

Abstract
Senile depression (SD) is a heterogeneous syndrome. Several clinical profiles are more likely to appear in SD than in early‐life depression, but it remains unclear whether the pathophysiology is different. The prevalence of dementia increases with aging, and the underlying pathophysiological processes in the preclinical phase begin even before cognitive deficits or neurological signs appear. SD may be either a risk factor for developing dementia or a prodromal stage of dementia. The inconsistent findings regarding the association between SD and incident dementia may be attributable to the neuropathological heterogeneity underlying SD. Most studies have focused on patients with the clinical diagnosis of Alzheimer disease (AD) as an outcome, but several clinicopathological studies suggest that primary age‐related tauopathy and argyrophilic grain disease may account for a proportion of cases clinically misdiagnosed as AD in the elderly population. Furthermore, most AD cases have additional neuropathologic changes such as cerebrovascular disease and Lewy body disease. Here, we review the neuropathological findings linking SD to incident dementia, focusing on common age‐related neuropathologies. In particular, the roles of disturbance of neural circuity, imbalance of monoaminergic systems, dysregulation of the hypothalamic–pituitary–adrenal axis, and elevated neuroinflammatory status are discussed. Finally, we review the current treatment of SD in the context of age‐related neuropathological changes.
Keywords: Alzheimer disease, Lewy body disease, neuropathology, senile depression, tau
Introduction
Overview of senile depression in the context of age‐related neuropathology
The prevalence of depression and depressive symptoms, known as senile depression (SD), is high in elderly populations, 1 , 2 and several expert consensus reports and guidelines on the diagnosis and treatment of SD have appeared in recent years. 3 , 4 SD is a heterogeneous syndrome; for example, the onset of depression in patients with dementia is later than in patients with depression alone, 5 and there may be various coexisting age‐related neuropathologies. Brain neuroimaging studies have uncovered significant abnormalities in patients with SD. 6 , 7 , 8 Cortical tau deposition identified using tau radioligand in geriatric patients with major depressive disorder (MDD) was significantly greater than in controls, whereas amyloid β (Aβ) accumulation was similar in the two groups. 6 Indeed, based on the Alzheimer's Disease Neuroimaging Initiative's Depression Project (ADNI‐D) database, the SD group showed less Aβ deposition than the nondepressed group. 8 A subset of patients with late‐onset depressive disorder share clinical features with Lewy body disease (LBD), including abnormal dopamine transporter (DAT) binding. 7 In the analysis of the National Alzheimer's Coordinating Center database, a longitudinal study revealed that neuropsychiatric symptoms in cognitively normal elderly individuals were useful in predicting incident dementia and its subtypes. 9 Risk estimates of affective symptoms or psychotic symptoms were different across subtypes of incident dementia during follow‐up. These findings suggest the presence of distinct subgroups, and may provide insights into the pathophysiology of SD. Therefore, it is important to understand SD in the context of coexisting age‐related neuropathologies.
The prevalence of dementia increases with aging, and the underlying pathophysiological process in the long predementia phase begins even before cognitive deficits or neurological signs appear. SD may be a risk factor for developing dementia or may represent a prodromal stage of dementia. These possibilities are not mutually exclusive, but many conflicting results have been reported, possibly attributable to the neuropathological heterogeneity underlying SD. Many previous studies have focused on Alzheimer disease (AD) as a possible outcome, but there are few data regarding specific diagnostic biomarkers or pathological verification. Several clinicopathological studies indicate that some primary age‐related tauopathy (PART) and argyrophilic grain disease (AGD) may have been clinically misdiagnosed as AD in elderly patients. 10 Furthermore, most patients with AD have complications such as cerebrovascular disease or LBD including dementia with Lewy bodies (DLB). 10 However, there has been little investigation of non‐AD subtypes as outcomes. This review highlights the potential neuropathological basis for linking SD to incident dementia based on the common age‐related neuropathologies. In particular, we focus on similarities in the neurobiological alterations in depression and dementia, such as disturbance of neural circuity, imbalance of monoaminergic systems, dysregulation of the hypothalamic–pituitary–adrenal (HPA) axis, and elevated neuroinflammatory events. Finally, we review the current treatment of SD in the context of age‐related neuropathological changes.
Clinical characteristics of SD
Although neither the United Nations nor the World Health Organization has clearly defined ‘senile’ or ‘late‐life’, it is generally defined as being 65 years of age or older. However, the age cutoffs of studies on SD have varied between 50 and 75 years. The frequency of SD in people 65 years and older in Japan is reported to be 4.5%. 11 Suicidal ideation, pessimism, psychomotor agitation, hypochondria, and somatic symptoms are more frequent 12 and psychotic symptoms are more likely to appear in SD than in early‐life depression. 13 SD is less responsive to antidepressant treatment 14 and has a higher recurrence rate. 15 , 16 There are significant differences between early‐onset and late‐onset SD. Late‐onset SD is associated with greater neurological changes, 17 , 18 worse cognitive performance, 17 , 19 , 20 , 21 less severe depression, 17 , 22 family history of mood disorder, 23 superior response to electroconvulsive therapy (ECT), 24 and higher incidence of dementia 22 compared with early‐onset SD. It has been shown that 45% of inpatients with SD have psychotic symptoms, 25 and patients with senile psychotic depression have later onset age than patients with nonpsychotic SD. 13 Psychotic symptoms in SD may be associated with the pathophysiology of aging and dementia. In a meta‐analysis, patients with senile psychotic depression showed greater deficits in all cognitive domains, except verbal fluency, compared with patients with nonpsychotic SD. 26 It is crucial that clinicians and researchers take cognitive deficits into account in senile psychotic depression. These data may also indicate that senile psychotic depression is associated with the pathophysiology of aging and dementia.
Biological Aspects of SD and Its Relation to Age‐Related Neuropathology
Overlapping of biological features between SD and dementia
SD often occurs in patients with dementia. For example, 15% of patients with AD 27 and 30% of patients with mild cognitive impairment (MCI) 28 were reported to have comorbid depression. Apathy, which is difficult to differentiate from depression, is also present in 50% of patients with AD, 29 and patients with DLB also frequently develop depression before the onset of core symptoms. 30 Apathy, generally lacking the features of sad mood or distress, might be related to frontal lobe pathology, including frontotemporal dementia. 3 A magnetic resonance imaging (MRI) study suggests that progression of apathy is associated with low baseline gray matter volume in the frontal and cingulate regions. 31
A large cohort study found an increase in depressive symptoms ≈10 years before the onset of dementia, 32 and 71% of patients with pseudodementia caused by SD progressed to dementia. 33 The comorbidity and clinical overlaps suggest a role of common pathophysiological process(es) in SD and dementia.
Figure 1 shows the pathophysiological relationships between depression and dementia. Depression is characterized by a decrease in monoamines and an increase in the HPA system, whereas dementia has characteristic pathological findings. Similarities between these disorders include neuroinflammation, elevated proinflammatory cytokines, decreased neurotrophic factors, abnormal synaptic function, deep white matter lesions, and increased brain atrophy. If depression is a prodrome of dementia, and if it is present 10 years before the onset of dementia, then several biomarkers may be useful for diagnosing SD. 34 , 35 , 36 , 37
Fig. 1.
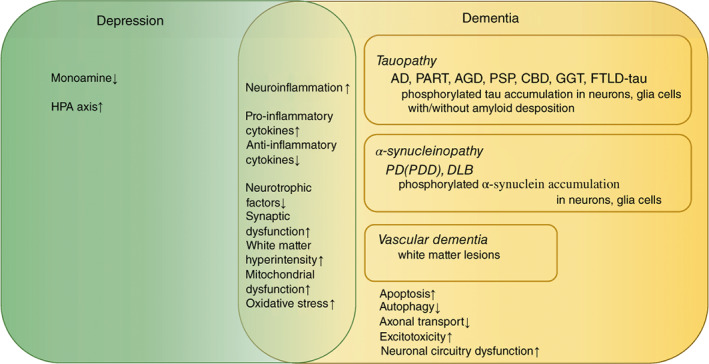
Pathophysiological relationships between depression and neurodegenerative disorders. AD, Alzheimer disease; AGD, Argyrophilic grain disease; CBD, corticobasal degeneration; DLB, dementia with Lewy bodies; FTLD, frontotemporal lobar degeneration; GGT, globular glial tauopathies; HPA, hypothalamic–pituitary–adrenal; PART, primary age‐related tauopathy; PD, Parkinson disease; PDD, Parkinson disease dementia; PSP, progressive supranuclear palsy.
Imbalance of monoaminergic systems
The monoamine hypothesis is the most explored putative explanation for the pathophysiology of MDD. Consequently, monoamine neurotransmitters, including serotonin, norepinephrine, and dopamine, play a pivotal role in the current pharmacological treatment of depression. These monoaminergic systems are altered or impaired in association with age‐related neuropathological changes. The monoaminergic neurons in the brainstem nuclei innervate a multitude of brain regions, and are involved in early disease process, such as accumulation of phosphorylated tau and α‐synuclein. Some recent clinicopathological studies have focused on the relationship of neurodegenerative changes in these nuclei to SD. Considering the progressive character of neurodegenerative disorders and the vulnerability of monoaminergic systems, such neuropathological changes may influence the clinical course, including recurrence of depressive episodes.
The recent development of neuroimaging techniques has enabled evaluation of changes in fibers arising from the brainstem nuclei, including white matter lesions. The current standardized antidepressant treatments rapidly increase the concentration of monoamine in synaptic clefts, but symptomatic relief in terms of an effect on mood takes several weeks. Therefore, subsequent studies have focused on modulating monoaminergic neurotransmission, including changes at the level of gene and protein expression. 37 , 38 , 39 Levels of receptors related to synaptic transmission have also been investigated by means of high‐resolution imaging with highly selective radiotracers, as well as histological methods. Serotonin transporter levels are decreased in the temporal cortical and limbic (amygdala and hippocampus) regions in SD, 38 and this may lead to a poor response to antidepressants in acute treatment. For example, a recent meta‐analysis showed that there was no difference in response between antidepressants and placebo in persons older than 65 years. 14 One reason for the poor response may be the pathophysiology of aging and dementia. For example, amyloid status may have significant effects on antidepressant response. 39 Interestingly, patients with amyloid‐negative SD showed a better response to antidepressant treatment than amyloid‐positive patients. However, further research is needed.
Disturbances in HPA system in SD
SD is associated with neuroendocrine changes. In the HPA pathway of SD or dementia, this axis is activated, with elevated corticotrophin‐releasing hormone and vasopressin production by cells of the hypothalamic paraventricular nucleus. 40 In the hypothalamus and pituitary, negative feedback regulation is impaired owing to decreased expression of corticosteroid receptors as well as upstream central nervous system (CNS) regulatory centers. 41 Increased glucocorticoid production may induce hippocampal atrophy via downregulation of glucocorticoid receptors. 40 HPA dysregulation induces chronic elevation of adrenal glucocorticoid production with impaired negative feedback and abnormal homeostatic regulation in MDD, 41 and postdexamethasone cortisol levels are increased. 42 In animal studies, the cortisol‐hippocampal link is associated with response to stress, and it was shown that high‐stress conditions or exogenous glucocorticoids can cause memory impairment based on hippocampal neuronal damage. 43 Human studies in the elderly have demonstrated that hippocampal volume is decreased in the setting of elevated glucocorticoids and in proportion to the duration of hypercortisolemia. 44
In various neuropathological processes, hypothalamic dysfunction caused by disease‐specific pathology affects body weight, sleep, and circadian rhythm. The affected cell types are still unknown, especially in humans, although some animal model studies have been reported. 45 It seems likely that glucocorticoid‐related derangement of hippocampal physiology may increase vulnerability to other pathophysiologic mechanisms such as accumulation of phosphorylated tau or cerebrovascular disease. 41
Neuroinflammation in SD related to dementia
Inflammatory processes are closely associated with multiple neurodegenerative pathways involved in depression or neurodegenerative diseases, 46 and inflammation‐induced cytokines play a role in dementia. 46 In the brain tissues, microglia are primarily responsible for inflammatory responses. 47 Furthermore, a strong peripheral inflammatory response to systemic lipopolysaccharide 48 or viral infection 49 leads to leukocyte infiltration of the CNS, resulting in neuroinflammation and neurodegeneration. Microglial activation follows, triggering the release of inflammatory mediators that promote blood–brain barrier (BBB) permeabilization. Subsequently, peripheral leukocytes, including T cells and macrophages, which share several functional features with microglia, infiltrate into the CNS.48
Possible reasons for the association between inflammation and SD include oxidative stress, elevated levels of the proinflammatory cytokines interleukin (IL) 6 and IL‐8, 50 deconjugation of endothelial nitric oxide synthase, and hyperglutaminergic activity. Thus, indirect evidence of neurovascular dysfunction has been found in MDD 51 in the form of elevated levels of peripheral inflammatory markers, depression, and severe psychiatric disorders associated with death by suicide. 52 Microglia are a major source and target of inflammatory cytokines in the CNS and have been implicated in the incidence and progression of MDD. 53 Meta‐analyses have shown that elevated levels of tumor necrosis factor α and IL‐6 in peripheral blood strongly correlated with MDD. 54 and depressed patients with AD had the highest levels of circulating IL‐6 and tumor necrosis factor α. 55 Furthermore, depressed patients with AD showed a positive correlation between cognitive impairment and cytokine levels. 55 Similarly, type 2 diabetes (T2D), which is associated with systemic inflammation, 56 significantly increases the risk of AD in depressed patients. An epidemiological study including data collected from all Danish citizens older than 50 years (≈14 million person‐years) showed that the incidence of AD is higher in patients with coexisting T2D and depression than in patients with either condition alone. 57 Nevertheless, T2D, depression, and AD are complex disorders, and, as discussed below, other mechanisms in addition to inflammation may be involved in their association.
As an example of neuroinflammation related to depression, viral infections such as coronavirus disease 2019 (COVID‐19) may lead to prolonged inflammation in the brain and induce brain atrophy. 58 The resulting neuropsychiatric symptoms, such as depression, brain fog, and anxiety, can lead to a variety of neurological disorders. Viral proteins are found in vascular endothelium, together with vascular inflammation and angiotensin‐converting enzyem 2 receptor expression, leading to activation of cytokine release and hypercoagulation pathways in the blood, and resulting in the formation of small‐ and large‐vessel occlusions in the brain. High levels of cytokines in the cerebral vessels can damage the BBB, and once infiltrated into the brain, can damage neurons and glia, resulting in seizures and/or encephalopathy. It may be possible to mitigate prolonged neuropsychiatric symptoms after COVID‐19 infection by preventing hypoxia and severe infection and maintaining cerebral blood circulation.
SD and Age‐Related Neuropathology
The Table 1 summarizes the neuropathological features in common age‐related pathology associated with depressive symptoms.
Table 1.
Summary of neuropathological features in common age‐related pathology associated with depressive symptoms
| Age‐related pathology | Typical affected brain regions | Neuropathological hallmarks | Candidate regions associated with comorbid depressive symptoms |
|---|---|---|---|
| Vascular pathology | Periventricular, deep white matter | Arteriosclerosis, dilated periventricular spaces, ischemia, perivascular demyelination, vascular ecstasia, lacunar infarction | Frontostriatal circuits; frontal cortex, paralimbic areas, and striatum |
| Alzheimer disease |
Medial temporal cortex (hippocampus) in early stage With progression, parietal and frontal cortex |
Senile plaques (amyloid β), neurofibrillary tangles (phosphorylated tau) with neuronal loss/astrogliosis in the affected neocortices 3 repeat/4 repeat tauopathy |
Limbic regions, various associated neocortical regions, as well as subcortical nuclei |
| Primary age‐related tauopathy | Limited regions in the medial temporal lobe, especially in hippocampus/hippocampal gyrus |
Neurofibrillary tangles including ghost tangles with neuronal loss/astrogliosis, lacking significant amyloid β plaques 3 repeat/4 repeat tauopathy |
Limbic regions, subcortical nuclei especially in the nucleus accumbens |
| Argyrophilic grain disease | Limbic regions including the hippocampus, entorhinal cortex, and the amygdala nucleus |
Neurofibrillary tangles with a characteristic massive occurrence of argyrophilic and tau‐positive grains 4 repeat tauopathy |
Limbic regions, including the amygdala |
| Lewy body disease | Brainstem‐predominant Lewy body disease; brainstem including substantianigra and locus coeruleus, transitional Lewy body disease; mainly in limbic regions, and diffuse neocortical Lewy body disease; neocortices | Lewy bodies and Lewy neurites in the central, peripheral, and autonomic nervous system. α‐synucleinopathy | Monoaminergic systems including the nigrostriatum, limbic regions |
Vascular pathology
Vascular changes appearing as white matter hyperintensities in the elderly
White matter hyperintensities (WMH), which are frequently seen on MRI, particularly T2‐weighted MRI 59 , 60 of elderly people, are associated with various geriatric disorders, such as cerebrovascular diseases, cardiovascular diseases, dementia (including mild cognitive impairments), and SD. White matter consists of myelinated axons, and the neuroimaging condition known as WMH neuropathologically reflects decreased axon number (Fig. 2a,b), smaller axons coupled with losses in the ependymal cell layer, reactive gliosis, and increased periependymal extracellular fluid content. 61 Several pathophysiological mechanisms may be involved, such as cerebrovascular risk factors, particularly hypertension, diabetes, history of myocardial infarction or coronary artery disease, and smoking. 62 WMH has been differentiated into periventricular WMH (PVWMH) and deep WMH (DWMH). 63 PVWMH is caused by leakage of cerebrospinal fluid into the periventricular regions, whereas DWMH is induced as a result of various degenerative processes including atherosclerosis, lacunar infarctions (Fig. 2c), atrophic demyelination, and arteriolar hyalinization (Fig. 2d). PVWMH occasionally shows continuity with DWMH. PVWMH has been mostly associated with cognitive impairment, whereas DWMH is linked more to mood disorders. 64 , 65 The Fazekas scale, one of the visual scales for WMH, is mostly used to assess the severity. 63 Recently, another scale has been introduced to differentiate the subcortical white matter and basal ganglia. 66
Fig. 2.
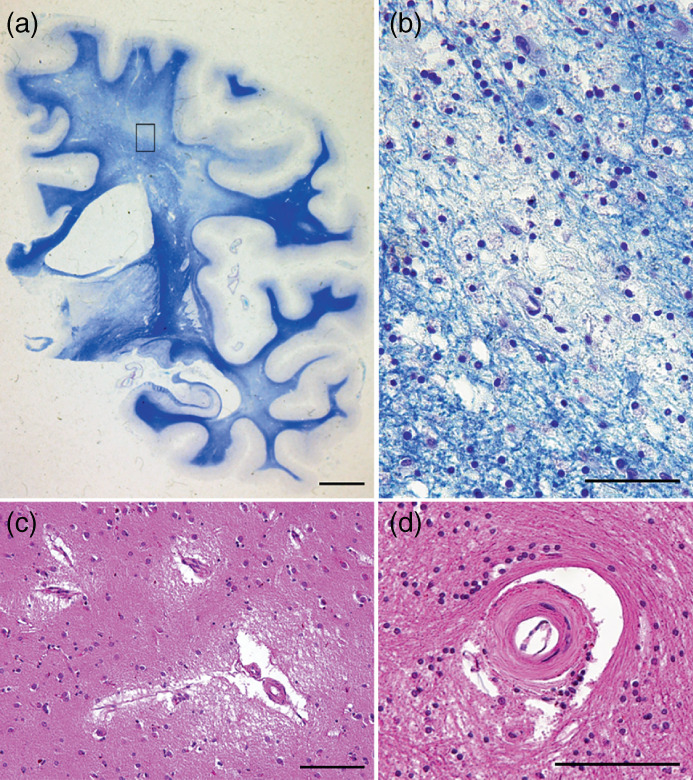
Vascular pathology. (a) Lacunar infarction in white matter hyperintensities. (b) Fibrohyalynosis with narrowing of the arteriolar lumen. (c) Demyelination of the cerebral white matter in periventricular regions and the white matter of frontoparietal lobes. Klüver‐Barrera stain. (d) Enlargement of the square frame in panel c. Myelin pallor is not homogeneous, but islands of decreased myelination are surrounded by normal tissue. Scale bars: (a) 100 μm; (a, d) 50 μm; (c) 2 cm.
Associations of WMH with vascular changes and SD
WMHs in patients with SD reflect a cerebrovascular change that predisposes individuals to the development of depression, 67 according to the well‐known hypothesis concerning vascular depression of late onset. 67 , 68 Some quantitative studies have found significant differences between early‐ and late‐onset depression, suggesting different etiological mechanisms, 59 although this remains controversial. 69 , 70
Recent neuropathological and imaging studies have highlighted the importance of WMH, especially in prefrontal regions that play a role in neural circuits related to depression. The pathology of PVWMH spreads from the subcortical regions, which can affect microvascular blood flow to the prefrontal regions. 71 Thus, PVWMH frequently leads to defects in psychomotor control and function. 72 DWMH correlates with defects in motivation, concentration, and decision‐making, as well as apathy, negative affect, and dysphoria. 73 As regards regional mechanisms of depression, frontostriatal circuits through the fiber tract connecting frontal cortex, paralimbic areas, and striatum, which are associated with depression, are disrupted. 67 , 74 Pathological studies indicate lower densities of oligodendroglial cells in MDD. 75 MRI studies of segmented brain white matter hyperintensities revealed that patients with SD had damage in white matter tracts, such as the superior longitudinal fasciculus, fronto‐occipital fasciculus, uncinate fasciculus, extreme capsule, and inferior longitudinal fasciculus, which are associated with episodic memory, poor processing speed, and poor executive function. 76 Some genetic studies support an association between WMH and mood disorders 77 , 78 Patients with bipolar disorder show more severe WMH than patients with MDD or healthy controls, 79 , 80 suggesting that patients with bipolar disorder may be more vulnerable to such changes. Further studies, especially by diffusion tensor imaging, are needed.
Tauopathies
Clinicopathological concepts of tauopathy
SD is frequent in patients with tauopathies, which are a broad class of neurodegenerative diseases characterized by pathological aggregation of tau protein, including AD, PART, AGD, progressive supranuclear palsy, corticobasal degeneration, Pick disease, frontotemporal dementia and parkinsonism linked to chromosome 17, globular glial tauopathies, chronic traumatic encephalopathy, and aging‐related tau astrogliopathy. 81 Tauopathies have been distinguished based on the ratio of 3 repeat (3R) and/or 4 repeat (4R) tau and major bands such as 60, 64, and 68 kDa. 82 Pick disease is classified into the 3R tauopathy group. The 4R tauopathies group consists of corticobasal degeneration, progressive supranuclear palsy, AGD, aging‐related tau astrogliopathy, and globular glial tauopathy, while the mixed 3R/4R group consists of neurofibrillary tangles (NFT)‐dementia, including AD and PART. Neuropathological diagnoses of these diseases are based on the molecular isoforms of abnormally accumulated tau, the pattern of accumulation in neurons and glial cells, and the regional distribution. As in all neurodegenerative diseases, tauopathies show characteristic vulnerability of specific brain regions. The regional distributions of tau and neuronal loss reflect different clinical features. The pathophysiological basis of tauopathies and depression with cognitive impairments remains unclear, but some recent studies suggest that the characteristic symptoms reflect specific regional neuronal degeneration. Next, we will consider typical tauopathies frequently presenting neuropsychiatric symptoms, including SD with mild cognitive decline.
Alzheimer disease
AD is a well‐known tauopathy, and its diagnostic hallmarks are the presence of both tau‐reactive NFTs (Fig. 3a) and Aβ deposits (called senile plaque [SP]) (Fig. 3b). Recent biomarker studies suggest that neuropsychiatric symptoms are driven by neuropathological changes in AD. 83 , 84 For evaluating progression of NFTs in the brain, the Braak staging system is highly reproducible. 85 NFTs mainly start in the hippocampal region and spread gradually throughout the brain. In Braak stages I/II, cortical NFTs start from the entorhinal/transentorhinal cortex, then spread to paralimbic cortices and other hippocampal regions in Braak stages III/IV, and finally reach the higher‐order association areas and primary neocortex in Braak stages V/VI85.
Fig. 3.
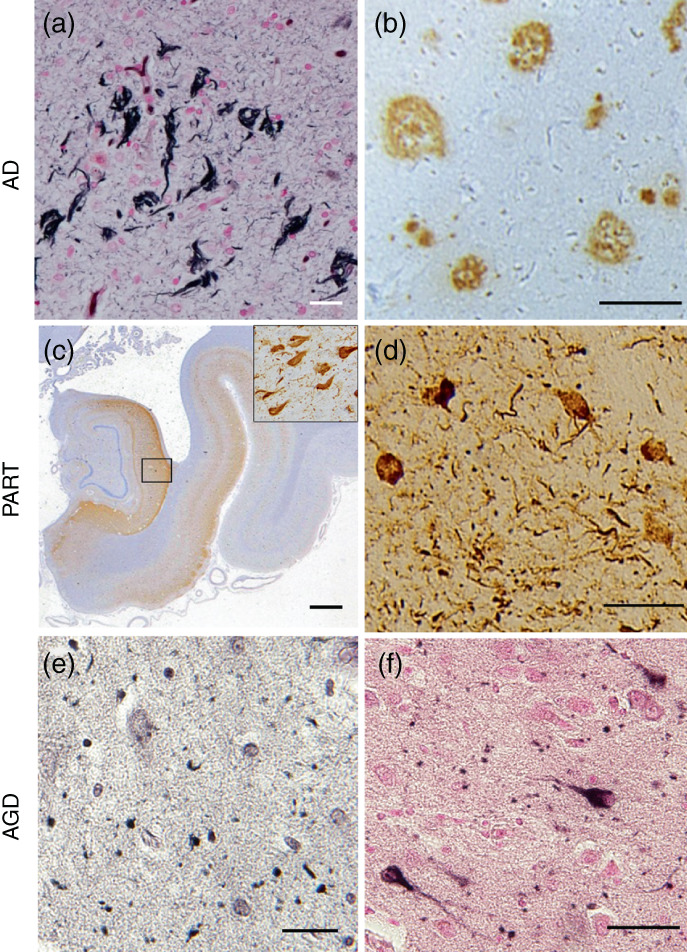
Pathology in tauopathies. (a, b) Alzheimer disease (AD) pathology. Neurofibrillary tangles in transenorhinal cortex (a, AT8 antibody) and senile plaques in temporal neocortex (b, 4G8 antibody) in AD. (c, d) Primary age‐related tauopathy (PART) pathology. Phosphorylated‐tau accumulation in hippocampus (c, AT8 antibody). The enlargement inserted in panel c shows massive neurofibrillary tangles (NFTs) in the transenorhinal cortex. Nucleus accumbens shows many NFTs stained with p‐tau antibody (d, AT8 antibody). (e, f) Argyrophilic grain disease (AGD) pathology. Gallyas‐Braak staining indicates massive grains in amygdala (e). Patients with AGD show pretangles in transenorhinal cortex (f, AT8 antibody). Scale bars: (a, d, e, f) 50 μm; (b) 100 μm; (c) 1 cm.
Pathological reports suggest a strong correlation between NFTs and cognitive impairment. 86 Braak stage III correlates poorly with cognitive decline, whereas Braak stages IV and above are consistently associated with at least mild dementia. 86 Interestingly, in terms of psychiatric symptoms, Braak stage I/II correlates with agitation, anxiety, appetite changes, and depression, while delusions are associated with Braak stage III/IV or higher. 87 In this context, Braak stage III or less might most frequently present with psychiatric symptoms. Other neuropathological studies have also supported the notion that deposition of phosphorylated tau may be accelerated in AD with psychosis. 88 , 89 The Braak staging system has recently been revised to include the onset of NFT pathology in subcortical nuclei as a precortical stage. In early‐stage AD, NFT pathology initially develops in the subcortical nuclei, such as the locus coeruleus, dorsal raphe nucleus, and perifornical nucleus of the hypothalamus, rather than in the hippocampus and neocortices. 90 Subcortical nuclei accumulate NFTs at Braak stage 0, in association with anxiety, depression, and sleep disturbances. Thus, neuropsychiatric symptoms might be part of the constellation of early clinical symptoms. Consequently, effective treatment of these symptoms might also be effective for AD pathology. Thus, it would be useful to identify neuropsychiatric symptoms precisely at the precognitive stage.
With the progression of AD, other neurological features gradually appear. The regional spread of phosphorylated tau and its intraneuronal accumulation affect distinct phenomena that could contribute in different ways to the disease phenotype. 88 Memory problems usually occur first, and agnosia, apraxia, or impaired speech will be present besides neuropsychiatric symptoms, reflecting the involvement of limbic regions and associative cortices. The NFT densities in the CA1 region of the hippocampus, the superior parietal cortex (Brodmann area 7), and the posterior cingulate cortex (Brodmann area 23) are linked to development of spatial and temporal disorientation. 91 NFT densities in the anterior cingulate cortex are associated with ideomotor responses and dressing apraxia, whereas constructional apraxia is linked to NFT densities in the superior parietal, posterior cingulate, and occipital cortex. 92 , 93 Acalculia and visuospatial dysfunction reflect the involvement of tau pathology in the parietal lobe. 94 Some atypical subtypes of AD are of clinical interest. For instance, postcortical atrophy presenting with visual symptomatology reflects a high NFT burden in the occipital–parietal–temporal junction and posterior cingulate cortex. 95 In frontal‐variant AD, psychomotor retardation or apathy is prominent. Overall, neuropsychiatric symptoms in tauopathy are comprehensively linked to the various associated neocortical regions, as well as subcortical nuclei. In a recent tau positron emission tomography study, the mean cortical [11C]PBB3 standardized uptake value ratio in patients with MDD was significantly higher than in age‐matched controls. 6 Thus, imaging tau accumulations may provide mechanistic insights into neuronal dysfunctions in tauopathies, underlining the importance of understanding the pathological background in SD.
Aβ deposition has been assumed to modify the clinical phenotype of AD, including psychiatric symptoms, less than NFTs pathology. 87 , 91 The fact that SPs are seen even in cognitively normal elderly persons supports this notion. 86 Whereas NFTs mainly start in the hippocampal region or even earlier in the subcortical nuclei, which are considered to be linked to the psychiatric symptoms, the progression of SPs precedes the appearance in the inferior temporal cortices. 96 There are some studies supporting the association between Aβ pathology and depression. SD is associated with decreased levels of Aβ‐42 in the cerebrospinal fluid. 97 Some early positron emission tomography studies provide support for Aβ pathology as a possible mechanism underlying the connection between SD and AD. Elevated cortical Aβ deposition in the elderly with current or lifetime major depression has been observed 98 and longitudinal cohort studies have reported positive associations between Aβ pathology and subsyndromal depressive symptoms in cognitively normal older adults. 83 , 99 However, few pathological studies support the notion. 100 Moreover, a recent pathological paper reported opposite findings, that 119 patients with SD showed lower Aβ deposition than nondepressed patients in the ADNI‐D database. 8 Thus, memory deficits and accelerated cognitive decline in SD may not be attributable to greater cortical Aβ accumulation. Overall, these apparently conflicting results concerning Aβ deposition suggest tremendous heterogeneity of depression syndromes occurring late in life. 101 Nevertheless, tau might be more important than Aβ deposition in SD.
Primary age‐related tauopathy
PART has recently been defined as a pathological continuum, ranging from localized hippocampal NFTs in elderly persons with cognitively normal or mildly impaired cognition to widespread NFTs in patients with tangle‐predominant dementia. 102 They lack significant Aβ deposits. PART has mainly been studied in autopsied brains, so its clinical aspects remain to be fully elucidated. Even though PART is assumed to be on the same continuum as AD pathology, 103 recent reports have proposed an etiology different from that of AD. Although there is a significant overlap in NFT distribution between PART and AD, there are significant genetic differences, such as a high frequency of the epsilon2 allele 104 and MAPT H1 haplotype 105 in PART.
In PART, AD‐type NFTs (3R4R), including ghost tangles, are mainly distributed in the hippocampus and to a limited extent in the medial temporal lobe, but not in neocortices (Fig. 3c). The pathology corresponds to Braak stages I to III.102 In addition to the hippocampus and cortex, NFTs are abundant in subcortical nuclei such as the locus coeruleus, amygdala, nucleus basalis of Meynert, nucleus accumbens, hypothalamus, thalamus, olfactory system, and dorsal raphe nucleus in PART. As mentioned above in connection with AD, these subcortical nuclei might modulate specific neuropsychiatric symptoms, including anxiety, sleep disturbances, and depression.
Clinically, patients with PART pathology are frequently diagnosed as having AD with mild cognitive impairments. They usually preserve cognitive functions and lack aphasia, apraxia, or agnosia, reflecting the lack of lobar degeneration. Some patients may be diagnosed as having SD or senile psychosis, since they frequently present with psychosis including delusion, depression, and agitation, 106 , 107 although delusion of persecution is also seen in AD. We previously reported abnormal tau accumulation in the subcortical nuclei, especially in the nucleus accumbens of demented patients with PART 106 (Fig. 3d). Those results suggest that abnormal tau aggregation would propagate via reward neural circuitry, which may be associated with emotion and psychiatric symptoms. Interestingly, retrospective studies of autopsied late‐onset schizophrenia or paraphrenia have revealed massive limbic tau pathology with sparse Aβ deposits, similar to PART. 108 Further studies are needed validate the PART concept and identify clinical biomarkers, as well as to disentangle the biological mechanisms in SD and to discover new biologic targets for the treatment of cognitive impairment in individuals with SD.
Argyrophric grain diseases
AGD is a sporadic 4R tauopathy presenting with NFTs and a characteristic massive occurrence of argyrophilic and tau‐positive grains in the neuropil as revealed by Gallyas‐Braak staining (Fig. 3e). The main affected regions are limbic regions including the hippocampus, entorhinal regions, and subsequently the amygdala nucleus, where mild to moderate tissue degeneration is also often observed. Usually, people at the most advanced age show such pathology as well as PART. Pathologically, grains start from the ambient gyrus and amygdala and progress anteriorly or posteriorly in medial temporal lobes. 109 Patients with AGD present with many pretangle‐like phosphorylated tau–positive inclusions in the transenorhinal cortex (Fig. 3f). They frequently show prominent neuropsychiatric features including delusion, irritability, agitation, and apathy besides amnesia, although other cognitive functions are relatively preserved. 110 Their personality change is frequently characterized by emotional disorders with aggression or ill‐temper. 111 Some patients with AGD have demonstrated depression with aggression and irritability, 110 , 111 although the frequency of the symptoms might be low. 112 Clinical features might be associated with the extensive accumulation of AGD pathology in limbic regions, 110 especially in the amygdala. However, the precise mechanisms are unknown. Notably, recent Japanese neuropathological studies found that patients with bipolar disorders or late‐onset schizophrenia presented with high frequencies of AGD pathology. 113 , 114 The relationship between AGD and SD remains to be fully established.
Lewy body disease
Clinicopathological concept of LBD
LBD is clinically defined as a chronic progressive neuropsychiatric disorder, which includes Parkinson disease (PD), PD dementia (PDD), and DLB. 115 In current PD diagnostic criteria, the motor syndrome is the core feature, as well as non‐motor manifestations such as depression, rapid eye movement sleep behavior disorder (RBD), dysautonomia, and hyposmia. 116 In the setting of well‐established PD, a minimum of 1 year with parkinsonism until onset of dementia is recommended to define PDD116. DLB is the second most common neurodegenerative dementia after AD. In the current DLB clinical diagnostic criteria, probable DLB requires two or more core features (fluctuating cognition, parkinsonism, visual hallucinations, or RBD) or one core feature plus one indicative biomarker (low striatal DAT uptake, reduced cardiac [123I]meta‐iodobenzylguanidine uptake, or rapid eye movement sleep without atonia on polysomnography). 117 Supportive clinical features of DLB include psychiatric symptoms, dysautonomia, and hyposmia. The psychiatric symptoms include depression, apathy, anxiety, hallucinations in other modalities, and systematized delusions.
Lewy bodies (LBs) and Lewy neurites in the central, peripheral, and autonomic nervous system are the histopathologic hallmark of LBD. In PD, the essential neuropathology is moderate to severe neuronal loss in the pars compacta of the substantia nigra associated with Lewy pathology, which is the neurobiological basis of the extrapyramidal motor features. 118 Brainstem‐type LBs have an eosinophilic core and a pale‐staining halo that is highly immunoreactive for α‐synuclein (Fig. 4a,c), while cortical‐type LBs have less well‐defined eosinophilic intraneuronal inclusions without a halo, and can be identified by α‐synuclein‐immunostaining (Fig. 4b,d). According to the pathological criteria of DLB, 119 LBD is pathologically classified into three subtypes: brainstem‐predominant LBD, transitional LBD, and diffuse neocortical LBD. In patients with LBD, parkinsonism is postulated to occur after a critical level of nigral neuronal loss is reached. 115 As cortical involvement of Lewy pathology is a key pathological basis in the development of dementia, transitional LBD or diffuse neocortical LBD is mandatory for inclusion in the high‐likelihood DLB category which is associated with the DLB clinical syndrome. 117 , 119 In contrast, most patients with a clinical diagnosis of PD without cognitive impairment typically have brainstem‐predominant LBD at autopsy. There is considerable pathological overlap between DLB and PDD, but the degree of cerebral β‐amyloid accumulation is significantly greater in DLB than PDD. Many clinicopathological studies have demonstrated that the degree of cerebral β‐amyloid accumulation is related to the cortical α‐synuclein burden and the timing of the onset of dementia relative to that of parkinsonism in LBD.
Fig. 4.
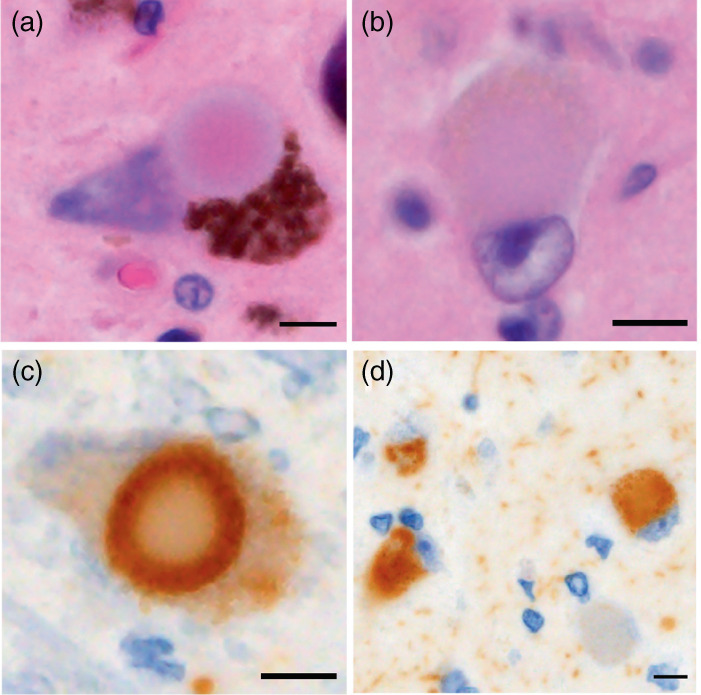
Lewy body (LB) pathology. LBs are eosinophilic intraneuronal inclusions stained with hematoxylin and eosin. The brainstem type of LBs has a pale‐staining halo (a), but the cortical type lacks the halo (b). Immunostaining for α‐synuclein reveals LBs in the brainstem nuclei (c) as well as in the cortices (d). Scale bar: 5 μm.
Depression is commonly observed even in prodromal stages of patients with LBD. In terms of neural circuity, it is unclear whether a specific pathological basis for the development of depression in patients with LBD is present or absent. Regarding the anatomical brain regions associated with depression, early clinicopathological studies in brains of patients with LBD reported inconsistent results. 112 , 120 , 121 As to monoaminergic systems, recent clinicopathological studies reveal the involvement of nigrostriatal degeneration with depression across the LBD spectrum. 122 , 123 , 124 Regarding the HPA axis, Lewy pathology is frequently identified in the hypothalamus, pituitary lobe, and adrenal gland, 125 , 126 , 127 and clinical studies found high levels of cortisol to be associated with depression in patients with PD. 128 However, there has been no clinicopathological investigation of the relationship of the neuroendocrine signaling system to depression. Regarding neuroinflammation in the brains of patients with LBD, increased numbers of microglia in several brain regions were detected immunohistochemically. 129 , 130 Some studies, however, failed to find significant microglia activation in brains of patients with DLB. 131 , 132 As changes in brain tissue may represent the end‐stage of disease, the temporal trajectories of inflammatory markers may reflect disease progression.
Incidental LBD and prodromal LBD
Lewy pathology is identified in the brains of deceased individuals with no history of parkinsonism or dementia while alive. This clinicopathological condition is termed incidental LBD, and occurs in 8% to 12% of elderly individuals aged 60 years and older as seen with routine histological methods. 118 Clinicopathological studies revealed that incidental LBD is associated with the presence of LB‐related symptoms, including RBD, depression, olfactory dysfunction, and constipation. 118 , 133 There are several neuropathological studies regarding late‐life depression in cognitively unimpaired individuals. In a community‐based study, 134 Tsopelas et al. found that late‐life depression was significantly associated with the presence of LBs in the substantia nigra and locus coeruleus in 153 nondemented individuals (mean age at death, 84.3 years). 135 Wilson et al. reported an association between brainstem aminergic nuclei and late‐life depressive symptoms in 124 nondemented community‐dwelling individuals (mean age at death, 87.7 years). 135 A higher level of depressive symptoms was associated with a higher density of LBs in the brainstem. Sweet et al. clinicopathologically followed up nine consecutive patients with late‐life major depression until autopsy, 136 and, of six patients who developed dementia, three were classified into a high‐likelihood category of DLB. One of the remaining three patients had incidental LBD. Nagao et al. pathologically compared 11 patients with depression (mean age at onset, 62.3 ± 8.8 years) to 71 normal controls, and found no difference in the prevalence of LBD between them (27.3% vs 11.3%). 114 However, when they focused on individuals aged 65 years and older, the prevalence of LBD was significantly higher in patients with late‐onset depression (60%) than in controls (10.7%).
Considering that LB‐related symptoms commonly precede the onset of LBD, the presence of LB‐related clinical features in patients with depression may help us to understand the pathophysiological process of LBD. 115 The clinical research criteria for prodromal PD/DLB have stimulated further clinical studies. 137 , 138 In the prodromal PD criteria, a diagnosis of depression is defined as a clinical nonmotor marker with a likelihood ratio of 1.8. In the prodromal DLB criteria, a psychiatric‐onset subtype is proposed as one of three prototypical forms. The most common presentation of psychiatric‐onset prodromal DLB is late‐onset depressive disorder.
Can we identify prodromal LBD in patients with late‐onset depression?
Clinical data regarding potential underlying pathophysiology of LBD in patients with late‐onset depression has been gradually accumulating. However, few studies have followed up the patients until conversion to PD/DLB. Two longitudinal follow‐up studies of older patients with idiopathic RBD who received psychiatric treatment have been reported. Wing et al. found that premorbid psychiatric disorders, especially depression, were associated with increased risk of PD after adjusting for sex, age at RBD diagnosis, and smoking status (hazard ratio, 7.0; 95% confidence interval, 1.3–38.3). 139 Postuma et al. reported that patients with RBD taking antidepressants (mean age, 64.1 years) had a lower risk of developing neurodegenerative disease than those without antidepressant use (5‐year risk = 22% vs 59%; risk ratio, 0.22 [95% confidence interval, 0.06–0.74]). 140 As for autonomic abnormality, diagnostic utility of the ventilatory response to hypercapnia for identification of prodromal DLB was reported. 141 Of 18 patients with late‐onset MDD who developed DLB during the observation period (mean age at diagnosis of MDD, 61.4 years), cardiac [123I]meta‐iodobenzylguanidine abnormality was seen in 11 patients (61.1%). Other autonomic findings were not different, but the frequency of hypersensitivity to psychotropics was significantly higher in converters compared with nonconverters. In this study, the period until conversion to DLB ranged from ≈5 to 10 years.
Recent studies have shown that a subset of patients with late‐onset depressive disorder share common clinical features with LBD. In a cross‐sectional study, 36 patients with late‐onset depressive disorder (mean age, 67.4 years) and 30 healthy controls underwent detailed assessment for clinical features of LBD. 7 Late‐onset depression was significantly associated with increased rates of both motor and nonmotor features of LBD. Of the 29 patients with late‐onset depression who underwent 123I‐ioflupane single‐photon emission computed tomography, seven with abnormal DAT binding showed significantly higher Unified Parkinson's Disease Rating Scale motor scores (mean, 4.6) than the 22 without abnormal DAT binding (mean, 2.2). There were, however, no differences in other examined clinical features between groups with and without DAT abnormality. Krüger et al. reported the association of olfaction deficit with neuroleptic‐induced parkinsonism. 142 In 79 patients with MDD, 15 with psychotic features who developed neuroleptic‐induced parkinsonism showed significantly lower olfactory scores than 44 patients with psychotic features without neuroleptic‐induced parkinsonism and 20 without psychotic features. Partial correlations controlling for age in 15 patients with neuroleptic‐induced parkinsonism showed that odor threshold and odor identification were significantly correlated with Unified Parkinson's Disease Rating Scale motor scores. A multicenter case‐control study found that the prevalence of depression and concomitant antidepressant use were significantly higher in patients with idiopathic RBD (mean age, 67.3 years) than in controls. 143 Although the link between antidepressant usage and subsequent RBD onset is a potential confounder, a clinic‐based two‐phase epidemiological study found that 8.77% of patients with MDD (mean age, 54.0 years) in the psychiatric outpatient service had video polysomnography–confirmed RBD (Wang et al. 144 ). In addition to these patients with MDD, several studies noted that middle‐aged and older patients with antidepressant‐associated RBD showed some clinical markers of underlying LBD. 116 , 133 , 139 A naturalistic follow‐up study reported that PSG‐confirmed RBD symptoms persisted despite discontinuing or switching antidepressants. 145 These findings suggest that antidepressant‐associated RBD may represent an early phase of LBD, but the temporal trajectories of the underlying LBD markers in patients with late‐onset depression remain to be established.
Although a direct biomarker of LB‐related pathology is not yet established for clinical diagnosis, detection of LB‐related pathology in tissue biopsies and surgical resections has emerged as a potential candidate. 146 Hall et al. also reported that α‐synuclein real‐time quaking‐induced conversion of cerebrospinal fluid samples is highly sensitive and specifc for identifying cases with autopsy‐confirmed LBD. 147 Considering the long‐term nature and diversity of LBD, it remains unclear whether patients with late‐onset depression who have LBD markers may fulfill the clinical criteria of PD/DLB during their lifetime. Further follow‐up studies with pathological verification are needed to determine the clinical significance of LBD markers in patients with late‐onset depression.
Treatment for Depression in the Context of Age‐Related Neuropathology
Overview of antidepressant treatment for SD
Nonpharmacological intervention is generally the treatment of choice, since elderly patients with dementia are more vulnerable to adverse effects of medications. The National Institute for Health and Care Excellence 2018 guideline recommends psychological treatments for mild to moderate SD in patients with mild to moderate dementia. 148 An earlier Cochrane review and meta‐analysis suggested that psychological treatments were effective in reducing depressive and anxiety symptoms in MCI and dementia patients. 149 A recent scoping review of 20 studies of randomized controlled trials on nonpharmacological interventions for SD and apathy with MCI or mild to moderate dementia instead found that effective interventions for depressive symptoms in single studies were mostly emotion‐oriented and/or stimulation‐oriented approaches. 150 Only a few of these studies set SD as the primary outcome of intervention. That is, dementia patients with depressive symptoms rather than syndromic SD were recruited. In addition, subtypes of dementia were often not specified. Another approach is to target modifiable factors on the premise that improvement of these factors by nonpharmacological interventions may benefit depressive symptoms as well. A systematic review identified pain, neuropsychiatric symptoms, cognitive decline, social isolation, and quality of life as five potentially modifiable factors associated with SD in community‐dwelling individuals. 151
The pharmacological intervention trials mainly recruited patients with either unspecified dementia or AD. As to antidepressants, a Cochrane review identified only four unconfounded, double‐blind, randomized trials comparing antidepressant drugs with placebo eligible for meta‐analysis, with a total of only 137 patients with SD and dementia. 152 Two of the four studies investigated tricyclic antidepressants, and the other two investigated selective serotonin reuptake inhibitors (fluoxetine and sertraline). Again, the meta‐analysis found that antidepressants were not more effective than placebo in terms of SD symptom rating scales, cognitive function or daily‐living activities, and were associated with more frequent adverse events and dropouts. Accordingly, the routine offer of antidepressants to manage mild to moderate SD in people living with mild to moderate dementia was discouraged. 148 This suggestion is supported by a recent systematic review and meta‐analysis, which found 10 interventions more effective than usual care for improving depressive symptoms in patients with dementia without the diagnosis of MDD; none of the 10 interventions involved a drug alone. However, in the real world, antidepressants are still commonly prescribed to patients with dementia. 153 One possible reason is that implementation of nonpharmacological intervention is not always feasible. The many comorbidities, mental and physical constraints, and even depressive symptoms per se may prevent patients with dementia from being engaged in physical activities. 154 There are individual trials showing that antidepressant treatment is more effective than placebo, 155 , 156 for example in the case of agomelatine, a newer antidepressant. 156 Overall, there is only limited evidence regarding pharmacotherapy for SD in patients with dementia, although it is still recommended by expert consensus and clinical guidelines. 148 , 157 , 158 , 159 Antidepressants with procognitive effects (duloxetine, tianeptine, and vortioxetine) and other dual‐action antidepressants (e.g. venlafaxine, desvenlafaxine, mirtazapine) may be preferable. 160
Cholinesterase inhibitors are standard treatment for cognitive decline in patients with AD, and are effective to ameliorate neuropsychiatric symptoms of dementia, including symptoms of SD. 161 , 162 , 163 , 164 Postmarketing surveillance of another cognitive enhancer, memantine, an NMDA receptor antagonist, showed improvement of affective symptoms in patients with moderately severe to severe AD at 20 mg/day for 6 months. 165 A double‐blind, randomized, placebo‐controlled trial in patients with late‐life SD with subjective memory complaints found a combination of memantine with escitalopram was more effective than escitalopram alone in improving cognitive measures but not depression outcomes at 12 months. 166
ECT is an invasive brain stimulation method that is highly effective for management of treatment‐resistant depression and SD. 167 , 168 Although ECT is associated with cognitive side effects such as temporary anterograde and/or retrograde amnesia, cognitive abnormalities related to ECT usually resolve within 3 days posttreatment and several cognitive domains appear to be better than baseline 15 days later. 169 However, no randomized controlled trials of ECT for treating SD in patients with dementia or MCI have been reported. Observational studies have supported the efficacy and tolerability of ECT, and cognitive side effects usually recover within 2 months. 170 , 171 , 172 , 173
Vascular depression
The response of vascular depression to antidepressant treatment is usually attenuated, and safety and tolerability further limit the use of antidepressants. 174 Since white matter hyperintensity is related to disease progression and poor prognosis, 175 , 176 management of vascular risk factors may be both preventive and therapeutic. An early double‐blind, randomized, controlled trial compared fluoxetine alone and fluoxetine augmented with nimodipine, a calcium channel blocker, in the treatment of vascular depression. Augmentation with nimodipine resulted in greater improvement of depressive symptoms, greater chance of remission, and less recurrence. 177 However, whether antihypertensive drugs, oral hypoglycemic medications, antihyperlipidemic drugs, or anticoagulant can improve acute or long‐term outcomes of vascular depression remains unclear. In addition to pharmacotherapy, brain stimulation by different neuromodulatory methods has been explored for treating vascular depression. A randomized sham‐controlled study found that repetitive transcranial magnetic stimulation delivered to the left dorsolateral prefrontal cortex was more effective than sham stimulation for patients who discontinued antidepressant therapy before brain stimulation. 178 Another double‐blind, randomized, sham‐controlled study revealed superior efficacy of add‐on of transcranial direct current stimulation to sertraline versus sertraline alone. 179
Lewy body disease
Although identification of SD as prodromal DLB is not established, trials for treatment of depression in patients with PD, potentially regarded as a predementia state of PDD, have already been performed. Specifically for SD in patients with PD, meta‐analyses supported better‐than‐placebo efficacy for cognitive‐behavioral therapy, antidepressants overall, type‐B selective monoamine oxidase inhibitors, selective serotonin reuptake inhibitors, and tricyclic antidepressants. 180 , 181 Another meta‐analysis found moderate though statistically insignificant effects of antidepressants in treating SD in patients with PD.181 Tricyclic antidepressants were more effective than selective serotonin reuptake inhibitors. 182 However, concerns regarding tolerability and unwanted anticholinergic effects make tricyclics less favorable choices, especially for patients with cardiac problems, angle‐closure glaucoma, urinary retention, and constipation. Serotonergic antidepressants may worsen motor symptoms through inhibition of dopaminergic neurons in the substantia nigra, and sertraline may be preferable in this regard. 157 , 183 Consensus guidelines also support the use of dual‐acting antidepressants such as serotonin‐norepinephrine reuptake inhibitors, vortioxetine, bupropion, and mirtazapine. 157 Among anti‐Parkinsonian medications, some evidence supports the use of pramipexole, a potent dopamine D2 agonist, in treating SD in patients with PD. 184 , 185 , 186
ECT seems to be an important therapeutic modality for SD in patients with Lewy body dementia, since there have been few randomized controlled trials examining the efficacy and safety of antidepressants. 187 A systematic review summarized several case reports or case series studies of ECT for SD treatment in patients with Lewy body dementia. 188 ECT was effective in five studies with a total sample size of <30, but it was unclear whether the improvements could be sustained. Considering the changes of dopaminergic transmission seen by neuroimaging in patients with MDD responding to ECT, progressive nigrostriatal neurodegeneration may affect the clinical course, such as the recurrence of depression in LBD. 189 A recent case series showed ultrabrief right unilateral ECT to be effective and well‐tolerated for the treatment of agitation and depressive symptoms in seven patients with Lewy body dementia. 190 Right unilateral ECT was used because it causes fewer cognitive side effects than traditional bitemporal ECT and is equally effective.
Conclusions and Future Directions
This review focuses on the current understanding of the pathophysiology of SD and its neuropathological features on the basis of established pathological diagnoses. Depression commonly occurs in patients with neurodegenerative disorders, despite different underlying pathological diagnoses. In late‐onset depression, tau and synuclein might be the most influential accumulated proteins. The pathophysiology of SD also involves cerebrovascular pathology, neuroinflammation and neurodegeneration, and depressive symptoms or senile psychiatric manifestations have been associated with vulnerability of specific brain regions, including limbic regions and the subcortical nuclei, rather than the nature of accumulated proteins. Shared pathophysiological mechanisms are considered to be involved in depression and neurodegenerative disorders. Disease‐modifying therapy targeting neurodegeneration‐linked proteins and inflammatory processes might improve the quality of life of depressive patients and their caregivers. In considering appropriate therapy for the elderly, comorbidity of depression and dementia, as well as the underlying distinct neuropathological features, should be taken into account. Pharmacological approaches based on the common affected neuronal circuits are a promising avenue for research, although SD is multifaceted, and psychogenic, environmental and physical factors must be considered. In terms of preventing the development of dementia, it remains important to seek alternatives to conventional antidepressant therapies, such as anti‐inflammatory therapy, based on common pathophysiological processes between SD and dementia.
Author contributions
Conception, design of the study, and first draft of the manuscript: I.K., J.I., S.T., Y.T.L., and H.F.; first draft of the figures: I.K., J.I., and H.F.; critical review and edits: I.K. and H.F.
Disclosure statement
The authors declare that they have no conflicts of interest.
Ethics Approval
This study was performed in accordance with the ethical standards outlined in the 1964 Declaration of Helsinki and its later amendments.
Acknowledgments
This research was supported by The Naito Foundation (to I.K.), JSPS KAKENHI grant number 20K16660 (to I.K.) and 22K07597 (to J.I.), and the Japan Agency for Medical Research and Development JP22dk0207053 (to J.I.)
Contributor Information
Ito Kawakami, Email: i.kawakami.vn@juntendo.ac.jp.
Hiroshige Fujishiro, Email: fujishiro17@hotmail.co.jp.
References
- 1. Djernes JK. Prevalence and predictors of depression in populations of elderly: A review. Acta Psychiatr. Scand. 2006; 113: 372–387. [DOI] [PubMed] [Google Scholar]
- 2. Blazer DG. Depression in late life: Review and commentary. J. Gerontol. A Biol. Sci. Med. Sci. 2003; 58: 249–265. [DOI] [PubMed] [Google Scholar]
- 3. Baba H, Kito S, Nukariya K et al. Guidelines for diagnosis and treatment of depression in older adults: A report from the Japanese society of mood disorders. Psychiatry Clin. Neurosci. 2022; 76: 222–234. [DOI] [PubMed] [Google Scholar]
- 4. Aguera‐Ortiz L, Claver‐Martin MD, Franco‐Fernandez MD et al. Depression in the elderly. Consensus statement of the Spanish psychogeriatric association. Front. Psych. 2020; 11: 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Alexopoulos GS, Young RC, Meyers BS. Geriatric depression: Age of onset and dementia. Biol. Psychiatry 1993; 34: 141–145. [DOI] [PubMed] [Google Scholar]
- 6. Moriguchi S, Takahata K, Shimada H et al. Excess tau PET ligand retention in elderly patients with major depressive disorder. Mol. Psychiatry 2021; 26: 5856–5863. [DOI] [PubMed] [Google Scholar]
- 7. Kazmi H, Walker Z, Booij J et al. Late onset depression: Dopaminergic deficit and clinical features of prodromal Parkinson's disease: A cross‐sectional study. J. Neurol. Neurosurg. Psychiatry 2021; 92: 158–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mackin RS, Insel PS, Landau S et al. Late‐life depression is associated with reduced cortical amyloid burden: Findings from the Alzheimer's disease neuroimaging initiative depression project. Biol. Psychiatry 2021; 89: 757–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liew TM. Neuropsychiatric symptoms in cognitively normal older persons, and the association with Alzheimer's and non‐Alzheimer's dementia. Alzheimers Res. Ther. 2020; 12: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer's disease. Mol. Neurodegener. 2019; 14: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hidaka S, Ikejima C, Kodama C et al. Prevalence of depression and depressive symptoms among older Japanese people: Comorbidity of mild cognitive impairment and depression. Int. J. Geriatr. Psychiatry 2012; 27: 271–279. [DOI] [PubMed] [Google Scholar]
- 12. Hegeman JM, Kok RM, van der Mast RC, Giltay EJ. Phenomenology of depression in older compared with younger adults: Meta‐analysis. Br. J. Psychiatry 2012; 200: 275–281. [DOI] [PubMed] [Google Scholar]
- 13. Jaaskelainen E, Juola T, Korpela H et al. Epidemiology of psychotic depression – systematic review and meta‐analysis. Psychol. Med. 2018; 48: 905–918. [DOI] [PubMed] [Google Scholar]
- 14. Mallery L, MacLeod T, Allen M et al. Systematic review and meta‐analysis of second‐generation antidepressants for the treatment of older adults with depression: Questionable benefit and considerations for frailty. BMC Geriatr. 2019; 19: 306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mitchell AJ, Subramaniam H. Prognosis of depression in old age compared to middle age: A systematic review of comparative studies. Am. J. Psychiatry 2005; 162: 1588–1601. [DOI] [PubMed] [Google Scholar]
- 16. Mueller TI, Kohn R, Leventhal N et al. The course of depression in elderly patients. Am. J. Geriatr. Psychiatry 2004; 12: 22–29. [PubMed] [Google Scholar]
- 17. Sachs‐Ericsson N, Corsentino E, Moxley J et al. A longitudinal study of differences in late‐ and early‐onset geriatric depression: Depressive symptoms and psychosocial, cognitive, and neurological functioning. Aging Ment. Health 2013; 17: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Janssen J, Hulshoff Pol HE, de Leeuw FE et al. Hippocampal volume and subcortical white matter lesions in late life depression: Comparison of early and late onset depression. J. Neurol. Neurosurg. Psychiatry 2007; 78: 638–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eraydin IE, Mueller C, Corbett A et al. Investigating the relationship between age of onset of depressive disorder and cognitive function. Int. J. Geriatr. Psychiatry 2019; 34: 38–46. [DOI] [PubMed] [Google Scholar]
- 20. Hashem AH, Nasreldin M, Gomaa MA, Khalaf OO. Late versus early onset depression in elderly patients: Vascular risk and cognitive impairment. Curr. Aging Sci. 2017; 10: 211–216. [DOI] [PubMed] [Google Scholar]
- 21. Mackin RS, Nelson JC, Delucchi KL et al. Association of age at depression onset with cognitive functioning in individuals with late‐life depression and executive dysfunction. Am. J. Geriatr. Psychiatry 2014; 22: 1633–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sachs‐Ericsson N, Moxley JH, Corsentino E et al. Melancholia in later life: Late and early onset differences in presentation, course, and dementia risk. Int. J. Geriatr. Psychiatry 2014; 29: 943–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grayson L, Thomas A. A systematic review comparing clinical features in early age at onset and late age at onset late‐life depression. J. Affect. Disord. 2013; 150: 161–170. [DOI] [PubMed] [Google Scholar]
- 24. Dols A, Bouckaert F, Sienaert P et al. Early‐ and late‐onset depression in late life: A prospective study on clinical and structural brain characteristics and response to electroconvulsive therapy. Am. J. Geriatr. Psychiatry 2017; 25: 178–189. [DOI] [PubMed] [Google Scholar]
- 25. Coryell W, Leon A, Winokur G et al. Importance of psychotic features to long‐term course in major depressive disorder. Am. J. Psychiatry 1996; 153: 483–489. [DOI] [PubMed] [Google Scholar]
- 26. Vermeulen T, Lauwers T, Van Diermen L, Sabbe BG, van der Mast RC, Giltay EJ. Cognitive deficits in older adults with psychotic depression: A meta‐analysis. Am. J. Geriatr. Psychiatry 2019; 27: 1334–1344. [DOI] [PubMed] [Google Scholar]
- 27. Asmer MS, Kirkham J, Newton H et al. Meta‐analysis of the prevalence of major depressive disorder among older adults with dementia. J. Clin. Psychiatry 2018; 79(5): 17r11772. [DOI] [PubMed] [Google Scholar]
- 28. Ismail Z, Elbayoumi H, Fischer CE et al. Prevalence of depression in patients with mild cognitive impairment: A systematic review and meta‐analysis. JAMA Psychiat. 2017; 74: 58–67. [DOI] [PubMed] [Google Scholar]
- 29. Kurzyna A, Goscik E, Gozdziewski A, Skotnicka B, Hassmann‐Poznanska E. Presentation, diagnosis and management of neck abscesses in children. Otolaryngol. Pol. 2015; 69: 1–8. [DOI] [PubMed] [Google Scholar]
- 30. Fujishiro H, Iseki E, Nakamura S et al. Dementia with Lewy bodies: Early diagnostic challenges. Psychogeriatrics 2013; 13: 128–138. [DOI] [PubMed] [Google Scholar]
- 31. Malpetti M, Jones PS, Tsvetanov KA et al. Apathy in presymptomatic genetic frontotemporal dementia predicts cognitive decline and is driven by structural brain changes. Alzheimers Dement. 2021; 17: 969–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Singh‐Manoux A, Dugravot A, Fournier A et al. Trajectories of depressive symptoms before diagnosis of dementia: A 28‐year follow‐up study. JAMA Psychiat. 2017; 74: 712–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Saez‐Fonseca JA, Lee L, Walker Z. Long‐term outcome of depressive pseudodementia in the elderly. J. Affect. Disord. 2007; 101: 123–129. [DOI] [PubMed] [Google Scholar]
- 34. Mori H, Funahashi Y, Yoshino Y et al. Blood CDKN2A gene expression in aging and neurodegenerative diseases. J. Alzheimers Dis. 2021; 82: 1737–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kumon H, Yoshino Y, Funahashi Y et al. PICALM mRNA expression in the blood of patients with neurodegenerative diseases and geriatric depression. J. Alzheimers Dis. 2021; 79: 1055–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ochi S, Iga JI, Funahashi Y et al. Identifying blood transcriptome biomarkers of Alzheimer's disease using transgenic mice. Mol. Neurobiol. 2020; 57: 4941–4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jack CR Jr, Knopman DS, Jagust WJ et al. Tracking pathophysiological processes in Alzheimer's disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013; 12: 207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Smith GS, Kuwabara H, Gould NF et al. Molecular imaging of the serotonin transporter availability and occupancy by antidepressant treatment in late‐life depression. Neuropharmacology 2021; 194: 108447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Taylor WD, Boyd BD, Elson D et al. Preliminary evidence that cortical amyloid burden predicts poor response to antidepressant medication treatment in cognitively intact individuals with late‐life depression. Am. J. Geriatr. Psychiatry 2021; 29: 448–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Byers AL, Yaffe K. Depression and risk of developing dementia. Nat. Rev. Neurol. 2011; 7: 323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Butters MA, Young JB, Lopez O et al. Pathways linking late‐life depression to persistent cognitive impairment and dementia. Dialogues Clin. Neurosci. 2008; 10: 345–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. O'Brien JT, Ames D, Schweitzer I, Colman P, Desmond P, Tress B. Clinical and magnetic resonance imaging correlates of hypothalamic‐pituitary‐adrenal axis function in depression and Alzheimer's disease. Br. J. Psychiatry 1996; 168: 679–687. [DOI] [PubMed] [Google Scholar]
- 43. Cereseto M, Reines A, Ferrero A, Sifonios L, Rubio M, Wikinski S. Chronic treatment with high doses of corticosterone decreases cytoskeletal proteins in the rat hippocampus. Eur. J. Neurosci. 2006; 24: 3354–3364. [DOI] [PubMed] [Google Scholar]
- 44. Lupien SJ, de Leon M, de Santi S et al. Cortisol levels during human aging predict hippocampal atrophy and memory deficits. Nat. Neurosci. 1998; 1: 69–73. [DOI] [PubMed] [Google Scholar]
- 45. Ishii M, Wang G, Racchumi G, Dyke JP, Iadecola C. Transgenic mice overexpressing amyloid precursor protein exhibit early metabolic deficits and a pathologically low leptin state associated with hypothalamic dysfunction in arcuate neuropeptide Y neurons. J. Neurosci. 2014; 34: 9096–9106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen WW, Zhang X, Huang WJ. Role of neuroinflammation in neurodegenerative diseases (review). Mol. Med. Rep. 2016; 13: 3391–3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Santos LE, Beckman D, Ferreira ST. Microglial dysfunction connects depression and Alzheimer's disease. Brain Behav. Immun. 2016; 55: 151–165. [DOI] [PubMed] [Google Scholar]
- 48. Noh H, Jeon J, Seo H. Systemic injection of LPS induces region‐specific neuroinflammation and mitochondrial dysfunction in normal mouse brain. Neurochem. Int. 2014; 69: 35–40. [DOI] [PubMed] [Google Scholar]
- 49. Zhou L, Miranda‐Saksena M, Saksena NK. Viruses and neurodegeneration. Virol. J. 2013; 10: 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Baune BT, Smith E, Reppermund S et al. Inflammatory biomarkers predict depressive, but not anxiety symptoms during aging: The prospective Sydney memory and aging study. Psychoneuroendocrinology 2012; 37: 1521–1530. [DOI] [PubMed] [Google Scholar]
- 51. Najjar S, Pearlman DM, Devinsky O, Najjar A, Zagzag D. Neurovascular unit dysfunction with blood‐brain barrier hyperpermeability contributes to major depressive disorder: A review of clinical and experimental evidence. J. Neuroinflammation 2013; 10: 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age‐of‐onset distributions of DSM‐IV disorders in the National Comorbidity Survey Replication. Arch. Gen. Psychiatry 2005; 62: 593–602. [DOI] [PubMed] [Google Scholar]
- 53. Yirmiya R, Rimmerman N, Reshef R. Depression as a microglial disease. Trends Neurosci. 2015; 38: 637–658. [DOI] [PubMed] [Google Scholar]
- 54. Dowlati Y, Herrmann N, Swardfager W et al. A meta‐analysis of cytokines in major depression. Biol. Psychiatry 2010; 67: 446–457. [DOI] [PubMed] [Google Scholar]
- 55. Khemka VK, Ganguly A, Bagchi D et al. Raised serum proinflammatory cytokines in Alzheimer's disease with depression. Aging Dis. 2014; 5: 170–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. De Felice FG, Ferreira ST. Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes 2014; 63: 2262–2272. [DOI] [PubMed] [Google Scholar]
- 57. Katon W, Pedersen HS, Ribe AR et al. Effect of depression and diabetes mellitus on the risk for dementia: A national population‐based cohort study. JAMA Psychiat. 2015; 72: 612–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Douaud G, Lee S, Alfaro‐Almagro F et al. SARS‐CoV‐2 is associated with changes in brain structure in UK biobank. Nature 2022; 604: 697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Herrmann LL, Le Masurier M, Ebmeier KP. White matter hyperintensities in late life depression: A systematic review. J. Neurol. Neurosurg. Psychiatry 2008; 79: 619–624. [DOI] [PubMed] [Google Scholar]
- 60. Thomas AJ, O'Brien JT, Barber R, McMeekin W, Perry R. A neuropathological study of periventricular white matter hyperintensities in major depression. J. Affect. Disord. 2003; 76: 49–54. [DOI] [PubMed] [Google Scholar]
- 61. Tham MW, Woon PS, Sum MY, Lee TS, Sim K. White matter abnormalities in major depression: Evidence from post‐mortem, neuroimaging and genetic studies. J. Affect. Disord. 2011; 132: 26–36. [DOI] [PubMed] [Google Scholar]
- 62. Kumar A, Miller D, Ewbank D et al. Quantitative anatomic measures and comorbid medical illness in late‐life major depression. Am. J. Geriatr. Psychiatry 1997; 5: 15–25. [PubMed] [Google Scholar]
- 63. Fazekas F, Chawluk JB, Alavi A, Hurtig HI, Zimmerman RA. MR signal abnormalities at 1.5 T in Alzheimer's dementia and normal aging. AJR Am. J. Roentgenol. 1987; 149: 351–356. [DOI] [PubMed] [Google Scholar]
- 64. Kim KW, MacFall JR, Payne ME. Classification of white matter lesions on magnetic resonance imaging in elderly persons. Biol. Psychiatry 2008; 64: 273–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Barber R, Scheltens P, Gholkar A et al. White matter lesions on magnetic resonance imaging in dementia with Lewy bodies, Alzheimer's disease, vascular dementia, and normal aging. J. Neurol. Neurosurg. Psychiatry 1999; 67: 66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wahlund LO, Barkhof F, Fazekas F et al. A new rating scale for age‐related white matter changes applicable to MRI and CT. Stroke 2001; 32: 1318–1322. [DOI] [PubMed] [Google Scholar]
- 67. Alexopoulos GS, Meyers BS, Young RC, Campbell S, Silbersweig D, Charlson M. 'Vascular depression' hypothesis. Arch. Gen. Psychiatry 1997; 54: 915–922. [DOI] [PubMed] [Google Scholar]
- 68. Krishnan KR, Hays JC, Blazer DG. MRI‐defined vascular depression. Am. J. Psychiatry 1997; 154: 497–501. [DOI] [PubMed] [Google Scholar]
- 69. Krishnan KR, McDonald WM, Doraiswamy PM et al. Neuroanatomical substrates of depression in the elderly. Eur. Arch. Psychiatry Clin. Neurosci. 1993; 243: 41–46. [DOI] [PubMed] [Google Scholar]
- 70. Lloyd AJ, Ferrier IN, Barber R, Gholkar A, Young AH, O'Brien JT. Hippocampal volume change in depression: Late‐ and early‐onset illness compared. Br. J. Psychiatry 2004; 184: 488–495. [DOI] [PubMed] [Google Scholar]
- 71. Hickie I, Naismith S, Ward PB et al. Vascular risk and low serum B12 predict white matter lesions in patients with major depression. J. Affect. Disord. 2005; 85: 327–332. [DOI] [PubMed] [Google Scholar]
- 72. Walther S, Hugli S, Hofle O et al. Frontal white matter integrity is related to psychomotor retardation in major depression. Neurobiol. Dis. 2012; 47: 13–19. [DOI] [PubMed] [Google Scholar]
- 73. Nebes RD, Vora IJ, Meltzer CC et al. Relationship of deep white matter hyperintensities and apolipoprotein E genotype to depressive symptoms in older adults without clinical depression. Am. J. Psychiatry 2001; 158: 878–884. [DOI] [PubMed] [Google Scholar]
- 74. Brown FW, Lewine RJ, Hudgins PA, Risch SC. White matter hyperintensity signals in psychiatric and nonpsychiatric subjects. Am. J. Psychiatry 1992; 149: 620–625. [DOI] [PubMed] [Google Scholar]
- 75. Uranova NA, Vostrikov VM, Orlovskaya DD, Rachmanova VI. Oligodendroglial density in the prefrontal cortex in schizophrenia and mood disorders: A study from the Stanley neuropathology consortium. Schizophr. Res. 2004; 67: 269–275. [DOI] [PubMed] [Google Scholar]
- 76. Sheline YI, Price JL, Vaishnavi SN et al. Regional white matter hyperintensity burden in automated segmentation distinguishes late‐life depressed subjects from comparison subjects matched for vascular risk factors. Am. J. Psychiatry 2008; 165: 524–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hashimoto R, Numakawa T, Ohnishi T et al. Impact of the DISC1 Ser704Cys polymorphism on risk for major depression, brain morphology and ERK signaling. Hum. Mol. Genet. 2006; 15: 3024–3033. [DOI] [PubMed] [Google Scholar]
- 78. Frodl T, Zill P, Baghai T et al. Reduced hippocampal volumes associated with the long variant of the tri‐ and diallelic serotonin transporter polymorphism in major depression. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2008; 147B: 1003–1007. [DOI] [PubMed] [Google Scholar]
- 79. Silverstone T, McPherson H, Li Q, Doyle T. Deep white matter hyperintensities in patients with bipolar depression, unipolar depression and age‐matched control subjects. Bipolar Disord. 2003; 5: 53–57. [DOI] [PubMed] [Google Scholar]
- 80. Dupont RM, Butters N, Schafer K, Wilson T, Hesselink J, Gillin JC. Diagnostic specificity of focal white matter abnormalities in bipolar and unipolar mood disorder. Biol. Psychiatry 1995; 38: 482–486. [DOI] [PubMed] [Google Scholar]
- 81. Kovacs GG. Invited review: Neuropathology of tauopathies: Principles and practice. Neuropathol. Appl. Neurobiol. 2015; 41: 3–23. [DOI] [PubMed] [Google Scholar]
- 82. Spillantini MG, Goedert M. Tau pathology and neurodegeneration. Lancet Neurol. 2013; 12: 609–622. [DOI] [PubMed] [Google Scholar]
- 83. Donovan NJ, Locascio JJ, Marshall GA et al. Longitudinal Association of Amyloid Beta and Anxious‐Depressive Symptoms in cognitively Normal older adults. Am. J. Psychiatry 2018; 175: 530–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Gatchel JR, Donovan NJ, Locascio JJ et al. Depressive symptoms and tau accumulation in the inferior temporal lobe and entorhinal cortex in cognitively normal older adults: A pilot study. J. Alzheimers Dis. 2017; 59: 975–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Braak H, Braak E. Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol. 1991; 82: 239–259. [DOI] [PubMed] [Google Scholar]
- 86. Gold G, Bouras C, Kovari E et al. Clinical validity of Braak neuropathological staging in the oldest‐old. Acta Neuropathol. 2000; 99: 579‐82; discussion 583‐4. [DOI] [PubMed] [Google Scholar]
- 87. Ehrenberg AJ, Suemoto CK, Franca Resende EP et al. Neuropathologic correlates of psychiatric symptoms in Alzheimer's disease. J. Alzheimers Dis. 2018; 66: 115–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Murray PS, Kirkwood CM, Gray MC et al. Hyperphosphorylated tau is elevated in Alzheimer's disease with psychosis. J. Alzheimers Dis. 2014; 39: 759–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Farber NB, Rubin EH, Newcomer JW et al. Increased neocortical neurofibrillary tangle density in subjects with Alzheimer disease and psychosis. Arch. Gen. Psychiatry 2000; 57: 1165–1173. [DOI] [PubMed] [Google Scholar]
- 90. Braak H, Del Tredici K. The pathological process underlying Alzheimer's disease in individuals under thirty. Acta Neuropathol. 2011; 121: 171–181. [DOI] [PubMed] [Google Scholar]
- 91. Giannakopoulos P, Gold G, Duc M, Michel JP, Hof PR, Bouras C. Neural substrates of spatial and temporal disorientation in Alzheimer's disease. Acta Neuropathol. 2000; 100: 189–195. [DOI] [PubMed] [Google Scholar]
- 92. Giannakopoulos P, Duc M, Gold G, Hof PR, Michel JP, Bouras C. Pathologic correlates of apraxia in Alzheimer disease. Arch. Neurol. 1998; 55: 689–695. [DOI] [PubMed] [Google Scholar]
- 93. Jadhav S, Avila J, Scholl M et al. A walk through tau therapeutic strategies. Acta Neuropathol. Commun. 2019; 7: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Elahi FM, Miller BL. A clinicopathological approach to the diagnosis of dementia. Nat. Rev. Neurol. 2017; 13: 457–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Crutch SJ, Schott JM, Rabinovici GD et al. Consensus classification of posterior cortical atrophy. Alzheimers Dement. 2017; 13: 870–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Thal DR, Rub U, Orantes M, Braak H. Phases of a beta‐deposition in the human brain and its relevance for the development of AD. Neurology 2002; 58: 1791–1800. [DOI] [PubMed] [Google Scholar]
- 97. Nascimento KK, Silva KP, Malloy‐Diniz LF, Butters MA, Diniz BS. Plasma and cerebrospinal fluid amyloid‐beta levels in late‐life depression: A systematic review and meta‐analysis. J. Psychiatr. Res. 2015; 69: 35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kumar A, Kepe V, Barrio JR et al. Protein binding in patients with late‐life depression. Arch. Gen. Psychiatry 2011; 68: 1143–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Krell‐Roesch J, Lowe VJ, Neureiter J et al. Depressive and anxiety symptoms and cortical amyloid deposition among cognitively normal elderly persons: The Mayo Clinic study of aging. Int. Psychogeriatr. 2018; 30: 245–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Rapp MA, Schnaider‐Beeri M, Grossman HT et al. Increased hippocampal plaques and tangles in patients with Alzheimer disease with a lifetime history of major depression. Arch. Gen. Psychiatry 2006; 63: 161–167. [DOI] [PubMed] [Google Scholar]
- 101. van Dyck CH, O'Dell RS, Mecca AP. Amyloid‐associated depression‐or not? Biol. Psychiatry 2021; 89: 737–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Crary JF, Trojanowski JQ, Schneider JA et al. Primary age‐related tauopathy (PART): A common pathology associated with human aging. Acta Neuropathol. 2014; 128: 755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Duyckaerts C, Braak H, Brion JP et al. PART is part of Alzheimer disease. Acta Neuropathol. 2015; 129: 749–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Ikeda K, Akiyama H, Arai T et al. A subset of senile dementia with high incidence of the apolipoprotein E epsilon2 allele. Ann. Neurol. 1997; 41: 693–695. [DOI] [PubMed] [Google Scholar]
- 105. Santa‐Maria I, Haggiagi A, Liu X et al. The MAPT H1 haplotype is associated with tangle‐predominant dementia. Acta Neuropathol. 2012; 124: 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Kawakami I, Hasegawa M, Arai T et al. Tau accumulation in the nucleus accumbens in tangle‐predominant dementia. Acta Neuropathol. Commun. 2014; 2: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Jellinger KA, Attems J. Neurofibrillary tangle‐predominant dementia: Comparison with classical Alzheimer disease. Acta Neuropathol. 2007; 113: 107–117. [DOI] [PubMed] [Google Scholar]
- 108. Casanova MF, Stevens JR, Brown R, Royston C, Bruton C. Disentangling the pathology of schizophrenia and paraphrenia. Acta Neuropathol. 2002; 103: 313–320. [DOI] [PubMed] [Google Scholar]
- 109. Saito Y, Ruberu NN, Sawabe M et al. Staging of argyrophilic grains: An age‐associated tauopathy. J. Neuropathol. Exp. Neurol. 2004; 63: 911–918. [DOI] [PubMed] [Google Scholar]
- 110. Togo T, Isojima D, Akatsu H et al. Clinical features of argyrophilic grain disease: A retrospective survey of cases with neuropsychiatric symptoms. Am. J. Geriatr. Psychiatry 2005; 13: 1083–1091. [DOI] [PubMed] [Google Scholar]
- 111. Ikeda K, Akiyama H, Arai T, Matsushita M, Tsuchiya K, Miyazaki H. Clinical aspects of argyrophilic grain disease. Clin. Neuropathol. 2000; 19: 278–284. [PubMed] [Google Scholar]
- 112. Frisina PG, Haroutunian V, Libow LS. The neuropathological basis for depression in Parkinson's disease. Parkinsonism Relat. Disord. 2009; 15: 144–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Shioya A, Saito Y, Arima K et al. Neurodegenerative changes in patients with clinical history of bipolar disorders. Neuropathology 2015; 35: 245–253. [DOI] [PubMed] [Google Scholar]
- 114. Nagao S, Yokota O, Ikeda C et al. Argyrophilic grain disease as a neurodegenerative substrate in late‐onset schizophrenia and delusional disorders. Eur. Arch. Psychiatry Clin. Neurosci. 2014; 264: 317–331. [DOI] [PubMed] [Google Scholar]
- 115. Fujishiro H, Nakamura S, Sato K, Iseki E. Prodromal dementia with Lewy bodies. Geriatr Gerontol Int 2015; 15: 817–826. [DOI] [PubMed] [Google Scholar]
- 116. Postuma RB, Berg D, Stern M et al. MDS clinical diagnostic criteria for Parkinson's disease. Mov. Disord. 2015; 30: 1591–1601. [DOI] [PubMed] [Google Scholar]
- 117. McKeith IG, Boeve BF, Dickson DW et al. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB consortium. Neurology 2017; 89: 88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Dickson DW, Braak H, Duda JE et al. Neuropathological assessment of Parkinson's disease: Refining the diagnostic criteria. Lancet Neurol. 2009; 8: 1150–1157. [DOI] [PubMed] [Google Scholar]
- 119. McKeith IG, Dickson DW, Lowe J et al. Diagnosis and management of dementia with Lewy bodies: Third report of the DLB consortium. Neurology 2005; 65: 1863–1872. [DOI] [PubMed] [Google Scholar]
- 120. Paulus W, Jellinger K. The neuropathologic basis of different clinical subgroups of Parkinson's disease. J. Neuropathol. Exp. Neurol. 1991; 50: 743–755. [DOI] [PubMed] [Google Scholar]
- 121. Samuels SC, Brickman AM, Burd JA, Purohit DP, Qureshi PQ, Serby M. Depression in autopsy‐confirmed dementia with Lewy bodies and Alzheimer's disease. Mt. Sinai J. Med. 2004; 71: 55–62. [PubMed] [Google Scholar]
- 122. Patterson L, Rushton SP, Attems J, Thomas AJ, Morris CM. Degeneration of dopaminergic circuitry influences depressive symptoms in Lewy body disorders. Brain Pathol. 2019; 29: 544–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Saari L, Heiskanen L, Gardberg M, Kaasinen V. Depression and Nigral neuron density in Lewy body spectrum diseases. Ann. Neurol. 2021; 89: 1046–1050. [DOI] [PubMed] [Google Scholar]
- 124. Fischer NM, Hinkle JT, Perepezko K et al. Brainstem pathologies correlate with depression and psychosis in Parkinson's disease. Am. J. Geriatr. Psychiatry 2021; 29: 958–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Fumimura Y, Ikemura M, Saito Y et al. Analysis of the adrenal gland is useful for evaluating pathology of the peripheral autonomic nervous system in lewy body disease. J. Neuropathol. Exp. Neurol. 2007; 66: 354–362. [DOI] [PubMed] [Google Scholar]
- 126. Homma T, Mochizuki Y, Mizutani T. Phosphorylated alpha‐synuclein immunoreactivity in the posterior pituitary lobe. Neuropathology 2012; 32: 385–389. [DOI] [PubMed] [Google Scholar]
- 127. Langston JW, Forno LS. The hypothalamus in Parkinson disease. Ann. Neurol. 1978; 3: 129–133. [DOI] [PubMed] [Google Scholar]
- 128. Soares NM, Pereira GM, Altmann V, de Almeida RMM, Rieder CRM. Cortisol levels, motor, cognitive and behavioral symptoms in Parkinson's disease: A systematic review. J. Neural Transm. (Vienna) 2019; 126: 219–232. [DOI] [PubMed] [Google Scholar]
- 129. Mackenzie IR. Activated microglia in dementia with Lewy bodies. Neurology 2000; 55: 132–134. [DOI] [PubMed] [Google Scholar]
- 130. Imamura K, Hishikawa N, Sawada M, Nagatsu T, Yoshida M, Hashizume Y. Distribution of major histocompatibility complex class II‐positive microglia and cytokine profile of Parkinson's disease brains. Acta Neuropathol. 2003; 106: 518–526. [DOI] [PubMed] [Google Scholar]
- 131. Halliday GM, Shepherd CE, McCann H et al. Effect of anti‐inflammatory medications on neuropathological findings in Alzheimer disease. Arch. Neurol. 2000; 57: 831–836. [DOI] [PubMed] [Google Scholar]
- 132. Amin J, Erskine D, Donaghy PC et al. Inflammation in dementia with Lewy bodies. Neurobiol. Dis. 2022; 168: 105698. [DOI] [PubMed] [Google Scholar]
- 133. Fujishiro H, Okuda M, Iwamoto K et al. Early diagnosis of Lewy body disease in patients with late‐onset psychiatric disorders using clinical history of rapid eye movement sleep behavior disorder and [(123) I]‐metaiodobenzylguanidine cardiac scintigraphy. Psychiatry Clin. Neurosci. 2018; 72: 423–434. [DOI] [PubMed] [Google Scholar]
- 134. Tsopelas C, Stewart R, Savva GM et al. Neuropathological correlates of late‐life depression in older people. Br. J. Psychiatry 2011; 198: 109–114. [DOI] [PubMed] [Google Scholar]
- 135. Wilson RS, Nag S, Boyle PA et al. Brainstem aminergic nuclei and late‐life depressive symptoms. JAMA Psychiat. 2013; 70: 1320–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Sweet RA, Hamilton RL, Butters MA et al. Neuropathologic correlates of late‐onset major depression. Neuropsychopharmacology 2004; 29: 2242–2250. [DOI] [PubMed] [Google Scholar]
- 137. Berg D, Postuma RB, Adler CH et al. MDS research criteria for prodromal Parkinson's disease. Mov. Disord. 2015; 30: 1600–1611. [DOI] [PubMed] [Google Scholar]
- 138. McKeith IG, Ferman TJ, Thomas AJ et al. Research criteria for the diagnosis of prodromal dementia with Lewy bodies. Neurology 2020; 94: 743–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Wing YK, Lam SP, Zhang J et al. Reduced striatal dopamine transmission in REM sleep behavior disorder comorbid with depression. Neurology 2015; 84: 516–522. [DOI] [PubMed] [Google Scholar]
- 140. Postuma RB, Gagnon JF, Tuineaig M et al. Antidepressants and REM sleep behavior disorder: Isolated side effect or neurodegenerative signal? Sleep 2013; 36: 1579–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Takahashi S, Mizukami K, Arai T et al. Ventilatory response to hypercapnia predicts dementia with Lewy bodies in late‐onset major depressive disorder. J. Alzheimers Dis. 2016; 50: 751–758. [DOI] [PubMed] [Google Scholar]
- 142. Kruger S, Haehner A, Thiem C, Hummel T. Neuroleptic‐induced parkinsonism is associated with olfactory dysfunction. J. Neurol. 2008; 255: 1574–1579. [DOI] [PubMed] [Google Scholar]
- 143. Frauscher B, Jennum P, Ju YE et al. Comorbidity and medication in REM sleep behavior disorder: A multicenter case‐control study. Neurology 2014; 82: 1076–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Wang J, Chau SWH, Lam SP, et al. Prevalence and correlates of REM sleep behaviour disorder in patients with major depressive disorder: a two‐phase study. J. Neurol. Neurosurg. 2022; 93(9): 1010–1017. [DOI] [PubMed] [Google Scholar]
- 145. Lam SP, Zhang J, Tsoh J et al. REM sleep behavior disorder in psychiatric populations. J. Clin. Psychiatry 2010; 71: 1101–1103. [DOI] [PubMed] [Google Scholar]
- 146. Zitser J, Gibbons C, Miglis MG. The role of tissue biopsy as a biomarker in REM sleep behavior disorder. Sleep Med. Rev. 2020; 51: 101283. [DOI] [PubMed] [Google Scholar]
- 147. Hall S, Orru CD, Serrano GE et al. Performance of alphaSynuclein RT‐QuIC in relation to neuropathological staging of Lewy body disease. Acta Neuropathol. Commun. 2022; 10: 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. National Institute for Health and Care Excellence . Dementia: Assessment, Management and Support for People Living with Dementia and their Carers. National Institute for Health and Care Excellence, London, 2018. [PubMed] [Google Scholar]
- 149. Orgeta V, Leung P, Del‐Pino‐Casado R, et al. Psychological treatments for depression and anxiety in dementia and mild cognitive impairment. Cochrane Database Syst Rev. 2022; 4(4): CD009125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Oba H, Kobayashi R, Kawakatsu S, Suzuki K, Otani K, Ihara K. Non‐pharmacological approaches to apathy and depression: A scoping review of mild cognitive impairment and dementia. Front. Psychol. 2022; 13: 815913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Kubo Y, Hayashi H, Kozawa S, Okada S. Relevant factors of depression in dementia modifiable by non‐pharmacotherapy: A systematic review. Psychogeriatrics 2019; 19: 181–191. [DOI] [PubMed] [Google Scholar]
- 152. Dudas R, Malouf R, McCleery J, Dening T. Antidepressants for treating depression in dementia. Cochrane Database Syst. Rev. 2018; 8: Cd003944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Hasan SS, Zaidi STR, Nirwan JS et al. Use of central nervous system (CNS) medicines in aged care homes: A systematic review and meta‐analysis. J. Clin. Med. 2019; 8(9): 1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Watts AS, Mortby ME, Burns JM. Depressive symptoms as a barrier to engagement in physical activity in older adults with and without Alzheimer's disease. PLoS One 2018; 13: e0208581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Lyketsos CG, DelCampo L, Steinberg M et al. Treating depression in Alzheimer disease: Efficacy and safety of sertraline therapy, and the benefits of depression reduction: The DIADS. Arch. Gen. Psychiatry 2003; 60: 737–746. [DOI] [PubMed] [Google Scholar]
- 156. Heun R, Ahokas A, Boyer P et al. The efficacy of agomelatine in elderly patients with recurrent major depressive disorder: A placebo‐controlled study. J. Clin. Psychiatry 2013; 74: 587–594. [DOI] [PubMed] [Google Scholar]
- 157. Aguera‐Ortiz L, Garcia‐Ramos R, Grandas Perez FJ et al. Focus on depression in Parkinson's disease: A Delphi consensus of experts in psychiatry, neurology, and geriatrics. Parkinsons Dis. 2021; 2021: 6621991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Ihl R, Frolich L, Winblad B et al. World Federation of Societies of biological psychiatry (WFSBP) guidelines for the biological treatment of Alzheimer's disease and other dementias. World J. Biol. Psychiatry 2011; 12: 2–32. [DOI] [PubMed] [Google Scholar]
- 159. Sorbi S, Hort J, Erkinjuntti T et al. EFNS‐ENS guidelines on the diagnosis and management of disorders associated with dementia. Eur. J. Neurol. 2012; 19: 1159–1179. [DOI] [PubMed] [Google Scholar]
- 160. Aguera‐Ortiz L, Garcia‐Ramos R, Grandas Perez FJ et al. Depression in Alzheimer's disease: A Delphi consensus on etiology, risk factors, and clinical management. Front. Psych. 2021; 12: 638651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Weiner MF, Martin‐Cook K, Foster BM, Saine K, Fontaine CS, Svetlik DA. Effects of donepezil on emotional/behavioral symptoms in Alzheimer's disease patients. J. Clin. Psychiatry 2000; 61: 487–492. [DOI] [PubMed] [Google Scholar]
- 162. Gauthier S, Feldman H, Hecker J et al. Efficacy of donepezil on behavioral symptoms in patients with moderate to severe Alzheimer's disease. Int. Psychogeriatr. 2002; 14: 389–404. [DOI] [PubMed] [Google Scholar]
- 163. Cummings JL, McRae T, Zhang R, Donepezil‐Sertraline Study G . Effects of donepezil on neuropsychiatric symptoms in patients with dementia and severe behavioral disorders. Am. J. Geriatr. Psychiatry 2006; 14: 605–612. [DOI] [PubMed] [Google Scholar]
- 164. Graipaspong N, Thaipisuttikul P, Vallipakorn SA. Cholinesterase inhibitors and Behavioral & Psychological Symptoms of Alzheimer's disease. J. Med. Assoc. Thail. 2016; 99: 433–440. [PubMed] [Google Scholar]
- 165. Clerici F, Vanacore N, Elia A et al. Memantine effects on behaviour in moderately severe to severe Alzheimer's disease: A post‐marketing surveillance study. Neurol. Sci. 2012; 33: 23–31. [DOI] [PubMed] [Google Scholar]
- 166. Lavretsky H, Laird KT, Krause‐Sorio B et al. A randomized double‐blind placebo‐controlled trial of combined Escitalopram and Memantine for older adults with major depression and subjective memory complaints. Am. J. Geriatr. Psychiatry 2020; 28: 178–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. van Rooij SJH, Riva‐Posse P, McDonald WM. The efficacy and safety of Neuromodulation treatments in late‐life depression. Curr. Treat. Opt. Psych. 2020; 7: 337–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Cappon D, den Boer T, Jordan C, Yu W, Metzger E, Pascual‐Leone A. Transcranial magnetic stimulation (TMS) for geriatric depression. Ageing Res. Rev. 2022; 74: 101531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Semkovska M, McLoughlin DM. Objective cognitive performance associated with electroconvulsive therapy for depression: A systematic review and meta‐analysis. Biol. Psychiatry 2010; 68: 568–577. [DOI] [PubMed] [Google Scholar]
- 170. Oudman E. Is electroconvulsive therapy (ECT) effective and safe for treatment of depression in dementia? A short review. J. ECT 2012; 28: 34–38. [DOI] [PubMed] [Google Scholar]
- 171. Hausner L, Damian M, Sartorius A, Frolich L. Efficacy and cognitive side effects of electroconvulsive therapy (ECT) in depressed elderly inpatients with coexisting mild cognitive impairment or dementia. J. Clin. Psychiatry 2011; 72: 91–97. [DOI] [PubMed] [Google Scholar]
- 172. Rao V, Lyketsos CG. The benefits and risks of ECT for patients with primary dementia who also suffer from depression. Int. J. Geriatr. Psychiatry 2000; 15: 729–735. [DOI] [PubMed] [Google Scholar]
- 173. Dybedal GS, Tanum L, Sundet K, Bjolseth TM. The role of baseline cognitive function in the neurocognitive effects of electroconvulsive therapy in depressed elderly patients. Clin. Neuropsychol. 2015; 29: 487–508. [DOI] [PubMed] [Google Scholar]
- 174. Aizenstein HJ, Baskys A, Boldrini M et al. Vascular depression consensus report – A critical update. BMC Med. 2016; 14: 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Hickie I, Scott E, Wilhelm K, Brodaty H. Subcortical hyperintensities on magnetic resonance imaging in patients with severe depression – a longitudinal evaluation. Biol. Psychiatry 1997; 42: 367–374. [DOI] [PubMed] [Google Scholar]
- 176. O'Brien J, Ames D, Chiu E, Schweitzer I, Desmond P, Tress B. Severe deep white matter lesions and outcome in elderly patients with major depressive disorder: Follow up study. BMJ 1998; 317: 982–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Taragano FE, Bagnatti P, Allegri RF. A double‐blind, randomized clinical trial to assess the augmentation with nimodipine of antidepressant therapy in the treatment of “vascular depression”. Int. Psychogeriatr. 2005; 17: 487–498. [DOI] [PubMed] [Google Scholar]
- 178. Jorge RE, Moser DJ, Acion L, Robinson RG. Treatment of vascular depression using repetitive transcranial magnetic stimulation. Arch. Gen. Psychiatry 2008; 65: 268–276. [DOI] [PubMed] [Google Scholar]
- 179. Zanardi R, Poletti S, Prestifilippo D, Attanasio F, Barbini B, Colombo C. Transcranial direct current stimulation: A novel approach in the treatment of vascular depression. Brain Stimul. 2020; 13: 1559–1565. [DOI] [PubMed] [Google Scholar]
- 180. Bomasang‐Layno E, Fadlon I, Murray AN, Himelhoch S. Antidepressive treatments for Parkinson's disease: A systematic review and meta‐analysis. Parkinsonism Relat. Disord. 2015; 21: 833–842 discussion 833. [DOI] [PubMed] [Google Scholar]
- 181. Mills KA, Greene MC, Dezube R, Goodson C, Karmarkar T, Pontone GM. Efficacy and tolerability of antidepressants in Parkinson's disease: A systematic review and network meta‐analysis. Int. J. Geriatr. Psychiatry 2018; 33: 642–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Troeung L, Egan SJ, Gasson N. A meta‐analysis of randomised placebo‐controlled treatment trials for depression and anxiety in Parkinson's disease. PLoS One 2013; 8: e79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Slaughter JR, Slaughter KA, Nichols D, Holmes SE, Martens MP. Prevalence, clinical manifestations, etiology, and treatment of depression in Parkinson's disease. J. Neuropsychiatry Clin. Neurosci. 2001; 13: 187–196. [DOI] [PubMed] [Google Scholar]
- 184. Leentjens AF, Koester J, Fruh B, Shephard DT, Barone P, Houben JJ. The effect of pramipexole on mood and motivational symptoms in Parkinson's disease: A meta‐analysis of placebo‐controlled studies. Clin. Ther. 2009; 31: 89–98. [DOI] [PubMed] [Google Scholar]
- 185. Barone P, Poewe W, Albrecht S et al. Pramipexole for the treatment of depressive symptoms in patients with Parkinson's disease: A randomised, double‐blind, placebo‐controlled trial. Lancet Neurol. 2010; 9: 573–580. [DOI] [PubMed] [Google Scholar]
- 186. Barone P, Scarzella L, Marconi R et al. Pramipexole versus sertraline in the treatment of depression in Parkinson's disease: A national multicenter parallel‐group randomized study. J. Neurol. 2006; 253: 601–607. [DOI] [PubMed] [Google Scholar]
- 187. Taylor JP, McKeith IG, Burn DJ et al. New evidence on the management of Lewy body dementia. Lancet Neurol. 2020; 19: 157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Connors MH, Quinto L, McKeith I et al. Non‐pharmacological interventions for Lewy body dementia: A systematic review. Psychol. Med. 2018; 48: 1749–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Saijo T, Takano A, Suhara T et al. Electroconvulsive therapy decreases dopamine D(2)receptor binding in the anterior cingulate in patients with depression: A controlled study using positron emission tomography with radioligand [(1)(1)C]FLB 457. J. Clin. Psychiatry 2010; 71: 793–799. [DOI] [PubMed] [Google Scholar]
- 190. Hermida AP, Sterina E, Schwab PP et al. Ultrabrief right unilateral electroconvulsive therapy for the treatment of the neuropsychiatric symptoms of dementia with Lewy bodies. J. ECT 2022; 38: 39–44. [DOI] [PubMed] [Google Scholar]