

Abstract
Capmatinib is a highly specific, potent, and selective mesenchymal–epithelial transition factor inhibitor predominantly eliminated by cytochrome P450 (CYP) 3A4 and aldehyde oxidase. Here, we investigated the effects of a strong CYP3A inhibitor (itraconazole) and a strong CYP3A inducer (rifampicin) on single‐dose pharmacokinetics of capmatinib. In addition, serum creatinine and cystatin C were monitored to assess the potential inhibition of renal transporters by capmatinib. This was an open‐label, 2‐cohort (inhibition and induction), 2‐period (capmatinib alone and inhibition/induction periods) study in healthy subjects. In the inhibition cohort, capmatinib (400 mg/day) was given alone, then with itraconazole (200 mg/day for 10 days, 5‐day lead‐in before coadministration). In the induction cohort, capmatinib (400 mg/day) was given alone, then with rifampicin (600 mg/day for 9 days, 5‐day lead‐in before coadministration). Fifty‐three subjects (inhibition cohort, n = 27; induction cohort, n = 26) were enrolled. Coadministration of itraconazole resulted in an increase of capmatinib area under the plasma concentration–time curve from time 0 to infinity by 42% (geometric mean ratio [GMR], 1.42; 90%CI, 1.33–1.52) with no change in maximum plasma concentration (GMR, 1.03; 90%CI, 0.866–1.22). Coadministration of rifampicin resulted in a reduction of capmatinib area under the plasma concentration–time curve from time 0 to infinity by 66.5% (GMR, 0.335; 90%CI, 0.300–0.374) and a decrease in maximum plasma concentration by 55.9% (GMR, 0.441; 90%CI, 0.387–0.502). After a single dose of capmatinib, a transient increase in serum creatinine was observed with no change in serum cystatin C concentration during the 3‐day monitoring period. In conclusion, coadministration of itraconazole or rifampicin resulted in clinically relevant changes in systemic exposure to capmatinib. The transient increase in serum creatinine without any increase in cystatin C suggests inhibition of renal transport by capmatinib.
Keywords: capmatinib, CYP3A, drug‐drug interaction, itraconazole, pharmacokinetics, renal transporter, rifampicin, serum creatinine
Mesenchymal–epithelial transition factor (MET) dysregulation, through gene amplification, mutation, and/or overexpression has been described in multiple tumor types, including lung; hepatocellular carcinoma; glioblastoma multiforme; papillary renal cell carcinoma; and breast, colon, and gastric cancers. 1 MET mutations have been identified in primary tumors as well as metastatic lesions of several cancers, including head and neck, renal, liver, ovarian, and non–small‐cell lung cancer 2 , 3 , 4 , 5 ; among MET mutations, those leading to MET exon 14 skipping have emerged as strong molecular drivers. 6 , 7
Capmatinib (INC280; molecular weight, 412.4) is an orally bioavailable, adenosine triphosphate–competitive, highly potent, and selective inhibitor of the MET receptor tyrosine kinase, with demonstrated in vitro and in vivo activities against preclinical cancer models with various types of MET activation. 8 , 9 , 10 Preliminary clinical data showed a manageable safety profile and promising efficacy for capmatinib in various solid tumors. 11 , 12 , 13 , 14 In the phase 2 GEOMETRY mono‐1 trial, capmatinib demonstrated clinically meaningful efficacy and a manageable toxicity profile in patients with advanced non–small‐cell lung cancer harboring MET exon 14 skipping mutations. 15
Human absorption, distribution, metabolism, and excretion study and in vitro enzyme phenotyping study results have shown that capmatinib is cleared via metabolism by cytochrome P450 (CYP) 3A4 and aldehyde oxidase. Coadministration of compounds that inhibit and/or induce CYP3A could impact the metabolic clearance of capmatinib in humans. 16 We report here the results from the phase I drug‐drug interaction (DDI) study, which investigated the effect of a strong CYP3A inhibitor (itraconazole) or a strong CYP3A inducer (rifampicin) on pharmacokinetics (PK) of capmatinib.
The major circulating metabolite (inactive) of capmatinib in human plasma is CMN288. 16 Although aldehyde oxidase is responsible for the formation of CMN288, inhibition or induction of CYP3A4 may affect the exposure of CMN288 through metabolic pathway switching. Hence, the effect of itraconazole and rifampicin on the PK of CMN288 was also investigated.
Mild and moderate elevated creatinine levels have been frequently reported with capmatinib 400 mg twice daily, and subsequently return to baseline level following the end of treatment. 17 Renal transporters are responsible for 10%–40% of creatinine clearance through renal tubular excretion in addition to glomerular filtration. 18 In vitro data indicated that capmatinib is an inhibitor of renal transporters multidrug and toxic compound extrusion (MATE) 1 and MATE2K (dissociation constant [Ki] value of 0.28 and 0.29 µM, respectively; Novartis data on file); therefore, we hypothesized that the transient elevation in serum creatinine concentration following treatment of capmatinib could be due to the inhibition of renal transporters MATE1 and MATE2K. 19 Cystatin C, a renal biomarker that is exclusively cleared through glomerular filtration, 20 was monitored in this study in comparison to serum creatinine concentration to evaluate the potential effect of capmatinib on renal transporters involved in active tubular secretion.
Methods
This clinical study was designed and implemented in accordance with the principles of the Declaration of Helsinki and the Good Clinical Practice guidelines of the International Conference on Harmonization, with applicable local regulations. The study protocol and all amendments were reviewed by the independent ethics committee or institutional review board at Landesamt für Gesundheit und Soziales Berlin, Ethik‐Kommission des Landes Berlin (Berlin, Germany). All subjects provided written informed consent before the screening.
Subjects
Adult healthy male and/or healthy sterile or postmenopausal female subjects (aged 18–55 years) with body mass index (BMI) of 18–29.9 kg/m2, and body weight 50–120 kg (at the time of informed consent) were eligible to participate in this study. Subjects had to have no clinically significant abnormalities as determined by past medical history, physical examination, vital signs, electrocardiogram, and clinical laboratory tests. Inclusion criteria also included adequate venous access for blood sampling. Subjects were excluded if they had any medical condition that could significantly alter the PK of drugs; history of malignancy of any organ system (other than localized basal cell carcinoma of the skin or in situ cervical cancer) ≤5 years, regardless of whether there was recurrence or metastases; concomitant administration of strong CYP3A4/5 inhibitors or inducers and/or medications that prolong the QT interval, ≤4 weeks before dosing; participation in a previous clinical study ≤30 days before dosing; or a history of drug or alcohol abuse ≤12 months before the first dose.
Study Design
This was a single‐center, open‐label, 2‐arm (cohort), fixed sequence, 2‐period, DDI, phase I study (Figure 1). The study was conducted at the Early Phase Clinical Unit Berlin, PAREXEL International GmbH (Berlin, Germany). The study consisted of a screening/baseline period of up to 21 days (from day –21 to day –1) and 2 treatment periods (capmatinib‐alone period and inhibition or induction period), followed by a 30‐day (±2 days) safety follow‐up. In the capmatinib‐alone period, a single oral dose of capmatinib was administered in the fasted state: 400 mg in the induction cohort and 200 mg in the inhibition cohort. A washout period of 4 days was used after the single capmatinib dose on day 1 before the start of the inhibition and induction periods. The inhibition and induction period started on day 5 with daily dosing of itraconazole 200 mg for 10 days (day 5 to day 14) and rifampicin 600 mg for 9 days (day 5 to day 13), respectively. For subjects assigned to the inhibition cohort, a single oral dose of capmatinib 200 mg was administered an hour after administration of the sixth dose of itraconazole on day 10, while for subjects assigned to the induction cohort, a single oral dose of capmatinib 400 mg was coadministered with the sixth dose of rifampicin on day 10. Serial PK samples were collected up to 96 hours in the inhibition cohort and up to 72 hours in the induction cohort following day 1 and day 10 capmatinib administration. In both cohorts, blood samples were collected at 0, 2, 4, 8, 24, 48, and 72 hours after the capmatinib administration on day 1 to evaluate cystatin C and creatinine concentration. Subjects were confined to the study facility until the completion of the PK sampling and the end of treatment evaluation.
Figure 1.
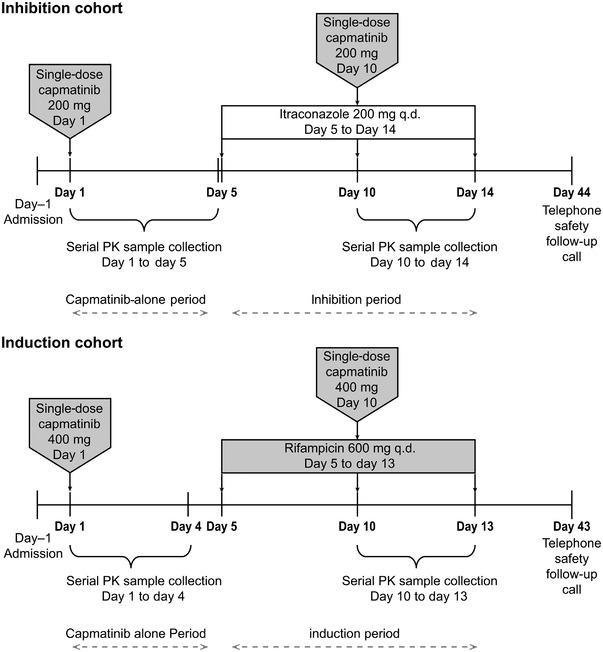
Study design. PK, pharmacokinetics; q.d. once daily.
Sample Size
The intrasubject coefficient of variation (CV%) for capmatinib in healthy subjects was 22.8% for area under the plasma concentration–time curve (AUC) and 29.9% for maximum plasma concentration (Cmax). Using the largest intra‐patient CV of 29.9% with a sample size of 20, the half‐width of the 90%CIs for test‐reference comparison in the log scale was 0.16 from the observed difference in means. The calculations were based on a paired t‐test with a 1‐sided α‐level of 0.05 and N‐1 degrees of freedom. It was also assumed that the variability of one particular PK parameter is the same across different treatments. Furthermore, no adjustments were made for multiple comparisons. Assuming a nonevaluability rate of 20% (including dropouts), the total number of patients enrolled was expected to be ≈25.
Bioanalytical Method
Serial blood samples for determination of plasma capmatinib and metabolite CMN288, itraconazole, and rifampicin concentrations were collected. Plasma was separated from whole blood by centrifugation and subsequently yielded plasma was transferred into polypropylene cryotubes. The plasma samples were frozen at <–70°C and stored until shipment to the bioanalytical laboratory, where the plasma concentrations of itraconazole, rifampicin, capmatinib, and CMN288 were determined using a validated liquid chromatography–tandem mass spectrometry assay with a lower limit of quantification of ≈5.0, 1.0, 1.0, and 1.0 ng/mL for itraconazole, rifampicin, capmatinib, and CMN288, respectively. Details pertaining to sample extraction and liquid chromatography–tandem mass spectrometry conditions for the analysis of capmatinib, CMN288, itraconazole, and rifampicin are described in the Supplemental Information.
The interday accuracy and precision of the 3 methods (capmatinib and CMN288, itraconazole, rifampicin) were evaluated as the mean bias and precision of quality control samples analyzed during at least 3 validation days. The bias (accuracy) was within ±15%, and the precisions (%CV) were within 15% at all concentrations in the validation of quality control samples. The selectivity of all methods was tested in at least 6 different lots of blank matrix (plasma) against the interference of endogenous components. There were no significant peaks detected at the mass transitions and expected retention times of the analytes (capmatinib, CMN288, itraconazole, rifampicin) or the corresponding internal standards that would interfere with quantitation in all the 6 lots tested. Furthermore, no significant interference of itraconazole at 2000 ng/mL and rifampicin at 1100 ng/mL to capmatinib and CMN288 was observed. Similarly, no significant interference of capmatinib at 5000 ng/mL to itraconazole and rifampicin was noted.
Pharmacokinetics
The primary PK parameters assessed were Cmax, AUC from time 0 to the last quantifiable concentration point (AUClast), AUC from time 0 to infinity (AUCinf), and time to reach maximum plasma drug concentration (tmax). The secondary PK parameters were apparent total body clearance of the drug from the plasma (CL/F), and terminal‐phase half‐life (t1/2). PK parameters were estimated using a noncompartmental method with Phoenix WinNonlin version 6.4 (Pharsight, Mountain View, California).
Safety Assessment
Safety was monitored by assessing vital signs, physical examination, electrocardiogram, hematology, blood chemistry, coagulation, urinalysis, lipid panel, cystatin C and creatinine, and thyroid hormones, as well as pregnancy assessment for women at site visits. Adverse events (AEs) were documented at every visit. Study drug dose adjustments and/or interruptions were not permitted. AEs were coded using the Medical Dictionary for Regulatory Activities version 20.0 and were graded using Common Terminology Criteria for AEs version 4.03. Each AE was evaluated for its relationship to the study treatment (reasonable possibility that AE is related: no, yes; investigational treatment, yes; the study treatment [non‐investigational], yes, both, and/or indistinguishable).
Statistical Analysis
The full analysis set and safety set included all subjects who received at least 1 dose of any study treatment (capmatinib, itraconazole, or rifampicin). The full analysis set and safety set in this study were identical. The PK analysis set for capmatinib in inhibition/induction cohorts included all subjects who provided an evaluable PK profile for at least 1 period.
Formal statistical analysis was performed for Cmax, AUClast, and AUCinf. A linear mixed‐effects model was fitted to the log‐transformed PK parameters (Cmax, AUClast, and AUCinf) to assess the effect of itraconazole and rifampicin on capmatinib 200 and 400 mg in inhibition and induction cohorts, respectively. The model included treatment as a fixed factor and subject as a random factor. For each of the comparisons, a point estimate and the corresponding 2‐sided 90%CI for the difference between means of test and reference treatment (test‐reference) were calculated. For this analysis, capmatinib 200 mg + itraconazole and capmatinib 400 mg + rifampicin were the test treatments, and capmatinib 200 mg and capmatinib 400 mg were the reference treatments for the inhibition and induction cohorts, respectively. Point estimates and the corresponding 2‐sided 90%CIs within each cohort were calculated and antilogged to obtain the point estimates and 90%CIs for the ratio of test versus reference on the original scale. The median and the range of the differences of tmax values of capmatinib were calculated for test versus reference. Descriptive statistics (mean, CV%, median, geometric mean, geometric CV%) were presented for PK parameters. Only median values and ranges were given for tmax. Cystatin C and creatinine concentration change from baseline values were summarized descriptively by time point.
Safety was based on all subjects in the study who received at least 1 dose of any study medication (capmatinib, itraconazole, or rifampicin). Categorical data were summarized by frequency counts and percentages. Continuous data were summarized by appropriate descriptive statistics. SAS® version 9.4 (SAS Institute, Cary, North Carolina) was used in all analyses.
Results
Subject Disposition
A total of 53 healthy subjects were enrolled in the study, of which 27 subjects were in the inhibition cohort and 26 in the induction cohort. In the inhibition cohort, 4 subjects (14.8%) discontinued study treatment due to AEs (n = 3; 11.1%) and physician decision (n = 1; 3.7%), while 2 subjects (7.7%) discontinued due to AEs (n = 1; 3.8%) and subject/guardian decision (n = 1; 3.8%) in the induction cohort. All subjects received at least 1 dose of any study treatment components and thus were included for safety assessment. One subject from each of the cohorts was not included in the PK analysis.
Subject Demographics
In the inhibition cohort (n = 27), the mean (standard deviation [SD]) age of the subjects was 40.4 (10.6) years, mean (SD) body weight was 84.1 (10.7) kg, mean (SD) BMI was 26.1 (3.1) kg/m2, all were men and most were White (92.6%). In the induction cohort (n = 26), the mean (SD) age of the subjects was 45.3 (8.4) years, mean (SD) body weight was 82.5 (10.1) kg, mean (SD) BMI was 25.6 (2.3) kg/m2, most were men (80.8%) and White (96.2%) (Table 1).
Table 1.
Subject Demographics (Full Analysis Set)
| Characteristic |
Inhibition Cohort N = 27 n (%) |
Induction Cohort N = 26 n (%) |
|---|---|---|
| Mean age, y (SD) | 40.4 (10.6) | 45.3 (8.4) |
| Sex, n (%) | ||
| Female | 0 | 5 (19.2) |
| Male | 27 (100) | 21 (80.8) |
| Race, n (%) | ||
| White | 25 (92.6) | 25 (96.2) |
| Black | 1 (3.7) | 0 |
| Asian | 1 (3.7) | 1 (3.8) |
| Mean weight (SD), kg | 84.1 (10.7) | 82.5 (10.1) |
| Mean height (SD), cm | 179.5 (6.5) | 179.4 (7.0) |
| Mean body mass index (SD), kg/m2 | 26.1 (3.1) | 25.6 (2.3) |
| Mean body surface area (SD), m2 | 2.05 (0.15) | 2.03 (0.16) |
| Mean serum creatinine (SD), mg/dL | 0.85 (0.11) | 0.80 (0.12) |
SD, standard deviation.
Effect of Itraconazole on the PK of Capmatinib (Inhibition Cohort)
The geometric mean and arithmetic mean (±SD) concentration‐time profiles for capmatinib are shown in Figure 2a. When capmatinib was administered with itraconazole, the geometric mean of AUCinf and AUClast increased by 42% (geometric mean ratio [GMR], 1.42; 90%CI, 1.33–1.52) and 41% (GMR, 1.41; 90%CI, 1.29–1.53), respectively, compared to capmatinib administered alone. There is no effect of itraconazole on Cmax (GMR, 1.03; 90%CI, 0.866–1.22) (Table 2, Figure S1). Tmax was 1 hour with capmatinib alone and 2 hours when administered in combination with itraconazole. The geometric mean of CL/F was slightly lower when capmatinib was administered with itraconazole (26.8 L/h) compared to capmatinib alone (37.9 L/h), which reflected the effect on AUC (Table 3). Despite the increase of AUC and decrease of CL/F, the t1/2 was shortened from 9.29 to 6.34 hours by itraconazole coadministration. The variability for capmatinib AUC and Cmax in the capmatinib‐alone period was similar to that for the capmatinib‐itraconazole coadministration period.
Figure 2.
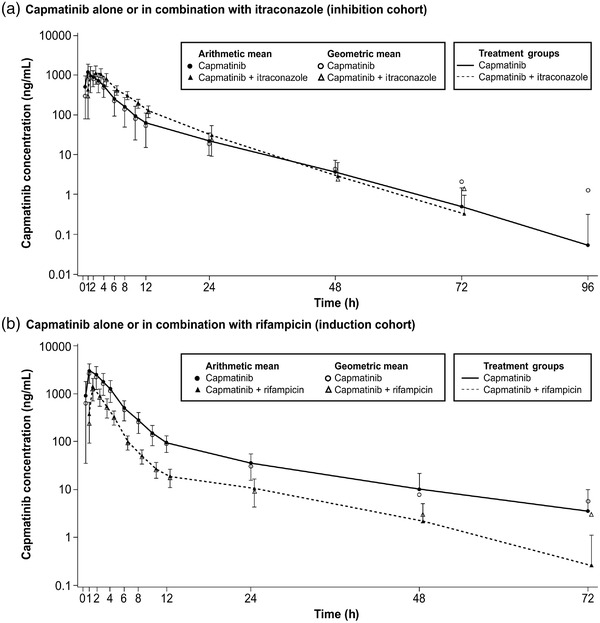
Geometric mean and arithmetic mean (standard deviation) concentration‐time profiles. Note: Zero concentrations at individual time points are excluded from geometric mean computation.
Table 2.
Summary of Statistical Analysis of Primary PK Parameters of Capmatinib Before and After Coadministration of Itraconazole or Rifampicin (PK Analysis Set)
| Treatment Comparison (Perpetrator Drug + Capmatinib/Capmatinib Alone) | |||
|---|---|---|---|
| PK Parameter | N a | Adjusted Geometric Mean | Geometric Mean Ratio (90%CI) |
| Capmatinib (perpetrator drug, itraconazole) | |||
| AUCinf (ng · h/mL) | |||
| Capmatinib alone | 25 | 5180 | 1.42 (1.33–1.52) |
| Perpetrator + capmatinib | 22 | 7360 | |
| AUClast (ng · h/mL) | |||
| Capmatinib alone | 26 | 5220 | 1.41 (1.29–1.53) |
| Perpetrator + capmatinib | 22 | 7340 | |
| Cmax (ng/mL) | |||
| Capmatinib alone | 26 | 1260 | 1.03 (0.866–1.22) |
| Perpetrator + capmatinib | 22 | 1300 | |
| Tmax (h) | |||
| Capmatinib alone | 26 | 1.00 | 0.489 (–3.02 to 3.00) |
| Perpetrator + capmatinib | 22 | 2.00 | |
| Capmatinib (perpetrator drug, rifampicin) | |||
| AUCinf (ng · h/mL) | |||
| Capmatinib alone | 24 | 11,500 | 0.335 (0.300–0.374) |
| Perpetrator + capmatinib | 18 | 3850 | |
| AUClast (ng · h/mL) | |||
| Capmatinib alone | 25 | 11,600 | 0.322 (0.294–0.353) |
| Perpetrator + capmatinib | 23 | 3720 | |
| Cmax (ng/mL) | |||
| Capmatinib alone | 25 | 3070 | 0.441 (0.387–0.502) |
| Perpetrator + capmatinib | 23 | 1350 | |
| tmax (h) | |||
| Capmatinib alone | 25 | 0.994 | –0.0317 (–1.05 to 1.01) |
| Perpetrator + capmatinib | 23 | 0.994 |
AUCinf, area under the plasma concentration–time curve from time 0 to infinity; AUClast, area under the plasma concentration–time curve from time 0 to the last quantifiable concentration point; Cmax, maximum plasma drug concentration; PK, pharmacokinetic; tmax, time to reach maximum plasma drug concentration.
For Tmax, median is presented under Adjusted Geometric Mean, median difference under Geometric Mean Ratio, and minimum and maximum differences under 90%CI.
aN = number of observations used for analysis.
Table 3.
Summary of PK Parameters of Capmatinib Before and After Coadministration of Itraconazole or Rifampicin
| AUCinf (ng · h/mL) | AUClast (ng · h/mL) | Cmax (ng/mL) | tmax a (h) | CL/F (L/h) | t1/2 (h) | |
|---|---|---|---|---|---|---|
| Inhibition cohort | ||||||
| Capmatinib | n = 25 | n = 26 | n = 26 | n = 26 | n = 25 | n = 25 |
| Mean (CV%) | 5500 (27.1) | 5430 (27.0) | 1380 (40.4) | 1.00 (0.505–4.00) | 39.6 (33.4) | 9.29 (46.8) |
| Geometric mean (geometric CV%) | 5280 (30.6) | 5220 (30.6) | 1260 (46.9) | 37.9 (30.6) | 8.29 (53.7) | |
| Capmatinib + itraconazole | n = 22 | n = 22 | n = 22 | n = 22 | n = 22 | n = 22 |
| Mean (CV%) | 7680 (24.3) | 7660 (24.5) | 1410 (41.5) | 2.00 (0.495–4.01) | 27.8 (28.1) | 6.34 (32.8) |
| Geometric mean (geometric CV%) | 7450 (26.4) | 7430 (26.5) | 1290 (48.6) | 26.8 (26.4) | 6.05 (31.3) | |
| Induction cohort | ||||||
| Capmatinib | n = 24 | n = 25 | n = 25 | n = 25 | n = 24 | n = 24 |
| Mean (CV%) | 11 600 (24.1) | 12 100 (34.5) | 3330 (41.4) | 0.994 (0.461–3.00) | 36.4 (23.7) | 11.2 (63.2) |
| Geometric mean (geometric CV%) | 11 300 (24.3) | 11 600 (30.3) | 3070 (43.0) | 35.4 (24.3) | 9.71 (55.9) | |
| Capmatinib + rifampicin | n = 18 | n = 23 | n = 23 | n = 23 | n = 18 | n = 18 |
| Mean (CV%) | 4050 (32.6) | 3860 (31.3) | 1460 (43.4) | 0.994 (0.478–2.00) | 107 (26.1) | 11.1 (35.2) |
| Geometric mean (geometric CV%) | 3890 (29.4) | 3720 (27.0) | 1350 (41.8) | 103 (29.4) | 10.3 (40.8) |
AUCinf, area under the plasma concentration–time curve from time 0 to infinity; AUClast, area under the plasma concentration–time curve from time 0 to the last quantifiable concentration point; CL/F, apparent total body clearance of drug from the plasma; Cmax, maximum plasma drug concentration; CV, coefficient of variation; PK, pharmacokinetic; t1/2, terminal‐phase half‐life; tmax, time to reach maximum plasma drug concentration; Vz/F, apparent volume of distribution during the terminal phase.
n = number of subjects with corresponding evaluable PK parameters.
Tmax data are shown as median (range).
The major circulating metabolite CMN288 was an inactive metabolite and was formed by aldehyde oxidase. The exposure of CMN288 was measured when capmatinib was given alone and in combination with itraconazole to evaluate the impact of inhibiting the CYP3A pathway on the formation of CMN288. The estimated metabolic ratios of CMN288 relative to capmatinib for AUCinf, AUClast, and Cmax were slightly lower with capmatinib + itraconazole versus capmatinib alone, that is, 0.554 versus 0.677, 0.552 versus 0.660, and 0.442 versus 0.506, respectively, indicating no significant impact on CMN288 formation by itraconazole (Table 4).
Table 4.
Summary of PK Parameters of CMN288 Before and After Coadministration of Itraconazole or Rifampicin With Capmatinib
| AUCinf (ng · h/mL) | AUClast (ng · h/mL) | Cmax (ng/mL) | tmax a (h) | MR (AUCinf) | MR (AUClast) | MR (Cmax) | |
|---|---|---|---|---|---|---|---|
| Inhibition cohort | |||||||
| Capmatinib | n = 26 | n = 26 | n = 26 | n = 26 | n = 25 | n = 26 | n = 26 |
| Mean (CV%) | 3870 (35.5) | 3810 (36.2) | 736 (47.0) | 1.01 (0.968–6.02) | 0.699 (25.4) | 0.685 (27.5) | 0.533 (30.5) |
| Geometric mean (geometric CV%) | 3640 (36.6) | 3580 (37.6) | 665 (48.7) | 0.677 (26.1) | 0.660 (29.1) | 0.506 (34.7) | |
| Capmatinib + itraconazole | n = 22 | n = 22 | n = 22 | n = 22 | n = 22 | n = 22 | n = 22 |
| Mean (CV%) | 4600 (35.7) | 4570 (36.0) | 682 (50.8) | 2.00 (0.983–4.01) | 0.572 (25.1) | 0.570 (25.6) | 0.462 (29.2) |
| Geometric mean (geometric CV%) | 4290 (42.3) | 4260 (43.2) | 590 (63.8) | 0.554 (26.2) | 0.552 (26.9) | 0.442 (31.6) | |
| Induction cohort | |||||||
| Capmatinib | n = 25 | n = 25 | n = 25 | n = 25 | n = 24 | n = 25 | n = 25 |
| Mean (CV%) | 7710 (30.1) | 7600 (30.2) | 1610 (39.2) | 0.994 (0.957–3.00) | 0.634 (25.2) | 0.627 (25.1) | 0.492 (30.8) |
| Geometric mean (geometric CV%) | 7420 (28.4) | 7310 (28.4) | 1500 (41.4) | 0.615 (25.5) | 0.609 (25.1) | 0.470 (31.1) | |
| Capmatinib + rifampicin | n = 23 | n = 23 | n = 23 | n = 23 | n = 18 | n = 23 | n = 23 |
| Mean (CV%) | 5360 (32.1) | 5320 (32.5) | 1690 (38.1) | 0.998 (0.924–3.00) | 1.37 (26.0) | 1.35 (25.0) | 1.17 (28.9) |
| Geometric mean (geometric CV%) | 5110 (31.9) | 5070 (32.3) | 1570 (39.8) | 1.33 (28.6) | 1.31 (26.4) | 1.13 (28.7) |
AUCinf, area under the plasma concentration–time curve from time 0 to infinity; AUClast, area under the plasma concentration–time curve from time 0 to the last quantifiable concentration point; Cmax, maximum plasma drug concentration; CV, coefficient of variation; MR, metabolite‐to‐parent drug ratio/metabolic ratio; PK, pharmacokinetic; tmax, time to reach maximum plasma drug concentration.
n = number of subjects with corresponding evaluable PK parameters.
Tmax data are shown as median (range).
Effect of Rifampicin on the PK of Capmatinib (Induction Cohort)
The geometric mean and arithmetic mean (±SD) concentration‐time profiles for capmatinib are shown in Figure 2b. When capmatinib was administered with rifampicin, the geometric mean of AUCinf, AUClast, and Cmax decreased by 66.5% (GMR, 0.335; 90%CI, 0.300–0.374), 67.8% (GMR, 0.322; 90%CI, 0.294–0.353), and 55.9% (GMR, 0.441; 90%CI, 0.387–0.502), respectively, compared to capmatinib alone (Table 2, Figure S2). Tmax was similar with capmatinib + rifampicin versus capmatinib alone (Table 2). The geometric mean of CL/F was higher with coadministration of capmatinib with rifampicin (103 L/h) compared to capmatinib alone (35.4 L/h), which reflected the effect of rifampicin on AUC (Table 3). No change in t1/2 was observed upon rifampicin coadministration despite the decrease of AUC and increase of CL/F. The variability for capmatinib AUC and Cmax in the capmatinib‐alone period were similar to that for the capmatinib‐rifampicin coadministration period.
When capmatinib was administrated with rifampicin, the geometric mean AUCinf for CMN288 was slightly lower compared to capmatinib alone (5110 vs 7420 ng · h/mL) with no difference in Cmax. The estimated metabolic ratios of CMN288 for AUCinf, AUClast, and Cmax were higher with capmatinib + rifampicin versus capmatinib alone, that is, 1.33 versus 0.615, 1.31 versus 0.609, and 1.13 versus 0.470, respectively, which mainly reflected the lower capmatinib exposure upon rifampicin treatment (Table 4).
Effect of Capmatinib on Serum Creatinine and Cystatin C Concentration
The mean creatinine and cystatin C concentration changes from baseline versus time profiles for the inhibition and induction cohorts are described in Figure 3a,b, respectively. In the inhibition cohort, upon administration of a single dose of 200 mg of capmatinib, the creatinine concentration slightly increased from baseline, reached the maximum concentration at 24 hours with an increase of 21.0% from baseline, and returned to baseline at 48 hours. In the induction cohort, upon administration of a single dose of 400 mg of capmatinib, the creatinine concentration slightly increased from baseline, reached the maximum concentration at 24 hours with an increase of 27.3% from baseline, and returned to baseline at 72 hours. Conversely, there was no change in the concentration of cystatin C throughout the time course evaluated in both cohorts.
Figure 3.
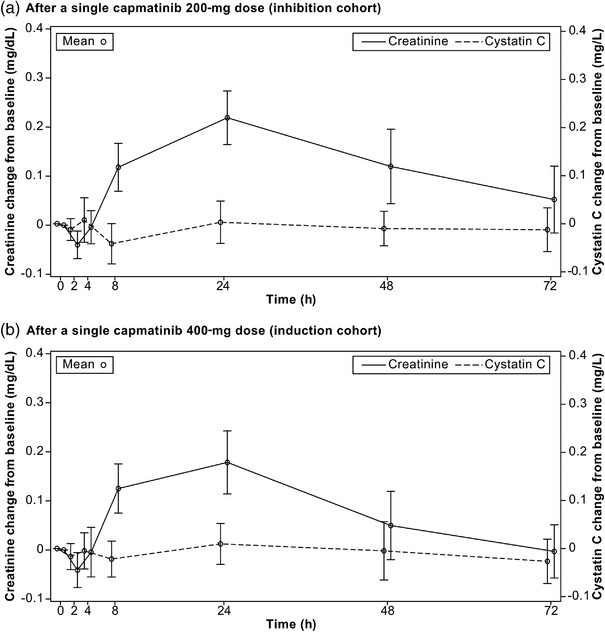
Mean (standard deviation) cystatin C and creatinine concentration change from baseline versus time profiles (safety set).
Safety
The safety analyses included only on‐treatment assessments (from date of first administration of study treatment to 30 days after date of last administration of study treatment). Overall, 19 subjects (70.4%) and 11 subjects (42.3%) experienced at least 1 AE (regardless of study drug relationship) in the inhibition and induction cohorts, respectively. Grade 3 AEs were reported in 3 subjects (11.1%) in the inhibition cohort, including hypertension (n = 2; 7.4%) and amylase increased (n = 1; 3.7%); and 1 subject (3.8%) had grade 3 hypertension in the induction cohort. No grade 4 or 5 AEs were observed in either of the cohorts (Table S1). AEs suspected to be related to the study treatment (any grade) occurred in 13 subjects (48.1%) and 10 subjects (38.5%) in the inhibition and induction cohorts, respectively. Data on most frequent AEs and discontinuations due to AEs are described in the Supplemental Information. No serious AEs were reported in either of the treatment cohorts. No clinically significant change in laboratory parameters was noted during the study. No deaths were reported in either of the treatment cohorts.
Discussion
With capmatinib being a substrate of CYP3A4, the study was designed to assess the effect of a potent CYP3A4 inhibitor (itraconazole) and inducer (rifampicin) on the PK of a single oral dose of capmatinib. The results could be used to guide the concomitant use of CYP3A inhibitors and inducers.
A daily dose of itraconazole 200 mg with a lead‐in of 5 days before coadministration of capmatinib, followed by continued itraconazole dosing for additional 4 doses during the PK sampling period was done to maximize the interaction by ensuring continued perpetrator exposure during capmatinib elimination. Clinical interaction studies with this regimen have shown profound increases in the exposure of sensitive CYP3A4 substrates such as midazolam (6‐ to 10‐fold), 21 , 22 triazolam (27‐fold), 23 and buspirone (15‐fold). 24 The increases in oral AUC observed after a lead‐in of 4 or 6 days of itraconazole were similar. 21 , 25 To maximize the exposure of itraconazole in this study, it was administered as an oral solution under fasted conditions. 26 In addition, on the day of coadministration, capmatinib was given 1 hour after the itraconazole administration to avoid the potential interference of hydroxypropyl‐β‐cyclodextrin in itraconazole oral solution on capmatinib absorption/PK and DDI evaluation. 27 Itraconazole has been proposed as a recommended replacement for ketoconazole in DDI studies after comparing CYP3A inhibition potency, specificity, safety profile, and extent of clinical experience among several candidates. 28
The dosing regimen of rifampicin used in this study, duration of 4 days at 600 mg once daily, was as reported in the literature 21 to have maximum induction potential. Besides the strong CYP3A induction effect, rifampicin is also an inhibitor of organic anion transporting polypeptide (OATP) 1B1/1B3. A staggered design with a separation of 12 hours between rifampicin and investigational drug has been proposed to avoid the confounding of the inhibition effect on the CYP3A induction evaluation. 29 However, capmatinib is not a substrate for OATP1B1/1B3, and the simultaneous use of capmatinib and rifampicin is not expected to impact the accurate evaluation of CYP3A induction effect on capmatinib exposure.
The plasma trough concentrations on day 10 for itraconazole (inhibition cohort) and rifampicin (induction cohort) were measured to ensure adequate exposure for inhibitor or inducer of CYP3A. The concentrations obtained in this study were comparable to steady‐state itraconazole and rifampicin concentrations reported in the literature following 200 mg/day itraconazole or 600 mg/day rifampicin administration (Novartis data on file). 30
Coadministration of itraconazole increased the AUCinf of capmatinib by 42% with no significant change in Cmax, compared to capmatinib administered alone. These results suggest that coadministration of CYP3A inhibitors may increase capmatinib exposure in patients and subsequently lead to increased incidence and severity of adverse reactions. Accordingly, we advise caution and close monitoring of patients who are on concomitant treatment with CYP3A inhibitors and capmatinib. The strong CYP3A inducer rifampicin led to a 65% decrease in capmatinib AUCinf and 55% decrease in Cmax. The reduction in capmatinib exposure can potentially lead to a decrease in antitumor activity of capmatinib; therefore, we suggest avoiding concomitant use of strong and moderate CYP3A inducers in patients treated with capmatinib. This extent of impact of itraconazole and rifampicin on capmatinib exposure is consistent with the fact that capmatinib is not a sensitive CYP3A4 substrate, as CYP3A4 contributes to 40%–50% of capmatinib metabolism.
The metabolite CMN288 is a major circulating inactive metabolite, formed by aldehyde oxidase. Exposure of CMN288 was monitored in this study to explore any impact on the formation of CMN288 through aldehyde oxidase pathway while inhibiting or inducing CYP3A by itraconazole or rifampicin, respectively. No significant difference in CMN288 exposure or metabolic ratio was observed when capmatinib was administered with or without itraconazole, indicating that itraconazole had no impact on the pathway. Upon coadministration with rifampicin, CMN288 exposure decreased modestly compared to capmatinib alone. The metabolic ratio of CMN288 to capmatinib increased from 0.615 to 1.33 upon coadministration with rifampicin. However, this increase was largely driven by the 67% decrease of capmatinib AUC by rifampicin. The metabolites of capmatinib formed by CYP3A4 were at low concentration in circulation. Among them, the metabolite M8 (hydroxylation metabolite) was at the highest concentration and accounted for only 5% of the circulating component. 16 Therefore, the metabolite formed by CYP3A4 was not monitored in this study due to its low plasma concentration.
A transient reversible increase in creatinine has been observed following a single dose of capmatinib in healthy subjects and after repeat dosing in patients (Novartis data on file). Renal transporters are responsible for 10%–40% of creatinine excretion in addition to glomerular filtration. 18 Lepist et al 31 have shown that inhibitors for organic anion transporters and MATEs could impact the active secretion of creatinine in renal tubule without affecting glomerular filtration. Additionally, there are several drugs and investigational new chemical entities that inhibit renal transporters and therefore increase serum creatinine without change in the estimated glomerular filtration rate or other renal biomarkers. 32 , 33 Serum creatinine has been proposed as an endogenous substrate to evaluate the renal transporter–related drug interaction. In vitro data indicated that capmatinib is an inhibitor of renal transporters MATE1 and MATE2K with a Ki value of 0.28 µM and 0.29 µM, respectively. Based on the steady‐state Cmax of 11.6 µM (0.464 µM unbound) at a therapeutic dose of 400 mg twice daily, capmatinib is likely to inhibit MATE 1 and MATE2K in humans. With creatinine being an endogenous substrate for renal transporters, it is likely that the transient increase in serum creatinine by capmatinib could be due to the reversible inhibition of MATE transporters. The renal biomarker cystatin C was selected in this study to compare with creatinine because it is solely cleared through glomerular filtration and is considered a better marker for risk determination based on renal function. 34 No change in the cystatin C concentration following capmatinib administration confirmed that renal function measured by glomerular filtration has not been impacted by capmatinib. The transient increase of serum creatinine could be explained by the reversible inhibition of renal transporters by capmatinib. A maximum 21% increase of serum creatinine was observed at 24 hours after administration of 200‐mg capmatinib, and a 27.3% increase of serum creatinine was observed at 24 hours after administration of 400 mg of capmatinib. While there seems to be a dose‐dependent increase in serum creatinine concentration following capmatinib administration, in general, the extent of increase from baseline in this study is consistent with the fact that a relatively smaller proportion of creatinine is excreted through renal tubular secretion, while most of the creatinine is excreted through glomerular filtration. This is also consistent with previously reported data that showed that a decrease in creatinine clearance or increase in serum creatinine concentration from baseline due to the inhibition of renal transporters was usually in the range of 5%–30%. 28 It is also worth mentioning that comparison of the time course of changes in renal biomarkers creatinine and cystatin C in this study allowed simultaneous evaluation of DDI interaction mediated by CYP3A and provides evidence on renal transporter inhibition due to capmatinib. These results have provided further understanding on the safety profile of patients receiving capmatinib who frequently experience a low‐grade and transient increase in serum creatinine.
Author Contributions
Xiaoming Cui, Xinhui Chen, Nathalie Pognan, and Monica Giovannini contributed to designing of the study, patient accrual, data analysis, data interpretation, manuscript writing, and approval. Tirtha Sengupta and Gholamreza Rahmanzadeh contributed to designing of the study, data analysis, data interpretation, manuscript writing, and approval. Ruediger Kornberger contributed to data analysis, data interpretation, manuscript writing, and approval.
Conflicts of Interest
Ruediger Kornberger is an employee of Parexel. Xiaoming Cui, Xinhui Chen, Nathalie Pognan, Tirtha Sengupta, Gholamreza Rahmanzadeh, and Monica Giovannini are employees of Novartis. Xiaoming Cui, Xinhui Chen, Nathalie Pognan, Tirtha Sengupta, Monica Giovannini, and Gholamreza Rahmanzadeh own Novartis stock.
Funding
This study was funded by Novartis Pharmaceuticals Corporation.
Principal Investigator Statement
The authors confirm that the Principal Investigator for this paper is Dr Ruediger Kornberger and that he had direct clinical responsibility for participants.
Supporting information
Supporting Information., Additional supplemental information can be found by clicking the Supplements link in the PDF toolbar or the Supplemental Information section at the end of the web‐based version of this article.
Supporting Information.
Supporting Information.
Supporting Information.
Acknowledgments
The authors thank Shilpa Garg and Varunkumar Pandey of Novartis Healthcare Pvt. Ltd., for providing medical writing assistance with this manuscript.
Clinical Trial Registration: This study was done in healthy subjects, and therefore the study was not registered in a clinical trial registry.
Data Availability Statement
Novartis is committed to sharing with qualified external researchers, access to patient‐level data and supporting clinical documents from eligible studies. These requests are reviewed and approved by an independent review panel on the basis of scientific merit. All data provided are anonymized to respect the privacy of patients who have participated in the trial in line with applicable laws and regulations. This trial data availability is according to the criteria and process described at www.clinicalstudydatarequest.com.
References
- 1. Christensen JG, Burrows J, Salgia R. c‐Met as a target for human cancer and characterization of inhibitors for therapeutic intervention. Cancer Lett. 2005;225(1):1‐26. [DOI] [PubMed] [Google Scholar]
- 2. Lee JH, Han SU, Cho H, et al. A novel germ line juxtamembrane Met mutation in human gastric cancer. Oncogene. 2000;19(43):4947‐4953. [DOI] [PubMed] [Google Scholar]
- 3. Lorenzato A, Olivero M, Patane S, et al. Novel somatic mutations of the MET oncogene in human carcinoma metastases activating cell motility and invasion. Cancer Res. 2002;62(23):7025‐7030. [PubMed] [Google Scholar]
- 4. Reungwetwattana T, Liang Y, Zhu V, Ou SI. The race to target MET exon 14 skipping alterations in non‐small cell lung cancer: the why, the how, the who, the unknown, and the inevitable. Lung Cancer. 2017;103:27‐37. [DOI] [PubMed] [Google Scholar]
- 5. Tong JH, Yeung SF, Chan AW, et al. MET amplification and Exon 14 splice site mutation define unique molecular subgroups of non‐small cell lung carcinoma with poor prognosis. Clin Cancer Res. 2016;22(12):3048‐3056. [DOI] [PubMed] [Google Scholar]
- 6. Sacco JJ, Clague MJ. Dysregulation of the Met pathway in non‐small cell lung cancer: implications for drug targeting and resistance. Transl Lung Cancer Res. 2015;4(3):242‐252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schrock AB, Frampton GM, Suh J, et al. Characterization of 298 patients with lung cancer harboring MET Exon 14 skipping alterations. J Thorac Oncol. 2016;11(9):1493‐1502. [DOI] [PubMed] [Google Scholar]
- 8. Baltschukat S, Engstler BS, Huang A, et al. Capmatinib (INC280) is active against models of non‐small cell lung cancer and other cancer types with defined mechanisms of MET activation. Clin Cancer Res. 2019;25(10):3164‐3175. [DOI] [PubMed] [Google Scholar]
- 9. Liu X, Wang Q, Yang G, et al. A novel kinase inhibitor, INCB28060, blocks c‐MET‐dependent signaling, neoplastic activities, and cross‐talk with EGFR and HER‐3. Clin Cancer Res. 2011;17(22):7127‐7138. [DOI] [PubMed] [Google Scholar]
- 10. Fujino T, Kobayashi Y, Suda K, et al. Sensitivity and resistance of MET Exon 14 mutations in lung cancer to eight MET tyrosine kinase inhibitors in vitro. J Thorac Oncol. 2019;14(10):1753‐1765. [DOI] [PubMed] [Google Scholar]
- 11. Bang Y‐J, Su W‐C, Nam D‐H, et al. Phase I study of the safety and efficacy of INC280 in patients with advanced MET‐dependent solid tumors. J Clin Oncol 2014;32(15_suppl):2520‐2520. [Google Scholar]
- 12. Shao‐Weng Tan D, Lee DH, Soo R, et al. P3.02b‐117 Phase Ib results from a study of capmatinib (INC280) + EGF816 in patients with EGFR‐mutant non‐small cell lung cancer (NSCLC):Topic: EGFR RES. J Thorac Oncol. 2017;12(1):S1264‐S1265. [Google Scholar]
- 13. Tanwandee T, Sukeepaisarnjaroen W, Chan SL, et al. A phase (Ph) II study of the efficacy and safety of the cMET inhibitor capmatinib (INC280) in patients (pts) with advanced hepatocellular carcinoma (HCC). J Clin Oncol. 2016;34(suppl_no.15):4074‐4074. [Google Scholar]
- 14. Wu YL, Zhang L, Kim DW, et al. Phase Ib/II study of capmatinib (INC280) plus gefitinib after failure of epidermal growth factor receptor (EGFR) inhibitor therapy in patients with EGFR‐mutated, MET factor‐dysregulated non‐small‐cell lung cancer. J Clin Oncol. 2018;36(31):3101‐3109. [DOI] [PubMed] [Google Scholar]
- 15. Wolf J, Seto T, Han JY, et al. Capmatinib in MET Exon 14‐mutated or MET‐amplified non‐small‐cell lung cancer. N Engl J Med. 2020;383(10):944‐957. [DOI] [PubMed] [Google Scholar]
- 16. Glaenzel U, Jin Y, Hansen R, et al. Absorption, distribution, metabolism, and excretion of capmatinib (INC280) in healthy male volunteers and in vitro aldehyde oxidase phenotyping of the major metabolite. Drug Metab Dispos. 2020;48(10):873‐885. [DOI] [PubMed] [Google Scholar]
- 17. Esaki T, Hirai F, Makiyama A, et al. Phase I dose‐escalation study of capmatinib (INC280) in Japanese patients with advanced solid tumors. Cancer Sci. 2019;110(4):1340‐1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Levey AS, Perrone RD, Madias NE. Serum creatinine and renal function. Annu Rev Med. 1988;39:465‐490. [DOI] [PubMed] [Google Scholar]
- 19. Mathialagan S, Rodrigues AD, Feng B. Evaluation of renal transporter inhibition using creatinine as a substrate in vitro to assess the clinical risk of elevated serum creatinine. J Pharm Sci. 2017;106(9):2535‐2541. [DOI] [PubMed] [Google Scholar]
- 20. Filler G, Bokenkamp A, Hofmann W, Le Bricon T, Martinez‐Bru C, Grubb A. Cystatin C as a marker of GFR–history, indications, and future research. Clin Biochem. 2005;38(1):1‐8. [DOI] [PubMed] [Google Scholar]
- 21. Backman JT, Kivistö KT, Olkkola KT, Neuvonen PJ. The area under the plasma concentration‐time curve for oral midazolam is 400‐fold larger during treatment with itraconazole than with rifampicin. Eur J Clin Pharmacol. 1998;54(1):53‐58. [DOI] [PubMed] [Google Scholar]
- 22. Olkkola KT, Backman JT, Neuvonen PJ. Midazolam should be avoided in patients receiving the systemic antimycotics ketoconazole or itraconazole. Clin Pharmacol Ther. 1994;55(5):481‐485. [DOI] [PubMed] [Google Scholar]
- 23. Varhe A, Olkkola KT, Neuvonen PJ. Oral triazolam is potentially hazardous to patients receiving systemic antimycotics ketoconazole or itraconazole. Clin Pharmacol Ther. 1994;56(6):601‐607. [DOI] [PubMed] [Google Scholar]
- 24. Kivistö KT, Lamberg TS, Kantola T, Neuvonen PJ. Plasma buspirone concentrations are greatly increased by erythromycin and itraconazole. Clin Pharmacol Ther. 1997;62(3):348‐354. [DOI] [PubMed] [Google Scholar]
- 25. Olkkola KT, Ahonen J, Neuvonen PJ. The effects of the systemic antimycotics, itraconazole and fluconazole, on the pharmacokinetics and pharmacodynamics of intravenous and oral midazolam. Anesth Analg. 1996;82(3):511‐516. [DOI] [PubMed] [Google Scholar]
- 26. Barone JA, Moskovitz BL, Guarnieri J, et al. Enhanced bioavailability of itraconazole in hydroxypropyl‐beta‐cyclodextrin solution versus capsules in healthy volunteers. Antimicrob Agents Chemother. 1998;42(7):1862‐1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen Y, Cabalu TD, Callegari E, et al. Recommendations for the design of clinical drug‐drug interaction studies with itraconazole using a mechanistic physiologically‐based Pharmacokinetic model. CPT Pharmacometrics Syst Pharmacol. 2019;8(9):685‐695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu L, Bello A, Dresser MJ, et al. Best practices for the use of itraconazole as a replacement for ketoconazole in drug‐drug interaction studies. J Clin Pharmacol. 2016;56(2):143‐151. [DOI] [PubMed] [Google Scholar]
- 29. Tornio A, Filppula AM, Niemi M, Backman JT. Clinical studies on drug‐drug interactions involving metabolism and transport: methodology, pitfalls, and interpretation. Clin Pharmacol Ther. 2019;105(6):1345‐1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ashbee HR, Barnes RA, Johnson EM, Richardson MD, Gorton R, Hope WW. Therapeutic drug monitoring (TDM) of antifungal agents: guidelines from the British Society for Medical Mycology. J Antimicrob Chemother. 2014;69(5):1162‐1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lepist EI, Zhang X, Hao J, et al. Contribution of the organic anion transporter OAT2 to the renal active tubular secretion of creatinine and mechanism for serum creatinine elevations caused by cobicistat. Kidney Int. 2014;86(2):350‐357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chu X, Bleasby K, Chan GH, Nunes I, Evers R. The complexities of interpreting reversible elevated serum creatinine levels in drug development: does a correlation with inhibition of renal transporters exist? Drug Metab Dispos. 2016;44(9):1498‐1509. [DOI] [PubMed] [Google Scholar]
- 33. Topletz‐Erickson AR, Lee AJ, Mayor JG, et al. Tucatinib inhibits renal transporters OCT2 and MATE without impacting renal function in healthy subjects. J Clin Pharmacol. 2021;61(4):461‐471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shlipak MG, Matsushita K, Arnlov J, et al. Cystatin C versus creatinine in determining risk based on kidney function. N Engl J Med. 2013;369(10):932‐943. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information., Additional supplemental information can be found by clicking the Supplements link in the PDF toolbar or the Supplemental Information section at the end of the web‐based version of this article.
Supporting Information.
Supporting Information.
Supporting Information.
Data Availability Statement
Novartis is committed to sharing with qualified external researchers, access to patient‐level data and supporting clinical documents from eligible studies. These requests are reviewed and approved by an independent review panel on the basis of scientific merit. All data provided are anonymized to respect the privacy of patients who have participated in the trial in line with applicable laws and regulations. This trial data availability is according to the criteria and process described at www.clinicalstudydatarequest.com.