

Abstract
Background and purpose
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease with limited treatment options. RNS60 is an immunomodulatory and neuroprotective investigational product that has shown efficacy in animal models of ALS and other neurodegenerative diseases. Its administration has been safe and well tolerated in ALS subjects in previous early phase trials.
Methods
This was a phase II, multicentre, randomized, double‐blind, placebo‐controlled, parallel‐group trial. Participants diagnosed with definite, probable or probable laboratory‐supported ALS were assigned to receive RNS60 or placebo administered for 24 weeks intravenously (375 ml) once a week and via nebulization (4 ml/day) on non‐infusion days, followed by an additional 24 weeks off‐treatment. The primary objective was to measure the effects of RNS60 treatment on selected biomarkers of inflammation and neurodegeneration in peripheral blood. Secondary objectives were to measure the effect of RNS60 on functional impairment (ALS Functional Rating Scale—Revised), a measure of self‐sufficiency, respiratory function (forced vital capacity, FVC), quality of life (ALS Assessment Questionnaire‐40, ALSAQ‐40) and survival. Tolerability and safety were assessed.
Results
Seventy‐four participants were assigned to RNS60 and 73 to placebo. Assessed biomarkers did not differ between arms. The mean rate of decline in FVC and the eating and drinking domain of ALSAQ‐40 was slower in the RNS60 arm (FVC, difference 0.41 per week, standard error 0.16, p = 0.0101; ALSAQ‐40, difference –0.19 per week, standard error 0.10, p = 0.0319). Adverse events were similar in the two arms. In a post hoc analysis, neurofilament light chain increased over time in bulbar onset placebo participants whilst remaining stable in those treated with RNS60.
Conclusions
The positive effects of RNS60 on selected measures of respiratory and bulbar function warrant further investigation.
Keywords: ALS, clinical trial, placebo‐controlled, randomized, treatment
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a progressive and fatal neurodegenerative disease that affects motor neurons in the spinal cord, brainstem and motor cortex resulting in muscle atrophy and paralysis. Respiratory complications are the most common cause of death and develop due to progressive paralysis of the diaphragm and the accessory muscles that support ventilation [1]. Riluzole [2] and edaravone [3] are the only two available disease‐modifying medications on the market and they both have only modest effects on disease outcomes. Thus, there is an urgent need to develop more treatments for people living with ALS.
In recent years, multiple lines of evidence have supported a critical role for neuroinflammation in ALS pathophysiology including in vivo imaging studies in mouse ALS models, ex vivo analyses revealing astrocyte and microglia activation even in pre‐symptomatic phases, and recent discoveries showing involvement of several genes directly linked to the immune response [4]. Defects in mitochondrial bioenergetics have also been implicated in the pathogenesis of ALS and are being considered as potential therapeutic targets [5, 6, 7].
RNS60 is an investigational product generated by using modified Taylor–Couette–Poiseuille flow under elevated oxygen pressure, which is hypothesized to generate oxygen‐filled charge‐stabilized nanostructures (O2 nanobubbles). Although at a molecular level the mechanism of action of RNS60 has not been fully elucidated yet, its immunomodulatory and cytoprotective properties have been demonstrated in animal models of neuroinflammation, neurodegeneration and brain injury [8, 9, 10, 11]. Importantly, in SOD1G93A transgenic mice, a widely used model of ALS, RNS60 administered at the onset of the disease delayed the neuromuscular deficit and paralysis by virtue of its effects on multiple disease mechanisms in motor neurons, glial cells and peripheral immune cells [8]. RNS60 promotes mitochondrial function and biogenesis [12, 13] and activates the PI3K‐Akt pro‐survival pathway in neurons [14]. Furthermore it reduces glial inflammation [9] and increases regulatory T cells (Tregs) in multiple sclerosis and ALS mouse models [8, 11, 15]. Moreover, RNS60 has been shown to increase neurotransmission and reduce fatigability of nerve stimulated muscles in a murine phrenic nerve diaphragm ex vivo preparation [16]. Clinically, long‐term RNS60 administration has been shown to be safe and well tolerated in a pilot open‐label trial in ALS patients. No serious adverse events (AEs) related to RNS60 occurred and no participant withdrew from the trial due to drug‐related AEs [17]. Here the results are reported of a phase II trial in people with ALS where the effects of RNS60 on a number of candidate biomarkers and clinical end‐points were investigated. Biomarkers measured in this trial included markers known to be modified by RNS60 in preclinical studies (interleukin‐17 [IL‐17], Tregs and protein nitration) [8, 11], markers implicated in ALS pathogenesis (monocyte chemoattractant protein‐1 [MCP‐1] and cyclophilin A/peptidyl prolyl isomerase A [PPIA]) [18, 19] and neurofilament light chain (NfL), an established biomarker of neuroaxonal degeneration [20].
METHODS
Trial design
This was a multicentre, randomized, double‐blind, placebo‐controlled, parallel group, add‐on trial.
The primary study objective was to measure the effect of RNS60 treatment on candidate markers of inflammation and neurodegeneration in the peripheral blood of people living with ALS. Candidate markers were MCP‐1, PPIA, tyrosine‐nitrated actin (actin‐NT), 3‐nitrotyrosine (3‐NT), IL‐17, NfL and Tregs (measured via FOXP3 and CD25 mRNA). Secondary objectives were to evaluate the effect of RNS60 on functional impairment as measured by the ALS Functional Rating Scale—Revised (ALSFRS‐R); the effect on respiratory function as measured by the forced vital capacity (FVC) (per cent predicted of normal value); the impact on quality of life as measured by ALS Assessment Questionnaire‐40 (ALSAQ‐40) (five domains: physical mobility, activities of daily living and independence, eating and drinking, communication, emotional reactions); the effect on self‐sufficiency defined as having a score of 3 or 4 on all three selected ALSFRS‐R questions (specifically swallowing, cutting food and handling utensils, and walking); the effect on survival; tolerability and safety as measured by the identification of AEs.
Study population
Trial participants were enrolled at 22 Italian Expert ALS Centres from May 2017 to January 2020. The trial enrolled adults (aged 18–80 years) with a diagnosis of definite, probable or probable laboratory‐supported ALS according to the revised El Escorial criteria [21] whose symptom onset had occurred 6 to 24 months prior to enrolment. Additional eligibility criteria included geographic access to the enrolling centre (so that they could go to the site on a weekly basis for intravenous infusions); self‐sufficiency (defined as having a score of 3 or 4 on all three selected ALSFRS‐R questions, specifically swallowing, cutting food and handling utensils, and walking); satisfactory respiratory function (FVC ≥ 80% of predicted normal value); documented progression of ALS symptoms in the 3 months prior to screening as measured by a decrease of at least 1 point in the ALSFRS‐R scale total score; stable treatment with riluzole at a dose of 50 mg twice a day.
Trial interventions and procedures
Participants were randomly assigned to receive either RNS60 or matching placebo whilst concomitantly taking riluzole. Treatment allocation was centrally managed using a computer‐generated, permuted block (with a block size of 4), 1:1 randomization scheme. Any concomitant treatment could be continued as per medical indication. RNS60 or placebo was administered intravenously (375 ml infused at a rate of 700 ml/h) once a week as well as inhaled via nebulization (4 ml RNS60 or placebo) every morning (6 days a week on non‐infusion days) for 24 weeks (on‐treatment period). The intravenous (IV) dose was scaled from animal studies using conventional formulae, although the dosing in animal studies (intraperitoneal route) was more frequent. To minimize the need for more frequent IV infusions, the earlier pilot trial had employed daily inhalations to bridge the interval between the higher volume weekly IV doses. As that combination dose had been well tolerated in the pilot trial, it was decided to use the same dose in this trial. Participants were also followed for an additional 24 weeks off‐treatment period (through week 48) to investigate whether RNS60 effects persist after suspension of the treatment and, in the case of a positive effect, to evaluate their duration. Adherence with the assigned home treatment was measured by counting the number of used/unused syringes returned by the participant at each visit.
Nebulizer machines, RNS60 and placebo for infusion and inhalation were supplied by Revalesio Corporation in identical IV bags and syringes, respectively. Both participants and study staff were blinded to the treatment assignment. Assigned box (infusion and nebulizer) treatment was shown by an interactive web response system at each visit making investigators blinded for the entire study.
Data collection
Blood samples for biomarker analyses (RNA and proteins; for detailed methods see Supplementary Material) were collected on day 1 (before the first infusion) and at weeks 4, 12, 24 (on‐treatment period) and 48 (off‐treatment). Safety and efficacy were assessed by way of physical examination, vital signs and AEs. Changes in function and quality of life were assessed using the ALSFRS‐R, FVC (on day 1 and at weeks 4, 12, 24, 36 and 48) and ALSAQ‐40 (on day 1 and at weeks 4, 24 and 48).
Statistical analysis
Separate analyses were performed for (1) all randomized participants (intention‐to‐treat [ITT] population, primary analysis); (2) study completers and compliers (which included participants who completed the 24‐week on‐treatment period and took at least 75% of the assigned dose); (3) participants who did not have any major protocol deviation (PP population).
Descriptive statistics were performed comparing the two treatment groups. Continuous variables were described using mean and standard deviation or median and interquartile range; categorical variables were described as counts and percentages.
Pre‐enrolment progression rate was calculated as follows: (48 – ALSFRS score at diagnosis)/(disease duration at diagnosis), where disease duration at diagnosis is the time from onset to diagnosis in months. Participants with a progression rate higher than or equal to 0.67 were classified as fast progressing, whilst those with a score lower than 0.67 were classified as slow progressing [22, 23].
Biomarker analyses
The mean changes in biomarker levels over the on‐treatment period (weeks 0–24) and the follow‐up, off‐treatment period (weeks 24–48) were analysed using repeated measures analysis of variance (ANOVA) with an unstructured variance–covariance matrix (see Supplementary Material for details). The variation of the biomarkers over time was tested by the time effect, the difference between treatment groups at specific visit times was tested by the treatment main effect, and the difference between groups in the variation over time was tested by the treatment*time interaction term. In all models, time was expressed in weeks.
Clinical outcome analyses
The mean changes in FVC (per cent predicted), ALSFRS‐R total score and the five domains of the ALSAQ‐40 scale were analysed over the on‐treatment period (weeks 0–24) and the follow‐up, off‐treatment period (weeks 24–48) using repeated measures linear mixed models with random intercept and slope and an unstructured variance–covariance matrix (see Supplementary Material for details). In these models, the treatment effect was measured by the difference between the slopes of the two treatment arms, which was tested by the treatment*time interaction term. Differences in baseline values between treatment groups were tested by the treatment main effect, whilst changes over time were tested by the time effect. In all models, time was expressed in weeks.
The cumulative proportions of participants remaining self‐sufficient and survival were calculated in both treatment arms at 4, 12, 24, 36 and 48 weeks using Kaplan–Meier survival curves. Differences between survival curves were assessed with the log‐rank test.
Safety
Total, mean number (with standard deviation) and the proportion of patients with at least one AE were reported separately for different types of AEs (mild, moderate, severe, serious AEs, AEs leading to treatment discontinuation and drug‐related AEs) at the end of the study (week 48) and during the on‐treatment period at weeks 4, 12 and 24, in each study group. Differences between treatment arms were tested with the Wilcoxon–Mann–Whitney test and Fisher's exact test. For classification of the different types of AEs, see the Supplementary Material.
Significance level was set at 5% for all analyses and all tests were two‐tailed. Missing data were handled including all available data, using maximum likelihood estimations in repeated measures ANOVA and mixed models. All analyses were performed with SAS 9.4 (SAS Institute, Cary, NC, USA).
Post hoc analyses
Forced expiratory volume 1 (FEV1) (a measure of the amount of air that a person can exhale within the first second of forced expiration) and the three ALSFRS‐R subscores (bulbar, motor, respiratory) were evaluated using the same statistical methods used for FVC and ALSFRS‐R.
Subgroup analyses were performed stratifying participants by site of onset (bulbar or spinal), pre‐enrolment progression rate (fast or slow) and NfL level at baseline (below the median or above the median). The outcomes analysed included all biomarkers, FVC and ALSFRS‐R. These analyses were performed using the same models as in the total sample with the addition of the stratification variable main effect (in this case onset and, separately, progression rate) and its interactions with all other effects included in the model. Least squares means were estimated separately for each category of the stratification variable (bulbar and spinal onset, fast and slow progressing patients) at each time point and in each treatment group. Differences between categories of the stratification variable were tested by the stratification variable main effect, the treatment*(stratification variable) and the time*(stratification variable) interaction terms. The difference in the treatment effect between categories of the stratification variable was tested by the treatment*time*(stratification variable) triple interaction.
Determination of sample size
The sample size was determined making different assumptions for each biomarker included as primary outcome. A total of 142 patients was required to detect with 80% power a 44% decrease in the rate of progression of PPIA at a two‐sided 5% level of significance, allowing for a 10% drop‐out rate over the entire study period. The number of patients required for other biomarkers ranged from 8 to 68. In addition, with this sample size the study was also powered (80% at a 5% two‐sided level of significance) to detect a 43% decrease in the rate of progression in ALSFRS‐R over 24 weeks, and to show an absolute 25% reduction in the proportion of patients becoming non self‐sufficient at 24 weeks assuming that this proportion in the placebo arm was 85%. See the Supplementary Material for a detailed explanation of sample size calculation and relative assumptions.
Ethical issues and quality assurance
The study and any amendments were approved by an independent ethics committee of each participating centre. This study was planned and performed according to the principles of good clinical practice (ICH‐GCP), the Declaration of Helsinki, and the national laws and regulations about clinical studies. Eligible patients were included in the study only after written IRB/IEC/REB‐approved informed consent or, if incapable of doing so, after approval by a legally acceptable representative. Data review and management were performed by Mario Negri IRCCS.
The study was registered at ClinicalTrials.gov number NCT03456882; EudraCT number 2016‐002382‐62.
RESULTS
A total of 147 participants were enrolled and randomly assigned to RNS60 (74 participants) or placebo (73 participants). Results are reported for the ITT population that included all randomized participants. The study flow‐chart is illustrated in Figure 1.
FIGURE 1.
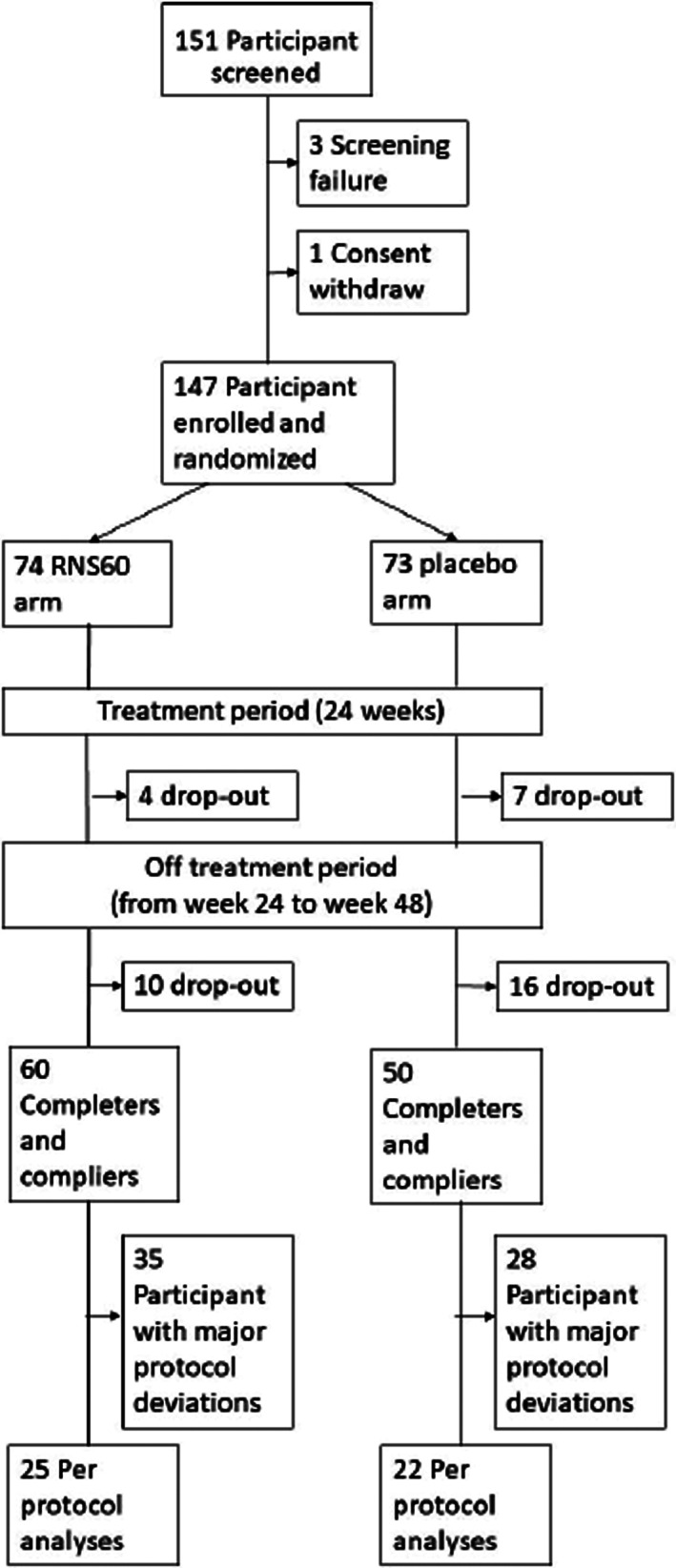
Study flow‐chart: screening, randomization and follow‐up
Demographic and other baseline characteristics
Baseline characteristics of the ITT population are shown in Table 1. The baseline characteristics of the two treatment groups were fairly similar.
TABLE 1.
Demographic and clinical characteristics of the trial population at baseline (ITT population)
| RNS60 (N = 74) | Placebo (N = 73) | p value | |||
|---|---|---|---|---|---|
| Mean | SD | Mean | SD | ||
| Age | 59.3 | 10.4 | 56.0 | 10.0 | 0.0534 |
| Disease duration (months) | 15.5 | 5.8 | 14.3 | 5.8 | 0.2172 |
| Diagnostic delay (months) | 9.8 | 5.1 | 8.8 | 4.9 | 0.2292 |
| BMI | 25.0 | 3.3 | 24.8 | 4.1 | 0.7878 |
| FVC (% of predicted normal value) | 102.7 | 18.2 | 103.3 | 16.1 | 0.8317 |
| ALSFRS‐R total score | 41.6 | 3.2 | 41.4 | 3.6 | 0.6871 |
| ALSAQ‐40 | |||||
| Physical mobility | 32.1 | 21.3 | 33.2 | 23.1 | 0.7540 |
| ADL and independence | 36.8 | 27.8 | 32.9 | 26.1 | 0.3762 |
| Eating and drinking | 14.5 | 23.9 | 13.5 | 23.9 | 0.8015 |
| Communication | 19.3 | 25.5 | 18.6 | 28.6 | 0.8742 |
| Emotional reactions | 33.6 | 18.5 | 33.1 | 22.2 | 0.8825 |
| n | % | n | % | ||
| Sex | 0.4467 | ||||
| Male | 52 | 70.3 | 47 | 64.4 | |
| Female | 22 | 29.7 | 26 | 35.6 | |
| Site of onset | 0.6778 | ||||
| Bulbar | 11 | 14.9 | 9 | 12.5 | |
| Spinal | 63 | 85.1 | 63 | 87.5 | |
| Pre‐enrolment progression rate | 0.7232 | ||||
| Fast | 18 | 27.3 | 18 | 27.3 | |
| Slow | 48 | 72.7 | 48 | 72.7 | |
| Unknown | 8 | 7 | |||
Abbreviations: ADL, activities of daily living; ALSAQ‐40, Amyotrophic Lateral Sclerosis Assessment Questionnaire‐40; ALSFRS‐R, Amyotrophic Lateral Sclerosis Functional Rating Scale—Revised; BMI, body mass index; FVC, forced vital capacity; ITT, intention to treat.
Biomarker levels
The levels of MCP‐1 (p = 0.0162), NfL (p = 0.0373) and FOXP3 mRNA (p = 0.0067) changed significantly over the course of study participation in both treatment arms. In both arms, MCP‐1 and NfL levels increased over time, whilst FOXP3 mRNA decreased, without significant differences between the two arms, as indicated by the treatment main effect and treatment*time interaction effect that were not statistically significant. PPIA, actin‐NT, 3‐NT, IL‐17 and CD25 mRNA did not significantly change over time, and no significant differences in their levels between treatment groups were observed. The mean estimates at each visit time for all analysed biomarkers and their graphical representation are shown in Table S1, Figures 2 and S1.
FIGURE 2.
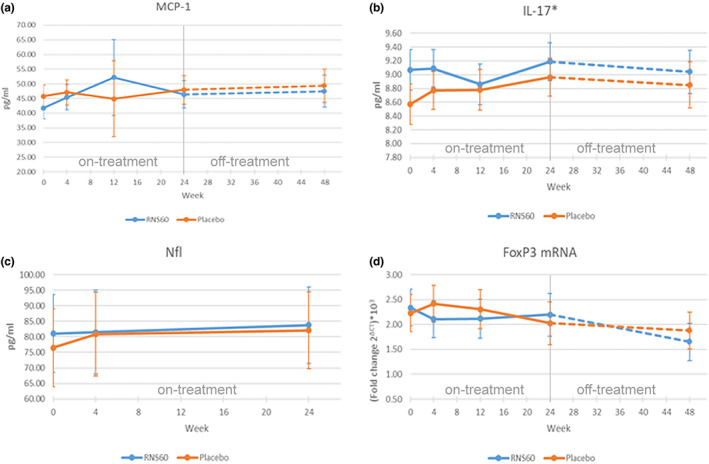
Effect plots for MCP‐1, IL‐17, NfL, FOXP3 mRNA at different visit times (in weeks) in each treatment arm (ITT population)
Clinical outcomes
Forced vital capacity and FEV1
The RNS60 and the placebo groups showed similar FVC values at baseline. FVC decreased significantly over time in both arms (p < 0.0001) (Table 2 and Figure 3a). During the on‐treatment period (baseline to week 24), the estimated mean rate of change was lower in the RNS60 arm (−0.46 per week) compared to the placebo arm (−0.87 per week) (p = 0.0101). During the off‐treatment follow‐up period (week 24 to week 48), no differences were observed (−0.45 per week for RNS60; −0.54 per week for placebo; p = 0.5924).
TABLE 2.
Global tests of fixed effects, means and contrasts for FVC, FEV1, ALSFRS‐R at different visit times in each treatment arm (ITT population)
| FVC | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Tests of fixed effects | |||||||||
| Effect | Week | Treatment | Week*treatment | ||||||
| p value | <0.0001 | 0.9928 | 0.0101 | ||||||
| Effect | Follow‐up | Follow‐up*treatment | |||||||
| p value | 0.1387 | 0.1734 | |||||||
| Estimated means and contrasts | |||||||||
| Visit time (week) | RNS60 | Placebo | Difference (RNS60 – placebo) | ||||||
| Mean | SE | p | Mean | SE | p | Mean | SE | p | |
| Treatment period | |||||||||
| 0 | 102.6 | 1.9 | <0.0001 | 102.6 | 2.0 | <0.0001 | ‐0.02 | 2.8 | 0.9928 |
| 4 | 100.7 | 2.1 | <0.0001 | 99.1 | 2.1 | <0.0001 | 1.6 | 2.9 | 0.5778 |
| 12 | 97.0 | 2.5 | <0.0001 | 92.1 | 2.5 | <0.0001 | 4.9 | 3.5 | 0.1647 |
| 24 | 91.4 | 3.5 | <0.0001 | 81.6 | 3.5 | <0.0001 | 9.8 | 4.9 | 0.0474 |
| Slope (treatment period) | −0.46 | 0.11 | <0.0001 | −0.87 | 0.1 | <0.0001 | 0.41 | 0.16 | 0.0101 |
| Follow‐up period (off‐treatment) | |||||||||
| 36 | 86.0 | 3.7 | <0.0001 | 75.1 | 3.8 | <0.0001 | 10.9 | 5.3 | 0.0394 |
| 48 | 80.6 | 4.4 | <0.0001 | 68.6 | 4.6 | <0.0001 | 12.0 | 6.3 | 0.0571 |
| Slope (follow‐up period) | −0.45 | 0.11 | <0.0001 | −0.54 | 0.13 | <0.0001 | 0.09 | 0.18 | 0.5924 |
| FEV1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Tests of fixed effects | |||||||||
| Effect | Week | Treatment | Week*treatment | ||||||
| p value | <0.0001 | 0.8723 | 0.0146 | ||||||
| Effect | Follow‐up | Follow‐up*treatment | |||||||
| p value | 0.4219 | 0.1148 | |||||||
| Estimated means and contrasts | |||||||||
| Visit time (week) | RNS60 | Placebo | Difference (RNS60 – placebo) | ||||||
| Mean | SE | p | Mean | SE | p | Mean | SE | p | |
| Treatment period | |||||||||
| 0 | 96.7 | 2.2 | <0.0001 | 97.2 | 2.2 | <0.0001 | −0.5 | 3.1 | 0.8723 |
| 4 | 95.2 | 2.2 | <0.0001 | 94.1 | 2.2 | <0.0001 | 1.1 | 3.1 | 0.7107 |
| 12 | 92.3 | 2.5 | <0.0001 | 87.9 | 2.5 | <0.0001 | 4.4 | 3.5 | 0.2128 |
| 24 | 87.9 | 3.4 | <0.0001 | 78.6 | 3.5 | <0.0001 | 9.3 | 4.9 | 0.0563 |
| Slope (treatment period) | −0.36 | 0.1 | 0.0022 | −0.77 | 0.1 | <0.0001 | 0.41 | 0.2 | 0.0146 |
| Follow‐up period (off‐treatment) | |||||||||
| 36 | 82.6 | 3.7 | <0.0001 | 72.3 | 3.8 | <0.0001 | 10.3 | 5.3 | 0.0522 |
| 48 | 77.2 | 4.3 | <0.0001 | 66.0 | 4.4 | <0.0001 | 11.2 | 6.1 | 0.0678 |
| Slope (follow‐up period) | −0.45 | 0.1 | <0.0001 | −0.52 | 0.1 | <0.0001 | 0.07 | 0.1 | 0.5758 |
| ALSFRS‐R | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Tests of fixed effects | |||||||||
| Effect | Week | Treatment | Week*treatment | ||||||
| p value | <0.0001 | 0.9076 | 0.5725 | ||||||
| Effect | Follow‐up | Treatment*follow‐up | |||||||
| p value | 0.3181 | 0.3363 | |||||||
| Estimated means and contrasts | |||||||||
| Week | RNS60 | Placebo | Difference (RNS60 – placebo) | ||||||
| Mean | SE | p | Mean | SE | p | Mean | SE | p | |
| Treatment period | |||||||||
| 0 | 41.4 | 0.4 | <0.0001 | 41.3 | 0.4 | <0.0001 | 0.1 | 0.6 | 0.9076 |
| 4 | 40.4 | 0.5 | <0.0001 | 40.2 | 0.5 | <0.0001 | 0.2 | 0.7 | 0.8010 |
| 12 | 38.3 | 0.6 | <0.0001 | 37.9 | 0.6 | <0.0001 | 0.4 | 0.9 | 0.6877 |
| 24 | 35.1 | 0.9 | <0.0001 | 34.5 | 1.0 | <0.0001 | 0.6 | 1.3 | 0.6280 |
| Slope (treatment period) | −0.26 | 0.03 | <0.0001 | –0.28 | 0.03 | <0.0001 | 0.02 | 0.04 | 0.5725 |
| Follow‐up period (off‐treatment) | |||||||||
| 36 | 32.0 | 1.1 | <0.0001 | 31.6 | 1.1 | <0.0001 | 0.4 | 0.6 | 0.8048 |
| 48 | 28.9 | 1.3 | <0.0001 | 28.8 | 1.3 | <0.0001 | 0.1 | 1.9 | 0.9493 |
| Slope (follow‐up period) | −0.26 | 0.03 | <0.0001 | –0.24 | 0.03 | <0.0001 | –0.02 | 0.04 | 0.5728 |
| ALSAQ‐40 (Eating and drinking) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Tests of fixed effects | |||||||||
| Effect | Week | Treatment | Week*treatment | ||||||
| p value | <0.0001 | 0.5022 | 0.0319 | ||||||
| Estimated means and contrasts | |||||||||
| Week | RNS60 | Placebo | Difference (RNS60 – placebo) | ||||||
| Mean | SE | p | Mean | SE | p | Mean | SE | p | |
| 0 | 15.9 | 2.8 | <0.0001 | 13.3 | 2.8 | <0.0001 | 2.6 | 3.9 | 0.5022 |
| 24 (end of treatment) | 20.4 | 3.2 | <0.0001 | 22.4 | 3.2 | <0.0001 | −2.0 | 4.5 | 0.6524 |
| 48 (follow‐up) | 24.9 | 4.1 | <0.0001 | 31.6 | 4.2 | <0.0001 | −6.7 | 5.8 | 0.2521 |
| Slope | 0.19 | 0.1 | 0.0032 | 0.38 | 0.1 | <0.0001 | −0.19 | 0.1 | 0.0319 |
Abbreviations: ALSAQ‐40, Amyotrophic Lateral Sclerosis Assessment Questionnaire‐40; ALSFRS‐R, Amyotrophic Lateral Sclerosis Functional Rating Scale—Revised; BMI, body mass index; FEV, forced expiratory volume; FVC, forced vital capacity; ITT, intention to treat.
FIGURE 3.
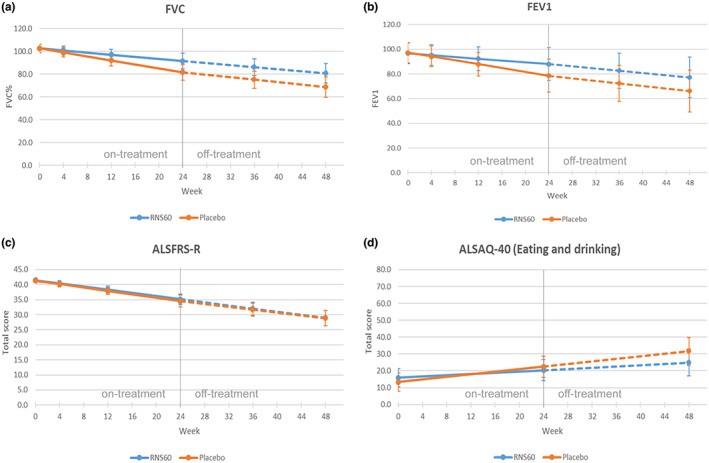
Effect plots for FVC, FEV1, ALSFRS‐R, ALSAQ40 (eating and drinking) at different visit times (in weeks) in each treatment arm (ITT population)
In a post hoc analysis, FEV1 was also evaluated. A significant difference in the rate of decrease of FEV1 between the two treatment groups was observed (−0.45 per week for RNS60; −0.52 per week for placebo; p = 0.0146) during the on‐treatment period, with the RNS60 group showing a slower decline, in line with the findings on FVC (Table 2 and Figure 3b). There were no significant differences between groups during the follow‐up, off‐treatment period.
ALSFRS‐R
The ALSFRS‐R scores were not significantly different between groups at baseline. The ALSFRS‐R total score declined significantly over time in both groups (p < 0.0001) (Table 2 and Figure 3c). The rate of change over time in ALSFRS‐R scores did not differ between treatment arms.
ALSAQ‐40
There were no significant differences between treatment groups at the baseline visit for any of the domains of the ALSAQ‐40 (Table S2). The mean scores for each domain increased significantly over time (p < 0.0001), corresponding to a decrease in each aspect of quality of life. The mean rate of change in the eating and drinking domains of the ALSAQ‐40 were significantly slower in the RNS60 arm (difference −0.19 per week, SE 0.1, p = 0.0319) (Figure 3d). No significant differences between treatment groups were detected for the other four domains of the ALSAQ‐40 scale.
Self‐sufficiency and survival
No significant differences in percentage of participants who remained self‐sufficient were seen in the treatment arms throughout the study period. The cumulative probability of remaining self‐sufficient in the RNS60 arm was 62% at 4 weeks, 29% at 12 weeks, 15% at 24 weeks and 7% at 48 weeks. The corresponding probabilities in the placebo group were 62%, 38%, 23%, 0% (Figure S2A). Survival was no different between treatment arms. The cumulative survival probability was 99% in both arms at 24 weeks, 88% in the RNS60 arm and 89% in the placebo arm at 48 weeks (Figure S2B).
Completers and compliers and PP population
Data for completers and compliers (n = 110, n = 60 in the RNS60 group; n = 50 in the placebo group) and PP populations (n = 47, n = 25 in the RNS60 group; n = 22 in the placebo group) are shown in the Supplementary Material.
Results of the biomarkers’ analyses are shown in Tables S3 and S4. For MCP‐1, FOXP3 mRNA and NfL a significant change over the course of the treatment and follow‐up periods was confirmed in completers and compliers. For MCP‐1 and FOXP3 mRNA this effect was confirmed also in the PP population. A significant treatment main effect, entirely explained by differences at the baseline visit, was found for IL‐17 only in the completers and compliers population. A significant difference in the evolution over time between treatment groups was found for PPIA only in the PP population. A change over time of borderline significance was found only in the PP population for actin‐NT. No other significant effects were found for any biomarker in the completers and compliers nor in the PP population.
The results of the analysis of clinical outcomes are shown in Tables S5 and S6. The evolution over time was consistently significant in all clinical continuous outcomes (decrease of FVC, decrease of ALSFRS‐R, increase of all five domains of the ALSAQ‐40), both in the completers and compliers and in the PP population.
The significant difference between treatment groups in the evolution over time found in the ITT population for FVC was not confirmed in completers and compliers, nor in the PP population. The difference between treatment groups in the evolution over time for the eating and drinking domain of the ALSAQ‐40 was consistently significant in completers and compliers and the PP population. A difference in the evolution over time for the emotional reactions domain of the ALSAQ‐40 was found only in the PP population, with the RNS60 group showing a slower increase. No other significant effects were found for the clinical outcomes in the completers and compliers nor in the PP population.
Adverse events
The total number of AEs and the proportion of participants experiencing at least one AE were similar in the two groups, except for a higher number and, respectively, proportion of moderate AEs and AEs leading to study discontinuation in the placebo group (Table 3). The list of all AEs in each treatment group is shown in Table S7.
TABLE 3.
Adverse events occurring during the on‐treatment period (up to week 24) and the entire study period (from baseline to week 48) (ITT population)
| Total number of AEs | Patients with at least one AE | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RNS60 | Placebo | p value | RNS60 | Placebo | p value | ||||||||
| Sum | Mean | SD | Sum | Mean | SD | n | % | n | % | ||||
| AEs occurring during the on‐treatment period (up to week 24) | |||||||||||||
| All AEs | 128 | 1.7 | 2.6 | 144 | 2.0 | 3.6 | 0.6477 | 42 | 56.8 | 47 | 64.4 | 0.3442 | |
| Mild AEs | 117 | 1.6 | 2.5 | 120 | 1.6 | 3.3 | 0.7427 | 42 | 56.8 | 40 | 54.8 | 0.8107 | |
| Moderate AEs | 6 | 0.1 | 0.3 | 18 | 0.2 | 0.5 | 0.0166 | 5 | 6.8 | 15 | 20.6 | 0.0147 | |
| Severe AEs | 3 | 0.04 | 0.2 | 6 | 0.1 | 0.3 | 0.4520 | 3 | 4.1 | 5 | 6.9 | 0.4940 | |
| SAEs | 5 | 0.1 | 0.3 | 5 | 0.1 | 0.3 | 0.9882 | 4 | 5.4 | 4 | 5.5 | 0.9842 | |
| AEs leading to study/treatment discontinuation | 0 | 0.0 | 0.0 | 3 | 0.04 | 0.2 | 0.0826 | 0 | 0.0 | 3 | 4.1 | 0.1199 | |
| Drug‐related AEs | 8 | 0.1 | 0.6 | 1 | 0.01 | 0.1 | 0.3140 | 3 | 4.0 | 1 | 1.4 | 0.6198 | |
| AEs occurring through week 48 | |||||||||||||
| All AEs | 142 | 1.9 | 2.8 | 158 | 2.1 | 3.6 | 0.6084 | 44 | 59.5 | 48 | 65.8 | 0.4304 | |
| Mild AEs | 128 | 1.8 | 2.8 | 129 | 1.8 | 3.4 | 0.8810 | 42 | 56.8 | 40 | 54.8 | 0.8107 | |
| Moderate AEs | 7 | 0.1 | 0.4 | 20 | 0.3 | 0.6 | 0.0101 | 5 | 6.8 | 16 | 21.9 | 0.0086 | |
| Severe AEs | 7 | 0.1 | 0.3 | 9 | 0.1 | 0.4 | 0.8563 | 7 | 9.5 | 6 | 8.2 | 0.7912 | |
| SAEs | 6 | 0.1 | 0.3 | 6 | 0.1 | 0.3 | 0.4122 | 5 | 6.8 | 5 | 6.9 | 0.9822 | |
| AEs leading to study/treatment discontinuation | 0 | 0 | 0 | 5 | 0.1 | 0.3 | 0.0243 | 0 | 0.0 | 5 | 6.9 | 0.0224 | |
| Drug‐related AEs | 10 | 0.1 | 0.7 | 1 | 0.01 | 0.1 | 0.1728 | 4 | 5.4 | 1 | 1.4 | 0.3664 | |
Abbreviations: AE, adverse event; ITT, intention to treat; SAE, serious adverse event.
Post hoc subgroup analyses
Bulbar and spinal onset subgroup analyses
A significant difference in the evolution over time of NfL was found between bulbar and spinal onset in different treatment arms (significant triple interaction) (Figure 4a, Table S8). When considering participants with spinal onset, the baseline levels and the change over time were similar between the two treatment arms. When considering participants with a bulbar onset, those in the RNS60 arm showed higher values than those in the placebo arm at baseline; NfL levels were stable over the 24 weeks of treatment in the RNS60 arm, whilst in the placebo arm the levels increased. However, the difference between treatment arms in the bulbar onset subgroup was not statistically significant due to small sample size. A significant difference between the bulbar and spinal onset subgroups was also found for FVC: bulbar onset participants had lower FVC values and a more rapid decrease over time compared to spinal onset participants. A slower decrease of FVC in the RNS60 arm compared with the placebo arm was also observed in both subgroups, particularly in bulbar onset; however, this difference was not statistically significant (Figure 4b, Table S8).
FIGURE 4.
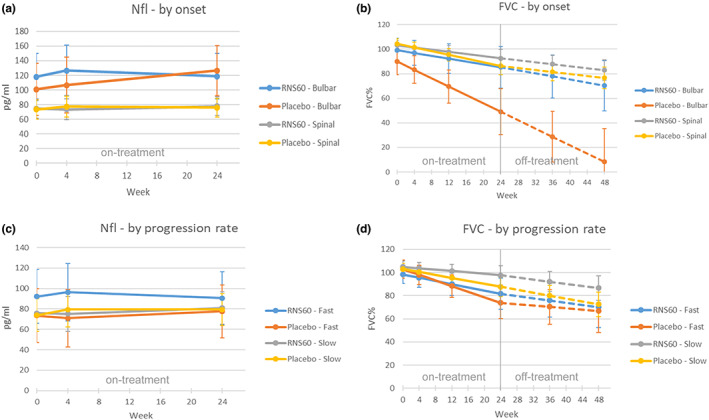
Effect plots for NfL and FVC at different visit times in each treatment arm by site of onset (ITT population)
Fast and slow progressing subgroups
A significant difference in the evolution over time of NfL was found between fast and slow progressing participants included in different treatment arms (significant triple interaction). When considering participants with a slow progression rate, the baseline values and the change over time (increase) was similar between the two treatment arms. When considering participants with a fast progression rate, those in the RNS60 arm showed values that were higher than those in the placebo arm, and the change over time was different between the two treatment groups: participants in the RNS60 arm showed an increase in the first 4 weeks that was followed by a decrease between week 4 and week 24; in the placebo arm a progressive increase between baseline and week 24 was observed (Figure 4c, Table S9).
A significant difference between fast and slow progressing participants was found in the rate of decrease of FVC, with fast progressing participants showing a more rapid decrease compared to slow participants, but without significant differences between treatment groups (Figure 4d, Table S9).
A significant difference between fast and slow participants was observed in the progression rate of the ALSFRS‐R score, with fast progressing patients showing lower values. However, no significant differences between treatment groups were found (data not shown).
Neurofilament light chain levels at baseline
A significant difference in the rate of change of FVC, ALSFRS‐R and all ALSAQ‐40 domains, with the exception of the emotional reaction domain, was found between patients with high (above the median) or low (below the median) baseline NfL levels. Participants with high baseline NfL levels showed a faster decline compared to patients with low baseline levels. However, no significant differences in RNS60 treatment effects were detected between the two subgroups (data not shown).
DISCUSSION
In this phase II randomized, double‐blind, placebo‐controlled trial, administration of RNS60 showed no effects on a number of biological markers of inflammation and neurodegeneration.
The biomarkers that were investigated in this trial were chosen based on previous preclinical data and/or relevance to ALS pathophysiology. IL‐17 was chosen because increased serum and cerebrospinal fluid levels of IL‐17 have been observed in ALS patients [24] and, in a mouse model of multiple sclerosis, RNS60 treatment was associated with reduced expression of IL‐17 [15]. Yet, measurement of IL‐17 is limited by the fact that there is no prior information about the longitudinal pattern of IL‐17 in ALS patients and by the very low levels of IL‐17 in the blood (<1 pg/ml). FOXP3 mRNA was measured because, in a mouse model of multiple sclerosis and in SOD1G93A mice, RNS60 upregulated FOXP3 mRNA [8, 15]. FOXP3 mRNA is low in rapidly progressing ALS patients and its levels inversely correlate with progression rates [25]. 3‐NT was evaluated because, in a previous study conducted in our laboratory, RNS60 reduced 3‐NT levels in the spinal cord of the SOD1G93A mouse model, suggesting a possible role of RNS60 in suppressing nitric oxide (NO) production [8]. Increased levels of 3‐NT and actin‐NT were found in peripheral blood mononuclear cells (PBMCs) of ALS patients [26, 27, 28], implicating inflammation and oxidative/nitrative stress in disease pathogenesis. Decreased levels of PPIA in the PBMCs of ALS patients have been associated with an early onset of the disease [29] and with a short disease duration [30] indicating that reduced chaperone/foldase activity may be at the basis of a more severe phenotype. Increased levels of MCP‐1 have been found in the central and peripheral nervous system of the SOD1G93A mouse model [19] and in serum and cerebrospinal fluid of ALS patients [31, 32, 33, 34], indicating central and peripheral immune activation. NfL levels were measured as there is growing evidence supporting their prognostic and pharmacodynamic potential [20]. In our study, NfL values were significantly higher in participants with bulbar onset, in agreement with recent studies showing an association between higher NfL serum levels and bulbar onset in ALS [35].
Although RNS60 did not affect NfL levels in the whole population of the study, a statistically different trajectory of NfL was found by the post hoc analysis in patients with bulbar onset, in which RNS60 seems to have a stabilizing effect.
Whilst RNS60 administration did not affect the levels of candidate biomarkers of neuroinflammation and neurodegeneration, encouraging results were obtained on selected clinical outcomes. Specifically, RNS60 administration was associated with slower rates of decline in respiratory function and in the bulbar domains of ALSAQ‐40 compared to placebo over a 24‐week treatment period. These effects were not accompanied by positive effects on global function or survival, although longer treatment periods may be needed to detect such changes. During the off‐treatment follow‐up period (weeks 24–48), the difference in the two treatment arms persisted, suggesting a protective effect of early treatment with the drug rather than a transient symptomatic effect. These findings were similar in post hoc analyses showing differences in FVC decline when comparing bulbar and spinal ALS onset, although differences did not attain statistical significance probably due to the small sample size of the subgroups. RNS60 treatment slowed the decline of FVC in patients with bulbar onset and stabilized levels of NfL. Indirect evidence of a possible beneficial effect of the active treatment comes from the difference in the drop‐out rate between active treatment and placebo (18.9% vs. 31.5%, respectively) over 48 weeks. The lower drop‐out rate in the RNS60 arm could be explained by possible subjective perception of a positive effect of the treatment. It is worth noting that there was a steep rise of drop‐outs in both treatment arms towards the end of follow‐up. This might reflect the absence of treatment and perhaps disease progression in the absence of treatment during the second semester of the study. Our study also confirmed the benign safety profile of RNS60 expanding on previous open‐label observations [17].
The positive effects of RNS60 on respiratory function are supported by an exploratory study on the functional consequences of tissue oxygenation with O2 nanobubbles in murine phrenic nerve diaphragm ex vivo preparations [16]. Alternatively, there may be a symptomatic effect of the nebulized treatment, which may be particularly evident in bulbar patients. Future trials focused on clinical efficacy of RNS60 may need to include additional measures of respiratory and bulbar function. Spirometry manoeuvres in the present study were performed in the seated position. Future studies may include supine FVC which may be a more sensitive measure of decline in diaphragm function. Measures of orthopnoea and sleep disruption may also be considered. Hence it is thought that a larger clinical trial focused on clinical efficacy of inhaled/nebulized administration of RNS60 with respiratory and functional end‐points in well‐selected populations of patients is necessary to conclude whether RNS60 can be used for the treatment of respiratory symptoms or for preventing and delaying disease progression [16]. Treatment with RNS60 for 24 weeks did not affect disease progression as measured by the ALSFRS‐R or mortality. However, another compound with anti‐inflammatory properties like palmitoylethanolamide was able to improve the pulmonary function in ALS patients without modifying the ALSFRS‐R scale; the effect was attributed to a direct action of palmitoylethanolamide on the neuromuscular junction of the diaphragmatic muscle which are the last muscles to be paralysed [36]. RNS60 may also have directly affected the diaphragm muscle.
These results are in contrast with the positive effects of RNS60 reported in the animal model of ALS; however, in those mice the drug was administrated intraperitoneally before overt symptom onset [8]. The motor deficit slowing effect observed in RNS60‐treated SOD1G93A mice was accompanied by partial prevention of lumbar spinal motor neuron loss and neuromuscular junction denervation of skeletal muscle. Interestingly, the clinical studies with tofersen (VALOR, NCT02623699, NCT02623699) [37, 38], an antisense oligonucleotide targeting SOD1, have shown that those who started tofersen earlier had better outcomes than those who started later (https://investors.biogen.com/static‐files/4e9393b8‐0f67‐47a3‐a331‐05ef3ac7fc63). A therapeutic intervention in earlier phases of ALS is under investigation in the ATLAS study (NCT04856982), in which tofersen is initiated in clinically pre‐symptomatic SOD1 variant carriers [39]. It should be noted that in the 6‐month phase 3 study there were positive results only with regard to neurofilaments and not to ALSFRS‐R and survival, as occurred in the present study. Moreover, a recent clinical trial on high caloric nutrition diets has yielded positive results for fast progressing patients only [40], possibly because the duration of the investigation was also too short to detect effects in slower progressing patients, which could have been the case in our study, too. Therefore, it is possible that earlier and/or longer treatment with RNS60 might have a better clinical benefit in people living with ALS, which warrants further investigation.
The strengths of the study include the analysis of multiple candidate biomarkers by a centralized laboratory at the Mario Negri Institute IRCCS using standard operation procedures, thereby limiting analytical variation. Thus, this trial provided an excellent opportunity to characterize the pattern and longitudinal behaviour of these markers in the context of a well‐defined trial population. Tertiary sites with broad national distribution were included to increase access and support the generalizability of study results. FVC measurements were performed using the same instrument at each site provided by the study sponsor. Finally, the study was sufficiently powered to detect clinically meaningful differences in measures of functional impairment.
The study was limited by the COVID‐19 outbreak that started in Italy in February 2020. The pandemic caused restrictions in terms of participant access to trial sites and limitations in the performance of respiratory function tests. This context led to missing visits and missing data.
Also, a total of 155 protocol deviations occurred during the study (71 RNS60 group, 84 placebo group) in 100 patients (49 RNS60 group, 51 placebo group), of which 22 (10 RNS60 group, 12 placebo group) were due to the COVID‐19 pandemic. The description of all protocol deviations that occurred during the study with number of occurrences in each treatment group is reported in Table S10. The third limit was the drug administration route that required weekly visits to the participating centre, with significant discomfort for disabled patients. For this reason, testing the efficacy of a full treatment via the inhalation route only at the patient's home would have been preferred.
CONCLUSION
In conclusion, administration of RNS60 to people living with ALS for 24 weeks showed no effects on candidate biomarkers of inflammation and neurodegeneration, ALSFRS‐R scores or survival. Positive effects of RNS60 on selected measures of respiratory and bulbar function warrant further investigation, which might include earlier treatment, longer follow‐up, additional measures of respiratory and bulbar function, and focus on dosage and route of administration.
AUTHOR CONTRIBUTIONS
See the Appendix A.
FUNDING INFORMATION
Funded by the ALS Association, Fondazione Banco Popolare Novara, Fondazione Comunità del Novarese, Get Out and URSLA Onlus. ALSFAC provided support to the ALS Association USA for this study. Revalesio Corporation donated active study drug and matching placebo.
CONFLICT OF INTEREST
E. Beghi reports grants from the Italian Ministry of Health, grants from SOBI, personal fees from Arvelle Therapeutics, outside the submitted work. E. Beghi reports grants from American ALS Association for the conduct of the present study. E. Pupillo reports grants from the Italian Ministry of Health, grants from AIFA, outside the submitted work. A. Sherman reports research grants from the ALS Association, ALS Finding a Cure and the NIH, outside the submitted work. S. Paganoni reports research grants from Amylyx Therapeutics, Revalesio Corporation, UCB/Ra Pharma, Biohaven, Clene, Prilenia, Seelos, the ALS Association, the American Academy of Neurology, ALS Finding a Cure, the Salah Foundation, the Spastic Paraplegia Foundation, the Muscular Dystrophy Association and reports personal consulting fees for advisory panels from Orion and Cytokinetics. All of them are outside the submitted work. M. Filosto reports serving on a scientific advisory board for Amicus, Sanofi, Sarepta; honoraria for speaking engagements for Sanofi, Alnylam. All of them are outside the submitted work.
A. Padovani serves on a scientific advisory board: Director of School of Neurology, University of Brescia; President‐elect of the National Society of Neurology SIN; GE Healthcare, Lilly and Actelion Ltd Pharmaceuticals. Travel funded by a commercial entity: Roche, Ely‐Lilly, Biogen, Zambon, Nutricia, Lundbeck. Serving as a journal editor, associate editor, or on an editorial advisory board: MDPI. Patents held or pending: Method of generation of a diagnostic index for Alzheimer's disease, electronic device for implementing the method and system, 2016. Honoraria for speaking engagements: He has received honoraria from Nutricia, PIAM, Langstone Technology, GE Healthcare, Lilly and Chiesi. Commercial research support: Advisory and served on the Scientific Advisory Board for GE Healthcare, Lilly and Actelion Ltd Pharmaceuticals. Government research support (including funding organization, grant number, and role): He has participated as PI in more than 30 research projects funded by the Ministry of Health and MIUR. Support from a non‐profit foundation or society: CHDI Foundation, Fondazione Cariplo. All of them are outside the submitted work.
G. Logroscino received funds from Roche, Amplifon; he received travel funds for participation in a congress as speaker. All of them are outside the submitted work. C. Lunetta received compensation for consulting services from Neuraltus, Cytokinetics, Mitsubishi Tanabe Pharma Europe and Italfarmaco and has received funds from ARISLA and Italian Ministry of Health. All of them are outside the submitted work. J. Mandrioli received research grants from Fondazione Italiana di Ricerca per la Sclerosi Laterale Amiotrofica (ARISLA), the Agenzia Italiana del Farmaco (AIFA), the Italian Ministry of Health, Emilia Romagna Regional Health Authority, Roche, Pfizer. All of them are outside the submitted work. F. Trojsi served as a journal editor, associate editor, or on an editorial advisory board: Associate Editor of Frontiers in Neuroscience/Frontiers in Neurology (section Neurodegeneration) (2017–today) and Orphanet Journal of Rare Diseases (BMC/ Springer Nature) (2021); Academic Editor of Brain Sciences (MDPI Ed.) (2021). Fee for consultant: Alnylam Italy Srl (2020). All of them are outside the submitted work.
All other authors report no disclosures.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
The authors express their gratitude to Professor Merit Cudkowicz at Massachusetts General Hospital (MGH) for her expert advice and support; to the Center for Innovation and BioInformatics at Neurological Clinical Research Institute at MGH for providing the PharmaENGINE Electronic Data Capture platform; to Dr Schoenfeld at MGH for providing input about the statistical analysis plan; to Serena Scozzari and Eliana Sammali at the Mario Negri Institute for helping with biomarker analyses. Open access funding provided by BIBLIOSAN.
APPENDIX A. Authors
| Name | Location | Contribution |
|---|---|---|
| Ettore Beghi, MD | Department of Neuroscience, Istituto di Ricerche Farmacologiche Mario Negri IRCCS, Milano | Study conception, project coordinator, first and final draft |
| Elisabetta Pupillo, PharmD, PhD | Department of Neuroscience, Istituto di Ricerche Farmacologiche Mario Negri IRCCS, Milano | Study conception, director of monitoring activities, study management, first and final draft |
| Elisa Bianchi, MSc | Department of Neuroscience, Istituto di Ricerche Farmacologiche Mario Negri IRCCS, Milano | Study conception, statistical analysis, first and final draft |
| Valentina Bonetto, PharmD, PhD |
Department of Biochemistry and Molecular Pharmacology, Istituto di Ricerche Farmacologiche Mario Negri IRCCS, Milano |
Centralized laboratory management and sample analyses, first and final draft |
| Silvia Luotti, BiolD | Department of Biochemistry and Molecular Pharmacology, Istituto di Ricerche Farmacologiche Mario Negri IRCCS, Milano | Centralized laboratory management and sample analyses |
| Laura Pasetto, BiolD, PhD |
Department of Biochemistry and Molecular Pharmacology, Istituto di Ricerche Farmacologiche Mario Negri IRCCS, Milano |
Centralized laboratory management and sample analyses |
| Caterina Bendotti, PharmD, PhD | Department of Neuroscience, Istituto di Ricerche Farmacologiche Mario Negri IRCCS, Milano | Centralized laboratory management and sample analyses, first and final draft |
| Massimo Tortarolo, PharmD, PhD | Department of Neuroscience, Istituto di Ricerche Farmacologiche Mario Negri IRCCS, Milano | Centralized laboratory management and sample analyses |
| Francesca Sironi, BiolD, PhD | Department of Neuroscience, Istituto di Ricerche Farmacologiche Mario Negri IRCCS, Milano | Centralized laboratory management and sample analyses |
| Laura Camporeale, BiolD | Department of Neuroscience, Istituto di Ricerche Farmacologiche Mario Negri IRCCS, Milano | Centralized laboratory management and sample analyses |
| Alexander V. Sherman, MSc | Neurological Clinical Research Institute, Massachusetts General Hospital, Harvard Medical School | Revision of first and final draft |
| Sabrina Paganoni, MD, PhD | Sean M. Healey & AMG Center for ALS at Mass General Hospital, Department of Neurology and Spaulding Rehabilitation Hospital, Department of PM&R, Harvard Medical School | Study conception, revision of first and final draft |
| Ada Scognamiglio, BiolD | ALS Expert Center ‘Maggiore della Carità’ Hospital and University of Piemonte Orientale, Novara | Data collection at study site |
| Fabiola De Marchi, MD | ALS Expert Center ‘Maggiore della Carità’ Hospital and University of Piemonte Orientale, Novara | Identification of patients, data collection at study site, revision of final draft |
| Paolo Bongioanni, MD, PhD | Spinal Cord Injuries Section, Azienda Ospedaliero‐Universitaria Pisana | Identification of patients, data collection at study site, revision of final draft |
| Renata Del Carratore, BiolD, PhD | Institute of Clinical Physiology, CNR‐Pisa | Biomarkers’ assays |
| Claudia Caponnetto, MD | IRCCS Ospedale Policlinico San Martino Genoa | Identification of patients, data collection at study site, revision of final draft |
| Luca Diamanti, MD, PhD | Neuroncology Unit, IRCCS Mondino Foundation, Pavia | Identification of patients, data collection at study site, revision of final draft |
| Daniele Martinelli, MD | Dept. of Brain and Behavioural Sciences, University of Pavia, Pavia, and Headache Science and Neurorehabilitation Center, IRCCS Mondino Foundation, Pavia, Italy | Identification of patients, data collection at study site, revision of final draft |
| Andrea Calvo, MD, PhD | Centro Regionale Esperto per la Sclerosi Laterale Amiotrofica Dipartimento di Neuroscienze ‘Rita Levi Montalcini’ Università degli Studi diTorino AOU Città della Salute e della Scienza di Torino | Identification of patients, data collection at study site, revision of final draft |
| Massimiliano Filosto, MD, PhD | Department of Clinical and Experimental Sciences, University of Brescia; NeMO‐Brescia Clinical Center for Neuromuscular Diseases, Brescia | Identification of patients, data collection at study site, revision of final draft |
| Alessandro Padovani, MD, PhD | Department of Clinical and Experimental Sciences, University of Brescia; Unit of Neurology, ASST Spedali Civili, Brescia | Identification of patients, data collection at study site, revision of final draft |
| Stefano Cotti Piccinelli, MD | Department of Clinical and Experimental Sciences, University of Brescia; Unit of Neurology, ASST Spedali Civili, Brescia | Identification of patients, data collection at study site, revision of final draft |
| Claudia Ricci, MS, PhD | Department of Medical, Surgical and Neurological Sciences, University of Siena, Siena | Data collection in study site |
| Stefania Dalla Giacoma, MD | Department of Medical, Surgical and Neurological Sciences, University of Siena, Siena | Identification of patients, data collection at study site, revision of final draft |
| Nicoletta De Angelis, MD | Department of Medical, Surgical and Neurological Sciences, University of Siena, Siena | Identification of patients, data collection at study site, revision of final draft |
| Maurizio Inghilleri, MD, PhD | Dipartimento di Neuroscienze Umane Università di Roma ‘Sapienza’ UOSD Malattie Neurodegenerative, Centro Malattie Rare‐ Neuromuscolari Policlinico Universitario Umberto I | Identification of patients, data collection at study site, revision of final draft |
| Rossella Spataro, MD, PhD | ALS Clinical Research Center, AOUP ‘P Giaccone’, University of Palermo, Palermo | Identification of patients, data collection at study site, revision of final draft |
| Vincenzo La Bella, MD, PhD | ALS Clinical Research Center, AOUP ‘P Giaccone’, University of Palermo, Palermo | Identification of patients, data collection at study site, revision of final draft |
| Giancarlo Logroscino, MD, PhD | Center for Neurodegenerative Diseases and the Aging Brain, Department of Clinical Research in Neurology of the University of Bari at‘Pia Fondazione Card G. Panico ‘Hospital Tricase; Department of Basic Medicine Neuroscience and Sense Organs, University Aldo Moro Bari | Identification of patients, data collection at study site, revision of final draft |
| Christian Lunetta, MD | ||
| Centro Clinico NeMO Milano, Fondazione Serena ONLUS | Identification of patients, data collection at study site, revision of final draft | |
| Claudia Tarlarini, BiolD | Centro Clinico NeMO Milano, Fondazione Serena ONLUS | Data collection at study site |
| Jessica Mandrioli, MD | Department of Biomedical, Metabolic and Neural Sciences, University of Modena and Reggio Emilia, Modena, Italy; Department of Neurosciences, Azienda Ospedaliero‐Universitaria Di Modena, Modena, Italy | Identification of patients, data collection at study site, revision of final draft |
| Ilaria Martinelli, MD | Department of Neurosciences, Azienda Ospedaliero‐Universitaria Di Modena, Modena, Italy; Clinical and Experimental Medicine PhD Program, University of Modena and Reggio Emilia, Modena, Italy. | Identification of patients, data collection at study site, revision of final draft |
| Cecilia Simonini, BiolD | Department of Neurosciences, Azienda Ospedaliero‐Universitaria Di Modena, Modena, Italy | Data collection at study site |
| Elisabetta Zucchi, MD |
Department of Neurosciences, Azienda Ospedaliero‐Universitaria Di Modena, Modena, Italy; Neuroscience PhD Program, University of Modena and Reggio Emilia, Modena, Italy |
Identification of patients, data collection at study site, revision of final draft |
| Maria Rosa Monsurrò, MD | Department of Advanced Medical and Surgical Sciences, University of Campania ‘Luigi Vanvitelli’, Naples | Identification of patients, data collection at study site, revision of final draft |
| Dario Ricciardi, MD | Department of Advanced Medical and Surgical Sciences, University of Campania ‘Luigi Vanvitelli’, Naples | Identification of patients, data collection at study site, revision of final draft |
| Francesca Trojsi, MD, PhD | Department of Advanced Medical and Surgical Sciences, University of Campania ‘Luigi Vanvitelli’, Naples | Identification of patients, data collection at study site, revision of final draft |
| Nilo Riva, MD, PhD | Neurology Unit, Neurorehabilitation Unit and Neurophysiology Unit, Vita‐Salute San Raffaele University and San Raffaele Scientific Institute, Milan, Italy | Identification of patients, data collection at study site, revision of final draft |
| Massimo Filippi, MD, FEAN, FAAN | Neurology Unit, Neurorehabilitation Unit and Neurophysiology Unit, Vita‐Salute San Raffaele University and San Raffaele Scientific Institute, Milan, Italy | Identification of patients, data collection at study site, revision of final draft |
| Isabella Laura Simone, MD | Neurology Unit, Department of Basic Medical Sciences, Neurosciences and Sense Organs, University of Bari | Identification of patients, data collection at study site, revision of final draft |
| Gianni Sorarù, MD, PhD | Motor Neuton Disease Center, Department of Neurosciences, Azienda Ospedale Università di Padova | Identification of patients, data collection at study site, revision of final draft |
| Cristina Spera, MD | Azienda Ospedaliera ‘S Maria’ Terni | Identification of patients, data collection at study site, revision of final draft |
| Lucia Florio, MD, PhD | Neurology Department, Fondazione IRCCS Casa Sollievo della Sofferenza, San Giovanni Rotondo (FG) | Identification of patients, data collection at study site, revision of final draft |
| Sonia Messina, MD, PhD | Department of Clinical and Experimental Medicine University of Messina | Identification of patients, data collection at study site, revision of final draft |
| Massimo Russo, MD, PhD | Department of Clinical and Experimental Medicine University of Messina | Identification of patients, data collection at study site, revision of final draft |
| Gabriele Siciliano, MD | Department of Clinical and Experimental Medicine, University of Pisa, Pisa, Italy; | Identification of patients, data collection at study site, revision of final draft |
| Amelia Conte, MD | Centro Clinico NEMO‐Fondazione Policlinico Universitario A. Gemelli IRCCS | Identification of patients, data collection at study site, revision of final draft |
| Maria Valeria Saddi, MD | Neurology Department, San Francesco Hospital, Nuoro | Identification of patients, data collection at study site, revision of final draft |
| Nicola Carboni, MD | Neurology Department, San Francesco Hospital, Nuoro | Identification of patients, data collection at study site, revision of final draft |
| Letizia Mazzini, MD | ALS Expert Center ‘Maggiore della Carità’ Hospital and University of Piemonte Orientale, Novara | Study conception, project coordinator, first and final draft |
All authors contributed to the revision of the manuscript and accepted the final version.
APPENDIX B. RNS60‐ALS Study Group
Gabriele Enia, Andrea Zucchella, Lorenzo Pinzani, Lorena De Giorgi, Celeste Nicoletti, Elisa Semprucci, Emilio Davide Arippol, Giorgia Giussani (Milano, Italy); Monica Graziani, Cinzia Ferrari, Roberto Cantello (Novara, Italy); Enrica Bersano (Milano, Italy); Cristina Dolciotti, Silvia Maria Masciandaro, Ilaria Da Prato (Pisa, Italy); Corrado Cabona, Giuseppe Meo (Genova, Italy); Mauro Ceroni, Beatrice Dal Fabbro (Pavia, Italy); Adriano Chiò, Cristina Moglia, Umberto Manera, Giuseppe Fuda, Federico Casale, Giovanni De Marco, Paolina Salamone (Torino, Italy); Fabio Giannini, Silvia Bocci (Siena, Italy); Marco Ceccanti, Chiara Cambieri, Laura Libonati, Federica Moret, Vittorio Frasca (Roma, Italy); Tiziana Coletti (Palermo, Italy); Rosanna Tortelli, Rosa Capozzo (Lecce, Italy); Francesca Gerardi, Valeria Sansone (Milano, Italy); Nicola Fini, Giulia Gianferrari, Annalisa Gessani (Modena, Italy); Yuri Falzone, Paride Schito, Laura Pozzi, Teuta Domi (San Raffaele, Milano, Italy); Eustachio D'Errico, Antonella Morea, Gianmarco Milella (Bari, Italy); Matteo Gizzi (Padova, Italy); Francesca Paci, Simonetta Ferracchiato (Terni, Italy); Michele Zarrelli, Nino Desina (Foggia, Italy); Alessandra Govoni, Erika Schirinzi, Costanza Simoncini, (Pisa, Italy); Mario Sabatelli, Daniela Bernardo, Giulia Bisogni (Roma, Italy); Giovanna Piras, Maria Monne, Elisabetta Manca (Nuoro, Italy).
Beghi E, Pupillo E, Bianchi E, et al. Effect of RNS60 in amyotrophic lateral sclerosis: a phase II multicentre, randomized, double‐blind, placebo‐controlled trial. Eur J Neurol. 2023;30:69‐86. doi: 10.1111/ene.15573
For RNS60‐ALS Study Group—see Appendix B.
Contributor Information
Elisabetta Pupillo, Email: elisabetta.pupillo@marionegri.it.
the RNS60‐ALS Study Group:
Gabriele Enia, Andrea Zucchella, Lorenzo Pinzani, Lorena De Giorgi, Celeste Nicoletti, Elisa Semprucci, Emilio Davide Arippol, Giorgia Giussani, Monica Graziani, Cinzia Ferrari, Roberto Cantello, Enrica Bersano, Cristina Dolciotti, Silvia Maria Masciandaro, Ilaria Da Prato, Corrado Cabona, Giuseppe Meo, Mauro Ceroni, Beatrice Dal Fabbro, Adriano Chiò, Cristina Moglia, Umberto Manera, Giuseppe Fuda, Federico Casale, Giovanni De Marco, Paolina Salamone, Fabio Giannini, Silvia Bocci, Marco Ceccanti, Chiara Cambieri, Laura Libonati, Federica Moret, Vittorio Frasca, Tiziana Coletti, Rosanna Tortelli, Rosa Capozzo, Francesca Gerardi, Valeria Sansone, Nicola Fini, Giulia Gianferrari, Annalisa Gessani, Yuri Falzone, Paride Schito, Laura Pozzi, Teuta Domi, Eustachio D’Errico, Antonella Morea, Gianmarco Milella, Matteo Gizzi, Francesca Paci, Simonetta Ferracchiato, Michele Zarrelli, Nino Desina, Alessandra Govoni, Erika Schirinzi, Costanza Simoncini, Mario Sabatelli, Daniela Bernardo, Giulia Bisogni, Giovanna Piras, Maria Monne, and Elisabetta Manca
DATA AVAILABILITY STATEMENT
Anonymised data are available from the corresponding author upon request.
REFERENCES
- 1. Beghi E, Mennini T, Bendotti C, et al. The heterogeneity of amyotrophic lateral sclerosis: a possible explanation of treatment failure. Curr Med Chem. 2007;14(30):3185‐3200. [DOI] [PubMed] [Google Scholar]
- 2. Miller RG, Mitchell JD, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev. 2012. Mar 14;2012(3):CD001447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Writing Group; Edaravone (MCI‐186) ALS 19 Study Group . Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double‐blind, placebo‐controlled trial. Lancet Neurol. 2017;16(7):505‐512. [DOI] [PubMed] [Google Scholar]
- 4. De Marchi F, Munitic I, Amedei A, et al. Interplay between immunity and amyotrophic lateral sclerosis: clinical impact. Neurosci Biobehav Rev 2021;127:958‐978. doi: 10.1016/j.neubiorev.2021.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Calió ML, Henriques E, Siena A, Bertoncini CRA, Gil‐Mohapel J, Rosenstock TR. Mitochondrial dysfunction, neurogenesis, and epigenetics: putative implications for amyotrophic lateral sclerosis neurodegeneration and treatment. Front Neurosci. 2020;14:679. doi: 10.3389/fnins.2020.00679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mehta AR, Walters R, Waldron FM, et al. Targeting mitochondrial dysfunction in amyotrophic lateral sclerosis: a systematic review and meta‐analysis. Brain Commun. 2019;1(1):fcz009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Granatiero V, Manfredi G. Mitochondrial transport and turnover in the pathogenesis of amyotrophic lateral sclerosis. Biology. 2019;8(2):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vallarola A, Sironi F, Tortarolo M, et al. RNS60 exerts therapeutic effects in the SOD1 ALS mouse model through protective glia and peripheral nerve rescue. J Neuroinflammation. 2018;15(1):65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Khasnavis S, Roy A, Ghosh S, Watson R, Pahan K. Protection of dopaminergic neurons in a mouse model of Parkinson's disease by a physically‐modified saline containing charge‐stabilized nanobubbles. J Neuroimmune Pharmacol. 2014;9(2):218‐232. [DOI] [PubMed] [Google Scholar]
- 10. Rangasamy SB, Ghosh S, Pahan K. RNS60, a physically‐modified saline, inhibits glial activation, suppresses neuronal apoptosis and protects memory in a mouse model of traumatic brain injury. Exp Neurol. 2020;328:113279. [DOI] [PubMed] [Google Scholar]
- 11. Mondal S, Rangasamy SB, Ghosh S, Watson RL, Pahan K. Nebulization of RNS60, a physically‐modified saline, attenuates the adoptive transfer of experimental allergic encephalomyelitis in mice: implications for multiple sclerosis therapy. Neurochem Res. 2017;42(5):1555‐1570. [DOI] [PubMed] [Google Scholar]
- 12. Rao VT, Khan D, Jones RG, et al. Potential benefit of the charge‐stabilized nanostructure saline RNS60 for myelin maintenance and repair. Sci Rep. 2016;6:30020. doi: 10.1038/srep30020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chandra G, Kundu M, Rangasamy SB, et al. Increase in mitochondrial biogenesis in neuronal cells by RNS60, a physically‐modified saline, via phosphatidylinositol 3‐kinase‐mediated upregulation of PGC1α. J Neuroimmune Pharmacol. 2018;13(2):143‐162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Modi KK, Jana A, Ghosh S, Watson R, Pahan K. A physically‐modified saline suppresses neuronal apoptosis, attenuates tau phosphorylation and protects memory in an animal model of Alzheimer's disease. PLoS One. 2014;9(8):e103606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mondal S, Martinson JA, Ghosh S, Watson R, Pahan K. Protection of Tregs, suppression of Th1 and Th17 cells, and amelioration of experimental allergic encephalomyelitis by a physically‐modified saline. PLoS One. 2012;7(12):e51869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ivannikov MV, Sugimori M, Llinás RR. Neuromuscular transmission and muscle fatigue changes by nanostructured oxygen. Muscle Nerve. 2017;55(4):555‐563. [DOI] [PubMed] [Google Scholar]
- 17. Paganoni S, Alshikho MJ, Luppino S, et al. A pilot trial of RNS60 in amyotrophic lateral sclerosis. Muscle Nerve. 2019;59(3):303‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pasetto L, Grassano M, Pozzi S, et al. Defective cyclophilin A induces TDP‐43 proteinopathy: implications for amyotrophic lateral sclerosis and frontotemporal dementia. Brain. 2021;144(12):3710‐3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Trolese M C, Scarpa C, Melfi V, et al. Boosting the peripheral immune response in the skeletal muscles improved motor function in ALS transgenic mice. Mol Ther 2022;30(8):2760‐2784. doi:10.1016/j.ymthe.2022.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zucchi E, Bonetto V, Sorarù G, et al. Neurofilaments in motor neuron disorders: towards promising diagnostic and prognostic biomarkers. Mol Neurodegener. 2020;15(1):58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brooks BR, Miller RG, Swash M, Munsat TL, World Federation of Neurology Research Group on Motor Neuron Diseases . El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(5):293‐299. [DOI] [PubMed] [Google Scholar]
- 22. Hamidou B, Marin B, Lautrette G, et al. Exploring the diagnosis delay and ALS functional impairment at diagnosis as relevant criteria for clinical trial enrolment. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(7‐8):519‐527. [DOI] [PubMed] [Google Scholar]
- 23. Kimura F, Fujimura C, Ishida S, et al. Progression rate of ALSFRS‐R at time of diagnosis predicts survival time in ALS. Neurology. 2006;66(2):265‐267. [DOI] [PubMed] [Google Scholar]
- 24. Rentzos M, Rombos A, Nikolaou C, et al. Interleukin‐17 and interleukin‐23 are elevated in serum and cerebrospinal fluid of patients with ALS: a reflection of Th17 cells activation? Acta Neurol Scand. 2010;122(6):425‐429. [DOI] [PubMed] [Google Scholar]
- 25. Henkel JS, Beers DR, Wen S, et al. Regulatory T‐lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol Med. 2013;5(1):64‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Casoni F, Basso M, Massignan T, et al. Protein nitration in a mouse model of familial amyotrophic lateral sclerosis: possible multifunctional role in the pathogenesis. J Biol Chem. 2005;280(16):16295‐16304. [DOI] [PubMed] [Google Scholar]
- 27. Nardo G, Pozzi S, Mantovani S, et al. Nitroproteomics of peripheral blood mononuclear cells from patients and a rat model of ALS. Antioxid Redox Signal. 2009;11(7):1559‐1567. [DOI] [PubMed] [Google Scholar]
- 28. Nardo G, Pozzi S, Pignataro M, et al. Amyotrophic lateral sclerosis multiprotein biomarkers in peripheral blood mononuclear cells. PLoS One. 2011;6(10):e25545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Filareti M, Luotti S, Pasetto L, et al. Decreased levels of foldase and chaperone proteins are associated with an early‐onset amyotrophic lateral sclerosis. Front Mol Neurosci. 2017;10:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Luotti S, Pasetto L, Porcu L, et al. Diagnostic and prognostic values of PBMC proteins in amyotrophic lateral sclerosis. Neurobiol Dis. 2020;139:104815. [DOI] [PubMed] [Google Scholar]
- 31. Baron P, Bussini S, Cardin V, et al. Production of monocyte chemoattractant protein‐1 in amyotrophic lateral sclerosis. Muscle Nerve. 2005;32(4):541‐544. [DOI] [PubMed] [Google Scholar]
- 32. Guo J, Yang X, Gao L, Zang D. Evaluating the levels of CSF and serum factors in ALS. Brain Behav. 2017;7(3):e00637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kuhle J, Lindberg RL, Regeniter A, et al. Increased levels of inflammatory chemokines in amyotrophic lateral sclerosis. European J Neurol. 2009;16(6):771‐774. [DOI] [PubMed] [Google Scholar]
- 34. Tateishi T, Yamasaki R, Tanaka M, et al. CSF chemokine alterations related to the clinical course of amyotrophic lateral sclerosis. J Neuroimmunol. 2010;222(1‐2):76‐81. [DOI] [PubMed] [Google Scholar]
- 35. Benatar M, Zhang L, Wang L, et al. Validation of serum neurofilaments as prognostic and potential pharmacodynamic biomarkers for ALS. Neurology. 2020;95(1):e59‐e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Palma E, Reyes‐Ruiz JM, Lopergolo D, et al. Acetylcholine receptors from human muscle as pharmacological targets for ALS therapy. Proc Natl Acad Sci U S A. 2016;113(11):3060‐3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Miller T, Cudkowicz M, Shaw PJ, et al. Phase 1–2 trial of antisense oligonucleotide tofersen for SOD1 ALS. N Engl J Med. 2020;383(2):109‐119. [DOI] [PubMed] [Google Scholar]
- 38. Miller T, Cudkowicz M. In American Neurological Association Annual Meeting (Virtual, 2021 Oct 17–19).
- 39. Benatar M, Wuu J, Andersen PM, et al. Design of a randomized, placebo‐controlled, phase 3 trial of tofersen initiated in clinically presymptomatic SOD1 variant carriers: the ATLAS Study. Neurotherapeutics. 2022. doi: 10.1007/s13311-022-01237-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ludolph AC, Dorst J, Dreyhaupt J, et al. Effect of high‐caloric nutrition on survival in amyotrophic lateral sclerosis. Ann Neurol. 2020;87(2):206‐216. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
Anonymised data are available from the corresponding author upon request.