

Abstract
Pseudouridine (Ψ) is an RNA base modification ubiquitously found in many types of RNAs. In humans, the isomerization of uridine is catalyzed by different stand‐alone pseudouridine synthases (PUS). Genomic mutations in the human pseudouridine synthase 3 gene (PUS3) have been identified in patients with neurodevelopmental disorders. However, the underlying molecular mechanisms that cause the disease phenotypes remain elusive. Here, we utilize exome sequencing to identify genomic variants that lead to a homozygous amino acid substitution (p.[(Tyr71Cys)];[(Tyr71Cys)]) in human PUS3 of two affected individuals and a compound heterozygous substitution (p.[(Tyr71Cys)];[(Ile299Thr)]) in a third patient. We obtain wild‐type and mutated full‐length human recombinant PUS3 proteins and characterize the enzymatic activity in vitro. Unexpectedly, we find that the p.Tyr71Cys substitution neither affect tRNA binding nor pseudouridylation activity in vitro, but strongly impair the thermostability profile of PUS3, while the p.Ile299Thr mutation causes protein aggregation. Concomitantly, we observe that the PUS3 protein levels as well as the level of PUS3‐dependent Ψ levels are strongly reduced in fibroblasts derived from all three patients. In summary, our results directly illustrate the link between the identified PUS3 variants and reduced Ψ levels in the patient cells, providing a molecular explanation for the observed clinical phenotypes.
Keywords: intellectual disorder, protein stability, pseudouridylation, PUS3, tRNA modification
The homodimer of pseudouridine synthase 3 converts uridines at position 39 in tRNAs to pseudouridine (ψ), which affects tRNA stability and regulates RNA–protein interactions. Several patient‐derived variants of PUS3 have been associated with neurodegenerative diseases. In this study, the authors describe the clinical features and biochemical consequences of the homozygotic variant Y71C and the compound heterozygotic variant Y71C/I299T.

1. INTRODUCTION
Intellectual disability (ID) is a neurodevelopment disease with a prevalence estimated between 1% and 3% in the general population (Patel et al., 2020). Individuals with ID display significant limitations in intellectual ability and adaptive function. Widely performed genomic sequencing studies have identified an increasing number of causative monogenic variants for ID. The relevant genes encode a diverse group of proteins and interestingly, pathogenic variants in tRNA modification enzymes are often found in individuals with autosomal recessive ID (De crécy‐Lagard et al., 2019; Ramos et al., 2019). The defective tRNA modification activity most probably affects neuronal development by inducing a proteome imbalance.
Pseudouridine (Ψ) is one of the most abundant posttranscriptional modifications in RNAs, including mRNAs, tRNAs, rRNAs, and other noncoding RNAs (Penzo et al., 2017). The modification is an isomerization product of uridine, where the canonical carbon‐nitrogen glycosidic bond (C1–N1) between the sugar and ribose of uridine is replaced by a carbon–carbon bond (C1–C5). The free N1‐H group of Ψ is able to engage in additional interactions with its surrounding molecules and can for instance form novel hydrogen bonds with the phosphate groups of neighboring RNA nucleotides (Durant & Davis, 1999). Hence, the presence of Ψ increases the stability of linear and folded RNA molecules and can enhance the formation of structured domains from single‐stranded and duplex regions of RNAs. Ψ are incorporated post‐translationally and the high evolutionary conservation of certain Ψ‐sites highlights their functional importance (Angelova et al., 2018; Carlile et al., 2014; Li et al., 2015; Martinez et al., 2022; Schwartz et al., 2014). For instance, the U2 small nuclear RNA (snRNA) requires Ψ for efficient mRNA splicing (Wu et al., 2011) and rRNA carries Ψ‐clusters in functionally important regions near the decoding center (Sloan et al., 2017). tRNAs are heavily decorated with various modifications (Krutyhołowa et al., 2019) and typically carry 2–3 Ψs per individual tRNA molecule. The incorporations of Ψ at positions 13, 38/39, and 55 are known to stabilize the three‐dimensional structure of tRNAs and thus have an impact on ribosomal protein translation (Klassen et al., 2016; Klassen & Schaffrath, 2017; Lecointe et al., 2002).
The detection of Ψ is based on the selective labeling by N3‐[N‐cyclohexyl‐N′‐β‐(4‐methylmorpholinium)ethylcarbodiimide (CMC), which can be identified by reverse transcription or next‐generation sequencing (NGS) techniques (T. Y. Lin et al., 2021). Recently it was shown, that nanopore sequencing might offer the possibility of a label‐free method to detect and quantify the incorporation of Ψ in different types of RNAs (Begik et al., 2021). Several transcriptome‐wide chemical mapping studies recently revealed the vast presence of Ψ in numerous cellular mRNAs, too (Carlile et al., 2014; Lovejoy et al., 2014; Schwartz et al., 2014).
Depending on the respective target RNA, Ψ formation can be catalyzed by two different mechanisms and classes of enzymes—(i) RNA‐guided ribonucleoprotein complexes consisting of one H/ACA snoRNA and four core proteins (DKC1, NHP2, NOP10, GAR1), which catalyze pseudouridylation mostly in rRNAs and snRNAs, and (ii) stand‐alone pseudouridine synthases (PUS), which introduce Ψ in all forms of RNAs (T. Y. Lin et al., 2021). More than 10 different stand‐alone PUS enzymes have been identified in eukaryotes, which share a highly similar protein fold and a conserved catalytic mechanism (Huang et al., 1998; Rintala‐Dempsey & Kothe, 2017). Despite their similarity, various PUS enzymes, namely PUS1‐10, recognize different subsets of RNA targets and modification patterns. Interestingly, their specific sites do not dramatically overlap. For instance, PUS7 (MIM# 616261) modifies Ψ13 in tRNAs and PUS10 (MIM# 612787) is responsible for Ψ54 (de Brouwer et al., 2018; Song et al., 2020). PUS1 (MIM# 608109) and PUS3 (MIM# 616283) belong to the same subclass of PUS enzymes (TruA), however PUS1 targets several sites in tRNAs (e.g., Ψ1 or Ψ26/27/28) whereas PUS3 is highly specific for Ψ38/39 (Rintala‐Dempsey & Kothe, 2017).
Several clinically relevant and potentially disease‐causing pathogenic/likely pathogenic variants have been identified in the genes encoding three human PUS enzymes, namely PUS1, PUS3, and PUS7. A biallelic missense mutation in PUS1 has been associated with mitochondrial myopathy and sideroblastic anemia (MLASA) (Bykhovskaya et al., 2004). Mutations in PUS7 have been identified in several individuals suffering from severe neurodevelopmental and growth delay (de Brouwer et al., 2018). Several pathogenic PUS3 variants (MIM# 617051), including homozygous and compound heterozygous variants, have been suggested to be causative for intellectual disorder syndromes, microcephaly, and severe growth deficiency (Bykhovskaya et al., 2004; Metodiev et al., 2015; Nøstvik et al., 2021; Shaheen et al., 2016, 2019). Most of the pathogenic/likely pathogenic variants were neither tested in vitro nor their effects in cells. For example, several individuals with ID were reported to carry a specific genomic substitution (c.212A>G, rs761792490), which leads a mutated PUS3 protein NP_112597.4:p.[(Tyr71Cys)] (Y71C) (Gulkovskyi et al., 2015; Nøstvik et al., 2021). However, the impacts of this single amino acid substitution on the activity of the PUS3 enzymes and the individual Ψ‐sites remain elusive.
During clinical diagnostic testing, we have identified three individuals with homozygous (NP_112597.4:p.[(Tyr71Cys)];[(Tyr71Cys)]) or heterozygous compound variants (NP_112597.4:p.[(Tyr71Cys)];[(Ile299Thr)]) of PUS3. Strikingly, the purified recombinant PUS3Y71C protein shows similar binding affinities and modification activities as the wild‐type PUS3 enzyme in vitro. However, the Y71C mutation compromises the thermostability of the protein and leads to almost complete depletion of PUS3 protein levels in patient‐derived fibroblasts. The strongly reduced levels of PUS3 in these cells are associated with a dramatic reduction of Ψ levels at known PUS3‐mediated sites, confirming the specificity of PUS3 in human cells and emphasizing the importance of PUS3 for neurodevelopmental diseases.
2. MATERIAL AND METHODS
2.1. Reagents
Libraries for exome sequencing (ES), including Roche HyperExome, SureSelectXT Human All Exon v5, and Twist Human Core Exome, were obtained from Roche, Agilent Technologies, and Twist Bioscience, respectively. Sequencing was performed using Illumina or 3500 Series Genetic Analyzer (Thermo Fisher Scientific). Reagents obtained from Thermo Fisher Scientific include Phusion™ High‐Fidelity DNA Polymerase (F530S), BigDye Terminator v3.1 Cycle Sequencing kit (4337455), PageRuler prestained protein ladder (26616), SuperScript III Reverse Transcriptase (18080044), SuperScript IV Reverse Transcriptase (18090010), RNasin (N8080119), pyrophosphatase (EF0221), and TaqMan Gene Expression Assays. Penicillin/streptomycin (P0781), fetal bovine serum (FBS; F9665), CMC with the methyl‐p‐toluenesulfonate salt (C106402), and primers for mutagenesis were obtained from Sigma‐Aldrich while Minimum Essential Medium (11095080), Dulbecco's modified phosphate‐buffered saline (DPBS) without calcium and magnesium (14190136), Dulbecco's Modified Eagle Medium (DMEM) with high glucose, GlutaMAXTM supplement and pyruvate (31966021), trypsin (15090046), and Hanks' balanced salt solution (14175129) were from Gibco. T75 vented flasks (I734‐2313) and culture dishes (15 cm, 734‐2322) were obtained from VWR. The Genomic Mini DNA isolation kit (116‐250) was purchased from A&A Biotechnology (Poland). DS‐11 Spectrophotometer was from DeNovix. ExoSAP‐IT™ PCR Product Cleanup (78250.40.UL) was purchased from Applied Biosystems. The protease inhibitor cocktail cOmplete EDTA‐free (11873580001), phosphatase inhibitors (4906845001), and real‐time PCR reagent (Light Cycler 480 Probes Master Mix, 04707494001) and LightCycler480 real‐time PCR system were from Roche. The Bioruptor® Plus sonication device was from Diagenode. The SDS‐PAGE stain‐free gel (4568046) was purchased from Bio‐Rad. PVDF membrane (IPVH00010) was obtained from Merck and the Trans‐Blot Turbo Blotting System was from Bio‐Rad. Antibodies were purchased from Abcam while HRP Chemiluminescent reagents, including VisiGlo™ Select HRP Chemiluminescent Substrate Kit (1B1583KIT100ML) and Amersham ECL Prime Western Blotting Detection Reagent (RPN2236), were from VWR or Cytiva, respectively. Reagents from Qiagen include RLT buffer (79216), RNeasy Mini Kit (74104), and RNase‐Free DNase Set (79254). The random hexamers (S300) were purchased from GeneON. The NucleoBond AX100 column was purchased from Bionovo. Bac‐to‐Bac® Baculovirus Expression System was obtained from Invitrogen and the ESF 921™ Insect Cell Culture Medium (protein free, 96‐001‐01) was from Expression Systems. All columns for protein and tRNA purifications as well as the fast protein liquid chromatography (FPLC) equipment and Amersham Imager 600 were obtained from Cytiva. The Zetasizer Nano ZS was obtained from Malvern Panalytical. Cy5‐cytosine (NU‐831‐CY5) was purchased from Jena Bioscience. The microscale thermophoresis assay (MST) and nano differential scanning fluorescence (DSF) instruments as well as the premium capillaries (MO‐K025) were obtained from NanoTemper Technologies.
2.2. ES
DNA from patients and their parents was extracted from whole blood using standard methods. To search for other variants which could possibly explain probands' symptoms, ES was performed on patient 1 and his parents using the same analysis pipeline. ES for patient 1 and his parents was performed using the Roche HyperExome protocol and sequencing was performed on Illumina NovaSeq 6000 (2× 150 cycles, 99.2, 99.1, and 99.1 of target covered at min. 20× and 99.4, 99.2, and 99.4 at 10×, values for patient, mother, and father, respectively). Genomic data processing was based on an in‐house developed pipeline (Dawidziuk et al., 2021). The presence of all identified variants in the PUS3 gene of probands and parents was confirmed by Sanger sequencing with BigDye Terminator v3.1 Cycle Sequencing kit according to the manufacturer's protocol. Capillary electrophoresis was performed using 3500 Series Genetic Analyzer. For patient 2 and patient 3, ES libraries were performed using SureSelectXT Human All Exon v5 (patient 2) and Twist Human Core Exome (patient 3), respectively. 2 × 100 bp paired‐end reads sequencing was performed on Illumina HiSeq 1500. The target coverage at min. 20× and 10× was 99.2, 99.6 and 93.0, 98.0, for patient 2 and patient 3, respectively. Bioinformatics analysis of raw ES data for patient 2 and patient 3 was performed as previously described (Śmigiel et al., 2020). For the parental ES (patients 2 and 3), a Twist Core Exome panel with RefSeq and mitochondrial spike‐in were used for target enrichment and sequencing was carried out on an Illumina NovaSeq 6000 sequencer. The target coverage at min. 20× and 10× was 99.5, 99.7 and 99.6, 99.7, for the mother and father of patient 2, and 99.5, 99.7 and 99.5, 99.8 for the mother and father of patient 3, respectively. Phenotypic and genotypic data have been submitted to the LOVD database (https://www.lovd.nl/3.0/home) with the following accession numbers: patient 1‐#00413320, patient 2‐#00413076, patient 3‐#00413230.
2.3. Primary cell culture
A skin biopsy was performed from the patients and healthy controls to generate fibroblasts and the culture procedure was followed as previously described (Villegas & McPhaul, 2005). In detail, the biopsy explants were placed in a biopsy culture medium (10% FBS, 1% penicillin/streptomycin in Minimum Essential Medium and washed three times with 10 ml DPBS without calcium and magnesium). Next, the biopsy was cut into pinhead‐size pieces and placed into a T25 tissue culture–treated flask, and the cells were grown until 90% confluency in fibroblast culture medium (FCM) containing 20% FBS and 1% penicillin/streptomycin in DMEM with high glucose, GlutaMAX™ supplement, and pyruvate. Confluent cultures were washed twice with 4 ml DPBS, detached using 1 ml of 0.25% trypsin in Hanks' balanced salt solution at 37°C, neutralized with 3 ml FCM and passaged into T75 vented flasks with 15 ml FCM. To maintain the cell line, the medium was partially replaced every 3 days and each confluent T75 flask (~1.5 × 106 cells) was passaged into three T75 flasks every 3 days. Alternatively, one T75 flask was passaged into one cell culture dish.
2.4. Sequencing of fibroblast‐derived DNA
Human fibroblasts from T75 flasks at 90% confluency were washed with DPBS, detached using trypsin as described above, and pelleted by centrifugation at 1200 g for 10 min. The cell pellets were washed with PBS and DNA was isolated using a Genomic Mini DNA isolation kit. DNA concentration was calculated from the absorbance at 260 nm using DS‐11 Spectrophotometer. Two different primer pairs were designed to amplify regions of DNA encompassing mutated sites. Primers were designed using Primer3 software (version 4.1.0). Following primers were used: PUS3Fw: AACCAGACTGAGAAGCTCCT, PUS3Rev: AACGGATCTCTTCAGCAGCA amplifying 571 bp including region encoding tyrosine 71 (rs761792490) and PUS3Fw2: AAGTACAGCTAGTGGGCCAG, PUS3Rev2: CCTATATTGAGGCTTTTGGGGA amplifying 212 bp including region encoding isoleucine 299 (rs141362244). Equal amounts of DNA samples (50 ng) were amplified using 1 U of Phusion™ High‐Fidelity DNA Polymerase, 0.5 µM of forward and reverse primer (PUSFw&PUS3Rev or PUSFw2&PUS3Rev2 depending on PCR reaction), 0.2 mM dNTP in HF buffer in the total 50 µl reaction. The same following cycling conditions were used for both PCR reactions: initial denaturation at 98°C for 30 s and 30 amplification cycles of 98°C/10 s, 63°C/30 s, 72°C/20 s, then final extension at 72°C/5 min. Each reaction (10 µl) was resolved on 1% agarose gel to verify the specific product size. Next, PCR reactions were cleaned up using ExoSAP‐IT™ PCR Product Cleanup. Briefly, 10 µl of each PCR reaction was mixed with 4 µl of ExoSAP‐IT and incubated at 37°C for 15 min. The enzyme was inactivated at 80°C for 15 min. The PCR products (14 µl) were mixed with 1 µl of 10 µM forward primer (PUS3Fw or PUS3Fw2). Sequencing was performed by Genomed SA. The resulting chromatograms were visualized and analyzed using Chromas software (version 2.6.6).
2.5. Western blot
Human fibroblasts from T75 flasks at 90% confluency were lysed in modified RIPA buffer (65 mM Tris, pH 7.4, 150 mM NaCl, 1% NP‐40, 0.25% sodium deoxycholate, 1 mM EDTA, 0.1% SDS, and 1% Triton‐X100) supplemented with protease inhibitor cocktail cOmplete EDTA‐free and phosphatase inhibitors by pipetting. The lysates were sonicated using Bioruptor® Plus for 10 cycles 20/20 s at “high” and centrifuged at 21,130 g for 10 min at 4°C to remove debris. The supernatant was collected, and protein concentration was measured using a Bradford assay. Protein samples were mixed with Laemmli loading dye with β‐mercaptoethanol and boiled for 5 min. Protein samples (30 µg) were resolved in 12% SDS‐PAGE stain‐free gel, PageRuler prestained protein ladder was used as a molecular weight marker. After electrophoresis, proteins in the gel were visualized using Bio‐Rad's ImageLab software (version 5.1) to verify the equal protein loading. Proteins were transferred to PVDF membranes (pore size 0.45 µm, Immobilon‐P,) using Trans‐Blot Turbo Blotting System. Membranes were blocked for 1 h at room temperature in 5% nonfat dry milk in PBS‐T (PBS with 0.01% Tween‐20), followed by overnight incubation at 4°C with primary antibodies (rabbit anti‐PUS3 C‐terminal domain, #ab211270; Abcam and rabbit anti‐HSP90, #ab13495; Abcam) in 5% milk in PBS‐T (1:500). Blots were washed 3 × 5 min with PBS‐T, incubated 1 h at room temperature with HRP‐conjugated secondary antibody (1:10,000 in 5% milk) and washed 3 × 5 min with PBS‐T. HRP signal was detected using VisiGlo™ Select HRP Chemiluminescent Substrate Kit for PUS3 or Amersham ECL Prime Western Blotting Detection Reagent for HSP90 on Amersham Imager 600.
2.6. RNA extraction and quantitative real‐time PCR (qRT‐PCR)
Human fibroblasts were plated onto six‐well culture plates and at 90% confluency were scraped in 350 µl of RLT buffer and lysed by pipetting. RNA was isolated using the RNeasy Mini Kit following the manufacturer's protocol. Optional on‐column digestion of DNA was performed using RNase‐Free DNase Set. RNA concentration was calculated from the absorbance at 260 nm, and the purity of the RNA was determined by the 260/280 nm absorbance ratio using DS‐11 Spectrophotometer.
Equal amounts of RNA (600 ng) were reverse‐transcribed using random hexamers and SuperScript IV Reverse Transcriptase according to the manufacturer's instructions. The cDNA was diluted 3× with H2O and 3 μl of each cDNA sample was amplified using a set of custom sequence‐specific primers and TaqMan MGB probes in a final reaction volume of 15 μl. The following TaqMan Gene Expression Assays were used: Hs00229938_m1 (PUS3, pseudouridylate synthase 3), Hs00939627_m1 (GUSB, glucuronidase beta). qRT‐PCRs were performed using Light Cycler 480 Probes Master Mix in a LightCycler480 real‐time PCR system according to the manufacturer's instructions. Following conditions were used: preincubation at 95°C for 10 min and 40 amplification cycles of 95°C/10 s, 60°C/50 s, 72°C/1 s.
Relative PUS3 mRNA levels in different cell lines were determined using the ∆Ct (where Ct is threshold cycle) relative quantification method. Values were normalized to GUSB endogenous control. Data were calculated as fold of control cells from a healthy individual without any PUS3 variants (CTRL).
2.7. Total tRNA extraction
Cells were cultured and collected from nine T75 flasks with 90% confluency. The cells were washed with PBS and lysed in 335 µl lysis buffer (10 mM Tris‐HCl pH 7.5, 100 mM NaCl, 10 mM MgCl2, 1% Triton X‐100, 0.5 mM DTT and 0.5% sodium deoxycholate). An equal volume of water was added to the lysate and the total RNA was isolated by three subsequent extractions of one volume of acid phenol‐chloroform (Acid‐Phenol:Chloroform, pH 4.5 (with IAA, 125:24:1)) and followed by final extraction with one volume of chloroform. During each extraction, the mixture was vortexed thoroughly and followed by centrifugation at 4500 g for 10 min at 4°C. The upper aqueous phase was transferred to a new tube and the extraction was performed again. The RNA, in the upper phase, was precipitated with 0.1 volume of 3 M sodium acetate (pH 6.3), 3 volumes of 96% EtOH, and 10 µg glycogen. The solution was incubated overnight at −80°C and RNA was spun down at 7100 g for 30 min at 4°C. The RNA pellet was washed in 70% EtOH and air‐dried for 2 min. The pellet was then dissolved in RNase‐free water and subjected to total tRNA isolation. A NucleoBond AX100 column was equilibrated with 10 ml equilibration buffer with Triton X‐100 (10 mM Bis‐Tris HCl, pH 6.3, 200 mM KCl, 15% EtOH, and 0.15% Triton X‐100). The total RNA was dissolved in 2 ml equilibration buffer without Triton X‐100 and applied to the column. The column was washed twice with 12 ml wash buffer (10 mM Bis‐Tris HCl, pH 6.3, 300 mM KCl, 15% EtOH). Bound tRNA was eluted with 12 ml elution buffer (10 mM Bis‐Tris HCl, pH 6.3, 750 mM KCl, 1% EtOH) and followed by precipitation using 2.5 volumes of 96% EtOH and 10 µg glycogen. The solution was incubated overnight at −80°C and tRNA was spun down at 7100 g for 30 min at 4°C. The pellet was washed twice in 70% EtOH and air‐dried for 2 min. The tRNA pellet was dissolved in 20 µl of RNase‐free water. The quality of the extracted tRNA was checked via resolving the samples in a 10% urea gel, staining by ethidium bromide solution, and visualizing with a Bio‐Rad ChemiDoc imaging system.
2.8. Recombinant protein expression and purification
The codon‐optimized sequence of a synthetic human PUS3 ORF fused with a glutathione‐S‐transferase (GST)‐tag was cloned into the pFastBac vector for expression in insect cells. The PUS3 mutants were generated using the standard mutagenesis PCR with primers that contain the mutation site sequence. The protein expression was performed using the Bac‐to‐Bac® Baculovirus Expression System (Kost et al., 2005) according to the manual. In brief, the ORF‐containing constructs were transformed into a bacterial Escherichia coli DH10Bac strain for the production of the recombinant bacmid DNA, which was transfected into sf9 insect cell lines. The recombinant baculoviruses were collected from sf9 cells and delivered to Hi5 cells to produce PUS3 recombinant proteins. The infected cells were cultured in media (ESF 921™ Insect Cell Culture Medium, protein free) with 0.5% FBS for 2 days, and then collected and lysed in buffer (50 mM HEPES, pH 7.5, 300 mM NaCl, 5 mM DTT, 5% glycerol, 5 mM MgCl2) containing protease inhibitors and DNase and with additional three cycles of freezing and thawing. The solution was sonicated and the soluble part was separated from cell debris by centrifugation at 80,000 g for 90 min. The cleared supernatant was subjected to a GSTPrep column and the column was washed with lysis buffer and washing buffer (same as lysis buffer but with 1 M NaCl) and the protein of interest was then eluted in the elution buffer which contained 18 mM reduced GSH. The elute was collected and incubated with GST‐fused tobacco etch virus (TEV) protease overnight at 4°C and followed by the removal of GST‐fused TEV and the cleaved GST‐tag via a second round of GST affinity chromatography. The flow through was concentrated and applied to a HiLoad Superdex 200 pg preparative gel filtration column, equilibrated with 20 mM HEPES, pH 7.5, 150 mM NaCl, 5 mM DTT, to obtain the pure and homogenous protein fractions. The purified protein was concentrated (3 g/L) and stored at −80°C.
2.9. Right‐angle light scattering (RALS) measurement
The dynamic light scattering (DLS) measurements were performed using a Zetasizer Nano ZS, equipped with a Superose 6 column. Proteins (500 µg) were injected into the Superose 6 column which was pre‐equilibrated with protein storage buffer (20 mM HEPES, pH 7.5, 5 mM DTT, 150 mM NaCl). The samples were measured with standard settings and the results were converted directly into the respective molecular weights.
2.10. Production of tRNAs
The DNA template of each tRNA was generated using the Primerize method (Tian et al., 2015) and the T7 promoter sequence was included for T7 RNA polymerase driven in vitro transcription (T.‐Y. Lin et al., 2019). An overnight T7‐driven transcription reaction was performed at 37°C with the following: 20 mM Tris, pH 8.0, 5 mM DTT, 150 mM NaCl, 8 mM MgCl2, 2 mM spermidine, 20 mM NTPs, linear DNA, RNasin, T7 RNA polymerase, and pyrophosphatase. RNase‐free DNase I (Thermo Fisher Scientific) was added to digest the DNA template and the reaction was stopped by the addition of EDTA. The reaction was subjected to a FPLC system using a DEAE weak anion exchange column (GE) and followed by temperature‐gradient‐based annealing. Refolding of RNA was carried out by heating the RNA solution to 80°C for 2 min and slowly cooling to room temperature. The annealed RNAs were further purified using a Superdex 75 Increase gel filtration column (GE) in the buffer (20 mM HEPES, pH 7.5, 5 mM DTT, 150 mM NaCl) and the fractions of interest were pooled, concentrated, and stored at −20°C.
2.11. MST
The RNA was labeled using Cy5‐cytosine directly during in vitro transcription. The tRNA (250 nM) was mixed with serial diluted PUS3 with equal volume in 20 mM HEPES, pH 7.5, 150 mM NaCl, 5 mM MgCl2, and 5 mM DTT. The mixture was incubated at 37°C for 30 min and then subjected to premium capillaries for measurement. The binding profiles were analyzed using MO. control software (NanoTemper Technologies) and the EC‐50s were calculated based on the measurement results (n = 3) using MO. AffinityAnalysis (NanoTemper Technologies) with Hill model fitting (T.‐Y. Lin et al., 2019).
2.12. DSF and DLS measurement
Recombinant protein (10 µg) was prepared in 10 µl buffer (20 mM HEPES, pH 7.5, 150 mM NaCl, and 5 mM DTT). The protein solution was subjected to a glass capillary and the measurement was performed using the Prometheus PANTA. PR. PantaControl software (NanoTemper Technologies) was used to control the experiment in modes: size analysis and thermal melting. Three independent measurements were performed, and data sets were analyzed and merged using the PR. PantaAnalysis software (NanoTemper Technologies).
2.13. Pseudouridylation assay and primer extension assay
PUS3 (6 µg) was mixed with tRNA substrates (8 µg) in a 25‐µl reaction volume in reaction buffer containing 100 mM ammonium acetate, 100 mM NaCl, 20 mM Tris, pH 8.0, 5 mM MgCl2, 5 mM DTT (Zhao et al., 2007). The reaction was performed at 37°C for 2 h and the tRNA was phenol‐chloroform extracted, precipitated in ethanol, and redissolved in H2O for the subsequent CMC treatment and reverse transcription. CMC with the methyl‐p‐toluenesulfonate salt (Sigma) was prepared at 0.2 M concentration in BEU buffer (50 mM bicine, pH 8.3, 4 mM EDTA, and 7 M urea) (Adachi et al., 2019). Twenty microliters of total tRNA extract (15 µg) or PUS3 treated tRNA (8 µg) were mixed with 100 µl of 0.2 M CMC at 37°C for 30 min to form the Ψ‐CMC conjugate. The negative control from the same sample but omitting CMC was also prepared. Samples were then precipitated and treated by an alkaline condition (50 mM sodium bicarbonate, pH 10.7, and 2 mM EDTA) at 37°C for 2 h to reverse the CMC conjugation on G and U nucleotides but not Ψ. The RNA was again precipitated and dissolved in H2O (15 µl). The obtained RNA was subjected to primer extension to detect the presence of Ψs.
Primer extension assays require BEU‐treated and CMC‐treated tRNA as the templates. The reverse transcription reaction was primed using a tRNA‐specific Cy5‐labeled primer. Each primer was designed to be complementary to the 3′‐end of each tRNA substrate (position 50‐76 of tRNA). All ingredients were prepared in the 10‐µl reaction volume. tRNA (1 µl) was first mixed with 1 µl 10 mM primer and 4.5 µl H2O and followed by heating at 80°C for 3 min and 65°C for 5 min and cooling down on the ice for 1 min. The sample mixture was then mixed with 2 µl of 5X reaction buffer, 0.5 µl of 100 mM DTT, and 0.5 µl SuperScriptIII. The reverse transcription reaction was carried out at 50°C for 1 h and followed by 85°C for 5 min to stop the reaction. To remove proteins in the reaction, proteinase K (>700 U) was added and the sample was incubated at 37°C for 30 min. The reverse‐transcribed products were resolved in a 15% urea denaturing gel run at 200 V for 60 min. Products with different sizes were visualized using a gel scanner.
2.14. Sequence and structural alignment
Homo sapiens PUS3 (NP_112597.4), Mus musculus Pus3 (NP_075781.3), Drosophila melanogaster Dmel (NP_611646.1), Saccharomyces cerevisiae S288C Deg1 (NP_116655.1), Arabidopsis thaliana Pus (NP_564438.1), Methanocaldococcus infernus TruA (WP_013099720.1), Methanocaldococcus jannaschii TruA (WP_010871199.1), Staphylococcus aureus TruA (WP_075109215.1), E. coli TruA (QCJ59352.1), H. sapiens PUS1 (NP_079491.2) were retrieved from NCBI database. These sequences were aligned using MUSCLE (Edgar, 2004). To predict the structure of human PUS3 the AlphaFold2 coupled with MMseq2 server (Mirdita et al., 2021) for automatic MSA generation was used. In detail, AlphaFold run in the unpaired mode with subsampling, which yielded five high‐quality models within three recycles. Models converged to identical predicted homodimer interfaces and their pLDDTs ranged from 79.25 to 80.91, while pTMscores were in the range of 0.7655–0.7861 (Jumper et al., 2021). We selected rank_1 as the representative model for presentation. The structural alignment was performed using Coot (Emsley et al., 2010) and the cartoon presentation was illustrated using PyMOL (DeLano, 2020).
2.15. Statistics
Data on the graph are expressed as means ± standard errors of the means (SEM) from three independent experiments. The statistical analysis of the data was performed with GraphPad Prism software (version 7.05) using statistical analysis performed with two‐way analysis of variance (α = 0.05) with a Tukey's multiple comparisons test. Statistically significant differences are indicated (**p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001).
3. RESULTS
3.1. Clinical description and genetic testing of three Polish individuals harboring PUS3 variants
We identified three probands from three unrelated families in Poland, presenting development delay, ID, and growth delay (Figure 1a and Table 1). Patient 1, a 9‐year‐old boy, has delayed psychomotor, speech development, and a moderate degree of ID. The physical examination performed at 8 years of age showed hypotonia, a disturbing aggressive behavioral phenotype, and mild facial dysmorphia. He had the first epileptic seizure at the age of 5 years, but the nuclear magnetic resonance (NMR) images of the brain were normal. He is intensively rehabilitated and is now under the care of the Genetic Clinic for developmental disorders and epilepsy. Patient 2, an 8‐year‐old boy, has a significant delay in psychomotor and speech development. He started to speak his first single words at the age of 8 years. He has a severe ID and abnormal behavioral phenotype. The EEG showed abnormal recordings and epilepsy was diagnosed. The first epileptic seizure occurred at the age of 4 years. NMR of the brain was normal. The cardiac ultrasound diagnosed an atrial septal defect (without surgical treatment) and the ophthalmological consultation diagnosed optic disc pallor and astigmatism. Physical examination was performed at the age of 8 years. It showed clear facial dysmorphia. Hands and feet were typical except for a single crease on the left hand. The hypotonia, excessive drooling, and scoliosis were also observed. In addition, the patient could not control urination and defecation. Patient 3, an 8‐year‐old boy has a delay in psychomotor, speech, and intellectual development as well as with behavioral abnormalities. The physical examination revealed moderate ID, speech delay, microcephaly, and distinctive facial dysmorphia. The ophthalmological exam showed nystagmus. There were no congenital defects. NMR of the brain was normal. The leading symptoms of the presented three patients are similar, namely developmental delay with severe speech delay as well as an abnormal behavioral phenotype and ID without MRI abnormalities. In addition, two of the patients suffer from seizures. Of note, the parents of the patients do not present any clinical abnormalities.
Figure 1.
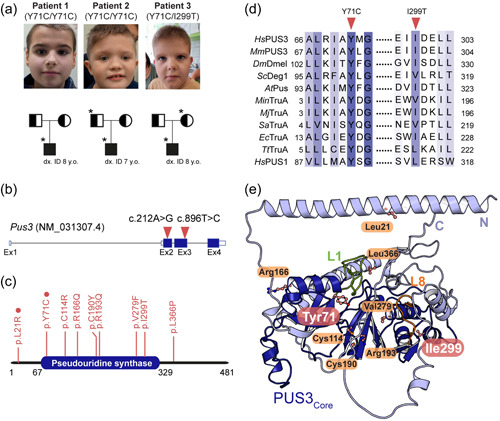
Identification of the Y71C and I299T substitutions. (a) Pedigrees of the three individuals and their families. *The available samples for fibroblasts culture and further studies. The carriers are denoted as a symbol with half color whereas the patients are marked as a symbol filled with black. The age of genetic diagnosis of each patient is as indicated. (b) Schematic presentation of the human PUS3 transcript (red triangles indicate the point mutations). (c) Domain structure of the full‐length human PUS3 protein (a.a. 1‐481) where the pseudouridine synthase domain is as indicated (a.a. 67‐329). The reported causative missense variants are indicated, and the homozygotic mutations are highlighted with red circles. (d) Schematic presentation of the partial sequence alignment in human PUS3 and its homologs around the tyrosine 71 or the isoleucine 299 residues (indicated as triangles). TruA proteins include HsPUS3 (NP_112597.3), Drosophila melanogaster Dmel (NP_611646.1), Mus musculus Pus3 (NP_075781.3), Saccharomyces cerevisiae Deg1 (NP_116655.1), Methanocaldococcus infernus TruA (WP_013099720.1), Methanocaldococcus jannaschii TruA (WP_010871199.1), Staphylococcus aureus TruA (WP_075109215.1), EcTruA (QCJ59352.1), Arabidopsis thaliana Pus (NP_564438.1), and Thermus thermophilus TruA (WP_024118717.1). Human PUS1 sequence (NP_079491.2) was also aligned. (e) The cartoon representation of the predicted human PUS3 structure using Alphafold2 where the core domain is colored in deep blue and the N‐ and C‐termini are in light blue. The equivalent tRNA binding finger motifs (L1 and L8) of EcTruA are annotated as L1 (green, residue 71–88) and L8 (orange, residue 227–238) motifs in human PUS3. The predicted positions of all reported pathogenic PUS3 substitutions are indicated in ball and sticks presentation while the tyrosine 71 residue and isoleucine 299 residue are highlighted in pink. Oxygen is labeled in red while nitrogen is in blue.
Table 1.
Medical history, development, and neurological assessment of patients
| Patient 1 | Patient 2 | Patient 3 | |
|---|---|---|---|
| Sex, age (years) | Male, 9 | Male, 8 | Male, 8 |
| Country (origin) | Poland | Poland | Poland |
| Variants | Homozygous variant | Homozygous variant | Heterozygous variant |
| c.212A>G (p.Tyr71Cys) | c.212A>G (p.Tyr71Cys) | c.212A>G (p.Tyr71Cys) | |
| c.896T>C (p.Ile299Thr) | |||
| Age of genetic diagnosis (years) | 8 | 7 | 8 |
| Family history /affected siblings | Unencumbered | Unencumbered | Unaffected |
| Delivery and term of delivery (weeks) | Normal, 39 | Normal, 39 | Normal, 40 |
| Developmental delay | Y | Y | Y |
| Sitting (age) | 10 months | 19 months | 9 months |
| Walking (age) | 22 months | 36 months | 20 months |
| Speech (age) | 5 years of age | 8 years of age, single words | 4 years of age, single words |
| Hypotonia | Y | Y | Y |
| ID | Moderate | Severe | Moderate |
| ABP | Y, aggressive behavior | Y | Y |
| Feeding difficulties | N | Y | N |
| Dysphagia | N | N | N |
| Dysmorphism | Mild | Distinctive | Distinctive |
| Microcephaly | 3rd–10th | 10th | <3rd |
| Gray sclerae | ‐ | + | + |
| Coarse face | ‐ | + | + |
| Wide nasal bridge | ‐ | + | + |
| Flat facial profile | + | + | + |
| Epicanthal folds | ‐ | + | + |
| Dysmorphic ears | + | + (protruding) | + |
| Seizures | Y | Y | N |
| Age of first seizure | 5 years of age | 4 years of age | ‐ |
| Treatment | Valproic acid, klobazam, etosuksymid, steroid (metyloprednizolon) | Valproic acid | ‐ |
| MRI imaging | Without abnormalities | Without abnormalities | Without abnormalities |
| Hearing loss sensorineural | N | N | N |
| Abnormality of vision | ‐ | Y, optic disc pallor, astigmatism | Y, nystagmus |
| Heart defects | N | Y, ASD, PDA | N |
| Scoliosis | N | Y | Y |
| Body mass/age | 28 kg (50th−75th), 8 years | 31 kg (75th–90th), 8 years | 31 kg (75th–90th), 8 years |
| Height/age | 130 cm (50th), 8 years | 122 cm (3rd−10th), 8 years | 119 cm (3rd), 8 years |
| OFC/age | 51 cm (3rd−10th), 8 years | 51 cm (3rd−10th), 8 years | 50 cm (<3rd), 8 years |
Note: Symptom code: developmental delay (HP: 0001263); hypotonia (generalized HP: 0001290; central HP: 0011398); ID (HP: 0001249); Abnormal behavioral phenotype (HP: 0000708); feeding difficulty (HP: 0011968); dysphagia (HP: 0002015); dysmorphism (HP: 0001999); microcephaly (HP: 0000252); gray sclerae (HP: 0000591); coarse face (HP: 0000280); wide nasal bridge (HP: 0000431); flat facial profile (HP: 0012368); epicanthal folds (HP: 0007930); seizures (HP: 0001250); hearing loss sensorineural (HP: 0000407); abnormality of vision (HP: 0000504); Strabismus (HP: 0000486); Optic disc pallor (HP 0000543); ASD (HP: 0001631); patent ductus arteriosus (HP: 0001643); scoliosis (HP: 0002650). NM_031307.4 and NP_112597.4 were used as reference sequences for variant description.
Abbreviations: ABP, abnormal behavioral phenotype; ASD, Atrial Septal Defect; ID, intellectual disorder; M, male; N, no; OFC, occipital frontal circumference; PDA, patent ductus arteriosus; Y, yes.
ES showed homozygous PUS3 c.212A>G (p.Tyr71Cys) variant in patient 1 and patient 2, and a compound heterozygosity for variants c.212A>G (p.Tyr71Cys) and c.896T>C (p.Ile299Thr) in PUS3 in patient 3 (Figure 1b and Supporting Information: Figure S1a). Both parents of all three patients were carriers of single PUS3 variants consistent with autosomal recessive inheritance. Although both variants in our study have very low frequency in the gnomAD population database (heterozygous frequency in gnomAD v3.1.2: c.212A>G, 6/152248 or 0,000039; c.896T>C: 1/152172 or 0.0000065; homozygous count for both variants: 0), their positions are strongly conserved as indicated by phyloP100way values of 7.57 and 8.82 (Richards et al., 2015), respectively. In addition, the majority of predictors for missense variants classify the substitutions as pathogenic, including meta‐predictors tools (BayesDel_addAF, DANN, EIGEN, FATHMM‐MKL, FATHMM‐XF, LIST‐S2, LRT, M‐CAP, MVP, MutationAssessor, MutationTaster, SIFT, PROVEAN, MetaLR, and MetaSVM). To exhaustively search for other genomic variants, which could have contributed to the phenotype of the patients, ES was performed in parents and the resulting variants were re‐analyzed and compared, accordingly. However, apart from the above‐mentioned PUS3 variants, no other variants could be regarded as disease‐relevant (Supporting Information: Table S1).
According to our internal database, the frequency of the PUS3 c.212A>G variant in the Polish population (15/8833 or 0.0018) is relatively high compared to gnomAD (6/152,248 or 0.000039), even when European (non‐Finnish) samples are exclusively considered (1/68,022 or 0.000015). The relatively high PUS3 c.212A>G prevalence in Poland might suggest a founder effect. To get more evidence, we analyzed the haplotypes on which PUS3 c.212A>G was present. In both patients homozygous for PUS3 c.212A>G, the variant was located in regions characterized by runs of homozygosity (ROH)—0.9 Mb (chr11: 125,087,907–126,003,744) and 7.2 Mb (chr11: 121,622,363–128,916,399), for patients 1 and 2, respectively. Inspection of single‐nucleotide variants (SNVs) in these regions indicated the presence of a shared haplotype. Whereas the majority of SNVs found in ROH had high population frequency (>0.5), we focused on a relatively low‐frequency marker, namely rs12274923 (chr11:126 001 820‐G>A, located 105,747 bp from the PUS3 c.212A>G, gnomAD derived allele frequency 24,260/152,022 or 0.16). We found that among 17 unrelated Polish individuals heterozygous for PUS3 c.212A>G in our database, 82.3% (14/17) also had the A allele at rs12274923, whereas among the remaining group the prevalence was lower—37.5% (1520/4049) with the difference being statistically significant—Yates corrected χ 2 = 12.6, p = 0.00038. This demonstrates the presence of linkage disequilibrium in our population between PUS3 c.212A>G and rs12274923. These results support the existence of a founder effect as the explanation of the relatively high frequency of the PUS3 c.212A>G in the Polish general population and in our patients.
The substituted tyrosine residue (Tyr71) is located in the highly conserved core pseudouridine synthase domain of PUS3 (Figure 1c). The amino acid is strictly conserved among the sequences of TruA subfamily members from bacteria, archaea to human and is even found in the human PUS1 protein (Figure 1d and Supporting Information: Figure S1b). To understand the precise position and functional impact of the Y71C substitution on the molecular level, we employed AlphaFold2 to predict the structure of full‐length human PUS3. The model of the core structure resembles the common PUS domain fold, whereas the additional unannotated N‐ and C‐ termini are predicted as long helices with a rather low model confidence (Figure 1e and Supporting Information: Figure S2a). Tyr71 is predicted to be in close proximity to the L1 loop, which is known to be involved in binding the anticodon stem loop (ASL) of tRNAs (Hur & Stroud, 2007). The substituted isoleucine residue (Ile299) is located in a helix residing in the core domain, but is not as highly conserved as Tyr71 (Figure 1d). We also mapped other recently identified heterozygous compound mutations (C114R, R166Q, C190R, R193Q, V279F, and L366P) as well as another homozygotic mutation (L21R) on the structure (Figure 1e) (Nøstvik et al., 2021). Some variants seem to be grouped into specific areas. For instance, Cys114, Cys190, Arg193, and Val279 locate to the tRNA binding region while Leu21 and Leu366 are found within long helices at the N‐ or C‐ termini, respectively.
3.2. The homodimer of full‐length PUS3 is not affected by Y71C
Among the clinically relevant variants, the Y71C substitution is the only variant identified in both, homozygotic and compound heterozygous forms. This prompted us to focus on investigating the role of the Y71C mutation on the enzymatic activity of PUS3. Therefore, we first produced the recombinantly expressed full‐length wild‐type PUS3 protein (PUS3WT) in insect cells and purified it to homogeneity. Of note, we also attempted to produce full‐length PUS3 and the truncated versions of the pseudouridine synthase domain of PUS3 in bacterial expression systems, which resulted in expressed but completely insoluble protein products. Next, we also used insect cells to produce the Y71C mutant (PUS3Y71C) as well as the D118A mutant (PUS3D118A), which substitutes the highly conserved aspartic acid residue 118 in the catalytic active site of PUS3. After consecutive affinity‐ and size exclusion‐chromatography steps, we obtained reasonable quantities of all recombinant proteins at very high purity (Figure 2a). In addition, the gel filtration profile suggested that the full‐length PUS3 protein exists as a homodimer in the solution. To confirm this observation, the purified PUS3 protein was treated with glutaraldehyde and the crosslinked sample exhibited a prominent band at approximately double the molecular mass of the monomer (~110 kDa) in denaturing SDS‐PAGE gels (Supporting Information: Figure S2b). Both purified mutants, namely PUS3Y71C and PUS3D118A, exhibited almost identical gel filtration profiles as the wild type. Furthermore, we used RALS measurements to determine the estimated molecular weight of PUS3WT (124.573 kDa) and PUS3Y71C (129.625 kDa), confirming the dimeric nature of the proteins. We also managed to express the I299T mutant (PUS3I299T) recombinantly in insect cells, but the expressed protein eluted unexpectedly early during size exclusion chromatography, indicating a large mass of the purified samples and suggesting it forms an aggregate of partially unfolded protein (Figure 2a). We used the mass spectrometry method to verify that PUS3 represents the main component of the aggregates. Thus, it is not feasible to determine further biochemical or biophysical parameters of this mutant.
Figure 2.
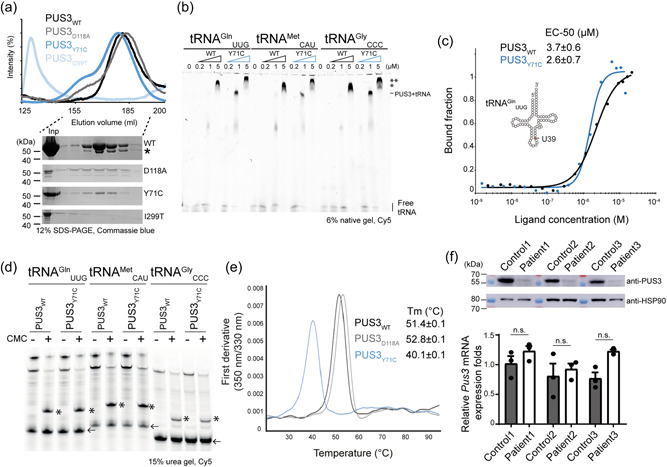
The biochemical and biophysical characterizations of PUS3 proteins. (a) Gel filtration profiles of the recombinant HsPUS3, including PUS3WT, PUS3Y71C, PUS3I299T, and PUS3D118A, and the SDS‐PAGE gels of the corresponding eluted fractions of all proteins. *The protein identity is verified by mass spectrometry as a shorter version of PUS3. Inp: input. (b) The EMSA characterization of tRNA‐PUS3 complex formation. The unbound and bound tRNA are as annotated whereas the two different bound states are indicated (*, ** possible higher orders of PUS3‐tRNA complex). (c) Characterization of tRNA binding abilities of PUS3 by MST assays. The bound and unbound states of the fluorescent‐labeled tRNAGln UUG were plotted against the protein titrations (PUS3WT or PUS3Y71C). The measurements were triplicates and the average EC‐50 (μM) is listed in the inset. (d) Detection of PUS3‐dependent Ψ39 formation on various in vitro transcribed tRNAs. The reverse‐transcribed cDNA products were resolved in a 15% urea gel and the CMC‐Ψ stops the reverse transcription and results in shorter fragments which are indicated by *. Each tRNA primer (labeled with Cy5) is indicated by an arrow. (e) The melting temperatures (Tm) of PUS3 proteins. The intrinsic absorption at 350 and 330 nm were recorded, and the first derivative was calculated as the melting points of proteins and listed in the inset. (f) Western blot analysis of PUS3 protein levels and real‐time PCR analysis of PUS3 mRNA expression levels in the patients and healthy control‐derived fibroblasts. The HSP90 is the internal loading control for western blot while GUSB is the internal control for real‐time PCR. The relative amount of PUS3 mRNA was normalized to GUSB mRNA and is presented as fold change relative to control cells. Protein samples as well as mRNA samples were prepared from three independent cultures. Statistical analysis was performed using one‐way ANOVA (α = 0.05) with Sidak's multiple comparisons test. Data are presented as means ± SEM (n.s.: not significant). ANOVA, analysis of variance; MST, Microscale thermophoresis assay.
Next, we employed Alphafold2 to predict the potential interface between two PUS3 molecules and to obtain a homo‐dimeric model of PUS3 (Supporting Information: Figure S2c,d). The model suggests with high confidence that the core domain of human PUS3 dimerizes in a very similar fashion as the prokaryotic PUS enzyme TruA (Supporting Information: Figure S2d,e). In addition, the model predicts the presence of long alpha helices in the N‐ and C‐terminal extensions of PUS3, which are absent in TruA and could additionally stabilize the dimer by enlarging the interaction interface (Supporting Information: Figure S2e). Our data shows that full‐length PUS3 forms a stable homodimer in solution and that neither Y71C substitution nor D118A affects dimerization.
3.3. Wild type and mutated full‐length PUS3 bind and modify tRNAs
Next, we measured the tRNA binding affinity of PUS3 using EMSA and MST assays. First, we analyzed PUS3‐tRNA complex formation under native conditions using EMSA with three in vitro transcribed human tRNAs, namely tRNAGln UUG, tRNAMet CAU, and tRNAGly CCC (Figure 2b). In addition, we tested yeast tRNASer UGA (Supporting Information: Figure S3a), which is known to get modified by yeast PUS3 (Lecointe et al., 1998). As increasing protein concentrations (PUS3WT and PUS3Y71C), the amount of free tRNA decreased while a clearly shifted band of the complex appeared at a protein concentration of approximately 1 µM. Intriguingly, we observed a super‐shifted second band appearing at higher protein concentration, suggesting that the stoichiometry changes and that the complex can consist of a PUS3 dimer bound to two tRNAs or a higher order assembly of tRNA‐bound PUS3 molecules. Of note, the PUS3Y71C‐tRNA complex seems to have another additional shift at higher protein concentration (5 µM). Furthermore, two in vitro transcribed tRNAs (human tRNAGln UUG and yeast tRNASer UGA) were used for further quantitative MST measurements. PUS3WT binds to both tRNAs with an EC50 of 3.7 ± 0.6 µM, which is in agreement with the EMSA result and within the comparable affinity range of the bacterial homolog (Figure 2c and Supporting Information: Figure S3b) (Hur & Stroud, 2007). Overall, the PUS3Y71C mutant shows almost identical binding parameters and an even slightly increased affinity (EC50 of 2.6 ± 0.7 µM). We further analyzed the pseudouridylation modification activity of the purified recombinant proteins in vitro. To detect the presence of Ψ, we employed selective chemical labeling with CMC and identified CMC‐Ψ sites using a site‐specific primer extension assay. This method allowed us to monitor the enzyme activity on different tRNA substrates and showed that the PUS3WT and the PUS3Y71C mutant can introduce Ψ39 on all tested tRNA transcripts very efficiently (Supporting Information: Figure S3c). The PUS3D118A mutant was included as a negative control and indeed completely loses the catalytic activity (Supporting Information: Figure S3c). We show that PUS3 can isomerize U39 into Ψ39 in tRNAGln UUG, tRNAMet CAU, and tRNAGly CCC, as well as yeast tRNASer UGA (Figure 2d). These observations are consistent with the existing structural models of tRNA binding to the PUS3 protein, which shows that the tyrosine residue position is located rather distant from the tRNA binding site, suggesting that Tyr71 is not directly involved in tRNA binding or catalytic reaction during the modification process (Supporting Information: Figure S3d). Our rather unexpected findings suggest that the PUS3Y71C mutation has no detrimental effect on dimerization, tRNA binding, and modification activity of the full‐length PUS3 enzyme, questioning the link between the clinical mutations and the onset of the observed clinical features.
3.4. Mutated full‐length PUS3 displays a strongly reduced protein stability
Next, we were wondering whether the Y71C mutation has an impact on the overall stability of the PUS3 enzyme. Hence, we measured the thermal unfolding rates, melting temperature, and aggregation propensity under isothermal conditions of PUS3WT, PUS3Y71C mutant, and PUS3D118A. Our nanoDSF analyses show that the introduction of the Y71C mutation strongly compromises the stability of the PUS3 protein, resulting in a melting temperature that is ~10°C lower than for PUS3WT. The PUS3D118A exhibits an even slightly elevated melting temperature, suggesting higher protein stability (Figure 2e). In addition, the Y71C mutant shows polydispersity while PUS3WT and PUS3D118A are monodisperse in solution (Supporting Information: Figure S3e,f). These results indicate that the PUS3Y71C mutation leads to reduced PUS3 protein levels in the cells. To investigate the impact of this Y71C mutation on protein stability in cells, we obtained fibroblast cells from all three affected individuals as well as respective healthy controls. We confirmed the presence of the respective variants in the cells via targeted Sanger sequencing. We analyzed the protein expression levels using Western blots, which showed that the endogenous protein level of PUS3 was significantly reduced in the patient cells (Figure 2f). The patients and the healthy controls expressed similar levels of PUS3 mRNAs, showing that the observed drop in protein levels is caused post‐transcriptionally (Supporting Information: Figure S3g). Of note, the mRNA and protein levels of PUS3 in patient 3 are very similar to other two patients, which supports our observation that PUS3I299T misbehaves during purification, indicating that the I299T mutation leads to a destabilized protein, which is degraded in human cells.
3.5. Patients lack PUS3‐specific Ψ in various tRNAs
To investigate whether the reduced cellular PUS3 protein levels affect the PUS3‐dependent tRNA modifications, we extracted total tRNA from the respective fibroblasts (Supporting Information: Figure S3h) and monitored the Ψ39 modification level on several tRNA transcripts using tRNA species‐specific primers (Figure 3a). First, we analyzed the Ψ profiles of tRNAGly CCC, tRNAGln UUG, tRNAMet CAU, tRNASer UGA, and tRNATyr GUA in the affected individuals. Our analyses show that the levels of Ψ39 dramatically decreased on all tested tRNA transcripts from the patient samples (Figure 3a). These tRNAs carry Ψs at several other positions, which are introduced by different PUS enzymes, namely PUS1, PUS3, and PUS7 (Boccaletto et al., 2018). The levels of these other Ψs in the patient samples did not decrease compared to the healthy controls. Furthermore, we have detected Ψ31 in tRNAMet CAU, although the predicted human PUS6 enzyme has not yet been identified. The counterintuitive increase of signal of other Ψ sites is a consequence of the higher read‐through rates when position 39 is not modified as Ψ. Of note, the detection of modifications on the TΨC loop, including Ψ54 and Ψ55, was not possible with the used primer, which is designed to pair to the 3' end of tRNA (from position 50 to 76). Our observations clearly indicate that in humans the level of Ψ39 on tRNAs is exclusively regulated by PUS3 (Figure 3b).
Figure 3.
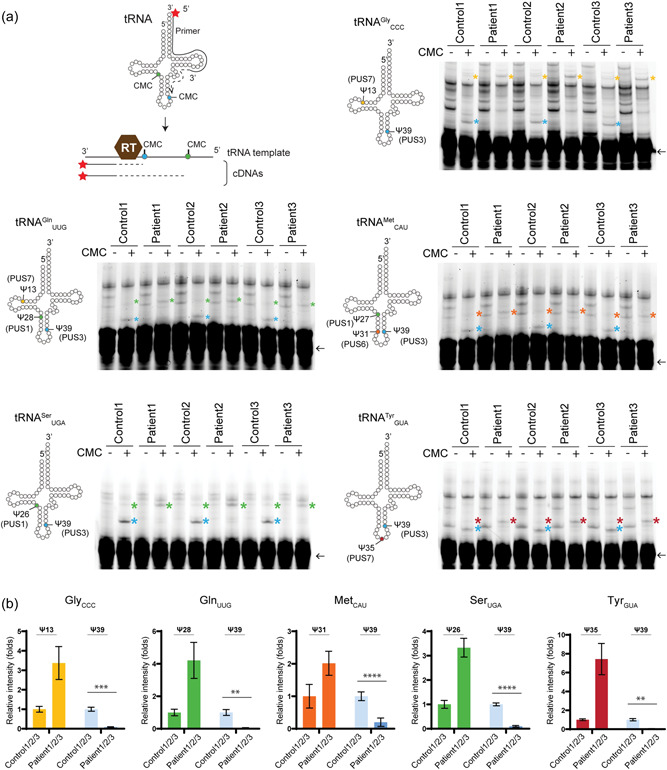
The Ψ profiles of various tRNAs in fibroblasts. (a) The scheme of detecting the presence of CMC‐Ψ using a reverse transcription method. The primer is labeled with Cy5 at its 5′ end. As the reverse transcriptase (RT) reverse transcribes the cDNA using the tRNA as a template, the presence of CMC‐Ψ causes RT to stop at the position before the CMC‐Ψ. As a result, various lengths of cDNA are generated according to the Ψ‐containing positions. The profile of Ψ sites based on the experimental validated MODOMICS database is presented in a cartoon presentation style (left to each gel). Each primer is base‐pairing at 3′ end of tRNA and it covers the Ψ55 position. Therefore, Ψ55 is undetectable in this assay and thus not included in the illustration. The reverse‐transcribed cDNA products were resolved in 15% urea gels and each PUS‐mediated CMC‐Ψ‐dependent fragments are indicated as * and colored coded (blue: Ψ39 by PUS3; green: Ψ26/27/28 by PUS1; yellow: Ψ13 by PUS7, orange: Ψ31 by PUS6, and red: Ψ35 by PUS1 or PUS7). Each tRNA primer is indicated by an arrow. (b) The triplicate measurements were performed. Statistical analysis was performed with two‐way ANOVA (α = 0.05) with Tukey's multiple comparisons test. Statistically significant differences are indicated (**p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001). Data represent mean ± SEM. Ψ, pseudouridine; ANOVA, analysis of variance.
Currently, only tRNAs are the known validated targets of human PUS3. Several studies have proposed that the steroid receptor RNA activator (SRA), a long noncoding RNA, contains several Ψ sites, which are potential PUS3 targeting sites (Li et al., 2015; Zhao et al., 2007). Among those Ψ sites, only Ψ206, located in Domain I of SRA, is confirmed to be PUS1‐dependent, while the rest are not associated with a specific PUS enzyme, yet. Therefore, we analyzed the pseudouridylation status on the SRA transcript in the patient fibroblasts and identified seven potent Ψ sites in Domain I and Domain III of SRA, including Ψ206. All these detected Ψ were equally present in the cells from healthy controls and the affected individuals. Moreover, we also monitored several known H/ACA snoRNA‐mediated Ψ sites in human 18s rRNA (Taoka et al., 2018; Xiao et al., 2009), showing that their Ψ profiles remain unchanged in PUS3 depleted cells (Supporting Information: Figure S4).
4. DISCUSSION
In this study, we identified the homozygous variant (c.212A>G (p.Tyr71Cys)) and two compound heterozygous variants in trans, including the c.212A>G (p.Tyr71Cys) and a novel c.896T>C (p.Ile299Thr), of the human PUS3 during clinical diagnostic testing in three unrelated patients. The patients had ID and a global developmental delay as well as epileptic seizures. Apart from this report on the three unrelated Polish individuals, there are four other individuals with moderate to severe ID who have the homozygous p.Tyr71Cys substitution in PUS3 gene (Gulkovskyi et al., 2015; Nøstvik et al., 2021). By extensive genetic analysis of the patients and their parents, the variants in PUS3 are the only plausible candidates to cause the observed disease. Taken together, the prevalence of the (p.Tyr71Cys) variant among patients is consistent, especially it has a relatively high frequency in the general population (0.0001 in Europeans from gnomAD Exome database https://gnomad.broadinstitute.org/) (Karczewski et al., 2020). In addition to this substitution, several reports have recently identified other homozygous or heterozygous variants in human PUS3 (De Paiva et al., 2019; Fang et al., 2020; Nøstvik et al., 2021; Shaheen et al., 2016). One affected individual from a Palestinian consanguineous family carried a homozygous null variant in PUS3 (p.(Ser394Cysfs*18)) and had microcephaly, severe hypotonia, and global developmental delay (Abdelrahman et al., 2018). This variant results in the loss of PUS3 transcript. Three patients from Saudi Arabia carried a nonsense homozygous mutation PUS3 p.(Arg435*) and displayed mental retardation, microcephaly, and growth delay (Shaheen et al., 2016). This nonsense mutation has been linked to the reduced Ψ levels in tRNAPhe in patient‐derived cells. A Chinese patient with severe epileptic encephalopathy and multiple malformations was reported to carry the compound heterozygous PUS3 mutations p.(Arg19*) and p.(Thr208Asnfs*14) (Fang et al., 2020). Two individuals from a nonconsanguineous Brazilian family carried compound heterozygous PUS3 variants for p.(Arg166Gln) and p.(Leu366Pro) and suffered from syndromic ID with dysmorphic features, white matter disease, and renal abnormalities (De Paiva et al., 2019). A recent study has reported even more PUS3 mutations among patients with ID, including missense variants (p.(Leu21Arg), p.(Cys190Tyr), p.(Cys114Arg), p.(Arg193Gln), p.(Val279Phe), p.(Tyr160*) and p.(Asn146Lysfs*5)), one nonsense variant (p.(Arg280*)), and one splice variant. The expression levels of proteins or mRNAs from these individuals have not been analyzed. According to VarSome (Kopanos et al., 2019), some of those PUS3 variants are classified as “uncertain significance/likely pathogenic” in the context of an autosomal recessive disease. Of note, the most common feature of these patients was ID despite the wide clinical spectrum of observed phenotypes. In summary, there is sufficient evidence that certain intellectual disorders can be caused by homozygous or compound heterozygous mutations of the PUS3 gene.
The affected tyrosine 71 residue is highly conserved among PUS3 homologs and when the substitution occurs on both alleles, the cells have nearly depleted PUS3 protein level and dramatically diminished abundance of Ψ39 in tRNAs. Conversely, the heterozygous parents of the affected individual who carry the Y71C substitution on one allele (WT/Y71C) had their PUS3 protein expression level and Ψ39 levels at comparable amounts to the healthy control (WT/WT). I299T locates in a distal region to the catalytic core and therefore is unlikely to affect either tRNA binding or the catalytic reaction. As described above, purified recombinant PUS3I299T protein forms soluble aggregates, suggesting that the I299T substitution in PUS3 decreases its stability and leads to strongly decreased protein levels in vivo, thus causing loss of PUS3 and PUS3‐dependent pseudouridine modification sites.
In addition to our efforts to characterize the molecular link between the specific mutations and the diseases, we provide several fundamental insights into the human PUS3 enzyme. Foremost, we successfully expressed and purified recombinant full‐length human PUS3 using an insect cell expression system. This allowed us to confirm that the human PUS3 forms a homo‐dimer (like E. coli TruA), displays similar tRNA binding affinity to its bacterial homolog proteins (Hur & Stroud, 2007) and employs the conserved catalytic residue to catalyze the Ψ39 modification in tRNAs. In addition, the human PUS3 can also modify the in vitro transcribed yeast tRNA, indicating a highly conserved selectivity of PUS3 across all domains of life. This adds to other studies showing that certain human mRNA targets can be modified by homologs human and yeast PUS enzymes (Carlile et al., 2019).
As the Ψ39 is residing in the stem regions of ASL of tRNAs, it plays an important role in maintaining the ASL structure which is proposed to be crucial for the protein production in the ribosome during translation (Han et al., 2015; Klassen & Schaffrath, 2017). This theory has been tested in some model organisms, such as E. coli (TruA) and S. cerevisiae (Deg1), and it has been shown that the loss of function of PUS3 homologs caused growth retardation under specific stress conditions (Han et al., 2015). The correlation between defective PUS enzymes and human neurodegenerative diseases has emerged and corroborated over the last couple of years (Jung & Goldman, 2018) but the underlying mechanisms are not fully understood. It is likely that in addition to the tRNA‐linked problems in translation regulation, other cellular mechanisms are affected and cause problems at different stages of gene expression. For instance, several global transcriptome‐wide studies have identified and mapped target sites outside tRNAs for several PUS enzymes, including PUS1, PUS4, and PUS7 (Carlile et al., 2014; Schwartz et al., 2014). Apart from the known Ψ sites in noncoding RNAs, the presence of a large number of Ψ sites on mRNAs suggests an additional role of Ψ in gene expression regulation. These additional targets might directly or indirectly contribute to the pathogenic impact of PUS mutations in diseases (de Brouwer et al., 2018). Several PUS3‐dependent Ψ sites on mRNAs have been reported in the yeast system, but human mRNAs still need to be investigated.
Finally, we noticed that the purified PUS3Y71C also forms aggregates with a distinct increase in hydrodynamic radius. This intrinsic propensity of PUS3Y71C aggregation could explain the slightly shifted band in the EMSA gels. PUS3WT and PUS3D118A mostly remain monodisperse during the nanoDSF and DLS measurements and do not form a significant aggregation profile as PUS3Y71C during the unfolding process in a thermal gradient. However, we did detect the appearance of a ladder of distinct higher molecular weight complexes of PUS3WT after applying mild crosslinking conditions. Hence, the formation of additional higher‐order assemblies of PUS3 that remain functional cannot be currently excluded. In addition, the higher molecular weight in PUS3 may explain the additional shift band observed in our EMSA results.
In summary, our work represents the first in‐depth characterization of human PUS3 in vitro. We use patient‐derived mutations and samples to establish a direct link between the protein levels of PUS3 and the neurodevelopmental disease. We confirm the PUS3‐dependent pseudouridylation profile in human cells and showed the pathogenic role of the amino acid substitution p.(Y71C) and p.(I299T) in the identified ID patients at the molecular level. Our results suggest that additional molecular and functional investigations on the brain and neuron development will be required to finally understand the underlying pathological mechanism. At this point, we suspect a key role for PUS3 in neurodevelopment, which is caused by the direct influence of PUS on the proteome of specific cells and cell types.
AUTHOR CONTRIBUTIONS
Robert Smigiel, Joanna Kosińska, Rafał Ploski, Monika Gos, Sylwia Rzonca‐Niewczas, and Mateusz Biela collected the clinical data (including ES) and performed fibroblast sampling; Izabela Laczmanska performed fibroblast culture. Bozena Kuzniewska and Joanna J. Chmielewska cultured the fibroblasts for experiments, Bozena Kuzniewska performed Western blotting on fibroblasts samples, extracted the RNA and performed the qRT‐PCR analysis, isolated DNA from fibroblasts, and amplified products for sequencing. Ting‐Yu Lin purified proteins with the help of Jakub Jeżowski, Anna Kościelniak, and Dominika Dobosz; Andrzej Chramiec‐Głąbik produced and purified tRNAs; Anna Biela and Dominika Dobosz extracted tRNAs from cell lines and performed RT‐PCR analyses; Ting‐Yu Lin performed biochemical and biophysical analyses with the help of Jakub Jeżowski and Jakub Nowak; Ting‐Yu Lin and Sebastian Glatt designed experiments, analyzed the results, prepared figures, and wrote the manuscript, with the input from all other authors. Ting‐Yu Lin, Magdalena Dziembowska, Rafał Ploski, and Sebastian Glatt secured funding and supervised the project.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Supplementary information.
Supplementary information.
ACKNOWLEDGEMENTS
We thank the members of the Glatt, Dziembowska, and Ploski laboratories for vivid discussion and suggestions. In particular, we thank Rostyslav Krutyholova for assistance with the structural modeling and we thank the Proteomics and Mass Spectrometry Core Facility from MCB for providing service for protein identity verification. This study was supported by the SONATA (2019/35/D/NZ1/02397 to T.‐Y. L.), the OPUS (2017/27/B/NZ5/02223 to R. S.), the PRELUDIUM (2017/27/N/NZ1/01381 to J. J. C.) and the GRIEG (2019/34/H/NZ3/00733 to M. D.) grants from the National Science Centre. This project has also received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement No 101001394 to S. G.). In addition, we thank the MCB structural biology core facility (supported by the TEAM TECH CORE FACILITY/2017‐4/6 grant from Foundation for Polish Science) for providing computational infrastructure and support.
Lin, T.‐Y. , Smigiel, R. , Kuzniewska, B. , Chmielewska, J. J. , Kosińska, J. , Biela, M. , Biela, A. , Kościelniak, A. , Dobosz, D. , Laczmanska, I. , Chramiec‐Głąbik, A. , Jeżowski, J. , Nowak, J. , Gos, M. , Rzonca‐Niewczas, S. , Dziembowska, M. , Ploski, R. , & Glatt, S. (2022). Destabilization of mutated human PUS3 protein causes intellectual disability. Human Mutation, 43, 2063–2078. 10.1002/humu.24471
Ting‐Yu Lin and Robert Smigiel are joint authors of this study.
Contributor Information
Magdalena Dziembowska, Email: m.dziembowska@cent.uw.edu.pl.
Rafał Ploski, Email: rafal.ploski@wum.edu.pl.
Sebastian Glatt, Email: sebastian.glatt@uj.edu.pl.
DATA AVAILABILITY STATEMENT
The study was approved of by the Ethics Committee of Wroclaw Medical University, Poland (430/2018). The variant description in this study has been submitted to the LOVD database (https://databases.lovd.nl/shared/genes/PUS3) with the following accession numbers: patient 1—#00413320, patient 2—#00413076, patient 3—#00413230. Data are available upon reasonable request to the corresponding authors.
REFERENCES
- Abdelrahman, H. A. , Al‐Shamsi, A. M. , Ali, B. R. , & Al‐Gazali, L. (2018). A null variant in PUS3 confirms its involvement in intellectual disability and further delineates the associated neurodevelopmental disease. Clinical Genetics, 94, 586–587. 10.1111/cge.13443 [DOI] [PubMed] [Google Scholar]
- Adachi, H. , DeZoysa, M. D. , & Yu, Y. T. (2019). Detection and quantification of pseudouridine in RNA. Methods in Molecular Biology, 1870, 219–235. 10.1007/978-1-4939-8808-2_17 [DOI] [PubMed] [Google Scholar]
- Angelova, M. T. , Dimitrova, D. G. , Dinges, N. , Lence, T. , Worpenberg, L. , Carré, C. , & Roignant, J. Y. (2018). The emerging field of epitranscriptomics in neurodevelopmental and neuronal disorders. Frontiers in Bioengineering and Biotechnology, 6, 46. 10.3389/fbioe.2018.00046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begik, O. , Lucas, M. C. , Pryszcz, L. P. , Ramirez, J. M. , Medina, R. , Milenkovic, I. , Cruciani, S. , Liu H., Vieira H. G. S., Sas‐Chen A., Mattick J. S., Schwartz S., Novoa E. M. (2021). Quantitative profiling of pseudouridylation dynamics in native RNAs with nanopore sequencing. Nature Biotechnology, 39(10), 1278–1291. 10.1038/s41587-021-00915-6 [DOI] [PubMed] [Google Scholar]
- Boccaletto, P. , MacHnicka, M. A. , Purta, E. , Piątkowski, P. , Bagiński, B. , Wirecki, T. K. , De crécy‐Lagard, V. , Ross, R. , Limbach, P. A. , Kotter, A. , Helm, M. , & Bujnicki, J. M. (2018). MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic Acids Research, 46(D1), D303–D307. 10.1093/nar/gkx1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Brouwer, A. P. M. , Abou Jamra, R. , Körtel, N. , Soyris, C. , Polla, D. L. , Safra, M. , Zisso, A. , Powell C. A., Rebelo‐Guiomar P., Dinges N., Morin V., Stock M., Hussain M., Shahzad M., Riazuddin S., Ahmed Z. M., Pfundt R., Schwarz F., de Boer L., … Schwartz S. (2018). Variants in PUS7 cause intellectual disability with speech delay, microcephaly, short stature, and aggressive behavior. The American Journal of Human Genetics, 103(6), 1045–1052. 10.1016/j.ajhg.2018.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bykhovskaya, Y. , Casas, K. , Mengesha, E. , Inbal, A. , & Fischel‐Ghodsian, N. (2004). Missense mutation in pseudouridine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic anemia (MLASA). The American Journal of Human Genetics, 74(6), 1303–1308. 10.1086/421530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlile, T. M. , Martinez, N. M. , Schaening, C. , Su, A. , Bell, T. A. , Zinshteyn, B. , & Gilbert, W. V. (2019). mRNA structure determines modification by pseudouridine synthase 1. Nature Chemical Biology, 15(10), 966–974. 10.1038/s41589-019-0353-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlile, T. M. , Rojas‐Duran, M. F. , Zinshteyn, B. , Shin, H. , Bartoli, K. M. , & Gilbert, W. V. (2014). Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature, 515(7525), 143–146. 10.1038/nature13802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawidziuk, M. , Gambin, T. , Bukowska‐olech, E. , Antczak‐marach, D. , Badura‐stronka, M. , Buda, P. , Budzynska, E. , Castaneda, J. , Chilarska, T. , Czyzyk, E. , Eckersdorf‐Mastalerz, A. , Fijak‐Moskal, J. , Gieruszczak‐Bialek, D. , Glodek‐Brzozowska, E. , Goszczanska‐Ciuchta, A. , Grzeszykowska‐Podymniak, M. , Gurda, B. , Jakubiuk‐Tomaszuk, A. , Jamroz, E. , … Wiszniewski, W. (2021). Exome sequencing reveals novel variants and expands the genetic landscape for congenital microcephaly. Genes, 12(12), 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano, W. L. (2020). The PyMOL molecular graphics system, version 2.3. Schrödinger LLC. [Google Scholar]
- De crécy‐Lagard, V. , Boccaletto, P. , Mangleburg, C. G. , Sharma, P. , Lowe, T. M. , Leidel, S. A. , & Bujnicki, J. M. (2019). Matching tRNA modifications in humans to their known and predicted enzymes. Nucleic Acids Research, 47(5), 2143–2159. 10.1093/nar/gkz011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durant, P. C. , & Davis, D. R. (1999). Stabilization of the anticodon stem‐loop of tRNALys,3 by an A+ ‐C base‐pair and by pseudouridine 1. Journal of Molecular Biology, 285(1), 115–131. 10.1006/jmbi.1998.2297 [DOI] [PubMed] [Google Scholar]
- Edgar, R. C. (2004). MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics, 5, 113. 10.1186/1471-2105-5-113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley, P. , Lohkamp, B. , Scott, W. G. , & Cowtan, K. (2010). Features and development of Coot. Acta Crystallographica Section D: Biological Crystallography, 66(4), 486–501. 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, H. , Zhang, L. , Xiao, B. , Long, H. , & Yang, L. (2020). Compound heterozygous mutations in PUS3 gene identified in a Chinese infant with severe epileptic encephalopathy and multiple malformations. Neurological Sciences, 41, 465–467. 10.1007/s10072-019-04049-1 [DOI] [PubMed] [Google Scholar]
- Gulkovskyi, R. V. , Chernushyn, S. Y. , & Livshits, L. A. (2015). Novel gene PUS3 c.A212G mutation in Ukrainian family with intellectual disability. Biopolymers and Cell, 31(2), 123–130. 10.7124/bc.0008D6 [DOI] [Google Scholar]
- Han, L. , Kon, Y. , & Phizicky, E. M. (2015). Functional importance of Ψ38 and Ψ39 in distinct tRNAs, amplified for tRNAGln(UUG) by unexpected temperature sensitivity of the s2U modification in yeast. RNA, 21(2), 188–201. 10.1261/rna.048173.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, L. , Pookanjanatavip, M. , Gu, X. , & Santi, D. V. (1998). A conserved aspartate of tRNA pseudouridine synthase is essential for activity and a probable nucleophilic catalyst. Biochemistry, 37(1), 344–351. 10.1021/bi971874+ [DOI] [PubMed] [Google Scholar]
- Hur, S. , & Stroud, R. M. (2007). How U38, 39, and 40 of many tRNAs become the targets for pseudouridylation by TruA. Molecular Cell, 26(2), 189–203. 10.1016/j.molcel.2007.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper, J. , Evans, R. , Pritzel, A. , Green, T. , Figurnov, M. , Ronneberger, O. , Tunyasuvunakool, K. , Bates, R. , Žídek, A. , Potapenko, A. , Bridgland, A. , Meyer, C. , Kohl, S. A. A. , Ballard, A. J. , Cowie, A. , Romera‐Paredes, B. , Nikolov, S. , Jain, R. , Adler, J. , … Hassabis, D. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873), 583–589. 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, Y. , & Goldman, D. (2018). Role of RNA modifications in brain and behavior. Genes, Brain and Behavior, 17, e12444. 10.1111/gbb.12444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski, K. J. , Francioli, L. C. , Tiao, G. , Cummings, B. B. , Alföldi, J. , Wang, Q. , Collins, R. L. , Laricchia, K. M. , Ganna, A. , Birnbaum, D. P. , Gauthier, L. D. , Brand, H. , Solomonson, M. , Watts, N. A. , Rhodes, D. , Singer‐Berk, M. , England, E. M. , Seaby, E. G. , Kosmicki, J. A. , … Macarthur, D. G. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans, Genome Aggregation Database Consortium. Nature, 581, 434–443. 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klassen, R. , Ciftci, A. , Funk, J. , Bruch, A. , Butter, F. , & Schaffrath, R. (2016). TRNA anticodon loop modifications ensure protein homeostasis and cell morphogenesis in yeast. Nucleic Acids Research, 44(22), 10946–10959. 10.1093/nar/gkw705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klassen, R. , & Schaffrath, R. (2017). Role of pseudouridine formation by Deg1 for functionality of two glutamine isoacceptor tRNAs. Biomolecules, 7(1), 8. 10.3390/biom7010008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopanos, C. , Tsiolkas, V. , Kouris, A. , Chapple, C. E. , Albarca Aguilera, M. , Meyer, R. , & Massouras, A. (2019). VarSome: The human genomic variant search engine. Bioinformatics, 35, 1978–1980. 10.1093/bioinformatics/bty897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kost, T. A. , Condreay, J. P. , & Jarvis, D. L. (2005). Baculovirus as versatile vectors for protein expression in insect and mammalian cells. Nature Biotechnology, 23, 567–575. 10.1038/nbt1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krutyhołowa, R. , Zakrzewski, K. , & Glatt, S. (2019). Charging the code—tRNA modification complexes. Current Opinion in Structural Biology, 55, 138–146. 10.1016/j.sbi.2019.03.014 [DOI] [PubMed] [Google Scholar]
- Lecointe, F. , Namy, O. , Hatin, I. , Simos, G. , Rousset, J. P. , & Grosjean, H. (2002). Lack of Pseudouridine 38/39 in the anticodon arm of yeast cytoplasmic tRNA decreases in vivo recoding efficiency. Journal of Biological Chemistry, 277(34), 30445–30453. 10.1074/jbc.M203456200 [DOI] [PubMed] [Google Scholar]
- Lecointe, F. , Simos, G. , Sauer, A. , Hurt, E. C. , Motorin, Y. , & Grosjean, H. (1998). Characterization of yeast protein Deg1 as pseudouridine synthase (Pus3) catalyzing the formation of Ψ38 and Ψ39 in tRNA anticodon loop. Journal of Biological Chemistry, 273(3), 1316–1323. 10.1074/jbc.273.3.1316 [DOI] [PubMed] [Google Scholar]
- Li, X. , Zhu, P. , Ma, S. , Song, J. , Bai, J. , Sun, F. , & Yi, C. (2015). Chemical pulldown reveals dynamic pseudouridylation of the mammalian transcriptome. Nature Chemical Biology, 11(8), 592–597. 10.1038/nchembio.1836 [DOI] [PubMed] [Google Scholar]
- Lin, T.‐Y. , Abbassi, N. E. H. , Zakrzewski, K. , Chramiec‐Głąbik, A. , Jemioła‐Rzemińska, M. , Różycki, J. , & Glatt, S. (2019). The elongator subunit Elp3 is a non‐canonical tRNA acetyltransferase. Nature Communications, 10, 625. 10.1038/s41467-019-08579-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, T.‐Y. , Mehta, R. , & Glatt, S. (2021). Pseudouridines in RNAs: Switching atoms means shifting paradigms. FEBS Letters, 595, 2310–2322. 10.1002/1873-3468.14188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovejoy, A. F. , Riordan, D. P. , & Brown, P. O. (2014). Transcriptome‐wide mapping of pseudouridines: Pseudouridine synthases modify specific mRNAs in S. cerevisiae . PLoS One, 9(10), e110799. 10.1371/journal.pone.0110799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez, N. M. , Su, A. , Burns, M. C. , Nussbacher, J. K. , Schaening, C. , Sathe, S. , Yeo, G. W. , & Gilbert, W. V. (2022). Pseudouridine synthases modify human pre‐mRNA co‐transcriptionally and affect pre‐mRNA processing. Molecular Cell, 82(3), 645–659. 10.1016/j.molcel.2021.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metodiev, M. D. , Assouline, Z. , Landrieu, P. , Chretien, D. , Bader‐Meunier, B. , Guitton, C. , Munnich, A. , & Rötig, A. (2015). Unusual clinical expression and long survival of a pseudouridylate synthase (PUS1) mutation into adulthood. European Journal of Human Genetics, 23(6), 880–882. 10.1038/ejhg.2014.192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirdita, M. , Steinegger, M. , Breitwieser, F. , Söding, J. , & Levy Karin, E. (2021). Fast and sensitive taxonomic assignment to metagenomic contigs. Bioinformatics, 37(18), 3029–3031. 10.1093/bioinformatics/btab184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nøstvik, M. , Kateta, S. M. , Schönewolf‐Greulich, B. , Afenjar, A. , Barth, M. , Boschann, F. , Doummar, D. , Haack T. B., Keren B., Livshits L. A., Mei D., Park J., Pisano T., Prouteau C., Umair M., Waqas A., Ziegler A., Guerrini R., Møller R. S., Tümer Z. (2021). Clinical and molecular delineation of PUS3‐associated neurodevelopmental disorders. Clinical Genetics, 100(5), 628–633. 10.1111/cge.14051 [DOI] [PubMed] [Google Scholar]
- De Paiva, A. R. B. , Lynch, D. S. , Melo, U. S. , Lucato, L. T. , Freua, F. , De Assis, B. D. R. , Barcelos, I. , Listik, C. , dos Santos, D. d. C. , Macedo‐Souza, L. I. , Houlden, H. , & Kok, F. (2019). PUS3 mutations are associated with intellectual disability, leukoencephalopathy, and nephropathy. Neurology: Genetics, 5, e306. 10.1212/NXG.0000000000000306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, D. R. , Cabral, M. D. , Ho, A. , & Merrick, J. (2020). A clinical primer on intellectual disability. Translational Pediatrics, 9, S23–S35. 10.21037/TP.2020.02.02 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzo, M. , Guerrieri, A. , Zacchini, F. , Treré, D. , & Montanaro, L. (2017). RNA pseudouridylation in physiology and medicine: For better and for worse. Genes, 8, 301. 10.3390/genes8110301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos, J. , Han, L. , Li, Y. , Hagelskamp, F. , Kellner, S. M. , Alkuraya, F. S. , Phizicky, E. M. , & Fu, D. (2019). Formation of tRNA wobble inosine in humans is disrupted by a millennia‐old mutation causing intellectual disability. Molecular and Cellular Biology, 39, e00203–e00219. 10.1128/MCB.00203-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , & Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17, 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rintala‐Dempsey, A. C. , & Kothe, U. (2017). Eukaryotic stand‐alone pseudouridine synthases—RNA modifying enzymes and emerging regulators of gene expression? RNA Biology, 14, 1185–1196. 10.1080/15476286.2016.1276150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz, S. , Bernstein, D. A. , Mumbach, M. R. , Jovanovic, M. , Herbst, R. H. , León‐Ricardo, B. X. , Engreitz, J. M. , Guttman M., Satija R., Lander E. S., Fink G., Regev A. (2014). Transcriptome‐wide mapping reveals widespread dynamic‐regulated pseudouridylation of ncRNA and mRNA. Cell, 159(1), 148–162. 10.1016/j.cell.2014.08.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen, R. , Han, L. , Faqeih, E. , Ewida, N. , Alobeid, E. , Phizicky, E. M. , & Alkuraya, F. S. (2016). A homozygous truncating mutation in PUS3 expands the role of tRNA modification in normal cognition. Human Genetics, 135(7), 707–713. 10.1007/s00439-016-1665-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen, R. , Tasak, M. , Maddirevula, S. , Abdel‐Salam, G. M. H. , Sayed, I. S. M. , Alazami, A. M. , Al‐Sheddi, T. , Alobeid E., Phizicky E. M., Alkuraya F. S. (2019). PUS7 mutations impair pseudouridylation in humans and cause intellectual disability and microcephaly. Human Genetics, 138(3), 231–239. 10.1007/s00439-019-01980-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan, K. E. , Warda, A. S. , Sharma, S. , Entian, K. D. , Lafontaine, D. L. J. , & Bohnsack, M. T. (2017). Tuning the ribosome: The influence of rRNA modification on eukaryotic ribosome biogenesis and function. RNA Biology, 14(9), 1138–1152. 10.1080/15476286.2016.1259781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Śmigiel, R. , Biela, M. , Szmyd, K. , Błoch, M. , Szmida, E. , Skiba, P. , Walczak, A. , Gasperowicz, P. , Kosińska, J. , Rydzanicz, M. , Stawiński, P. , Biernacka, A. , Zielińska, M. , Gołębiowski, W. , Jalowska, A. , Ohia, G. , Głowska, B. , Walas, W. , Królak‐Olejnik, B. , … Płoski, R. (2020). Rapid whole‐exome sequencing as a diagnostic tool in a neonatal/pediatric intensive care unit. Journal of Clinical Medicine, 9(7), 2220. 10.3390/jcm9072220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, J. , Zhuang, Y. , Zhu, C. , Meng, H. , Lu, B. , Xie, B. , Peng, J. , Li, M. , & Yi, C. (2020). Differential roles of human PUS10 in miRNA processing and tRNA pseudouridylation. Nature Chemical Biology, 16(2), 160–169. 10.1038/s41589-019-0420-5 [DOI] [PubMed] [Google Scholar]
- Taoka, M. , Nobe, Y. , Yamaki, Y. , Sato, K. , Ishikawa, H. , Izumikawa, K. , Yamauchi, Y. , Hirota, K. , Nakayama, H. , Takahashi, N. , & Isobe, T. (2018). Landscape of the complete RNA chemical modifications in the human 80S ribosome. Nucleic Acids Research, 46(18), 9289–9298. 10.1093/nar/gky811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, S. , Yesselman, J. D. , Cordero, P. , & Das, R. (2015). Primerize: Automated primer assembly for transcribing non‐coding RNA domains. Nucleic Acids Research, 43(W1), W522–W526. 10.1093/nar/gkv538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villegas, J. , & McPhaul, M. (2005). Establishment and culture of human skin fibroblasts. In Ausubel F. M., Brent R., Kingston R. E., Moore D. D., Seidman J. G., Smith J. A., & Struhl K. (Eds.), Current protocols in molecular biology. Chapter 28 (pp. 1–9). 10.1002/0471142727.mb2803s71 [DOI] [PubMed] [Google Scholar]
- Wu, G. , Xiao, M. , Yang, C. , & Yu, Y. T. (2011). U2 snRNA is inducibly pseudouridylated at novel sites by Pus7p and snR81 RNP. The EMBO Journal, 30(1), 79–89. 10.1038/emboj.2010.316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, M. , Yang, C. , Schattner, P. , & Yu, Y. T. (2009). Functionality and substrate specificity of human box H/ACA guide RNAs. RNA, 15(1), 176–186. 10.1261/rna.1361509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, X. , Patton, J. R. , Ghosh, S. K. , Fischel‐Ghodsian, N. , Shen, L. , & Spanjaard, R. A. (2007). Pus3p‐ and Pus1p‐dependent pseudouridylation of steroid receptor RNA activator controls a functional switch that regulates nuclear receptor signaling. Molecular Endocrinology, 21(3), 686–699. 10.1210/me.2006-0414 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information.
Supplementary information.
Data Availability Statement
The study was approved of by the Ethics Committee of Wroclaw Medical University, Poland (430/2018). The variant description in this study has been submitted to the LOVD database (https://databases.lovd.nl/shared/genes/PUS3) with the following accession numbers: patient 1—#00413320, patient 2—#00413076, patient 3—#00413230. Data are available upon reasonable request to the corresponding authors.