

Abstract
Congenital disorders of glycosylation are genetic disorders that occur due to defects in protein and lipid glycosylation pathways. A deficiency of N‐glycanase 1, encoded by the NGLY1 gene, results in a congenital disorder of deglycosylation. The NGLY1 enzyme is mainly involved in cleaving N‐glycans from misfolded, retro‐translocated glycoproteins in the cytosol from the endoplasmic reticulum before their proteasomal degradation or activation. Despite the essential role of NGLY1 in deglycosylation pathways, the exact consequences of NGLY1 deficiency on global cellular protein glycosylation have not yet been investigated. We undertook a multiplexed tandem mass tags‐labeling‐based quantitative glycoproteomics and proteomics analysis of fibroblasts from NGLY1‐deficient individuals carrying different biallelic pathogenic variants in NGLY1. This quantitative mass spectrometric analysis detected 8041 proteins and defined a proteomic signature of differential expression across affected individuals and controls. Proteins that showed significant differential expression included phospholipid phosphatase 3, stromal cell‐derived factor 1, collagen alpha‐1 (IV) chain, hyaluronan and proteoglycan link protein 1, and thrombospondin‐1. We further detected a total of 3255 N‐glycopeptides derived from 550 glycosylation sites of 407 glycoproteins by multiplexed N‐glycoproteomics. Several extracellular matrix glycoproteins and adhesion molecules showed altered abundance of N‐glycopeptides. Overall, we observed distinct alterations in specific glycoproteins, but our data revealed no global accumulation of glycopeptides in the patient‐derived fibroblasts, despite the genetic defect in NGLY1. Our findings highlight new molecular and system‐level insights for understanding NGLY1‐CDDG.
Keywords: congenital disorder of deglycosylation, congenital disorder of glycosylation, glyco, mass spectrometry, NGLY1, proteomics, proteomics
1. INTRODUCTION
Congenital disorders of glycosylation (CDG) are genetic disorders that arise due to a defect in the synthesis of oligosaccharides, failure to add glycans onto the proteins/lipids, or defective maturation of glycoproteins and glycolipids in the cells. 1 CDG are monogenic disorders with predominant autosomal inheritance, besides autosomal and X‐linked inheritance. 1 , 2 Broadly, the CDG are classified into four groups—N‐linked glycosylation, O‐linked glycosylation, combined N‐ and O‐linked glycosylation, and glycosylphosphatidylinositol anchor and glycolipid biosynthesis defects. 1 , 3
More than 160 different types of CDG have been described to date. Besides lysosomal disorders, NGLY1‐CDDG is a congenital disorder of deglycosylation. This autosomal recessive disease was initially. 4 , 5 , 6 , 7 , 8 This autosomal recessive disease was initially described in 2012 following whole‐exome sequencing in a patient suspected of a CDG. 8 NGLY1 codes for the enzyme peptide‐N(4)‐(N‐acetyl‐beta‐glucosaminyl)asparagine amidase (PNGase), also known as N‐glycanase 1, a highly conserved enzyme of the endoplasmic reticulum‐associated degradation (ERAD) pathway, which catalyzes the deglycosylation of misfolded N‐glycoproteins. The enzyme NGLY1 catalyzes the hydrolysis of proximal β‐aspartylglucosamine attached to the asparagine residue, with subsequent release of glycans and misfolded proteins for proteasome degradation. 9 , 10 , 11 In addition to its role in ERAD pathway, a role of NGLY1 has also been described in cellular signaling. For example, it has been shown that NGLY1 promotes the bone morphogenic protein 4 (BMP4) signaling pathway by removing N‐glycans from misfolded BMP4. 12 Several studies have also demonstrated the role of NGLY1 in activation of nuclear factor erythroid 2 like‐1 (NEF2L1, also known as Nrf1), which is a known transcription factor. Nrf1 regulates various lipid and amino acid metabolic pathways, bone formation, and expression of genes required for proteasomal subunit complex. 13 , 14 , 15 The transcriptional functionality of Nrf1 is dependent on the activity of NGLY1, where NGLY1 deglycosylates Nrf1 before it is translocated into the nucleus for the expression of proteasome subunits. 16 , 17 , 18 Additionally, NGLY1 is also believed to regulate the expression of aquaporins at the transcription level through Atf1/Creb1 signaling pathway. 19 It has also been shown that NGLY1, along with cytosolic O‐GlcNAcylation, plays a key role in maintaining gut homeostasis by regulating proliferation and apoptosis. 20
Clinically, NGLY1 deficiency presents with a multisystem phenotype with global developmental delay and intellectual disability. 8 , 21 Since its recent discovery, more than 100 patients have been reported. 8 , 22 , 23 Most NGLY1 patients present with, hypo‐ or alacrima, transient elevation in serum transaminases and a complex hyperkinetic movement disorder. Seizures, failure to thrive due to feeding difficulties and peripheral neuropathy are also common. 21 , 24 , 25 In the absence of NGLY1, a cytosolic enzyme, endo‐β‐N‐acetylglucosaminidase (ENGase) catalyzes the deglycosylation of misfolded glycoproteins in the cytosol and generates N‐GlcNAc proteins, which is lethal to cells. 9 , 10 , 26 Defects in NGLY1 show accumulation of undegraded protein aggregates that may damage cells in the brain, eyes, and liver. 9 , 27 , 28 , 29
The measurement of NGLY1 activity by using a BODIPY‐labeled asialo glycopeptide has been developed and NGLY1 mutants show reduced activity. 30 However, when the endogenous NGLY1 activity was measured using glycosylated hepta‐cyclopeptide, the test lacked specificity. 30 The routinely used diagnostics in a suspected NGLY1 patient is the mass spectrometry‐based measurement of oligosaccharides (NeuAc1Hex1GlcNAc1‐Asn) in urine sample as a biomarker. 31 More recent studies have demonstrated the detection of N‐acetylglucosamine‐asparagine (GlcNAc‐Asn) residue in cell lines, rodents (urine, cerebrospinal fluid, plasma, and tissues), and patients (plasma and urine) 32 including dried blood spots. 33 Despite these developments, whole‐exome sequencing is employed in most cases for the diagnosis of NGLY1‐CDDG. Recently transcriptomics and proteomics studies were performed in patient‐derived NGLY1 fibroblasts and lymphoblastoid cells and reported changes in gene and protein expression which are involved in various cellular pathways. 34 These studies and case reports have proved to be invaluable tools to understand the biology and molecular mechanism of NGLY1; however, its effect on cellular glycosylation remains largely elusive. In order to understand this, combined glycoproteomics and proteomics studies are necessary to evaluate glycan composition, glycan heterogeneity, their site of attachment to the protein and site occupancy on the proteins.
The essential role of NGLY1 in deglycosylation pathways has led investigators in the field to suggest a global accumulation of cellular N‐linked glycoproteins. To test this hypothesis, we performed quantitative N‐glycoproteomics in patient‐derived NGLY1‐deficient fibroblasts and age‐matched control fibroblasts to detect any differences in site‐specific N‐glycosylation. In parallel, global proteomics analysis was performed to detect alterations at the protein level.
2. MATERIALS AND METHODS
2.1. Patient information
The four patient‐derived fibroblasts were included in the in vitro proteomics, glycoproteomics, and expression studies under an approved IRB protocol (IRB: 16‐004682). The patients carried different biallelic pathogenic variants (p.R401X/p.Q631Sfs, p.R401X/p.R401X, p.Q208X/p.G310G, and p.R321X/p.Q631Sfs*6) in NGLY1.
All pediatric patients were males, and their age was between 4 and 10 years. They presented with classic NGLY1‐related clinical features; development and speech delay, early muscle hypotonia, hypo/alacrima, progressive dystonia, and elevated transaminases, which spontaneously improved after the age of 6.
2.2. Cell culture
Four patient‐derived NGLY1‐deficient fibroblasts with different biallelic pathogenic variants in NGLY1 and four age‐matched control fibroblasts (GM5381, GM5400, GM5565, and GM5757; Coriell Institute) were cultured in Minimum Essential Media (MEM; Gibco; 1 g/L glucose) supplemented with 10% Fetal Bovine Serum (FBS; Gibco), 10% antibiotic‐antimycotic with gentamicin, and 10% non‐essential amino acids maintained in an incubator at 37°C, 5% CO2 environment. Cells were cultured and harvested at 90% confluence by scraping in phosphate‐buffered saline (PBS, pH 7.4) and centrifuged at 2000 rpm at 4°C for 10 min.
2.3. Sample preparation for proteomics and glycoproteomics
Harvested cells were lysed as described previously. 35 Briefly, modified RIPA lysis buffer (50 mM Tris–HCl, pH 8.0, with 150 mM sodium chloride, 1.0% NP‐40, 0.25% sodium deoxycholate, and 0.1% sodium dodecyl sulfate, Sigma‐Aldrich) supplemented with protease inhibitors (Sigma‐Aldrich) was added to the cell pellet. Cells were then sonicated with a tip sonicator at 40% amplitude for 3 cycles of 10 s each. Protein quantities in the harvested cells obtained were estimated by BCA assay as per the manufacturer's instructions (Thermo Fisher Scientific). Equal amounts of protein from both patient and control fibroblasts were first precipitated by adding chilled acetone to proteins in a 1:5 ratio for 2 h at −20°C. The proteins were pelleted down from the solution by centrifugation at 14000g for 30 min. The protein pellet was solubilized in 8 M urea (in 100 mM TEAB buffer). These solubilized proteins were reduced with 10 mM dithiothreitol (Sigma‐Aldrich) at 37°C with mild shaking on thermomixer, followed by alkylated with 40 mM iodoacetamide (Sigma‐Aldrich) at room temperature and samples were incubated for 30 min in the absence of light. The proteins were then digested with trypsin (Worthington) at 37°C overnight with mild shaking on thermomixer. Resulting peptides were cleaned up using C18 tips (Glygen) and labeled with tandem mass tags (TMTs) (Thermo Fisher Scientific) for a 16‐plex mass spectrometry experiment as per the manufacturer's protocol.
2.4. Basic pH reversed‐phase liquid chromatography
The TMT‐labeled samples were pooled and subsequently split into two aliquots. One aliquot was resuspended in solvent A (5 mM ammonium formate, pH 9) and fractionated by basic pH reversed‐phase liquid chromatography (bRPLC) on a reversed‐phase C18 column (5 μm, 4.6 × 100 mm2 column, Waters) using an increasing gradient of solvent B (5 mM ammonium formate, pH 9, in 90% acetonitrile) on the Ultimate 3000 UHPLC system. About 96 fractions for a total run time of 120 min were collected and subsequently concatenated into 24 fractions. These concatenated 24 fractions were dried down in a SpeedVac system and resuspended in 0.1% formic acid for liquid chromatography–tandem mass spectrometry (LC–MS/MS) analysis.
2.5. Size exclusion chromatography
The other aliquot was resuspended in 100 μl of 0.1% formic acid and injected into Superdex peptide 10/300 column (GE Healthcare) as described previously with some modifications. 36 , 37 The peptides were separated using an isocratic flow of 0.1% formic acid for 130 min and 24 early fractions were collected starting at 10 min after injection. The fractions were dried and resuspended in 0.1% formic acid for LC–MS/MS analysis.
2.6. Liquid chromatography–tandem mass spectrometry
The LC–MS/MS analysis of fractionated samples was carried essentially as described. 35 , 38 Briefly, the samples were analyzed on an Orbitrap Exploris 480 mass spectrometer (Thermo Fisher Scientific) connected to Ultimate 3000 liquid chromatography system (Thermo Fisher Scientific). The peptides/glycopeptides were loaded onto a trap column (2.7 μm) at a flow rate of 10 μl/min using 0.1% formic acid and separated on an analytical column (EasySpray 75 μm × 50 cm, C18 2 μm, 100 Å; Thermo Fisher Scientific), with a flow rate of 300 nl/min with a linear gradient of 3%–50% solvent B (100% ACN, 0.1% formic acid) for proteomics and 3%–32% solvent B for glycoproteomics over a 160 min gradient. Precursor ions were acquired in the Orbitrap mass analyzer in m/z range of 350–1800 for proteomics and 350–2000 for glycoproteomics at a resolution of 120 000 (at m/z 200). Precursor fragmentation was carried out using higher‐energy collisional dissociation (HCD) method using normalized collision energy (NCE) of 34 for proteomics or stepped HCD (15, 25, and 45) for glycoproteomics. The fragment ions were acquired at a resolution of 30000 (at m/z 200). The scans were acquired in top‐speed method with 3 s cycle time between MS and MS/MS. Ionization of eluting peptides was performed using an EASY‐Spray source kept at an electric potential of 1.9 kV. For internal mass calibration, lock mass option was enabled with polysiloxane ion (m/z, 445.120025) from ambient air.
2.7. Data analysis
The data analysis was performed as described previously. 39 The proteomics data were searched using Sequest node in Proteome Discoverer 2.5 against the UniProt Human Reviewed protein sequences (20 432 entries). We searched the glycoproteomics data using the publicly available software pGlyco version 2.2.2. with an in‐built glycan database containing 8092 entries. 40 Two missed cleavages were allowed for both proteomics and glycoproteomics analysis. Error tolerance for precursor and fragment ions was set to 10 ppm and 0.02 Da, respectively, for proteomics and 5 ppm and 20 ppm for fragment ions, respectively, for glycoproteomics. Cysteine carbamidomethylation was set as fixed modification, whereas oxidation of methionine and deamidation of asparagine and glutamine as variable modification. False discovery rate (FDR) was set to 1% at the peptide‐spectrum matches (PSMs), peptide, protein, and glycopeptides levels. For proteomics, quantitation of peptides across patients and controls was done using TMT reporter ion intensities as surrogates using “reporter ion quantifier” node. In the case of glycopeptide quantitation, reporter ion quantification was performed in Proteome Discoverer version 2.5 and glycopeptide IDs obtained from pGlyco were matched with quantitation on a scan‐to‐scan basis (MS/MS). Statistical analysis was performed using the publically available MetaboAnalyst and Perseus computational platforms. The TMT reporter ion intensities were analyzed using two‐sample Student's t‐test to identify differentially expressed proteins and glycopeptides in NGLY1‐CDDG patients.
2.8. Western blotting
To perform western blotting, 30 μg proteins from both patient and control fibroblasts were separated onto a Novex NuPAGE 10% Bis‐Tris Gel (Thermo Fisher Scientific) according to the manufacturer's instructions. After electrophoresis, the proteins were transferred to nitrocellulose membrane using transfer stacks in the iBlot2 Gel Transfer Device (Invitrogen). The membranes were blocked for 1 h in either sea blocker or milk at room temperature or 4°C. Immunodetection was performed using 1:250 ratio of Rabbit polyclonal anti‐human phospholipid phosphatase 3 (Thermo Fisher Scientific; Cat# PA5‐90665), Rabbit polyclonal anti‐human podocalyxin (Proteintech; Cat# 18150‐1‐AP), Rabbit polyclonal anti‐NGLY1 (Sigma‐Aldrich; Cat# HPA036825) and Goat polyclonal anti‐human hyaluronan and proteoglycan link protein 1 (R&D System; Cat# AF2608) and 1:20 000 ratio of monoclonal mouse anti‐GAPDH (Invitrogen; Cat# AM4300) as an internal control diluted in blocking buffer and the membrane was rocked 48 h at 4°C. Following this, the membrane was incubated with donkey anti‐mouse cross‐absorbed secondary antibody (for GAPDH), DyLight 800 conjugate (1:5000, Thermo Fisher Scientific; Cat# SA5‐10082) and donkey anti‐rabbit cross‐adsorbed secondary antibody, DyLight 680 conjugate (1:5000, Thermo Fisher Scientific; Cat# SA5‐10042) for 1 h at room temperature or 4°C. For HAPLN1, anti‐Donkey HRP (Santa Cruz Biotechnology, Cat# sc‐2020) was used. The membrane was visualized on a Licor Odyssey CLx Infrared Imaging System (LI‐COR Biosciences). Signal intensity was quantified offline by Odyssey software (version 2.0). All measured intensities of protein bands were normalized with GAPDH intensity. One of the control cell lines became compromised after proteomics and glycoproteomics analysis; therefore, western blotting was performed using three controls and four patient fibroblasts.
3. RESULTS
3.1. Study population and experiment pipeline
The four patient‐derived fibroblasts as well as four age‐matched control fibroblasts were analyzed by deep multiplexed glycoproteomics and proteomics (Table S1). Protein was extracted from fibroblasts and subjected to in‐solution proteolytic digestion using trypsin. The resulting peptides from each sample were then labeled with TMT. The pooled labeled sample was split into two aliquots. One aliquot was fractionated using bRPLC into 24 fractions for proteomics. Glycopeptides were enriched from second aliquot using SEC into 24 fractions for glycoproteomics study. All these fractions were then analyzed by LC–MS/MS on an Orbitrap Exploris 480 mass spectrometer. Schematic of the optimized workflow for the multiplexed proteomics and glycoproteomics is presented in Figure 1.
FIGURE 1.
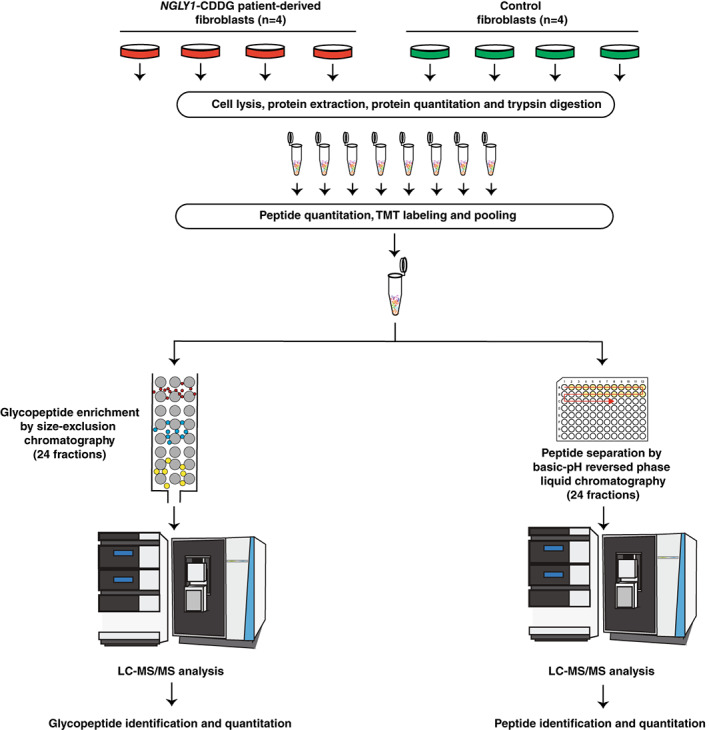
A schematic workflow for glycoproteomic and proteomic analyses of NGLY1‐deficient fibroblasts. The cells from NGLY1‐CDDG (n = 4) and control (n = 4) fibroblasts were lysed, proteins were extracted and quantified using BCA assay. Equal amounts of protein were trypsin digested and peptides were quantified. Equal amounts of peptides were labeled with tandem mass tags (TMTs) prior to pooling. Pooled TMT labeled sample was then split into two aliquots, where one aliquot was subjected to basic reversed‐phase liquid chromatography (bRPLC) for proteomic analysis and the other part was used to enrich glycopeptides using size exclusion chromatography (SEC) for glycoproteomic analysis. Mass spectrometry analysis was done on an Orbitrap Exploris 480 mass spectrometer for both bRPLC and SEC fractions.
3.2. Proteomic alterations in NGLY1‐deficient fibroblasts
Multiplexed proteomics study identified a total of 8041 proteins with 100 092 peptides. No dramatic changes in global protein expression were observed in patient‐derived fibroblasts (Figure 2A). For example, only two proteins (phospholipid phosphatase 3 (PLPP3) and stromal cell‐derived factor 1 (CXCL12)) showed a significant upregulation (>2‐fold; log2 fold‐change>1), while 16 proteins showed a significant decrease in expression in patients‐derived fibroblasts (log2 fold‐change < 1) which included hyaluronan and proteoglycan link protein 1 (HAPLN1), retinal dehydrogenase 2 (ALDH1A2), NGLY1, and synaptopodin‐2. Among proteins that showed a log2 fold change of >0.378 (30% change) and <0.05 p‐value, podocalyxin (PODXL), fibrinogen gamma chain (FGG), plasminogen activator inhibitor 2 (SERPINB2), palmitoyltransferase ZDHHC9, integrin alpha‐4, and raftlin were upregulated while collagen alpha‐1 (IV) chain, leiomodin‐1, latexin, and myosin regulatory light polypeptide 9 were downregulated in patient‐derived NGLY1‐deficient fibroblasts.
FIGURE 2.
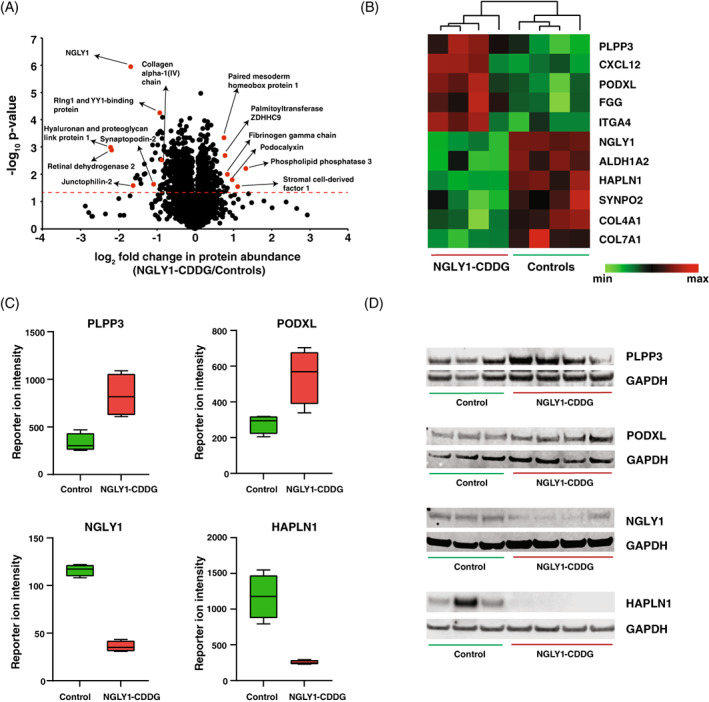
Quantitative proteomic changes in patient‐derived NGLY1‐deficient fibroblasts. (A) Volcano plot depicting the differentially abundant proteins in patient‐derived NGLY1‐deficient fibroblasts. X‐axis is log2 fold change (NGLY1‐CDDG/controls) and Y‐axis is the negative logarithm of p‐value of t‐test for significance. The horizontal dashed red line marks the cutoff for significance (<0.05). Some of the changing proteins are marked in red circles and protein names are provided. Statistical analysis was performed by two‐sample t‐test. (B) Heatmap of 12 abundant proteins showing the differential expression (p < 0.05) in NGLY1‐CDDG patient fibroblasts. The pattern is color coded and protein names are given for each glycopeptide. (C) Box plots showing reporter ion intensities of two upregulated proteins (PLPP3 and PODXL) and two downregulated proteins (NGLY1 and HAPLN1) in NGLY1‐CDDG patient fibroblasts. Y‐axis is the reporter ion intensity of TMT channels. Each dot in the plots represents the individual control or patient sample. (D) Western blots showing the upregulation of PLPP3 and PODXL and the downregulation of NGLY1 and HAPLN1 in patient‐derived NGLY1‐deficient fibroblasts. GAPDH was used as an internal control for each protein.
Complete data with all quantified proteins for each sample is provided in Table S2. The expression pattern of several of the most upregulated and downregulated proteins across both group of samples are shown in a heatmap (Figure 2B). The distributions for PLPP3, PODXL, HALPN1, and NGLY1 are shown in Figure 2C as box plots, where the expression pattern of these four proteins showed no overlap between the NGLY1‐CDDG and controls.
To validate our results obtained by proteomic analysis, we assessed the relative abundance of PODXL, PLPP3, NGLY1, and HAPLN1 in fibroblasts of individuals of NGLY1 and controls by western blotting (Figure 2D). The relative abundance of PLPP3 and PODXL, bands appropriate for the predicted size of the proteins, were both upregulated in NGLY1 individuals compared to controls. The NGLY1 Western blot showed one band corresponding to the most prevalent NGLY1 isoform. The relative NGLY1 protein abundance in individuals with NGLY1 was lower in all affected individuals compared to controls. The relative HAPLN1 protein abundance was decreased in NGLY1 individuals compared to the controls. The western blot analysis of these proteins confirms the mass spectrometry findings.
3.3. NGLY1‐deficient fibroblasts exhibit distinct glycosylation remodeling
We enriched glycopeptides from TMT‐labeled pooled peptides by size exclusion chromatography using the parameters described previously. 36 First 24 fractions were analyzed using LC–MS/MS and data were analyzed by pGlyco 2.2.2. In our global glycoproteomics study of fibroblasts, we identified and quantified 3255 individual N‐glycopeptides with 172 unique glycan compositions on 550 glycosylation sites of 407 glycoproteins. In terms of glycan subtypes, complex glycans were present on >50% of glycosylation sites, followed by high‐mannose (33%) and hybrid glycans (9.5%) (Figure 3A). In addition, about 60% of the glycosylation sites were either fucosylated or sialylated, or both, while the remaining 40% of sites did not have either (Figure 3B). Nearly one‐third of identified N‐glycopeptides (33%) contained high‐mannose glycans, of which ~50% were either Man8 or Man9 types of glycans (Figure 3C). In terms of glycosylation sites, out of 407 identified glycoproteins, 66% glycoproteins had only one glycosylation site and 16% glycoproteins possessed two glycosylation sites, rest of the glycoproteins had three or more glycosylation sites (Figure 3D).
FIGURE 3.
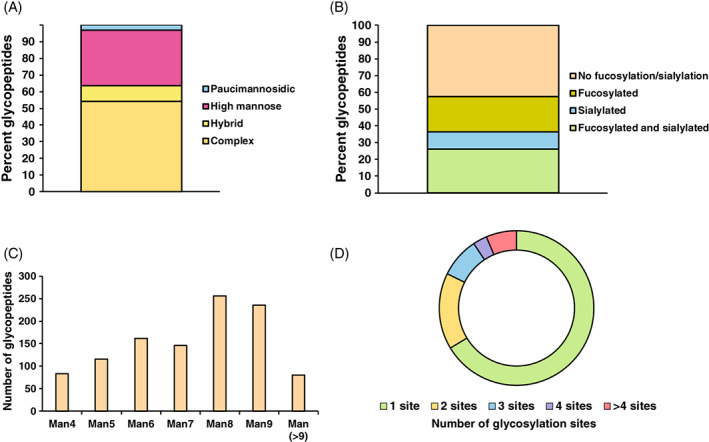
Global view of fibroblast glycosylation. (A) All quantified glycopeptides were classified according to their category of glycans. Y‐axis represents the percent glycopeptides in each category. (B) Percentages are shown for fucosylated, sialylated, both fucosylated and sialylated and not fucosylated or sialylated glycan containing glycopeptides. (C) High‐mannose glycans containing glycopeptides are shown with numbers representing glycopeptides found with increasing hexose units. (D) All glycoproteins are divided into category of number of glycosylation sites detected per glycoprotein
We recently reported an N‐glycoproteomics analysis of patient‐derived phosphomannomutase 2 (PMM2) deficient fibroblasts where a significant reduction of global N‐glycosylation was observed. 39 Similarly, the expectation in this study was that glycopeptides in samples from individuals with NGLY1 deficiency would result in accumulation of glycopeptides. However, we did not observe any global accumulation of glycopeptides in patient‐derived NGLY1‐deficient fibroblasts. Indeed, quantitation of glycopeptide levels revealed subtle changes (Figure 4A) with only ~1% (36 glycopeptides) being significantly upregulated and <3% (89 glycopeptides) significantly downregulated (an absolute log2 fold change of >1 as a cutoff and p < 0.05) in patient‐derived NGLY1‐deficient fibroblasts. Among others, the top upregulated glycopeptides included a high‐mannose glycan containing glycopeptide that belonged to aggrecan core protein and hybrid glycan containing glycopeptides belonged to neprilysin. The top downregulated glycopeptides included a high‐mannose glycan containing glycopeptide thyrotropin‐releasing hormone‐degrading ectoenzyme and hybrid glycan containing glycopeptide belonged to HAPLN1. Partial Least Squares Discriminant Analysis (PLS‐DA) revealed separation of NGLY1‐CDDG and control fibroblasts at the glycopeptide level (Figure 4B).
FIGURE 4.
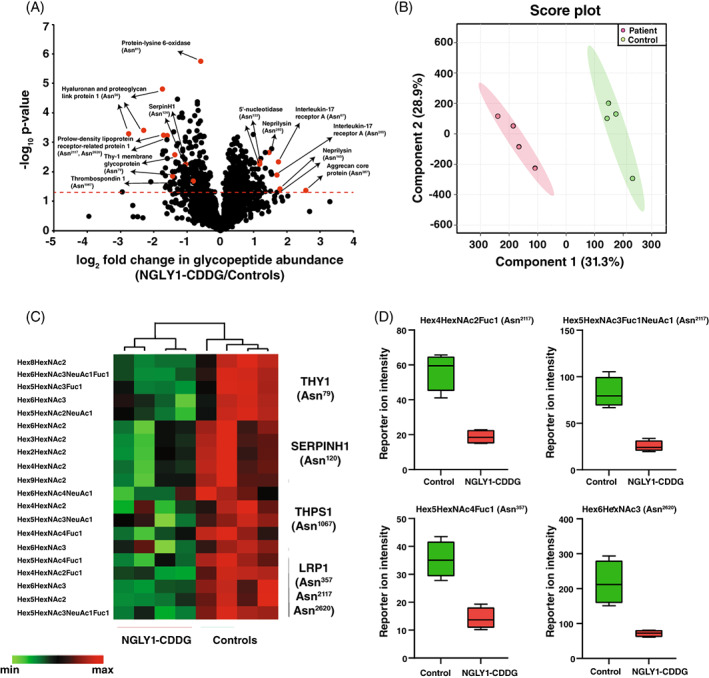
Glycoproteome alterations in patient‐derived NGLY1‐deficient fibroblasts. (A) Volcano plot of global glycoproteomics of patient‐derived NGLY1‐deficient fibroblasts and controls. X‐axis is log2 fold change (NGLY1‐CDDG/controls) and Y‐axis is the negative logarithm of p‐value of t‐test for significance. The horizontal dashed red line marks the cutoff for significance (<0.05). Some of the changing glycopeptides are marked in red circles and glycoproteins' names and glycosylation sites are drawn. Statistical analysis was performed by two‐sample t‐test. (B) Partial Least Squares Discriminant Analysis (PLS‐DA) based on reporter ion intensities for all identified glycopeptides of NGLY1‐CDDG patients and controls. The percentage of total variance associated with each component is shown in brackets with the axis label. (C) Heatmap of selected 20 glycopeptides illustrating downregulation (p < 0.05) in NGLY1‐CDDG patient fibroblasts from four different glycoproteins; thy‐1 membrane glycoprotein (THY1), Serpin H1 (SERPINH1), thrombospondin 1 (THBS1), and prolow‐density lipoprotein receptor‐related protein 1 (LRP1). The pattern is color‐coded and protein names with site/s of glycosylation are given for each glycopeptide. The composition of each glycan for their respective glycopeptide is also given. (D) Box plots showing downregulation in four representative glycopeptides of prolow‐density lipoprotein receptor‐related protein 1 (LRP1). Y‐axis is the reporter ion intensity of TMT channels. The glycan compositions on each glycosylation site are also shown on the top of box plots. Fuc, Fucose; Hex, Hexose; HexNAc, N‐acetylhexosamine; NeuAc, N‐acetylneuraminic acid.
After imposing a cutoff value of 30% change, 261 glycopeptides were found to be upregulated, whereas 247 were downregulated. The glycopeptides that exhibited the greatest increase in patient fibroblasts were derived from neprilysin (NEP; 11 glycopeptides from two glycosylation sites), interleukin‐17 receptor A (IL17RA; 6 glycopeptides from two glycosylation sites), laminin subunit alpha‐4 (LAMA4; 5 glycopeptides from two glycosylation sites), integrin alpha‐4 (ITA4; 5 glycopeptides from two glycosylation sites), 5′‐nucleotidase (5NTD; 20 glycopeptides from three glycosylation sites), CD166 (CD166; 13 glycopeptides from six glycosylation sites), and CD109 (CD109; 10 glycopeptides from four glycosylation sites). The expression of IL17RA, LAMA4, and CD109 was not altered at the protein level. The glycopeptides with the greatest reduction in patient fibroblasts were derived from hyaluronan and proteoglycan link protein 1 (HAPLN1; 10 glycopeptides from one glycosylation site), lactadherin (MFGM; 7 glycopeptides from one glycosylation site), prolow‐density lipoprotein receptor‐related protein 1 (LRP1; 19 glycopeptides from six glycosylation sites), thy‐1 membrane glycoprotein (THY1; 62 glycopeptides from two glycosylation sites), thrombospondin‐1 (THBS1; 10 glycopeptides from two glycosylation sites), serpin H1 (SERPINH1; 9 glycopeptides from one glycosylation site) and lysosome‐associated membrane glycoprotein 1 (LAMP1; 18 glycopeptides from two glycosylation sites). The expression of LRP1, THY1, SERPINH1, and LAMP1 was not altered at the protein level, while the abundance of other proteins was also accompanied by a reduction at the protein level. Some of the significantly downregulated glycopeptides are shown in Figure 4C. The reduced abundance of four different glycopeptides derived from LRP1 is shown as boxplots in Figure 4D. A list of top 20 upregulated and downregulated glycopeptides with the attached glycan composition and glycosylation site information is provided in Tables 1 and 2, respectively. A list of all identified glycopeptides with their glycan composition and quantitation values is given in Table S3.
TABLE 1.
Top significantly upregulated glycopeptides (p < 0.05) in patient‐derived NGLY1‐deficient fibroblasts. Protein names, gene symbols, the peptide sequence, the site of glycosylation, glycan composition, and log2 fold change of these glycopeptides are given.
| Protein name | Gene symbol | Peptide | Glycosylation site in protein | Glycan composition | log2 fold change (NGLY1/Control) |
|---|---|---|---|---|---|
| Aggrecan core protein | PGCA | NITEGEAR | 387 | Hex9HexNAc2 | 2.57 |
| Neprilysin | NEP | EIANATAKPEDR | 285 | Hex4HexNAc3 | 1.79 |
| Neprilysin | NEP | EIANATAKPEDR | 285 | Hex5HexNAc4NeuAc1 | 1.78 |
| Interleukin‐17 receptor A | IL17RA | NLTPSSPK | 67 | Hex9HexNAc2 | 1.74 |
| Interleukin‐17 receptor A | IL17RA | SNVTLTLR | 265 | Hex8HexNAc2 | 1.70 |
| Interleukin‐17 receptor A | IL17RA | NLTPSSPK | 67 | Hex8HexNAc2 | 1.54 |
| Neprilysin | NEP | VMELEKEIANATAKPEDR | 285 | Hex5HexNAc4NeuAc1Fuc1 | 1.52 |
| Neprilysin | NEP | SCINESAIDSR | 145 | Hex5HexNAc4 | 1.50 |
| Neprilysin | NEP | EIANATAKPEDR | 285 | Hex4HexNAc3Fuc1 | 1.46 |
| Interleukin‐17 receptor A | IL17RA | NLTPSSPK | 67 | Hex6HexNAc2 | 1.35 |
| Laminin subunit alpha‐4 | LAMA4 | LITEEANR | 742 | Hex9HexNAc2 | 1.35 |
| Neprilysin | NEP | SCINESAIDSR | 145 | Hex4HexNAc3 | 1.34 |
| Interleukin‐17 receptor A | IL17RA | NLTPSSPK | 67 | Hex7HexNAc2 | 1.32 |
| Neprilysin | NEP | SCINESAIDSR | 145 | Hex8HexNAc2 | 1.26 |
| Integrin alpha‐4 | ITA4 | QPGENGSIVTCGHR | 138 | Hex4HexNAc4Fuc1 | 1.26 |
| Interleukin‐17 receptor A | IL17RA | SNVTLTLR | 265 | Hex9HexNAc2 | 1.22 |
| 5′‐nucleotidase | 5NTD | IKLDNYSTQELGK | 333 | Hex3HexNAc3Fuc1 | 1.20 |
| Integrin alpha‐4 | ITA4 | QPGENGSIVTCGHR | 138 | Hex5HexNAc4Fuc1 | 1.19 |
| Neprilysin | NEP | SCINESAIDSR | 145 | Hex5HexNAc4NeuAc1 | 1.18 |
| Integrin alpha‐4 | ITA4 | QPGENGSIVTCGHR | 138 | Hex4HexNAc3Fuc1 | 1.17 |
Abbreviations: Fuc, Fucose; Hex, Hexose; HexNAc, N‐acetylhexosamine; NeuAc, N‐acetylneuraminic acid.
TABLE 2.
Top significantly downregulated glycopeptides (p < 0.05) in patient‐derived NGLY1‐deficient fibroblasts. Protein names, gene symbols, the peptide sequence, the site of glycosylation, glycan composition, and log2 fold change of these glycopeptides are given.
| Protein name | Gene symbol | Peptide | Glycosylation site in protein | Glycan composition | log2 fold change (NGLY1/Control) |
|---|---|---|---|---|---|
| Thyrotropin‐releasing hormone‐degrading ectoenzyme | TRHDE | GGNGSLPGSAR | 89 | Hex8HexNAc2 | −2.95 |
| Hyaluronan and proteoglycan link protein 1 | HAPLN1 | GGNVTLPCK | 56 | Hex5HexNAc4Fuc1 | −2.76 |
| Hyaluronan and proteoglycan link protein 1 | HAPLN1 | GGNVTLPCK | 56 | Hex5HexNAc4NeuAc1Fuc1 | −2.31 |
| Dickkopf‐related protein 3 | DKK3 | GSNGTICDNQR | 204 | Hex9HexNAc2 | −2.09 |
| Hyaluronan and proteoglycan link protein 1 | HAPLN1 | GGNVTLPCK | 56 | Hex9HexNAc2 | −1.75 |
| Lactadherin | MFGM | NNSIPDK | 238 | Hex7HexNAc2 | −1.75 |
| Platelet‐derived growth factor receptor beta | PGFRB | NVSETR | 371 | Hex4HexNAc3Fuc1 | −1.74 |
| Lactadherin | MFGM | NNSIPDK | 238 | Hex6HexNAc2 | −1.72 |
| Prolow‐density lipoprotein receptor‐related protein 1 | LRP1 | THANGSIK | 2117 | Hex5HexNAc3NeuAc1Fuc1 | −1.71 |
| Lactadherin | MFGM | NNSIPDK | 238 | Hex6HexNAc3 | −1.66 |
| Basement membrane‐specific heparan sulfate proteoglycan core protein | PGBM | NLHQSNTSR | 1755 | Hex3HexNAc3Fuc1 | −1.65 |
| Prolow‐density lipoprotein receptor‐related protein 1 | LRP1 | DGTCIGNSSR | 2620 | Hex5HexNAc2 | −1.65 |
| Lactadherin | MFGM | NNSIPDK | 238 | Hex8HexNAc2 | −1.65 |
| C‐type mannose receptor 2 | MRC2 | NASLAYVPDPYTQAFLTQAAR | 1157 | Hex5HexNAc4Fuc1 | −1.64 |
| Hyaluronan and proteoglycan link protein 1 | HAPLN1 | GGNVTLPCK | 56 | Hex5HexNAc4Fuc2 | −1.60 |
| Prolow‐density lipoprotein receptor‐related protein 1 | LRP1 | DGTCIGNSSR | 2620 | Hex6HexNAc3 | −1.60 |
| Prolow‐density lipoprotein receptor‐related protein 1 | LRP1 | THANGSIK | 2117 | Hex4HexNAc2Fuc1 | −1.59 |
| Basement membrane‐specific heparan sulfate proteoglycan core protein | PGBM | NLHQSNTSR | 1755 | Hex4HexNAc3Fuc1 | −1.53 |
| CD276 antigen | CD276 | DQGSAYANR | 91 | Hex5HexNAc4Fuc2 | −1.52 |
| Platelet‐derived growth factor receptor beta | PGFRB | NVSETR | 371 | Hex5HexNAc4NeuAc1Fuc1 | −1.50 |
Abbreviations: Fuc, Fucose; Hex, Hexose; HexNAc, N‐acetylhexosamine; NeuAc, N‐acetylneuraminic acid.
3.4. Glycosylation alterations in glycoproteins implicated in several extracellular matrix proteins and intercellular adhesion molecules
We identified several differentially expressed extracellular matrix proteins and intracellular adhesion molecules with widespread site‐specific aberrant glycosylation pattern in patient‐derived NGLY1‐deficient fibroblasts. The top increased glycopeptide (~6‐fold) containing a high‐mannose glycan (Hex9HexNAc2) belonged to aggrecan core protein, a proteoglycan which is a major component of extracellular matrix (ECM) and aggregates with hyaluronan. 41 Aggrecan also plays an important role in the organization of the neural extracellular space by binding and organizing hyaluronan to the cell surface. 42 The abundance of aggrecan core protein was also increased at the protein level (2.8‐fold); however, the difference was not significant (p = 0.11). Another glycoprotein with a global increase in glycosylation was neprilysin. Among the top 20 upregulated glycopeptides in patient fibroblasts, 8 glycopeptides belonged to NEP on two different glycosylation sites (Asn145 and Asn285). The relative abundance of four glycopeptides from NEP is showed as boxplots in Figure 5A. The glycosylation of laminin subunit alpha‐4 (LAMA4) was significantly increased for five glycopeptides on three glycosylation sites (Asn215, Asn742, and Asn787), while the protein levels of LAMA4 were not altered between NGLY1‐CDDG patients and controls. All the increased five glycopeptides were high‐mannose containing peptides. The expression of 10 glycopeptides of CD109 on four different sites was increased in patients; out of which, 6 glycopeptides were fucosylated and 4 were both fucosylated and sialylated. Interestingly, the protein level of LAMA4, CD109, and NEP was not significantly changed in patient fibroblasts. Another cell membrane protein, CD166 also showed increase in glycosylation. We observed increase in 13 unique glycopeptides on six different glycosylation sites. Surprisingly, all the glycopeptides were fucosylated, out of which eight glycopeptides were both fucosylated and sialylated. The relative abundance of four glycopeptides from NEP is showed as boxplots in Figure 5B.
FIGURE 5.
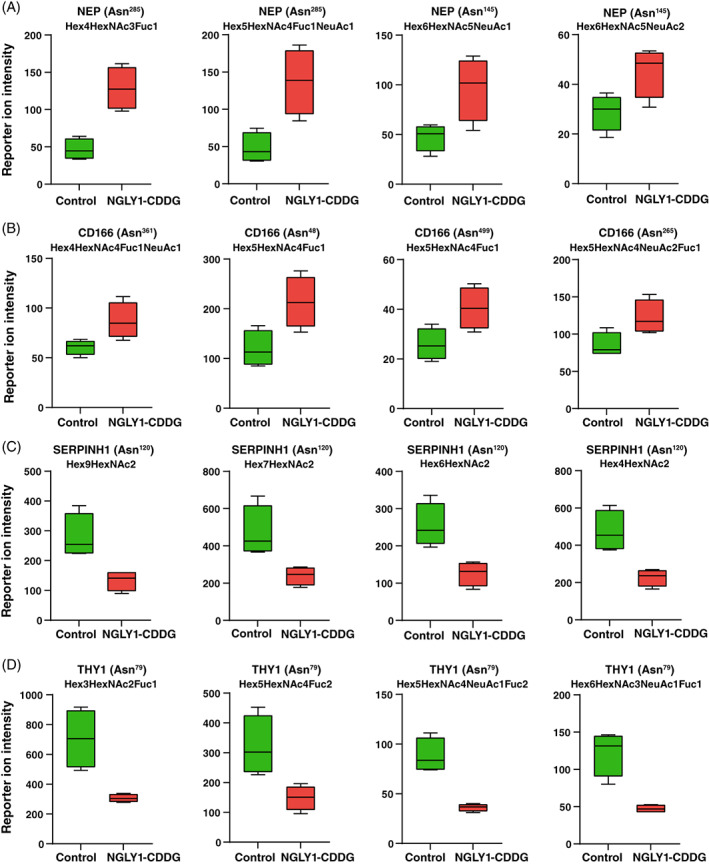
Distinct glycosylation in extracellular matrix and cell adhesion proteins. Box plots showing reporter ion intensities of individual glycopeptides in controls and NGLY1‐CDDG patients for different extracellular matrix and cell adhesion proteins; (A) CD166, (B) neprilysin (NEP), (C) Serpin H1 (SERPINH1), and (D) thy‐1 membrane glycoprotein (THY1). NGLY1‐CDDG samples reflected the increased glycosylation in CD166 and NEP, whereas reduction in glycosylation was observed in SERPINH1 and THY1. Protein names, glycosylation sites, and glycan compositions are marked on the top of box plots and X‐axis depicts sample type while Y‐axis is reporter ion intensity at the glycopeptide level. Fuc, Fucose; Hex, Hexose; HexNAc, N‐acetylhexosamine; NeuAc, N‐acetylneuraminic acid.
We also identified several other extracellular matrix proteins and intracellular adhesion molecules which were significantly downregulated in the patient fibroblasts. One of the ECM‐related glycoproteins with the reduced glycosylation was serpin H1 (SERPINH1). We identified 15 unique glycopeptides for SERPINH1 on a single glycosylation site (Asn120), out of which expression of 9 glycopeptides was reduced in patient fibroblasts while expression of remaining 6 glycopeptides was not altered. Of the glycopeptides that were reduced in abundance, six were high‐mannose containing glycopeptides ranging from Man5 to Man9, whereas two glycopeptides were truncated (Hex2HexNAc2 and Hex3HexNAc2). The relative abundance of four glycopeptides from SERPINH1 is showed as boxplots in Figure 5C. The other downregulated protein was HAPLN1, where the expression of 10 glycopeptides was reduced on one glycosylation site (Asn56), of which 7 glycopeptides were fucosylated. Two glycans with the composition Hex5HexNAc4Fuc1 and Hex5HexNAc4NeuAc1Fuc1 were reduced in patients with >2‐fold‐change. The glycosylation on thy‐1 membrane glycoprotein (THY1) was dramatically reduced in patients, where we observed decrease in 62 unique glycopeptides on two different glycosylation sites (Asn42 and Asn79). Of note, the expression of THY1 was not altered at the protein level in patient fibroblasts. The site Asn79 was occupied with 53 unique glycopeptides, while Asn42 was occupied with 9 unique glycopeptides. Out of these 62 glycopeptides, 19 were reduced by>2‐fold and 30 glycopeptides were reduced by > 1.5‐fold. Notably, 43 glycopeptides were fucosylated, while 28 glycopeptides were sialylated. There were 19 glycopeptides which were both fucosylated and sialylated. Only three glycopeptides of THY1 contained high‐mannose sugar chains. The relative abundance of four glycopeptides from THY1 is showed as boxplots in Figure 5D.
4. DISCUSSION
The NGLY1 enzyme is mainly involved in cleaving the N‐glycans from misfolded, retro‐translocated glycoproteins in the cytosol from the endoplasmic reticulum before their proteasomal degradation or activation. Despite the hypothesis that NGLY1 deficiency should lead to accumulation of misfolded glycoproteins, the exact consequences of NGLY1 deficiency on global cellular protein glycosylation have not yet been investigated.
We performed multiplexed glycoproteomics and proteomics in four patient‐derived fibroblast cell lines carrying different biallelic pathogenic variants in NGLY1. We found no significant accumulation of glycoproteins in the NGLY1‐deficient fibroblasts. However, we detected distinct changes in a subset of glycoproteins. To the best of our knowledge, this is the first report describing profiling of the glycoproteins in patient‐derived NGLY1‐deficient fibroblasts which provides evidence of alteration in glycosylation on fibroblast glycoproteins in NGLY1‐CDDG.
Global proteomics analysis identified only a few proteins that were altered in patient‐derived NGLY1‐CDDG fibroblasts. The most upregulated protein PLPP3 is a magnesium‐independent transmembrane glycoprotein that catalyzes the dephosphorylation of phosphates of sphingolipids and glycerolipids. 43 PLPP3 has been demonstrated to influence cell signaling, cell migration, adhesion, and survival. 44 , 45 PODXL is also cell membrane‐associated glycoprotein with high levels of sialic acids that are expressed abundantly in kidney glomeruli. 46 It was shown that PODXL regulates the cell adhesion through integrin‐dependent activity and the expression of PODXL has enhanced the migration of cells onto extracellular ligands and intracellular interactions with vascular endothelial cells. 47 Both PLPP3 and PODXL were increased in NGLY1‐CDDG fibroblast. The level of HAPLN1 was reduced in NGLY1‐CDDG. It is a linker protein, which links proteoglycans and hyaluronan., 48 potentially imparting the cells with compression resistance. HAPLN1 stabilizes the proteoglycan‐hyaluronan structures 49 and its loss in transgenic animals leads to chondrodysplasia and loss of perineuronal nets (PNNs), particularly at calyx of held, and persistent plasticity. 50 , 51 , 52 PNNs have been shown to be important in synaptic transmission of neurons, including those at calyx of held. Interestingly, auditory neuropathy has been described in NGLY1‐CDDG patients, which leads to neurotransmission‐related dysfunctions. 53 We identified several differentially expressed extracellular matrix proteins and intracellular adhesion molecules with widespread site‐specific aberrant glycosylation pattern in patient‐derived NGLY1‐deficient fibroblasts. Proteins exhibiting upregulation of glycosylation included NEP, LAMA4, and CD109 in individuals with NGLY1 deficiency. NEP is an integral membrane zinc metallopeptidase with the active site facing the ECM. 54 NEP is known to cleave extracellular amyloid‐β (Aβ) and it has been shown that the expression of NEP is reduced in Alzheimer's disease. 55 , 56 The transport of NEP to cell surface and its activity has been shown to be significantly impacted by glycosylation alterations. 57 LAMA4 is a part of ECM and plays a significant role in promoting proliferation, migration, differentiation, and survival of various cell types. 58 CD109 is a glycosylphosphatidylinositol (GPI)‐anchored protein that is present on the cell surface and differentially regulates several ECM proteins. 59 , 60 Several other proteins also showed the downregulation at their glycosylation levels such as SERPINH, THY1, and HAPLN1. SERPINH1, normally resides in the endoplasmic and plays a role in collagen biosynthesis and its folding as a molecular chaperone. 61 , 62 The protein level of SERPINH1 was not altered. THY1, also known as CD90, is a heavily glycosylated protein with a GPI anchor, which is present on the cell membrane of several cell types including fibroblasts. 63 Several biological functions have been ascribed to THY1 including a role in T cell function, intracellular adhesion, and extracellular signaling. 64 , 65 HAPLN1 is an important ECM component that stabilizes other ECM proteins. 66 HAPLN1 is proven to be key component of the brain ECM and for neuronal function and plasticity. 67 The expression of HAPLN1 was also reduced to ~80% at the protein level.
The extracellular matrix is composed of various glycoproteins and proteoglycans which are essential for cell–cell interaction, cell proliferation, migration, and differentiation. ECM proteins are known to be important for maintaining homeostasis and regulating skeletal muscle. 68 , 69 ECM proteins are involved in the development of skeletal muscles in different ages. 69 The deficiency of NGLY1 leads to deformities in skeletal muscle and develops abnormalities in central and peripheral nervous system, 70 , 71 additionally cells undergo abnormal glycosylation. Therefore, in the absence of NGLY1, the changes in ECM protein glycosylation may modulate phenotypic characteristics in the cells. In a recent study, transcriptomics and proteomics study was performed in patient‐derived NGLY1‐deficient fibroblasts and lymphoblastoid cells. 34 In their study, NGLY1 protein was not detected in fibroblasts cell, although it was downregulated from 2.5‐ to 9.5‐fold in lymphoblastoid cells. They also found that several proteasomal subunits, proteins associated with ERAD and ribosomal mRNA processing were differentially regulated in patients. In our study, we found downregulation of several proteasomal subunits such as PSMD3, PSMD6, PSMD11, PSMC2, and PSMB5, although the magnitude of change was only ~10%–20%. Similar to their study, we also observed a small decrease (~15%) in ERAD‐associated proteins, NPLOC4, and VCP patient fibroblasts. Their study reported a small increase in proteins associated with ribosomal mRNA processing and small decrease in proteins associated with glutathione metabolism in patient fibroblasts. In our data, the levels of these proteins were comparable and no significant difference was observed.
The diagnosis is challenging as the common diagnostic test; the transferrin glycoform analysis (also called carbohydrate‐deficient transferrin; CDT analysis), used in glycosylation disorders in blood does not show any changes in this group of patients. Clinical differential diagnosis is often challenging and CDDG is often misdiagnosed as a mitochondrial disease. So far, most of the reported patients have been diagnosed by whole exome sequencing, and frequent genetic variants were confirmed based on loss of NGLY1 function, 30 however, whole‐exome sequencing lacks specificity. 30
Similar to other studies, 19 , 72 we also detected reduced levels of NGLY1 protein in fibroblasts, which could be used in as an additional diagnostic tool to confirm the disease in skin fibroblasts. Mueller et al. have recently reported elevated N‐acetylglucosamine‐asparagine (GlcNAc‐Asn; GNA) levels that can differentiate patients from controls, are stable over time, and correlate with changes in NGLY1 activity. 32 GNA as a biomarker has shown a good potential to identify patients with NGLY1 deficiency. In clinical practice, however in individuals with variants of uncertain significance or carrying likely pathogenic variants, the lack of a broadly available clinical test still significantly hampers diagnostic success.
5. CONCLUSIONS
In summary, we provide evidence for alterations in protein and glycoprotein expression in fibroblasts from NGLY1‐CDDG patients. Contrary to expectations, we could not observe any significant accumulation of glycoproteins in patient fibroblasts suggesting NGLY1‐CDDG may be more complex than currently believed. This study provides evidence that deglycosylation may not be essential for the proteasomal degradation of retro‐translocated glycoproteins from the endoplasmic reticulum. Interestingly, patient‐derived NGLY1‐deficient fibroblasts possess aberrant glycosylation pattern for several extracellular matrix proteins and cell adhesion molecules while the expression of most of these proteins was comparable between patients and controls. The differentially expressed proteins and glycan compositions on different glycopeptides could be further pursued to identify additional predictive biomarkers that can be used for early diagnosis and treatment of NGLY1‐CDDG. Additionally, the presented blueprint of quantitative cellular glycosylation will increase our general understanding NGLY1‐CDDG and lead to new lines of investigation and targeted therapies.
AUTHOR CONTRIBUTIONS
Rohit Budhraja, Tamas Kozicz, Akhilesh Pandey, and Eva Morava were involved in the planning of the research and designing of the study. Rohit Budhraja, Diederik De Graef, and Wasantha Ranatunga performed the experiments. Jehan Mousa worked on the clinical data of all patients. Rohit Budhraja, Mayank Saraswat, Akhilesh Pandey, and Eva Morava analyzed the data and interpreted the data. Rohit Budhraja, Mayank Saraswat, Madan G. Ramarajan, and Diederik De Graef drafted the manuscript. All authors were involved in involved in the reviewing and editing the manuscript.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ETHICS STATEMENT
This study was approved by the Institutional Review Board (IRB) of Mayo Clinic. Studies in fibroblasts were done under an IRB‐approved study (IRB: 16‐004682).
Supporting information
TABLE S1 This table demonstrates the different pathogenic variants in NGLY1‐CDDG patients
TABLE S2 All identified proteins with their quantitation value with TMT channel name, fold changes and p‐values, and their logarithmic values
TABLE S3 LC–MS/MS data for all glycopeptides showing their glycan composition, gene name, glycosylation site, quantitation values for each sample with TMT channel name, fold changes and p‐values, and their logarithmic values
ACKNOWLEDGMENTS
RB, EM, TK, and WR received funding from the grant titled Frontiers in Congenital Disorders of Glycosylation (1U54NS115198‐01) from the National Institute of Neurological Diseases and Stroke (NINDS), National Institute of Child Health and Human Development (NICHD) and the National Center for Advancing Translational Sciences (NCATS), and the Rare Disorders Clinical Research Network (RDCRN), at the National Institute of Health. The authors also thank the Mayo Clinic DERIVE Office and Mayo Clinic Center for Biomedical Discovery for financial support. DDG was supported by a Fellowship of the Belgian American Educational Foundation.
Budhraja R, Saraswat M, De Graef D, et al. N‐glycoproteomics reveals distinct glycosylation alterations in NGLY1‐deficient patient‐derived dermal fibroblasts. J Inherit Metab Dis. 2023;46(1):76‐91. doi: 10.1002/jimd.12557
Communicating Editor: Jaak Jaeken
Funding information National Institute of Neurological Disorders and Stroke, Grant/Award Number: 1U54NS115198‐01; Belgian American Educational Foundation
Contributor Information
Akhilesh Pandey, Email: pandey.akhilesh@mayo.edu.
Eva Morava, Email: morava-kozicz.eva@mayo.edu.
DATA AVAILABILITY STATEMENT
The mass spectrometry proteomics data have been deposited with the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD034364.
REFERENCES
- 1. Chang IJ, He M, Lam CT. Congenital disorders of glycosylation. Ann Transl Med. 2018;6(24):477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cirnigliaro L, Bianchi P, Sturiale L, et al. COG6‐CDG: novel variants and novel malformation. Birth Defects Res. 2022;114(5‐6):165‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jaeken J, Hennet T, Matthijs G, Freeze HH. CDG nomenclature: time for a change! Biochim Biophys Acta. 2009;1792(9):825‐826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ferreira CR, Rahman S, Keller M, Zschocke J. An international classification of inherited metabolic disorders (ICIMD). J Inherit Metab Dis. 2021;44(1):164‐177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Freeze HH, Chong JX, Bamshad MJ, Ng BG. Solving glycosylation disorders: fundamental approaches reveal complicated pathways. Am J Hum Genet. 2014;94(2):161‐175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Francisco R, Marques‐da‐Silva D, Brasil S, et al. The challenge of CDG diagnosis. Mol Genet Metab. 2019;126(1):1‐5. [DOI] [PubMed] [Google Scholar]
- 7. Lipiński P, Bogdańska A, Tylki‐Szymańska A. Congenital disorders of glycosylation: prevalence, incidence and mutational spectrum in the Polish population. Mol Genet Metab Rep. 2021;27:100726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Need AC, Shashi V, Hitomi Y, et al. Clinical application of exome sequencing in undiagnosed genetic conditions. J Med Genet. 2012;49(6):353‐361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huang C, Harada Y, Hosomi A, et al. Endo‐β‐N‐acetylglucosaminidase forms N‐GlcNAc protein aggregates during ER‐associated degradation in Ngly1‐defective cells. Proc Natl Acad Sci U S A. 2015;112(5):1398‐1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Maynard JC, Fujihira H, Dolgonos GE, Suzuki T, Burlingame AL. Cytosolic N‐GlcNAc proteins are formed by the action of endo‐β‐N‐acetylglucosaminidase. Biochem Biophys Res Commun. 2020;530(4):719‐724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Suzuki T, Huang C, Fujihira H. The cytoplasmic peptide: N‐glycanase (NGLY1)—structure, expression and cellular functions. Gene. 2016;577(1):1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Galeone A, Adams JM, Matsuda S, et al. Regulation of BMP4/Dpp retrotranslocation and signaling by deglycosylation. eLife. 2020;9:e55596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tsujita T, Peirce V, Baird L, et al. Transcription factor Nrf1 negatively regulates the cystine/glutamate transporter and lipid‐metabolizing enzymes. Mol Cell Biol. 2014;34(20):3800‐3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim J, Xing W, Wergedal J, Chan JY, Mohan S. Targeted disruption of nuclear factor erythroid‐derived 2‐like 1 in osteoblasts reduces bone size and bone formation in mice. Physiol Genomics. 2010;40(2):100‐110. [DOI] [PubMed] [Google Scholar]
- 15. Lee CS, Lee C, Hu T, et al. Loss of nuclear factor E2‐related factor 1 in the brain leads to dysregulation of proteasome gene expression and neurodegeneration. Proc Natl Acad Sci U S A. 2011;108(20):8408‐8413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tomlin FM, Gerling‐Driessen UI, Liu Y‐C, et al. Inhibition of NGLY1 inactivates the transcription factor Nrf1 and potentiates proteasome inhibitor cytotoxicity. ACS Cent Sci. 2017;3(11):1143‐1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Owings KG, Lowry JB, Bi Y, Might M, Chow CY. Transcriptome and functional analysis in a drosophila model of NGLY1 deficiency provides insight into therapeutic approaches. Hum Mol Genet. 2018;27(6):1055‐1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang K, Huang R, Fujihira H, Suzuki T, Yan N. N‐glycanase NGLY1 regulates mitochondrial homeostasis and inflammation through NRF1. J Exp Med. 2018;215(10):2600‐2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tambe MA, Ng BG, Freeze HH. N‐Glycanase 1 transcriptionally regulates aquaporins independent of its enzymatic activity. Cell Rep. 2019;29(13):4620‐4631.e4624. [DOI] [PubMed] [Google Scholar]
- 20. Na HJ, Abramowitz LK, Hanover JA. Cytosolic O‐GlcNAcylation and PNG1 maintain Drosophila gut homeostasis by regulating proliferation and apoptosis. PLoS Genet. 2022;18(3):e1010128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lipari Pinto P, Machado C, Janeiro P, et al. NGLY1 deficiency—a rare congenital disorder of deglycosylation. JIMD Rep. 2020;53(1):2‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pandey A, Jafar‐Nejad H. Tracing the NGLY1 footprints: insights from drosophila. J Biochem. 2022;171(2):153‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dabaj I, Sudrié‐Arnaud B, Lecoquierre F, et al. NGLY1 deficiency: a rare newly described condition with a typical presentation. Life. 2021;11(3):187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lam C, Wolfe L, Need A, Shashi V, Enns G. NGLY1‐related congenital disorder of deglycosylation. GeneReviews. 2018. https://www.ncbi.nlm.nih.gov/books/NBK481554/. Accessed Feburary 8, 2018. [Google Scholar]
- 25. Heeley J, Shinawi M. Multi‐systemic involvement in NGLY1‐related disorder caused by two novel mutations. Am J Med Genet A. 2015;167(4):816‐820. [DOI] [PubMed] [Google Scholar]
- 26. Fujihira H, Masahara‐Negishi Y, Tamura M, et al. Lethality of mice bearing a knockout of the Ngly1‐gene is partially rescued by the additional deletion of the Engase gene. PLoS Genet. 2017;13(4):e1006696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lipiński P, Cielecka‐Kuszyk J, Socha P, Tylki‐Szymańska A. Liver involvement in NGLY1 congenital disorder of deglycosylation. Pol J Pathol. 2020;71(1):66‐68. [DOI] [PubMed] [Google Scholar]
- 28. Rios‐Flores IM, Bonal‐Pérez MÁ, Castellanos‐González A, et al. Acute liver failure in a male patient with NGLY1‐congenital disorder of deglycosylation. Eur J Med Genet. 2020;63(8):103952. [DOI] [PubMed] [Google Scholar]
- 29. Stuut T, Popescu O, Oviedo A. N‐Glycanase 1 deficiency is a rare cause of pediatric neurodegeneration with neuronal inclusions and liver steatosis. Cureus. 2021;13(10):e19126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hirayama H, Tachida Y, Seino J, Suzuki T. A method for assaying peptide: N‐glycanase/N‐glycanase 1 activities in crude extracts using an N‐glycosylated cyclopeptide. Glycobiology. 2022;32(2):110‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hall PL, Lam C, Alexander JJ, et al. Urine oligosaccharide screening by MALDI‐TOF for the identification of NGLY1 deficiency. Mol Genet Metab. 2018;124(1):82‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mueller WF, Zhu L, Tan B, et al. GlcNAc‐Asn is a biomarker for NGLY1 deficiency. J Biochem. 2022;171(2):177‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Haijes HA, Prinsen HC, Willems AP, et al. Aspartylglycosamine is a biomarker for NGLY1‐CDDG, a congenital disorder of deglycosylation. Mol Genet Metab. 2019;127(4):368‐372. [DOI] [PubMed] [Google Scholar]
- 34. Rauscher B, Mueller WF, Clauder‐Münster S, et al. Patient‐derived gene and protein expression signatures of NGLY1 deficiency. J Biochem. 2022;171(2):187‐199. [DOI] [PubMed] [Google Scholar]
- 35. Mun DG, Renuse S, Saraswat M, et al. PASS‐DIA: a data‐independent acquisition approach for discovery studies. Anal Chem. 2020;92(21):14466‐14475. [DOI] [PubMed] [Google Scholar]
- 36. Saraswat M, Garapati K, Mun DG, Pandey A. Extensive heterogeneity of glycopeptides in plasma revealed by deep glycoproteomic analysis using size‐exclusion chromatography. Mol Omics. 2021;17(6):939‐947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Saraswat M, Mangalaparthi KK, Garapati K, Pandey A. TMT‐based multiplexed quantitation of N‐glycopeptides reveals glycoproteome remodeling induced by oncogenic mutations. ACS Omega. 2022;7(13):11023‐11032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chavan S, Mangalaparthi KK, Singh S, et al. Mass spectrometric analysis of urine from COVID‐19 patients for detection of SARS‐CoV‐2 viral antigen and to study host response. J Proteome Res. 2021;20(7):3404‐3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ligezka AN, Radenkovic S, Saraswat M, et al. Sorbitol is a severity biomarker for PMM2‐CDG with therapeutic implications. Ann Neurol. 2021;90(6):887‐900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu MQ, Zeng WF, Fang P, et al. pGlyco 2.0 enables precision N‐glycoproteomics with comprehensive quality control and one‐step mass spectrometry for intact glycopeptide identification. Nat Commun. 2017;8(1):438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kiani C, Chen L, Wu YJ, Yee AJ, Yang BB. Structure and function of aggrecan. Cell Res. 2002;12(1):19‐32. [DOI] [PubMed] [Google Scholar]
- 42. Morawski M, Brückner G, Arendt T, Matthews RT. Aggrecan: beyond cartilage and into the brain. Int J Biochem Cell Biol. 2012;44(5):690‐693. [DOI] [PubMed] [Google Scholar]
- 43. Brindley DN, Waggoner DW. Mammalian lipid phosphate phosphohydrolases. J Biol Chem. 1998;273(38):24281‐24284. [DOI] [PubMed] [Google Scholar]
- 44. Touat‐Hamici Z, Weidmann H, Blum Y, et al. Role of lipid phosphate phosphatase 3 in human aortic endothelial cell function. Cardiovasc Res. 2016;112(3):702‐713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Long J, Darroch P, Wan KF, et al. Regulation of cell survival by lipid phosphate phosphatases involves the modulation of intracellular phosphatidic acid and sphingosine 1‐phosphate pools. Biochem J. 2005;391(Pt 1):25‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kerjaschki D, Vernillo AT, Farquhar MG. Reduced sialylation of podocalyxin—the major sialoprotein of the rat kidney glomerulus—in aminonucleoside nephrosis. Am J Pathol. 1985;118(3):343‐349. [PMC free article] [PubMed] [Google Scholar]
- 47. Larrucea S, Butta N, Arias‐Salgado EG, et al. Expression of podocalyxin enhances the adherence, migration, and intercellular communication of cells. Exp Cell Res. 2008;314(10):2004‐2015. [DOI] [PubMed] [Google Scholar]
- 48. Hardingham TE, Muir H. The specific interaction of hyaluronic acid with cartillage proteoglycans. Biochim Biophys Acta. 1972;279(2):401‐405. [DOI] [PubMed] [Google Scholar]
- 49. Oegema TR Jr, Laidlaw J, Hascall VC, Dziewiatkowski DD. The effect of proteoglycans on the formation of fibrils from collagen solutions. Arch Biochem Biophys. 1975;170(2):698‐709. [DOI] [PubMed] [Google Scholar]
- 50. Czipri M, Otto JM, Cs‐Szabó G, et al. Genetic rescue of chondrodysplasia and the perinatal lethal effect of cartilage link protein deficiency. J Biol Chem. 2003;278(40):39214‐39223. [DOI] [PubMed] [Google Scholar]
- 51. Carulli D, Pizzorusso T, Kwok JC, et al. Animals lacking link protein have attenuated perineuronal nets and persistent plasticity. Brain. 2010;133(Pt 8):2331‐2347. [DOI] [PubMed] [Google Scholar]
- 52. Nojima K, Miyazaki H, Hori T, Vargova L, Oohashi T. Assessment of possible contributions of hyaluronan and proteoglycan binding link protein 4 to differential perineuronal net formation at the calyx of held. Front Cell Dev Biol. 2021;9:730550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lam C, Ferreira C, Krasnewich D, et al. Prospective phenotyping of NGLY1‐CDDG, the first congenital disorder of deglycosylation. Genet Med. 2017;19(2):160‐168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Turner AJ, Tanzawa K. Mammalian membrane metallopeptidases: NEP, ECE, KELL, and PEX. FASEB J. 1997;11(5):355‐364. [DOI] [PubMed] [Google Scholar]
- 55. Shirotani K, Tsubuki S, Iwata N, et al. Neprilysin degrades both amyloid beta peptides 1‐40 and 1‐42 most rapidly and efficiently among thiorphan‐ and phosphoramidon‐sensitive endopeptidases. J Biol Chem. 2001;276(24):21895‐21901. [DOI] [PubMed] [Google Scholar]
- 56. Maruyama M, Higuchi M, Takaki Y, et al. Cerebrospinal fluid neprilysin is reduced in prodromal Alzheimer's disease. Ann Neurol. 2005;57(6):832‐842. [DOI] [PubMed] [Google Scholar]
- 57. Oh‐hashi K, Ohkubo K, Shizu K, et al. Biosynthesis, processing, trafficking, and enzymatic activity of mouse neprilysin 2. Mol Cell Biochem. 2008;313(1–2):103‐111. [DOI] [PubMed] [Google Scholar]
- 58. DeHahn KC, Gonzales M, Gonzalez AM, et al. The alpha4 laminin subunit regulates endothelial cell survival. Exp Cell Res. 2004;294(1):281‐289. [DOI] [PubMed] [Google Scholar]
- 59. Man XY, Finnson KW, Baron M, Philip A. CD109, a TGF‐β co‐receptor, attenuates extracellular matrix production in scleroderma skin fibroblasts. Arthritis Res Ther. 2012;14(3):R144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vorstenbosch J, Nguyen CM, Zhou S, et al. Overexpression of CD109 in the epidermis differentially regulates ALK1 versus ALK5 signaling and modulates extracellular matrix synthesis in the skin. J Invest Dermatol. 2017;137(3):641‐649. [DOI] [PubMed] [Google Scholar]
- 61. Cai H, Sasikumar P, Little G, et al. Identification of HSP47 binding site on native collagen and its implications for the development of HSP47 inhibitors. Biomol Ther. 2021;11(7):983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ito S, Nagata K. Biology of Hsp47 (serpin H1), a collagen‐specific molecular chaperone. Semin Cell Dev Biol. 2017;62:142‐151. [DOI] [PubMed] [Google Scholar]
- 63. Barboni E, Gormley AM, Pliego Rivero FB, Vidal M, Morris RJ. Activation of T lymphocytes by cross‐linking of glycophospholipid‐anchored Thy‐1 mobilizes separate pools of intracellular second messengers to those induced by the antigen‐receptor/CD3 complex. Immunology. 1991;72(4):457‐463. [PMC free article] [PubMed] [Google Scholar]
- 64. Rege TA, Hagood JS. Thy‐1 as a regulator of cell‐cell and cell‐matrix interactions in axon regeneration, apoptosis, adhesion, migration, cancer, and fibrosis. FASEB J. 2006;20(8):1045‐1054. [DOI] [PubMed] [Google Scholar]
- 65. Hagood JS. Thy‐1 as an integrator of diverse extracellular signals. Front Cell Dev Biol. 2019;7:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Long KR, Newland B, Florio M, et al. Extracellular matrix components HAPLN1, lumican, and collagen I cause hyaluronic acid‐dependent folding of the developing human neocortex. Neuron. 2018;99(4):702‐719.e706. [DOI] [PubMed] [Google Scholar]
- 67. Oohashi T, Edamatsu M, Bekku Y, Carulli D. The hyaluronan and proteoglycan link proteins: organizers of the brain extracellular matrix and key molecules for neuronal function and plasticity. Exp Neurol. 2015;274(Pt B):134‐144. [DOI] [PubMed] [Google Scholar]
- 68. Gillies AR, Lieber RL. Structure and function of the skeletal muscle extracellular matrix. Muscle Nerve. 2011;44(3):318‐331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ahmad K, Shaikh S, Ahmad SS, Lee EJ, Choi I. Cross‐talk between extracellular matrix and skeletal muscle: implications for myopathies. Front Pharmacol. 2020;11:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Asahina M, Fujinawa R, Nakamura S, et al. Ngly1−/− rats develop neurodegenerative phenotypes and pathological abnormalities in their peripheral and central nervous systems. Hum Mol Genet. 2020;29(10):1635‐1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mesika A, Nadav G, Shochat C, et al. NGLY1 deficiency zebrafish model manifests abnormalities of the nervous and musculoskeletal systems. Front Cell Dev Biol. 2022;10:902969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. He P, Grotzke JE, Ng BG, et al. A congenital disorder of deglycosylation: biochemical characterization of N‐glycanase 1 deficiency in patient fibroblasts. Glycobiology. 2015;25(8):836‐844. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1 This table demonstrates the different pathogenic variants in NGLY1‐CDDG patients
TABLE S2 All identified proteins with their quantitation value with TMT channel name, fold changes and p‐values, and their logarithmic values
TABLE S3 LC–MS/MS data for all glycopeptides showing their glycan composition, gene name, glycosylation site, quantitation values for each sample with TMT channel name, fold changes and p‐values, and their logarithmic values
Data Availability Statement
The mass spectrometry proteomics data have been deposited with the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD034364.