference in the number of indels detected in tumors from each pigmentation group (Figure 2c).
UV‐induced CPDs promote the formation of “UV signature” C>T transition mutations (Cadet & Douki, 2018; Mohania et al., 2017). Therefore, we compared the types of mutations present in black and albino TpN melanomas. As expected, C>T transitions were the most numerous and frequent mutation type in tumors from both pigmentation groups (Figure 2d). However, the overall number and frequency of C>T transitions among all tumor mutations was similar in black and albino melanomas (Figure 2d). We also examined the trinucleotide context of alterations within our tumors using both lego and linear plots, since UV‐signature C>T transitions occur predominantly at dipyrimidine sites (Cadet & Douki, 2018; Mohania et al., 2017) (Figure 3). C>T transitions were frequently located within a dipyrimidine site in both the black and albino melanomas, and there were no differences in the trinucleotide contexts of these transitions (Figure 3b,c, red bars). C>A (or G>T) transversions often result from the UV‐dependent oxidation of guanine to 8‐oxoG (de Gruijl et al., 2001; Markkanen et al., 2012) and were enriched at TCT sites in both UV‐accelerated black and albino tumors (Figure 3b,c). Overall, these data highlight the prevalence of similar mutation types (C>T and C>A) in black and albino TpN tumors, suggesting that melanocytic melanin is not a major determinant of the location of tumorigenic alterations resulting from acute UV irradiation.
FIGURE 3.
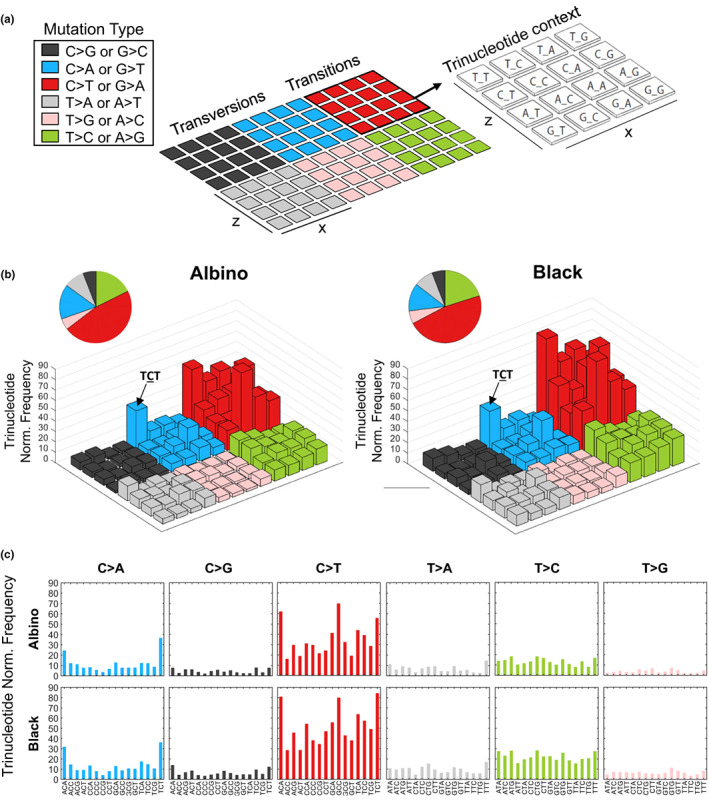
Trinucleotide context of genetic alterations in UV‐induced black and albino TpN melanomas. (a) Each colored, 4 × 4 square within the example lego plot represents a single mutation type. The nucleotides surrounding each mutation type are indicated by the bar's position within the 4 × 4 grid. The height of each bar denotes the average frequency of mutations located within the indicated trinucleotide context. Mutation frequencies were normalized to the relative occurrence of each trinucleotide sequence in the mouse exome. (b) Lego plots depicting the average frequency of mutations identified by WES in melanomas from UV‐irradiated albino and black mice. Pie charts show the total frequency of each mutation type in the experimental group. Statistical significance was determined using multiple t‐tests with a two‐stage, 5% FDR approach. (c) Separate bar graphs depicting the average frequency of mutations identified by WES in melanomas from UV‐irradiated albino and black mice.
We next examined whether different gene sets were altered in black and albino TpN tumors. Deleterious mutations in the sequenced tumors were compared to genes commonly mutated in ≥15% of human melanomas in the TCGA provisional dataset (Figure S4). Many of the mutated genes from our TpN tumors overlapped with those found in human cancer. However, no specific gene was preferentially mutated in black or albino tumors.
3.3. Cell‐intrinsic melanin does not enhance initial UVB damage in melanocytes
A possible explanation for why SNVs are more frequent in black than albino TpN tumors is that melanocytic melanin production augments UV damage. We tested this idea by breeding TpN mice heterozygous for Tyr c to generate experimental litters containing black and albino littermates. The resulting pups were treated with tamoxifen and UVB irradiated using the same schema as our tumorigenesis studies. One hour after irradiation, the mice were euthanized, and their dorsal skin collected. Dot blots of whole skin DNA showed no difference in the staining intensity of CPD or 8‐oxoG lesions in black and albino skin, suggesting that melanocytic melanin alone, cannot protect the skin from UV lesions (Figure 4a,b).
FIGURE 4.
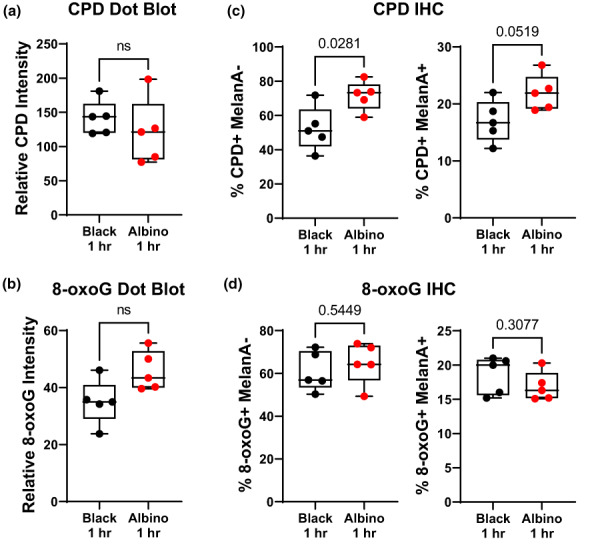
Cell‐intrinsic melanin does not enhance initial UV damage in melanocytes. (a, b) Box and whisker plots of CPD (“a”) and 8‐oxoG (“b”) staining intensity in dot blots of DNA isolated from the dorsal skin of black and albino TpN mice 1 h after UV irradiation. (c, d) Quantification of immunofluorescent co‐staining of CPD (“c”) or 8‐oxoG (“d”) and MelanA (melanocyte maker) in formalin‐fixed skin sections from black and albino TpN mice 1 h after UV irradiation. (a–d) Dots represent biological replicates. Boxes span the 25th–75th percentile with whiskers from the minimum to maximum value. Statistical significance was determined using Welch's t‐tests.
Keratinocytes constitute the bulk of skin tissue and are poorly melanized on postnatal day three (Figure S2). Therefore, the ability of cell intrinsic melanin to protect melanocytes from UV damage may be difficult to detect in dot blots of whole skin DNA. We addressed this experimental pitfall by performing immunofluorescence staining of CPDs and 8‐oxoG in whole‐skin sections from the same black and albino TpN mice used for our dot blot analyses. Black and albino skin had the same number of 8‐oxoG+ cells (Figure 4c). However, there were fewer CPD+ pigmented (MelanA+), and non‐pigmented (MelanA−) cells in the skin of black mice (Figure 4d). These data establish that melanocytic melanin production may protect the skin from some forms of initial UV damage even in the absence of epidermal pigmentation.
3.4. Cell‐intrinsic melanin does not protect melanocytes from post‐UVB DNA damage
Recent evidence indicates that hours after exposure, UV‐induced reactive melanin species cause DNA damage in the form of “dark” CPDs (Premi et al., 2015, 2019). Therefore, we examined whether the mutational similarities between albino and black melanomas might arise from late evolving, dark CPDs. Dorsal skin from black and albino TpN mice was harvested prior to, and 6, 24, or 48 h, after UVB irradiation. Skin samples were divided in half and probed for markers of DNA damage (CPDs, 8‐oxoG) using either dot blots or immunofluorescence staining. CPD and 8‐oxoG staining intensity was similar in DNA dot blots from black and albino samples (Figure 5a,b). The only difference observed was an enrichment of 8‐oxoG lesions in black skin samples taken at the 24‐h time point (p < .01). By contrast, immunofluorescent staining revealed that the number of CPD+ melanocytes, but not keratinocytes, remained higher throughout the time course, reaching statistical significance at 48 h (Figure 5d). These results suggest that melanocytic melanin production does not protect the skin from DNA lesions arising days after UVB exposure.
FIGURE 5.
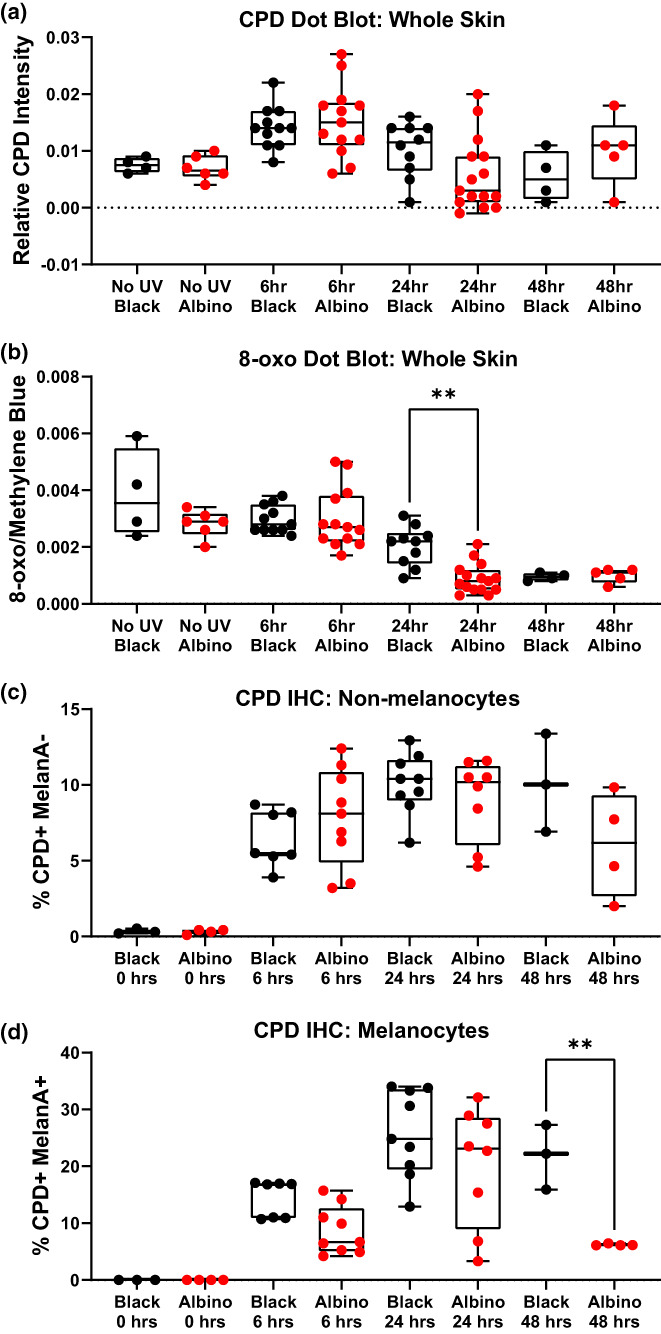
Cell‐intrinsic melanin does not protect melanocytes or keratinocytes from post‐UV DNA damage. (a, b) Box and whisker plots show the amount of CPD (“a”) and 8‐oxoG (“b”) lesions detected in dot blots of dorsal skin harvested from black or albino TpN mice prior to, and 6, 24, or 48 h after UVB irradiation. CPD and 8‐oxoG signals were normalized to methylene blue staining. (c, d) Box and whisker plots showing the amount of immunofluorescent CPD staining in dorsal skin samples from black or albino TpN mice harvested prior to, 6, 24, or 48 h after UV irradiation. Co‐staining with MelanA+ was used to identify keratinocytes (“c”; Melan‐A−) and melanocytes (“d”; Melan‐A+), respectively. (a–d) Each dot represents one biological sample. Boxes span the 25th–75th percentile with whiskers from the minimum to maximum value. Statistical significance was determined using ANOVA with a Dunnett's T3 multiple comparisons test.
4. DISCUSSION
Here, we used NRAS‐mutant mouse models (TpN) to dissect the cell‐intrinsic role of melanin in the prevention of melanocyte mutagenesis and tumorigenesis. We irradiated our mice at a developmental stage in which melanin is present in melanocytes, but not keratinocytes (Figure S2). These experiments revealed that in the absence of epidermal pigmentation, black TpN mice are better protected from UVB‐accelerated tumor formation than albino TpN mice (Figure 1c,d). However, UVB‐accelerated melanomas from black and albino mice had similar growth rates, mutational burdens, and percentages of C>T transitions (Figures 2 and 3). Examination of DNA lesions 1 h after UVB irradiation showed similar, if not more CPD lesions in albino than black skin (Figure 4c). This difference was reversed at later time points in which DNA lesions were more numerous in black than albino melanocytes (Figure 5d). These data are consistent with the idea that cell‐intrinsic melanin does not protect melanocytes from mutagenesis, but rather limits UVB‐stimulated, non‐mutational events that promote melanoma progression.
How melanocytic melanin influences UVB‐dependent, non‐mutational events is unclear. UV is known to regulate proteomic, transcriptomic, epigenomic, structural, and immunologic processes associated with tumorigenesis (Shen et al., 2017). UVB is also an important inducer of melanin synthesis. To this end, it is important to note that melanocytes from C57BL/6 mice are largely eumelanotic and produce little pheomelanin (Ito & Wakamatsu, 2003). By contrast, human melanocytes from all ethnicities produce similar levels of pheomelanin (Ito & Wakamatsu, 2003). The ratio of eumelanin to pheomelanin metabolism could have significant effects on UV mutagenesis (Wakamatsu et al., 2021). For example, eumelanin is considered photoprotective due to its ability to absorb UV irradiation and reactive oxygen species (Kobayashi et al., 1998; Smit et al., 2001). Some studies also suggest that the eumelanin precursor, 5,6‐dihydroxyindole‐2‐carboxylic acid (DHICA), can limit cellular proliferation, promote differentiation, and protect skin cells and melanocytes from oxidative stress (Kovacs et al., 2012; Novellino et al., 1999). DHICA may also enhance melanocyte immunogenicity to promote the clearance of tumorigenic precursors (Liu et al., 2011). While melanocytes from wild‐type C57BL/6J mice produce and secrete DHICA, albino mice lack this key metabolite (Ito & Wakamatsu, 2003). This deficit could limit anti‐tumorigenic cellular and microenvironmental events independent of UV‐mutagenesis. Of note, we saw no difference in the rate of tumor onset in our non‐irradiated black and albino mice (Figure 1c). Whether the protective action of DHICA or other eumelanin metabolites can only be seen in the context of a stressor remains to be tested.
Melanin synthesis also requires a significant energetic investment and impacts glutathione synthesis, resulting in altered melanocyte cellular metabolism and reducing potential (Wakamatsu et al., 2021). In particular, pheomelanin production generates ROS and consumes amino acids important for ROS neutralization (Wakamatsu et al., 2021). However, the limited production of pheomelanin in TpN mice is unlikely to influence tumor mutational burden. Melanin production may also alter the levels of cellular chromophores and metabolites (membrane lipids, tryptophan, vitamin D precursors) known to play a role in UV‐dependent immunosuppression (Bernard et al., 2019). Along with our data, these observations support the need for more extensive studies characterizing differences in the metabolome and immune microenvironment of melanocytes with distinct melanin metabolism.
Another important implication of this work is that cell‐intrinsic melanin is not sufficient to protect melanocytes from UVB mutagenesis. We find that early after UVB irradiation, black mice have equal or fewer DNA lesions that albino mice (Figure 4c,d). However, this may be a melanin‐independent phenomenon as amelanotic cell types in the skin of black mice are also protected from CPD formation (Figure 4c). Differences in the rate of CPD repair in black and albino mice are unlikely to explain this observation, as the half‐life of CPD resolution is mouse skin is ~6 h (Birger et al., 2003). At later timepoints after UVB irradiation, black melanocytes have higher CPD and 8‐oxoG levels than albino melanocytes (Figure 5b,d). These dark lesions presumably arise as a consequence of the sustained activity of enzymes such as inducible nitric oxide synthase (iNOS) and NADPH oxidase, which promote the formation of oxidant bioproducts capable of inducing CPDs (Angele‐Martinez et al., 2022). Consistent with the idea that one of the major oxidant byproducts is formed from melanin itself, post‐UV CPD levels are higher in black than albino melanocytes (Figure 5d). What is interesting, however, is that although melanomas from black mice had a higher tumor mutational burden than white tumors, C>T transitions were not specifically enriched among the black samples (Figures 2b,d and 3).
One caveat of our model is that it cannot recapitulate all aspects of human UV exposure and melanoma formation. The mice in this study received a single, neonatal dose of UV, while humans are repeatedly exposed to UV. Therefore, it is unclear how the 35% of black TpN mice that escaped the effects of UV would translate to human risk over the course of a lifetime. Moreover, the role of epidermal melanin in protecting melanocytes from UV carcinogenesis is likely significant, possibly negating the detrimental effects of cell‐intrinsic pigment production. In prior experiments using the TpN mouse model, only one of 14 black mice (7%) “escaped” the effects of UV, whereas here, six of 17 black (35%) TpN mice showed delayed melanoma onset after irradiation (Bowman et al., 2021; Figure 1c). These “escapees” originated from different litters but could have arisen due to movement of the neonates during UV irradiation or the co‐inheritance of a select set of polymorphisms along with the Tyr c‐2j albino allele. However, it is more likely that differences in our light sources contributed to this effect. In this study we used a solar simulator equipped with a UVB bandpass filter, which produces a defined spectrum (Figure S3B). Our prior studies used a hand‐held device with UVB bulbs (Bowman et al., 2021; Hennessey et al., 2017). The spectrum of these bulbs contained ~8% UVC and ~50% UVA, which may promote further carcinogenicity and a higher penetrance of accelerated melanoma formation.
In conclusion, our data help to clarify the role of cell‐intrinsic melanin in melanocyte mutagenesis and melanoma formation. Consistent with Noonan et al., we find that cell‐intrinsic melanin does not directly protect melanocytes from UVB mutagenesis, nor does it promote tumorigenesis in GEMMs (Noonan et al., 2012). Our results are also consistent with those from a therapeutic TpN model in which daily aspirin administration was found to protect against UVB‐induced DNA lesions in melanocytes but did not affect melanoma development (Rahman et al., 2021). Melanin production may increase the number of DNA lesions arising after UVB exposure in TpN melanocytes, but the resulting mutational pattern in spontaneous melanomas is no different from that observed in albino tumors. In the absence of UV, black TpN mice develop tumors at the same rate as their albino counterparts, which is contrary to findings in the HGF‐transgenic model (Noonan et al., 2012), but similar to that observed in a BRAF‐mutant GEMM (Mitra et al., 2012). Noonan et al. hypothesized that deregulated melanin aggregation was the cause of tumor promotion in black HGF‐transgenic mice (Noonan et al., 2012). Similar aggregates may not occur in BRAF‐ and NRAS‐driven tumors in which HGF is not overexpressed. Our study suggests new avenues for exploration in the field, including examination of the non‐genomic, tumorigenic events blocked by melanin metabolism, and the importance of epidermal melanosome positioning in melanoma prevention.
CONFLICT OF INTEREST
The authors declare no conflicts of interest related to this work.
Supporting information
Figure S1
Figure S2
Figure S3
Figure S4
ACKNOWLEDGMENTS
This work was supported by the Melanoma Research Alliance (309669 to C.E.B.), Damon Runyon Foundation (38‐16 to C.E.B., Pelotonia to E.R.C.), The National Institutes of Health (R01 CA237213 to C.E.B and V.P., R01 AR077664 to J.H.Z. and P30 CA016058 to The Ohio State University Comprehensive Cancer Center), and the Department of Dermatology at the University of Utah and the Huntsman Cancer Foundation (D.G.).
Weiss, T. J. , Crawford, E. R. , Posada, V. , Rahman, H. , Liu, T. , Murphy, B. M. , Arnold, T. E. , Gray, S. , Hu, Z. , Hennessey, R. C. , Yu, L. , D’Orazio, J. A. , Burd, C. J. , Zippin, J. H. , Grossman, D. , & Burd, C. E. (2023). Cell‐intrinsic melanin fails to protect melanocytes from ultraviolet‐mutagenesis in the absence of epidermal melanin. Pigment Cell & Melanoma Research, 36, 6–18. 10.1111/pcmr.13070
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are openly available in NCBI Sequence Read Archive (SRA) at https://www.ncbi.nlm.nih.gov/bioproject/PRJNA591179/, reference number PRJNA591179.
REFERENCES
- Angele‐Martinez, C. , Goncalves, L. C. P. , Premi, S. , Augusto, F. A. , Palmatier, M. A. , Amar, S. K. , & Brash, D. E. (2022). Triplet‐energy quenching functions of antioxidant molecules. Antioxidants (Basel), 11(2). 10.3390/antiox11020357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer, J. , Curtin, J. A. , Pinkel, D. , & Bastian, B. C. (2007). Congenital melanocytic nevi frequently harbor NRAS mutations but no BRAF mutations. The Journal of Investigative Dermatology, 127(1), 179–182. 10.1038/sj.jid.5700490 [DOI] [PubMed] [Google Scholar]
- Bernard, J. J. , Gallo, R. L. , & Krutmann, J. (2019). Photoimmunology: How ultraviolet radiation affects the immune system. Nature Reviews. Immunology, 19(11), 688–701. 10.1038/s41577-019-0185-9 [DOI] [PubMed] [Google Scholar]
- Birger, Y. , West, K. L. , Postnikov, Y. V. , Lim, J. H. , Furusawa, T. , Wagner, J. P. , Laufer, C. S. , Kraemer, K. H. , & Bustin, M. (2003). Chromosomal protein HMGN1 enhances the rate of DNA repair in chromatin. The EMBO Journal, 22(7), 1665–1675. 10.1093/emboj/cdg142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosenberg, M. , Muthusamy, V. , Curley, D. P. , Wang, Z. , Hobbs, C. , Nelson, B. , Nogueira, C. , Horner, J. W. , Depinho, R. , & Chin, L. (2006). Characterization of melanocyte‐specific inducible Cre recombinase transgenic mice. Genesis, 44(5), 262–267. 10.1002/dvg.20205 [DOI] [PubMed] [Google Scholar]
- Bowman, R. L. , Hennessey, R. C. , Weiss, T. J. , Tallman, D. A. , Crawford, E. R. , Murphy, B. M. , Webb, A. , Zhang, S. , La Perle, K. M. D. , Burd, C. J. , Levine, R. , Shain, A. H. , & Burd, C. E. (2021). UVB mutagenesis differs in Nras‐ and Braf‐mutant mouse models of melanoma. Life Science Alliance, 4(9). 10.26508/lsa.202101135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner, M. , & Hearing, V. J. (2008). The protective role of melanin against UV damage in human skin. Photochemistry and Photobiology, 84(3), 539–549. 10.1111/j.1751-1097.2007.00226.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd, C. E. , Liu, W. , Huynh, M. V. , Waqas, M. A. , Gillahan, J. E. , Clark, K. S. , Fu, K. , Martin, B. L. , Jeck, W. R. , Souroullas, G. P. , Darr, D. B. , Zedek, D. C. , Miley, M. J. , Baguley, B. C. , Campbell, S. L. , & Sharpless, N. E. (2014). Mutation‐specific RAS oncogenicity explains NRAS codon 61 selection in melanoma. Cancer Discovery, 4(12), 1418–1429. 10.1158/2159-8290.CD-14-0729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadet, J. , & Douki, T. (2018). Formation of UV‐induced DNA damage contributing to skin cancer development. Photochemical & Photobiological Sciences, 17(12), 1816–1841. 10.1039/c7pp00395a [DOI] [PubMed] [Google Scholar]
- Cadet, J. , Sage, E. , & Douki, T. (2005). Ultraviolet radiation‐mediated damage to cellular DNA. Mutation Research, 571(1–2), 3–17. 10.1016/j.mrfmmm.2004.09.012 [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network . (2015). Genomic classification of cutaneous melanoma. Cell, 161(7), 1681–1696. 10.1016/j.cell.2015.05.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cripps, D. J. (1981). Natural and artificial photoprotection. The Journal of Investigative Dermatology, 77(1), 154–157. 10.1111/1523-1747.ep12479359 [DOI] [PubMed] [Google Scholar]
- de Gruijl, F. R. , van Kranen, H. J. , & Mullenders, L. H. (2001). UV‐induced DNA damage, repair, mutations and oncogenic pathways in skin cancer. Journal of Photochemistry and Photobiology B: Biology, 63(1–3), 19–27. 10.1016/s1011-1344(01)00199-3 [DOI] [PubMed] [Google Scholar]
- Dobin, A. , Davis, C. A. , Schlesinger, F. , Drenkow, J. , Zaleski, C. , Jha, S. , Batut, P. , Chaisson, M. , & Gingeras, T. R. (2013). STAR: Ultrafast universal RNA‐seq aligner. Bioinformatics, 29(1), 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulkes, W. D. , Flanders, T. Y. , Pollock, P. M. , & Hayward, N. K. (1997). The CDKN2A (p16) gene and human cancer. Molecular Medicine, 3(1), 5–20. [PMC free article] [PubMed] [Google Scholar]
- Gu, Z. , Eils, R. , & Schlesner, M. (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics, 32(18), 2847–2849. 10.1093/bioinformatics/btw313 [DOI] [PubMed] [Google Scholar]
- Hennessey, R. C. , Holderbaum, A. M. , Bonilla, A. , Delaney, C. , Gillahan, J. E. , Tober, K. L. , Oberyszyn, T. M. , Zippin, J. H. , & Burd, C. E. (2017). Ultraviolet radiation accelerates NRas‐mutant melanomagenesis: A cooperative effect blocked by sunscreen. Pigment Cell & Melanoma Research, 30(5), 477–487. 10.1111/pcmr.12601 [DOI] [PubMed] [Google Scholar]
- Ito, S. , & Wakamatsu, K. (2003). Quantitative analysis of eumelanin and pheomelanin in humans, mice, and other animals: A comparative review. Pigment Cell Research, 16(5), 523–531. 10.1034/j.1600-0749.2003.00072.x [DOI] [PubMed] [Google Scholar]
- Johnson, R. E. , Washington, M. T. , Prakash, S. , & Prakash, L. (2000). Fidelity of human DNA polymerase eta. The Journal of Biological Chemistry, 275(11), 7447–7450. 10.1074/jbc.275.11.7447 [DOI] [PubMed] [Google Scholar]
- Kim, S. , Scheffler, K. , Halpern, A. L. , Bekritsky, M. A. , Noh, E. , Kallberg, M. , Chen, X. , Kim, Y. , Beyter, D. , Krusche, P. , & Saunders, C. T. (2018). Strelka2: Fast and accurate calling of germline and somatic variants. Nature Methods, 15(8), 591–594. 10.1038/s41592-018-0051-x [DOI] [PubMed] [Google Scholar]
- Kiprono, S. K. , Chaula, B. M. , & Beltraminelli, H. (2014). Histological review of skin cancers in African albinos: A 10‐year retrospective review. BMC Cancer, 14, 157. 10.1186/1471-2407-14-157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi, N. , Nakagawa, A. , Muramatsu, T. , Yamashina, Y. , Shirai, T. , Hashimoto, M. W. , Ishigaki, Y. , Ohnishi, T. , & Mori, T. (1998). Supranuclear melanin caps reduce ultraviolet induced DNA photoproducts in human epidermis. The Journal of Investigative Dermatology, 110(5), 806–810. 10.1046/j.1523-1747.1998.00178.x [DOI] [PubMed] [Google Scholar]
- Koboldt, D. C. , Zhang, Q. , Larson, D. E. , Shen, D. , McLellan, M. D. , Lin, L. , Miller, C. A. , Mardis, E. R. , Ding, L. , & Wilson, R. K. (2012). VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Research, 22(3), 568–576. 10.1101/gr.129684.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs, D. , Flori, E. , Maresca, V. , Ottaviani, M. , Aspite, N. , Dell'Anna, M. L. , Panzella, L. , Nappolitano, A. , Picardo, M. , & d'Ischia, M. (2012). The eumelanin intermediate 5,6‐dihydroxyindole‐2‐carboxylic acid is a messenger in the cross‐talk among epidermal cells. The Journal of Investigative Dermatology, 132(4), 1196–1205. 10.1038/jid.2011.457 [DOI] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with burrows‐wheeler transform. Bioinformatics, 25(14), 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X. M. , Zhou, Q. , Xu, S. Z. , Wakamatsu, K. , & Lei, T. C. (2011). Maintenance of immune hyporesponsiveness to melanosomal proteins by DHICA‐mediated antioxidation: Possible implications for autoimmune vitiligo. Free Radical Biology & Medicine, 50(9), 1177–1185. 10.1016/j.freeradbiomed.2011.01.017 [DOI] [PubMed] [Google Scholar]
- Love, M. I. , Huber, W. , & Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biology, 15(12), 550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markkanen, E. , Hubscher, U. , & van Loon, B. (2012). Regulation of oxidative DNA damage repair: The adenine:8‐oxo‐guanine problem. Cell Cycle, 11(6), 1070–1075. 10.4161/cc.11.6.19448 [DOI] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , Garimella, K. , Altshuler, D. , Gabriel, S. , Daly, M. , & DePristo, M. A. (2010). The genome analysis Toolkit: A MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Research, 20(9), 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren, W. , Gil, L. , Hunt, S. E. , Riat, H. S. , Ritchie, G. R. , Thormann, A. , Flicek, P. , & Cunningham, F. (2016). The Ensembl variant effect predictor. Genome Biology, 17(1), 122. 10.1186/s13059-016-0974-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra, D. , Luo, X. , Morgan, A. , Wang, J. , Hoang, M. P. , Lo, J. , Guerrero, C. R. , Lennerz, J. K. , Mihm, M. C. , Wargo, J. A. , Robinson, K. C. , Devi, S. P. , Vanover, J. C. , D'Orazio, J. A. , McMahon, M. , Bosenberg, M. W. , Haigis, K. M. , Haber, D. A. , Wang, Y. , & Fisher, D. E. (2012). An ultraviolet‐radiation‐independent pathway to melanoma carcinogenesis in the red hair/fair skin background. Nature, 491(7424), 449–453. 10.1038/nature11624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohania, D. , Chandel, S. , Kumar, P. , Verma, V. , Digvijay, K. , Tripathi, D. , Choudry, K. , Mitten, S. K. , & Shah, D. (2017). Ultraviolet radiations: Skin defense‐damage mechanism. Advances in Experimental Medicine and Biology, 996, 71–87. 10.1007/978-3-319-56017-5_7 [DOI] [PubMed] [Google Scholar]
- Monahan, K. B. , Rozenberg, G. I. , Krishnamurthy, J. , Johnson, S. M. , Liu, W. , Bradford, M. K. , Horner, J. , DePinho, R. A. , & Sharpless, N. E. (2010). Somatic p16(INK4a) loss accelerates melanomagenesis. Oncogene, 29(43), 5809–5817. 10.1038/onc.2010.314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay, P. , Ferguson, B. , Muller, H. K. , Handoko, H. Y. , & Walker, G. J. (2016). Murine melanomas accelerated by a single UVR exposure carry photoproduct footprints but lack UV signature C>T mutations in critical genes. Oncogene, 35(25), 3342–3350. 10.1038/onc.2015.386 [DOI] [PubMed] [Google Scholar]
- Noonan, F. P. , Zaidi, M. R. , Wolnicka‐Glubisz, A. , Anver, M. R. , Bahn, J. , Wielgus, A. , Cadet, J. , Douki, T. , Mouret, S. , Tucker, M. A. , Popratiloff, A. , Merlino, G. , & De Fabo, E. C. (2012). Melanoma induction by ultraviolet a but not ultraviolet B radiation requires melanin pigment. Nature Communications, 3, 884. 10.1038/ncomms1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novellino, L. , Napolitano, A. , & Prota, G. (1999). 5,6‐Dihydroxyindoles in the Fenton reaction: A model study of the role of melanin precursors in oxidative stress and hyperpigmentary processes. Chemical Research in Toxicology, 12(10), 985–992. 10.1021/tx990020i [DOI] [PubMed] [Google Scholar]
- Premi, S. (2020). Role of melanin chemiexcitation in melanoma progression and drug resistance. Frontiers in Oncology, 10, 1305. 10.3389/fonc.2020.01305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premi, S. , Han, L. , Mehta, S. , Knight, J. , Zhao, D. , Palmatier, M. A. , Kornacker, K. , & Brash, D. E. (2019). Genomic sites hypersensitive to ultraviolet radiation. Proceedings of the National Academy of Sciences of the United States of America, 116(48), 24196–24205. 10.1073/pnas.1907860116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premi, S. , Wallisch, S. , Mano, C. M. , Weiner, A. B. , Bacchiocchi, A. , Wakamatsu, K. , Bechara, E. J. , Halaban, R. , Douki, T. , & Brash, D. E. (2015). Photochemistry. Chemiexcitation of melanin derivatives induces DNA photoproducts long after UV exposure. Science, 347(6224), 842–847. 10.1126/science.1256022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman, H. , Kumar, D. , Liu, T. , Okwundu, N. , Lum, D. , Florell, S. R. , Burd, C. E. , Boucher, K. M. , vanBrocklin, M. W. , & Grossman, D. (2021). Aspirin protects melanocytes and keratinocytes against UVB‐induced DNA damage in vivo. The Journal of Investigative Dermatology, 141(1), 132–141.e133. 10.1016/j.jid.2020.06.003 [DOI] [PubMed] [Google Scholar]
- Rees, J. L. (2004). The genetics of sun sensitivity in humans. American Journal of Human Genetics, 75(5), 739–751. 10.1086/425285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robles‐Espinoza, C. D. , Roberts, N. D. , Chen, S. , Leacy, F. P. , Alexandrov, L. B. , Pornputtapong, N. , Halaban, R. , Krauthammer, M. , Cui, R. , Timothy Bishop, D. , & Adams, D. J. (2016). Germline MC1R status influences somatic mutation burden in melanoma. Nature Communications, 7, 12064. 10.1038/ncomms12064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh, M. R. , Eliades, P. , Gupta, S. , & Tsao, H. (2015). Genetics of melanocytic nevi. Pigment Cell & Melanoma Research, 28(6), 661–672. 10.1111/pcmr.12412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, Y. , Stanislauskas, M. , Li, G. , Zheng, D. , & Liu, L. (2017). Epigenetic and genetic dissections of UV‐induced global gene dysregulation in skin cells through multi‐omics analyses. Scientific Reports, 7, 42646. 10.1038/srep42646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit, N. P. , Vink, A. A. , Kolb, R. M. , Steenwinkel, M. J. , van den Berg, P. T. , van Nieuwpoort, F. , Roza, L. , & Pavel, S. (2001). Melanin offers protection against induction of cyclobutane pyrimidine dimers and 6‐4 photoproducts by UVB in cultured human melanocytes. Photochemistry and Photobiology, 74(3), 424–430. [DOI] [PubMed] [Google Scholar]
- Swope, V. B. , & Abdel‐Malek, Z. A. (2018). MC1R: Front and Center in the Bright Side of dark eumelanin and DNA repair. International Journal of Molecular Sciences, 19(9). 10.3390/ijms19092667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend, D. , Witkop, C. J., Jr. , & Mattson, J. (1981). Tyrosinase subcellular distribution and kinetic parameters in wild type and C‐locus mutant C57BL/6J mice. The Journal of Experimental Zoology, 216(1), 113–119. 10.1002/jez.1402160112 [DOI] [PubMed] [Google Scholar]
- Trucco, L. D. , Mundra, P. A. , Hogan, K. , Garcia‐Martinez, P. , Viros, A. , Mandal, A. K. , Macagno, N. , Gaudy‐Marqueste, C. , Allan, D. , Baenke, F. , Cook, M. , & Marais, R. (2019). Ultraviolet radiation‐induced DNA damage is prognostic for outcome in melanoma. Nature Medicine, 25(2), 221–224. 10.1038/s41591-018-0265-6 [DOI] [PubMed] [Google Scholar]
- Viros, A. , Sanchez‐Laorden, B. , Pedersen, M. , Furney, S. J. , Rae, J. , Hogan, K. , Ejiama, S. , Girotti, M. R. , Cook, M. , Dhomen, N. , & Marais, R. (2014). Ultraviolet radiation accelerates BRAF‐driven melanomagenesis by targeting TP53. Nature, 511(7510), 478–482. 10.1038/nature13298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakamatsu, K. , Zippin, J. H. , & Ito, S. (2021). Chemical and biochemical control of skin pigmentation with special emphasis on mixed melanogenesis. Pigment Cell & Melanoma Research, 34(4), 730–747. 10.1111/pcmr.12970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf Horrell, E. M. , Boulanger, M. C. , & D'Orazio, J. A. (2016). Melanocortin 1 receptor: Structure, function, and regulation. Frontiers in Genetics, 7, 95. 10.3389/fgene.2016.00095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakubu, A. , & Mabogunje, O. A. (1993). Skin cancer in African albinos. Acta Oncologica, 32(6), 621–622. 10.3109/02841869309092440 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Figure S4
Data Availability Statement
The data that support the findings of this study are openly available in NCBI Sequence Read Archive (SRA) at https://www.ncbi.nlm.nih.gov/bioproject/PRJNA591179/, reference number PRJNA591179.