

Abstract
Objectives
To report the final results of the 2‐year TAURUS study, assessing weekly prophylaxis dosing regimens of octocog alfa (Kovaltry®/BAY 81–8973) used in standard clinical practice in patients with moderate‐to‐severe haemophilia A.
Methods
TAURUS (NCT02830477) is a phase 4, multinational, prospective, non‐interventional, single‐arm study in patients of any age with moderate or severe haemophilia A (≤5% factor [F]VIII activity). TAURUS was designed to primarily investigate weekly prophylaxis dosing regimens used in standard clinical practice. Annualised bleeding rates (ABRs), treatment satisfaction and adherence, and safety were also assessed.
Results
Of 302 patients included in the full analysis set, 84.4% (n = 255) maintained their octocog alfa prophylaxis baseline regimen throughout the study, with a majority of patients (76.5%, n = 231) on two times or three times weekly regimens at the end of the observation period (≥1–≤2 years). ABRs, treatment satisfaction, and adherence remained stable during the observation period. Octocog alfa was well tolerated and there were no new or unexpected adverse events.
Conclusions
These data show that a smooth transition is observed when switching to octocog alfa from a previous FVIII treatment, with no safety issues and stable bleeding rates in a real‐world setting of patients with moderate‐to‐severe haemophilia A.
Keywords: drug switching, haemophilia A, octocog alfa, prophylaxis, prospective study
What is the NEW aspect of your work?
The study reports the final results of the phase 4 prospective, observational trial TAURUS, which investigated weekly prophylaxis dosing regimens of octocog alfa (BAY 81–8973) used in standard clinical practice in patients with moderate‐to‐severe haemophilia A.
What is the CENTRAL finding of your work?
In a real‐world setting, patients switching from previous FVIII treatment to octocog alfa had stable annualised bleeding rates, treatment satisfaction, and adherence over a period of 1–2 years with no new or unexpected safety concerns.
What is (or could be) the SPECIFIC clinical relevance of your work?
Patients with moderate‐to‐severe haemophilia A can potentially have a smooth transition from their previous FVIII treatment to octocog alfa with no safety issues and stable bleeding rates.
1. INTRODUCTION
Haemophilia A is an X‐linked, genetic bleeding disorder characterised by a deficiency of coagulation factor VIII (FVIII), 1 and has an estimated annual incidence of approximately 1 in 5000 live male births. 2 , 3 Prophylaxis with FVIII replacement is considered the standard of care for management of haemophilia A, as it has been shown to reduce complications from repeated bleeds, particularly joint outcomes. 1 , 4 , 5 Octocog alfa (Kovaltry® [BAY 81–8973]; Bayer) is an unmodified full‐length recombinant blood coagulation factor VIII (rFVIII) product 6 , 7 indicated for the treatment of haemophilia A.
In the LEOPOLD trials, octocog alfa demonstrated efficacy for treatment of bleeds, perioperative management, and prophylaxis administered as two‐ or three‐times‐weekly dosing regimens. 8 , 9 Octocog alfa was well tolerated with no significant safety concerns 8 , 9 and displayed a superior pharmacokinetic (PK) profile compared with sucrose‐formulated rFVIII (rFVIII‐FS/Kogenate®) 10 as well as an antihaemophilic factor (recombinant) plasma/albumin‐free method (rAHF‐PFM/Advate®). 11
The primary objective of the phase 4, open‐label TAURUS study was to investigate weekly prophylaxis dosing regimens of octocog alfa used in standard clinical practice to treat patients with moderate‐to‐severe haemophilia A. An interim TAURUS analysis has previously shown that octocog alfa demonstrated effective prophylaxis in the real world without compromising patient satisfaction or adherence. 12 Patients who received octocog alfa three or more times per week were shown to be younger and with a longer history of prophylaxis than those treated two or fewer times per week, suggesting successful individualisation of the prophylaxis regimen by physicians. 12 After switching to octocog alfa, the majority of patients remained on the same individualised prophylaxis treatment regimen for a year, maintaining treatment satisfaction and good adherence. 12 TAURUS was completed in March 2021, and the final results are presented here.
2. MATERIALS AND METHODS
2.1. Patients
Male patients of any age diagnosed with moderate‐to‐severe haemophilia A (≤5% FVIII:C [factor VIII coagulant activity]), with or without a history of inhibitors but without evidence of FVIII inhibitor at baseline, and previously treated with any FVIII product for ≥50 exposure days (EDs) were eligible for inclusion in the study. Key exclusion criteria included patients participating in an investigational programme with interventions outside of routine clinical practice, patients on immune tolerance induction treatment at the time of enrolment, and diagnosis of any bleeding disorder other than haemophilia A. All patients prescribed octocog alfa for a medically appropriate use, fulfilling the selection criteria, and consenting to participate were eligible for enrolment into the study.
2.2. Study design
TAURUS was a multinational, open‐label, prospective, non‐interventional, single‐arm phase 4 study (ClinicalTrials.gov identifier: NCT02830477). The study was conducted at 25 study locations throughout Asia, Europe, and the USA. A prospective cohort design was chosen to reflect real‐world characterisation of the prophylaxis dosing regimen used in children and adults with moderate‐to‐severe haemophilia A, enabling accurate measurement of exposure variables and multiple outcomes as defined by the primary and secondary endpoint measures.
Patients were followed for a minimum of 1 year and up to approximately 2 years or until the end of treatment with octocog alfa. The full analysis set (FAS) was defined as patients who fulfilled all inclusion and no exclusion criteria with documented initial dose of prophylaxis treatment with octocog alfa and documented end of observation. A patient was included in the safety analysis set (SAF) if they had received at least one dose of octocog alfa (documented by physician or entered in patient diary within study observational period).
Due to the coronavirus disease 2019 (COVID‐19) pandemic, the study was halted prematurely in all countries, except Italy. However, this decision had no impact on the safety or the physical or mental wellbeing of the study participants. The impact on the primary objective was considered minor since all patients could still be included in the analysis. The actual observation period of the prematurely discontinued patients was considered of sufficient length to enable a meaningful interpretation of the statistical analysis. The study was approved by the Institutional Review Board at each study site and was conducted in compliance with the principles of the Declaration of Helsinki, and Good Clinical Practice guidelines. All patients or their guardians provided written informed consent prior to enrolment in the study.
2.3. Outcomes and assessments
The primary objective of the study was to investigate weekly prophylaxis dosing regimens used in standard clinical practice; the primary outcome measure was the proportion of patients on two times weekly (2×/W) and three times weekly (3×/W) prophylaxis at the end of the observation period.
Secondary outcome measures included: reported annualised (joint) bleeding rates (A[J]BR); prophylaxis dosing by age group and country; change in prophylaxis dosing frequency and reason for change (from study start to the end of the observation period); total annualised FVIII consumption; occurrence of adverse events (AEs) and serious adverse events (SAEs); treatment satisfaction; and treatment adherence. Treatment satisfaction was assessed from baseline to the end of the observation period (1 and 2 years) 9 using the haemophilia‐specific treatment satisfaction questionnaire (Hemo‐SAT). 13 , 14 For this assessment, adult patients or parents/caregivers answered the respective Hemo‐SAT questionnaire versions (adults: Hemo‐SATA with 34 items, or parents: Hemo‐SATP with 35 items) related to the following six dimensions: ease and convenience, efficacy, burden, specialist/nurse, centre/hospital, and general satisfaction. Sub‐scores and total scores ranged from 0 (highest satisfaction) to 100 (highest dissatisfaction). Treatment adherence was assessed from baseline to 6 months and the end of the observation period (1 and 2 years) using the self‐/parent‐reported Validated Haemophilia Regimen Treatment Adherence Scale‐Prophylaxis (VERITAS‐Pro). 15 The VERITAS‐Pro consists of 24 questions on the following six subscales: time, dose, plan, remember, skip, and communicate. Patients who self‐infuse or parents/caregivers completed the questionnaire at baseline and 6 months, 1 year, and 2 years after baseline, and the total score ranged from 24 to 120, where 24 equalled the highest adherence.
Patient clinical information was documented at the time of initial visit and thereafter during routine clinic visits according to local clinical practice. Additionally, patients entered data on injections and bleeds in a patient diary.
2.4. Safety
Throughout the study, patients were closely monitored at each study visit for incidence of AEs and SAEs. The duration, severity, relationship to study drug, and outcomes were documented. FVIII inhibitor development was defined as a Nijmegen‐modified Bethesda assay measured titre of ≥0.6 Bethesda units (BU) and confirmed in a second plasma sample. The incidence of AEs and treatment‐emergent AEs (TEAEs) were assessed, with TEAEs defined as any event arising or worsening after the start of treatment with octocog alfa until 7 days after the last treatment.
2.5. Statistical analyses
All variables were analysed descriptively with appropriate statistical methods: categorical variables by frequency tables (absolute and relative frequencies) and continuous variables by sample statistics. All analyses were performed for the total study population and stratified by age group (0 to <6 years, 6 to <12 years, 12 to <18 years, 18 years and above), baseline prophylaxis dosing regimen (≤2.5 times weekly [≤2.5×/W], >2.5 times weekly [>2.5×/W]), and haemophilia severity at initial diagnosis (FVIII:C, 0 to <1%, 1% to 5%), as well as the combination between the two latter parameters. Chi‐square tests were used to assess numerical differences seen between these groups. Reported p‐values are exploratory; no confirmatory tests were done.
While the primary outcome measure was proportion of patients on 2×/W and 3×/W prophylaxis at the end of the observation period, the data were split into ≤2.5×/W and >2.5×/W owing to the different regimens patients were on at baseline (Figure 1). Statistical evaluation was performed by using the software package SAS release 9.2 (SAS Institute Inc., Cary, NC, USA).
FIGURE 1.
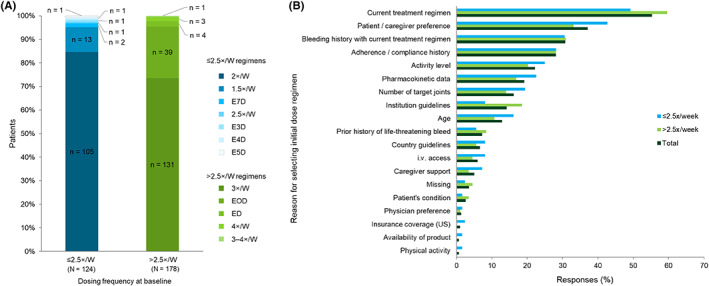
(A) Octocog alfa regimen at baseline and (B) reasons for selecting initial dosing regimen (dosage and dose frequency) for octocog alfa. 1.5×/W, 1.5 times per week; 2×/W, 2 times per week; 2.5×/W, 2.5 times per week; 3×/W, 3 times per week; 3–4×/W, 3–4 times per week; E3D, every 3 days; E4D, every 4 days; E5D, every 5 days, ED, every day; EOD, every other day; i.v., intravenous.
3. RESULTS
3.1. Patients
A total of 320 patients were screened for this study, of whom 318 were enrolled (99.4%) to receive octocog alfa. Data for 16 patients were not included in the FAS and the most frequent reason for non‐inclusion was “later violation of inclusion/exclusion criterion” (n = 10/16), followed by “no end of observation documented” (n = 9/16). For the majority of patients (71.5%), the main reason for the end of observation was “regular end of study”. The most common main reasons for not completing the study were “switch to other therapy” (11.9%) and “premature termination by sponsor due to COVID‐19 pandemic” (9.3%).
3.1.1. Safety analysis set
For safety evaluations, data were available for 313 out of 318 enrolled patients (98.4%). Five patients who had no documented dose of octocog alfa were excluded from the SAF. In total, 132 (42.2%) of 313 patients in the SAF had concomitant diseases at baseline, which included musculoskeletal and connective tissue disorders (n = 53, 16.9%), infections (n = 38, 12.1%), vascular disorders (n = 24, 7.7%), and metabolism and nutrition disorders (n = 18, 5.8%). Of the 53 patients who had musculoskeletal and connective tissue disorders at baseline, 25 had haemophilic arthropathy. Concomitant diseases were reported for 56 patients (42.4%) and 75 patients (41.9%) in the ≤2.5×/W and >2.5×/W baseline prophylaxis dosing regimen groups, respectively (baseline regimen data were missing for one patient noted to have concomitant disease). No major differences were found in concomitant diseases in the SAF compared with the FAS.
3.1.2. Full analysis set
Of the 318 enrolled patients, 302 (95.0%) patients were included in the FAS. Enrolled patients were from 12 countries (Belgium, n = 13; Canada, n = 14; Colombia, n = 17; France, n = 25; Germany, n = 41; Greece, n = 15; Italy, n = 60; Netherlands, n = 30; Slovenia, n = 5; Spain, n = 35; Taiwan, n = 20; and USA, n = 27; Figure S1).
Baseline demographics and clinical characteristics of the overall population (FAS) are shown in Table 1. All patients in this study were male and had a median (Q1; Q3) age of 23.5 (14.0; 39.0) years. The mean age for initiating prophylaxis therapy was 13.2 years (median [range] 6.0 [0–64] years) with 46.4% of patients aged ≥2–≤18 years. The median (range) length of continuous regular prophylaxis treatment prior to entry into this study was 10.0 (0–49.0) years, and 129 (42.7%) out of 302 patients in the FAS had concomitant diseases at baseline. The median (range) observation period for all patients (FAS) in the study was 1.1 (0.1–2.2) years. Corresponding values for the ≤2.5×/W and >2.5×/W groups were 1.1 (0.2–2.1) years and 1.1 (0.1–2.2) years, respectively.
TABLE 1.
Baseline patient demographics and clinical characteristics by octocog alfa prophylaxis regimen (FAS)
| ≤2.5×/W (n = 124) | >2.5×/W (n = 178) | Total (N = 302) | |
|---|---|---|---|
| Age, years, median (Q1; Q3) | 27 (15.5; 41.5) | 21.5 (13.0; 38.0) | 23.5 (14.0; 39.0) |
| Race, n (%) | |||
| White | 85 (68.5) | 138 (77.5) | 223 (73.8) |
| Asian | 14 (11.3) | 13 (7.3) | 27 (8.9) |
| Black or American African | 4 (3.2) | 5 (2.8) | 9 (3.0) |
| American Indian or Alaska native | 0 | 1 (0.6) | 1 (0.3) |
| Missing | 17 (13.7) | 8 (4.5) | 25 (8.3) |
| Not reported | 4 (3.2) | 13 (7.3) | 17 (5.6) |
| Length of pre‐study prophylaxis, years, median (Q1; Q3) | 8.0 (3.5; 14.0) | 12.0 (6.7; 17.0) | 10.0 (5.0; 16.0) |
| Patients with > 150 exposure days to FVIII, n (%) | 111 (89.5) | 165 (92.7) | 276 (91.4) |
| Severe haemophilia (FVIII <1%), n (%) | 98 (79.0) | 157 (88.2) | 255 (84.4) |
| Patients with at least one target joint at baseline, n (%) | 56 (45.2) | 71 (39.9) | 127 (42.1) |
| Total bleeds in 6 months pre‐study a , median (Q1; Q3) | 0.0 (0.0; 2.0) | 0.0 (0.0; 1.0) | 0.0 (0.0; 2.0) |
| Joint bleeds in 6 months pre‐study a , median (Q1; Q3) | 0.0 (0.0; 1.0) | 0.0 (0.0; 1.0) | 0.0 (0.0; 1.0) |
| Patients with positive inhibitor test, n (%) b | 9 (7.3) | 24 (13.5) | 33 (10.9) |
Information collected retrospectively by physician.
Median (range) titre for all patients with a history of inhibitors was 4.000 (0.02–64.0) BU, and all patients had resolution of last positive inhibitor (median 9.2 [range, 1.4–21.3] years) prior to baseline readings. 2.5×/W, 2.5 times per week; BU, Bethesda units; FAS, full analysis set; FVIII, factor VIII; Q, quartile.
3.2. Prior FVIII treatment
All 302 patients in the FAS had received prior FVIII treatment, with the majority (n = 228, 75.5%) treated with rFVIII‐FS pre‐study. A median (range) time of 6.41 (0.07–29.75) years from the start and 2.0 (−2 to 646) days from the end of the most recent FVIII treatment prior to initiation of octocog alfa was observed. The median duration of the most recent FVIII treatment prior to octocog alfa prophylaxis initiation was 6.5 years and the mean total weekly dose of the most recent FVIII treatment prior to octocog alfa was 71.41 IU/kg.
Most patients received regular FVIII prophylaxis (n = 289; 95.7%) with the dose frequency of the most recent FVIII prophylaxis regimen prior to octocog alfa being ≤2.5×/W in 107 patients (37.0%) and >2.5×/W in 181 patients (62.6%). Dose‐frequency data for one patient were missing.
3.3. Octocog alfa regimen at baseline and reasons for selection
At baseline, 124 patients were treated with octocog alfa ≤2.5×/W and 178 patients were treated with octocog alfa >2.5×/W (Figure 1A). The most frequent reasons for selection of initial dose and dosing frequency of octocog alfa in the overall population (FAS) were “current treatment regimen” (55.3%), “patient/caregiver preference” (37.1%), “bleeding history with current treatment regimen” (30.8%), “adherence/compliance history” (28.1%), “activity level” (22.2%), “pharmacokinetic data” (19.2%), “number of target joints” (16.2%), “institution guidelines” (14.2%), and “age” (12.9%) (Figure 1B).
The same reasons as those from the overall population were most frequently reported in both prophylaxis dosing regimen subgroups. Frequency of reports on “adherence/compliance history” was similar between the prophylaxis dose regimen subgroups (≤2.5×/W: 28.2%, n = 35; >2.5×/W: 28.1%, n = 50). “Patient/caregiver preference” was reported more frequently in the ≤2.5×/W group (42.7%, n = 53) than in the >2.5×/W group (33.1%, n = 59; p = .09), whereas “Current treatment regimen” was reported more frequently in the >2.5×/W group (59.6%, n = 106) than in the ≤2.5×/W group (49.2%, n = 61; p = .07).
The same reasons for selection of initial dose and dosing frequency as those of the overall population were most frequently reported for patients ≥12 years old (n = 245). In patients <12 years old, “Intravenous (i.v.) access” and “Prior history of life‐threatening bleeds” were among the most frequently mentioned reasons, while “Number of target joints” and “Pharmacokinetic data” were reported less frequently. Median (range) therapy duration based on prophylaxis regimen at baseline was 19.9 (2.5–102.1) months for the overall population. The corresponding values for the ≤2.5×/W and >2.5/W regimen subgroups were 21.6 (2.5–78.9) months and 18.3 (3.2–102.1) months, respectively.
3.4. Octocog alfa regimen switching and consumption
In the overall population (FAS), a total of 255 (84.4%) patients had no change to their octocog alfa regimen during the entire course of the study, whereas 47 patients had switched at least once during the study. At the end of observation, 21 patients had switched back to their baseline prophylaxis regimen.
Of the 124 patients who were receiving a ≤ 2.5×/W prophylaxis dosing regimen at baseline, 113 patients (91.1%) had remained in the same regimen category by the end of observation, while 11 patients (8.9%) had switched to >2.5×/W. At the end of observation, the majority of patients (75.8%, n = 94) in the ≤2.5×/W baseline prophylaxis subgroup were receiving the 2×/W octocog alfa regimen.
Of the 178 patients who were receiving a > 2.5×/W prophylaxis dosing regimen at baseline, 163 patients (91.6%) had remained in the same regimen category by the end of observation, while 15 patients (8.4%) had switched to ≤2.5×/W. At the end of observation, the majority of patients (65.2%, n = 116) in the >2.5×/W baseline prophylaxis subgroup were receiving the 3×/W octocog alfa regimen.
At the end of observation, 128 patients in the overall population (FAS) (42.4%) were receiving ≤2.5×/W prophylaxis and 174 patients (57.6%) were receiving >2.5×/W prophylaxis. Of 302 patients in the FAS, the majority of patients (76.5%, n = 231) were receiving either 2×/W or 3×/W regimens at the end of the observation period. A comparative representation of the prophylaxis regimens at baseline and end of observation is provided in Figure 2.
FIGURE 2.
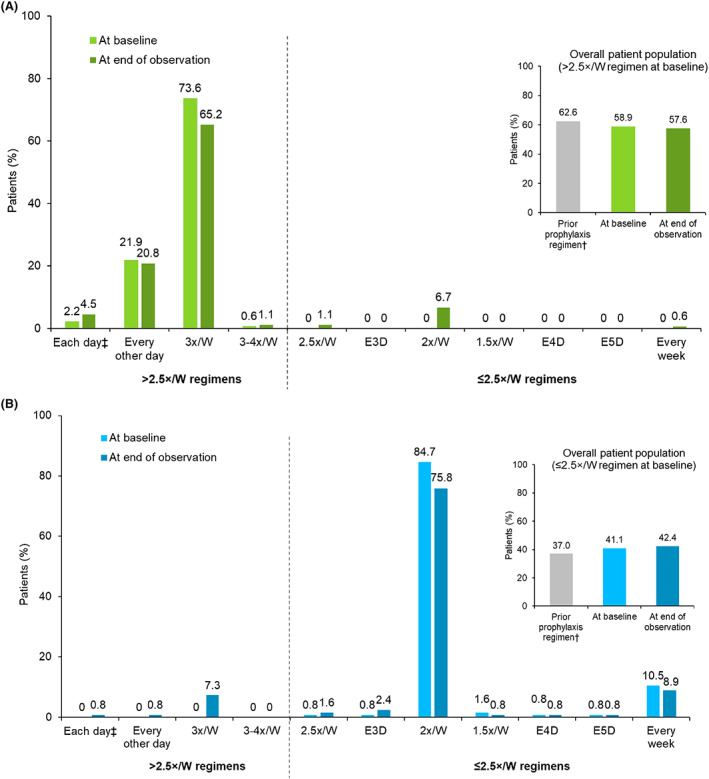
Octocog alfa prophylaxis dose frequency for individual regimens at study baseline and at the end of the observation period by octocog alfa baseline prophylaxis regimen. (A) Patients with >2.5×/W octocog alfa regimen at baseline, n = 178. (B) Patients with ≤2.5×/W octocog alfa regimen at baseline, n = 124. †Missing patients, n (%): prior prophylaxis, 1 (0.3%). Inset figures show the total prophylaxis dose frequency of prior FVIII treatment, octocog alfa treatment at study baseline and the at end of the observation period by octocog alfa baseline prophylaxis regimen. ‡Few patients ended the study on an increased treatment frequency due to adverse event, surgery, or bleed treatment. 1.5×/W, 1.5 times per week; 2×/W, 2 times per week; 2.5×/W, 2.5 times per week; 3×/W, 3 times per week; 3–4×/W, 3–4 times per week; E3D, every 3 days; E4D, every 4 days; E5D, every 5 days.
As for the overall population, a clear majority of patients in the <12 years (78.9%) and ≥ 12 years (85.7%) subgroups did not switch prophylaxis dosing regimen during the period from baseline to end of observation. However, an increase of prophylaxis dosing frequency was noted in 14.0% of patients aged <12 years and 7.3% of patients aged ≥12 years. Similar proportions of patients in these subgroups had a decrease of prophylaxis dosing frequency from baseline to end of observation (7.0% and 6.9%, respectively). In all countries, most patients remained on the same prophylaxis dosing regimen at the end of observation as at baseline (Figure S2).
Median (Q1; Q3) weekly doses of the most recent FVIII product prior to octocog alfa treatment in the ≤2.5×/W and >2.5×/W regimen groups at baseline were 53.2 (36.0; 75.5) and 75.0 (46.9; 100.0) IU/kg, respectively; for the overall population, the corresponding median (Q1; Q3) weekly dose was 66.7 (43.2; 86.1) IU/kg. The median (Q1; Q3) weekly doses for octocog alfa prophylaxis during the study in the ≤2.5×/W and >2.5×/W regimen groups at baseline were 55.6 (40.0; 75.0) and 75.1 (51.3; 105.3) IU/kg, respectively. The corresponding median (Q1; Q3) weekly dose during the study for the overall population was 69.8 (46.1; 89.9) IU/kg.
Median (Q1; Q3) total annualised FVIII consumption based on available data was 3923 (2860; 4797) IU/kg/year (n = 266). When stratified by age, median (range) of prescribed octocog alfa doses for patients <12 years old (n = 54) and ≥ 12 years old (n = 228) were 75.56 (29.41–276.32) IU/kg and 66.67 (11.90–228.26) IU/kg, respectively. The mean changes in prescribed weekly octocog alfa dose per kg from baseline to end of observation were 2.45 IU/kg (n = 293 patients), 2.69 IU/kg (n = 120), and 2.29 IU/kg (n = 173) in the total, ≤2.5×/W, and >2.5×/W at baseline prophylaxis groups, respectively.
3.5. Bleeding outcomes
Patient diary documentation was available for 268 patients with a median (range) documentation period of 368.5 (1.00–789) days. Patients in the FAS had a mean (standard deviation; SD) number of 1.7 (3.5) bleeds (median = 0.0) in the last 6 months prior to baseline and 3.4 (7.0) bleeds (median = 0.0) in the last 12 months prior to baseline.
The annualised number of reported bleeds and actual number of reported bleeds stratified by dosing frequency and age group are shown in Figure 3 and Table S1, respectively.
FIGURE 3.
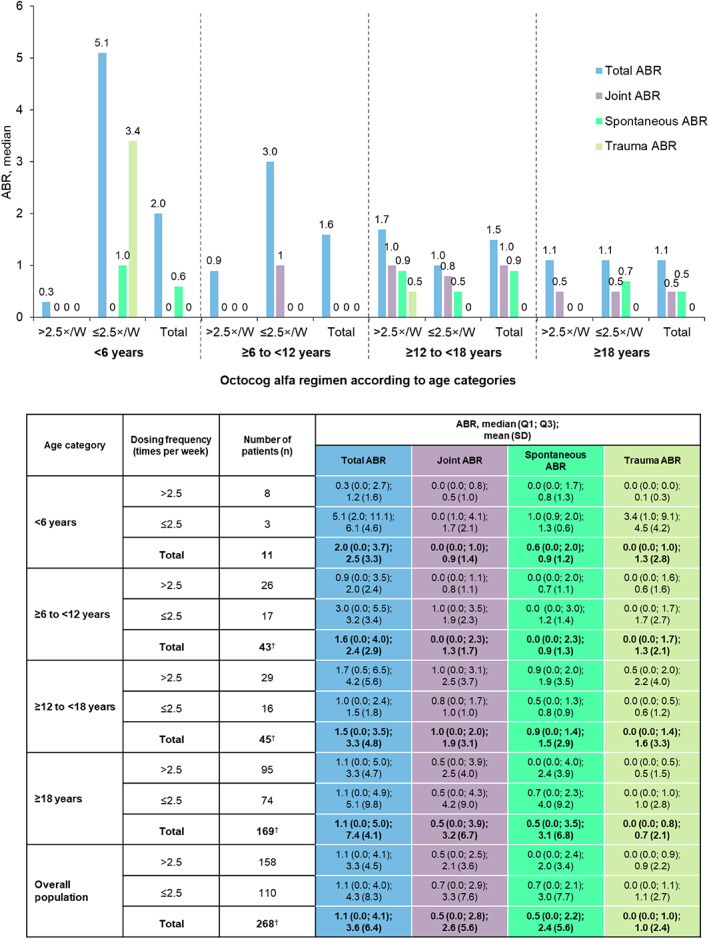
Annualised bleeding rates of patients receiving octocog alfa regular prophylaxis in the ≤2.5×/W and >2.5×/W octocog alfa regimen groups at baseline stratified by age group. †Number of patients with missing data: ≥6 to <12 years, n = 3; ≥12 to <18 years, n = 8; ≥18 years, n = 23; total = 34. 2.5×/W, 2.5 times per week; ABR, annualised bleeding rate; Q, quartile; SD, standard deviation.
3.6. Patient‐reported outcomes among patients completing the observation period
Treatment satisfaction among patient/caregivers in the FAS, as measured by Hemo‐SATA/Hemo‐SATP total scores at baseline, 1 year and 2 years after the baseline visit, and at last post‐baseline assessment, remained similar from baseline to last post‐baseline assessment within both prophylaxis groups, and overall (Table 2a). The total Hemo‐SATA scores for the overall population were 10.3 and 12.3 at 1 and 2 years from baseline, respectively, and Hemo‐SATP scores were 10.7 and 9.6 at 1 and 2 years from baseline, respectively (Table 2a).
TABLE 2.
(a) Treatment satisfaction for patients with 1 year or 2 years of follow‐up data, assessed by Hemo‐SATA and Hemo‐SATP and (b) treatment adherence for patients with 6 months, 1 year, or 2 years of follow‐up data, assessed by VERITAS‐PRO
| (a) Hemo‐SATA and Hemo‐SATP total scores | |||
|---|---|---|---|
| Hemo‐SATA total score, [n] median (Q1; Q3) | |||
| ≤2.5 times/week (n = 94) | >2.5 times/week (n = 124) | All patients (n = 218) | |
| Baseline a | [76] 10.7 (5.2; 19.9) | [95] 11.8 (5.9; 19.9) | [171] 11.0 (5.9; 19.9) |
| One year after baseline a | [43] 8.8 (3.0; 22.8) | [44] 11.8 (5.2; 16.5) | [87] 10.3 (4.4; 16.9) |
| Two years after baseline a | [18] 13.2 (8.8; 19.1) | [14] 7.5 (4.4; 14.7) | [32] 12.3 (4.8; 14.7) |
| Last post‐baseline assessment (≥300 days after baseline) | [62] 10.7 (4.4; 19.1) | [64] 11.4 (5.2; 16.4) | [126] 11.0 (4.4; 16.9) |
| Hemo‐SATP total score, [n] median (Q1; Q3) | |||
|---|---|---|---|
| ≤2.5 times/week (n = 27) | >2.5 times/week (n = 59) | All patients (n = 86) | |
| Baseline a | [26] 10.0 (5.7; 19.3) | [54] 11.1 (4.3; 23.6) | [80] 10.0 (5.0; 21.4) |
| One year after baseline a | [10] 10.0 (3.6; 16.4) | [25] 10.7 (6.4; 19.3) | [35] 10.7 (4.3; 19.3) |
| Two years after baseline a | [8] 9.6 (4.3; 12.5) | [6] 11.8 (7.9; 21.4) | [14] 9.6 (6.4; 15.0) |
| Last post‐baseline assessment (≥300 days after baseline) | [18] 5.4 (3.6; 12.9) | [35] 10.7 (6.4; 19.3) | [53] 9.3 (4.3; 15.0) |
| (b) VERITAS‐PRO total score | |||
|---|---|---|---|
| VERITAS‐PRO total score, [n] median (Q1; Q3) | |||
| ≤2.5 times/week (n = 119) | >2.5 times/week (n = 169) | All patients (n = 288) | |
| Baseline b | [108] 35.0 (31.0; 42.5) | [156] 35.0 (29.0; 45.7) | [264] 35.0 (30.0; 44.2) |
| Six months after baseline b | [80] 36.0 (29.5; 42.5) | [95] 36.0 (29.0; 45.0) | [175] 36.0 (29.0; 44.0) |
| One year after baseline b | [68] 33.5 (27.0; 41.0) | [96] 35.0 (30.0; 43.0) | [164] 34.0 (29.5; 42.0) |
| Two years after baseline b | [27] 33.0 (30.0; 47.0) | [24] 33.0 (28.0; 39.0) | [51] 33.0 (28.0; 39.0) |
| Last post‐baseline assessment (≥300 days after baseline) | [88] 33.0 (27.0; 42.0) | [119] 35.0 (29.0; 42.0) | [207] 34.0 (28.0; 42.0) |
FAS subgroup of adult patients with at least one documented Hemo‐SATA questionnaire at analysis timepoints. Baseline questionnaire assessment is defined as up to 30 days after initial visit, 1‐year assessment as between 300 and 420 days after initial visit, 2‐year assessment as between 660 and 780 days after initial visit; total score ranges from 0 (highest satisfaction) to 100 (highest dissatisfaction). Hemo‐SATA/Hemo‐SATP, haemophilia‐specific treatment satisfaction questionnaires for adult patients (Hemo‐SATA) and parents/carers of children with haemophilia A (Hemo‐SATP); Q, quartile.
FAS subgroup of patients with at least one documented VERITAS‐PRO questionnaire at analysis timepoints. Baseline questionnaire assessment is defined as up to 30 days after initial visit, 6‐month assessment as between 120 and 240 days, 1‐year assessment as between 300 and 420 days, 2‐year assessment as between 660 and 780 days after initial visit. Total score ranges from 24 (most adherent) to 120 (least adherent). VERITAS‐PRO, Validated Haemophilia Regimen Treatment Adherence Scale‐Prophylaxis; Q, quartile.
Treatment adherence among patients in the FAS at 6 months, 1 year, and 2 years after initial visit remained relatively stable and no major differences were observed between the subgroups by baseline prophylaxis dosing regimen (Table 2b). Mean (SD) changes in total score for VERITAS‐PRO questionnaires 6 months after baseline in the ≤2.5×/W (n = 72) and >2.5×/W (n = 89) dosing regimen groups were 0.11 (6.78) and − 0.93 (6.47), respectively. The mean change for VERITAS‐PRO questionnaires 1 year after baseline was −0.84 (8.48) in the ≤2.5×/W group (n = 60) and − 1.00 (7.53) in the >2.5×/W (n = 89) dosing regimen group, and 2 years after baseline was 0.48 (6.41) in 25 patients and − 2.86 (5.76) in 21 patients for these groups, respectively.
3.7. Safety
Of 313 patients in the SAF, 96 (30.7%) experienced an AE (Table 3). Serious TEAEs were observed in 31 patients (9.9%). The most common serious TEAEs were gastrointestinal disorders (n = 7 [2.2%]), infections (n = 6 [1.9%]), and injury, poisoning and procedural complications (n = 6 [1.9%]). Three patients experienced a drug‐related TEAE: nausea (n = 1), arthralgia (n = 1), and pruritus (n = 1); nausea and pruritus both led to discontinuation of octocog alfa treatment in their respective patients (n = 2, 0.6%). Two fatal TEAEs were observed in this study (0.6%). One patient was a 59‐year‐old male diagnosed with osmotic demyelination syndrome and the other patient was a 55‐year‐old male diagnosed with metastatic pancreatic carcinoma. The causality of both TEAEs was not related to treatment with octocog alfa. No inhibitor development or positive inhibitor measurement was observed.
TABLE 3.
Treatment‐emergent adverse events (TEAEs) following administration of octocog alfa, stratified by age group
| <6 years (n = 12) | ≥6–<12 years (n = 46) | ≥12–<18 years (n = 54) | >18 years (n = 201) | Total (N = 313) | |
|---|---|---|---|---|---|
| Any TEAE, n (%) | 6 (50%) | 17 (37%) | 14 (25.9%) | 59 (29.4%) | 96 (30.7%) |
| Serious TEAE, n (%) | 3 (25%) | 9 (19.6%) | 3 (5.6%) | 16 (8.0%) | 31 (9.9%) |
| Fatal TEAE, n (%) | 0 | 0 | 0 | 2 (1%) | 2 a (0.6%) |
| Drug‐related TEAE, n (%) | 0 | 0 | 0 | 3 (1.5%) | 3 b (1%) |
| Serious drug‐related TEAE, n (%) | 0 | 0 | 0 | 0 | 0 |
| Discontinuation due to TEAE, n (%) | 0 | 0 | 0 | 2 (1%) | 2 c (0.6%) |
One patient had osmotic demyelination syndrome and the other patient had metastatic pancreatic carcinoma (none related to octocog alfa treatment).
Nausea, n = 1; arthralgia, n = 1; pruritus, n = 1.
Nausea, n = 1; pruritus, n = 1.
4. DISCUSSION
Severity and burden of haemophilia A can vary markedly from person to person; however, when combined, patients with moderate‐to‐severe disease represent approximately 75% of the patient population with haemophilia A. 3 Via enrolment of eligible patients with moderate‐to‐severe disease, with or without other comorbidities, TAURUS was representative of the real‐world situation in patients with haemophilia A. This prospective, open‐label, non‐interventional, single‐arm, phase 4 study provided an opportunity to collect real‐life data on safety and effectiveness in children and adults with moderate‐to‐severe haemophilia A (≤5% FVIII:C) who were treated with octocog alfa.
Most patients in this study continued their baseline octocog alfa regimen throughout the study, without any dose or regimen changes. While patients did switch their prophylaxis dosing frequency between baseline and end of observation, many of these switches were temporary. At baseline and end of study observation, patients were most frequently treated with the 3×/W regimen followed by the 2×/W regimen.
ABRs also remained stable during the observation period compared with those prior to study entry or octocog alfa initiation. Treatment satisfaction and adherence at 1 and 2 years after initial visit were also similar over time, and no major differences were observed between the baseline prophylaxis dosing regimen subgroups. However, these results should be interpreted with caution due to low numbers of documented Hemo‐SATA, Hemo‐SATP, and VERITAS‐PRO questionnaires at later time points in both subgroups.
Octocog alfa was well tolerated during the observation period, during which no new or unexpected TEAEs or SAEs occurred, and no patients developed FVIII inhibitors. Three patients experienced a drug‐related AE, none of which were serious, and two patients discontinued the study due to drug‐related AEs (nausea and pruritus). Two fatal AEs occurred, although the causality of both AEs was not related to the study treatment. Overall, safety data suggest the benefit–risk analysis for octocog alfa prophylaxis in patients with haemophilia A is favourable.
The limitations of this study are those inherent to a real‐world, observational study design. TAURUS is a single‐arm study without a comparison group, performed in different countries and centres. Basic statistical analyses were applied to the data, and, therefore, no significant differences in efficacy between subgroups or study time periods could be established. The data collected in this study may potentially suffer from bias, either by systematic differences in data recording or different interpretations of information on exposure or outcome for different patients, as well as reporting and selection bias. Adherence to treatment is prone to be biased by adherence to documentation, with 237 patients with missing values for the VERITAS‐PRO questionnaire 2 years after baseline. Additionally, around one‐third of patients had an incomplete diary. Non‐compliance to documentation observed here is reflective of the real‐world scenario in which patients fail to complete their diaries regularly, 16 but could be useful in future for developing further initiatives on improving patient record‐keeping.
Considering that TAURUS is an observational study, the registry of PK parameters was optional and aligned with routine clinical practice; therefore, reported PK data were limited. Less than half of the patients included in this analysis had their PK assessments carried out since the start of their treatment with octocog alfa and one‐stage assay‐based PK assessments were performed more frequently than chromogenic assay‐based assessments. Since a robust PK analysis cannot be performed due to limited data availability, PK assessment data have not been included in this report. PK‐guided prophylaxis could be an area for improvement with the availability of new technologies and globally accessible online tools such as Web‐based Application for the Population Pharmacokinetic Service (WAPPS‐Hemo), particularly to assist with individualisation of prophylaxis dose and regimen. 17
These data highlight that patients with moderate‐to‐severe haemophilia A who switched from a previous FVIII treatment to octocog alfa had a smooth transition, with no safety issues and stable bleeding rates. Patients' treatment satisfaction and treatment adherence also remained unchanged. This confirms and extends clinical trial results, demonstrating effective protection from bleeds with octocog alfa prophylaxis in a real‐world setting, and no safety concerns. 8 , 9
AUTHOR CONTRIBUTIONS
C. Santoro, B. Fuh, P. H. Le, P. Maes, R. Berrueco, E. M. Mingot‐Castellano, S. von Mackensen, and M. Wang are principal investigators of the TAURUS trial, treated patients with study drug, and contributed to data acquisition and interpretation. C. Tueckmantel and J. F. Cabre‐Marquez were involved in the study design, analysis, and the interpretation of data. All authors contributed to the development of the manuscript, reviewed and commented on each draft, and approved the final draft.
CONFLICT OF INTEREST
C. Santoro has received honoraria for consulting or lecturing from Amgen, Bayer, CSL Behring, Novartis, Novo Nordisk, Pfizer, Roche, Sobi, and Takeda, and speaker's fees from Pfizer, Takeda, and Novartis. B. Fuh has received honoraria for consulting or lecturing from Bayer, Micelle Biopharma, Novartis, and Pfizer. P. Q. Le has no disclosures. P. Maes has received grants/research support from Bayer, CSL Behring, Shire, Sobi, and Pfizer, and honoraria for consulting or lecturing from Bayer, CSL Behring, Novo Nordisk, Pfizer, Shire, Sobi, and Roche. R. Berrueco has received grants/research support from Sobi and honoraria for consulting or lecturing from Bayer, CSL Behring, Novartis, Novo Nordisk, and Sobi. E. M. Mingot‐Castellano has received honoraria for consulting or lecturing from Amgen, Bayer, Baxter, Novartis, Novo Nordisk Janssen, Roche, and Pfizer, and speaker's fees from Amgen, Alexion, Bayer, Grifols, Leo Pharma, Novartis, Novo Nordisk, Pfizer, Shire, Sobi, and Roche. S. von Mackensen is a consultant for Bayer. C. Tueckmantel is an employee of Bayer. J. F. Cabre‐Marquez is an employee of Bayer. M. Wang has received honoraria for consulting/advisory from Bayer, Takeda, Novo Nordisk, BioMarin, CSL Behring, Genetech/Roche, and Sanofi/Bioverativ.
Supporting information
Table S1 Non‐annualised bleeding outcomes prospectively collected in patient‐reported bleeding diaries stratified by octocog alfa regimen groups at baseline and age group.
Figure S1 Patient inclusion by country.
Figure S2 Octocog alfa dosing frequency at baseline and end of observation stratified by country (N = 302).
ACKNOWLEDGEMENTS
The TAURUS study was funded by Bayer. We are grateful for the contributions of all TAURUS study investigators and site staff. The authors thank Sreerekha S. Pillai, PhD, of Darwin Healthcare Communications (Oxford, England) for providing medical writing support, which was fully funded by Bayer, in accordance with Good Publication Practice (GPP3) guidelines.
Santoro C, Fuh B, Le PQ, et al. Efficacy and safety in patients with haemophilia A switching to octocog alfa (BAY 81–8973): Final results of the global real‐world study, TAURUS . Eur J Haematol. 2023;110(1):77‐87. doi: 10.1111/ejh.13876
Funding information Bayer
DATA AVAILABILITY STATEMENT
Availability of the data underlying this publication will be determined later according to Bayer's commitment to the EFPIA/PhRMA “Principles for responsible clinical trial data sharing”. This pertains to scope, time point, and process of data access. As such, Bayer commits to sharing upon request from qualified scientific and medical researchers patient‐level clinical trial data, study‐level clinical trial data, and protocols from clinical trials in patients for medicines and indications approved in the United States (US) and European Union (EU) as necessary for conducting legitimate research. This applies to data on new medicines and indications that have been approved by the EU and US regulatory agencies on or after January 01, 2014. Interested researchers can use www.clinicalstudydatarequest.com to request access to anonymized patient‐level data and supporting documents from clinical studies to conduct further research that can help advance medical science or improve patient care. Information on the Bayer criteria for listing studies and other relevant information are provided in the Study sponsors section of the portal. Data access will be granted to anonymized patient‐level data, protocols, and clinical study reports after approval by an independent scientific review panel. Bayer is not involved in the decisions made by the independent review panel. Bayer will take all necessary measures to ensure that patient privacy is safeguarded.
REFERENCES
- 1. Srivastava A, Santagostino E, Dougall A, et al. WFH guidelines for the management of hemophilia, 3rd edition. Haemophilia. 2020;26(Suppl 6):1‐158. [DOI] [PubMed] [Google Scholar]
- 2. Orphanet . Hemophilia 2009. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=448.
- 3. National Hemophilia Foundation . 2021. https://www.hemophilia.org/bleeding-disorders-az/overview/fast-facts.
- 4. Manco‐Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357(6):535‐544. [DOI] [PubMed] [Google Scholar]
- 5. Gringeri A, Lundin B, von Mackensen S, et al. A randomized clinical trial of prophylaxis in children with hemophilia A (the ESPRIT Study). J Thromb Haemost. 2011;9(4):700‐710. [DOI] [PubMed] [Google Scholar]
- 6. FDA.gov . Kovaltry prescribing information. 2016. https://www.fda.gov/media/96221/download
- 7. EMA.eu . Kovaltry summary of product characteristics. 2016. https://www.ema.europa.eu/en/documents/product‐information/kovaltry‐epar‐product‐information_en.pdf
- 8. Kavakli K, Yang R, Rusen L, et al. Prophylaxis vs. on‐demand treatment with BAY 81‐8973, a full‐length plasma protein‐free recombinant factor VIII product: results from a randomized trial (LEOPOLD II). J Thromb Haemost. 2015;13(3):360‐369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ljung R, Kenet G, Mancuso ME, et al. BAY 81‐8973 safety and efficacy for prophylaxis and treatment of bleeds in previously treated children with severe haemophilia A: results of the LEOPOLD Kids Trial. Haemophilia. 2016;22(3):354‐360. [DOI] [PubMed] [Google Scholar]
- 10. Solms A, Lalezari S, Shah A, Kenet G. Population pharmacokinetic (PopPK) modelling indicates that patients switching to BAY 81‐8973 from rFVIII‐FS can continue their dosing schedule with improved protection. Haemophilia. 2020;26(3):e145‐e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shah A, Solms A, Garmann D, et al. Improved Pharmacokinetics with BAY 81‐8973 Versus Antihemophilic Factor (Recombinant) Plasma/Albumin‐Free Method: A Randomized Pharmacokinetic Study in Patients with Severe Hemophilia A. Clin Pharmacokinet. 2017;56(9):1045‐1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Santoro C, Fuh B, le PQ, et al. BAY 81‐8973 prophylaxis and pharmacokinetics in haemophilia A: Interim results from the TAURUS study. Eur J Haematol. 2020;105(2):164‐172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. von Mackensen S, Gringeri A, Mantovani L. Development and validation of the first treatment satisfaction scale for adult haemophiliacs (Hemo‐SatA). Haemophilia. 2004;10(Suppl 3):126.15479385 [Google Scholar]
- 14. von Mackensen S, Gringeri A. Chapter 215: quality of life in hemophilia. In: Preedy V, Watson R, eds. Handbook of Disease Burdens and Quality of Life Measures. Springer; 2010:1895‐1920. [Google Scholar]
- 15. Duncan N, Kronenberger W, Roberson C, Shapiro A. VERITAS‐Pro: a new measure of adherence to prophylactic regimens in haemophilia. Haemophilia. 2010;16(2):247‐255. [DOI] [PubMed] [Google Scholar]
- 16. Dolan G, Iorio A, Jokela V, Juusola K, Lassila R. Haemophilia in a real‐world setting: the value of clinical experience in data collection. Eur J Haematol. 2016;96(Suppl 82):3‐9. [DOI] [PubMed] [Google Scholar]
- 17. Hermans C, Dolan G. Pharmacokinetics in routine haemophilia clinical practice: rationale and modalities‐a practical review. Therap Adv Hematol. 2020;11:2040620720966888. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Non‐annualised bleeding outcomes prospectively collected in patient‐reported bleeding diaries stratified by octocog alfa regimen groups at baseline and age group.
Figure S1 Patient inclusion by country.
Figure S2 Octocog alfa dosing frequency at baseline and end of observation stratified by country (N = 302).
Data Availability Statement
Availability of the data underlying this publication will be determined later according to Bayer's commitment to the EFPIA/PhRMA “Principles for responsible clinical trial data sharing”. This pertains to scope, time point, and process of data access. As such, Bayer commits to sharing upon request from qualified scientific and medical researchers patient‐level clinical trial data, study‐level clinical trial data, and protocols from clinical trials in patients for medicines and indications approved in the United States (US) and European Union (EU) as necessary for conducting legitimate research. This applies to data on new medicines and indications that have been approved by the EU and US regulatory agencies on or after January 01, 2014. Interested researchers can use www.clinicalstudydatarequest.com to request access to anonymized patient‐level data and supporting documents from clinical studies to conduct further research that can help advance medical science or improve patient care. Information on the Bayer criteria for listing studies and other relevant information are provided in the Study sponsors section of the portal. Data access will be granted to anonymized patient‐level data, protocols, and clinical study reports after approval by an independent scientific review panel. Bayer is not involved in the decisions made by the independent review panel. Bayer will take all necessary measures to ensure that patient privacy is safeguarded.