

Abstract
Polynucleotides, DNA and RNA (mRNA and non‐coding RNAs) are critically involved in the molecular pathways of disease. Small molecule binding interactions with polynucleotides can modify functional polynucleotide topologies and/or their interactions with proteins. Current approaches to library design (lead‐like or fragment‐like libraries) are based on protein‐ligand interactions and often include careful consideration of the 3‐dimensional orientation of binding motifs and exclude π‐rich compounds (polyfused aromatics) to avoid off‐target R/DNA interactions. In contrast to proteins, where π,π‐interactions are weak, polynucleotides can form strong π,π‐interactions with suitable π‐rich ligands. To assist in designing a polynucleotide‐biased library, a scaffold‐divergent synthesis approach to polyfused aromatic scaffolds has been undertaken. Initial screening hits that form moderately stable polynucleotide‐ligand‐protein ternary complexes can be further optimized through judicious incorporation of substituents on the scaffold to increase protein‐ligand interactions. An example of this approach is given for topoisomerase‐1 (TOP1), generating a novel TOP1 inhibitory chemotype.
Keywords: alkynes, electrophilic cyclization, polynucleotides, RNA/DNA, topoisomerase
The targeting of polynucleotide (RNA and DNA) topologies and their protein complexes by small molecules has enormous potential in the discovery of new therapeutics across a broad range of diseases (infectious, chronic and congenital). Polynucleotides are very different structures to proteins and have a much greater capacity to form strong π,π‐interactions with suitably π‐rich small molecules. To exploit this difference, we have developed a diversity‐oriented synthesis approach to π‐rich scaffolds from a common set of substrates. The utility of this approach has been exemplified in the discovery of a novel topoisomerase‐1 inhibitor.
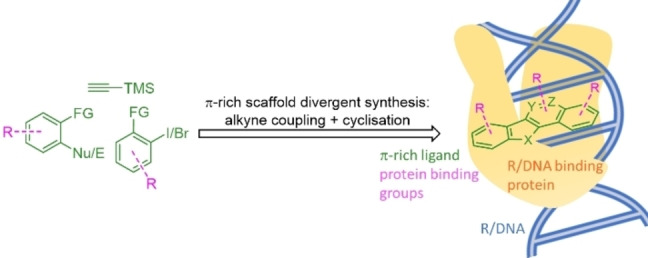
Introduction
Polynucleotides, DNA and RNA, play critical roles in protein expression and are involved in effectively all molecular pathways of disease.[ 1 , 2 , 3 , 4 , 5 , 6 , 7 ] While most small‐molecule drug discovery efforts are directed to the design of ligands for the encoded protein products of DNA and RNA, significant potential lies in the direct targeting of polynucleotides and the protein‐polynucleotide complexes involved in the decoding process (transcription and translation) and/or in epigenetic modifications to the code.[ 1 , 2 , 3 , 4 , 5 , 6 , 7 ] Over the last twenty years, diversity‐oriented synthesis (DOS) and fragment‐based drug discovery (FBDD) have emerged as successful methods for accessing suitable screening sets for phenotypic and target‐based drug discovery.[ 8 , 9 , 10 , 11 , 12 , 13 ] The library design principles employed in these DOS and FBDD efforts, such as fraction‐sp3 (Fsp3) and lead‐likeness, have been principally developed with protein targets in mind.[ 8 , 9 , 10 , 11 , 12 , 13 ] In contrast to proteins, where π,π‐interactions are weak, the binding of small molecules to polynucleotides often involves strong π,π‐interactions, favoring sp2‐rich molecules.[ 1 , 14 , 15 ] This is reflected in nature, where a diverse array of sp2‐rich bioactive secondary metabolites has been identified that make strong π,π‐interactions with polynucleotides, for example, DNA intercalators camptothecin 1 and berberine 2 (Figure 1).[ 16 , 17 , 18 ] Natural products 1 and 2 and their synthetic analogues, such as ARC111 3 and indenoisoquinolines LMP744 4, target DNA‐topoisomerase I (TOP1) cleavage complexes (TOP1ccs), disrupting DNA replication and transcription.[ 16 , 17 , 18 , 19 , 20 , 21 ] Transcriptional modification has also been achieved through the targeting of other DNA‐protein complexes (e. g., DNA complexes with transcription factors, RNA polymerases and epigenetic modulators) or of functional DNA topologies (e. g., Z‐DNA and G‐quadraplexes).[ 22 , 23 , 24 , 25 , 26 , 27 ] These DNA‐small molecule binding events can lead to changes in the expression of mRNAs and of non‐coding RNAs (e. g., micro‐RNAs), leading to down‐stream changes in protein expression and cellular phenotype. Direct targeting of RNAs with small molecules is also an area of intense interest. [28] A notable example is the recently approved drug for spinal muscular atrophy, risdiplam 5, that binds to the mRNA encoding the dysfunctional survival motor neuron 2 (SMN2) protein and promotes read‐through of a stop codon to give more functional SMN protein.[ 29 , 30 ] Another example is the screening hit 6, which selectively binds to a G‐quadruplex within the mRNA encoding the oncogenic N‐Ras protein, suppressing its translation. [31] These and the many other examples of sp2‐rich compounds targeting polynucleotides indicate that DOS approaches directed to diverse sets of sp2‐rich scaffolds could prove useful in the discovery of new therapies based on targeting polynucleotides (DNA, mRNA, micro‐RNA and other non‐coding RNAs).
Figure 1.

Polynucleotide targeting agents.
In this study, we describe a scaffold‐divergent approach to heteroacenes based on electrophilic cyclization of alkynes (Scheme 1A). The divergent methods employed in this work are complemented by several recent studies that we have undertaken to attain other heteroacene scaffolds from the same substrates (Scheme 1B).[ 32 , 33 , 34 , 35 ] By analogy with “fragment‐growth” approaches in FBDD,[ 36 , 37 , 38 , 39 , 40 ] we anticipate that by initially establishing positive π,π‐interactions with nucleotide bases, the polynucleotide binding fragment can be “grown” through SAR‐ and/or structure‐guided approaches to make additional interactions with a protein binding partner (Scheme 1C). To exemplify this possibility we have biased our new scaffolds towards TOP1 inhibitory activity through incorporation of N,N‐dimethylaminoethylene group [Scheme 1A, R=−(CH2)2NMe2] present in a number of non‐camptothecin TOP1 inhibitors, such as 3 and have identified a novel TOP1 inhibitor class. [19] Moreover, we anticipate this DOS method could be employed in the discovery of new polynucleotide targeting agents with novel modes of action.
Scheme 1.

Scaffold‐morphing approach to access sp2‐rich scaffolds.[ 32 , 33 , 34 , 35 ]
Results and Discussion
The electrophilic cyclization of alkynes has emerged as a functional group tolerant method of synthesis for a range of aromatic heterocycles and carbocycles.[ 41 , 42 , 43 , 44 , 45 , 46 , 47 , 48 ] In this work, we have sought to achieve a diversification of a discrete set of substrates by modifying the nature of the nucleophile (Nu) or electrophile (E) and X (X=halide, amide, or ester) (Scheme 1A). For reactions proceeding through diazonium and nitrilium intermediates one‐step bicyclization methods have been developed (Scheme 1). For those proceeding through a dihalide intermediate (I/Br or Br/Br), second ring closure can potentially be achieved through various methods.[ 49 , 50 , 51 ] In this work we have employed an Ullmann coupling cyclization (UCC) 7→8 and a Pd‐mediated carboxyamidation cyclization (PdCC) sequence 7→9 a,b→10 a,b (Schemes 1 and 2). The PdCC can be achieved in a single operation using catalytic Pd(OAc)2 and CO(g) (PdCC1), however, in cases where this stalls at the amide 9 a,b, Ullmann conditions (UCC) are employed to complete cyclization to 10 a,b (PdCC2). The regioselectivity of this process can be controlled based on the relative reactivity of I and Br to Pd‐insertion, i. e., 7 (X=I, Y=Br) gives lactam 10 a and 7 (X=Br, Y=I) gives lactam 10 b. In this work, all UCC and PdCC reactions have been performed with 1,1‐dimethylethylenediamine (DMD), to give 8–10 R=(CH2)2NMe2, so as to bias the product towards TOP1 inhibition. This R‐group can be further diversified in a broader screening set.
Scheme 2.

Late‐stage ring closure of dihalides 7. Ullmann coupling cyclization (UCC): R‐NH2, CuI 20–40 mol %, K3PO4, n‐BuOH, ethylene glycol. Pd‐mediated carboxyamidation cyclization‐1 (PdCC1): R‐NH2, Pd(OAc)2 10 mol %, PPh3, CO(g), Et3N, NMP. Pd‐mediated carboxyamidation cyclization‐2 (PdCC2): as for PdCC1 then UCC
Our first series of heteroacenes incorporated a strategy to optionally diversify the positioning of a carbonyl atom in related scaffolds 16 a,b, 17 a,b, 19 a,b, 23 and 27 (Scheme 3, Part A). Sequential Sonogashira coupling of terminal alkynes 12 a,b (accessed from 11 a,b) with either 1,2‐diiodobenzene or 1‐bromo‐2‐iodobenzene furnished substrates 13 a,b and 14 a,b in good to excellent yields (64 %–100 %). Iodocyclization of bromides 13 a,b with molecular iodine furnished iodo‐bromo compounds 15 a,b (78 %–95 %). The bromocyclization of iodides 14 a,b required greater experimentation, though the best yields were obtained using CuBr2 for the methylsulfide 14 a and N‐methylpyrrolidin‐2‐one hydrotribromide (MPHT) for the methyl ether 14 b to give corresponding bromo‐iodo compounds 18 a,b (42 %–81 %). [52] UCC and PdCC1/2 of 15 a,b with DMD gave pyrroles 16 a,b (29 %–42 %) and lactams 17 a,b (43 %–46 %), respectively. Attempted formation of the regioisomeric lactams 19 a,b through PdCC2 of 18 a,b with DMD was successful for the thiopheno system 18 a→19 a (63 %) but stalled at the amide stage for furano system 18 b (amide not shown), which could not be ring‐closed to 19 b, reflecting a limitation in the method for scaffold 19 (Nu=O).
Scheme 3.

Preparation of 16 a,b, 17 a,b, 19 a,b, 23, 27, 30 and 35 [53]
Further transposing of the carbonyl was achieved in the synthesis of scaffold analogues 23 and 27 (attempted for Nu=SMe only). For 23 this involved reaction of lithiated alkyne 12 a with Weinreb amide 20 to give propynone 21 (71 %), which underwent efficient iodocyclization to 22 (100 %) and UCC with DMD to give 23 (36 %) in modest yield. [53] For 27, reaction of lithiated 11 a (Li for I exchange) with propynamide 24 afforded propynone 25 (71 %), that underwent iodocyclization to 26 (52 %) and UCC with DMD to give 27 (78 %). These syntheses required two recent innovations in iodocyclization chemistry.[ 53 , 54 ] Firstly, the iodocyclization of alkynes with unfavourable electronic bias 21→22, using high iodine concentrations at elevated temperatures.[ 53 , 54 ] Secondly, endo/exo control in the iodocyclization of 25, where more polar iodonium sources (ICl in CH3CN) favour 6‐endo iodocyclization and iodine in CH2Cl2 favors 5‐exo cyclization. [54] It should be noted that the iodocyclization of alkynes bearing the carbonyl on the carbon undergoing the nucleophilic attack, as in 21→22, cannot be achieved for furans and indoles (i. e., where SMe is replaced with OMe and NMe2). [53] However, 6‐endo iodocyclization is highly favoured related substrates to 25, where SMe is replaced with OMe or NMe2,[ 55 , 56 ] suggesting plausible access to chromanone and quinolone equivalents to 27.
In Scheme 3(Part B), we exemplified two other modes of divergent heteroacene synthesis. Firstly, the 1,2‐dihalobenzene used to access 13–14 a,b can be replaced with 2,3‐dibromothiophene (and potentially other dihaloheterocycles) to progress through the Sonogashira coupling (28, 58 %), iodocyclization (29, 97 %) and PdCC2 sequence to give the thiophene analogue of 17 a, 30 (44 %). In the second example, another latent nucleophile (SMe) is introduced onto the alkyne 12 a to give 31 (96 %), which enables a sequence of iodocyclization (32, 91 %), Sonogashira coupling and iodocyclization (34, 48 %), followed by PdCC2 to give 35 (57 %). [35]
We next investigated the construction of a series of equivalent pyridyl analogues 40, 41, and 43 (Scheme 4, Part A). This approach centered on the halocyclization of imines 38 a,b. The synthesis of 38 a,b involved Sonogashira coupling of 2‐iodobenzaldehyde 36 with bromoethynylbenzene 33 to give 37 (92 %) followed by Schiff base condensation with MeONH2 (Method A) to give 38 a (94 %) or t‐BuNH2 (Method B) to give 38 b (not isolated). Bromocyclization of 38 a was achieved using the method previously described by Yu et al. [57] employing CuBr2 in DMA at 100 °C, giving 39 a (34 %). The yield of this reaction was limited by a competing oxidative‐cyclization to give lactam 39 b (44 %) as the major by‐product. [58] Oxime 38 a could not be iodocyclized, though the corresponding t‐Bu‐aldimine 38 b could be by employing ICl in CH3CN with a weak base (NaOAc) to give product 42 (51 %). [59] UCC of dibromide 39 a with DMD gave the heterotetracene 40 (53 %). PdCC1 of dibromide 39 a with DMD proved surprisingly regioselective, favoring lactam 41 (44 %) as the major product (no regioisomeric lactam could be detected). [60] A possible explanation for this regioselectivity is that under the thermal reaction conditions (80 °C in N‐methyl‐2‐pyrrolidone) nucleophilic aromatic substitution of the bromo group on the isoquinoline precedes Pd‐mediated carbonylative ring closure onto the bromophenyl ring. This regioselectivity is reversed in the PdCC2 of the iodobromo substrate 42 with DMD, giving 43 (45 %). In this case, Pd‐mediated carboxyamidation with DMD precedes ring closure onto the bromophenyl, in a separate UCC step.
Scheme 4.

Preparation of 40, 41, 43, 48, 49, and 51. [53]
In Scheme 4(Part B), alkyne 12 a was converted into 3‐iodobenzo[b]thiophene‐2‐carbaldehyde (44) by formylation and iodocyclization. Iodoaldehyde 44 was then subject to a related series of reactions to those used in Part A to generate a series of thiopheno‐fused systems 48, 49 and 51. [53]
In earlier work, we had demonstrated the utility of triazenes to operate as masked diazoniums that could be unmasked by acid in the presence of a nucleophile Nu (tethered or untethered) to give a cinnoline (Scheme 5 Box). [34] In this study, we exploited this chemistry in the rapid assembly of a series of cinnolines 56 a–d from 2‐iodoaniline 52 (Scheme 5). Terminal alkyne 53 was prepared in three steps, involving diazotisation and triazene formation, followed by Sonogashira coupling with TMS‐acetylene and deprotection. A Cu‐free Sonogashira coupling was employed to couple alkyne 53 to iodobenzenes 54 a–d, giving tolans 55 a–d (42 %–96 %). Treatment of tolans 55 a–c with MeSO3H unmasked the diazonium cation and induced electrophilic co‐cyclization to give 56 a–c. Treatment of the ester 55 d with MeSO3H in the presence of tetraethylammonium chloride gave a chlorocinnoline 57 (unpurified). Reaction of 57 with DMD at elevated temperature afforded 56 d (a previously described TOP1 inhibitor) [61] through a domino nucleophilic aromatic substitution/lactamization sequence in excellent yield (95 %).
Scheme 5.

Preparation of 56 a–d [34]
Given the success of the diazonium cyclizations to give cinnolines, we proposed to explore the related cyclization on nitrilium ion 62 to give 63 and 64 (Scheme 6). Sonogashira coupling of 2‐iodophenylformamide 58 to alkynes 59 and 33 gave tolans 60 a and 60 b, respectively (66–67 %). Reaction of 60 a with Burgess reagent and of 60 b with POCl3 and diisopropylethylamine (DIPEA) gave rise to the isonitriles 61 a and 61 b, respectively. Both isocyanides 61 a,b were stable in solution (1H NMR), but reverted to the formamides 60 a,b upon attempted extractive work up, consequently, they were not isolated but used directly in the next reaction. Attempted protonation and cyclization of 61 a and 61 b to quinolines 63 and 64 respectively, via nitrilium ion 62 failed. Rather, 61 a gave the regioisomeric quinoline 67 (21 % from 60 a) and 61 b reverted to the formamide 60 b. Bromocyclization of 61 b to give 69 (72 %) was achieved upon addition of n‐Bu4N.Br without acid, in a process previously described by Mitamura et al. [62] This involves nucleophilic cyclization of a bromide adduct ion 68 with concomitant protonation by residual diisopropylethylammonium ion (from isonitrile formation). Ring closure of dibromide 69 under UCC and PdCC2 conditions gave 70 (52 %) and 71 (32 %), respectively.
Scheme 6.

Preparation of 67, 70 and 71.
Finally, since 19 a (Scheme 3) proved to be active as a TOP1 inhibitor (see below), we also prepared an analogue 77 (Scheme 7) that bears the additional TOP1 protein binding methoxy and methylenedioxy groups seen in 3 and 4 (Scheme 1). [17] Sonogashira coupling of aryliodide 72 and arylalkyne 73 afforded tolan 74 (89 %). Iodocyclization of 74 proceeded chemoselectively through the methylsulfide (and not the ester) to give benzo[b]thiophene 75 (94 %). The ester was efficiently converted to the amide 76 (92 % over 3 steps) and cyclized under Buchwald‐Hartwig conditions to furnish the target compound 77 (51 %).
Scheme 7.

Preparation of fully decorated compound 77.
Topoisomerase I inhibitory activity
TOP1 plays a key role modifying and maintaining DNA topology during cellular replication and transcription.[ 17 , 63 ] TOP1 inhibitors, such as 1–4, exert their cytotoxic effect on cancer cells by binding to TOP1/DNA cleavage complexes (TOP1cc), forming stable ternary complexes that collide with replication forks leading to DNA damage and apoptosis.[ 17 , 21 ] TOP1 inhibitors also influence transcription, for example, in hypoxic cancer cells compounds 1 and 2 selectively suppress the expression of hypoxia inducible factor HIF‐1α, which is a driver of tumour progression.[ 64 , 65 , 66 ] In this scenario, inhibition of TOP1 increases in the transcription of micro‐RNAs, miR‐17‐5p and miR‐155, that promote selective degradation of HIF‐1α mRNA. [65]
While the purpose of this study has been to develop a DOS of heteroacenes using electrophilic alkyne activation, several scaffolds generated in this work are reminiscent of DNA intercalators that inhibit TOP1, such as 2–4 (Figure 1).[ 17 , 67 ] To further bias these scaffolds to interact with TOP1cc we included the N,N‐dimethylaminoethylene group in ARC111 to many of the synthesised compounds.
All new scaffolds (Figure 2) were tested for TOP1 inhibition at 100, 10, 1, and 0.1 μM in a TOP1‐mediated DNA cleavage assay. [68] This assay uses 3’‐radiolabeled DNA substrates to identify compounds that stabilise TOP1ccs. Active TOP1 inhibitors were also tested for cytotoxicity towards prostate cancer PC3 cells. Two of the scaffolds tested, 78 and 79, were generated using our previously described double‐electrophilic cyclization chemistry, which complements this work (Scheme 1Bii).[ 32 , 33 ] Of all the scaffolds studied only 19 a and 56 d showed significant TOP1 inhibition in a dose‐dependent manner (Figure 3). [69]
Figure 2.

Compounds evaluated for TOP1 activity. Active compounds from this work shown in blue (see also Figure 3), known actives shown in brown.[ 19 , 21 , 32 , 33 , 72 ]
Figure 3.
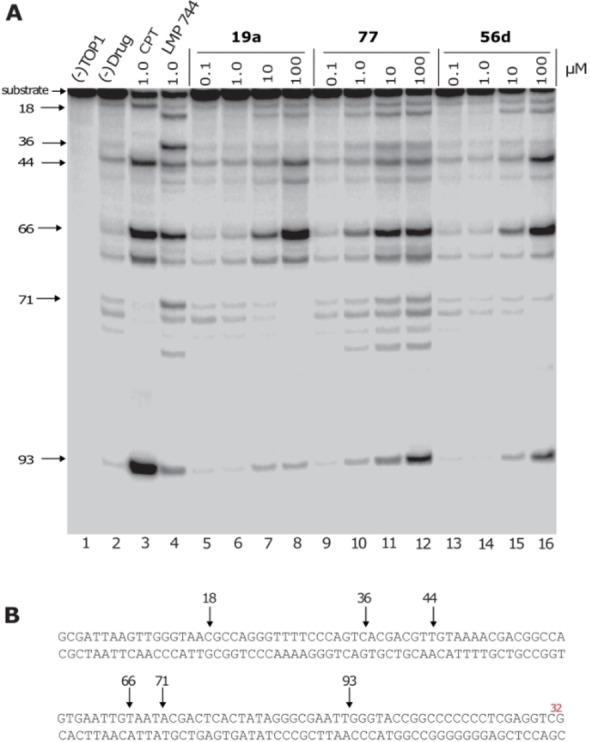
A. Representative gel of the TOP1‐mediated DNA cleavage assay. From left to right: Lane 1, DNA alone; lane 2, DNA and TOP1 without drug; lane 3, DNA and TOP1 with CPT (1 μM); lane 4, DNA and TOP1 with LMP744 (1 μM); lanes 5–16, DNA and TOP1 with the tested compounds at 0.1, 1.0, 10, and 100 μM concentrations, respectively. The arrows and numbers at left indicate the cleavage site positions. LMP744 is the positive non‐camptothecin indenoisoquinoline control. B. Sequence of the 3′‐[32P]‐labelled 117‐bp DNA (labeled Guanine in red) with the indicated TOP1 cleavage site positions. [68]
Aspects of the scaffold SAR are quite steep. For example, replacement of one nitrogen in cinnoline 56 d for a CH‐group in isoquinoline 41 led to a complete loss in activity, as did a swap in the location to the carbonyl in 19 a vs. 17 a. The carbonyl and ring heteroatoms of other related TOP1 inhibitors are known to facilitate protein‐binding within the TOP1cc. Crystal structures of camptothecin and non‐camptothecin ligands bound to the TOP1cc reflect a similar scenario to that depicted in Scheme 1C. where the π‐rich TOP1 inhibitor is sandwiched (intercalated) between two sets of DNA base pairs through π,π‐interactions and the ring‐heteroatoms, carbonyls and other substituents at the “edge” of this sandwich make important interactions with the TOP1 protein amino‐acid sidechains.[ 70 , 71 ] Accordingly, while the polyaromatic core of 56 d, 41, 19 a and 17 a makes important π,π‐interactions with DNA in the Top1cc, small differences in the location of the edge‐groups has significant impact on overall stability of the ternary complex and associated potency. Other substituents attached to this core also impact potency, presumably through ligand‐protein interactions. The inactivity of 56 c compared to 56 d suggests that the ethylene linked dimethylamino group may also make important protein interactions in the ternary TOP1cc, as do the methylenedioxy and/or methoxy groups present in 3, which is approximately 10‐fold more potent than 19 a in terms of TOP1 inhibition. [70] The relative potency of 19 a, 56 d, and 77 as TOP1 inhibitors is reflected in their inhibition of the PC3 cancer cell growth: 19 a≈56b<77 (Figure 4).
Figure 4.

PC3 cell viability was measured using an MTS colorimetric assay at eight concentrations (10−8–10−4 M, n=3 per concentration) and is given as the concentration required to inhibit 50 % of cell growth (IC50). [73]
Conclusion
A scaffold‐divergent synthesis strategy for the generation of a sp2‐rich polynucleotide‐biased fragment library has been devised based on the electrophilic cyclization of alkynes (Scheme 2). Scaffold modifications include the use of intermolecular and intramolecular electrophiles and variations in the nature of the second (dihalide) ring closure (Schemes 2, 3, 4, 5, 6). The iterative use of halocyclization further extends the range heteroacene scaffolds that can be accessed (Scheme 3 Part B and Scheme 4 Part B). These methods are yet further complemented by our other heteroacene syntheses using electrophilic cyclization (Scheme 1B).[ 32 , 33 , 34 , 35 ] The methods are also applicable to the generation of more substituted systems for further library diversification and/or lead optimisation (Scheme 7). The small library of scaffolds generated to date has proven useful in identifying novel TOP1 inhibitors, targeting the TOP1cc. Our group is currently engaged in further characterizing the interactive capacity of the new scaffolds for polynucleotides and further diversifying the library for target‐based and phenotypic screening.
Experimental Section
General procedure A (Sonogashira coupling)
For the synthesis of alkynes 13 a,b, 14 a,b, 28, 37, 45, 60 a,b, and 74: The respective 2‐iodobenzene was dissolved in Et3N (0.2 M) in a dry round‐bottom flask (RBF), followed by addition of CuI (4–6 mol %) and Pd(PPh3)2Cl2 (2–3 mol %). The RBF was then degassed and backfilled with N2(g) three times. Finally, a solution of the terminal alkyne (1.2 equiv.) in Et3N (1 M) was added dropwise under an N2(g) atmosphere. The reaction mixture was stirred at rt to 60 °C overnight. On completion, the suspension was filtered through Celite® and rinsed with Et2O. The organic extract was washed with H2O twice and with brine twice, dried over anhydrous MgSO4, filtered and concentrated under reduced pressure. The crude product obtained was purified by flash column chromatography to yield the desired alkyne.
General procedure B (Cu‐free Sonogashira coupling)
For the synthesis of alkynes 55 a–d: 54 a–d was dissolved in pyrrolidine (0.5 M), followed by addition of Pd(PPh3)4 (5 mol %). The RBF was degassed and backfilled with N2(g) for three times. Finally, a solution of 53 (1.5 equiv.) in pyrrolidine (3 M) was added dropwise under N2(g) atmosphere. The reaction mixture was heated at 60 °C for 4–16 h. On completion, the suspension was filtered through Celite® and rinsed with EtOAc. The organic extract was washed with H2O three times, dried over anhydrous MgSO4, filtered and concentrated under reduced pressure. The crude product obtained was purified by flash column chromatography (1 : 1 hexanes:EtOAc) to yield the desired alkynes 55 a–d.
General procedure C (Sonogashira Coupling‐Desilylation)
For the synthesis of alkynes 53, 59, and 73: The respective 2‐iodobenzene was dissolved in Et3N (0.2 M) in a dry round‐bottom flask (RBF), followed by addition of CuI (4–6 mol %) and Pd(PPh3)2Cl2 (2–3 mol %). The RBF was then degassed and backfilled with N2(g) three times. Finally, trimethylsilylacetylene (1.2 equiv.) was added dropwise under an N2(g) atmosphere. The reaction mixture was stirred at rt overnight. On completion, the suspension was filtered through Celite® and extracted with Et2O twice and washed with H2O twice and with brine twice. The combined organic extracts were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by a silica plug (100 % hexanes) to yield the TMS‐protected terminal alkyne, which was then dissolved in MeOH/Et2O (2 : 1, 0.2 M), followed by addition of K2CO3 (1–2 equiv.). The reaction mixture was stirred at rt overnight. On completion, the mixture was concentrated to a residue, taken up in Et2O, washed with H2O twice and with brine twice. The organic extract was dried over anhydrous MgSO4, filtered, and concentrated to yield the desired terminal alkyne, which was directly used in the next step without further purification.
General procedure D (Iodocyclization)
For the synthesis of iodides 15 a,b, 22, 29, 34, 44, 75: I2 (1.2–3 equiv.) was added to a stirred solution of the respective alkyne substrate in dry CH2Cl2 (0.2 M) under an N2(g) atmosphere. The reaction mixture was stirred at rt for 1–18 h. On completion, the reaction mixture was quenched with saturated Na2S2O3 solution and extracted with CH2Cl2 twice. The combined organic extracts were washed with H2O twice and with brine twice, dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to yield the desired iodocyclized product.
General UCC procedure
For the final ring closure of 15 a,b, 22, 26, 39 a, 47, 69: In a dry RBF, the respective dihalide was dissolved in dry n‐butanol or DMF (0.1–0.2 M). K3PO4 (4 equiv.), ethylene glycol (12 equiv.), 1,1‐dimethylethane‐1,2‐diamine (DMD) (15 equiv.) and CuI (10–40 mol %) were added sequentially into the flask. The RBF was degassed and backfilled with N2(g) three times, and the reaction mixture was heated at 80–110 °C. On completion, the reaction mixture was cooled down to rt, quenched with saturated NH4Cl solution and extracted with EtOAc twice. The combined organic extracts were washed with H2O three times and with brine twice, dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude product obtained was purified by flash column chromatography to yield the desired alkyne.
General PdCC1 procedure
For the final ring closure of 15 a, 39 a, 47: The respective dihalide, Pd(OAc)2 (10 mol %), CuI (10 mol %), PPh3 (1.5 equiv.), DMD (15 equiv.), Et3N (2 equiv.) and dry NMP (0.1–0.15 M) was added to a dry RBF. The RBF was degassed and backfilled with CO(g) for three times, the reaction mixture was then heated at 80 °C for 15–49 h under CO(g) atmosphere. On completion, the reaction mixture was cooled down to rt, quenched with saturated NH4Cl solution and extracted with EtOAc twice. The combined organic extracts were washed with H2O three times and with brine twice, dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude product obtained was purified by flash column chromatography to yield the desired final product.
General PdCC2 procedure
For the final ring closure of 15 b, 18 a, 29, 34, 42, 50, 69: Step 1: use General PdCC1 Procedure to form the secondary amide; step 2: use a modified UCC Procedure to close the ring and form final products (use N,N,N′,N′‐tetramethylethane‐1,2‐diamine (TMD) in lieu of DMD).
Supporting Information
TOP1‐mediated DNA cleavage assay gel of tested compounds, PC3 cell viability assays, Experimental procedures and characterization data, 1H and 13C NMR Spectral Data
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
Open Access publishing facilitated by Monash University, as part of the Wiley ‐ Monash University agreement via the Council of Australian University Librarians.
Chen S., Priebbenow D. L., Somkhit J., Scullino C. V., Agama K., Pommier Y., Flynn B. L., Chem. Eur. J. 2022, 28, e202201925.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Sheng J., Gan J., Huang Z., Med. Res. Rev. 2013, 33, 1119–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Breaker R. R., Joyce G. F., Chem. Biol. 2014, 21, 1059–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Connelly C. M., Moon M. H., Schneekloth J. S., Cell Chem. Biol. 2016, 23, 1077–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Warner K. D., Hajdin C. E., Weeks K. M., Nat. Rev. Drug Discovery 2018, 17, 547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Alnuqaydan A. M., Am. J. Transl. Res. 2020, 12, 3531–3556. [PMC free article] [PubMed] [Google Scholar]
- 6. Shao Y., Zhang Q. C., Essays Biochem. 2020, 64, 955–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Falese J. P., Donlic A., Hargrove A. E., Chem. Soc. Rev. 2021, 50, 2224–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tan D. S., Nat. Chem. Biol. 2005, 1, 74–84. [DOI] [PubMed] [Google Scholar]
- 9. Lenci E., Baldini L., Trabocchi A., Bioorg. Med. Chem. 2021, 41, 116218. [DOI] [PubMed] [Google Scholar]
- 10. Gerry C. J., Schreiber S. L., Nat. Rev. Drug Discovery 2018, 17, 333–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pavlinov I., Gerlach E. M., Aldrich L. N., Org. Biomol. Chem. 2019, 17, 1608–1623. [DOI] [PubMed] [Google Scholar]
- 12. Yi S., Varun B. V., Choi Y., Park S. B., Front. Chem. 2018, 6, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mortensen K. T., Osberger T. J., King T. A., Sore H. F., Spring D. R., Chem. Rev. 2019, 119, 10288–10317. [DOI] [PubMed] [Google Scholar]
- 14. Martinez R., Chacon-Garcia L., Curr. Med. Chem. 2012, 12, 127–151. [DOI] [PubMed] [Google Scholar]
- 15. Portugal J., Barceló F., Curr. Med. Chem. 2016, 23, 4108–4134. [DOI] [PubMed] [Google Scholar]
- 16. Hertzberg R. P., Caranfa M. J., Hecht S. M., Biochemistry 1989, 28, 4629–4638. [DOI] [PubMed] [Google Scholar]
- 17. Pommier Y., Nat. Rev. Cancer 2006, 6, 789–802. [DOI] [PubMed] [Google Scholar]
- 18. Kuo C. L., Chou C. C., Yung B. Y. M., Cancer Lett. 1995, 93, 193–200. [DOI] [PubMed] [Google Scholar]
- 19. Li T. K., Houghton P. J., Desai S. D., Daroui P., Liu A. A., Hars E. S., Ruchelman A. L., LaVoie E. J., Liu L. F., Cancer Res. 2003, 63, 8400–8407. [PubMed] [Google Scholar]
- 20. Pommier Y., Cushman M., Mol. Cancer Ther. 2009, 8, 1008–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thomas A., Pommier Y., Clin. Cancer Res. 2019, 25, 6581–6589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hagenbuchner J., Ausserlechner M. J., Biochem. Pharmacol. 2016, 107, 1–13. [DOI] [PubMed] [Google Scholar]
- 23. Ferreira R., Schneekloth J. S., Panov K. I., Hannan K. M., Hannan R. D., Cells 2020, 9, 266–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xiao W., Zhou Q., Wen X., Wang R., Liu R., Wang T., Shi J., Hu Y., Hou J., Front. Pharmacol. 2021, 12, 702360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang M., Yu Y., Liang C., Lu A., Zhang G., Int. J. Mol. Sci. 2016, 17, 779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. del Mundo I. M. A., Vasquez K. M., Wang G., Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nasiri H. R., Bell N. M., Mc Luckie K. I. E., Husby J., Abell C., Neidle S., Balasubramanian S., Chem. Commun. 2014, 50, 1704–1707. [DOI] [PubMed] [Google Scholar]
- 28. Fan R., Xiao C., Wan X., Cha W., Miao Y., Zhou Y., Qin C., Cui T., Su F., Shan X., RNA Biol. 2019, 16, 707–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Naryshkin N. A., Weetall M., Dakka A., Narasimhan J., Zhao X., Feng Z., Ling K. K. Y., Karp G. M., Qi H., Woll M. G., Chen G., Zhang N., Gabbeta V., Vazirani P., Bhattacharyya A., Furia B., Risher N., Sheedy J., Kong R., Ma J., Turpoff A., Lee C.-S., Zhang X., Moon Y.-C., Trifillis P., Welch E. M., Colacino J. M., Babiak J., Almstead N. G., Peltz S. W., Eng L. A., Chen K. S., Mull J. L., Lynes M. S., Rubin L. L., Fontoura P., Santarelli L., Haehnke D., McCarthy K. D., Schmucki R., Ebeling M., Sivaramakrishnan M., Ko C.-P., Paushkin S. v., Ratni H., Gerlach I., Ghosh A., Metzger F., Science. 2014, 345, 688–693. [DOI] [PubMed] [Google Scholar]
- 30. Wang J., Schultz P. G., Johnson K. A., Proc. Natl. Acad. Sci. USA 2018, 115, E4604–E4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Katsuda Y., Sato S. I., Asano L., Morimura Y., Furuta T., Sugiyama H., Hagihara M., Uesugi M., J. Am. Chem. Soc. 2016, 138, 9037–9040. [DOI] [PubMed] [Google Scholar]
- 32. Gupta A., Flynn B. L., Org. Lett. 2017, 19, 1939–1941. [DOI] [PubMed] [Google Scholar]
- 33. Gupta A., Flynn B. L., J. Org. Chem. 2016, 81, 4012–4019. [DOI] [PubMed] [Google Scholar]
- 34. Goeminne A., Scammells P. J., Devine S. M., Flynn B. L., Tetrahedron Lett. 2010, 51, 6882–6885. [Google Scholar]
- 35. Aurelio L., Volpe R., Halim R., Scammells P. J., Flynn B. L., Adv. Synth. Catal. 2014, 356, 1974–1978. [Google Scholar]
- 36.Fragment-growth refers to the incorporation of additional protein-binding groups to a fragment in order to increase affinity and potency for the target, see for examples in references 37–40.
- 37. Tsai J., Lee J. T., Wang W., Zhang J., Cho H., Mamo S., Bremer R., Gillette S., Kong J., Haass N. K., Sproesser K., Li L., Smalley K. S. M., Fong D., Zhu Y. L., Marimuthu A., Nguyen H., Lam B., Liu J., Cheung I., Rice J., Suzuki Y., Luu C., Settachatgul C., Shellooe R., Cantwell J., Kim S. H., Schlessinger J., Zhang K. Y. J., West B. L., Powell B., Habets G., Zhang C., Ibrahim P. N., Hirth P., Artis D. R., Herlyn M., Bollag G., Proc. Natl. Acad. Sci. USA 2008, 105, 3041–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang C., Ibrahim P. N., Zhang J., Burton E. A., Habets G., Zhang Y., Powell B., West B. L., Matusow B., Tsang G., Shellooe R., Carias H., Nguyen H., Marimuthu A., Zhang K. Y. J., Oh A., Bremer R., Hurt C. R., Artis D. R., Wu G., Nespi M., Spevak W., Lin P., Nolop K., Hirth P., Tesch G. H., Bollag G., Proc. Natl. Acad. Sci. USA 2013, 110, 5689–5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Davies T. G., Wixted W. E., Coyle J. E., Griffiths-Jones C., Hearn K., McMenamin R., Norton D., Rich S. J., Richardson C., Saxty G., Willems H. M. G., Woolford A. J. A., Cottom J. E., Kou J. P., Yonchuk J. G., Feldser H. G., Sanchez Y., Foley J. P., Bolognese B. J., Logan G., Podolin P. L., Yan H., Callahan J. F., Heightman T. D., Kerns J. K., J. Med. Chem. 2016, 59, 3991–4006. [DOI] [PubMed] [Google Scholar]
- 40. Murray C. W., Newell D. R., Angibaud P., MedChemComm 2019, 10, 1509–1511. [Google Scholar]
- 41. Larock R. C., in Acetylene Chemistry: Chemistry, Biology, and Material Science (Eds.: Diederich F., Stang P. J., Tykwinski R. R.), 2005, pp. 51–99. [Google Scholar]
- 42. Rodríguez F., Fañanás F. J., Handbook of Cyclization Reactions, Vol. 2 (Ed.: Ma S.), Wiley-VCH, 2010, p. 951–990. [Google Scholar]
- 43. Gilmore K., Alabugin I. v., Chem. Rev. 2011, 111, 6513–6556. [DOI] [PubMed] [Google Scholar]
- 44. Godoi B., Schumacher R. F., Zeni G., Chem. Pharm. Bull. 2011, 111, 2937–2980. [DOI] [PubMed] [Google Scholar]
- 45. Fang G., Bi X., Chem. Soc. Rev. 2015, 44, 8124–8173. [DOI] [PubMed] [Google Scholar]
- 46. Aggarwal T., Kumar S., Verma A. K., Org. Biomol. Chem. 2016, 14, 7639–7653. [DOI] [PubMed] [Google Scholar]
- 47. Sonawane A. D., Sonawane R. A., Ninomiya M., Koketsu M., Adv. Synth. Catal. 2020, 362, 1–32. [Google Scholar]
- 48. Pandey A. R., Tiwari D. K., Prakhar A., Mishra D. P., Monatsh. Chem. 2022, 153, 383–407. [Google Scholar]
- 49. Martín R., Larsen C. H., Cuenca A., Buchwald S. L., Org. Lett. 2007, 9, 3379–3382. [DOI] [PubMed] [Google Scholar]
- 50. Kicková A., Horváth B., Kerner L., Putala M., Chem. Pap. 2013, 67, 101–109. [Google Scholar]
- 51. Xue W., Xu H., Liang Z., Qian Q., Gong H., Org. Lett. 2014, 16, 4984–4987. [DOI] [PubMed] [Google Scholar]
- 52. Jacubert M., Tikad A., Provot O., Hamze A., Brion J. D., Alami M., Eur. J. Org. Chem. 2010, 2010, 4492–4500. [Google Scholar]
- 53. Chen S., Flynn B. L., Aust. J. Chem. 2021, 74, 65–76. [Google Scholar]
- 54. Volpe R., Aurelio L., Gillin M. G., Krenske E. H., Flynn B. L., Chem. Eur. J. 2015, 21, 10191–10199. [DOI] [PubMed] [Google Scholar]
- 55. Hessian K. O., Flynn B. L., Org. Lett. 2006, 8, 243–246. [DOI] [PubMed] [Google Scholar]
- 56. Zhou C., Dubrovsky A. v., Larock R. C., J. Org. Chem. 2006, 71, 1626–1632. [DOI] [PubMed] [Google Scholar]
- 57. Yu X., Wu J., J. Comb. Chem. 2009, 11, 895–899. [DOI] [PubMed] [Google Scholar]
- 58. Zhang H. P., Li H. Y., Xiao H. F., J. Chem. Res. 2013, 37, 556–558. [Google Scholar]
- 59. Huang Q., Hunter J. A., Larock R. C., J. Org. Chem. 2002, 67, 3437–3444. [DOI] [PubMed] [Google Scholar]
- 60.See the Supporting Information for the structural assignment of this compound using 2d nmr experiments.
- 61. Ruchelman A. L., Singh S. K., Ray A., Wu X., Yang J. M., Zhou N., Liu A., Liu L. F., LaVoie E. J., Bioorg. Med. Chem. 2004, 12, 795–806. [DOI] [PubMed] [Google Scholar]
- 62. Mitamura T., Nomoto A., Sonoda M., Ogawa A., Bull. Chem. Soc. Jpn. 2010, 83, 822–824. [Google Scholar]
- 63. Pommier Y., Nussenzweig A., Takeda S., Austin C., Nat. Rev. Mol. Cell Biol. 2022, DOI 10.1038/s41580-022-00452-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Meng F., Nguyen X., Cai X., Duan J., Matteucci M., Hart C. P., Anti-Cancer Drugs 2007, 18, 435–445. [DOI] [PubMed] [Google Scholar]
- 65. Rapisarda A., Uranchimeg B., Sordet O., Pommier Y., Shoemaker R. H., Melillo G., Cancer Res. 2004, 64, 1475–1482. [DOI] [PubMed] [Google Scholar]
- 66. Schovanek J., Bullova P., Tayem Y., Giubellino A., Wesley R., Lendvai N., Nölting S., Kopacek J., Frysak Z., Pommier Y., Kummar S., Pacak K., Endocrinology 2015, 156, 4094–4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pommier Y., Cushman M., Mol. Cancer Ther. 2009, 8, 1008–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dexheimer T. S., Pommier Y., Nat. Protoc. 2008, 3, 1736–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. See the Supporting Information for the Biological Data of Test Compounds, n.d.
- 70. Marchand C., Antony S., Kohn K. W., Cushman M., Ioanoviciu A., Staker B. L., Burgin A. B., Stewart L., Pommier Y., Mol. Cancer Ther. 2006, 5, 287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Pommier Y., Kiselev E., Marchand C., Bioorg. Med. Chem. Lett. 2015, 25, 3961–3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Satyanarayana M., Feng W., Cheng L., Liu A. A., Tsai Y. C., Liu L. F., LaVoie E. J., Bioorg. Med. Chem. 2008, 16, 7824–7831. [DOI] [PubMed] [Google Scholar]
- 73. Aurelio L., Scullino C. v., Pitman M. R., Sexton A., Oliver V., Davies L., Rebello R. J., Furic L., Creek D. J., Pitson S. M., Flynn B. L., J. Med. Chem. 2016, 59, 965–984. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.