

Abstract
Sulfate is the fourth most abundant anion in human plasma but is not measured in clinical practice and little is known about the consequences of sulfate deficiency. Nevertheless, sulfation plays an essential role in the modulation of numerous compounds, including proteoglycans and steroids. We report the first patient with a homozygous loss‐of‐function variant in the SLC13A1 gene, encoding a renal and intestinal sulfate transporter, which is essential for maintaining plasma sulfate levels. The homozygous (Arg12Ter) variant in SLC13A1 was found by exome sequencing performed in a patient with unexplained skeletal dysplasia. The main clinical features were enlargement of joints and spondylo‐epi‐metaphyseal radiological abnormalities in early childhood, which improved with age. In addition, autistic features were noted. We found profound hyposulfatemia due to complete loss of renal sulfate reabsorption. Cholesterol sulfate was reduced. Intravenous N‐acetylcysteine administration temporarily restored plasma sulfate levels. We conclude that loss of the SLC13A1 gene leads to profound hypersulfaturia and hyposulfatemia, which is mainly associated with abnormal skeletal development, possibly predisposing to degenerative bone and joint disease. The diagnosis might be easily missed and more frequent.
Keywords: acetylcysteine, autistic disorder, cholesterol, chondroitin, joint diseases, proteoglycans, skeletal dysplasia, sulfation
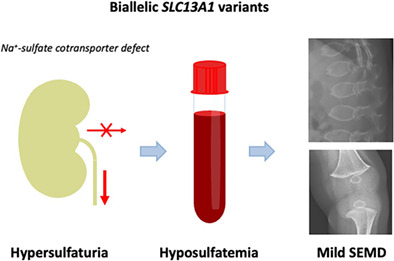
We describe the first patient with biallelic loss‐of‐function variants in the SLC13A1 gene, encoding a renal and intestinal sulfate transporter. The consequential loss of renal sulfate reabsorption leads to hypersulfaturia and hyposulfatemia which is associated with a mild Spondylo‐Epi‐Metaphyseal skeletal Dysplasia.
1. INTRODUCTION
Sulfate is the fourth most abundant anion in human plasma (at approximately 0.3 mM), 1 , 2 , 3 and sulfation modulates the activity of numerous compounds, including proteoglycans, steroids, neurotransmitters and exogenous chemicals and drugs. 2 However, sulfate is not currently measured in clinical practice and little is known about the consequences of sulfate deficiency.
Sulfate is derived both from the diet as inorganic sulfate and from the degradation of sulfur‐containing amino acids, cysteine and methionine, and from other thiols. 1 Dedicated sulfate transporters mediate the influx of hydrophilic sulfate across lipid cell membranes. The first, essential step in all sulfation reactions is the activation of inorganic sulfate to 3′‐phosphoadenosine‐5′‐phosphosulfate (PAPS) by PAPS synthases, while sulfotransferases catalyze final sulfation reactions. 1
The list of genetic disorders affecting sulfation is growing and phenotypes depend on the expression pattern and substrate specificity of the affected transporter/enzyme. Most disorders, such as defects of SLC26A2 (OMIM 606718), PAPSS2 (OMIM 603005), BPNT2 (OMIM 614010), CHST3 (OMIM 603799), CHST11 (OMIM 610128) and CHST14 (OMIM 608429) involve skeletal dysplasias (reviewed by Paganini) 4 but a few are associated with other conditions, such as hyperandrogenism (PAPSS2), intellectual disability (NDST1, OMIM 600853; EXT2, OMIM 608210; HS6ST2, OMIM 300545), ichthyosis (SULT2B1, OMIM 604125), corneal dystrophy (CHST6, OMIM 605294) or hypogonadotrophic hypogonadism (HS6ST1, OMIM 604846).
Plasma sulfate levels are maintained by reabsorption of the majority of filtered sulfate in the renal proximal tubule. 5 This requires the SLC13A1 (Solute Carrier Family 13 Member 1) encoded Na+‐sulfate cotransporter (NaS1) in the (apical) brush‐border membrane, together with the SLC26A1‐encoded sulfate‐anion exchange transporter in the contraluminal basolateral membrane. 5 , 6 SLC13A1 Na‐sulfate cotransport is believed to be the rate‐limiting step 5 and loss of this transporter may deplete the circulating sulfate pool and negatively affect general sulfation capacity. Here, we report the first patient with biallelic loss‐of‐function variants in the SLC13A1 gene (OMIM 606193).
2. MATERIALS AND METHODS
2.1. Patient study
We investigated a patient with unexplained skeletal dysplasia. Both the patient and his parents gave written informed consent for publication. This case study was approved by the institutional review board of VU University Medical Center (protocol number: METC VUmc 2021.0577).
2.2. Genetic testing
Genomic DNA isolated from peripheral blood lymphocytes of the patient and his parents was subjected to exome capture using SeqCap EZ Human Exome Library v3.0 kit (NimbleGen; 454 Life Sciences, Roche, Branford, CT). Sequencing was performed on an Illumina HiSeq2500 HTv4 (Illumina, San Diego, CA, USA) with paired‐end, 125‐bp reads. Read alignment to hg19 and variant calling were done with a pipeline based on BWA‐MEM0.7 and GATK 3.3.0. The median coverage of the captured target region was at least 85× with >90% 30× coverage of the captured region. Variant annotation and prioritizing were done using Cartagenia Bench Lab NGS (Agilent Technologies). We excluded variants located outside the exons and intron/exon boundaries, and variants with a minor allele frequency (MAF) of >1% in control databases, including dbSNP137 (http://www.ncbi.nlm.nih.gov/projects/SNP), 1000 Genomes Project (release of February 2012), and Exome Variant Server (EVS), NHLBI Exome Sequencing Project National Heart, Lung, and Blood Institute GO Exome Sequencing Project (ESP6500 release) (http://evs.gs.washington.edu/EVS/) and our in‐house exome controls. Variants that fitted with a de novo or recessive mode of inheritance were further analyzed.
2.3. Sulfate analysis
Free sulfate levels in plasma and urine of the patient, his parents and controls using an AB Sciex 4000 QTrap or 5000 liquid chromatography–tandem mass spectrometry (LC–MS/MS, Applied Biosystems, Foster City, CA, USA) system operating in the negative ionization mode. Samples were introduced via flow‐injection analysis, and the transitions m/z 97 > 80, and m/z 99 > 82 were monitored for sulfate (SO4) and 34SO4 (serving as internal standard, mass shift is +2), respectively.
Urinary samples were prepared by an initial 100‐fold dilution of the original urine with distilled water. A total of 20 μl of the diluted urine was mixed with 2 nmol of 34SO4, and the obtained mixture was purified through a 10 kDa filter (Amicon Ultra, Millipore, Merck, Darmstadt, Germany) by centrifugation. From the final filtrate, 10 μl were introduced to the mass spectrometer by flow injection analysis.
Plasma samples were prepared by mixing 20 μl of plasma with 2 nmol of 34SO4 and 180 μl of purified water, followed by a deproteinization step using a 10 kDa filter (Amicon Ultra) by centrifugation. From the final filtrate, 1 μl was introduced to the mass spectrometer by flow injection analysis (10 mM formic acid adjusted to pH 8.75 with ammonia). Aqueous calibrators with known amounts of sulfate were used for quantification. The obtained peak‐area ratios of the trace of sulfate related to the trace of the internal standard were then used for the estimation of the free sulfate levels in the examined body fluids. Plasma measurements were performed in triplicate.
2.4. Glycosaminoglycan sulfation analysis
Morning urine samples of the patient and his parents were stored at −20°C and shipped on dry ice, together with two Dutch age‐matched control samples. Three additional (Italian) lab control samples were included in the analysis. The sulfation level of urinary glycosaminoglycans (GAGs) was analyzed by high performance liquid chromatography (HPLC) as previously described. 7 , 8 Briefly, 1.8 ml of urine were clarified by centrifugation and GAGs were precipitated with 0.2% cetylpyridinium chloride overnight at 4°C. After centrifugation, the pellets were washed three times with 10% potassium acetate in 96% ethanol and three times with 96% ethanol. Samples were digested by 30 mU of chondroitinase ABC (AMSBIO) and 30 mU of chondroitinase ACII (Sigma‐Aldrich) in 0.1 M ammonium acetate, pH 7.35, overnight at 37°C and then lyophilized. Lyophilized disaccharides were derivatized in 40 μl of 12.5 mM 2‐aminoacridone (Life Technologies) in 85:15 (v/v) dimethyl sulfoxide: glacial acetic acid, incubated in the dark for 15 min and 40 μl of 1.25 M NaBH3CN (Sigma‐Aldrich) were added. Then samples were incubated overnight at 37°C in the dark and stored at −20°C until analysis. Disaccharides analysis was carried out by HPLC using a LiChroCART 250–4 Superspher 100 RP‐18 endcapped column (Merck) as previously reported. 9
2.5. Steroid analysis
Serum and urine of the patient was analyzed together with 5 age‐matched Dutch controls.
The serum steroid sulfatome was analyzed by LC–MS/MS as described previously. 10 Briefly, after incubation of serum (300 μl) with internal standards, protein precipitation was conducted using a solution containing 80% acetonitrile and 20% aqueous ZnSO4 (89 g/L). Methanol was utilized to extract sulfated steroids by solid phase extraction (SepPak C18 cartridges, Waters Associates, Milford, MA). The methanolic eluent was evaporated and reconstituted with 250 μl of a diluent (79.75% water, 10% MeOH, 10% ACN, and 0.25% ammonium hydroxide). An aliquot (10 μl) was injected into the LC–MS/MS system. Separation of steroid sulfates was achieved on an Accucore Phenyl‐X column (100 × 2.1 mm, 2.6 μm, Thermo Fisher Scientific). A triple quadrupole mass spectrometer (Quantum Ultra, Thermo Scientific) with an electrospray probe was used for the detection and quantification of steroid sulfates.
Urine steroid metabolome analysis was performed using gas chromatography–mass spectrometry (GC–MS) as described elsewhere. 11 In brief, free and conjugated steroids were extracted from a 5‐ml aliquot of a 24‐h urine by solid phase extraction (Sep‐Pak C18 cartridge), and the conjugates were enzymatically hydrolyzed (Helix pomatia, Sigma‐Aldrich Corp., St. Louis, MO). After re‐extraction and addition of internal standards, methyloxime‐trimethylsilyl ethers were formed as derivatives. GC was performed on an Optima‐1 fused silica column (Macherey‐Nagel, Dueren, Germany) using helium as carrier gas. The GC (Agilent 6890 series GC, Agilent 7683 Series Injector, Agilent Technologies, Waldbronn, Germany) was directly interfaced to a mass selective detector (Agilent 5973 N MSD, Agilent Technologies) operated in selected ion monitoring mode.
2.6. Intravenous N‐acetylcysteine treatment
Since degradation of cysteine is an alternative source of sulfate, we aimed to restore plasma sulfate levels by intravenous NAC administration. During the test the patient was on a protein restricted diet. A bolus of 133 mg/kg NAC (Fluimucil®) was administered intravenously in 60 min, followed by 133 mg/kg in 8 h in analogy to the treatment of acetaminophen intoxication. Blood samples were taken before start, after 1, 4, 9, and 24 h.
3. RESULTS
3.1. Clinical features
The patient, a boy, was born after a spontaneous pregnancy. He presented at 4 months with enlarged knees that could not fully extend, while his length (−0.5 SDS) and head circumference (+0.5 SDS) were normal. Skeletal survey at 6 months of age revealed generalized skeletal abnormalities, particularly ovoid‐shaped vertebral bodies and spiky widening of the metaphyses but also irregular tarsal bones, slight flaring of the ribs and somewhat smaller epiphyses which became more noticeable at 4 years (Figure 1A,B). No abnormalities of skull and pelvis were found. Serum levels of calcium, phosphate, alkaline phosphatase, parathyroid hormone, and 25‐hydroxyvitamin D3 were normal. Over the years, his growth was normal and the only complaint was a slightly abnormal gait and an inability to run easily. At 8 years of age, he sustained a forearm fracture that healed well. The radiological abnormalities ameliorated and at 11 years, only flattening of the cervical vertebral bodies and irregular lumbar vertebral endplates remained (Figure 1C). At 12 years of age, he exhibited enlarged knees and elbows, reduced movement in the lumbar spine and knees, winged scapulae, hypermobility of the fingers and flat feet. Although height was normal (+1 SDS with target height at +1 SDS) his trunk was relatively short, with a sitting height/height ratio of −2.0 SDS. Dual‐energy x‐ray absorptiometry (DEXA) at 13 years showed bone mineral density Z‐scores of 0.2 in the lumbar spine and −2.1 in the proximal femur.
FIGURE 1.
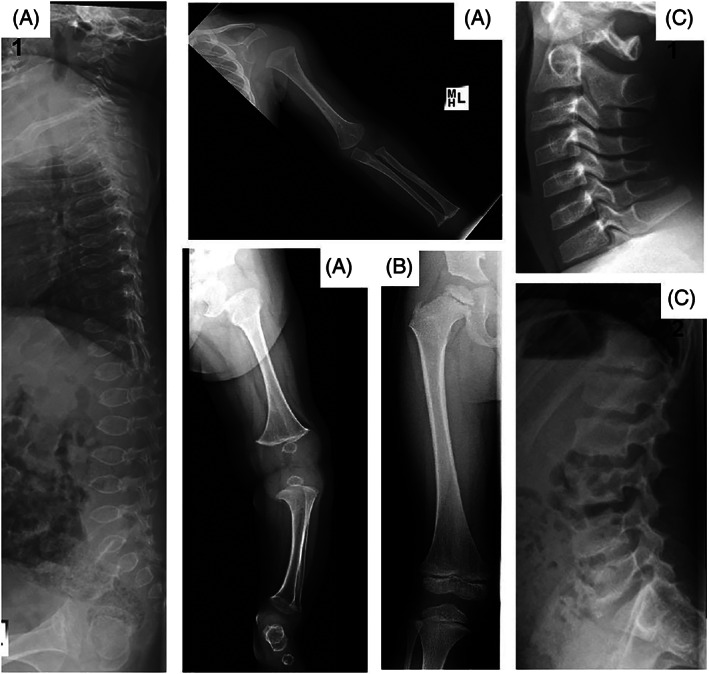
Skeletal radiographs. Panel A shows general abnormalities including ovoid‐shaped vertebral bodies, 1 spiky widening of all metaphyses and slightly delayed epiphyseal ossification in upper limb 2 and lower limb 3 and irregular tarsal bones 3 at 6 months of age. Panel B shows that at 4 years the metaphyseal abnormalities have improved but that the epiphyses are small and somewhat irregular. Panel C shows that at 11 years, flattening of the cervical vertebral bodies 1 and irregular lumbar vertebral endplates 2 remain. The metaphyses and epiphyses normalized (not shown).
3.1.1. Puberty
Pubertal onset (i.e., a testicular volume of 4 ml) occurred at age 11.5 years, the median age for Dutch boys. At this time, his bone age was more than 1 year advanced. A year later his Tanner stage was G3‐4Ph3 and his testicular volume was 8–10 ml.
3.1.2. Neurodevelopment
While the patients cognitive development was above average, autistic features such as difficulty with social communication, restricted patterns of behavior, interest and activities, an elevated sensitivity to changes in his environment and rigid thinking patterns were frequently leading to panic attacks and school absenteeism.
3.1.3. Other symptoms
The patient experienced no joint pain, no visual or hearing complaints, and no skin abnormalities. Persistent fatigue and chronic cough were ascribed to cystic fibrosis (CF), an incidental finding of the exome sequencing which was confirmed by a sweat chloride measurement of 64 mmol/L. Subsequently, he was treated for respiratory infections with oral antibiotics once to twice yearly. He had stable lung function (FEV1 85% predicted), no Pseudomonas aeruginosa infections and sufficient pancreatic function.
3.1.4. Family history
The patient was the only child of non‐consanguineous healthy Dutch parents with normal heights. The mother and the maternal grandmother had symptoms suggestive of polycystic ovary syndrome (POCS). Osteoarthritis occurred in the maternal grandmother and the maternal grandmothers father.
3.2. Genetic testing
A homozygous g.122839967G>A (Chr7[GRCh37]), c.34C>T, p.(Arg12Ter) variant was identified in the first exon of the SLC13A1 gene (NM_022444.3). This variant causes a gain of stop predicted to result in a truncated protein and has previously been shown to cause total loss‐of‐function in Xenopus laevis oocytes. 12 In addition, we found a known pathogenic homozygous g.117188849C>A (Chr7[GRCh37]), c.1364C>A p.(Ala455Glu) variant in the cystic fibrosis transmembrane conductance regulator (CFTR) gene (NM_000492.3). The patient had a 20 Mb region‐of‐homozygosity on chromosome 7q31.32 containing both the SLC13A1 and the CFTR gene, while his parents were heterozygous for both variants, suggesting a common ancestor.
3.3. Sulfate analysis
Plasma sulfate levels were severely reduced in the patient and also below the control range in both parents (Figure 2 and Table S1). Reference values for Fractional Sulfate Excretion (FEsulfate) range from 13% to 34%, 13 whereas the patient's FEsulfate was close to 100%, while his mother had an FEsulfate of 45% (Figure 3 and Table S1).
FIGURE 2.
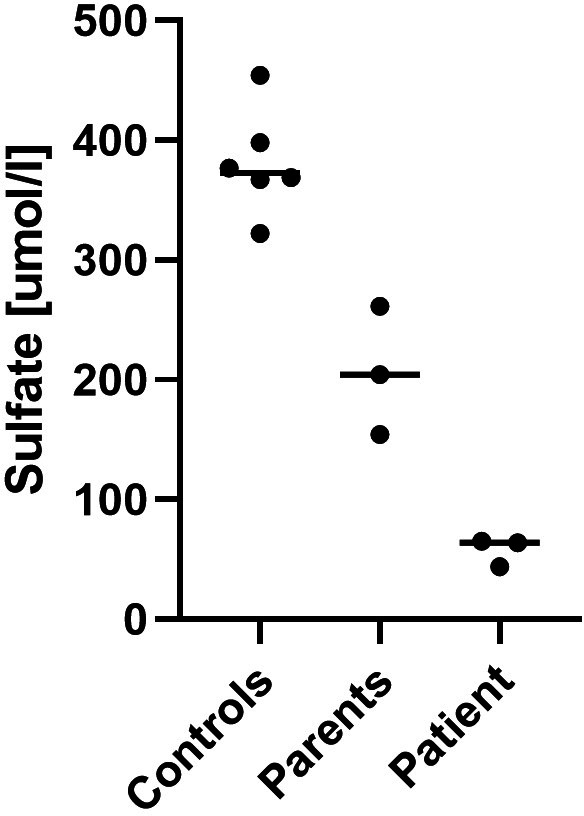
Plasma sulfate measurements. Plasma levels in the patient compared to his parents and lab controls.
FIGURE 3.
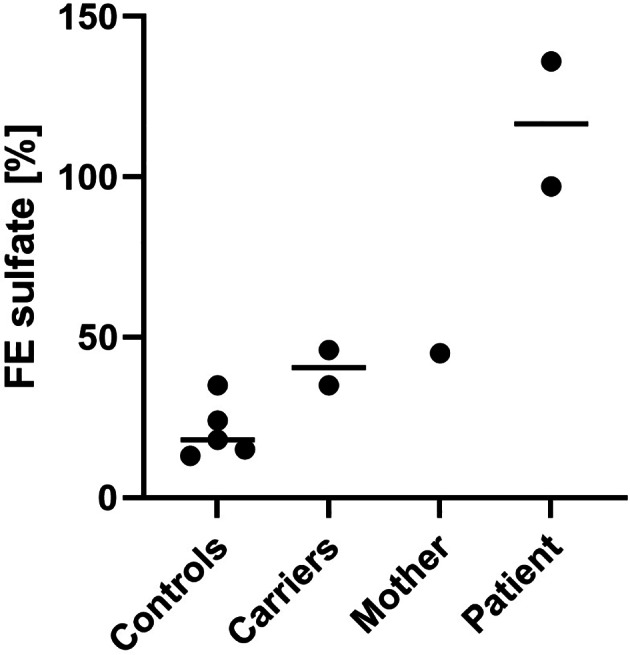
Urinary sulfate excretion. Calculated FEsulfate in the patient and his mother is compared to SLC13A1 Arg12Ter heterozygotes and wild‐type controls from Bowling et al 2013. 13 The FEsulfate is expressed in % and calculated as 100 × [plasma creatinine × urine sulfate]/[urine creatinine × plasma sulfate].
3.4. Glycosaminoglycan sulfate analysis
The percentage chondroitin sulfate disaccharides in urinary glycosaminoglycans did not differ significantly in the patient, the parents and the controls (Table S2).
3.5. Steroid analysis
Serum cholesterol sulfate was reduced in the patient (443 ng/ml; reference range 500–1236 ng/ml), while other serum steroid sulfates were within the reference range (Table S3).
Steroid metabolome analysis in 24 h‐urine showed an age‐appropriate pattern of C21‐ and C19‐steroids.
3.6. Intravenous N‐acetylcysteine treatment
Plasma sulfate concentrations normalized after bolus injection of NAC and during maintenance infusion but returned to baseline 15 h after stop of the infusion (Figure 4).
FIGURE 4.
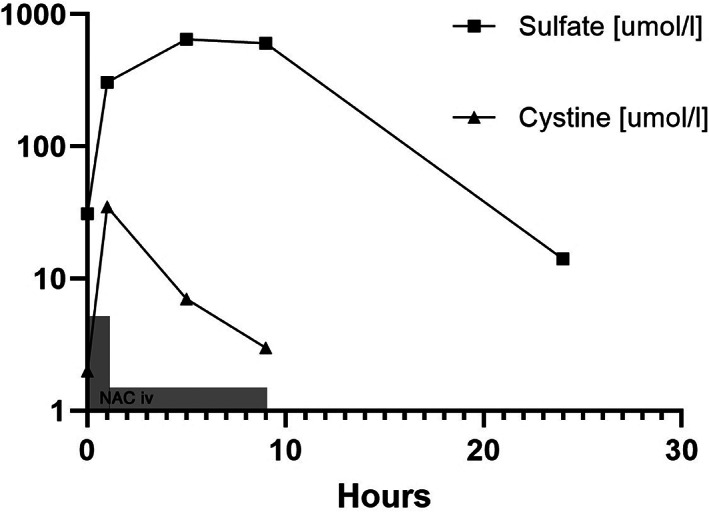
intravenous N‐Acetylcysteine treatment. Panel C shows plasma levels of sulfate and cystine (oxidized form of cysteine) on a logarithmic scale during and after NAC infusion. The gray rectangles indicate NAC administration (not to scale). The concentration cystine peaked earlier than sulfate as expected from the metabolism of the drug.
The patient developed erythema during the first bolus infusion. This disappeared after administration of 2 mg clemastine intravenously. No other adverse effects were noted.
4. DISCUSSION
Here we describe the first patient with a homozygous loss‐of‐function variant in the SLC13A1 gene, encoding a Na+‐sulfate cotransporter which is primarily expressed in the renal proximal tubule and intestinal brush‐border membrane and which is essential to the maintenance of plasma sulfate levels. 5
We found an FEsulfate close to 100%, consistent with complete loss of renal sulfate reabsorption in our patient, as well as an over 80% reduction in plasma sulfate levels. An existing SLC13A1 knockout mouse model showed similarly reduced plasma sulfate levels and hypersulfaturia, 14 while previously described individuals with heterozygous loss‐of‐function variants in SLC13A1 show a 27% decrease in serum sulfate levels, 15 as well as elevated FEsulfate (35%–46% vs. 13%–34% in non‐carriers). 13 Our patient's heterozygous parents showed similar values.
The patient presented with abnormal skeletal development evidenced by extensive spondylo‐epi‐metaphyseal radiological abnormalities early in life. However, apart from enlarged joints and some stiffness, he had few complaints and no growth delay. Remarkably, homozygous loss‐of‐function variants in SLC13A1 have occurred naturally in two animal breeds, the Texel sheep 16 and the miniature Poodle, 17 both with evident skeletal dysplasia, including stunted growth, abnormal stance and locomotion, enlarged joints, joint stiffness and early‐onset osteoarthritis. 16 , 17 Serum sulfate levels in the miniature Poodle were below the limit of detection. 17 The SLC13A1−/− mouse shows general growth retardation but no gross histological bone abnormalities. 14
The patients skeletal phenotype resembles other genetic disorders of sulfation such as PAPSS2 deficiency, which is associated with (subclinical) brachyolmia to overt spondyloepimetaphyseal dysplasia (SEMD), 18 and SLC26A2 deficiency, which causes a broad spectrum of skeletal dysplasias ranging from neonatal lethal atelosteogenesis type II and achondrogenesis Ib to diastrophic dysplasia (DTD) with short‐limbed short stature and mild recessive multiple epiphyseal dysplasia (rMED) with joint pain but usually normal stature. 4 , 19 Also other sulfation disorders primarily affect skeletal development. 4 The addition of the negatively charged sulfate group to glycosaminoglycan chains (i.e., chondroitin, dermatan, keratan, and/or heparin) of proteoglycans is essential not only in tissue hydration and elasticity but also in the organization of the extracellular matrix and interactions with signaling pathways. 4 Reduced chondroitin sulfation is associated with disorganized collagen fibers and diminished Indian Hedgehog signaling, which is essential in chondrocyte proliferation and differentiation. 20 , 21 The observation that deficiency of SLC26A2, encoding the Na+‐independent sulfate/chloride antiporter in chondrocytes, leads to abnormal skeletal development with reduced chondroitin sulfation, implies that chondrocytes mainly rely on inorganic sulfate from the extracellular space. 22 Significantly reduced chondroitin 4‐sulfate was also observed in the cartilage of SLC13A1‐deficient Texel sheep. 16 Unfortunately, we were unable to confirm decreased chondroitin sulfate in our patient's urine, which might have otherwise provided a useful biomarker for glycosaminoglycan sulfation defects. 7
The patient had reduced serum cholesterol sulfate, while other steroid sulfates were normal. Cholesterol sulfate is the most abundant steroid sulfate in human plasma, 23 perhaps explaining why this was most affected. Cholesterol sulfate is primarily involved in keratinocyte differentiation 23 and both an absence due to SULT2B1 cholesterol sulfotransferase deficiency and cholesterol sulfate accumulation due to X‐linked steroid sulfatase deficiency result in congenital ichthyosis, 24 which was not apparent in the patient. However, cholesterol has also been linked to chondrocyte differentiation. 25 Reduced cholesterol sulfation is further implicated in cholesterol accumulation and may be essential in atherosclerosis. 26
DHEAS is the second most abundant steroid sulfate in human plasma 23 and reduced sulfation increases androgenic activity, as evidenced by premature adrenarche and/or PCOS in PAPSS2 deficiency. 18 , 27 Although the patient had an age‐appropriate onset of puberty and normal sulfated androgen levels, the long‐term implications for his androgen profile might be more subtle. Interestingly, both his (heterozygous) mother and (not tested) maternal grandmother experienced symptoms suggestive of PCOS.
The patient showed normal cognitive development without epilepsy but autistic features were noted. This could not be attributed to the concomitant diagnosis of CF, which is associated with an increased risk of depression and anxiety 28 but has not been related to autism. A link between reduced sulfation capacity and autism has previously been suggested. 13 , 29 The SLC13A1−/− mouse also exhibits behavioral abnormalities, 30 impaired memory 31 and reduced brain serotonin levels 32 in addition to spontaneous clonic seizures. 14 Sulfated proteoglycans are important components of the extracellular matrix in the brain, regulating synaptic development and neuronal plasticity. 33 , 34 , 35 Other sulfated compounds in the brain include sphingolipids (e.g., cerebroside), neurosteroids, neurotransmitters and thyroid hormone. 2
Since sulfation is important in the clearance of xenobiotics such as acetaminophen, the patient might be at increased risk for hepatotoxicity, as noted in the SLC13A1−/− mouse model. 12 Heterozygous carriers of SLC13A1 loss‐of‐function variants reportedly have higher transaminase levels compared to non‐carriers, suggestive of subclinical liver damage. 15 Although the patient's transaminase levels were normal on multiple occasions, he has been strongly advised to avoid acetaminophen.
Early administration of NAC is known to be very effective in preventing liver injury in acetaminophen overdose, 36 and due to its role as an alternative sulfation source in cartilage, 7 , 37 NAC is also seen as a possible treatment for SLC26A2‐related skeletal dysplasia. 9 , 38 Intravenous NAC administration in our patient restored plasma sulfate levels, demonstrating that NAC is indeed a source of free sulfate, including extracellular sulfate, and perhaps explaining its efficacy in preventing acetaminophen hepatotoxicity, an action currently attributed solely to the replenishment of anti‐oxidant glutathione. 36 Bearing in mind the low bioavailability of about 10% 39 in combination with the renal sulfate loss in the patient it remains to be demonstrated whether oral NAC treatment can restore sulfate levels as well.
Although hyposulfatemia restricts general sulfation capacity, catabolism of sulfur‐containing amino acids may partially compensate in the cytoplasm, 9 , 37 perhaps explaining the relatively mild symptoms seen in our patient. In fact, two SLC13A1 loss‐of‐function variants, Arg12Ter and Trp48Ter, have a relatively high global allele frequencies (0.20% and 0.05%, respectively) and homozygosity for the Arg12Ter SLC13A1 variant is reported once in the Genome Aggregation Database (http://gnomad.broadinstitute.org/) 40 which notably excludes severe pediatric diseases. We hypothesize that biallelic SLC13A1 loss‐of‐function variants are more common and may also present later in life with degenerative bone and joint disease but that mild presentations escape diagnosis unless radiological evaluation is performed in childhood. In a recent genome‐wide association study, back pain due to intervertebral disc disorders was strongly associated with heterozygous loss‐of‐function variants in SLC13A1. 41 In addition, hyposulfatemia might be a risk factor for other health issues, such as hyperandrogenism, autism, atherosclerosis, and drug‐induced hepatotoxicity. Hyposulfatemia might also result from loss of the SLC26A1 gene, encoding the sulfate‐anion exchange transporter in the renal basolateral membrane as has been confirmed in a knockout mouse model. 42
Our patient had in addition a homozygous Ala455Glu variant in the CFTR gene, located 5.6 Mb centromeric to the SLC13A1 gene in a 20 Mb region‐of‐homozygosity. This CFTR variant is a presumably relatively recent founder variant mainly detected in the Netherlands and a specific French Canadian population. 43 Although the parents were unaware of consanguinity they both had ancestors in the Dutch province Zeeland where the Ala455Gly variant is most prevalent. 44 The variant is associated with a less severe CF phenotype, 45 probably explaining the mild CF symptoms in our patient. We considered an interaction between sulfate levels and CF phenotype. Altered glycoproteins sulfation in mucus of CF patients has been reported but the effect and mechanism hereof is uncertain. Sulfation of mucins was found to be independent of CRTR expression. 46
In conclusion, homozygous loss of the SLC13A1 gene resulted in complete loss of renal sulfate reabsorption and depletion of the plasma sulfate pool, a biochemical phenotype associated with abnormal skeletal development, evidenced by radiological abnormalities in early childhood. Autistic features and reduced cholesterol sulfate might be associated. It is important to be aware of a potential increased risk for acetaminophen‐induced hepatotoxicity. Intravenous NAC administration temporarily restored plasma sulfate levels.
We suggest that more extensive measurement of plasma sulfate will improve our understanding of the importance of sulfation in human health and disease.
AUTHOR CONTRIBUTIONS
Jiddeke M van de Kamp, Arend Bökenkamp, and Martijn JJ Finken designed the study; Jiddeke M van de Kamp and Martijn JJ Finken investigated the patient; Jiddeke M van de Kamp managed the data generation and data analysis and wrote the manuscript; Arend Bökenkamp and Martijn JJ Finken contributed to the analysis and interpretation of the data and reviewed the manuscript; Erwin EW Jansen, Eduard A Struys, Desiree EC Smith and Mirjam MC Wamelink developed and performed the plasma and urine sulfate analysis; Quinten Waisfisz performed the genetic analysis; Marieke Verkleij provided the psychological assessment. Chiara Paganini and Antonio Rossi performed the GAG sulfate analysis; Stefan A Wudy, Michaela F Hartmann, and Rong Wang performed the steroid analysis.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
Supporting information
Appendix S1 Supporting Information.
ACKNOWLEDGMENTS
We would like to thank the patient and his parents for their cooperation. This study was supported by grants from MIUR “Dipartimenti di Eccellenza 2018–2022” to Antonio Rossi.
van de Kamp JM, Bökenkamp A, Smith DEC, et al. Biallelic variants in the SLC13A1 sulfate transporter gene cause hyposulfatemia with a mild spondylo‐epi‐metaphyseal dysplasia. Clinical Genetics. 2023;103(1):45‐52. doi: 10.1111/cge.14239
Funding information MIUR “Dipartimenti di Eccellenza 2018–2022”
DATA AVAILABILITY STATEMENT
All data described in this study are provided within the article and supplementary material. Raw sequencing data and de‐identified clinical data is available from the corresponding authors upon request.
REFERENCES
- 1. Dawson PA. Role of sulphate in development. Reproduction. 2013;146(3):R81‐R89. [DOI] [PubMed] [Google Scholar]
- 2. Langford R, Hurrion E, Dawson PA. Genetics and pathophysiology of mammalian sulfate biology. J Genet Genomics. 2017;44(1):7‐20. [DOI] [PubMed] [Google Scholar]
- 3. Cole DE, Evrovski J. Quantitation of sulfate and thiosulfate in clinical samples by ion chromatography. J Chromatogr A. 1997;789(1–2):221‐232. [DOI] [PubMed] [Google Scholar]
- 4. Paganini C, Gramegna Tota C, Superti‐Furga A, Rossi A. Skeletal dysplasias caused by sulfation defects. Int J Mol Sci. 2020;21(8):1‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee A, Beck L, Markovich D. The human renal sodium sulfate cotransporter (SLC13A1; hNaSi‐1) cDNA and gene: organization, chromosomal localization, and functional characterization. Genomics. 2000;70(3):354‐363. [DOI] [PubMed] [Google Scholar]
- 6. Markovich D. Na+‐sulfate cotransporter SLC13A1 . Pflugers Arch. 2014;466(1):131‐137. [DOI] [PubMed] [Google Scholar]
- 7. Monti L, Paganini C, Lecci S, et al. N‐acetylcysteine treatment ameliorates the skeletal phenotype of a mouse model of diastrophic dysplasia. Hum Mol Genet. 2015;24(19):5570‐5580. [DOI] [PubMed] [Google Scholar]
- 8. Hendrickx G, Danyukova T, Baranowsky A, et al. Enzyme replacement therapy in mice lacking arylsulfatase B targets bone‐remodeling cells, but not chondrocytes. Hum Mol Genet. 2020;29(5):803‐816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Paganini C, Gramegna Tota C, Monti L, et al. Improvement of the skeletal phenotype in a mouse model of diastrophic dysplasia after postnatal treatment with N‐acetylcysteine. Biochem Pharmacol. 2021;185:114452. [DOI] [PubMed] [Google Scholar]
- 10. Sanchez‐Guijo A, Oji V, Hartmann MF, Traupe H, Wudy SA. Simultaneous quantification of cholesterol sulfate, androgen sulfates, and progestagen sulfates in human serum by LC‐MS/MS. J Lipid Res. 2015;56(9):1843‐1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wudy SA, Schuler G, Sanchez‐Guijo A, Hartmann MF. The art of measuring steroids: principles and practice of current hormonal steroid analysis. J Steroid Biochem Mol Biol. 2018;179:88‐103. [DOI] [PubMed] [Google Scholar]
- 12. Lee S, Dawson PA, Hewavitharana AK, Shaw PN, Markovich D. Disruption of NaS1 sulfate transport function in mice leads to enhanced acetaminophen‐induced hepatotoxicity. Hepatology. 2006;43(6):1241‐1247. [DOI] [PubMed] [Google Scholar]
- 13. Bowling FG, Heussler HS, McWhinney A, Dawson PA. Plasma and urinary sulfate determination in a cohort with autism. Biochem Genet. 2013;51(1–2):147‐153. [DOI] [PubMed] [Google Scholar]
- 14. Dawson PA, Beck L, Markovich D. Hyposulfatemia, growth retardation, reduced fertility, and seizures in mice lacking a functional NaSi‐1 gene. Proc Natl Acad Sci USA. 2003;100(23):13704‐13709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tise CG, Perry JA, Anforth LE, et al. From genotype to phenotype: nonsense variants in SLC13A1 are associated with decreased serum sulfate and increased serum aminotransferases. G3 (Bethesda). 2016;6(9):2909‐2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhao X, Onteru SK, Piripi S, et al. In a shake of a lamb's tail: using genomics to unravel a cause of chondrodysplasia in Texel sheep. Anim Genet. 2012;43(1):9‐18. [DOI] [PubMed] [Google Scholar]
- 17. Neff MW, Beck JS, Koeman JM, et al. Partial deletion of the sulfate transporter SLC13A1 is associated with an osteochondrodysplasia in the miniature poodle breed. PLoS One. 2012;7(12):e51917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Oostdijk W, Idkowiak J, Mueller JW, et al. PAPSS2 deficiency causes androgen excess via impaired DHEA sulfation–in vitro and in vivo studies in a family harboring two novel PAPSS2 mutations. J Clin Endocrinol Metab. 2015;100(4):E672‐E680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bonafe L, Mittaz‐Crettol L, Ballhausen D, Superti‐Furga A. Multiple epiphyseal dysplasia, recessive. 2002 Aug 29 [Updated 2014 Jan 23]. In: Adam MP, Everman DB, Mirzaa GM, et al., eds. GeneReviews([R]); Seattle, WA: University of Washington, Seattle;1993‐2022. [Google Scholar]
- 20. Gualeni B, Facchini M, De Leonardis F, et al. Defective proteoglycan sulfation of the growth plate zones causes reduced chondrocyte proliferation via an altered Indian hedgehog signaling. Matrix Biol. 2010;29(6):453‐460. [DOI] [PubMed] [Google Scholar]
- 21. Cortes M, Baria AT, Schwartz NB. Sulfation of chondroitin sulfate proteoglycans is necessary for proper Indian hedgehog signaling in the developing growth plate. Development. 2009;136(10):1697‐1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Satoh H, Susaki M, Shukunami C, Iyama K, Negoro T, Hiraki Y. Functional analysis of diastrophic dysplasia sulfate transporter. Its involvement in growth regulation of chondrocytes mediated by sulfated proteoglycans. J Biol Chem. 1998;273(20):12307‐12315. [DOI] [PubMed] [Google Scholar]
- 23. Strott CA, Higashi Y. Cholesterol sulfate in human physiology: what's it all about? J Lipid Res. 2003;44(7):1268‐1278. [DOI] [PubMed] [Google Scholar]
- 24. Heinz L, Kim GJ, Marrakchi S, et al. Mutations in SULT2B1 cause autosomal‐recessive congenital ichthyosis in humans. Am J Hum Genet. 2017;100(6):926‐939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Papathanasiou I, Anastasopoulou L, Tsezou A. Cholesterol metabolism related genes in osteoarthritis. Bone. 2021;152:116076. [DOI] [PubMed] [Google Scholar]
- 26. Seneff S, Davidson RM, Lauritzen A, Samsel A, Wainwright G. A novel hypothesis for atherosclerosis as a cholesterol sulfate deficiency syndrome. Theor Biol Med Model. 2015;12:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Noordam C, Dhir V, McNelis JC, et al. Inactivating PAPSS2 mutations in a patient with premature pubarche. N Engl J Med. 2009;360(22):2310‐2318. [DOI] [PubMed] [Google Scholar]
- 28. Quittner AL, Goldbeck L, Abbott J, et al. Prevalence of depression and anxiety in patients with cystic fibrosis and parent caregivers: results of the international depression epidemiological study across nine countries. Thorax. 2014;69(12):1090‐1097. [DOI] [PubMed] [Google Scholar]
- 29. Pagan C, Benabou M, Leblond C, et al. Decreased phenol sulfotransferase activities associated with hyperserotonemia in autism spectrum disorders. Transl Psychiatry. 2021;11(1):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dawson PA, Steane SE, Markovich D. Behavioral abnormalities of the hyposulphataemic Nas1 knockout mouse. Behav Brain Res. 2004;154(2):457‐463. [DOI] [PubMed] [Google Scholar]
- 31. Dawson PA, Steane SE, Markovich D. Impaired memory and olfactory performance in NaSi‐1 sulphate transporter deficient mice. Behav Brain Res. 2005;159(1):15‐20. [DOI] [PubMed] [Google Scholar]
- 32. Lee S, Kesby JP, Muslim MD, et al. Hyperserotonaemia and reduced brain serotonin levels in NaS1 sulphate transporter null mice. Neuroreport. 2007;18(18):1981‐1985. [DOI] [PubMed] [Google Scholar]
- 33. Kamimura K, Maeda N. Glypicans and Heparan sulfate in synaptic development, neural plasticity, and neurological disorders. Front Neural Circuits. 2021;15:595596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sharmin S, Pradhan J, Zhang Z, Bellingham M, Simmons D, Piper M. Perineuronal net abnormalities in SLC13A4(+/−) mice are rescued by postnatal administration of N‐acetylcysteine. Exp Neurol. 2021;342:113734. [DOI] [PubMed] [Google Scholar]
- 35. Zhang Z, Jhaveri D, Sharmin S, et al. Cell‐extrinsic requirement for sulfate in regulating hippocampal neurogenesis. Biol Open. 2020;9(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McGill MR, Jaeschke H. Metabolism and disposition of acetaminophen: recent advances in relation to hepatotoxicity and diagnosis. Pharm Res. 2013;30(9):2174‐2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pecora F, Gualeni B, Forlino A, et al. In vivo contribution of amino acid sulfur to cartilage proteoglycan sulfation. Biochem J. 2006;398(3):509‐514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rossi A, Cetta G, Piazza R, Bonaventure J, Steinmann B, Supereti‐Furga A. In vitro proteoglycan sulfation derived from sulfhydryl compounds in sulfate transporter chondrodysplasias. Pediatr Pathol Mol Med. 2003;22(4):311‐321. [DOI] [PubMed] [Google Scholar]
- 39. Teder K, Maddison L, Soeorg H, Meos A, Karjagin J. The pharmacokinetic profile and bioavailability of enteral N‐Acetylcysteine in intensive care unit. Medicina (Kaunas). 2021;57(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Karczewski KJ, Francioli LC, Tiao G, et al. Genome aggregation database C. the mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434‐443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bjornsdottir G, Stefansdottir L, Thorleifsson G, et al. Rare SLC13A1 variants associate with intervertebral disc disorder highlighting role of sulfate in disc pathology. Nat Commun. 2022;13(1):634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dawson PA, Russell CS, Lee S, et al. Urolithiasis and hepatotoxicity are linked to the anion transporter Sat1 in mice. J Clin Invest. 2010;120(3):706‐712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. de Vries HG, van der Meulen MA, Rozen R, et al. Haplotype identity between individuals who share a CFTR mutation allele "identical by descent": demonstration of the usefulness of the haplotype‐sharing concept for gene mapping in real populations. Hum Genet. 1996;98(3):304‐309. [DOI] [PubMed] [Google Scholar]
- 44. Collee JM, de Vries HG, Scheffer H, Halley DJ, ten Kate LP. Relative frequencies of cystic fibrosis mutations in The Netherlands as an illustration of significant regional variation in a small country. Hum Genet. 1998;102(5):587‐590. [DOI] [PubMed] [Google Scholar]
- 45. Gan KH, Veeze HJ, van den Ouweland AM, et al. A cystic fibrosis mutation associated with mild lung disease. N Engl J Med. 1995;333(2):95‐99. [DOI] [PubMed] [Google Scholar]
- 46. Leir SH, Parry S, Palmai‐Pallag T, et al. Mucin glycosylation and sulphation in airway epithelial cells is not influenced by cystic fibrosis transmembrane conductance regulator expression. Am J Respir Cell Mol Biol. 2005;32(5):453‐461. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supporting Information.
Data Availability Statement
All data described in this study are provided within the article and supplementary material. Raw sequencing data and de‐identified clinical data is available from the corresponding authors upon request.