

Abstract
In 2001 the molecular genetic basis of so‐called “ivermectin sensitivity” in herding breed dogs was determined to be a P‐glycoprotein deficiency caused by a genetic variant of the MDR1 (ABCB1) gene often called “the MDR1 mutation.” We have learned a great deal about P‐glycoprotein's role in drug disposition since that discovery, namely that P‐glycoprotein transports many more drugs than just macrocyclic lactones that P‐glycoprotein mediated drug transport is present in more places than just the blood brain barrier, that some cats have a genetic variant of MDR1 that results in P‐glycoprotein deficiency, that P‐glycoprotein dysfunction can occur as a result of drug–drug interactions in any dog or cat, and that the concept of P‐glycoprotein “inhibitors” versus P‐glycoprotein substrates is somewhat arbitrary and artificial. This paper will review these discoveries and discuss how they impact drug selection and dosing in dogs and cats with genetically mediated P‐glycoprotein deficiency or P‐glycoprotein dysfunction resulting from drug–drug interactions.
Keywords: ABCB1, cat, dog, MDR1, P‐glycoprotein
1. INTRODUCTION
Drug metabolizing enzymes and drug transporters can greatly influence drug disposition (absorption, metabolism, distribution, elimination). Accordingly, guidance from the FDA (2020) convey the importance of identifying the enzymes responsible for metabolizing human drug candidates and whether a drug molecule is a substrate for key drug transporters, notably P‐glycoprotein (P‐gp) (Akamine et al., 2019). P‐gp is a drug transporter that significantly impacts the distribution and excretion of a wide variety of drugs in humans, dogs, and cats (Mealey, 2004; Mealey et al., 2019; Zhou, 2008). Impaired P‐gp function can result from intrinsic (genetic polymorphisms) and/or acquired (drug–drug interactions) mechanisms, predisposing affected veterinary patients to potentially life‐threatening adverse drug reactions (Martinez et al., 2008). Consequently, human drug labels generally indicate whether a particular drug molecule is a substrate for human P‐gp. The corresponding information is rarely available for veterinary drugs. However, some veterinary drug classes such as the macrocyclic lactones require additional safety studies in “avermectin sensitive” collies to assess potential adverse effects in dogs with deficient P‐gp function.
P‐gp is expressed on the luminal surface of many mammalian tissues including brain capillary endothelial cells, biliary canaliculi, intestines, and renal tubular cells where it functions to actively efflux substrate drugs (Ginn, 1996). In this capacity, and depending on the species, P‐gp limits oral absorption, enhances biliary excretion, and restricts central nervous system entry of substrate drugs (Coelho et al., 2009; Ginn, 1996; Mealey, Greene, et al., 2008). P‐gp's evolutionary function is presumed to be a protective one, minimizing exposure of mammalian organisms from potentially toxic xenobiotics encountered in the environment. Its protective role is made devastatingly evident in animals that lack functional P‐gp such as abcb1 knockout mice (Borst & Schinkel, 2013), dogs with ABCB1‐1Δ (MDR1 mutation) and cats with ABCB11930_1931del TC (Mealey & Burke, 2015). When treated with a P‐gp substrate like ivermectin, these animals experience neurological toxicity at doses more than 10‐fold lower than for animals with normal P‐gp function. In dogs, it has been demonstrated that biliary excretion of a P‐gp substrate is undetectable in dogs with ABCB1‐1Δ while biliary excretion of that same P‐gp substrate proceeds vigorously in dogs with normal P‐gp function (Coelho et al., 2009).
While genetic polymorphisms that create a P‐gp null phenotype appear to be rare in people (Chandler, 2018), they have been well‐described in both dogs and cats, with the canine mutation discovered in 2001 (Mealey et al., 2001) and confirmed by a separate group 2 years later (Roulet et al., 2003) and the feline mutation discovered in 2015 (Mealey et al., 2015) and confirmed by a second group 7 years later (Nurnberger et al., 2022). Consequently, veterinarians are more likely to encounter P‐gp‐mediated adverse drug reactions in genetically susceptible patients than are physicians. Genetically susceptible dogs and cats can be identified by readily available genetic tests. What may be less understood in veterinary medicine relative to human medicine, and therefore remains largely unrecognized, is so‐called “acquired” (reversible) P‐gp dysfunction. While veterinarians are aware that inhibitors of drug metabolizing enzymes can lead to enhanced drug toxicity, there is a paucity of information on the potential consequences of P‐gp inhibition. Many commonly administered drugs can inhibit P‐gp function and depending on dose may result in adverse drug reactions when a P‐gp substrate with a narrow therapeutic index is administered concurrently (Mealey & Fidel, 2015). In fact, any drug that is a P‐gp substrate can serve as a competitive P‐gp inhibitor. Therefore, administering two P‐gp substrate drugs concurrently increases the risk of adverse effects for both drugs. It is important that veterinarians understand the potential mechanisms of P‐glycoprotein‐mediated adverse drug reactions and drug–drug interactions to prevent their occurrence.
The purpose of this review is three‐fold. First, the intent is to demonstrate how knowledge of a veterinary drug's P‐gp substrate status can improve drug safety for dogs and cats. Second, this article will review the physiologic and pharmacological consequences of impaired P‐gp function and the pathophysiological mechanisms of drug toxicity resulting from P‐gp dysfunction. Finally, this article provides veterinarians with foundational concepts to guide decisions on whether a P‐gp substrate should be used in a canine or feline patient with intrinsic or acquired P‐gp deficiency at a lower than generally recommended dose or if it should be avoided altogether.
2. P‐GP SUBSTRATES AND “INHIBITORS”
2.1. P‐gp substrates
One of the truly unique characteristics of P‐gp relative to most drug‐protein interactions is its wide substrate specificity. P‐glycoprotein can bind and transport a diverse array of structurally dissimilar drugs. Literally hundreds of compounds are known to be P‐gp substrates (Aller et al., 2009; Ford & Hait, 1993; Fromm, 2004). This feat is possible because P‐gp has a binding pocket with multiple, overlapping binding sites (Aller et al., 2009) rather than a single drug binding site. The binding pocket does not appear to be able to accommodate multiple drugs at any given time but will accommodate binding of a single drug molecule that can be of various shape, size, or charge (Chufan et al., 2015). It is important to note that the amino acid composition of P‐gp varies between species, including the domains comprising the binding pocket (Aller et al., 2009). Consequently, it should not be surprising that while there is substantial overlap of P‐gp substrates between species, there are also differences. Species differences in P‐gp substrates have not been studied extensively but based on available data it is wrong to assume that a P‐gp substrate in one species would necessarily be a P‐gp substrate in another species (Schinkel et al., 1996; Zolnerciks et al., 2011). This underscores the need for establishing protocols for assessing drugs as canine or feline P‐gp substrates and not relying on information derived from human P‐glycoprotein assays.
Several methods can be used to assess the P‐gp status of a drug compound, from in vivo studies involving animals with P‐gp null phenotypes to cell culture‐based transport assays (Feng et al., 2008). Drug compounds can be tested in P‐gp deficient dogs as has been done experimentally (Mealey, Greene, et al., 2008) and is currently required by some regulatory agencies (i.e., FDA, EMA) during the veterinary drug development process for certain drug classes intended for dogs in order to assess their therapeutic index (Table 1). An alternative mouse model has been described in an effort to decrease the use of large animal (canine) models during the drug development process. An Abcb1a knock‐in/Abcb1b knock‐out mouse model expressing the ABCB1‐1Δ canine gene was engineered and shown to be phenotypically similar to P‐gp deficient dogs (Swain et al., 2013). Cell culture systems for identifying P‐gp substates are an attractive way to minimize animal use in the drug development process. Two cell lines commonly used for human P‐gp substrate assays are CACO‐2 (human) cells and a genetically modified MDCK (canine kidney) cell line that expresses human, not canine or feline, P‐glycoprotein. Two different canine cell lines, an osteosarcoma cell line and a kidney cell line, have been used to identify canine P‐gp substrates (West & Mealey, 2007: Mealey et al., 2017). The authors are unaware of any reports of cell lines used to assess feline P‐gp substrates. Examples of canine and feline P‐gp substrates that have been assessed by species‐specific methods are provided in Tables 2 and 3, respectively. To date, data exist only for eprinomectin and ivermectin as substrates for feline P‐gp (Mealey et al., 2015; Mealey et al., 2021; Nurnberger et al., 2022). However, it is reasonable to assume that many canine P‐gp substrates are substrates for feline P‐gp also, but additional research is necessary to identify and confirm feline P‐gp substrates.
TABLE 1.
Approximate breed frequency of the MDR1 mutation in dogs
| Breed | Approximate frequency |
|---|---|
| Collie | 70% |
| Longhaired whippet | 65% |
| Australian shepherd dog | 50% |
| Mini Australian shepherd dog | 50% |
| McNab | 30% |
| Silken windhound | 30% |
| English shepherd dog | 15% |
| Shetland sheepdog | 15% |
| German shepherd dog | 10% |
| Herding breed cross | 10% |
| Mixed breed | 5% |
| Old English sheepdog | 5% |
| Border collie | <5% |
| New to the list | |
| Black Mouth Cur | 8/26 dogs tested |
| Boxer a | <1% |
| Chinook | 4/13 dogs tested |
| Carolina Dog | 4/13 dogs tested |
| Siberian Husky a | <1% |
Determined by pedigree and DNA breed analysis to be purebred.
TABLE 2.
List of drugs for which there is some canine‐specific evidence supporting their status as canine P‐gp substrates
| Canine P‐gp substrate | In vitro data | In vivo data |
|---|---|---|
| Acepromazine | Genotyped dogs; prospective study | |
| Apomorphine | Genotyped dog; case report | |
| Butorphanol | Genotyped dogs; anecdotal (KLM) | |
| Cyclosporine | Canine cell line; flow cytometry assay | Genotyped dog; case report |
| Doxorubicin | Genotyped dogs; anecdotal (KLM) | |
| Emodepside | Genotyped dog; case report | |
| Grapiprant | Genotyped dogs; prospective study | |
| Ivermectin | Canine cell line; flow cytometry assay | Genotyped dogs; retrospective study; target animal safety study |
| Loperamide | Canine cell line; Flow cytometry assay | Genotyped dogs; 2 prospective studies |
| Maropitant | Canine cell line; flow cytometry assay | Genotyped dogs; anecdotal (KLM) |
| Milbemycin oxime | Genotyped dogs; prospective study, target animal safety study | |
| Moxidectin |
Canine Abcb1a knock‐in/Abcb1b knock‐out mice; prospective study Genotyped dogs; target animal safety study |
|
| Selamectin | Canine cells; flow cytometry assay | Genotyped dogs; target animal safety study |
| Vincristine* | Canine cell line; flow cytometry assay | Genotyped dogs; prospective study |
Note: Barbet et al., 2009; Campbell et al., 2017; Deshpande et al., 2016; Gaens et al., 2019; Griffin et al, 2005; Heit et al., 2021; Mackin et al., 2020; Mealey et al., 2001; Mealey et al., 2017; Mealey, Fidel, et al., 2008; Mealey, Greene, et al., 2008; Meyers et al., 2015; Swain et al., 2013; West & Mealey, 2007; Zhu et al., 2016.
Anecdotal information also exists for vinblastine and vinorelbine.
TABLE 3.
List of drugs for which there is some feline‐specific evidence supporting their status as feline P‐gp substrates
| Feline P‐gp substrate | In vitro data | In vivo data |
|---|---|---|
| Eprinomectin | Genotyped cats; case reports | |
| Ivermectin | Genotyped cats; case reports | |
2.2. P‐gp “inhibitors”
Many drugs that are commonly administered to veterinary patients can inhibit P‐gp which has the potential to cause serious drug interactions. Similar to the actions of antagonists at drug receptors, P‐gp inhibiting drugs may inhibit P‐gp by various mechanisms. For example, a drug may compete with another P‐gp substrate for the P‐gp substrate binding pocket (Marchetti et al., 2007). Another way P‐gp function can be inhibited is by abrogating ATP hydrolysis since P‐gp transport is dependent on ATP. High concentrations of phytic acid inhibit P‐gp by this mechanism (Li et al., 2018). Finally, P‐gp function can be inhibited by compounds that alter the integrity of cell membrane lipids. Practically speaking, though, competitive inhibition is far and away the most important mechanism for P‐gp inhibition in veterinary patients. Thus, any drug that is a P‐gp substrate must be considered a potential P‐gp inhibitor if it is administered concurrently with another P‐gp substrate drug. Ketoconazole and cyclosporine have frequently been labeled P‐gp inhibitors, but in fact they are also P‐gp substrates (Anglicheau et al., 2006; Elsby et al., 2008).
Why do some drugs have a greater reputation as a P‐gp inhibitor than a P‐gp substrate? It is the authors' collective opinion that over the years, drugs (e.g., ivermectin, vincristine, loperamide) that cause acute adverse effects in P‐gp deficient animals are easily understood to be P‐gp substrates, particularly for those that may result in neurological adverse events. When co‐administered with ketoconazole or cyclosporine, for example, they are often considered the victim of the drug–drug interaction. Conversely, drugs that have less acute adverse effects (e.g., ketoconazole, cyclosporine) are “blamed” as the perpetrator of the drug–drug interaction when co‐administered with a P‐gp substrate such as vincristine or ivermectin. The authors have been guilty of this assumption and have reenforced this stereotype in multiple publications. The authors heretofore propose that competitive P‐gp substrates not be classified separately as P‐gp inhibitors. Instead, we propose that all P‐gp substrates be listed as such and advise veterinarians to consider the potential consequences when co‐administering two P‐gp substrates. If the therapeutic index or safety margin of one or both drugs is narrow, veterinarians should consider dose reductions or alternative therapeutic choices. The phenomenon of competitive P‐gp inhibition, which has resulted in serious and even fatal adverse drug reactions in veterinary patients, underscores the importance of knowing the P‐gp substrate status of veterinary drugs.
3. P‐GP AND DRUG DISPOSITION
As previously noted, mammalian P‐gp is expressed at strategically important tissue barriers where it may function to limit both systemic exposure (e.g., enterocytes, biliary canaliculi, and proximal tubule) and exposure of sensitive tissues (e.g., brain capillary endothelial cells, and placenta) to potentially toxic xenobiotics (Ginn, 1996; Conrad et al., 2001; Van Der Heyden et al., 2009). There is a great deal of data from pharmacokinetic studies in humans and mice regarding P‐gp's role in drug disposition. In dogs, there are data from a few pharmacokinetic studies in P‐gp deficient compared to wildtype dogs but there are no pharmacokinetic studies comparing P‐gp deficient to wildtype cats. While it is tempting to extrapolate human or mouse pharmacokinetic data and apply it to dogs and cats, there is evidence of important species differences. A comparative description of P‐gp's effect on drug absorption, distribution, and excretion is provided.
3.1. P‐gp and the blood brain barrier
Numerous studies in mdr1a knockout or other P‐gp deficient mouse models have illustrated how dramatically P‐gp limits brain penetration of many different P‐gp substrates (Kalvass et al., 2004; Schinkel et al., 1994). The brain to plasma concentration ratio of the P‐gp substrate ivermectin was 87 times higher in P‐gp deficient mice than in wildtype mice. The authors could identify only one study in human subjects that illustrated enhanced penetration of a P‐gp substrate (loperamide) in P‐gp deficient subjects compared with subjects with normal P‐gp function (Gunn et al., 2012). There is evidence of increased brain penetration of the P‐gp substrates loperamide (Mealey, Greene, et al., 2008), Tc99m‐sestamibi (Mealey, Greene, et al., 2008) as can be appreciated in Figure 1, acepromazine (Deshpande et al., 2016), milbemycin (Barbet et al., 2009) and ivermectin (Sherman et al., 2010) in P‐gp deficient dogs compared with dogs with normal P‐gp function. The authors are unaware of any prospective studies investigating brain penetration of P‐gp substrates in cats with P‐gp deficiency. However, a series of cases provide indirect evidence that cats with P‐gp deficiency are more susceptible to neurological adverse effects of the P‐gp substrates ivermectin, milbemycin oxime, and eprinomectin (Jenkins et al., 2019; Mealey et al, 2015; Mealey et al., 2021).
FIGURE 1.
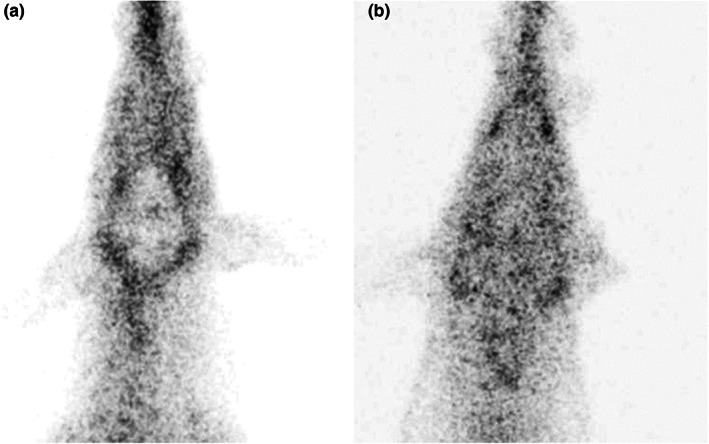
Nuclear scans after intravenous administration of the P‐gp substrate 99mTc‐MIBI to an MDR1 wildtype dog (a) and a dog homozygous for ABCB1‐1Δ (b). The images demonstrate diminished radioactivity in the brain of the wildtype dog due to P‐gp efflux of 99mTc‐MIBI, while the radioactivity of the brain P‐gp deficient dog cannot be distinguished from surrounding tissues.
3.2. P‐gp and oral pharmacokinetics
In humans, P‐gp expressed on enterocytes limits oral drug absorption such that oral bioavailability of P‐gp substrates such as cyclosporine (Benet, 2009), loperamide (Cha et al., 2013), and docetaxel (Malingre et al., 2001) is greater in subjects with P‐gp deficiency than in subjects with normal P‐gp function. It has been proposed that an interplay between human P‐gp and CYP 3A within enterocytes is responsible for the low oral bioavailability of compounds that are substrates of both CYP3A and P‐gp, but this has yet to be proven (Benet, 2009). In collies, the oral absorption of some P‐gp substrates (loperamide, cyclosporine, quinidine, nelfinavir) did not differ between dogs with normal P‐gp function compared with those with P‐gp deficiency (Kitamura et al., 2008; Mealey et al., 2010). In a different study, oral absorption of the P‐gp substrate fexofenadine was reported to be greater in P‐gp deficient dogs compared to normal dogs (Myers et al., 2018). Another group (Kitamura et al., 2008) that investigated the pharmacokinetics of orally administered fexofenadine, quinidine, and loperamide in P‐gp deficient in wildtype collies identified no statistical differences in Cmax or AUC of any of those P‐gp substrates. The group identified a significant difference in plasma fexofenadine concentrations at two of 6 time points (Kitamura et al., 2008), but no significant differences for the remaining 4 time points for fexofenadine, nor for any of the time points for quinidine or loperamide. There are reports of oral pharmacokinetic interactions when 2 P‐gp substrates were administered concurrently in dogs. Spinosad was shown to significantly increase the oral exposure of ivermectin when the drugs were dosed concurrently, presumably by inhibiting intestinal and/or hepatic P‐gp functions. However, the plasma concentrations of spinosad were unaffected by ivermectin (Dunn et al., 2011). The data thus far are insufficient to allow a clear resolution of P‐gp's role in oral pharmacokinetics in dogs. What can be said is that P‐gp effects on drug disposition in one species (i.e., humans) do not necessarily apply to other species and that further research on these effects in dogs and cats is necessary. The authors are unaware of any studies investigating oral bioavailability of P‐gp substrates in cats with P‐gp deficiency.
3.3. P‐gp and biliary excretion
The importance of P‐gp‐mediated efflux of drugs into bile has been demonstrated in rodent models (Hendrikx et al., 2013; Kong et al., 2016). Although many review articles state that P‐gp plays a key role in biliary excretion of P‐gp substrates in human subjects, actual data from human subjects are lacking (Taskar et al., 2022). In dogs, biliary excretion of the P‐gp substrate Tc99m sestamibi is nonexistent in dogs lacking P‐gp and is severely blunted in dogs with acquired P‐gp deficiency, as well as dogs that are heterozygous for ABCB1‐1Δ as illustrated in Figure 2. The lack of P‐gp‐mediated biliary excretion is considered to the be mechanism responsible for non‐neurological adverse effects associated with P‐gp substrates (e.g., doxorubicin and vinca alkaloids) when doses intended for “normal” dogs are administered to dogs with P‐gp deficiency (Mealey, Fidel, et al., 2008). Lack of biliary excretion may also contribute to neurological adverse effects due to decreased excretion of the P‐gp substrate, prolonged plasma concentrations, and the potential for greater brain penetration. The authors are unaware of any studies investigating biliary excretion of P‐gp substrates in cats with P‐gp deficiency.
FIGURE 2.
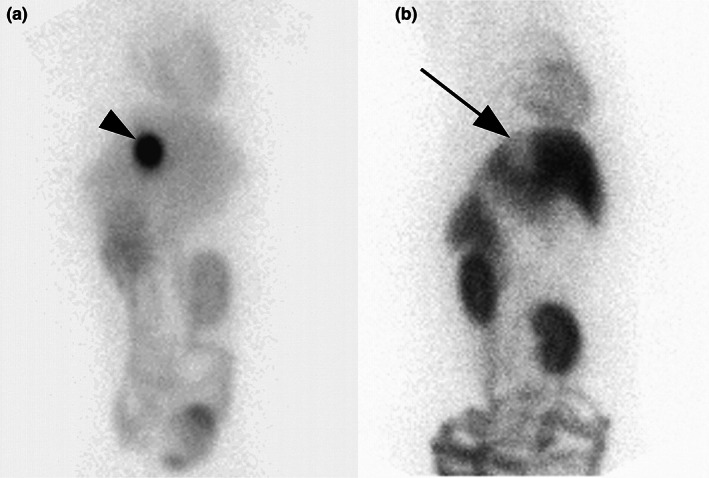
Ventral images of the abdomen 2 h after intravenous administration of the P‐gp substrate 99mTc‐MIBI in an ABCB1 wildtype dog (a) and a dog homozygous for ABCB1‐1Δ (b). The arrowhead indicates a high concentration of 99mTc‐MIBI within the gallbladder in the wildtype dog while the arrow indicates a gallbladder essentially devoid of 99mTc‐MIBI. Reprinted with permission (Coelho et al., 2009).
3.4. P‐gp and renal excretion
Despite the fact that P‐gp is expressed on renal tubules, and that P‐gp mediated efflux has been demonstrated in renal tubule cell culture studies, the authors could not identify a single study in human subjects that demonstrated an important role for P‐gp in renal drug excretion. In fact, the International Transporter Consortium recently concluded that there is little clinical risk of drug–drug interactions based on P‐gp inhibition of renal excretion (Taskar et al., 2022). The role of P‐gp in renal drug excretion in the dog and cat has not been investigated.
4. P‐GP DEFICIENCY AND DYSFUNCTION
4.1. Intrinsic P‐gp deficiency
A common misconception is that only herding breed dogs are at risk for P‐gp deficiency. The fact is that many dogs and cats can experience P‐gp deficiency and any dog or cat can experience P‐gp dysfunction. Veterinarians must consider both intrinsic P‐gp deficiency and acquired P‐gp dysfunction to avoid causing P‐gp mediated adverse drug reactions. Intrinsic P‐gp deficiency results from genetic variations in the MDR1 (ABCB1) gene such as the well characterized ABCB1‐1Δ in dogs (Mealey et al., 2001) and more recently described ABCB11930_1931del TC in cats (Mealey & Burke, 2015). ABCB1‐1Δ is present in 50% or more of the population in some dog breeds, including collies, Australian shepherds, and long‐haired whippets but is present in other breeds also (Table 4). A breed predilection for ABCB11930_1931del TC has not been detected but the frequency has been estimated at about 4% of cats. Both canine ABCB1‐1Δ and feline ABCB11930_1931del TC are frame shifting deletions that create premature stop codons. P‐gp synthesis is truncated and nonfunctional in dogs or cats homozygous for these mutations. Heterozygotes, dogs, or cats with one copy of the mutant ABCB1 allele and one copy of the normal ABCB1 allele, also experience increased susceptibility to adverse drug reactions but to a lesser extent than dogs or cats with two copies of the mutant ABCB1 allele (Coelho et al., 2009; Mealey, Fidel, et al., 2008).
TABLE 4.
US approved canine drugs that have undergone safety studies in P‐gp deficient dogs and their reported adverse reactions
| Drug | Active ingredient | Route | Safety information in P‐gp deficient dogs on the product label | Warnings or precautions related to safety risk in P‐gp deficient dogs on the product label |
|---|---|---|---|---|
|
Advantage Multi™ for Dogs NADA 141–251 |
Imidacloprid, moxidectin | Topical |
Dermal Safety Study in Ivermectin‐Sensitive collies: Advantage Multi™ for Dogs was administered topically at 3 and 5 times the recommended dose every 28 days for 3 treatments to Collies which had been prescreened for avermectin sensitivity. No clinical abnormalities were observed. Oral Safety Study in Ivermectin‐Sensitive Collies: Advantage Multi™ for Dogs was administered orally to 5 pre‐screened ivermectin‐sensitive collies. The collies were asymptomatic after ingesting 10% of the minimum labeled dose. At 40% of the minimum recommended topical dose, 4 of the dogs experienced neurological signs indicative of avermectin toxicity including depression, ataxia, mydriasis, salivation, muscle fasciculation, and coma, and were euthanized |
For the first 30 min after application: Ensure that dogs cannot lick the product from application sites on themselves or other treated dogs, and separate treated dogs from one another and from other pets to reduce the risk of accidental ingestion. Ingestion of this product by dogs may cause serious adverse reactions including depression, salivation, dilated pupils, incoordination, panting, and generalized muscle tremors. In avermectin sensitive dogs, the signs may be more severe and may include coma and death. Some dogs are more sensitive to avermectins due to a mutation in the MDR1 gene. Dogs with this mutation may develop signs of severe avermectin toxicity if they ingest this product. The most common breeds associated with this mutation include collies and collie crosses. Although there is no specific antagonist for avermectin toxicity, even severely affected dogs have completely recovered from avermectin toxicity with intensive veterinary supportive care |
|
Bravecto® NADA: 141–532 |
Fluralaner | Oral | No studies in avermectin‐sensitive collies are described on the product insert. However, a safety study in MDR1 (−/−) collies at 3 times the recommended dose was published. No adverse events were seen in at 3 times the maximum recommended therapeutic dose (Walther et al., 2014) | None |
|
Heartgard® NADA# 140–886 |
Ivermectin | Oral | Studies with ivermectin indicate that certain dogs of the collie breed are more sensitive to the effects of ivermectin administered at elevated dose levels (more than 16 times the target use level) than dogs of other breeds. At elevated doses, sensitive dogs showed adverse reactions which included mydriasis, depression, ataxia, tremors, drooling, paresis, recumbency, excitability, stupor, coma, and death. Heartgard® demonstrated no signs of toxicity at 10 times the recommended dose (60 mcg/kg) in sensitive collies. Results of these trials support the safety of Heartgard® products in dogs, including collies, when used as recommended | None |
|
Heartgard® Plus NADA# 140–971 |
Ivermectin, pyrantel pamoate | Oral | Studies with ivermectin indicate that certain dogs of the collie breed are more sensitive to the effects of ivermectin administered at elevated dose levels (more than 16 times the target use level) than dogs of other breeds. At elevated doses, sensitive dogs showed adverse reactions which included mydriasis, depression, ataxia, tremors, drooling, paresis, recumbency, excitability, stupor, coma, and death. Heartgard® demonstrated no signs of toxicity at 10 times the recommended dose (60 mcg/kg) in sensitive collies. Results of these trials and bioequivalency studies, support the safety of Heartgard® products in dogs, including collies, when used as recommended | None |
|
Interceptor® NADA: 140–915 |
Milbemycin oxime | Oral | A rising‐dose safety study conducted in rough‐coated collies, manifested a clinical reaction consisting of ataxia, pyrexia, and periodic recumbency, in one of fourteen dogs treated with milbemycin oxime at 12.5 mg/kg (25 times the monthly use rate). Prior to receiving the 12.5 mg/kg dose (25 times the monthly use rate) on day 56 of the study, all animals had undergone an exaggerated dosing regimen consisting of 2.5 mg/kg milbemycin oxime (5 times the monthly use rate) on Day 0, followed by 5.0 mg/kg (10 times the monthly use rate) on Day 14, and 10.0 mg/kg (20 times the monthly use rate) on Day 32. No adverse reactions were observed in any of the collies treated with this regimen up through the 10.0 mg/kg (20 times the monthly use rate) dose | None |
|
Interceptor® Plus NADA: 141–338 |
Milbemycin oxime, praziquantel | Oral | A rising‐dose safety study conducted in rough‐coated Collies resulted in ataxia, pyrexia, and periodic recumbency in one of fourteen dogs administered milbemycin oxime at 12.5 mg/kg (5 times the maximum exposure dose of Interceptor Plus). Prior to receiving the 12.5 mg/kg dose on day 56 of the study, all animals had undergone a dosing regimen consisting of 2.5 mg/kg milbemycin oxime on Day 0, followed by 5.0 mg/kg on Day 14, and 10.0 mg/kg on Day 32. No adverse reactions were observed in any of the collies treated with doses less than 12.5 mg/kg. No studies with avermectin‐sensitive dogs with praziquantel are described on the product label | None |
|
NexGard® NDAD: 141–406 |
Afoxolaner | Oral | No studies in avermectin‐sensitive collies are described on the product label. However, a safety study in MDR1 (−/−) collies evaluated afoxolaner and a combination of afoxolaner and milbemycin oxime (NexGard® Spectra; not currently approved in US). Afoxolaner was evaluated at 3.8 times the maximum recommended dose and the afoxolaner /milbemycin oxime combination was evaluated at 4.7 times the recommended dose. No significant adverse reactions were noted in either group (Drag et al., 2022) | None |
|
ProHeart®6 NADA# 141–189 |
moxidectin | Injectable | ProHeart®6 administered up to 5 times the recommended dose in ivermectin‐sensitive collies did not cause any adverse reactions | None |
|
ProHeart®12 NADA# 141–519 |
moxidectin | Injectable | In a laboratory study, 15 ivermectin‐sensitive collie dogs in three treatment groups were administered one dose of saline and one dose of ProHeart®12, 21 days apart. Each dog served as its own control and the order of administration of the saline and ProHeart®12 varied by treatment group. ProHeart®12 was dosed at 0.5 mg/kg body weight (recommended dose, five dogs), 1.5 mg/kg body weight (3 times the recommended dose, five dogs), or 2.5 mg/kg body weight (5 times the recommended dose, five dogs). No clinical signs of moxidectin toxicity were observed during the 42‐day study | None |
|
Revolution® NADA# 141–152 |
Selamectin | Topical | In a pre‐clinical study selamectin was dosed orally to ivermectin‐sensitive collies. The final market formulation was not used in this study. Oral administration of 2.5, 10, and 15 mg/kg in this dose escalating study did not cause any adverse reactions; however, eight hours after receiving 5 mg/kg orally, one dog became ataxic for several hours, but did not show any other adverse reactions after receiving subsequent doses of 10 and 15 mg/kg orally. In a topical safety study conducted at 1, 3, and 5 times the recommended dose (6 mg/kg) salivation was observed in all treatment groups, including the vehicle control | None |
|
Sentinel® Flavor Tabs NADA: 141–084 |
Lufenuron, milbemycin oxime | Oral | A rising‐dose safety study conducted in rough‐coated collies manifested a clinical reaction consisting of ataxia, pyrexia and periodic recumbency in one of fourteen dogs treated with milbemycin oxime at 12.5 mg/kg (25 times the recommended minimum dose). Prior to receiving the 12.5 mg/kg dose on day 56 of the study, all animals had undergone an exaggerated dosing regimen consisting of 2.5 mg/kg milbemycin oxime (5 times the recommended minimum dose) on Day 0, followed by 5.0 mg/kg (10 times the recommended minimum dose) on Day 14, and 10.0 mg/kg (20 times the recommended minimum dose) on Day 32. No adverse reactions were observed in any of the collies treated with this regimen up through the 10.0 mg/kg (20 times the recommended minimum dose) dose. No studies with avermectin‐sensitive dogs with lufenuron are described on the product label | None |
|
Sentinel® Spectrum® NADA: 141–333 |
Milbemycin oxime, lufenuron, praziquantel | Oral | A rising‐dose safety study conducted in rough‐coated collies resulted in ataxia, pyrexia, and periodic recumbency in one of fourteen dogs administered milbemycin oxime at 12.5 mg/kg (5 times the maximum exposure dose of Sentinel® Spectrum® Chews). Prior to receiving the 12.5 mg/kg dose on day 56 of the study, all animals had undergone a dosing regimen consisting of 2.5 mg/kg milbemycin oxime on Day 0, followed by 5.0 mg/kg on Day 14, and 10.0 mg/kg on Day 32. No adverse reactions were observed in any of the collies treated with doses less than 12.5 mg/kg. No studies with avermectin‐sensitive dogs with lufenuron or praziquantel are described on the product label | None |
|
Simparica Trio® NADA: 141–521 |
Sarolaner, moxidectin, pyrantel | Oral | Simparica Trio® was administered orally once at 1, 3, and 5 times the maximum recommended dose to collies that had been pre‐screened for avermectin sensitivity. Clinical signs (ataxia, muscle fasciculations, mydriasis) associated with avermectin sensitivity were observed in the 5X group. All dogs were completely recovered by the third day of the study | None |
|
Trifexis™ NADA: 141–321 |
Spinosad, milbemycin oxime | Oral | In an avermectin‐sensitive collie dog study, Trifexis™ was administered orally at 1, 3, and 5 times the upper half of the recommended dose band every 28 days. No signs of avermectin sensitivity were observed after administration of Trifexis™ during the study period to avermectin‐sensitive collie dogs. The adverse reactions observed in the treatment groups were vomiting and diarrhea. Body weights in all treatment groups were comparable with the control group. Hematology and clinical chemistry parameters showed no clinically significant changes from study start to end, and all dogs were considered healthy throughout the study | None |
Note: US approved label language found on https://dailymed.nlm.nih.gov/dailymed/index.cfm. Only the original approved products (pioneer approvals) are included. Numerous generic approvals exist for many of the listed products.
4.2. “Acquired” (reversible) P‐gp dysfunction
Acquired P‐gp dysfunction results when an animal is treated with a drug or other product such as a dietary supplement (“nutraceutical”) that inhibits P‐gp function (Coelho et al., 2009; Mealey & Fidel, 2015). It must be emphasized that P‐gp dysfunction is a type of drug–drug interaction that can occur in any dog or cat, regardless of breed. Many drugs commonly used to treat dogs and cats can competitively inhibit P‐gp and may cause severe, even fatal, adverse drug reactions if administered concurrently with another P‐gp substrate drug. Ketoconazole is a strong P‐gp substrate (Coelho et al., 2009) and has resulted in severe adverse drug reactions mediated by competitively inhibiting P‐gp. A dog treated with ketoconazole and the P‐gp substrate vinblastine experienced severe neutropenia and gastrointestinal signs and eventually succumbed to sepsis (Mealey & Fidel, 2015). One of the authors (KLM) is aware of neurological toxicosis in several dogs treated with ivermectin (300 μl/kg) concurrently with ketoconazole. The dogs were all genotyped and determined to be homozygous for the normal MDR1 allele. It should be noted that ketoconazole inhibits many canine cytochrome P450 enzymes (Aidasani et al., 2008) so it might be responsible for delayed metabolism as well as dysfunctional P‐gp transport of some drugs. Whether the well‐characterized and exploited effect of ketoconazole to increase cyclosporine plasma concentrations (Myre et al., 1991) is due to P‐gp inhibition, cytochrome P450 inhibition, or a combination of both is not known.
The key takeaway is that most drugs that inhibit P‐gp are actually P‐gp substrates—they function as competitive inhibitors. Two P‐gp substrates administered concurrently compete for the P‐gp binding pocket preventing efflux of the other drug from brain capillary endothelial cells and biliary canalicular cells resulting in increased drug concentrations in the brain and decreased biliary drug excretion.
5. POTENTIAL CONSEQUENCES OF ADMINISTERING P‐GP SUBSTRATES TO ANIMALS WITH P‐GP DEFICIENCY OR DYSFUNCTION
5.1. Neurologic adverse effects
P‐gp is an integral component of the blood brain barrier. Expressed on the luminal side of brain capillary endothelial cells, P‐gp actively effluxes substrate drugs back into the capillary lumen. Brain concentrations of P‐gp substrates such as ivermectin, loperamide, vinblastine, and ondansetron are 100‐fold, 13‐fold, 3‐fold, and 4‐fold greater in mdr1a knockout mice (P‐gp deficient) than in their wildtype counterparts, respectively (Schinkel et al., 1994, 1996). Canine P‐gp functions similarly as can be readily visualized using imaging studies with the radiolabeled P‐gp substrate technetium 99 m sestamibi (MIBI) as shown in Figure 1. If a drug exerts neurological pharmacological effects (i.e., interacts with receptors located in the CNS), those effects will be more pronounced in animals with P‐gp dysfunction compared to “normal” animals that receive the same dose. The P‐gp substrate loperamide serves as a great example. Loperamide is an opiate that does not induce typical opioid neurological effects because its access to the brain is restricted by P‐gp. When the same dose of loperamide is administered to dogs with normal P‐gp and P‐gp efficient dogs, neurological clinical signs are observed only in the P‐gp deficient dogs (Mealey & Burke, 2015; Mealey, Greene, et al., 2008). In both of the cited studies, there was wide intersubject variability in plasma loperamide concentrations and AUC, particularly in P‐gp deficient dogs. What is particularly striking is the fact that at doses of 0.01, 0.05, and 0.2 mg/kg, the AUC of loperamide in wildtype dogs versus P‐gp deficient (MDR1 mutant/mutant) dogs was not statistically significant different (Mealey & Burke, 2015; Mealey, Greene, et al., 2008) despite the incongruent effects on the central nervous system. At the 0.1 mg/kg dose rate, the loperamide AUC was greater in P‐glycoprotein deficient dogs than in wildtype dogs. Loperamide traverses the blood brain barrier to bind opioid receptors in the brain of P‐gp deficient dogs but is unable to do so in dogs with normal P‐gp function. Thus, neurological manifestations of P‐gp substrate drugs in P‐gp deficient animals appear to be primarily a function of enhanced penetration across the blood brain barrier rather than greater systemic exposure. These neurological effects were seen in all MDR1 mutant/mutant dogs treated with loperamide at the 0.2 mg/kg dose and in the majority of MDR1 mutant/mutant dogs treated at the 0.1 mg/kg dose (Mealey & Burke, 2015; Mealey, Greene, et al., 2008; Zhu et al., 2016). As might be expected, dogs with an intermediate P‐gp phenotype (MDR1 mutant/normal), loperamide‐induced neurological clinical signs are milder than in dogs with a P‐gp null phenotype (MDR1 mutant/mutant).
If all P‐gp substrates achieve higher concentrations in the brain of animals with P‐gp dysfunction animals compared with animals with normal P‐gp function, then why do not all P‐gp substrates cause neurological toxicity? This depends on the drug's pharmacology—if the drug acts on receptors in the CNS, there will be greater potential for exacerbated pharmacological effects which may result in CNS toxicity. For example, the P‐gp substrates loperamide and apomorphine bind to opioid receptors which are present in the brain. Both drugs cause neurological clinical signs in dogs with the MDR1 mutation (Mealey, Greene, et al., 2008, Campbell et al., 2017). Similarly, GABA gated chloride channels are present in the brain of mammals so macrocyclic lactones that gain access to the brain due to P‐gp deficiency will bind to these receptors and cause neurological clinical signs (Mealey et al., 2001, 2021; Nurnberger et al., 2022). By comparison, the P‐gp substrate cyclosporine, which primarily binds cyclophilin receptors in T‐lymphocytes (Matsuda & Koyasu, 2000) does not appear to cause any neurological clinical signs even at accelerated doses [NADA 141‐218(fda.gov)]. Similarly, vincristine does not typically cause neurological clinical signs in dogs with the MDR1 mutation (Mealey, Fidel, et al., 2008). However, animals with P‐gp dysfunction experience increased susceptibility to non‐neurological adverse effects of both cyclosporine (despite plasma concentrations within the therapeutic range) and vincristine (Mackin et al., 2020; Mealey, Fidel, et al., 2008) at doses considered to be therapeutic in dogs with normal P‐gp function. Dose reductions and supportive therapy are often required to manage these adverse effects.
5.2. Non‐neurologic adverse effects
Dogs homozygous for the MDR1 mutation are incapable of excreting P‐gp substrates into bile (Coelho et al., 2009). This is illustrated in Figure 2. The same would be expected for cats homozygous for ABCB11930_1931del TC. Because of P‐gp's role in biliary drug excretion, clearance of P‐gp substrates in animals with P‐gp dysfunction would be expected to be prolonged, potentially resulting in increased overall drug exposure. The clearance of the P‐gp substrate galliprant from the central compartment of dogs with P‐gp dysfunction (homozygous for ABCB1‐1Δ) was 71% lower than that of dogs with normal P‐gp function (Heit et al., 2021). Dogs with P‐gp dysfunction were also more likely to experience gastrointestinal adverse effects than dogs with normal P‐gp function (Heit et al., 2021), but no neurological adverse effects were observed. It is important to note that gastrointestinal adverse effects were observed in toxicology studies of grapiprant in beagle dogs (Galliprant™ label). The P‐gp substrate vincristine is significantly more likely to cause bone marrow suppression, manifested by neutropenia and thrombocytopenia, in dogs with P‐gp dysfunction (heterozygous or homozygous for ABCB1‐1Δ) than in dogs with normal P‐gp function (Mealey, Fidel, et al., 2008). A similar drug, vinblastine, caused severe bone marrow suppression, and gastrointestinal toxicity in a dog with acquired P‐gp dysfunction (Mealey & Fidel, 2015). Cyclosporine has also been documented to cause an exaggerated pharmacological response in dogs with P‐gp dysfunction (Mackin et al., 2020). In each of these examples, the adverse events are consistent with those expected after an excessive drug dose (i.e., an exaggerated pharmacological response). It is reasonable to deduce that blunted or nonexistent biliary clearance in dogs or cats with P‐gp dysfunction (Figure 3) results in greater overall exposure to P‐gp substrate drugs, thereby increasing the likelihood of adverse effects. MDR1 genotyping to assess intrinsic P‐gp dysfunction and drug‐interaction screening to assess acquired P‐gp dysfunction should be performed to identify at‐risk dogs or cats prior to treatment with P‐gp substrate drugs. If a patient is determined to have intrinsic or acquired P‐gp dysfunction, several therapeutic options can be considered.
FIGURE 3.
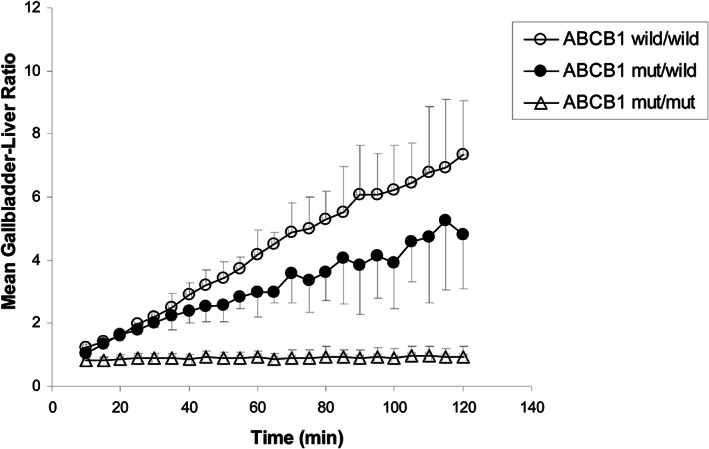
Graph depicting accumulation of the P‐gp substrate 99mTc‐MIBI in gall bladder over time in dogs homozygous for wildtype ABCB1 (open circles), heterozygote dogs (closed circles), or dogs homozygous for ABCB1‐1Δ (triangles). Note that there is no detectable biliary excretion of the P‐gp substrate in dogs homozygous for ABCB1‐1Δ. While heterozygotes have an intermediate level of biliary excretion of the P‐gp substrate. Reprinted with permission (Coelho et al., 2009).
5.3. Role of therapeutic index
An important concept to consider when dosing any drug is an understanding the safety profile of the drug, including the type of adverse effect that is most commonly seen and the dose where toxicity would be expected in normal animals. This is particularly true when administering P‐gp substrates, especially for drugs with CNS effects as brain levels of these substrates may increase markedly in P‐gp deficient animals.
As Paracelsus, the Father of Toxicology, famously said: “What is there that is not poison? All things are poison and nothing is without poison. Solely the dose determines that a thing is not a poison.” Many drugs manifest toxicity through an exaggerated pharmacological effect where higher doses result in prolonged or enhanced receptor activation. As mentioned earlier, loperamide toxicity in P‐gp deficient animals is an example of an exaggerated pharmacological effect due to altered distribution into the CNS resulting in neurological effects mediated through opioid receptors. Knowledge of a drug's potential for toxicity and the dose at which toxicity is expected is important when considering administering P‐gp substrates. Drugs with a narrow therapeutic index will have inherently greater risk profiles in P‐gp deficient animals than those with a wider therapeutic window.
Information on therapeutic index can be easily found on an approved drug's label (Dailymed: National Library of Medicine) and freedom of information summary provide by the FDA's Center for Veterinary Medicine (Freedom of Information (FOI) Summaries for Approved New Animal Drugs | FDA). Drug sponsors are required to evaluate the therapeutic index in target animal safety studies where the recommended dose and overdoses are given for extended periods of time. These studies often use dose levels of 1, 3, and 5 times the drug's recommended dose over a period of 3 times the expected duration of administration. In cases where the drug is to be given chronically, that is, greater than 3 consecutive months, these studies may be 6 months in duration. The data from these studies are summarized in detail and include any effects of the drug relative to placebo on clinical signs, clinical pathology, gross pathology, and histopathology (CVM GFI #185 (VICH GL43) Target Animal Safety for Veterinary Pharmaceutical Products | FDA). Addition safety information is also summarized from well controlled clinical trials in diverse patient populations representing many different dog breeds across a wide geographic area. Unfortunately, human drug labels (i.e., doxorubicin and vinblastine) do not provide safety information for dogs or cats; so, caution must be exercised when using human drugs off label for treating dogs or cats.
As macrocyclic lactone drugs have been associated with neurotoxicology in P‐gp deficient animals including CNS depression, ataxia, tremors, salivation, mydriasis, and in severe cases, coma and death, many regulatory bodies require drug sponsors of new chemical entities in this class to assess the margin of safety in P‐gp deficient dogs that have been shown to be sensitive to avermectin neurotoxicity. These studies are typically conducted at 1, 3, and 5 times the label dose. These important safety data in P‐gp deficient dogs are also summarized on the drug's label and detailed in the freedom of information summary. Information from these controlled safety and efficacy studies along with precaution or warning statements on the approved drug's label can be very helpful to assist the veterinarian to evaluate the risk to benefit ratio of using a P‐gp substrate in their specific patients.
6. THERAPEUTIC OPTIONS FOR P‐GP DEFICIENT DOGS AND CATS
Currently, treatment recommendations for dogs and cats with P‐gp dysfunction are based on experiential knowledge, some data from experimental P‐gp deficient animals, and in vitro data. Certainly, more research in this area is necessary as additional canine and feline P‐gp substrates are identified, but for now the options for treating P‐gp deficient animals are to identify alternative drugs that are not P‐gp substrates or to decrease doses of P‐gp substrate drugs in animals that are P‐gp deficient by either intrinsic or acquired mechanisms.
6.1. Alternative drug choices
For some, but not all, disease conditions for which a P‐gp substrates drug is a first line treatment option, there are sound alternative drug options that are not P‐gp substrates. That has not always been the case. For dogs with demodectic mange, for example, extralabel use of ivermectin administered daily at doses 50–100 times higher than the FDA approved heartworm prevention dose were routinely recommended (Mueller, 2004). These doses of ivermectin are often fatal in dogs with P‐gp deficiency unless the patient receives substantial medical care including ventilatory support (Merola et al., 2009). Alternative drug treatment for demodectic mange is necessary. Drugs such as amitraz or, more recently, off‐label use of the isooxazoline flea/tick preventives such as sarolaner or lotilaner (Perego et al., 2019) may be used instead. Two isoxazolines, afoxalaner, and fluralaner, have been studied in P‐glycoprotein deficient dogs and did not cause adverse effects (Drag et al., 2022; Walther et al., 2014). Currently, there are no data to support recommendations for safe and effective dose reductions of loperamide, for diarrhea, or apomorphine, for inducing emesis, in dogs with the MDR1 mutation; so, alternative drugs should be employed. Both P‐gp substrates cause CNS depression in dogs with the MDR1 mutation but are tolerated quite well in dogs with normal P‐gp function. Similarly, the authors do not recommend using emodepside‐containing products in dogs or cats with P‐gp deficiency because neurological adverse effects occurred in with a P‐gp deficient Australian shepherd treated with the label dose (Gaens et al., 2019). Alternative antiparasitic drugs should be employed. For cats with P‐gp deficiency, there are no data to support safe and effective dose reductions for commercial formulations of eprinomectin‐containing antiparasitic products, therefore alternative drugs should be employed. Serious neurological adverse effects have been reported when the products have been used at label doses in cats with P‐gp deficiency (Mealey et al., 2021).
6.2. Dose modification
It is important to remember that not all P‐gp substrates require dose modifications in P‐gp deficient animals. P‐gp substrates that do not have the potential to cause neurological adverse (i.e., those that do not interact with receptors in the CNS) may not require a dose reduction. Similarly, P‐gp substrates that have a wide margin of safety (wide therapeutic index) may not require a dose reduction. For example, some cephalosporins are substrates for human P‐gp and have been used at label doses in dogs with P‐gp dysfunction with no reports of adverse effects. However, many P‐gp substrates have been documented to cause serious adverse effects in dogs with P‐gp dysfunction and should not be used at generally recommended or label doses. One of the authors (KLM) has worked with veterinarians, pet owners, and/or industry experts on an individual basis to identify reasonable dose reductions for safe and effective use of the P‐gp substrates acepromazine, butorphanol, doxorubicin, vinca alkaloids, grapiprant, and cyclosporine. The dose reductions proposed are based on data from studies of a radiolabeled P‐gp substrate in dogs with normal P‐gp function, acquired P‐gp deficiency, and intrinsic P‐gp deficiency (Mealey, Greene, et al., 2008; Coelho et al., 2009), in vitro studies assessing drugs as P‐gp substrates (Mealey et al., 2017), pharmacokinetic and pharmacodynamic data (Heit et al., 2021; Mealey, Greene, et al., 2008; Mealey, Fidel, et al., 2008; Campbell et al., 2017; Mackin et al., 2020; Deshpande et al., 2016), and personal experience working with a colony of dogs with ABCB1‐1Δ. Individual cases will vary depending on concurrent disease conditions, their severity, and concurrent medications or nutritional supplements the animal is receiving but general recommendations can be used as a starting point. The general recommendation is to decrease the dose of P‐gp substrates by 25% in dogs heterozygous for ABCB1‐1Δ and by 50% in dogs homozygous for ABCB1‐1Δ. Evidence to date suggests that dogs with acquired P‐gp deficiency are phenotypically more similar to ABCB1‐1Δ heterozygotes than to ABCB1‐1Δ homozygotes (Coelho et al., 2009). Subsequent doses can be increased at 10% intervals if it has been determined that a higher dose is needed and if the patient has tolerated the previous dose well. Further refinement of this general recommendation is desirable but will require a substantial commitment of time, funding, and the collective expertise of primary care veterinarians, specialty veterinarians (oncologists, internists, and clinical pharmacologists) and pet owners. Unfortunately, therapeutic drug monitoring for P‐gp substrate drugs may not be helpful since plasma concentrations may not be significantly different in dogs with and without P‐gp deficiency (Kitamura et al., 2008; Mealey, Greene, et al., 2008). Pharmacodynamic monitoring, as may be available for cyclosporine, might prove to be a more reliable dosing guide than therapeutic drug monitoring (Mackin et al., 2020).
7. CONCLUSION
Serious adverse drug reactions can be prevented by knowing if a dog or cat has genetically mediated P‐gp deficiency or drug‐interaction “acquired” P‐gp dysfunction and whether the drug(s) being administered are a P‐gp substrates. Because there are species differences in P‐gp amino acid sequences, one should not assume that a human P‐gp substrate is also a canine or feline P‐gp substrate. Although the labels of many human drug products often include whether the drug is a P‐gp substrate, the corresponding species‐specific information is not available for canine or feline drug products with the exception of the macrocyclic lactone class of anti‐parasitics. Knowing the species‐specific P‐gp substrate status of drugs is important for dogs and cats since intrinsic P‐gp deficiency is more common in dogs and cats than in people. In humans, P‐gp substrate information is included in drug labels primarily to prevent adverse reactions resulting from acquired (drug interaction) P‐gp dysfunction, which can also occur in dogs and cats. While neurological manifestations are the most well‐known type of adverse reactions associated with P‐gp deficiency, non‐neurological adverse reactions are also possible and manifest as a relative over‐dosage of the P‐gp substrate in animals with P‐gp deficiency or dysfunction compared to an animal with normal P‐gp function. Lastly, it is important to note that many P‐gp substrates, those with a high therapeutic index and that do not bind to receptors in the CNS, are no more likely to cause adverse effects in animals with P‐gp deficiency or dysfunction than in animals with normal P‐gp function. A strategic, collaborative effort is needed to characterize the P‐gp status of the hundreds of drugs used to treat canine and feline patients, preferably prior to a drug being marketed, so that serious and potentially fatal adverse drug reactions in dogs and cats can be prevented.
8. AUTHOR CONTRIBUTIONS
Each author wrote individual sections of the article and each author contributed to editing and approving the final version.
9. ACKNOWLEDGMENTS
None.
CONFLICT OF INTEREST
“How Knowing a drug's P‐glycoprotein status can prevent adverse reactions in dogs and cats.” One of the authors (KLM) receives royalties for intellectual property owned by Washington State University related to MDR1 genotyping for dogs and cats, and assessment of drugs for their canine P‐glycoprotein substrate status. The other authors declare no conflicts of interest.
10. ANIMAL WELFARE AND ETHICS STATEMENT
This is a review article so no animals were used.
Mealey, K. L. , Owens, J. G. , & Freeman, E. (2023). Canine and feline P‐glycoprotein deficiency: What we know and where we need to go. Journal of Veterinary Pharmacology and Therapeutics, 46, 1–16. 10.1111/jvp.13102
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- Aidasani, D. , Zaya, M. J. , Malpas, P. B. , & Locuson, C. W. (2008). In vitro drug‐drug interaction screens for canine veterinary medicines: Evaluation of cytochrome P450 reversible inhibition. Drug Metabolism and Disposition, 36(8), 1512–1518. [DOI] [PubMed] [Google Scholar]
- Akamine, Y. , Yasui‐Furukori, N. , & Uno, T. (2019). Drug‐drug interactions of P‐gp substrates unrelated to CYP metabolism. Current Drug Metabolism, 20(2), 124–129. [DOI] [PubMed] [Google Scholar]
- Aller, S. G. , Yu, J. , Ward, A. , Weng, Y. , Chittaboina, S. , Zhio, R. , Harrell, P. M. , Trinh, Y. T. , Zhang, Q. , Urbatxch, I. L. , & Chang, G. (2009). Structure of P‐glycoprotein reveals a molecular basis for poly‐specific drug binding. Science, 323(5922), 1718–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglicheau, D. , Pallet, N. , Rabant, M. , Marquet, P. , Cassinat, B. , Meria, P. , Beaune, P. , Legendre, C. , & Thervet, E. (2006). Role of P‐glycoprotein in cyclosporine cytotoxicity in the cyclosporine‐sirolimus interaction. Kidney International, 70(6), 1019–1025. [DOI] [PubMed] [Google Scholar]
- Barbet, J. L. , Snook, T. , Gay, J. M. , & Mealey, K. L. (2009). ABCB1‐1Delta (MDR1‐1Delta) genotype is associated with adverse reactions in dogs treated with milbemycin oxime for generalized demodicosis. Veterinary Dermatology, 20(2), 111–114. [DOI] [PubMed] [Google Scholar]
- Benet, L. Z. (2009). The drug transport‐metabolism alliance: Uncovering and defining the interplay. Molecular Pharmacology, 6(6), 1631–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst, P. , & Schinkel, A. H. (2013). P‐glycoprotein ABCB1: A major player in drug handling by mammals. Journal of Clinical Investigation, 123(10), 4131–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, O. , de Lorimier, L.‐P. , & Mealey, K. L. (2017). Adverse reaction to apomorphine in a collie homozygous for the ABCB1‐1Δ (MDR1) mutation. Journal of Small Animals Practice, 58(2), 119. [DOI] [PubMed] [Google Scholar]
- Cha, Y. J. , Lee, H. , Gu, N. , Kim, T. E. , Lim, K. S. , Yoon, S. H. , Chung, J. Y. , Jang, I. J. , Shin, S. G. , Yu, K. S. , & Cho, J. Y. (2013). Sustained increase in the oral bioavailability of loperamide after a single oral dose of HM30181, a P‐glycoprotein inhibitor, in healthy male participants. Basic and Clinical Pharmacology and Toxicology, 113(6), 419–424. [DOI] [PubMed] [Google Scholar]
- Chandler, R. E. (2018). Serious neurological adverse events after ivermectin‐do they occur beyond the indication of onchocerciasis? American Journal of Topical Medicine and Hygeine, 98, 382–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chufan, E. E. , Sim, H.‐M. , & Ambudkar, S. V. (2015). Molecular basis of the polyspecificity of P‐glycoprotein (ABCB1): Recent biochemical and structural studies. Advances in Cancer Research, 125, 71–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho, J. C. , Tucker, R. , Mattoon, J. , Roberts, G. , Waiting, D. K. , & Mealey, K. L. (2009). Biliary excretion of technetium‐99m‐sestamibi in wild‐type dogs and in dogs with intrinsic (ABCB1‐1Delta mutation) and extrinsic (ketoconazole treated) P‐glycoprotein deficiency. Journal of Veterinary Pharmacology and Therapeutics, 32, 417–421. [DOI] [PubMed] [Google Scholar]
- Conrad, S. , Viertelhaus, A. , Orzechowski, A. , Hoogstraate, J. , Gjellan, K. , Schrenk, D. , & Kauffmann, H. M. (2001). Sequencing and tissue distribution of the canine MRP2 compared with MRP1 and MDR1. Toxicology, 156(2–3), 81–91. [DOI] [PubMed] [Google Scholar]
- Deshpande, D. , Hill, K. E. , Mealey, K. L. , Chambers, J. P. , & Gieseg, M. A. (2016). The effect of the canine ABCB1‐1Δ mutation on sedation after intravenous administration of acepromazine. Journal of Veterinary Internal Medicine, 30(2), 636–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drag, M. , Tielemans, E. , & Mitchell, E. (2022). Safety of oral afoxolaner formulated with or without milbemycin oxime in homozygous MDR1‐deficient collie dogs. Journal of Veterinary Pharmacology and Therapeutics, 45(4), 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn, S. T. , Hedges, L. , Sampson, K. E. , Lai, Y. , Mahabir, S. , Balogh, L. , & Locuson, C. W. (2011). Pharmacokinetic interaction of the antiparasitic agents ivermectin and Spinosad in dogs. Drug Metabolism and Disposition, 39(5), 789–795. [DOI] [PubMed] [Google Scholar]
- Elsby, R. , Surry, D. D. , Smith, V. N. , & Gray, A. J. (2008). Validation and application of Caco‐2 assays for the in vitro evaluation of development candidate drugs as substrates or inhibitors of P‐glycoprotein to support regulatory submissions. Xenobiotica, 38(7–8), 1140–1164. [DOI] [PubMed] [Google Scholar]
- Feng, B. , Mille, J. B. , Davidson, R. E. , Mireles, R. J. , Janiszerski, J. S. , Troutman, M. D. , & de Morais, S. M. (2008). In vitro P‐glycoprotein assays to predict the in vivo interactions of P‐glycoprotein with drugs in the central nervous system. Drug Metabolism and Disposition, 36(2), 268–275. [DOI] [PubMed] [Google Scholar]
- Ford, J. M. , & Hait, W. N. (1993). Pharmacologic circumvention of multidrug resistance. Cytotechnology, 12, 171–212. [DOI] [PubMed] [Google Scholar]
- Fromm, M. F. (2004). Importance of P‐glycoprotein at blood tissue barriers. Trends in Pharmacological Sciences, 25(8), 423–429. [DOI] [PubMed] [Google Scholar]
- Gaens, D. , Leithauser, C. , Hamann, M. , & Geyer, J. (2019). Advere drug reactions after administration of emodepside/praziquantel (Profender®) in an MDR1‐mutant Australian shepherd dog: Case report. Frontiers in Veterinary Science, 6, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginn, P. E. (1996). Immunohistochemical detection of P‐glycoprotein in formalin‐fixed and paraffin‐embedded normal and neoplastic canine tissue. Veterinary Pathology, 33(5), 533–541. [DOI] [PubMed] [Google Scholar]
- Gunn, R. N. , Summerfield, S. G. , Salinas, C. A. , Read, K. D. , Guo, Q. , Searle, G. E. , Parker, C. A. , Jeffrey, P. , & Laruelle, M. (2012). Combining PET biodistribution and equilibrium dialysis assays to asses the free brain concentration and BBB transport of CNS drugs. Journal of Cerebral Blood Flow and Metabolism, 32(5), 874–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin, J. , Fletcher, N. , Clemence, R. , Blanchflower, S. , Brayden, D.J. , (2005). Selamectin is a potent substrate and inhibitor of human and canine P‐glycoprotein. Journal of Veterinary Pharmacology and Therapeutics, 28(3), 257–265. [DOI] [PubMed] [Google Scholar]
- Heit, M. C. , Mealey, K. L. , & King, S. B. (2021). Tolerance and pharmacokinetics of Galliprant™ administered orally to collies homozygous for MDR1‐1Δ. Journal of Veterinary Pharmacology and Therapeutics, 44(5), 705–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrikx, J. J. M. A. , Lagas, J. S. , Rosing, H. , Schellens, J. H. M. , Beijnen, J. H. , & Schinkel, A. H. (2013). P‐glycoprotein and cytochrome P450 3A acto together in restricting the oral bioavailability of paclitaxel. International Journal of Cancer, 132(10), 2439–2447. [DOI] [PubMed] [Google Scholar]
- Jenkins, E. L. , DeSouza, N. J. , Beatty, J. A. , & Barrs, V. R. D. (2019). Suspected adverse drug interaction between Spinosad and milbemycin oxime in a cat. Journal of Feline Medicine and Surgery Open Reports, 5(1), 205511691882073. 10.1177/2055116918820733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalvass, J. C. , Graff, C. L. , & Pollack, G. M. (2004). Use of loperamide as a phenotypic probe of mdr1a status in CF‐1 mice. Pharmaceutical Research, 21(10), 1867–1870. [DOI] [PubMed] [Google Scholar]
- Kitamura, Y. , Koto, H. , Matsura, S. , Kawarata, T. , Tsuchiya, H. , Kusuhara, H. , Tsujimoto, H. , & Sugiyama, Y. (2008). Modest effect of impaired P‐glycoprotein on the plasma concentrations of fexofenadine, quinidine, and loperamide following oral administration in collies. Drug Metabolism and Disposition, 36, 807–810. [DOI] [PubMed] [Google Scholar]
- Kong, L. L. , Shen, G. L. , Want, Z. Y. , Zhuang, X. M. , Xiao, W. B. , Yuan, M. , Gong, Z. H. , & Li, H. (2016). Inhibition of P‐glycoprotein and multidrug resistance associated protein 2 regulates he hepatobiliary excretion and plasma exposure of thienorphine and its glucuronide conjugate. Frontiers in Pharmacology, 7, 242. 10.3389/fphar.2016.00242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L. , Fu, Q. , Xia, M. , Xin, L. , Shen, H. , Li, G. , Ji, G. , Meng, Q. , & Xie, Y. (2018). Inhibition of P‐glycoprotein mediated efflux in Caco‐2 cells by phytic acid. Journal of Agriculture and Food Chemistry, 66(4), 988–998. [DOI] [PubMed] [Google Scholar]
- Mackin, A. J. , Riggs, C. , Beatty, T. , Mealey, K. , Boothe, D. , & Archer, T. (2020). Excessive cyclosporine‐associated immunosuppression in a dog heterozygous for the MDR1 (ABCB1‐1delta) mutation. Journal of the American Animal Hospital Association, 56(3), 190. [DOI] [PubMed] [Google Scholar]
- Malingre, M. M. , Richel, D. J. , Beijnen, J. H. , Rosing, H. , Koopman, F. J. , Bokkel Huinink, W. W. , Schot, M. E. , & Schellens, J. H. (2001). Coadministration of cyclosporine strongly enhances the oral bioavailability of docetaxel. Journal of Clinical Oncology, 19(4), 1160–1166. [DOI] [PubMed] [Google Scholar]
- Marchetti, S. , Mazzanti, R. , Beijnen, J. H. , & Schellens, J. H. M. (2007). Concise review: Clinical relevance of drug‐drug and herb‐drug interactions mediated by the ABC transporter ABCB1 (MDR1, P‐glycoprotein). The Oncologist, 12, 927–941. [DOI] [PubMed] [Google Scholar]
- Martinez, M. , Modric, S. , Sharkey, M. , Troutman, L. , Walker, L. , & Mealey, K. (2008). The pharmacogenomics of P‐glycoprotein and its role in veterinary medicine. Journal of Veterinary Pharmacology and Therapeutics, 31(4), 285–300. [DOI] [PubMed] [Google Scholar]
- Matsuda, S. , & Koyasu, S. (2000). Mechanisms of action of cyclosporine. Immunopharmacology, 47(2–3), 19–125. [DOI] [PubMed] [Google Scholar]
- Mealey, K. L. (2004). Therapeutic implications of the MDR1 gene. Journal of Veterinary Pharmacology and Therapeutics, 27(5), 257–264. [DOI] [PubMed] [Google Scholar]
- Mealey, K. L. , Bentjen, S. A. , Gay, J. , & Cantor, G. H. (2001). Ivermectin sensitivity in collies is associated with a deletion mutation of the mdr1 gene. Pharmacogenetics, 11, 727–733. [DOI] [PubMed] [Google Scholar]
- Mealey, K. L. , & Burke, N. S. (2015). Identification of a nonsense mutation in feline ABCB1. Journal of Veterinary Pharmacology and Therapeutics, 38(5), 429–433. [DOI] [PubMed] [Google Scholar]
- Mealey, K. L. , Burke, N. S. , & Connors, R. L. (2021). Role of an ABCB11930_1931del TC gene mutation in a temporal cluster of macrocyclic lactone‐induced neurologic toxicosis in cats associated with products labeled for companion animal use. Journal of the American Veterinary Medical Association, 259, 72–76. [DOI] [PubMed] [Google Scholar]
- Mealey, K. L. , Dassanayake, S. , & Burke, N. S. (2017). Establishment of a cell line for assessing drugs as canine P‐gp substrates: Proof of principle. Journal of Veterinary Pharmacology and Therapeutics, 40(5), 545–551. [DOI] [PubMed] [Google Scholar]
- Mealey, K. L. , & Fidel, J. (2015). P‐glycoprotein‐mediated drug interactions in oncology patients. Journal of Veterinary Internal Medicine, 29(1), 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mealey, K. L. , Fidel, J. , Gay, J. M. , Impellizeri, J. , Clifford, C. , & Bergman, P. J. (2008). ABCB1‐1Δ polymorphism can predict hematologic toxicity in dogs treated with vincristine. Journal of Veterinary Internal Medicine, 22(4), 996–1000. [DOI] [PubMed] [Google Scholar]
- Mealey, K. L. , Greene, S. , Bagley, R. , Gay, J. , Tucker, R. , Gavin, P. , Schmidt, K. , & Nelson, F. (2008). P‐glycoprotein contributes to the blood‐brain, but not blood‐cerebrospinal fluid, barrier in a spontaneous canine p‐glycoprotein knockout model. Drug Metabolism and Disposition, 36, 1073–1079. [DOI] [PubMed] [Google Scholar]
- Mealey, K. L. , Martinez, S. E. , Villarino, N. F. , & Court, M. H. (2019). Personalized medicine: Going to the dogs? Human Genetics, 138(5), 467–481. [DOI] [PubMed] [Google Scholar]
- Mealey, K. L. , Waiting, D. , Raunig, D. L. , Schmidt, K. R. , & Nelson, F. R. (2010). Oral bioavailability of P‐glycoprotein substrate drugs do not differ between ABCB1‐1Δ and ABCB1 wildtype dogs. Journal of Veterinary Pharmacology and Therapeutics, 36(6), 1073–1079. [DOI] [PubMed] [Google Scholar]
- Merola, V. A. , Khan, S. , & Gwaltney‐Brant, S. (2009). Ivermectin toxicosis in dogs: A retrospective study. Journal of the American Animal Hospital Association, 45(3), 106–111. [DOI] [PubMed] [Google Scholar]
- Meyers, M. J. , Martinez, M. , Li, H. , Qiu, J. , Troutman, L. , Sharkey, M. , & Yancy, H. F. (2015). Influence of ABCB1 genotype in collies on the pharmacokinetics and pharmacodynamics of loperamide in a dose escalation study. Drug Metabolism and Disposition, 43(9), 1392–1407. [DOI] [PubMed] [Google Scholar]
- Mueller, R. S. (2004). Treatment protocols for demodicosis: An evidence‐based review. Veterinary Dermatology, 15(2), 75–89. [DOI] [PubMed] [Google Scholar]
- Myers, M. J. , Martinez, M. , Lif, F. , Howard, K. , Yancy, H. F. , Troutman, L. , & Sharkey, M. (2018). Impact of ABCB1 genotype in Collies on teh pharmacokinetics of R‐ and S‐fexofenadine. Journal of Veterinary Pharmaclogy and Therapeutics, 41(6), 805–814. [DOI] [PubMed] [Google Scholar]
- Myre, S. A. , Schoeder, T. J. , Gruen, V. R. , Wandstrat, T. L. , Nicely, P. G. , Pesce, A. J. , & First, M. R. (1991). Critical ketoconazole dosage range for ciclosporin clearance inhibition in the dog. Pharmacology, 43(5), 233–241. [DOI] [PubMed] [Google Scholar]
- Nurnberger, D. , Wagner, L. , Muller, S. F. , Leiting, S. , Leidolf, R. , Alber, J. , Hamann, M. , & Geyer, J. (2022). Detection of the ABCB11930_1931del TC mutation in two suspected ivermectin‐sensitive cats and their relatives by a novel TaqMan allelic discrimination assay. Frontiers in Veterinary Science, 8, 808392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perego, R. , Spada, E. , Foppa, C. , & Proverbio, D. (2019). Critically appraised topic for the most effective and safe treatment for canine generalized demodicosis. BMC Veterinary Research, 15, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulet, A. , Puel, O. , Gesta, S. , Lepage, J. , Drag, M. , Soll, M. , Alvinerie, M. , & Pineau, T. (2003). MDR1‐deficient genotype in collie dogs hypersensitive to the P‐glycoprotein substrate ivermectin. European Journal of Pharmacology, 460, 85–91. [DOI] [PubMed] [Google Scholar]
- Schinkel, A. H. , Smit, J. J. , van Tellingen, O. , Beijnen, J. H. , Wagenaar, E. , van Deemter, L. , Mol, C. A. , van der Valk, M. A. , Robanus‐Maandag, E. C. , te Riele, H. P. , Berns, A. J. M. , & Borst, P. (1994). Disruption of the mouse mdr1a P‐glycoprotein gene leads to a deficiency in the blood‐brain barrier and to increased sensitivity to drugs. Cell, 77(4), 491–502. [DOI] [PubMed] [Google Scholar]
- Schinkel, A. H. , Wagenaar, E. , Mol, C. A. A. M. , & van Deemter, L. (1996). P‐glycoprotein in the blood‐brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. Journal of Clinical Investigation, 97(11), 2517–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman, J. G. , Paul, A. J. , & Firkins, L. D. (2010). Evaluation of the safety of Spinosad and milbemycin 5‐oxime orally administered to collies with the MDR1 gene mutation. American Journal of Veterinary Research, 71(1), 115–119. [DOI] [PubMed] [Google Scholar]
- Swain, M. D. , Orzechowski, K. L. , Swaim, H. L. , Jones, Y. L. , Robl, M. G. , Tinaza, C. A. , Myers, M. J. , Jhingory, M. V. , Buckely, L. E. , Lancaster, V. A. , & Yancy, H. F. (2013). P‐gp substrate‐induced neurotoxicity in an Abcb1a knock‐in/Abcb1b knock‐out mouse model with a mutated canine ABCB1 targeted insertion. Research in Veterinary Science, 94(3), 656–661. [DOI] [PubMed] [Google Scholar]
- Taskar, K. S. , Yang, X. , Neuhoff, S. , Patel, M. , Yoshida, K. , Paine, M. F. , Brouwer, K. L. R. , Chu, X. , Sugiyama, Y. , Cook, J. , Polli, J. W. , Hanna, I. , Lai, Y. , & Zamek‐Gliszcyzynski, J. (2022). Clinical relevance of hepatic and renal P‐gp/BCRP inhibition of drugs: An international transporter consortium perspective. Clinical Pharmacology and Therapeutics, 112, 573–592. 10.1002/cpt.2670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Heyden, S. , Chiers, K. , & Ducatelle, R. (2009). Tissue distribution of P‐glycoprotein in cats. Anatomia Histologia Embryologia Journal of Veterinary Medicine, 38, 455–460. [DOI] [PubMed] [Google Scholar]
- Walther, F. M. , Paul, A. J. , Allan, M. J. , Roepke, R. K. A. , & Nuemberger, M. C. (2014). Safety of fluralaner, a novel systemic antiparasitic drug, in MDR1(−/−) collies after oral administration. Parasites & Vectors, 7, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West, C. L. , & Mealey, K. L. (2007). Assessment of antiepileptic drugs as substrates for canine P‐glycoprotein. American Journal of Veterinary Research, 68(10), 1106–1110. [DOI] [PubMed] [Google Scholar]
- Zhou, S. F. (2008). Structure, function and regulation of P‐glycoprotein and its clinical relevance in drug disposition. Xenobiotica, 38(7–8), 802–832. [DOI] [PubMed] [Google Scholar]
- Zhu, M. , Yancy, H. F. , Deaver, C. , Jones, Y. L. , & Myers, M. J. (2016). Loperamide‐induced expression of immune and inflammatory genes in collies associated with ivermectin sensitivity. Journal of Veterinary Pharmacology and Therapeutics, 39(2), 131–137. [DOI] [PubMed] [Google Scholar]
- Zolnerciks, J. K. , Booth‐Genthe, C. L. , Gupta, A. , Harris, J. , & Unadkat, J. D. (2011). Substrate‐ and species‐dependent inhibition of P‐glycoprotein‐mediated transport: Implications for predicting in vivo drug interactions. Journal of Pharmaceutical Sciences, 100(8), 3055–3061. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.