

Abstract
BACKGROUND
Targeting programmed cell death protein 1 (PD‐1) and indoleamine 2,3‐dioxygenase (IDO1) pathways is an appealing option for cancer treatment.
METHODS
The open‐label, phase 1/2 ECHO‐203 study evaluated the safety, tolerability, and efficacy of the IDO1 inhibitor epacadostat in combination with durvalumab, a human anti–PD‐L1 monoclonal antibody in adult patients with advanced solid tumors.
RESULTS
The most common treatment‐related adverse events were fatigue (30.7%), nausea (21.0%), decreased appetite (13.1%), pruritus (12.5%), maculopapular rash (10.8%), and diarrhea (10.2%). Objective response rate (ORR) in the overall phase 2 population was 12.0%. Higher ORR was observed in immune checkpoint inhibitor (CPI)‐naïve patients (16.1%) compared with patients who had received previous CPI (4.1%). Epacadostat pharmacodynamics were evaluated by comparing baseline kynurenine levels with those on therapy at various time points. Only the 300‐mg epacadostat dose showed evidence of kynurenine modulation, albeit unsustained.
CONCLUSIONS
Epacadostat plus durvalumab was generally well tolerated in patients with advanced solid tumors. ORR was low, and evaluation of kynurenine concentration from baseline to cycle 2, day 1, and cycle 5, day 1, suggested >300 mg epacadostat twice daily is needed to ensure sufficient drug effect.
Clinical trial information
A study of epacadostat (INCB024360) in combination with durvalumab (MEDI4736) in subjects with selected advanced solid tumors (ECHO‐203) (NCT02318277).
Keywords: durvalumab, epacadostat, kynurenine, neoplasms, PD‐1
Short abstract
Epacadostat plus durvalumab was generally well tolerated in patients with advanced solid tumors. Objective response rate was low, and evaluation of kynurenine concentration from baseline to cycle 2, day 1, and cycle 5, day 1, suggested that >300 mg twice daily of epacadostat is needed to ensure sufficient drug effect.
INTRODUCTION
Indoleamine 2,3‐dioxygenase (IDO1), a potential therapeutic target for cancer treatment, catalyzes the first and rate‐limiting step of tryptophan degradation in the kynurenine (KYN) pathway. 1 IDO1 is overexpressed by an array of human tumor and dendritic cells. 2 Increased IDO1 expression in tumor cells is associated with reduced overall survival in patients with melanoma, ovarian, colorectal, and pancreatic cancers. 3 , 4 , 5 , 6 , 7 , 8 Epacadostat is a potent and highly selective IDO1 enzyme inhibitor in both tumor and dendritic cells that reduces conversion of tryptophan to KYN. 9
In a phase 1 study of advanced solid tumors, epacadostat monotherapy was well tolerated, but no objective responses were reported. 10 Preclinical data demonstrated synergy between immune checkpoint inhibitors (CPIs) and IDO1 inhibitors, 11 and IDO1 and CPI programmed death‐ligand 1 (PD‐L1) are often coexpressed in tumor microenvironments. 12 Furthermore, anti–programmed cell death protein 1 (PD‐1) treatment can induce interferon γ production, which can induce IDO1 expression. 13 Thus, targeting PD‐1 and IDO pathways is an attractive option for cancer treatment. Durvalumab, a human anti–PD‐L1 monoclonal antibody that inhibits binding of PD‐L1 to PD‐1, 14 is approved to treat unresectable stage III non–small cell lung cancer (NSCLC) and extensive stage small cell lung cancer. 15 , 16 The phase 1/2 ECHO‐203 (NCT02318277) study evaluated safety, tolerability, and efficacy of epacadostat in combination with durvalumab across multiple advanced solid tumor types.
MATERIALS AND METHODS
Study population
ECHO‐203 was an open‐label, phase 1/2 study of epacadostat plus durvalumab in patients with histologically confirmed advanced melanoma, NSCLC, pancreatic cancer (phase 1 only), squamous cell carcinoma of the head and neck (SCCHN), triple‐negative breast cancer (TNBC), gastric or gastroenterologic cancer, or bladder cancer (phase 2 only). Patients were aged ≥18 years with an Eastern Cooperative Oncology Group performance status of 0 or 1 for whom one or more previous treatment regimen for locally advanced or metastatic disease had failed. Patients with metastatic melanoma were required to have a known V600E‐activating BRAF mutation status. In phase 1, patients who were BRAF mutation positive must have received previous treatment with a BRAF inhibitor with or without a MEK inhibitor. Patients with NSCLC who had an EGFR mutation or ALK fusion gene must have received targeted therapy and might have received a prior anti–PD‐1 target agent. Patients with pancreatic cancer were required to have an exocrine pancreatic neoplasm. Patients with SCCHN must have received prior platinum‐based therapy. Patients with gastroenterologic cancer were required to have a known HER2/neu status and have progressed after treatment with platinum or fluoropyrimidine and a HER2‐targeted agent, if appropriate. Patients were excluded for chronic use of systemic steroids at doses of ≥10 mg/day, untreated central nervous system metastases or carcinomatous meningitis, interstitial lung disease or noninfectious pneumonitis, clinically significant cardiac disease, known HIV infection, pregnancy, or receipt of monoamine oxidase inhibitors within 3 weeks or radiation within 2 weeks before initial study treatment.
Study design and treatment
Phase 1 was an open‐label dose escalation study to identify the maximum tolerated dose (MTD) or pharmacologically active dose of epacadostat in combination with durvalumab using a 3 + 3 dose‐escalation design. Patients received 25 mg of oral epacadostat twice daily in combination with 3 mg/kg of intravenous durvalumab every 2 weeks on day 1 of a 14‐day cycle, or 25, 50, 75, 100, or 300 mg of oral epacadostat twice daily in combination with 10 mg/kg of intravenous durvalumab every 2 weeks on day 1 of a 14‐day cycle for up to 12 months. A minimum of three patients were treated in each cohort and observed for a minimum of 42 days before enrolling a subsequent cohort. Epacadostat dosing was escalated if none of three evaluable patients in the previous cohort experienced a dose‐limiting toxicity (DLT). If a DLT occurred, the cohort was expanded to include three additional patients for treatment at that dose level. If ≥2 of either three or six enrolled patients experienced a DLT, the prior dose level was considered the MTD. Dose interruption or dose reduction (epacadostat only) was allowed in patients experiencing protocol‐defined adverse events (AEs). Hematologic DLT was defined as grade 4 thrombocytopenia; grade ≥3 neutropenia lasting >5 days; grade 4 anemia, febrile neutropenia, thrombotic thrombocytopenic purpura, hemolytic uremic syndrome, disseminated intravascular coagulation, or idiopathic thrombocytopenic purpura; or grade ≥3 hemolysis. Nonhematologic DLT included any grade ≥3 drug‐related or immune‐related toxicity or any grade ≥3 aspartate aminotransferase, alanine aminotransferase, or total bilirubin elevation. Additionally, patients who experienced >2‐week delay in starting cycle 4 or were unable to receive 75% of epacadostat or three doses of durvalumab during the DLT observation period because of a treatment‐related toxicity were classified as DLT even if DLT toxicity criteria were not met.
In phase 2, patients received two dose schedules of epacadostat (100 or 300 mg twice daily) plus durvalumab 10 mg/kg every 2 weeks. Enrollment in the melanoma, TNBC, and gastric cohorts was initiated once epacadostat 100 mg twice daily in combination with durvalumab was determined to be safe. On completion of dose escalation and determining the safety of epacadostat 300 mg twice daily in this combination, further enrollment into these and additional cohorts proceeded with this dose. After 12 months of treatment, patients discontinued both drugs and entered the safety follow‐up period. The initial version of the protocol allowed patients to continue treatment for up to an additional 12 months if their physician‐investigator determined they were experiencing clinical benefit. This option was removed by amendment during the study. Safety follow‐up visits were conducted 42 and 90 days after treatment cessation.
Epacadostat pharmacokinetic (PK) samples were collected in phase 1 on cycle 1, day 1 (C1D1), cycle 1, day 8, and cycle 2, day 1 (C2D1), and in phase 2 on C1D1 and C2D1. Durvalumab PK samples were collected in phase 1 on day 1 of cycles 1, 2, 5, 9, and 13 and every 8 weeks thereafter, at the end of treatment, and at the 90‐day safety follow‐up visit. For patients in phase 1, immunogenicity sampling occurred at cycles 5, 13, and 25. For patients in phase 2, immunogenicity and soluble PD‐L1 sampling occurred at cycles 7, 13, and 25.
This study was conducted in accordance with the provisions of the Declaration of Helsinki, as described in the International Council for Harmonization Guidelines for Good Clinical Practice, and was approved by the institutional review board at each participating institution. All patients provided informed consent before treatment initiation.
End Points
The primary endpoint for phase 1 was to determine the MTD or pharmacologically active dose. The primary endpoint for phase 2 was the objective response rate (ORR) per modified Response Evaluation Criteria in Solid Tumors (RECIST; version 1.1), in which confirmation of progressive disease was required by repeated, consecutive assessment no less than 4 weeks from first documentation. Tumor imaging occurred every 8–12 weeks during treatment and within 7 days after the last dose during treatment discontinuation. Stable disease (SD) was defined as meeting SD criteria at least once after study entry at a minimum interval of 56 (±7) days. Subjects who fail to meet these criteria will have best response of progressive disease (PD) if the next available RECIST evaluation after the initial scan indicates PD or not evaluable if there are no additional RECIST evaluations available. ORR by prior CPI status and PD‐L1 status were determined by investigator‐reported assessment using RECIST criteria.
Secondary endpoints assessed the safety and tolerability of epacadostat and durvalumab combination therapy, progression‐free survival (PFS), durvalumab and epacadostat PK, and the prevalence of anti‐durvalumab antibodies.
Safety and tolerability were assessed by frequency and severity of AEs as defined by Common Terminology Criteria for Adverse Events (version 4.03). AEs of special interest (AESI) were assessed using a predefined list associated with durvalumab monotherapy.
Statistical analysis
Safety analysis included all patients who received at ≥1 dose of epacadostat and durvalumab. Efficacy analysis included all enrolled patients (intent‐to‐treat population). The PK‐evaluable population included patients who had received ≥1 epacadostat dose and provided ≥1 postdose plasma sample. Median PFS was estimated using the nonparametric Kaplan–Meier method. Epacadostat PK was estimated by noncompartmental analysis, population PK modeling, or both. SAS software (version 9.1) (SAS Institute Inc., Cary, NC, USA) was used to generate all tables, graphs, and statistical analyses.
RESULTS
Patient disposition
Thirty‐four patients with NSCLC, SCCHN, pancreatic cancer, and melanoma were enrolled in phase 1. Patients received epacadostat plus durvalumab across six dosing cohorts as described in the Materials and Methods (Table 1). At the data cutoff date of August 28, 2019, five patients (14.7%) had completed 12 months of treatment, and all 34 patients had discontinued combination treatment. The reasons for treatment discontinuation were disease progression (n = 23; 67.6%), completion of 12‐month treatment (n = 5; 14.7%), AEs (n = 3; 8.8%), physician decision (n = 2; 5.9%), and death (n = 1; 2.9%). One patient was ongoing in the study, and 33 patients had been discontinued. The reasons for study discontinuation were death (n = 24; 70.6%), study termination by the sponsor (n = 4; 11.8%), consent withdrawn (n = 3; 8.8%), and loss to follow‐up (n = 2; 5.9%).
TABLE 1.
Patient disposition
| Disposition status, No. (%) | Phase 1 | Phase 2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 3 mg/kg Durva | 10 mg/kg Durva | Total (N = 34) | 10 mg/kg Durva | Total (N = 142) | ||||||
| 25 mg Epa (n = 6) | 25 mg Epa (n = 3) | 50 mg Epa (n = 4) | 75 mg Epa (n = 4) | 100 mg Epa (n = 8) | 300 mg Epa (n = 9) | 100 mg Epa (n = 49) | 300 mg Epa (n = 93) | |||
| Patients who completed 12 mo of combination treatment | 0 | 1 (33.3) | 1 (25.0) | 0 | 1 (12.5) | 2 (22.2) | 5 (14.7) | 4 (8.2) | 8 (8.6) | 12 (8.5) |
| Patients who discontinued combination treatment | 6 (100.0) | 3 (100.0) | 4 (100.0) | 4 (100.0) | 8 (100.0) | 9 (100.0) | 34 (100.0) | 49 (100.0) | 93 (100.0) | 142 (100.0) |
| Primary reason for treatment discontinuation | 6 (100.0) | 1 (33.3) | 2 (50.0) | 4 (100.0) | 4 (50.0) | 6 (66.7) | 23 (67.6) | 41 (83.7) | 69 (74.2) | 110 (77.5) |
| Disease progression adverse event | 0 | 1 (33.3) | 1 (25.0) | 0 | 1 (12.5) | 0 | 3 (8.8) | 1 (2.0) | 10 (10.8) | 11 (7.7) |
| Death | 0 | 0 | 0 | 0 | 1 (12.5) | 0 | 1 (2.9) | 0 | 1 (1.1) | 1 (0.7) |
| Consent withdrawn | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (2.0) | 2 (2.2) | 3 (2.1) |
| Lost to follow‐up | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Abbreviations: Durva, durvalumab; Epa, epacadostat.
In phase 2, 142 patients with NSCLC, SCCHN, TNBC, melanoma, bladder cancer, and gastric cancer were treated with epacadostat 100 mg twice daily (n = 49) and 300 mg twice daily (n = 93) in combination with 10 mg/kg of durvalumab (Table 1). As of August 28, 2019, 12 patients (8.5%) had completed 12 months of treatment, and all 142 patients had discontinued combination treatment. Reasons for combination treatment discontinuation were disease progression (n = 110; 77.5%), completion of 12 months of treatment (n = 12; 8.5%), AEs (n = 11; 7.7%), physician decision (n = 4; 2.8%), death (n = 1; 0.7%), and other (n = 1; 0.7%). All 142 patients had discontinued the study because of death (n = 92; 64.8%), study termination by the sponsor (n = 21; 14.8%), consent withdrawn (n = 20; 14.1%), loss to follow‐up (n = 7; 4.9%), physician decision (n = 1; 0.7%), or other (n = 1; 0.7%).
Baseline characteristics
Baseline demographics and disease characteristics for all patients are shown in Table 2. Among patients in phase 1, two patients with NSCLC had an EGFR mutation, two had a KRAS mutation, and one an ALK rearrangement; nine patients had adenocarcinoma and one had squamous NSCLC; one patient had received a prior tyrosine kinase inhibitor. Two patients with SCCHN were positive for human papilloma virus; 1 each had an EGFR mutation and a p53 mutation. Two patients had received a prior CPI (ipilimumab, n = 1; ipilimumab and nivolumab, n = 1). At initial diagnosis, four patients with SCCHN had a primary oral cavity tumor, two in the larynx, one in the oropharynx, and one in other. Five SCCHN patients had a poorly differentiated tumor, one had an intermediately differentiated tumor, and one had an undifferentiated tumor. Among patients with pancreatic cancer, six had a Whipple procedure (pancreatoduodenectomy), four had a pancreatectomy, and six had a biliary stent.
TABLE 2.
Patient demographics and disease characteristics at baseline
| Baseline characteristics, No. (%) | Phase 1 | Phase 2 | ||
|---|---|---|---|---|
| Total a (N = 34) | 100 mg Epa b (n = 49) | 300 mg Epa b (n = 93) | Total (N = 142) | |
| Age, median (range), y | 68 (46–84) | 60 (31‒85) | 65 (29‒87) | 64 (29‒87) |
| Age ≥65 y | 22 (64.7) | 17 (34.7) | 49 (52.7) | 66 (46.5) |
| Male | 21 (61.8) | 30 (61.2) | 68 (73.1) | 98 (69.0) |
| Race | ||||
| White | 33 (97.1) | 41 (83.7) | 83 (89.2) | 124 (87.3) |
| Black or African American | 1 (2.9) | 5 (10.2) | 5 (5.4) | 10 (7.0) |
| ECOG PS | ||||
| 0 | 6 (17.6) | 6 (12.2) | 20 (21.5) | 26 (18.3) |
| 1 | 28 (82.4) | 43 (87.8) | 72 (77.4) | 115 (81.0) |
| ≥2 | 0 | 0 | 1 (1.1) | 1 (0.7) |
| Tumor type | ||||
| Pancreatic | 15 (44.1) | 0 | 0 | 0 |
| TNBC | 0 | 13 (26.5) | 0 | 13 (9.2) |
| NSCLC | 10 (29.4) | 9 (18.4) | 28 (30.1) | 37 (26.1) |
| SCCHN | 8 (23.5) | 7 (14.3) | 31 (33.3) | 38 (26.8) |
| Melanoma | 1 (2.9) | 7 (14.3) | 8 (8.6) | 15 (10.6) |
| Gastric | 0 | 9 (18.4) | 0 | 9 (6.3) |
| Bladder | 0 | 4 (8.2) | 26 (28.0) | 30 (21.1) |
| Prior treatments for advanced/metastatic disease | ||||
| 0 | 1 (2.9) | 8 (16.3) | 15 (16.1) | 23 (16.2) |
| 1 | 11 (32.4) | 10 (20.4) | 27 (29.0) | 37 (26.1) |
| ≥2 | 22 (64.7) | 31 (63.3) | 51 (54.8) | 82 (57.7) |
| Prior use of checkpoint inhibitors | ||||
| Yes | 2 (5.9) | 9 (18.4) | 40 (43.0) | 49 (34.5) |
| No | 32 (94.1) | 40 (81.6) | 53 (57.0) | 93 (65.5) |
Abbreviations: ECOG PS, Eastern Cooperative Oncology Group performance status; Epa, epacadostat; NSCLC, non–small cell lung cancer; SCCHN, squamous cell carcinoma of the head and neck; TNBC, triple‐negative breast cancer.
Plus durvalumab 3 mg/kg every 2 weeks or 10 mg/kg every 2 weeks.
Plus durvalumab 10 mg/kg every 2 weeks.
In phase 2, one patient with NSCLC had an EGFR mutation, five had a KRAS mutation, and one an ALK rearrangement; nine patients had received a prior tyrosine kinase inhibitor. Histologically typed NSCLC samples showed that 26 patients had adenocarcinoma, one had large cell carcinoma, six had squamous NSCLC, two had adenosquamous carcinoma (mixed), and two had other types of NSCLC. Sixteen patients with SCCHN were human papilloma virus positive; three patients had an EGFR mutation, and five had a p53 mutation. At initial diagnosis, nine patients had a primary tumor in the oral cavity, five had a tumor in the larynx, 15 a tumor in the oropharynx, two a tumor in the hypopharynx, and seven a tumor in other. Fifteen patients with SCCHN had poorly differentiated tumors, six had intermediately differentiated tumors, four had well‐differentiated tumors, and 13 had undifferentiated tumors. Four patients with melanoma were positive for BRAF‐V600. Three patients with gastric cancer had tumors that were well or moderately differentiated, and six had tumors that were poorly differentiated or undifferentiated. Histologic classification of gastric cancer samples showed that three patients had indeterminate type, four had other types, and two had unknown types of gastric cancer. No patient was positive for Epstein‐Barr virus; one was positive for Helicobacter pylori. Of 30 patients with bladder cancer, 28 had transitional cell carcinoma, and two had carcinomas of other type. PD‐L1 status was positive in one patient, negative in two, and missing in 27. Of 13 patients with TNBC, five were BRCA negative, two were PD‐L1 negative, and one was MYC rearrangement positive. Forty‐nine patients had received a prior CPI (ipilimumab, n = 8; nivolumab, n = 24; pembrolizumab, n = 16; atezolizumab, n = 10; avelumab, n = 1; enoblituzumab, n = 1).
Safety and tolerability
In phase 1, median duration of exposure to epacadostat was 84.5 days (range, 14‐1339). The most common (occurring in ≥5% of patients) treatment‐related AEs (TRAEs) were fatigue (32.4%), pruritus (17.6%), nausea (11.8%), and diarrhea (11.8%) (Table 3). Grade ≥3 TRAEs were observed in seven patients (20.6%). The only grade ≥3 or higher TRAEs that occurred in >1 patient were fatigue (n = 3; 8.8%) and rash maculopapular (n = 2; 5.9%). No TRAEs leading to death were reported. One DLT of grade 3 rash requiring systemic steroids was reported in the epacadostat 300 mg twice daily plus durvalumab 10 mg/kg every‐2‐week cohort. The MTD was not reached, and epacadostat 100 mg twice daily and 300 mg twice daily were further evaluated in phase 2. AESIs included immune‐mediated AEs and serotonin syndrome. The most common (occurring in ≥5% of patients) immune‐mediated AESIs were pruritus (5.9%) and maculopapular rash (5.9%). No hepatic function abnormalities were reported. Two instances of pneumonitis (grade 2 and grade 1) occurred in one patient and resulted in treatment discontinuation; serotonin syndrome (grade 2) occurred in one patient who recovered but discontinued treatment because of disease progression.
TABLE 3.
Treatment‐related adverse events occurring in ≥5% of patients in phase 1 and phase 2
| Adverse event, No. (%) | Phase 1 | Phase 2 | ||||||
|---|---|---|---|---|---|---|---|---|
| Total (N = 34) | 100 mg Epa a (n = 49) | 300 mg Epa a (n = 93) | Total (N = 142) | |||||
| Any grade | Grade 3/4 | Any grade | Grade 3/4 | Any grade | Grade 3/4 | Any grade | Grade 3/4 | |
| Total | 27 (79.4) | 7 (20.6) | 35 (71.4) | 3 (6.1) | 79 (84.9) | 23 (24.7) | 114 (80.3) | 26 (18.3) |
| Fatigue | 11 (32.4) | 3 (8.8) | 12 (24.5) | 0 | 31 (33.3) | 4 (4.3) | 43 (30.3) | 4 (2.8) |
| Nausea | 4 (11.8) | 0 | 11 (22.4) | 0 | 22 (23.7) | 1 (1.1) | 33 (23.2) | 1 (0.7) |
| Decreased appetite | 3 (8.8) | 0 | 5 (10.2) | 0 | 15 (16.1) | 0 | 20 (14.1) | 0 |
| Maculopapular rash | 2 (5.9) | 2 (5.9) | 8 (16.3) | 1 (2.0) | 9 (9.7) | 5 (5.4) | 17 (12.0) | 6 (4.2) |
| Pruritus | 6 (17.6) | 0 | 3 (6.1) | 0 | 13 (14.0) | 0 | 16 (11.3) | 0 |
| Diarrhea | 4 (11.8) | 0 | 4 (8.2) | 0 | 10 (10.8) | 0 | 14 (9.9) | 0 |
| Vomiting | 0 | 0 | 3 (6.1) | 1 (2.0) | 10 (10.8) | 1 (1.1) | 13 (9.2) | 2 (1.4) |
| Increased AST | 1 (2.9) | 0 | 2 (4.1) | 0 | 9 (9.7) | 2 (2.2) | 11 (7.7) | 2 (1.4) |
| Rash | 1 (2.9) | 0 | 2 (4.1) | 0 | 9 (9.7) | 0 | 11 (7.7) | 0 |
| Increased ALP | 1 (2.9) | 0 | 0 | 0 | 9 (9.7) | 2 (2.2) | 9 (6.3) | 2 (1.4) |
| Chills | 1 (2.9) | 0 | 4 (8.2) | 0 | 5 (5.4) | 0 | 9 (6.3) | 0 |
| Headache | 1 (2.9) | 0 | 2 (4.1) | 0 | 7 (7.5) | 0 | 9 (6.3) | 0 |
| Pyrexia | 2 (5.9) | 0 | 6 (12.2) | 0 | 2 (2.2) | 0 | 8 (5.6) | 0 |
| Increased ALT | 0 | 0 | 1 (2.0) | 0 | 7 (7.5) | 2 (2.2) | 8 (5.6) | 2 (1.4) |
| Dizziness | 2 (5.9) | 0 | 2 (4.1) | 0 | 3 (3.2) | 1 (1.1) | 5 (3.5) | 1 (0.7) |
| Tumor flare | 3 (8.8) | 0 | 1 (2.0) | 0 | 3 (3.2) | 0 | 4 (2.8) | 0 |
| Constipation | 2 (5.9) | 0 | 1 (2.0) | 0 | 3 (3.2) | 2 (2.2) | 4 (2.8) | 2 (1.4) |
| Dyspnea | 2 (5.9) | 1 (2.9) | 3 (6.1) | 0 | 1 (1.1) | 0 | 4 (2.8) | 0 |
| Influenza‐like illness | 2 (5.9) | 0 | 0 | 0 | 4 (4.3) | 0 | 4 (2.8) | 0 |
| Anxiety | 2 (5.9) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Bone pain | 2 (5.9) | 0 | 2 (4.1) | 0 | 1 (1.1) | 0 | 3 (2.1) | 0 |
| Dry mouth | 2 (5.9) | 0 | 0 | 0 | 3 (3.2) | 0 | 3 (2.1) | 0 |
| Hyponatremia | 2 (5.9) | 0 | 1 (2.0) | 0 | 1 (1.1) | 1 (1.1) | 2 (1.4) | 1 (0.7) |
| Decreased weight | 2 (5.9) | 0 | 0 | 0 | 2 (2.2) | 0 | 2 (1.4) | 0 |
| Cough | 2 (5.9) | 0 | 0 | 0 | 1 (1.1) | 0 | 1 (0.7) | 0 |
Abbreviations: ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; Epa, epacadostat.
Plus durvalumab 10 mg/kg every 2 weeks.
In phase 2, median duration of exposure to epacadostat was 62.0 days (range, 5–839). The most common (occurring in ≥5% of patients) TRAEs were fatigue (30.3%), nausea (23.2%), decreased appetite (14.1%), maculopapular rash (12.0%), and pruritus (11.3%) (Table 3). Grade ≥3 TRAEs were observed in 26 patients (18.3%). No TRAEs leading to death were reported. The most common (occurring in ≥5% of patients) immune‐mediated AESIs were maculopapular rash (7.7%) and pruritus (5.6%); no other AESIs were reported in phase 2.
Efficacy
In the overall population, 17 patients (12.0%) had objective responses per RECIST (95% CI, 7.1–18.5); three patients (2.1%) had a complete response (CR), and 14 patients (9.9%) had a partial response (PR). A higher ORR was observed in the CPI‐naïve population than in the CPI‐experienced population. ORR in CPI‐naïve patients (n = 93) was 16.1% (95% CI, 9.3–25.2) (Table 4); three patients (3.2%) had a CR and 12 patients (12.9%) had a PR. Among CPI‐naïve patients, those with melanoma had the best ORR (n = 5; 80.0%; 95% CI, 28.4–99.5) in comparison with patients with other tumor types, including two CRs and two PRs. ORR in CPI‐experienced patients (n = 49) was 4.1% (95% CI, 0.5–14.0); no patient had a CR, and two patients (4.1%) had a PR. One patient with NSCLC previously received pembrolizumab for approximately 6 months with a best response of stable disease before disease progression. The other patient had melanoma and previously received nivolumab for approximately 2 months with a best response of progressive disease. Treatment in the current trial for the NSCLC and melanoma patients began 4 months and 1 month after discontinuing prior anti–PD‐1 treatment, respectively. Both PRs with previous CPI treatment received epacadostat 300 mg twice daily and durvalumab 10 mg/kg every 2 weeks in the current study. No responses were observed in patients with gastric cancer (0%; 95% CI, 0.0–33.6) in either category. Treatment responses by dose in CPI‐naïve individual patients enrolled in phase 2 are presented in Figure 1. There was no clear relationship between doses of epacadostat (100 mg twice daily vs 300 mg twice daily) and tumor responses. Median PFS was 1.9 months (range, 1.8–3.6) in CPI‐naïve patients and 2.0 months (95% CI, 1.9–2.7) in all phase 2 patients.
TABLE 4.
Best ORR by RECIST in CPI‐naïve patients
| NSCLC (N = 20) | SCCHN (N = 27) | Melanoma (N = 5) | Bladder cancer (N = 19) | Gastric cancer (N = 9) | TNBC (N = 13) | Total (N = 93) | |
|---|---|---|---|---|---|---|---|
| ORR, % (95% CI) | 15.0 (3.2–37.9) | 14.8 (4.2–33.7) | 80.0 (28.4–99.5) | 15.8 (3.4–39.6) | 0 (0.0–33.6) | 7.7 (0.2–36.0) | 16.1 (9.3–25.2) |
| CR, No. (%) | 0 | 0 | 2 (40.0) | 1 (5.3) | 0 | 0 | 3 (3.2) |
| PR, No. (%) | 3 (15.0) | 4 (14.8) | 2 (40.0) | 2 (10.5) | 0 | 1 (7.7) | 12 (12.9) |
| SD, No. (%) | 7 (35.0) | 11 (40.7) | 0 | 0 | 0 | 3 (23.1) | 21 (22.6) |
| PD, No. (%) | 7 (35.0) | 7 (25.9) | 1 (20.0) | 8 (42.1) | 6 (66.7) | 7 (53.8) | 36 (38.7) |
| NE, No. (%) | 0 | 0 | 0 | 0 | 0 | 1 (7.7) | 1 (1.1) |
| Missing, No. (%) | 3 (15.0) | 5 (18.5) | 0 | 8 (42.1) | 3 (33.3) | 1 (7.7) | 20 (21.5) |
Abbreviations: CPI, checkpoint inhibitor; CR, complete response; NE, not evaluable; NSCLC, non–small cell lung cancer; ORR, objective response rate; PD, progressive disease; PR, partial response; RECIST, Response Evaluation Criteria in Solid Tumors; SCCHN, squamous cell carcinoma of head and neck cancer; SD, stable disease; TNBC, triple negative breast cancer.
FIGURE 1.
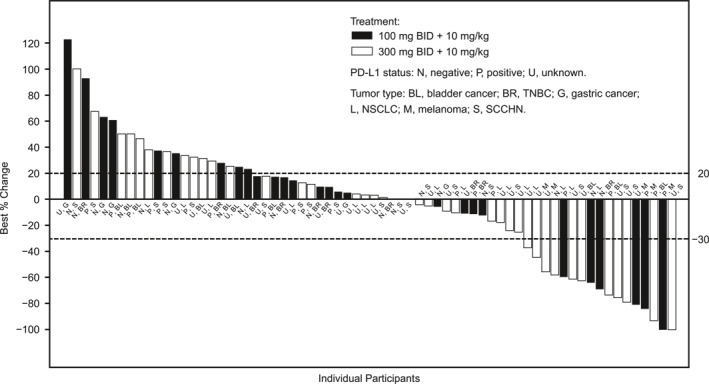
Change in target lesion from baseline (best response) in CPI‐naïve patients enrolled in phase 2 of the study. BID indicates twice daily; CPI, checkpoint inhibitor; NSCLC, non–small cell lung cancer; PD‐L1, programmed death ligand‐1; SCCHN, squamous cell carcinoma of the head and neck; TNBC, triple‐negative breast cancer.
Pharmacokinetics
Epacadostat exposure was generally consistent with previous reports. Peak exposures (Cmax±SD) of 100 mg epacadostat twice daily and 300 mg epacadostat twice daily doses were 960 ± 499 nM and 2500 ± 1190 nM, respectively. The areas under the concentration‐time curves (AUC±SD) for 100 mg epacadostat twice daily and 300 mg epacadostat twice daily doses were 4130 ± 1960 h/nM and 12,200 ± 5950 h/nM, respectively. Both 100 mg epacadostat twice daily and 300 mg epacadostat twice daily doses exhibited similar half‐lives (t1/2±SD, 4.10 h ± 1.69 h and 4.09 h ± 1.99 h, respectively), which were also consistent with historical data.
Patients with pancreatic cancer had lower peak exposures (Cmax). When dose‐normalized to 100 mg twice daily, patients who had a Whipple procedure demonstrated a lower AUC and Cmax (n = 5; 31.7 ± 22.2 h/nM/mg and 5.76 ± 6.43 nM/mg, respectively) in comparison to those who did not have a Whipple procedure (n = 101; 41.2 ± 20.2 h/nM/mg and 8.78 ± 4.39 nM/mg, respectively), suggesting a slight difficulty in absorbing epacadostat. These exposures are similar to those reported for epacadostat 50 mg twice daily. 13
Pharmacodynamics
The PD activity of epacadostat is demonstrated by the dose‐dependent decrease of KYN in plasma. Dose‐dependent PD change of plasma KYN (in microns) is shown in Figure 2. The 25‐mg group showed no decrease in KYN from cycle 1 to cycle 2. In fact, six of eight participants from this group demonstrated a slight increase in KYN level. This is presumably from the increased IDO1 activity from anti–PD‐L1 (durvalumab) therapy. By paired t test, the 300‐mg group was the only dose group with a significant decrease in KYN between C1D1 and C2D1.
FIGURE 2.
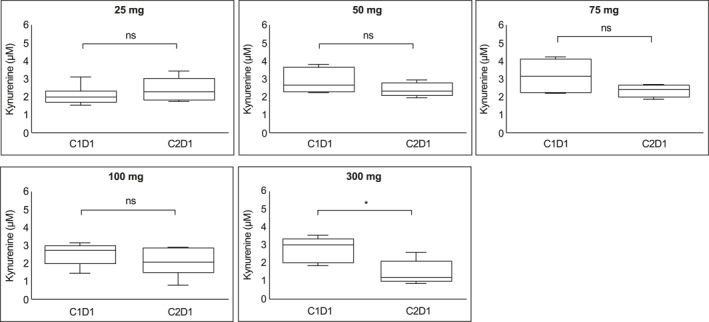
Dose‐dependent pharmacodynamic change of kynurenine. Kynurenine level at trough on C1D1 and C2D1 are shown in box graphs. Significance of difference from C1D1 to C2D1 was calculated using paired t test. Median levels are indicated. C1D1 indicates cycle 1 day 1; C2D1, cycle 2 day 1; ns, not significant. *p < .05.
To further evaluate the PD effect of epacadostat 300 mg BID, we compared plasma KYN concentration at steady state on C2D1 with the pretreatment level on C1D1 in patients with potentially immune responsive tumor types (SCCHN and NSCLC). Notably, there was a lack of consistent KYN reduction at C2D1 in responders treated with epacadostat 300 mg twice daily in CPI‐naïve patients (data on file). Further analysis revealed a rebound effect in mean KYN concentration at cycle 5, day 1, in patients with SCCHN and melanoma.
DISCUSSION
As a key regulator of immune escape, 17 IDO1 continues to be a valid target for cancer treatment. Previous studies have shown IDO1 is often overexpressed in cancer patients, and IDO1 overexpression is correlated with higher mortality. Additionally, studies with 1‐methyl‐tryptophan demonstrate IDO1 inhibition can significantly increase chemotherapy efficacy without increased toxicity. 17 Together, these results suggest IDO1 inhibitors are promising agents to study for the treatment of advanced solid tumors in combination with chemotherapeutics or immunotherapies.
In this phase 1/2 study in patients with advanced solid tumors, we show that epacadostat plus durvalumab was generally well tolerated, and epacadostat's safety profile was consistent with previous reports of durvalumab monotherapy. 18 Epacadostat exposure was generally consistent with previous reports, except in patients with pancreatic cancer, in whom lower peak exposures were observed (potentially because of Whipple procedures). After the phase 3 study of epacadostat in combination with pembrolizumab (ECHO‐301/KEYNOTE‐252) failed to meet its primary endpoint, enrollment in the current study was stopped. 19 However, dose exposure differed between the studies; ECHO‐301 provided patients with epacadostat 100 mg twice daily, whereas in phase 2 of this study, patients received two dose schedules of epacadostat (100 or 300 mg twice daily). Our pharmacokinetic and pharmacodynamic data suggest that even 300 mg epacadostat twice daily was insufficient to induce sustained target inhibition. Thus, the limited clinical activity observed in this study and in previously reported randomized phase 3 trials may be due to inadequate epacadostat exposure. Consistent with this hypothesis, a retrospective pooled analysis of several studies in combination with PD‐1 inhibition has shown epacadostat doses of ≥600 mg twice daily may be required to suppress kynurenine production to levels reported in healthy individuals. 20
Thus, the modest response rate observed is unsurprising. Overall ORR per RECIST criteria was 12.0%. Although ORR was higher among CPI‐naïve patients (16.1%) and highest among CPI‐naïve patients with melanoma (80.0%), the nominal response rates appear similar to what might be expected with PD‐L1 monotherapy, 21 , 22 although sample sizes in disease‐specific cohorts were limited. No baseline disease characteristics predictive of response were identified. Data interpretations in the current study are limited by several factors including small patient numbers, uncertainty of the correlation between plasma KYN and intratumoral KYN, correlation of changes in plasma KYN, and other predictors of response to CPI therapy.
In summary, epacadostat with durvalumab was well tolerated, but epacadostat exposure in this and other recent studies may have been subtherapeutic. Future studies should evaluate higher doses of epacadostat and potential predictive biomarkers, including baseline expression of intratumoral IDO1 expression, to identify patients with tumors likely to respond to epacadostat combination therapies.
AUTHOR CONTRIBUTIONS
Aung Naing: Conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript, and accountability for all aspects of the work. Alain P. Algazi: Collection and assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript, and accountability for all aspects of the work. Gerald S. Falchook: Collection and assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript, and accountability for all aspects of the work. Benjamin C. Creelan: Collection and assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript, and accountability for all aspects of the work. John Powderly: Collection and assembly of data, manuscript writing, final approval of manuscript, and accountability for all aspects of the work. Seth Rosen: Collection and assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript, and accountability for all aspects of the work. Minal Barve: Collection and assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript, and accountability for all aspects of the work. Niharika B. Mettu: Collection and assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript, and accountability for all aspects of the work. Pierre L. Triozzi: Collection and assembly of data; manuscript writing, final approval of manuscript, and accountability for all aspects of the work. John Hamm: Collection and assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript, and accountability for all aspects of the work. Gongfu Zhou: Data analysis and interpretation, manuscript writing, final approval of manuscript, and accountability for all aspects of the work. Chris Walker: Conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript, and accountability for all aspects of the work. Zhiwan Dong: Data analysis and interpretation, manuscript writing, final approval of manuscript, and accountability for all aspects of the work. Manish R. Patel: Collection and assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript, and accountability for all aspects of the work.
CONFLICT OF INTEREST
Aung Naing has received research funding from NCI, EMD Serono, MedImmune, Healios Onc. Nutrition, Atterocor/Millendo, Amplimmune, ARMO BioSciences, Karyopharm Therapeutics, Incyte Corporation, Novartis, Regeneron, Merck, Bristol‐Myers Squibb, Pfizer, CytomX Therapeutics, Neon Therapeutics, Calithera Biosciences, TopAlliance Biosciences, Eli Lilly, Kymab, PsiOxus, Arcus Biosciences, NeoImmuneTech, ImmuneOncia, and Surface Oncology; has been an advisory board member for CytomX Therapeutics, Novartis, Genome & Company, OncoSec KEYNOTE‐695, Kymab, and STCube Pharmaceuticals; and has received travel and accommodation expenses from ARMO BioSciences; spouse has received research funding from Immune Deficiency Foundation, Jeffery Modell Foundation and Chao Physician‐Scientist, and Baxalta; and spouse has been an advisory board member for Takeda, CSL, Behring, Horizon, and Pharming. Alain Algazi has received research support and has been an advisory board member, consultant, shareholder, and honorarium recipient for OncoSec; an advisory board member and stock shareholder for Valitor Biosciences; and an advisory board member and honorarium recipient for Regeneron and Array and has received research support from Acerta, Amgen, AstraZeneca, BMS, Dynavax, Genentech, Idera, Incyte Corporation, ISA, LOXO, Merck, Novartis, Sensei, and Tessa. Gerald S. Falchook has received speaking fees from Total Health Conferencing and Rocky Mountain Oncology Society; royalties from Wolters Kluwer; travel expenses from Bristol‐Myers Squibb, EMD Serono, Fujifilm, Millennium, and Sarah Cannon Research Institute; and research funding from 3‐V Biosciences, Abbisko, AbbVie, ADC Therapeutics, Aileron, American Society of Clinical Oncology, Amgen, ARMO/Eli Lilly, AstraZeneca, BeiGene, Bioatla, Bioinvent, Biothera, Bicycle, Celldex, Celgene, Ciclomed, Curegenix, Curis, Cyteir, Daiichi, DelMar, eFFECTOR, Eli Lilly, EMD Serono, Epizyme, Exelixis, Fujifilm, Genmab, GlaxoSmithKline, Hutchison MediPharma, IGM Biosciences, Ignyta, ImmunoGen/MacroGenics, Incyte Corporation, Jacobio, Jounce, Kolltan, Loxo/Bayer, MedImmune, Millennium, Merck, miRNA Therapeutics, National Institutes of Health, Navire, Novartis, OncoMed, Oncorus, Oncothyreon, Poseida, Precision Oncology, Prelude, PureTech, Regeneron, Rgenix, Ribon, Sapience, Silicon, Strategia, Syndax, Synthorx/Sanofi, Taiho, Takeda, Tarveda, Tesaro, Tocagen, Turning Point Therapeutics, University of Texas MD Anderson Cancer Center, Vegenics, and Xencor and has served in an advisory role for Fujifilm, Silicon, Navire, and EMD Serono. Benjamin Creelan has received speaking fees from AstraZeneca plc, ARIAD Pharmaceuticals, Inc., and F. Hoffmann‐La Roche AG; consultant fees from Xilio Therapeutics, Inc., Achilles plc, E.R. Squibb & Sons, LLC, F. Hoffmann‐La Roche AG, AstraZeneca plc, AbbVie, KSQ Therapeutics, Inc., GlaxoSmithKline plc, Gilead Sciences, Inc., and Celgene Corp; and research funding from NeoGenomics Laboratories and Adaptive Biotechnologies Corp and holds a patent application WO2020263919A1. John Powderly has received research funding to institution from Nucleus Inc, MT Group, STEMCELL, BMS, Roche, AstraZeneca‐MedImmune, EMD Serono, Lilly, Macrogenics, Incyte Corporation, Top Alliance BioScience, Seattle Genetics, AbbVie, Corvus, Curis, RAPT Therapeutics, Alkermes, Arcus BioSciences, Tempest Therapeutics, Calico Life Sciences, Apros, Jounce, Atreca, BioBank Online, Sequenom, Replimmune, Engineered Pharma, CEM Corporation, Therapeutic BrainPower LLC, Merck, Conjupro, and Repertoire Medicines and consulting fees from TerumoBCT; holds joint IP with institution and Atreca, Inc; has developed IP for institution and BioCytics; has served on data safety monitoring board for AbbVie; and has received stock or stock options in BioCytics, Inc. and Carolina BioOncology Institute PLLC. Niharika B. Mettu has received research grant/funding to institution from Amphivena, Incyte Corporation, Erytech Pharma, AstraZeneca plc, Bristol‐Myers Squibb, Genentech, Amgen, Repare, and BioMed Valley Discoveries and travel expenses from Genentech. John Hamm has received research funding to institution from Incyte Corporation. Gongfu Zhou declares employment with and stock ownership in Incyte Corporation. Chris Walker declares employment with and stock ownership in Incyte Corporation. Zhiwan Dong declares employment with and stock ownership in Incyte Corporation. Manish R. Patel has received honoraria for speakers' bureau/advisory boards from Genentech, Exelixis, Pfizer, EMD Serono, Bayer, Celgene, Phamacyclics, Janssen, and AbbVie and research funding to institution from Acerta Pharma, ADC Therapeutics, Agenus, Aileron Therapeutics, AstraZeneca, Bicycle Therapeutics, BioNTech, Boehringer Ingelheim, Calithera, Celgene, Checkpoint Therapeutics, Ciclomed, Clovis, Curis, Cyteir Therapeutics, Daiichi Sankyo, Effector Therapeutics, Eli Lilly, EMD Serono, Evelo Biosciences, Forma Therapeutics, Genentech/Roche, Gilead, GlaxoSmithKline, H3 Biomedicine, Hengrui, Hutchinson MediPharma, Ignyta, Incyte, Jacobio, Janssen, Jounce Therapeutics, Klus Pharma, Kymab, Loxo Oncology, LSK Biopartners, Lycera, Macrogenics, Merck, Millennium Pharmaceuticals, Mirati Therapeutics, ModernaTX, Pfizer, Phoenix Molecular Designs, Placon Therapeutics, Portola Pharmaceuticals, Prelude Therapeutics, Qilu Puget Sound Biotherapeutics, Revolution Medicines, Ribon Therapeutics, Seven and Eight Biopharmaceuticals, Syndax, Synthorx, Stemline Therapeutics, Taiho, Takeda, Tesaro, TopAlliance, Vedanta, Verastem, Vigeo, and Xencor. The other authors made no disclosures.
ACKNOWLEDGMENTS
This study was funded by Incyte Corporation (Wilmington, Delaware, USA) in collaboration with AstraZeneca (Cambridge, UK). The funding body was involved in study design, data analysis, and data interpretation. Medical writing and editorial assistance were provided by Michelle Mancher, MPH, and Celia Nelson of Ashfield MedComms, an Inizio Company, and was funded by Incyte Corporation. Stephanie Vadasz, PhD, a former employee of Ashfield Healthcare Communications (now Ashfield MedComms), also provided medical writing support.
Naing A, Algazi AP, Falchook GS, et al. Phase 1/2 study of epacadostat in combination with durvalumab in patients with metastatic solid tumors. Cancer. 2023;129(1):71‐81. doi: 10.1002/cncr.34512
Funded by Incyte Corporation (Wilmington, Delaware, USA) in collaboration with AstraZeneca (Cambridge, UK).
See editorial on pages 11–4, this issue.
REFERENCES
- 1. Moon YW, Hajjar J, Hwu P, Naing A. Targeting the indoleamine 2, 3‐dioxygenase pathway in cancer. J Immunother Cancer. 2015;3(1):51. doi: 10.1186/s40425-015-0094-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Uyttenhove C, Pilotte L, Théate I, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2, 3‐dioxygenase. Nat Med. 2003;9(10):1269‐1274. doi: 10.1038/nm934 [DOI] [PubMed] [Google Scholar]
- 3. Okamoto A, Nikaido T, Ochiai K, et al. Indoleamine 2, 3‐dioxygenase serves as a marker of poor prognosis in gene expression profiles of serous ovarian cancer cells. Clin Cancer Res. 2005;11(16):6030‐6039. doi: 10.1158/1078-0432.ccr-04-2671 [DOI] [PubMed] [Google Scholar]
- 4. Brandacher G, Perathoner A, Ladurner R, et al. Prognostic value of indoleamine 2, 3‐dioxygenase expression in colorectal cancer: effect on tumor‐infiltrating T cells. Clin Cancer Res. 2006;12(4):1144‐1151. doi: 10.1158/1078-0432.ccr-05-1966 [DOI] [PubMed] [Google Scholar]
- 5. Ino K, Yoshida N, Kajiyama H, et al. Indoleamine 2, 3‐dioxygenase is a novel prognostic indicator for endometrial cancer. Br J Cancer. 2006;95(11):1555‐1561. doi: 10.1038/sj.bjc.6603477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nakamura T, Shima T, Saeki A, et al. Expression of indoleamine 2, 3‐dioxygenase and the recruitment of Foxp3‐expressing regulatory T cells in the development and progression of uterine cervical cancer. Cancer Sci. 2007;98(6):874‐881. doi: 10.1111/j.1349-7006.2007.00470.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Witkiewicz A, Williams TK, Cozzitorto J, et al. Expression of indoleamine 2, 3‐dioxygenase in metastatic pancreatic ductal adenocarcinoma recruits regulatory T cells to avoid immune detection. J Am Coll Surg. 2008;206(5):849‐854. discussion 854‐6. doi: 10.1016/j.jamcollsurg.2007.12.014 [DOI] [PubMed] [Google Scholar]
- 8. Iga N, Otsuka A, Hirata M, et al. Variable indoleamine 2, 3‐dioxygenase expression in acral/mucosal melanoma and its possible link to immunotherapy. Cancer Sci. 2019;110(11):3434‐3441. doi: 10.1111/cas.14195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu X, Shin N, Koblish HK, et al. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood. 2010;115(17):3520‐3530. doi: 10.1182/blood-2009-09-246124 [DOI] [PubMed] [Google Scholar]
- 10. Beatty GL, O'Dwyer PJ, Clark J, et al. First‐in‐human phase I study of the oral inhibitor of indoleamine 2, 3‐dioxygenase‐1 epacadostat (INCB024360) in patients with advanced solid malignancies. Clin Cancer Res. 2017;23(13):3269‐3276. doi: 10.1158/1078-0432.ccr-16-2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of tumor rejection with doublets of CTLA‐4, PD‐1/PD‐L1, or IDO blockade involves restored IL‐2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J Immunother Cancer. 2014;2(1):3. doi: 10.1186/2051-1426-2-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Krähenbühl L, Goldinger SM, Mangana J, et al. A longitudinal analysis of IDO and PDL1 expression during ommune‐ or targeted therapy in advanced melanoma. Neoplasia. 2018;20(2):218‐225. doi: 10.1016/j.neo.2017.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Banzola I, Mengus C, Wyler S, et al. Expression of indoleamine 2, 3‐dioxygenase induced by IFN‐γ and TNF‐α as potential biomarker of prostate cancer progression. Front Immunol. 2018;9:1051. doi: 10.3389/fimmu.2018.01051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. De Sousa Linhares A, Battin C, Jutz S, et al. Therapeutic PD‐L1 antibodies are more effective than PD‐1 antibodies in blocking PD‐1/PD‐L1 signaling. Sci Rep. 2019;9(1):11472. doi: 10.1038/s41598-019-47910-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.IMFINZI (durvalumab) Injection [prescribing information]. AstraZeneca Pharmaceuticals LP; March 2020; revised February 2021. Accessed 21 April 2021. https://www.azpicentral.com/imfinzi/imfinzi.pdf
- 16. U.S. Food and Drug Administration . FDA approves durvalumab for extensive‐stage small cell lung cancer; March 2020. Accessed October 9, 2020. https://www.fda.gov/drugs/resources‐information‐approved‐drugs/fda‐approves‐durvalumab‐extensive‐stage‐small‐cell‐lung‐cancer
- 17. Muller AJ, DuHadaway JB, Donover PS, Sutanto‐Ward E, Prendergast GC. Inhibition of indoleamine 2, 3‐dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med. 2005;11(3):312‐319. doi: 10.1038/nm1196 [DOI] [PubMed] [Google Scholar]
- 18. Gibney GT, Hamid O, Lutzky J, et al. Phase 1/2 study of epacadostat in combination with ipilimumab in patients with unresectable or metastatic melanoma. J Immunother Cancer. 2019;7(1):80. doi: 10.1186/s40425-019-0562-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Long GV, Dummer R, Hamid O, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO‐301/KEYNOTE‐252): a phase 3, randomised, double‐blind study. Lancet Oncol. 2019;20(8):1083‐1097. doi: 10.1016/s1470-2045(19)30274-8 [DOI] [PubMed] [Google Scholar]
- 20. Smith M, Newton R, Owens S, et al. Retrospective pooled analysis of epacadostat clinical studies identifies doses required for maximal pharmacodynamic effect in anti‐PD‐1 combination studies. J Immunother Cancer. 2020;8(Suppl 3):A15‐A16. [Google Scholar]
- 21. Antonia SJ, Villegas A, Daniel D, et al. Durvalumab after chemoradiotherapy in stage III non‐small‐cell lung cancer. N Engl J Med. 2017;377(20):1919‐1929. doi: 10.1056/nejmoa1709937 [DOI] [PubMed] [Google Scholar]
- 22. Antonia SJ, Villegas A, Daniel D, et al. Overall survival with durvalumab after chemoradiotherapy in stage III NSCLC. N Engl J Med. 2018;379(24):2342‐2350. doi: 10.1056/nejmoa1809697 [DOI] [PubMed] [Google Scholar]