

Abstract
Aim
We aimed to analyse the association of clonal haematopoiesis of indeterminate potential (CHIP) with incident heart failure (HF) in a European population cohort.
Methods and results
From the prospective Prevention of Renal and Vascular End‐stage Disease (PREVEND) cohort, we included all 374 participants with incident HF and selected 1:1 age‐ and sex‐matched control subjects. Peripheral blood samples of 705 individuals were successfully analysed by error‐corrected next generation sequencing for acquired mutations at a variant allele frequency ≥2% in 27 CHIP driver genes. The median age of the study population was 65 years (interquartile range 58–70) and 35.6% were female. CHIP mutations positively correlated with age, smoking, hypertension and cardiovascular biomarkers including N‐terminal pro‐B‐type natriuretic peptide and mid‐regional pro‐A‐type natriuretic peptide, but the frequency of CHIP was comparable in individuals with incident HF and in control participants (18.4% vs. 17.3%; p = 0.69). In multivariable Cox regression models, CHIP was not significantly associated with incident HF (hazard ratio [HR] 1.24, 95% confidence interval [CI] 0.93–1.65; p = 0.144). This association, however, was modified by age (p for CHIP–age interaction = 0.002). Among people younger than 65 years, CHIP mutations were more frequently detected in the case cohort compared to the control cohort (14.2% vs. 5.8%; p = 0.009), and were significantly associated with new‐onset HF (HR 2.07, 95% CI 1.30–3.29; p = 0.002).
Conclusion
Clonal haematopoiesis of indeterminate potential correlates with HF risk factors and biomarkers, and is associated with incident HF in subjects <65 years of age.
Keywords: CHIP, Clonal haematopoiesis, Heart failure, Risk factors, Biomarkers
Association of clonal haematopoiesis of indeterminate potential (CHIP) with incident heart failure (HF).
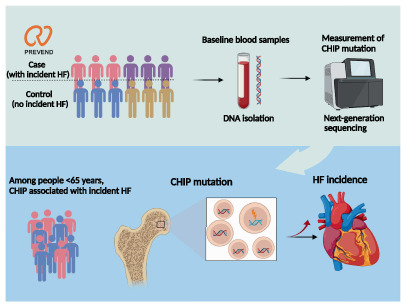
Introduction
Somatic mutations occur and gradually accumulate among a variety of healthy tissues, including skin and blood, potentially contributing to aging and tumorigenesis. 1 , 2 Expansion of haematopoietic stem or progenitor cells carrying somatic mutations in certain genes implicated in haematologic malignancies results in detectable, mutated clones of cells in the peripheral blood and bone marrow. These mutations can also be detected in individuals without haematologic malignancy, which has been defined as clonal haematopoiesis of indeterminate potential (CHIP). 3 , 4 Among all mutations, DNMT3A, TET2 and ASXL1 are three predominant driver genes of CHIP, involved in the epigenetic regulation of inflammation. 3 , 5
The prevalence of CHIP increases with age, and associates with incident cancer and mortality. 6 , 7 Interestingly, recent data demonstrated that CHIP also increases the risks of incident coronary heart disease, myocardial infarction (MI) and stroke. 3 , 8 , 9 In mechanistic studies, loss of TET2 potentiates atherosclerosis in low‐density lipoprotein receptor (Ldlr) knockout mice, and worsens cardiac remodelling and function in mice with heart failure (HF) by upregulating CXC chemokines and cytokines such as interleukin (IL)‐6 and IL‐1β. 10 , 11
As such, CHIP has been positioned as a link between cancer and cardiovascular disease (CVD), as a potential common genetic risk factor in the cardio‐oncology field. 12 , 13 In the CVD spectrum, HF in particular has emerged as a potential precipitant for incident cancer, and several pathophysiological factors are shared between HF and cancer. 12 , 13 , 14
Recent clinical data have shown that in patients with chronic HF, CHIP is significantly associated with all‐cause mortality and HF hospitalization, best characterized by DNMT3A and TET2 mutations. 15 , 16 Similar associations were reported among HF patients with reduced ejection fraction (HFrEF). 17 Moreover, a large American cohort study including over 50 000 participants found that CHIP correlated with a 25% increased risk of new‐onset HF. 18
However, the effects of CHIP mutations on HF risk factors and biomarkers, and HF incidence among the European population, and whether the effects vary between HF phenotypes remain unexplored. On this basis, we designed a case–control study to assess the associations of CHIP mutations with incident HF and HF subtypes (heart failure with preserved ejection fraction [HFpEF] and HFrEF) in a well‐characterized general European population cohort (the Prevention of Renal and Vascular End‐stage Disease [PREVEND] study). 19 , 20
Methods
Study population
For the present study data and biomaterials were used from the PREVEND cohort, a prospective population‐based cohort which has been fully described in detail before. 19 , 21 In brief, from 1997 to 1998, all residents (n = 85 421) in Groningen, the Netherlands, within an age range of 25 to 75 years, were asked to send in a first‐morning urine sample and complete a short questionnaire about demographics and CVD history. In total 47.8% participants (n = 40 856) responded. From those, 7786 subjects with urinary albumin excretion (UAE) >10 mg/L and 3395 randomly selected control individuals with a UAE <10 mg/L were invited to an outpatient clinic for a detailed assessment of cardiovascular and renal risk factors. After excluding pregnant women, subjects with insulin‐dependent diabetes mellitus, or unable or unwilling to participate, a total of 8592 subjects completed the initial screening. The PREVEND study complied with the Declaration of Helsinki and was approved by institutional medical ethics committee. Written informed consent was provided by the participants enrolled in the study.
Subsequently, 374 individuals developed new‐onset HF during a median 11.5 year follow‐up, of whom 241 (66%) people were diagnosed with HFrEF and 125 (34%) subjects developed a HFpEF phenotype, based on the European Society of Cardiology (ESC) guidelines for diagnosis of new‐onset HF (left ventricular ejection fraction [LVEF] ≤40% for HFrEF or ≥50% for HFpEF, respectively); the adjudication of HF events has been published in detail. 20 We selected all individuals with incident HF as a case cohort, and a 1:1 age and sex‐matched control cohort. The median age for both control and case cohorts was 65 years (interquartile range [IQR] 57–69; p = 1.0) and 64.4% were male (p = 1.0). The sample matching is shown in online supplementary Figure S1 .
Sample preparation and error‐corrected next generation sequencing
DNA samples at baseline were isolated from peripheral blood with Qiamp DNA extraction kits (Qiagen). A custom panel of single‐molecule‐tagged molecular inversion probes (smMIP) was designed, and a pooled smMIP‐DNA library was prepared for error‐corrected next generation sequencing with a standard protocol. 22 , 23 The panel targeted regions in 27 driver genes associated with haematological malignancies (online supplementary Table S1 ). Sequencing was performed for 300 cycles on a NovaSeq 6000 or NextSeq500 instrument (Illumina, San Diego, CA, USA), resulting in 2 × 150 bp paired‐end reads. Somatic variants were called and included in our analysis with a variant allele frequency (VAF) ≥2% and ≥ 10 mutant unique smMIP reads after being manually inspected and curated to exclude recurrent artifacts and polymorphisms. Variants with VAF >45% were further excluded, as we could not determine whether these are from germline or somatic origin. The median number of error‐corrected consensus reads for the entire cohort was 6274 and the median coverage was 3231, with a coverage >500× for 97.8% of all targeted regions (online supplementary Figure S2 ). Details regarding the design of sequencing panel, library preparation and variant calling are described in online supplementary Appendix S1 .
Study endpoints
We considered HF incidence, incident HFrEF and incident HFpEF as the endpoints.
Statistical analyses
Categorical variables are presented as numbers with percentages (%) and were compared using the Chi‐squared test. Continuous variables are displayed as medians with IQR and were compared using Student's independent t‐test if variables were normally distributed, whereas skewed variables were compared using Mann–Whitney U test. The distribution of CHIP was observed according to age quartiles.
A stepwise logistic regression model was used to identify clinical correlates of CHIP, and the final model included all clinical covariates with a p‐value <0.05. Odds ratios (ORs) with 95% confidence intervals (CIs) were calculated to estimate the correlations of CHIP with clinical factors. Linear regression models were conducted to analyse the associations of CHIP occurrence with CVD biomarkers including N‐terminal pro‐B‐type natriuretic peptide (NT‐proBNP), mid‐regional pro‐A‐type natriuretic peptide (MR‐proANP), troponin T, C‐reactive protein (CRP), mid‐regional pro‐adrenomedullin (MR‐proADM), C‐terminal pro‐endothelin‐1, procalcitonin, plasminogen activator inhibitor (PAI), galectin‐3, cystatin C and UAE. 24 , 25 , 26 All biomarker measurements were natural log‐transformed prior to analyses to obtain normal distribution. Univariable analyses with p < 0.05 for all biomarkers associated with CHIP were adjusted for age and sex, which were further adjusted for smoking, diabetes, hypertension, body mass index (BMI), cholesterol, history of MI, atrial fibrillation (AF) and cerebrovascular accident. Since age is a strong risk factor for incident HF and significantly associated with CHIP, an interaction test of age to determine the effects of CHIP on new‐onset HF was performed.
Univariable and multivariable Cox proportional hazards regression analyses were performed to evaluate associations between the presence of CHIP or specific single gene mutation(s) or clone size and outcomes. Age, sex, smoking, diabetes, hypertension, BMI, cholesterol, history of MI, AF and cerebrovascular accident were included as covariates in multivariable models. For each single gene analysis, the control group included individuals without CHIP mutations and people without variants in the specific single gene who may carry mutations in other CHIP driver genes. HRs are reported with 95% CIs. A 2‐tailed p‐value <0.05 was considered to denote statistically significant differences. All data analyses were performed with Stata15.1 (StataCorp, 2017, College Station, TX, USA, StataCorp LLC).
Results
Baseline characteristics
In the PREVEND cohort, all 374 individuals who developed HF were included together with 374 age‐ and sex‐matched control subjects. After excluding missing DNA of 28 people, four variant carriers with VAFs >45% and 11 samples failing to be sequenced, the final sequencing results were obtained for 347 controls and 358 cases, as illustrated in online supplementary Figure S3 . In the case cohort, 238 (66.5%) participants developed HFrEF and 120 people (33.5%) developed HFpEF. The baseline characteristics of the study population are presented in Table 1 . The median age of all participants was 65 (IQR 58–70) and 35.6% were female. Compared to the control cohort, people with incident HF showed higher baseline levels of BMI, and CVD biomarkers including NT‐proBNP, MR‐proANP, troponin T, CRP, MR‐proADM, PAI‐1, cystatin C and UAE, accompanied by greater prevalence of MI, AF, diabetes and hypertension.
Table 1.
Baseline characteristics of the study population
| Factor | Control (n = 347) (no HF) | Case (n = 358) (incident HF) | p‐value |
|---|---|---|---|
| CHIP, n (%) | 60 (17.3) | 66 (18.4) | 0.69 |
| Clinical characteristics | |||
| Age, years, median (IQR) | 65 (58–70) | 65 (58–70) | 0.95 |
| Female sex, n (%) | 123 (35.4) | 128 (35.8) | 0.95 |
| BMI, kg/m2, median (IQR) | 26.6 (24.8–29.3) | 27.9 (25.4–30.8) | <0.001 |
| Medical history, n (%) | |||
| Diabetes | 19 (5.5) | 46 (12.9) | <0.001 |
| Hypertension | 210 (60.5) | 277 (77.6) | <0.001 |
| Myocardial infarction | 38 (11.0) | 91 (25.4) | <0.001 |
| Atrial fibrillation | 7 (2.0) | 22 (6.1) | 0.006 |
| Cerebrovascular accident | 7 (2.0) | 11 (3.1) | 0.37 |
| Malignancy | 14 (4.0) | 25 (7.0) | 0.087 |
| Smoking | 0.12 | ||
| Never | −88 (25.4) | −77 (21.6) | |
| Past | −153 (44.2) | −145 (40.7) | |
| Current | −105 (30.3) | −134 (37.6) | |
| Laboratory, median (IQR) | |||
| Cholesterol, mmol/L | 5.9 (5.2–6.5) | 5.9 (5.3–6.7) | 0.14 |
| Telomere length, T/S | 0.91 (0.77–1.11) | 0.93 (0.77–1.13) | 0.78 |
| NT‐proBNP, ng/L | 50.8 (25.2–104.1) | 104.8 (43.0–284.7) | <0.001 |
| MR‐proANP, pmol/L | 57.8 (41.6–83.2) | 73.4 (50.3–111.7) | <0.001 |
| Troponin T, μg/L | 4 (2.5–8) | 7 (4–10) | <0.001 |
| CRP, mg/L | 1.9 (1.0–3.7) | 2.5 (1.2–4.8) | 0.006 |
| MR‐proADM, nmol/L | 0.45 (0.34–0.54) | 0.47 (0.37–0.59) | 0.006 |
| CT‐proET‐1, pmol/L | 38.4 (27.4–47.3) | 41.6 (29.3–52.2) | 0.048 |
| Procalcitonin, ng/L | 0.017 (0.014–0.022) | 0.018 (0.015–0.023) | 0.017 |
| PAI‐1, mg/L | 84.1 (52.5–124.7) | 93.3 (57.9–160.2) | 0.005 |
| Galectin‐3, mg/L | 12.2 (10.2–14.4) | 12.4 (10.4–14.7) | 0.5 |
| Cystatin C, mg/L | 0.95 (0.87–1.07) | 1.02 (0.90–1.14) | <0.001 |
| UAE, mg/24 h | 11.3 (6.9–26.2) | 19.5 (9.3–53.9) | <0.001 |
BMI, body mass index; CHIP, clonal haematopoiesis of indeterminate potential; CRP, C‐reactive protein; CT‐proET‐1, C‐terminal pro‐endothelin‐1; HF, heart failure; IQR, interquartile range; MR‐proADM, mid‐regional pro‐adrenomedullin; MR‐proANP, mid‐regional pro‐A‐type natriuretic peptide; NT‐proBNP, N‐terminal pro‐B‐type natriuretic peptide; PAI, plasminogen activator inhibitor; UAE, urine albumin excretion.
Distribution of CHIP mutations
CHIP mutations were detected in 60 (17.3%) people in the control cohort and 66 (18.4%) individuals from the case cohort. Details of the identified CHIP variants were listed in online supplementary Table S2 . All participants were categorized according to age quartiles as depicted in Figure 1A,B . The prevalence of CHIP mutations increased from 6.3% among people with ages ≤58 (11 out of 176 persons) to 29.7% in elderly people above 71 years (52 out of 175 persons). In total, 153 mutations were identified among 126 individuals, of which 106 people (84%) had only one mutation, and 14 and 6 persons were found carrying 2 or >2 mutations, respectively (Figure 1C ). The majority of the variants occurred in three genes: DNMT3A (58.8%), TET2 (22.9%) and ASXL1 (10.5%), as presented in Figure 1E . However, there were no significant differences observed between the case and control cohorts regarding the CHIP frequency, detected number of mutations, mutational spectrum and maximum VAF (Figure 1B, D, F ; online supplementary Figure S4 ).
Figure 1.
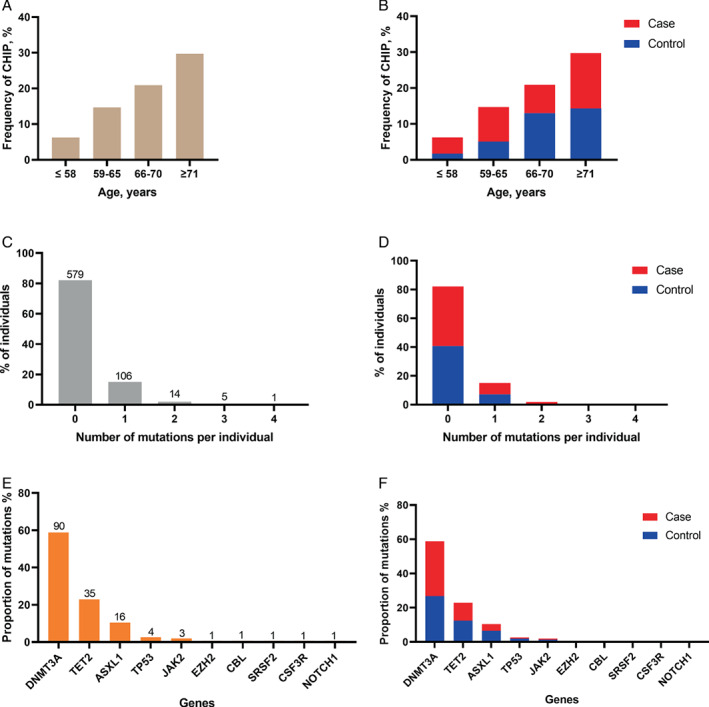
Distribution of clonal haematopoiesis of indeterminate potential (CHIP) mutations. Frequency of CHIP mutations according to age quartiles in (A) the entire cohort, and (B) control and case cohort. Number of mutations per participant in (C) the entire cohort, and (D) control and case cohort. Proportion of mutations according to gene spectrum in (E) the entire cohort, and (F) control and case cohort.
CHIP is associated with cardiovascular risk factors and biomarkers
As displayed in online supplementary Table S3 , 126 participants carrying CHIP mutations were older than the non‐carriers, and had significantly higher baseline levels of NT‐proBNP and MR‐proANP, which are biomarkers for incident HF.
Linear regression analyses in Table 2 showed that CHIP significantly correlated with 20.7% and 19.5% increases of log‐standardized NT‐proBNP (p = 0.019) and MR‐proANP (p = 0.034) levels, respectively, after adjustment for age, sex, smoking, diabetes, hypertension, BMI, cholesterol, history of MI, AF and cerebrovascular accident. Logistic regression model (online supplementary Table S4 ) demonstrated that an increase of age by 10 years strongly correlated with the presence of CHIP (OR 2.30, 95% CI 1.66–3.18; p < 0.001). Women had 66% higher chance to carry CHIP mutations than men. Additionally, CHIP was positively associated with past smoking (OR 2.11, 95% CI 1.16–3.83; p = 0.015) and hypertension (OR 1.83, 95% CI 1.06–3.19; p = 0.031), and negatively correlated with BMI (OR 0.72, 95% CI 0.54–0.95; p = 0.022) and prevalent MI (OR 0.54, 95% CI 0.30–0.95; p = 0.033).
Table 2.
Correlation analyses of clonal haematopoiesis of indeterminate potential with cardiovascular biomarkers
| Univariable | Multivariable1 | Multivariable2 | ||||
|---|---|---|---|---|---|---|
| Sβ (SE) | p‐value | Sβ (SE) | p‐value | Sβ (SE) | p‐value | |
| NT‐proBNP | +0.411 (0.098) | 0.000 | +0.222 (0.096) | 0.021 | +0.207 (0.086) | 0.019 |
| MR‐proANP | +0.443 (0.102) | 0.000 | +0.200 (0.096) | 0.038 | +0.195 (0.092) | 0.034 |
| Troponin T | +0.200 (0.099) | 0.043 | +0.015 (0.092) | 0.867 | ||
| CRP | −0.027 (0.100) | 0.785 | ||||
| MR‐proADM | +0.140 (0.103) | 0.177 | ||||
| CT‐proET‐1 | + 0.016 (0.104) | 0.881 | ||||
| Procalcitonin | −0.073 (0.104) | 0.484 | ||||
| PAI‐1 | −0.155 (0.099) | 0.117 | ||||
| Galectin‐3 | +0.127 (0.099) | 0.2 | ||||
| Cystatin C | +0.170 (0.102) | 0.096 | ||||
| UAE | −0.024 (0.098) | 0.804 | ||||
CRP, C‐reactive protein; CT‐proET‐1, C‐terminal pro‐endothelin‐1; MR‐proADM, mid‐regional pro‐adrenomedullin; MR‐proANP, mid‐regional pro‐A‐type natriuretic peptide; NT‐proBNP, N‐terminal pro‐B‐type natriuretic peptide; PAI, plasminogen activator inhibitor; Sβ, standardized regression coefficient; SE, standard error; UAE, urine albumin excretion.
Multivariable model 1 was adjusted for age and sex.
Multivariable model 2 was further adjusted for smoking, diabetes, hypertension, body mass index, cholesterol, history of myocardial infarction, atrial fibrillation and cerebrovascular accident.
Association of CHIP with incident heart failure and heart failure subtypes
There were 43 people with incident HFrEF detected with CHIP variants, and 23 individuals with incident HFpEF carrying CHIP mutations. Multivariable Cox regression models were performed to determine the associations of CHIP with incident HF and HF subtypes (online supplementary Table S5 ). However, the presence of CHIP was not significantly associated with overall HF incidence (HR 1.24, 95% CI 0.93–1.65; p = 0.144), HFrEF (HR 1.20, 95% CI 0.84–1.71; p = 0.321) or HFpEF (HR 1.09, 95% CI 0.67–1.77; p = 0.733). Among individual genes, DNMT3A variants were significantly associated with new‐onset HF with a HR of 1.52 (95% CI 1.10–2.10; p = 0.01). This association between DNMT3A and incident HF remained significant after further adjustment for NT‐proBNP (HR 1.40, 95% CI 1.01–1.94; p = 0.041). No associations were observed between HF incidence and variants in TET2 (HR 0.97, 95% CI 0.54–1.74; p = 0.919) or ASXL1 (HR 0.74, 95% CI 0.30–1.80; p = 0.507).
CHIP frequency is higher in younger individuals with incident heart failure
There was a significant interaction between CHIP and age for incident HF (p for CHIP–age interaction = 0.002). Therefore, we stratified our analyses by age categories <65 years and ≥65 years (65 years being the median age).
In total 35 (10.1%) individuals were detected with CHIP variants from 348 people younger than 65 years, while 91 (25.5%) out of 357 participants ≥65 years carried CHIP mutations, as shown in Table 3 . Interestingly, among individuals <65 years CHIP mutations were more frequently detected in the case cohort (n = 25, 14.2%) compared to the control subjects (n = 10, 5.8%; p = 0.009), whereas in people ≥65 years the prevalence of CHIP was comparable between case and control cohorts (n = 41, 22.5% in cases vs. n = 50, 28.6% in controls; p = 0.19).
Table 3.
Clonal haematopoiesis of indeterminate potential and baseline characteristics in individuals <65 and ≥65 years of age
| Factor | Individuals <65 years (n = 348) | Individuals ≥65 years (n = 357) | ||||
|---|---|---|---|---|---|---|
| Control (n = 172) (no HF) | Cases (n = 176) (incident HF) | p‐value | Control (n = 175) (no HF) | Cases (n = 182) (incident HF) | p‐value | |
| CHIP, n (%) | 10 (5.8) | 25 (14.2) | 0.009 | 50 (28.6) | 41 (22.5) | 0.19 |
| Clinical characteristics | ||||||
| Age, years, median (IQR) | 57 (51–62) | 58 (51–62) | 0.91 | 69 (67–73) | 70 (67–73) | 0.99 |
| Female sex, n (%) | 63 (36.6) | 64 (36.4) | 0.96 | 60 (34.3) | 64 (35.2) | 0.86 |
| BMI, kg/m2, mdian (IQR) | 26.9 (24.9–29.5) | 27.9 (25.6–31.1) | 0.017 | 26.2 (24.6–29.1) | 27.9 (25.2–30.3) | 0.003 |
| Medical history, n (%) | ||||||
| Diabetes | 8 (4.7) | 21 (12) | 0.015 | 11 (6.3) | 25 (13.7) | 0.019 |
| Hypertension | 75 (43.6) | 118 (67.4) | <0.001 | 135 (77.1) | 159 (87.4) | 0.011 |
| Myocardial infarction | 14 (8.1) | 38 (21.6) | <0.001 | 24 (13.7) | 53 (29.1) | <0.001 |
| Atrial fibrillation | 1 (0.6) | 8 (4.5) | 0.02 | 6 (3.4) | 14 (7.7) | 0.08 |
| Cerebrovascular accident | 3 (1.7) | 5 (2.8) | 0.49 | 4 (2.3) | 6 (3.3) | 0.56 |
| Malignancy | 5 (2.9) | 7 (4) | 0.58 | 9 (5.1) | 18 (9.9) | 0.09 |
| Smoking | 0.25 | 0.68 | ||||
| Never | −40 (23.4) | −33 (18.9) | −48 (27.4) | −44 (24.3) | ||
| Past | −69 (40.4) | −59 (33.7) | −84 (48) | −86 (47.5) | ||
| Current | −62 (36.3) | −83 (47.4) | −43 (24.6) | −51 (28.2) | ||
| Laboratory, median (IQR) | ||||||
| Cholesterol, mmol/L | 5.9 (5.2–6.4) | 6.1 (5.4–6.8) | 0.047 | 5.9 (5.2–6.5) | 5.8 (5.2–6.6) | 0.88 |
| Telomere length, T/S | 0.93 (0.78–1.16) | 0.95 (0.80–1.18) | 0.43 | 0.90 (0.77–1.10) | 0.90 (0.76–1.08) | 0.66 |
| NT‐proBNP, ng/L | 32.2 (15.9–60.8) | 69.4 (27.5–171.7) | <0.001 | 84.9(36.3–153.5) | 163.5 (77.5–427.4) | <0.001 |
| MR‐proANP, pmol/L | 48.1 (36.1–63.8) | 55.6 (42.2–82.6) | 0.002 | 74.0 (50.9–101.3) | 91.3 (63.2–131.8) | <0.001 |
| Troponin T, μg/L | 3 (2.5–5) | 5 (2.5–8) | <0.001 | 6 (3–10) | 8 (6–12) | <0.001 |
| CRP, mg/L | 1.8 (0.9–3.3) | 2.3 (1.1–4.7) | 0.022 | 1.9 (1.1–3.9) | 2.5 (1.2–4.9) | 0.15 |
| MR‐proADM, nmol/L | 0.39 (0.32–0.51) | 0.42 (0.35–0.52) | 0.12 | 0.50 (0.40–0.57) | 0.54 (0.40–0.63) | 0.012 |
| CT‐proET‐1, pmol/L | 36.9 (26.0–45.3) | 37.7 (25.4–48.7) | 0.33 | 40.3 (30.8–51.4) | 44.7 (33.0–55.3) | 0.1 |
| Procalcitonin, ng/L | 0.017 (0.014–0.022) | 0.018 (0.015–0.023) | 0.20 | 0.018 (0.015–0.022) | 0.019 (0.016–0.023) | 0.04 |
| PAI‐1, mg/L | 92.6 (55.5–164.0) | 100.4 (60.4–162.1) | 0.25 | 75.1 (50.4–113.8) | 89.3 (56.1–156.6) | 0.005 |
| Galectin‐3, mg/L | 11.4 (9.7–13.8) | 11.8 (9.4–13.1) | 0.77 | 13 (11–14.8) | 13.3 (11.4–15.9) | 0.17 |
| Cystatin C, mg/L | 0.9 (0.82–0.99) | 0.94 (0.85–1.03) | 0.016 | 1 (0.91–1.17) | 1.1 (1.01–1.21) | <0.001 |
| UAE, mg/24 h | 9.8 (6.4–20.5) | 18.0 (8.6–52.9) | <0.001 | 12.2 (7.5–29.9) | 22.3 (10.7–53.9) | <0.001 |
BMI, body mass index; CHIP, clonal haematopoiesis of indeterminate potential; CRP, C‐reactive protein; CT‐proET‐1, C‐terminal pro‐endothelin‐1; HF, heart failure; IQR, interquartile range; MR‐proADM, mid‐regional pro‐adrenomedullin; MR‐proANP, mid‐regional pro‐A‐type natriuretic peptide; NT‐proBNP, N‐terminal pro‐B‐type natriuretic peptide; PAI, plasminogen activator inhibitor; UAE, urine albumin excretion.
CHIP is associated with cardiovascular biomarkers and new‐onset heart failure among people <65 years of age
Linear regression models in online supplementary Table S6 showed that previously observed associations between CHIP and natriuretic peptides including NT‐proBNP and MR‐proANP in the entire cohort were mainly driven by individuals younger than 65 years. In people ≥65 years, no correlations of cardiovascular biomarkers with CHIP were observed.
Multivariable Cox regression models in Table 4 demonstrated that in people younger than 65 years, CHIP was significantly associated with HF incidence (HR 2.07, 95% CI 1.30–3.29; p = 0.002) and incident HFpEF (HR 2.11, 95% CI 1.01–4.41; p = 0.048) but not HFrEF (HR 1.45, 95% CI 0.79–2.66; p = 0.233). Those associations were mainly driven by DNMT3A mutations. After further adjusting for NT‐proBNP, the associations stayed significant between incident HF and CHIP (HR 1.72, 95% CI 1.08–2.76; p = 0.023) or DNMT3A mutations (HR 1.73, 95% CI 1.01–2.97; p = 0.045). There were no correlations between CHIP or DNMT3A variants with incident HF or HF subtypes among people older than 65 years.
Table 4.
Associations of clonal haematopoiesis of indeterminate potential with incident heart failure, heart failure subtypes, according to age categories
| Individuals <65 years | Individuals ≥65 years | |||||||
|---|---|---|---|---|---|---|---|---|
| CHIP HR (95% CI) | p‐value | DNMT3A HR (95% CI) | p‐value | CHIP HR (95% CI) | p‐value | DNMT3A HR (95% CI) | p‐value | |
| Incident HF | 2.07 (1.30–3.29) | 0.002 | 2.13 (1.25–3.61) | 0.005 | 0.95 (0.66–1.36) | 0.773 | 1.29 (0.86–1.95) | 0.220 |
| Incident HFrEF | 1.45 (0.79–2.66) | 0.233 | 1.35 (0.66–2.76) | 0.416 | 1.01 (0.65–1.58) | 0.957 | 1.23 (0.74–2.06) | 0.425 |
| Incident HFpEF | 2.11 (1.01–4.41) | 0.048 | 2.26 (1.01–5.04) | 0.047 | 0.78 (0.41–1.49) | 0.447 | 1.10 (0.54–2.23) | 0.789 |
CHIP, clonal haematopoiesis of indeterminate potential; CI, confidence interval; HF, heart failure; HFpEF, heart failure with preserved ejection fraction; HFrEF, heart failure with reduced ejection fraction; HR, hazard ratio.
Cox regression models were adjusted for age, sex, smoking, diabetes, hypertension, body mass index, cholesterol, history of myocardial infarction, atrial fibrillation and cerebrovascular accident.
Associations of clone size with heart failure incidence and heart failure subtypes
To assess whether larger clone size could increase the risk of incident HF or HF subtypes, we categorized highest VAFs into two subgroups: VAF 2–5% and VAF >5%. As shown in Table 5 , in multivariate analyses with adjustment for HF risk factors, we found only in people <65 years VAF 2–5% was significantly associated with incident HF (HR 1.98, 95% CI 1.15–3.41; p = 0.014), larger clone size (VAF >5%) increased the risk of HF events (HR 2.30, 95% CI 1.05–5.02; p = 0.037) and was associated with incident HFpEF (HR 4.74, 95% CI 1.76–12.74; p = 0.002).
Table 5.
Associations of highest variant allele frequencies with incident heart failure and its subtypes
| Entire cohort | Individuals <65 years | Individuals ≥65 years | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| VAF 2–5% HR (95% CI) | p‐value | VAF >5% HR (95% CI) | p‐value | VAF 2–5% HR (95% CI) | p‐value | VAF >5% HR (95% CI) | p‐value | VAF 2–5% HR (95% CI) | p‐value | VAF >5% HR (95% CI) | p‐value | |
| Incident HF | 1.29 (0.91–1.82) | 0.147 | 1.16 (0.75–1.80) | 0.506 | 1.98 (1.15–3.41) | 0.014 | 2.30 (1.05–5.02) | 0.037 | 1.04 (0.66–1.63) | 0.864 | 0.84 (0.49–1.42) | 0.510 |
| Incident HFrEF | 1.42 (0.94–2.14) | 0.092 | 0.87 (0.48–1.57) | 0.637 | 1.67 (0.87–3.22) | 0.126 | 0.85 (0.20–3.49) | 0.817 | 1.29 (0.75–2.19) | 0.357 | 0.74 (0.38–1.44) | 0.370 |
| Incident HFpEF | 0.83 (0.44–1.54) | 0.549 | 1.67 (0.87–3.22) | 0.123 | 1.32 (0.50–3.51) | 0.573 | 4.74 (1.76–12.74) | 0.002 | 0.62 (0.27–1.41) | 0.254 | 1.10 (0.46–2.64) | 0.837 |
CI, confidence interval; HF, heart failure; HFpEF, heart failure with preserved ejection fraction; HFrEF, heart failure with reduced ejection fraction; HR, hazard ratio; VAF, variant allele frequency.
Cox regression models were adjusted for age, sex, smoking, diabetes, hypertension, body mass index, cholesterol, history of myocardial infarction, atrial fibrillation and cerebrovascular accident.
We additionally studied the potential association between the extent of clonal expansion and incident HF by lowering the cutoff of CHIP mutation calling to VAF ≥1%, which led to the identification of 124 additional mutations in 73 people. We categorized VAFs into three groups: 1–2%, 2–5%, and >5%. However, CHIP with lower threshold (VAF ≥1%) was not associated with HF incidence (HR 1.03, 95% CI 0.81–1.31, p = 0.831), nor was larger clonal size (VAF >5%; HR 1.13, 95% CI 0.72–1.75, p = 0.591) in the entire cohort after adjustment for HF risk factors (online supplementary Table S7 ). Among the younger individuals, the association between CHIP with new‐onset HF was observed only in groups with VAF ≥2%.
Discussion
In this study we investigated the prevalence and associations of CHIP mutations for incident HF by error‐corrected targeted next generation sequencing. Firstly, our results confirm previous reports that CHIP mutations are more prevalent in older individuals, among which DNMT3A, TET2 and ASXL1 are three main driver mutations. 3 , 15 , 18 We found that the frequency of CHIP was comparable between subjects who developed incident HF and those who did not (control subjects). Next, we verified that CHIP correlated with cardiovascular risk factors, including age, smoking and hypertension, and was significantly associated with HF biomarkers including NT‐proBNP and MR‐proANP. Lastly, we demonstrated that the presence of CHIP did not associate with incident HF in the entire cohort but among people younger than 65 years. Larger clone size increased the risk of incident HF only in the younger individuals (Graphical Abstract).
Yu's study in five cohorts which collectively enrolled 56 597 participants showed that CHIP mutations detected by whole exome or genome sequencing (WES/WGS) were associated with a 25% increased risk for incident HF. 18 In our study, error‐corrected targeted next generation sequencing was used to detect CHIP mutations, which provides better sensitivity to reliably capture CHIP variants with VAF 2–5% than WES or WGS. 27 Compared to Yu's finding, our study further suggests the association of CHIP with HF incidence varies among individuals with different ages. We only found a significant correlation between CHIP and new‐onset HF in the relatively young subjects, while it was absent in people older than 65 years. This association was mainly driven by DNMT3A mutations instead of TET2 or ASXL1 variants observed in Yu's study, 18 which is in line with earlier basic research that inactivating mutations in gene DNMT3A promoted inflammation and cardiac hypertrophy, 28 and with clinical settings that DNMT3A variants significantly correlated with HF adverse outcomes. 15 , 17 Consistent with previous findings that large clone size associated with higher risk of incident HF, all‐cause mortality and HF hospitalization, 17 , 18 we observed clonal expansion (VAF >5%) increased the risk of HF events but only in the younger population.
This phenomenon could be explained as age is a strong confounder, highly involved in modifying the effects of HF risk factors. The attributable risk conferred by a risk factor in younger population might be greater and more easily exposed than that in older people whose baseline risk for HF is higher, and where multiple risk factors weigh in. For example, known risk factors such as hypertension, diabetes, obesity and current smoking history have been reported to confer greater relative HF risk in younger compared with older population. 29 , 30 , 31 In line with this, CHIP mutations at a young age may afford a potential risk for incident HF, despite a lower incidence and absolute risk of HF among younger compared with older people.
Cross‐sectional analyses showed that CHIP is associated with age, smoking, hypertension and CVD biomarkers. After multivariable adjustment, NT‐proBNP and MR‐proBNP remained significantly associated with CHIP, which is driven by the younger individuals. There is no clear evidence on whether the mutation(s) can directly affect NT‐proBNP production. Inflammation could be one of the possible mechanisms explaining the link between somatic mutations and natriuretic peptide release. For instance, experimental models for TET2, JAK2, and DNMT3A mutations demonstrated similar accelerating effects on cardiac dysfunction, hypertrophy and fibrosis. 11 , 28 , 32 Inactivating TET2 promoted the expression of IL‐1β, IL‐6, and chemokine C‐C motif ligand 5 (Ccl5), whereas DNMT3A deactivation increased the expression of CXC ligand 1 (Cxcl1), Cxcl2, IL‐6 and Ccl5. 28 In clinical settings, NT‐proBNP levels positively correlate with the levels of inflammatory markers in HF patients. 33 , 34 A possible mechanism linking these two is that cytokines such as IL‐6, IL‐1β, IL‐18 and tumour necrosis factor‐α can stimulate the expression and secretion of natriuretic peptides. 33 , 35 , 36
Classical risk factors including age, sex, smoking, hypertension, obesity, diabetes, smoking, lifestyle and pathophysiological pathways such as inflammation and oxidative stress, show strongly predictive value for incident CVD and cancer. 12 , 37 However, current risk models are only able to ascertain 53–75% of the individual's risk, and there is a substantial residual risk of genetics that remains unaccounted for. 29 Accumulated data support that CHIP has become a clinical entity with genetic susceptibility to incident HF, cancer and adverse outcomes, independent of environmental risk factors. 3 , 8 , 15 , 16 , 17 , 18 In our study, the greater attributable risk of CHIP at a young age for HF highlights the importance of early detection for CHIP and preventive intervention. Recently, the Canakinumab Anti‐inflammatory Thrombosis Outcome Study (CANTOS) verified that the IL‐1β neutralizing antibody canakinumab can reduce recurrent cardiovascular events, HF hospitalization, and elevated CRP levels in patients with stable coronary artery disease. 38 , 39 A CANTOS exploratory analysis further suggestively showed that patients with TET2 mutations have an improved response to canakinumab. 40 Whether CVD patients carrying CHIP mutations especially with young ages, are more sensitive to anti‐inflammatory therapy, which might be specific for mutated genes, should be further explored in large clinical trials.
Limitations
Our study suggesting that the presence of CHIP is associated with incident HF mainly in a relatively young population should be further validated in additional larger cohort studies. Although our cohort is clinically well annotated, due to the relative low number of cases, our study lacks sufficient statistical power to analyse the associations of specific gene mutations or number of mutations with incident HF or HF phenotypes. Similarly, correlations between CHIP and additional inflammatory markers such as IL‐6 or IL‐1β cannot be studied due to data unavailability of the PREVEND cohort. Subjects of <75 years were enrolled in the PREVEND cohort. Therefore, our results cannot be extrapolated to subjects of older than 75 years. Furthermore, we used the classic definition of CHIP with a VAF ≥2% to make this study comparable with previous data. 15 , 17 , 18 In Assmus's study, 41 lower VAF thresholds for TET2 and DNMT3A have shown significant associations with HF outcomes. Whether certain cutoffs of VAF (either <2% or ≥2%) for specific genes might confer a significant association with new‐onset HF remain to be explored in large cohort studies.
Conclusions
The current results support the notion that the somatic mutations in clonal haematopoiesis are associated with HF incidence mainly in individuals younger than 65 years.
Supporting information
Appendix S1. Supporting information.
Acknowledgements
We would like to thank Sietske N. Zijlstra, Susanne Feringa and Silke Oberdorf‐Maass for their laboratory assistance.
Funding
This work was supported by grants from the Dutch Heart Foundation (CVON SHE‐PREDICTS‐HF; grant 2017‐21; CVON RED‐CVD; grant 2017‐11; CVON PREDICT2; grant 2018‐30; CVON DOUBLE DOSE; grant 2020B005), by a grant from the Leducq Foundation Cure PhosphoLambaN induced Cardiomyopathy (Cure‐PLaN), and by a grant from the European Research Council (ERC CoG 818715; SECRETE‐HF). Canxia Shi is supported by a scholarship from the China Scholarship Council (CSC number: 201806170057).
Conflict of interest: The UMCG, which employs Dr. de Boer has received research grants and/or fees from AstraZeneca, Abbott, Boehringer Ingelheim, Cardior Pharmaceuticals Gmbh, Ionis Pharmaceuticals, Inc., Novo Nordisk, and Roche. Dr. de Boer received speaker fees from Abbott, AstraZeneca, Bayer, Novartis, and Roche. All other authors have nothing to disclose.
References
- 1. Martincorena I, Campbell PJ. Somatic mutation in cancer and normal cells. Science. 2015;349:1483–9. [DOI] [PubMed] [Google Scholar]
- 2. Yizhak K, Aguet F, Kim J, Hess JM, Kübler K, Grimsby J, et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science. 2019;364:eaaw0726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age‐related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Libby P, Ebert BL. CHIP (clonal hematopoiesis of indeterminate potential): potent and newly recognized contributor to cardiovascular risk. Circulation. 2018;138:666–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Calvillo‐Argüelles O, Jaiswal S, Shlush LI, Moslehi JJ, Schimmer A, Barac A, et al. Connections between clonal hematopoiesis, cardiovascular disease, and cancer: a review. JAMA Cardiol. 2019;4:380–7. [DOI] [PubMed] [Google Scholar]
- 7. Saiki R, Momozawa Y, Nannya Y, Nakagawa MM, Ochi Y, Yoshizato T, et al. Combined landscape of single‐nucleotide variants and copy number alterations in clonal hematopoiesis. Nat Med. 2021;27:1239–49. [DOI] [PubMed] [Google Scholar]
- 8. Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377:111–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bhattacharya R, Zekavat SM, Haessler J, Fornage M, Raffield L, Uddin MM, et al. Clonal hematopoiesis is associated with higher risk of stroke. Stroke. 2022;53:788–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355:842–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sano S, Oshima K, Wang Y, MacLauchlan S, Katanasaka Y, Sano M, et al. Tet2‐mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL‐1β/NLRP3 inflammasome. J Am Coll Cardiol. 2018;71:875–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. de Boer RA, Hulot JS, Tocchetti CG, Aboumsallem JP, Ameri P, Anker SD, et al. Common mechanistic pathways in cancer and heart failure. A scientific roadmap on behalf of the Translational Research Committee of the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). Eur J Heart Fail. 2020;22:2272–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Aboumsallem JP, Moslehi J, de Boer RA. Reverse cardio‐oncology: cancer development in patients with cardiovascular disease. J Am Heart Assoc. 2020;9:e013754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Koelwyn GJ, Aboumsallem JP, Moore KJ, de Boer RA. Reverse cardio‐oncology: exploring the effects of cardiovascular disease on cancer pathogenesis. J Mol Cell Cardiol. 2022;163:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dorsheimer L, Assmus B, Rasper T, Ortmann CA, Ecke A, Abou‐El‐Ardat K, et al. Association of mutations contributing to clonal hematopoiesis with prognosis in chronic ischemic heart failure. JAMA Cardiol. 2019;4:25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Abplanalp WT, Mas‐Peiro S, Cremer S, John D, Dimmeler S, Zeiher AM. Association of clonal hematopoiesis of indeterminate potential with inflammatory gene expression in patients with severe degenerative aortic valve stenosis or chronic postischemic heart failure. JAMA Cardiol. 2020;5:1170–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pascual‐Figal DA, Bayes‐Genis A, Díez‐Díez M, Hernández‐Vicente Á, Vázquez‐Andrés D, de la Barrera J, et al. Clonal hematopoiesis and risk of progression of heart failure with reduced left ventricular ejection fraction. J Am Coll Cardiol. 2021;77:1747–59. [DOI] [PubMed] [Google Scholar]
- 18. Yu B, Roberts MB, Raffield LM, Zekavat SM, Nguyen NQH, Biggs ML, et al. Supplemental association of clonal hematopoiesis with incident heart failure. J Am Coll Cardiol. 2021;78:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pinto‐Sietsma SJ, Janssen WM, Hillege HL, Navis G, De Zeeuw D, De Jong PE. Urinary albumin excretion is associated with renal functional abnormalities in a nondiabetic population. J Am Soc Nephrol. 2000;11:1882–8. [DOI] [PubMed] [Google Scholar]
- 20. Brouwers FP, de Boer RA, van der Harst P, Voors AA, Gansevoort RT, Bakker SJ, et al. Incidence and epidemiology of new onset heart failure with preserved vs. reduced ejection fraction in a community‐based cohort: 11‐year follow‐up of PREVEND. Eur Heart J. 2013;34:1424–31. [DOI] [PubMed] [Google Scholar]
- 21. Suthahar N, Meijers WC, Ho JE, Gansevoort RT, Voors AA, van der Meer P, et al. Sex‐specific associations of obesity and N‐terminal pro‐B‐type natriuretic peptide levels in the general population. Eur J Heart Fail. 2018;20:1205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van Zeventer IA, de Graaf AO, Wouters H, van der Reijden BA, van der Klauw MM, de Witte T, et al. Mutational spectrum and dynamics of clonal hematopoiesis in anemia of older individuals. Blood. 2020;135:1161–70. [DOI] [PubMed] [Google Scholar]
- 23. Neveling K, Mensenkamp AR, Derks R, Kwint M, Ouchene H, Steehouwer M, et al. BRCA testing by single‐molecule molecular inversion probes. Clin Chem. 2017;63:503–12. [DOI] [PubMed] [Google Scholar]
- 24. Brouwers FP, van Gilst WH, Damman K, van den Berg MP, Gansevoort RT, Bakker SJ, et al. Clinical risk stratification optimizes value of biomarkers to predict new‐onset heart failure in a community‐based cohort. Circ Heart Fail. 2014;7:723–31. [DOI] [PubMed] [Google Scholar]
- 25. de Boer RA, Nayor M, de Filippi CR, Enserro D, Bhambhani V, Kizer JR, et al. Association of cardiovascular biomarkers with incident heart failure with preserved and reduced ejection fraction. JAMA Cardiol. 2018;3:215–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Suthahar N, Lau ES, Blaha MJ, Paniagua SM, Larson MG, Psaty BM, et al. Sex‐specific associations of cardiovascular risk factors and biomarkers with incident heart failure. J Am Coll Cardiol. 2020;76:1455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bick AG, Weinstock JS, Nandakumar SK, Fulco CP, Bao EL, Zekavat SM, et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature. 2020;586:763–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sano S, Oshima K, Wang Y, Katanasaka Y, Sano M, Walsh K. CRISPR‐mediated gene editing to assess the roles of Tet2 and Dnmt3a in clonal hematopoiesis and cardiovascular disease. Circ Res. 2018;123:335–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tromp J, Paniagua SMA, Lau ES, Allen NB, Blaha MJ, Gansevoort RT, et al. Age dependent associations of risk factors with heart failure: pooled population based cohort study. BMJ. 2021;372:n461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rapsomaniki E, Timmis A, George J, Pujades‐Rodriguez M, Shah AD, Denaxas S, et al. Blood pressure and incidence of twelve cardiovascular diseases: lifetime risks, healthy life‐years lost, and age‐specific associations in 1.25 million people. Lancet. 2014;383:1899–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sattar N, Rawshani A, Franzén S, Rawshani A, Svensson AM, Rosengren A, et al. Age at diagnosis of type 2 diabetes mellitus and associations with cardiovascular and mortality risks. Circulation. 2019;139:2228–37. [DOI] [PubMed] [Google Scholar]
- 32. Sano S, Wang Y, Yura Y, Sano M, Oshima K, Yang Y, et al. JAK2 (V617F)‐mediated clonal hematopoiesis accelerates pathological remodeling in murine heart failure. JACC Basic Transl Sci. 2019;4:684–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fish‐Trotter H, Ferguson JF, Patel N, Arora P, Allen NB, Bachmann KN, et al. Inflammation and circulating natriuretic peptide levels. Circ Heart Fail. 2020;13:e006570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jensen J, Ma LP, Fu ML, Svaninger D, Lundberg PA, Hammarsten O. Inflammation increases NT‐proBNP and the NT‐proBNP/BNP ratio. Clin Res Cardiol. 2010;99:445–52. [DOI] [PubMed] [Google Scholar]
- 35. de Bold AJ. Cardiac natriuretic peptides gene expression and secretion in inflammation. J Invest Med. 2009;57:29–32. [DOI] [PubMed] [Google Scholar]
- 36. Di Somma S, Pittoni V, Raffa S, Magrini L, Gagliano G, Marino R, et al. IL‐18 stimulates B‐type natriuretic peptide synthesis by cardiomyocytes in vitro and its plasma levels correlate with B‐type natriuretic peptide in non‐overloaded acute heart failure patients. Eur Heart J Acute Cardiovasc Care. 2017;6:450–61. [DOI] [PubMed] [Google Scholar]
- 37. Koene RJ, Prizment AE, Blaes A, Konety SH. Shared risk factors in cardiovascular disease and cancer. Circulation. 2016;133:1104–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al.; CANTOS Trial Group . Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–31. [DOI] [PubMed] [Google Scholar]
- 39. Everett BM, Cornel JH, Lainscak M, Anker SD, Abbate A, Thuren T, et al. Anti‐inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation. 2019;139:1289–99. [DOI] [PubMed] [Google Scholar]
- 40. Svensson EC, Madar A, Campbell CD, He Y, Sultan M, Healey ML, et al. TET2‐driven clonal hematopoiesis and response to canakinumab: an exploratory analysis of the CANTOS randomized clinical trial. JAMA Cardiol. 2022;7:521–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Assmus B, Cremer S, Kirschbaum K, Culmann D, Kiefer K, Dorsheimer L, et al. Clonal haematopoiesis in chronic ischaemic heart failure: prognostic role of clone size for DNMT3A‐ and TET2‐driver gene mutations. Eur Heart J. 2021;42:257–65. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting information.