

Abstract
Background and Purpose
Multiple sclerosis (MS) is an unpredictable disease characterised by a highly variable disease onset and clinical course. Three main clinical phenotypes have been described. However, distinguishing between the two progressive forms of MS can be challenging for clinicians. This article examines how the diagnostic definitions of progressive MS impact clinical research, the design of clinical trials and, ultimately, treatment decisions.
Methods
We carried out an extensive review of the literature highlighting differences in the definition of progressive forms of MS, and the importance of assessing the extent of the ongoing inflammatory component in MS when making treatment decisions.
Results
Inconsistent results in phase III clinical studies of treatments for progressive MS, may be attributable to differences in patient characteristics (e.g., age, clinical and radiological activity at baseline) and endpoint definitions. In both primary and secondary progressive MS, patients who are younger and have more active disease will derive the greatest benefit from the available treatments.
Conclusions
We recommend making treatment decisions based on the individual patient's pattern of disease progression, as well as functional, clinical and imaging parameters, rather than on their clinical phenotype. Because the definition of progressive MS differs across clinical studies, careful selection of eligibility criteria and study endpoints is needed for future studies in patients with progressive MS.
Keywords: diagnosis, multiple sclerosis, primary progressive, secondary progressive, treatment
INTRODUCTION
Multiple sclerosis (MS) is an unpredictable disease characterised by a highly variable disease onset and clinical course [1]. Three main phenotypes have been described: (1) relapsing–remitting MS (RRMS) is defined by acute relapses interspersed with periods of full or partial recovery and stable clinical status. At disease onset, most patients have relapses with focal neurological deficits followed by complete or partial remission [2]. RRMS is the most common phenotype, affecting 85% of all patients [3]; (2) primary progressive MS (PPMS), which progresses continuously from onset of clinically manifest disease, typically with no relapses but possible periods of plateauing, and affects around 15% of MS patients [3]; and (3) secondary progressive MS (SPMS), characterised by disease progression, with or without acute relapses, accumulating disability in ambulatory, autonomic and cognitive functions [4]. Up to 5% of patients may convert from RRMS to SPMS per year [5].
Distinguishing between progressive phenotypes of MS can be challenging for clinicians [6, 7]. PPMS is difficult to discern at the onset of neurological symptoms because it often requires several visits to a physician to establish the continuous worsening over time. Conversely, with SPMS it can be difficult to distinguish a permanent relapse‐related deficit from bona fide progression; futhermore, patients can progress independently of relapses [8, 9]. This can lead to long periods of uncertainty as patients transition from RRMS to SPMS [1].
The pathophysiological mechanisms associated with disease progression are probably present from disease onset in RRMS patients, but are often silent [9]. Growing data suggest that a MS‐related disability increase may occur not only as relapse‐associated worsening (RAW), but also as progression independent of relapse activity (PIRA) [10]. We carried out an extensive review of the published literature (PubMed records containing relevant key words) to explore the definitions of progressive MS in order to determine how they impact clinical research and, ultimately, treatment decisions.
DEFINITIONS OF PROGRESSIVE MS
The definitions of progressive MS according to the US National Multiple Sclerosis Society (NMSS) Advisory Committee on Clinical Trials in Multiple Sclerosis and the revised 2017 McDonald criteria [11] are shown in Table 1. To identify early predictors of the revised classification of MS phenotypes [2] among patients with increasing disability, Tomassini and colleagues conducted a longitudinal real‐world study to examine the proportion of patients who reached an Expanded Disability Status Scale (EDSS) of ≥6 over a mean follow‐up period of 6.3 years [12]. They found that this milestone was reached by 64% of patients with confirmed clinical or magnetic resonance imaging (MRI) activity, and by only 36% of patients with no disease activity. The study showed that the risk of reaching the disability outcome was higher in patients older than 45 years, age being the only predictor of an increasing EDSS in the absence of clinical and MRI activity [12].
TABLE 1.
Criteria used to define progressive multiple sclerosis phenotypes
| Source | Definition |
|---|---|
| US NMSS [2] |
Progressive disease (primary or secondary) Clinical: steadily increasing objectively documented neurological dysfunction/disability without unequivocal recovery (fluctuations and phases of stability may occur) Imaging (MRI): imaging measures of progression are not established or standardised and not (yet) useful as phenotype descriptors for individual patients. Under consideration are increasing number and volume of T1‐hypointense lesions, brain volume loss and changes in magnetic transfer imaging and diffusion tensor imaging |
| Lorscheider et al. 2016 [14] | SPMS
|
| 2017 McDonald criteria [11] | PPMS
|
Abbreviations: CSF, cerebrospinal fluid; EDSS, Expanded Disability Status Scale; FS, functional status; IgG, immunoglobulin G; MRI, magnetic resonance imaging; MS, multiple sclerosis; PPMS, primary progressive multiple sclerosis; SPMS, secondary progressive multiple sclerosis; US NMSS, United States National Multiple Sclerosis Society.
No distinction is made between symptomatic and asymptomatic MRI lesions.
More recently, Prosperini and colleagues [13] estimated the proportion of patients with RRMS who experienced RAW or PIRA in a median follow‐up time of 12 years. Their study showed that the proportion of patients with RAW slightly exceed those with PIRA. RAW was associated with focal neuroinflammation parameters such as the presence of >9 T2 brain lesions and contrast enhancing lesions on baseline MRI scans, whereas the risk of PIRA was mainly related to advancing age and spinal cord lesions. However, data derived from randomised clinical trials showed that PIRA events comprised approximately 70%–90% of all disability accrual events in a follow‐up time of 2–10 years [9, 10]. Differences in the study design, in the EDSS calculation and in the definition of disability accrual (changes of EDSS alone or using a composite scores) may account for such discrepancies.
To develop a validated definition of SPMS, Lorscheider and colleagues [14] examined a range of different definitions for the magnitude of progression and based on the performance characteristics of these definitions, chose one with three strata (see Table 1). Compared with a physician diagnosis, which was based on a retrospective assessment, using this definition on the MSBase dataset was more sensitive (89% sensitivity), although less specific (86% specificity), and resulted in a higher proportion of patients being diagnosed with SPMS (18% compared with 14% by physician diagnosis) [14].
Other more recent studies have shown conflicting results. Iaffaldano and colleagues [15] demonstrated that patients with SPMS identified by data‐driven algorithms were older, more disabled and with a faster progression to severe disability than those identified by a neurologist. These findings contrast with those reported by Kopp and colleagues [16]. They identified an additional 20% patients with possible SPMS who were younger, with shorter disease duration, more disease activity and slightly less disability than patients with a clinically assigned SPMS diagnosis. However, the SPMS assessment by clinicians may differ between countries and could be influenced by drug prescription guidelines, reimbursement issues and other societal factors [17].
THE MS CONTINUUM
Part of the difficulty in defining the progressive phenotypes is that MS is considered a single disease [18], in which the phenotypes represent different stages in the continuum rather than distinct forms.
While PPMS and SPMS may present differently, their differences are more relative than absolute; they are part of the same disease spectrum and do not seem different at a pathophysiological level [2]. There are no qualitative differences in the disease activity, lesion morphology or immunopathology between SPMS and PPMS. In addition, there are no biomarkers that differentiate SPMS from PPMS. Genome‐wide association studies have failed to find any genetic variants that differentiate the two phenotypes of progressive MS.
After the onset of progression, the rate of progression and the age at which disability milestones are reached are similar in both SPMS and PPMS [19, 20], with age being the most significant predictor of disability accumulation [20]. In addition to age there are other factors implicated in the progression of MS, with some worsening MS prognosis, such as smoking habit [21], or improving it, such as vitamin D supplementation [22].
The pathogenesis of MS involves both inflammatory and neurodegenerative components, although the relative importance of these components changes over the course of the disease [23]. The coexistence of these two aspects is indeed quite specific to MS, considering that progression is not a common feature in other central nervous system (CNS) demyelinating diseases such as neuromyelitis optica spectrum disorder (NMOSD) and myelin oligodendrocyte glycoprotein antibody disorders (MOGAD) [24, 25]. Neurodegeneration plays a greater role with increasing age and disease duration than focal inflammatory processes [23].
MS becomes clinically progressive when the magnitude of the lesions/injury outstrips the CNS's ability to functionally compensate, leading to continuous neurological disability [26, 27]. However, there is no point at which inflammatory processes cease to exert an influence, since both inflammation and neurodegeneration are present in all lesions at all phases of the disease [28].
Studies using MRI have shown that slowly expanding lesions (SELs) correlate with the pathology of MS disease progression in progressive MS, and are a marker of chronic, active MS lesions [29]. In 2020, a pathological study reported that the molecular and immune profile of SELs in progressive MS is different from that of the active demyelinating lesions that occur in the relapsing phase [30]. In particular, SELs were completely demyelinated and had a rim of macrophages/microglia at the lesion edge, which was absent in chronic inactive lesions. Interestingly, only a few T cells and B cells were detected at the margins of the lesion rim, indicating that the adaptive immune system does not play a key role in SEL. In vivo positron emission tomography (PET) imaging studies using translocator protein (TSPO)‐binding radioligands similarly demonstrated microglia activation at the SELs’ edges [31]. In addition, both neuropathological studies and TSPO‐PET studies demonstrate widespread microglial activation also outside chronic lesions, in the normal‐appearing white matter in progressive MS [28, 32].
Although SELs are present both in RRMS and SPMS, lesions in SPMS were significantly more destructive, reflecting increased changes ascribed to secondary axonal degeneration in the periplaque white matter [33].
In addition, meningeal inflammation exhibited as either diffuse infiltrates or inflammatory aggregates containing T, B and plasma cells (ectopic lymphoid follicles) correlated with subpial cortical demyelination and neurodegeneration [34]. Interestingly, the ectopic lymphoid follicles described in patients with SPMS [35] are already present in the early stages of MS and gradually increase with disease duration and patient age.
Cortical pathology predominates in progressive MS, with several areas of subpial cortical demyelination [36]. Autopsy studies indicate a medial cortical lesion area of 11%–13% in SPMS compared to 2%–3% in RMS [37].
Diffuse white matter injury and axonal pathology measured by axonal spheroid density are also characteristic hallmarks of PPMS and SPMS injury [36]. These changes can be detected by MRI. Although more profound in patients with SPMS, these alterations are also observed in RRMS [38].
Remyelination is more pronounced in early MS [39], but robust active remyelination can be observed in MS patients of all ages [40].
There are no demonstrable differences in MRI findings between patients with PPMS and SPMS [26]; different MRI phenotypes can be seen in patients with the same clinical phenotype, and the same MRI phenotype can be seen in patients with different clinical phenotypes. Tauhid and colleagues defined four different MRI phenotypes of MS based on the severity and volume of the T2 lesion [41]. Type I was characterised by low lesion volume and mild atrophy, type II by a high lesion volume and mild atrophy, type III by low lesion volume and moderate to severe atrophy and, finally, type IV was characterised by a high lesion volume and moderate to severe atrophy. Whilst most patients with SPMS have the type IV MRI phenotype, there are individuals with SPMS in all MRI phenotype groups [41, 42]. In addition, patients with type II MS showed the greatest rate of brain atrophy over a period of 5 years, whereas patients with type IV MS showed the greatest progression in EDSS score over 5 years compared with type I [42].
TREATMENT OF PROGRESSIVE MS
The treatment options for patients with progressive forms of MS are generally limited, although the recent availability of novel treatments has expanded the disease‐modifying treatment (DMT) options for these MS phenotypes [43]. The general approach to the treatment of SPMS has been to continue DMTs in patients with active inflammation or ongoing relapses, but discontinue them in all others [44].
The availability of ocrelizumab, a monoclonal antibody against the CD20 antigen on B cells, and siponimod, a selective modulator of the sphingosine‐1‐phosphate receptor, has expanded the MS treatment options, and most guidelines now recommend ocrelizumab for patients with PPMS [44, 45]. Some guidelines also include the option of ocrelizumab (guidelines for Europe and Middle East/North Africa) or siponimod (Canada and Middle East/North Africa) in patients with active SPMS [44, 45]. For active SPMS, the European guidelines also include the option of cladribine, interferon (IFN) or mitoxantrone [45].
This inconsistency between guideline recommendations partly reflects the differences in prescribing procedures between countries. For instance, siponimod is indicated in the US for the treatment of relapsing forms of MS in adults, including clinically isolated syndrome, RRMS and active SPMS [46], whereas in Europe, siponimod is indicated for adult patients with SPMS and active disease shown by relapses or imaging features of inflammatory activity [47]. In Europe, regulators define disease activity as relapses or new MRI lesions, whereas US regulators define disease activity as clinical relapses without mentioning MRI, a problem that the International Advisory Committee on Clinical Trials in Multiple Sclerosis noticed and explained in a paper meant to clarify this situation [48].
Clinical studies in patients with PPMS
In randomised, placebo‐controlled studies, IFN‐β‐1a, IFN‐β‐1b, rituximab or glatiramer acetate had no significant effect on slowing disability progression compared with placebo in the overall study populations of patients with PPMS (Table 2) [49, 50, 51, 52]. Although there were subtle differences in the definition of sustained disability progression, the studies generally used a definition of ≥1.0 point increase in patients with EDSS of up to 5.0 or 5.5 at baseline or ≥0.5 point increase in those with EDSS of >5.5, sustained for 3 months.
TABLE 2.
Randomised, placebo‐controlled studies conducted in patients with primary progressive multiple sclerosis prior to 2020 [49, 50, 51, 52]
| Study | Comparators | N | Follow‐up duration | Primary endpoint | Primary efficacy occurrence | HR (95% CI) p‐value |
|---|---|---|---|---|---|---|
| Leary et al. 2003 [50] | Placebo | 20 | 2 years | 3‐month CDP a | NR |
NR No difference between groups |
| IFN‐β‐1a 30 or 60 μg | 30 | |||||
| Montalban et al. 2009 [51] | Placebo | 37 | 2 years | 3‐month CDP b | 56.8% |
NR p = 0.3135 |
| IFN‐β‐1b 8 MU | 36 | 65.8% | ||||
| Wolinsky et al. 2007 [52] | Placebo | 316 |
3 years (planned) 2 years (completed c ) |
3‐month CDP d | 45.2% |
0.87 (0.71–1.07) p = 0.1753 |
| Glatiramer acetate 20 mg | 627 | 39.6% | ||||
| OLYMPUS study [49] | Placebo | 147 | 96 weeks | 12‐week CDP e | 38.5% |
0.77 p = 0.1442 |
| Rituximab 1000 mg | 292 | 30.2% |
Abbreviations: CDP, confirmed disability progression; CI, confidence interval; EDSS, Expanded Disability Status Scale; HR, hazard ratio; IFN, interferon; MU, million units; NR, not reported.
A sustained increase in EDSS of ≥1.0 points from baseline if the baseline EDSS was ≤5.5 or an increase of ≥0.5 points if the baseline EDSS was >5.5.
A sustained increase in EDSS of ≥1.0 points from baseline if the baseline EDSS was <5.5 or an increase of 0.5 points if the baseline EDSS was >5.5.
Study stopped at the 2‐year interim analysis for futility.
A sustained increase in EDSS of ≥1.0 points from baseline if the baseline EDSS was 3.0–5.0 (inclusive) or an increase of ≥0.5 points if the baseline EDSS was 5.5–6.5.
A sustained increase in EDSS of ≥1.0 points from baseline if the baseline EDSS was 2.0–5.5 (inclusive) or an increase of ≥0.5 points if the baseline EDSS was >5.5, and when the change could not be attributed to another aetiology.
While no significant differences were seen between placebo and active treatment in the overall study populations, the two larger studies showed a significant improvement in confirmed disability progression (CDP) versus placebo in some subgroups. Glatiramer acetate significantly slowed CDP versus placebo in men (HR 0.71, 95% CI 0.53–0.95, p = 0.0193) [52] and rituximab significantly slowed CDP versus placebo in patients aged <51 years with ≥1 Gd+ lesion at baseline (HR 0.33, 95% CI 0.14–0.79, p = 0.0088) [49].
These data indicate that in patients with PPMS, those who are younger and have more active disease will derive the greatest benefit from treatment. This is consistent with the known association of age with disability accumulation in MS [53, 54].
The INFORMS study on fingolimod [55] and the ORATORIO study on ocrelizumab [56] have investigated the safety and efficacy of these agents compared with placebo in patients with PPMS. The primary endpoint (sustained disability progression) in INFORMS was defined as the occurrence of any of the following: 3‐month sustained increase of ≥20% from baseline in the 25‐feet walk (T25FW) test; 3‐month sustained increase from baseline in the EDSS score (1 point in patients with a baseline EDSS score of 3.5–5.0 or 0.5 point in patients with a baseline EDSS score of 5.5 or 6.0); or 3‐month sustained increase of ≥20% from baseline in the 9‐hole peg test (9HPT). There was no significant difference between fingolimod and placebo groups in the occurrence of the primary endpoint (HR 0.95, 95% CI 0.80–1.12, p = 0.689) or in the key secondary endpoint of a sustained improvement in EDSS (HR 0.88, 95% CI 0.72–1.08, p = 0.315) [55]. While fingolimod significantly decreased the number of new/newly enlarging T2 lesions and the number of Gd+ T1 lesions on MRI relative to placebo, there was no significant difference between groups in brain volume loss on MRI [55]. In a separate study, fingolimod did slightly reduce microglial activation within focal lesions, but not in the normal‐appearing white matter [56].
In contrast, the ORATORIO study found that ocrelizumab significantly delayed 12‐week CDP, the primary endpoint (defined as 12‐week increase in EDSS of ≥1 point in patients with baseline EDSS of ≤5.5 or 0.5 point in patients with baseline EDSS score of >5.5), compared with placebo (HR 0.76, 95% CI 0.59–0.68, p = 0.03) [57]. In addition, ocrelizumab significantly delayed the onset of 24‐week CDP (HR 0.75, 95% CI 0.58–0.98, p = 0.04) compared with placebo [57]. Unlike the INFORMS study, the ORATORIO study showed a significant improvement in MRI parameters (i.e., brain volume and total volume of T2‐weighted lesions) with ocrelizumab versus placebo (p < 0.0001) [57]. A small proportion of patients (29.9% with ocrelizumab and 9.4% with placebo) had no evidence of progression using EDSS, 9HPT or T25FW test, no MRI signs of active inflammation (i.e., new/enlarging T2 lesions and Gd+ T1 lesions) and had no relapses over 120 weeks of treatment [58]. Upon completion of the double‐blind phase of this study, patients in the placebo group crossed over to open‐label treatment with ocrelizumab; throughout 6.5 years of follow‐up, the rate of progression to 24‐week CDP was significantly slower in the group that had received ocrelizumab from the start of the study [59].
What would explain the difference in findings between the INFORMS and the ORATORIO studies? The key difference appears to be that the ORATORIO study included a slightly younger group of patients, with a higher proportion of patients having active inflammation, and a greater burden of brain inflammation, as shown by the volume of T2‐weighted lesions on MRI (Table 3). Age has emerged as an important factor in study recruitment. Indeed, every large study presented in this review with a mean patient age at baseline >48 years (e.g. PROMISE [glatiramer] [52], OLYMPUS [rituximab] [49] and INFORMS [fingolimod] [55]) produced negative results.
TABLE 3.
Key baseline characteristics of patients with primary progressive multiple sclerosis receiving active treatment in the INFORMS (fingolimod) and ORATORIO (ocrelizumab) studies [55, 57]
| Fingolimod arm in the INFORMS study (n = 336) [55] | Ocrelizumab arm in the ORATORIO study (n = 488) [57] | |
|---|---|---|
| Males, n (%) | 173 (51.4) | 251 (51.4) |
| Age, years | ||
| Mean (SD) | 48.5 (8.6) | 44.7 (7.9) |
| Median (range) | 49 (42.5–55.0) | 46 (20–56) |
| Time since diagnosis of PPMS, mean (SD), years | 2.8 (2.6) | 2.9 (3.2) |
| EDSS score | ||
| Mean (SD) | 4.7 (1.0) | 4.7 (1.2) |
| Median (range) | 4.5 (4–6) | 4.5 (2.5–7) |
| Gd+ lesions present on T1‐weighted MRI, n (%) | 46 (13.7) | 133/484 (27.5) |
| Total volume of lesions on T2‐weighted MRI, mean (SD), cm3 | 9.4 (10.2) | 12.7 (15.1) |
| Normalised brain volume, mean (SD), cm2 | 1490.9 (86.5) | 1469.9 (88.7) |
Abbreviations: Gd+, gadolinium‐enhancing; MRI, magnetic resonance imaging; PPMS, primary progressive multiple sclerosis; SD, standard deviation.
Clinical studies in SPMS
Early studies in patients with SPMS focused on treatment with mitoxantrone and IFN‐β. Mitoxantrone 12 mg/m2 significantly improved outcomes compared with placebo in a phase III study in patients with SPMS (48%) or worsening RRMS (52%) [60]. While this study showed significant benefit in the treated group across a number of clinical parameters, mitoxantrone can be associated with significant adverse events (including nausea/vomiting, alopecia, amenorrhoea, leukopenia, leukaemia, cardiotoxicity and congenital anomalies), hence a careful benefit:risk assessment is recommended [61].
In two studies (IMPACT and SPECTRIMS), IFN‐β‐1a had no significant effect on slowing disability progression by EDSS compared with placebo [62, 63]. However, the primary endpoint in IMPACT was not EDSS, but rather a composite measure comprising quantitative tests of ambulation, arm function and cognition [62]. There was a significant improvement in this composite score with IFN‐β‐1a compared with placebo, driven mainly by improvements in arm function and cognition [62].
In SPECTRIMS, IFN‐β‐1a was associated with significantly slower progression in EDSS only in patients who were having relapses at baseline [63, 64]. In addition, IFN‐β‐1a was associated with significantly less MRI activity compared with placebo [64], with benefits more marked in those patients with relapses at baseline.
IFN‐β‐1b was compared with placebo in two studies in patients with SPMS—one conducted in Europe and one in North America (NA) [65, 66]. In the European study, IFN‐β‐1b significantly slowed CDP, which was defined as an increase in the patient's EDSS score of 1.0 point over a sustained period of ≥3 months or a 0.5 point increase in patients with an EDSS score of 6.0–6.5 at baseline [66]. However, in the NA study, which had a slightly different definition of CDP, there was no significant difference in sustained progression between the IFN‐β‐1b and the placebo groups [66]. When the two studies were compared using the same definition of CDP (the one from the NA study), their results were similar, showing a significant improvement between treatment arms in the European study but not in the NA study (Figure 1) [67].
FIGURE 1.
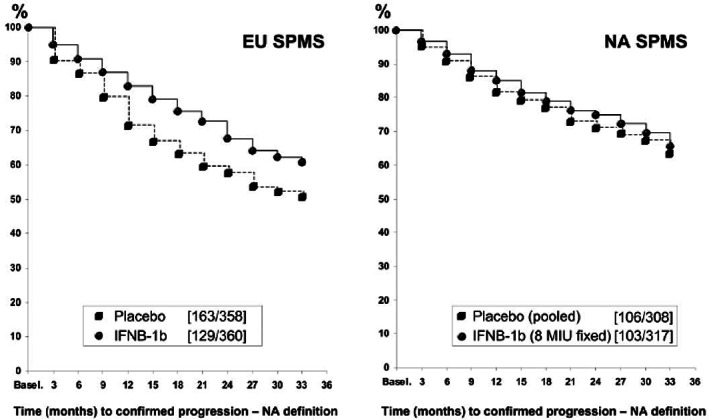
Time to confirmed disability progression in the North American (NA) and European (EU) studies with interferon β‐1b (IFNB‐1b) in patients with secondary progressive multiple sclerosis (SPMS) [67]. Confirmed progression was defined as time to a ≥1.0 point increase in Expanded Disability Status Scale (EDSS) sustained for ≥6 months or 0.5 point increase in patients with baseline EDSS of 6.0–6.5. [Kappos L, Weinshenker B, Pozzilli C, et al. Interferon beta‐1b in secondary progressive MS: a combined analysis of the two trials. Neurology. 2004;63(10):1779–1787. doi:10.1212/01.wnl.0000145561.08973.4f, with permission.]
In addition to the more stringent definition of CDP in the NA study, the probable reason for the discordant results between these studies lies in the patients' demographics at baseline. Compared with patients in the European study, those in the NA study were older (mean age 46.8 years vs. 41.0 years in the European study), had experienced fewer relapses in the previous 2 years (0.8 vs. 1.7, respectively) and had a lower mean number of enhancing lesions (1.5 vs. 2.6, respectively). The number of patients who were relapse‐free in the 2 years before study entry was also significantly higher in the NA than the European study (55% vs. 30%, respectively) [67]. Interestingly, similar treatment effects in the two studies were seen for endpoints that are thought to reflect the inflammatory aspects of MS, such as relapse rate and enhancing lesions.
The European Study on Intravenous Immunoglobulin (IVIG) in MS aimed to investigate IVIG in the secondary progressive phase of the disease [68]. The primary outcome was CDP, defined as the time to first confirmed progression on the EDSS. IVIG treatment had no beneficial effect on the time to confirmed EDSS progression, and the annual relapse rate was 0.46 in both groups. No significant differences were found in any of the other secondary outcome measures or in the change of T2‐lesion load over time between the treated group versus placebo [68].
The ASCEND study of natalizumab in patients with SPMS used a multicomponent primary endpoint, similar to the one used in the IMPACT study [62], comprising EDSS, the timed T25FW test and 9HPT [69]. No significant improvement in the combined endpoint was noted during 2 years of treatment compared with placebo, primarily because there was no significant change in EDSS or T25FW. A small but significant improvement in the 9HPT was noted with natalizumab versus placebo [69]. The patient cohort in the ASCEND study had an advanced disease stage; the mean time since the diagnosis of SPMS was 4.7 years, median baseline EDSS was 6.0, 63% of patients had a baseline EDSS score of 6.0–6.5, and 71% had no relapses in the preceding 2 years. A TSPO‐PET study including a subpopulation of ASCEND patients demonstrated a modest reduction in microglial activation following natalizumab treatment [70].
In a generally similar SPMS patient population, the EXPAND study demonstrated a significant 21% reduction in the risk of 3‐month CDP (the primary efficacy outcome, defined as a 1‐point increase in EDSS if baseline score was 3.0–5.0 and 0.5 point increase if baseline EDSS was 5.5–6.5 confirmed ≥3 months later) with siponimod compared with placebo [71]. Such improvements were seen in all patient subgroups based on age, gender, relapse history, Gd+ T1 lesion burden, previous treatments and MS severity, although there was a trend towards a more favourable response in younger patients [71]. In addition, siponimod significantly reduced the risk of 6‐month CDP by 26% and the time to first confirmed relapse by 46%, and significantly improved MRI parameters, including T2‐weighted lesion volume, and the number of new or enhancing T2‐weighted lesions and Gd+ T1 lesions.
While the EXPAND patient population was similar to that in the ASCEND study in terms of age, mean/median EDSS and time since conversion to progressive MS, the EXPAND study population was larger (n = 1651 vs. n = 887 in ASCEND) and included a lower proportion of patients with EDSS ≥6.0 at baseline (56% vs. 63%) and Gd+ lesions at baseline (21% vs. 24%; Table 4) [69, 71]. Furthermore, the proportion of patients receiving placebo in the EXPAND study who had on‐study relapses was lower than in the ASCEND study (19% vs. 27%).
TABLE 4.
Comparisons between overall patient characteristics in the EXPAND (siponimod) and ASCEND (natalizumab) studies in secondary progressive multiple sclerosis [69, 71]
| EXPAND [71] | ASCEND [69] | |
|---|---|---|
| Patients, n | 1651 | 887 |
| Age, years (mean) | 48 | 47 |
| Time since onset, years (mean) | 17 | 17 |
| Time since MS diagnosis, years (mean) | 13 | 12 |
| Time since PMS diagnosis, years (mean) | 4 | 5 |
| Patients without previous DMT, % patients | 22% | NR |
| Relapse‐free for prior 2 years, % patients | 64% | 71% |
| EDSS (median) | 6.0 | 6.0 |
| EDSS ≥6.0 (proportion) | 56% | 63% |
| Presence of T1 Gd lesions (proportion) | 21% | 24% |
| T2 lesion load, mm2 or cm3 (mean) | 15 cm3 | 17 cm3 |
| On‐study relapses, % patients in the placebo group | 19% | 27% |
| On‐study ARR in the placebo group | 0.16 | 0.17 |
Abbreviations: ARR, annualised relapse rate; DMT, disease‐modifying treatment; EDSS, Expanded Disability Status Scale; Gd, gadolinium; MS, multiple sclerosis; NR, not reported; PMS, progressive multiple sclerosis.
Clinical studies in patients with progressive MS
Some patients with progressive MS (either PPMS or SPMS) can achieve stable disease during treatment, with no progression in clinical or MRI variables and no relapses, which raises the question—is it possible to reverse progressive disability in this population? This question was addressed in the MS‐SPI study with biotin, a product being developed to target energy metabolism and myelin repair [72]. In this study, the primary efficacy endpoint was disability reversal at month 9, which was confirmed at month 12. Disability reversal was defined as a decrease in EDSS of ≥1 point in patients with baseline EDSS <6, and of ≥0.5 point in those with a baseline EDSS of 6–7. Overall, 13 patients in the biotin group (12.6%) and none in the placebo group achieved the sustained disability reversal endpoint (p = 0.005). More recently, a phase III study (SPI2) evaluated the safety and efficacy of biotin 100 mg three times daily compared with placebo in 642 patients with progressive MS without recent relapses, also called non‐active MS. The primary endpoint was the reversal of functional disability (defined as a decrease in EDSS of ≥1 point in patients with baseline EDSS <6, and of ≥0.5 point in those with a baseline EDSS of 6–7) or decreased T25FW over a period of 12 months, confirmed on month 15. The secondary endpoints included the relative risk of disability progression (time to 12‐week confirmed EDSS progression), global impression scales and mean changes from baseline in T25FW. The primary endpoint was not met [73]. Similarly, there was no significant difference between groups in the time to 12‐week confirmed EDSS progression (HR 0.97, 95% CI 0.680–1.385, p = 0.4348).
POSITION STATEMENT
The reviewed studies in patients with progressive forms of MS have provided valuable lessons that can be incorporated into future research, particularly in relation to patient selection criteria and study endpoints.
Patient selection
Based on these studies, the MS Experts of the ParadigMS Foundation recommend focusing clinical recruitment on the patients who are most likely to benefit from treatment.
In the case of anti‐inflammatory agents, younger patients with active inflammation are more likely to benefit from such treatments. In older patients with long‐term disease duration, degeneration predominates and inflammation has a smaller role in MS progression. Therefore, the capacity for repair, remyelination and other functions required to stabilise a relapsing–remitting course decline in progressive disease [74]. This could help to explain why younger patients with RRMS have better recovery from relapses than older patients [75], for every 10 years of age, there is a 1.33‐fold increase in the odds of EDSS not improving (p < 0.0001).
It has been shown that statistical predictors of patients reaching EDSS ≥6 within 10 years of symptom onset are age >35 years at symptom onset, EDSS ≥3 in the first year, and the presence of pyramidal signs in the first year [76].
Age is also a critical factor in determining treatment safety. For example, older patients (>45 years) are at significantly higher risk of developing neoplasms during treatment with depletive DMTs compared with younger patients [77].
Other demographic and clinical parameters needed to define disease subtype are sex, severity of previous relapses with incomplete remission, frequency of relapses in the first years, length of intervals between relapses, and early accrual of disability with superimposed attacks [78].
The selection of patients to be included in a trial should also consider cognitive dysfunction. De Meo and colleagues identified five cognitive phenotypes, including preserved cognition, mild‐verbal memory/semantic fluency, mild‐multidomain, severe‐executive/attention and severe‐multidomain involvement [79]. Using MRI, they were able to identify distinct underlying neuroanatomical substrates, supporting data‐driven cognitive findings with a biological basis. Future clinical studies should also consider stratifying MS patients based on MRI evidence of lesion load and brain atrophy, given that those with more severe brain damage are likely to show EDSS deterioration over the next several years [42, 80].
Cortical atrophy is accelerated in progressive MS compared to RRMS [81]. However, only the atrophy rate in the deep grey matter seems to be associated with disability accumulation [82]. By using a machine learning approach, Eshaghi and colleagues [83] divided MS into four phenotypes based on the earliest abnormalities on brain MRI. They suggest that MRI‐based subtypes predict MS disability progression and response to treatment and may be used to define groups of patients in interventional trials. In addition, the presence of enhancing lesions at baseline has been related to MS disease activity and it also seems to be age dependent [84].
More recently, it has been shown that SELs significantly predict clinical progression, evaluated by EDSS, T25FW and 9HPT, and patients with multiple SELs have more aggressive disease and reach higher motor and cognitive disability at a younger age [85, 86].
Spinal cord lesion load, which is higher in progressive forms of MS than in RRMS, is significantly correlated with physical disability and predicts the risk of disability worsening [87, 88]. Moreover, spinal MRI lesions are clinically relevant even when they are asymptomatic [89]. Spinal cord volume loss is another marker of disease progression that is evident at the onset of progressive MS and seems partially independent of the number of spinal cord lesions [90]. Higher spinal cord atrophy rates and lesion load increase predicted higher future EDSS score worsening over time in SPMS [91].
Among easily accessible biomarkers enabling the identification of MS patients who will accumulate irreversible disability in the long term are serum neurofilaments (NfLs). Elevated NfLs levels at early stages of MS were associated with an increased risk of reaching sustained disability worsening; although, the risk of reaching sustained EDSS score 6.0 and conversion to SPMS has not been demonstrated [92].
More recently, however, Uphaus and colleagues reported that NfL levels at baseline predict relapse‐free disability progression in a prospective longitudinal cohort study 6 years later, supporting the early identification of patients at risk for later SPMS conversion [93]. Another potential marker for progression‐related CNS pathology in MS is glial fibrillary acidic protein [94].
Clinical outcome measures for progressive MS trials
Measures other than EDSS, such as T25W, 9HPT and patient‐reported outcomes (PROs), should be considered when measuring treatment‐related improvement. EDSS may be a relatively blunt instrument for measuring disease progression in patients with progressive MS, particularly in those with baseline EDSS scores close to 5. Studies have shown that T25FW and 9HPT are associated with fewer measurement errors and are more reliable than EDSS for measuring patients' improvement [95].
In addition, studies like IMPACT and ASCEND, which used composite endpoints, have demonstrated that most of the treatment‐related improvement was achieved in measures of non‐ambulatory function, such as arm function and cognition [62, 69]. The ASCEND investigators noted that improvement in only one functional system could be clinically and functionally significant for patients, and that upper limb function is an important parameter for patients with MS [69].
PROs are increasingly used in MS research and are important during the development and approval process for new drugs. PROs are captured directly from patients and include symptoms, function, health status, and health‐related quality of life. Strong predictors of progression are gradual worsening of symptoms including worsening ambulation, cognition, balance, muscle weakness, visual symptoms, bladder symptoms, fatigue and unemployment [96].
Further research on the reliablity of the available tools to measure patients' progression is required to improve clinical study design and to evaluate patient progression more accurately.
Imaging outcome measures for progressive MS trials
The use of semi‐automatic programmes for quantifying SELs can help with their identification [25, 97]. While the increase in lesion burden in the CNS and the increase in the number of gadolinium‐enhancing lesions and T2 lesions reflect acute lesion activity, SELs reflect chronic lesion activity [98] making them good MRI endpoints in progressive MS. TSPO‐PET is another imaging modality with automated analysis methods to sensitively measure changes in the burden of innate immune system activation within the CNS [32].
MRI brain volume measures are widely used as outcome measures in phase III clinical trials; however, their abilities to measure disease progression in trials over only 2 years of follow‐up are limited [99, 100].
Since deep grey matter is associated with disability accumulation [82], measures of thalamus volume as well as spinal cord volume are potentially new biomarkers for testing the efficacy of new drugs in progressive MS [90, 91].
There are a number of promising imaging techniques with different strengths and limitations, which can be now selected for clinical trial settings [101]. Advanced MRI techniques, such the the magnetization transfer ratio (MTR), which measures tissue integrity in apparently normal white matter and can predict severe impairment and disability, should be used in clinical trials to study tissue remyelination in patients with SPMS [99].
CONCLUSIONS
Data on the treatment of progressive MS highlight the importance of assessing the extent of the ongoing inflammatory component of the disease, and the patient's position on that continuum when making treatment decisions. Rather than trying to define a patient's phenotype and the moment of transition from RRMS to SPMS, we recommend basing the eligibility criteria for studies in the progressive MS population on similar baseline characteristics of the patients (i.e., age and MRI findings). Patients with ongoing active inflammation are likely to derive the greatest benefit from anti‐inflammatory treatments.
In designing new trials, physicians should also consider the functional abilities that are important for patients. For example, patients may be less concerned about maintaining ambulatory function than upper limb functionality or cognitive function.
Future trial designs in MS should give more importance to the PROs and to surrogate measures that can serve as predictive markers of treatment response. The large number of patients and the long duration required for phase III trials in progressive MS have also stimulated researchers to design accelerated clinical trials. Examples of such designs are the adaptive, multi‐arm and multi‐stage trials used to evaluate multiple agents simultaneously and the use of Simon (two‐stage) trial designs to screen compounds for non‐futility.
Novel treatments, which target both the adaptive and innate immune system, such as tyrosine kinase inhibitors that selectively target mast cell and microglia activity, are also becoming available, and these may be good candidates for patients with progressive MS. Several phase III trials with oral Bruton's tyrosine kinase (BTK) inhibitors, ibudilast, masitinib and statins are ongoing. One day it may also be possible to reverse the disability progression in some patients with progressive MS with neuroprotective and remyelinating therapies. A discussion of all potential targets that impact myelin repair is outside of the scope of this article; however, remyelinating therapies continue to advance through the pipeline, and phase II studies are underway in both relapsing and progressive MS patients [102]. Future clinical studies should include endpoints that can demonstrate this effect.
AUTHOR CONTRIBUTIONS
Conceptualization, Carlo Pozzilli and Maura Pugliatti. Methodology, Carlo Pozzilli and Maura Pugliatti. Validation, Carlo Pozzilli, Maura Pugliatti, Patrick Vermersch, Nikolaos Grigoriadis, Mona Alkhawajah, Laura Airas, Celia Oreja‐Guevara. Investigation, Carlo Pozzilli, Maura Pugliatti, Patrick Vermersch, Nikolaos Grigoriadis, Mona Alkhawajah, Laura Airas, Celia Oreja‐Guevara. Writing—original draft preparation, Carlo Pozzilli and Maura Pugliatti. Writing—review and editing, Carlo Pozzilli, Maura Pugliatti, Patrick Vermersch, Nikolaos Grigoriadis, Mona Alkhawajah, Laura Airas, Celia Oreja‐Guevara. Funding acquisition, Carlo Pozzilli, Maura Pugliatti, Celia Oreja‐Guevara. All authors have read and agreed to the published version of the manuscript.
FUNDING INFORMATION
ParadigMS activities are co‐funded by Sanofi, Roche and Merck for logistical and organizational purposes relating to meetings and publications of the Foundation.
CONFLICT OF INTEREST
Carlo Pozzilli has served on scientific advisory boards for Novartis, Merck, Biogen, Sanofi‐Genzyme, Roche, Janssen, Alexion, has received funding for travel and speaker honoraria from Merck, Serono, Biogen, Sanofi‐Genzyme, Roche, Almirall, Janssen, Alexion and Novartis, and receives research support from Merck, Biogen, Novartis and Almirall. Maura Pugliatti has served on scientific advisory boards for Merck Serono, Biogen and Mylan, has received funding for travel and speaker honoraria from Merck Serono, Biogen, Sanofi‐Genzyme, Teva and Almirall, and has received financial support for research and scientific events from Biogen Idec and Sanofi‐Genzyme. Patrick Vermersch has received honoraria and consulting fees from Biogen, Sanofi‐Genzyme, Novartis, Teva, Merck, Roche, Imcyse, AB Science and Celgene, and research support from Novartis, Sanofi‐Genzyme and Roche. Nikolaos Grigoriadis has received honoraria, travel support, consultancy fees and research grants from Biogen Idec, Biologix, Genesis Pharma, Novartis, TEVA, Bayer, Merck Serono, Sanofi – Genzyme, ROCHE, Celgene and ELPEN. Mona Alkhawajah has received speaker honorarium, consultation fees and/or educational travel support from Roche, Merck, Biogen, Novartis, SAJA, Hikma, Actelion and/or Sanofi. Laura Airas has served on scientific advisory boards for Novartis, Merck Serono, Biogen, Sanofi‐Genzyme and Roche, and has received institutional research support from Merck Serono and Sanofi‐Genzyme. Celia Oreja‐Guevara has received speaker and consultation fees from Biogen Idec, Celgene, Sanofi‐Genzyme, Novartis, Roche, Merck and Teva.
ACKNOWLEDGMENTS
We would like to thank Catherine Rees, Alma Orts‐Sebastian and Monica Hoyos of Springer Healthcare Communications who wrote the first draft of this manuscript. This medical writing assistance was funded by ParadigMS Foundation. The authors acknowledge the financial and operational support of the ParadigMS Foundation that made it possible to produce this article. ParadigMS Foundation is a group of European, Middle Eastern and North African experts in multiple sclerosis. The content of this publication is based upon in‐depth discussions on this topic by the group members at Expert Meetings. The current list of group members can be consulted at ParadigMS's website.
The views expressed are based on the group members' opinions and do not represent the views of ParadigMS's co‐founders of the Expert Meetings: Sanofi‐Genzyme and Roche.
Pozzilli C, Pugliatti M, Vermersch P, et al. Diagnosis and treatment of progressive multiple sclerosis: A position paper. Eur J Neurol. 2023;30:9‐21. doi: 10.1111/ene.15593
Carlo Pozzilli and Maura Pugliatti contributed equally to the article.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Krieger SC, Sumowski J. New insights into multiple sclerosis clinical course from the topographical model and functional reserve. Neurol Clin. 2018;36(1):13‐25. [DOI] [PubMed] [Google Scholar]
- 2. Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278‐286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Antel J, Antel S, Caramanos Z, Arnold DL, Kuhlmann T. Primary progressive multiple sclerosis: part of the MS disease spectrum or separate disease entity? Acta Neuropathol. 2012;123(5):627‐638. [DOI] [PubMed] [Google Scholar]
- 4. Rovaris M, Confavreux C, Furlan R, Kappos L, Comi G, Filippi M. Secondary progressive multiple sclerosis: current knowledge and future challenges. Lancet Neurol. 2006;5(4):343‐354. [DOI] [PubMed] [Google Scholar]
- 5. Vukusic S, Confavreux C. Primary and secondary progressive multiple sclerosis. J Neurol Sci. 2003;206(2):153‐155. [DOI] [PubMed] [Google Scholar]
- 6. Correale J, Gaitán MI, Ysrraelit MC, Fiol MP. Progressive multiple sclerosis: from pathogenic mechanisms to treatment. Brain. 2017;140(3):527‐546. [DOI] [PubMed] [Google Scholar]
- 7. Ontaneda D, Fox RJ, Chataway J. Clinical trials in progressive multiple sclerosis: lessons learned and future perspectives. Lancet Neurol. 2015;14(2):208‐223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. University of California San Francisco MS‐EPIC Team , Cree BA, Hollenbach JA, et al. Silent progression in disease activity–free relapsing multiple sclerosis. Ann Neurol. 2019;85(5):653‐666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cree BAC, Mares J, Hartung HP. Current therapeutic landscape in multiple sclerosis: an evolving treatment paradigm. CurrOpin Neurol. 2019;32(3):365‐377. [DOI] [PubMed] [Google Scholar]
- 10. Kappos L, Wolinsky JS, Giovannoni G, et al. Contribution of relapse‐independent progression vs relapse‐associated worsening to overall confirmed disability accumulation in typical relapsing multiple sclerosis in a pooled analysis of 2 randomized clinical trials. JAMA Neurol. 2020;77(9):1132‐1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17(2):162‐173. [DOI] [PubMed] [Google Scholar]
- 12. Tomassini V, Fanelli F, Prosperini L, Cerqua R, Cavalla P, Pozzilli C. Predicting the profile of increasing disability in multiple sclerosis. Mult Scler. 2019;25(9):1306‐1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Prosperini L, Ruggieri S, Haggiag S, Tortorella C, Pozzilli C, Gasperini C. Prognostic accuracy of NEDA‐3 in long‐term outcomes of multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. 2021;8(6):e1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lorscheider J, Buzzard K, Jokubaitis V, et al. Defining secondary progressive multiple sclerosis. Brain. 2016;139(9):2395‐2405. [DOI] [PubMed] [Google Scholar]
- 15. Iaffaldano P, Lucisano G, Patti F, et al. Transition to secondary progression in relapsing‐onset multiple sclerosis: definitions and risk factors. Mult Scler. 2021;27(3):430‐438. [DOI] [PubMed] [Google Scholar]
- 16. Kopp TI, Bramow S, Illes Z, et al. Application of definitions for conversion to secondary progressive MS in a Danish nationwide population. Mult Scler Relat Disord. 2021;56:103319. [DOI] [PubMed] [Google Scholar]
- 17. Hillert J, Forsberg L, Manouchehrinia A, et al. Ongoing Disease Modifying Treatment Associated with Mis‐classification of Secondary Progressive as Relapsing‐Remitting Multiple Sclerosis. PS05.04. presented at. ECTRIMS; 2020. [Google Scholar]
- 18. Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol. 2012;8(11):647‐656. [DOI] [PubMed] [Google Scholar]
- 19. Confavreux C, Vukusic S, Moreau T, Adeleine P. Relapses and progression of disability in multiple sclerosis. N Engl J Med. 2000;343(20):1430‐1438. [DOI] [PubMed] [Google Scholar]
- 20. Harding KE, Wardle M, Moore P, et al. Modelling the natural history of primary progressive multiple sclerosis. J Neurol Neurosurg Psychiatry. 2015;86(1):13‐19. [DOI] [PubMed] [Google Scholar]
- 21. Rosso M, Chitnis T. Association between cigarette smoking and multiple sclerosis: a review. JAMA Neurol. 2020;77(2):245‐253. [DOI] [PubMed] [Google Scholar]
- 22. Sintzel MB, Rametta M, Reder AT. Vitamin D and multiple sclerosis: a comprehensive review. Neurol Ther. 2018;7(1):59‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mahad DH, Trapp BD, Lassmann H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015;14(2):183‐193. [DOI] [PubMed] [Google Scholar]
- 24. Sellner J, Boggild M, Clanet M, et al. EFNS guidelines on diagnosis and management of neuromyelitis optica. Eur J Neurol. 2010;17(8):1019‐1032. [DOI] [PubMed] [Google Scholar]
- 25. Jurynczyk M, Geraldes R, Probert F, et al. Distinct brain imaging characteristics of autoantibody‐mediated CNS conditions and multiple sclerosis. Brain. 2017;140(3):617‐627. [DOI] [PubMed] [Google Scholar]
- 26. Schumacher A‐M, Mahler C, Kerschensteiner M. Pathology and pathogenesis of progressive multiple sclerosis: concepts and controversies. Neurol Int Open. 2017;1(3):E171‐E181. [Google Scholar]
- 27. Schoonheim MM, Geurts JJ, Barkhof F. The limits of functional reorganization in multiple sclerosis. Neurology. 2010;74(16):1246‐1247. [DOI] [PubMed] [Google Scholar]
- 28. Frischer JM, Bramow S, Dal‐Bianco A, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain. 2009;132(5):1175‐1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Elliott C, Wolinsky JS, Hauser SL, et al. Slowly expanding/evolving lesions as a magnetic resonance imaging marker of chronic active multiple sclerosis lesions. Mult Scler. 2019;25(14):1915‐1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jäckle K, Zeis T, Schaeren‐Wiemers N, et al. Molecular signature of slowly expanding lesions in progressive multiple sclerosis. Brain. 2020;143(7):2073‐2088. [DOI] [PubMed] [Google Scholar]
- 31. Airas L, Nylund M, Rissanen E. Evaluation of microglial activation in multiple sclerosis patients using positron emission tomography. Front Neurol. 2018;9:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Airas L, Yong VW. Microglia in multiple sclerosis—pathogenesis and imaging. Curr Opin Neurol. 2022;35(3):299‐306. [DOI] [PubMed] [Google Scholar]
- 33. Dal‐Bianco A, Grabner G, Kronnerwetter C, et al. Long‐term evolution of multiple sclerosis iron rim lesions in 7 T MRI. Brain. 2021;144(3):833‐847. [DOI] [PubMed] [Google Scholar]
- 34. Magliozzi R, Howell O, Vora A, et al. Meningeal B‐cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain. 2007;130(4):1089‐1104. [DOI] [PubMed] [Google Scholar]
- 35. Howell OW, Reeves CA, Nicholas R, et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain. 2011;134(9):2755‐2771. [DOI] [PubMed] [Google Scholar]
- 36. Kutzelnigg A, Lucchinetti CF, Stadelmann C, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain. 2005;128(11):2705‐2712. [DOI] [PubMed] [Google Scholar]
- 37. Haider L, Simeonidou C, Steinberger G, et al. Multiple sclerosis deep grey matter: the relation between demyelination, neurodegeneration, inflammation and iron. J Neurol Neurosurg Psychiatry. 2014;85(12):1386‐1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Preziosa P, Rocca MA, Mesaros S, et al. Intrinsic damage to the major white matter tracts in patients with different clinical phenotypes of multiple sclerosis: a voxelwise diffusion‐tensor MR study. Radiology. 2011;260(2):541‐550. [DOI] [PubMed] [Google Scholar]
- 39. Patrikios P, Stadelmann C, Kutzelnigg A, et al. Remyelination is extensive in a subset of multiple sclerosis patients. Brain. 2006;129(12):3165‐3172. [DOI] [PubMed] [Google Scholar]
- 40. Chang A, Staugaitis SM, Dutta R, et al. Cortical remyelination: a new target for repair therapies in multiple sclerosis. Ann Neurol. 2012;72(6):918‐926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tauhid S, Neema M, Healy BC, Weiner HL, Bakshi R. MRI phenotypes based on cerebral lesions and atrophy in patients with multiple sclerosis. J Neurol Sci. 2014;346(1–2):250‐254. [DOI] [PubMed] [Google Scholar]
- 42. Hemond CC, Healy BC, Tauhid S, et al. MRI phenotypes in MS: longitudinal changes and miRNA signatures. Neurol Neuroimmunol Neuroinflamm. 2019;6(2):e530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sørensen PS, Fox RJ, Comi G. The window of opportunity for treatment of progressive multiple sclerosis. Curr Opin Neurol. 2020;33(3):262‐270. [DOI] [PubMed] [Google Scholar]
- 44. Freedman MS, Devonshire V, Duquette P, et al. Treatment optimization in multiple sclerosis: Canadian MS Working Group recommendations. Can J Neurol Sci. 2020;47(4):437‐455. [DOI] [PubMed] [Google Scholar]
- 45. Montalban X, Gold R, Thompson AJ, et al. ECTRIMS/EAN Guideline on the pharmacological treatment of people with multiple sclerosis. Mult Scler. 2018;24(2):96‐120. [DOI] [PubMed] [Google Scholar]
- 46. Food and Drug Administration . Mayzent (Siponimod) Tablets, for Oral Use: Prescribing Information. 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/209884s000lbl.pdf [Google Scholar]
- 47. European Medicines Agency . Siponimod Summary of Product Characteristics. 2020. https://www.ema.europa.eu/en/documents/product‐information/mayzent‐epar‐product‐information_en.pdf [Google Scholar]
- 48. Lublin FD, Coetzee T, Cohen JA, Marrie RA, Thompson AJ. International Advisory Committee on Clinical Trials in MS. The 2013 clinical course descriptors for multiple sclerosis: a clarification. Neurology. 2020;94(24):1088‐1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hawker K, O'Connor P, Freedman MS, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double‐blind placebo‐controlled multicenter trial. Ann Neurol. 2009;66(4):460‐471. [DOI] [PubMed] [Google Scholar]
- 50. Leary SM, Miller DH, Stevenson VL, Brex PA, Chard DT, Thompson AJ. Interferon β‐1a in primary progressive MS: an exploratory, randomized, controlled trial. Neurology. 2003;60(1):44‐51. [DOI] [PubMed] [Google Scholar]
- 51. Montalban X, Sastre‐Garriga J, Tintoré M, et al. A single‐center, randomized, double‐blind, placebo‐controlled study of interferon beta‐1b on primary progressive and transitional multiple sclerosis. Mult Scler. 2009;15(10):1195‐1205. [DOI] [PubMed] [Google Scholar]
- 52. Wolinsky JS, Narayana PA, O'Connor P, et al. Glatiramer acetate in primary progressive multiple sclerosis: results of a multinational, multicenter, double‐blind, placebo‐controlled trial. Ann Neurol. 2007;61(1):14‐24. [DOI] [PubMed] [Google Scholar]
- 53. Signori A, Schiavetti I, Gallo F, Sormani MP. Subgroups of multiple sclerosis patients with larger treatment benefits: a meta‐analysis of randomized trials. Eur J Neurol. 2015;22(6):960‐966. [DOI] [PubMed] [Google Scholar]
- 54. Weideman AM, Tapia‐Maltos MA, Johnson K, Greenwood M, Bielekova B. Meta‐analysis of the age‐dependent efficacy of multiple sclerosis treatments. Front Neurol. 2017;8:577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lublin F, Miller DH, Freedman MS, et al. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): a phase 3, randomised, double‐blind, placebo‐controlled trial. Lancet. 2016;387(10023):1075‐1084. [DOI] [PubMed] [Google Scholar]
- 56. Sucksdorff M, Rissanen E, Tuisku J, et al. Evaluation of the effect of fingolimod treatment on microglial activation using serial PET imaging in multiple sclerosis. J Nucl Med. 2017;58(10):1646‐1651. [DOI] [PubMed] [Google Scholar]
- 57. Montalban X, Hauser SL, Kappos L, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. 2017;376(3):209‐220. [DOI] [PubMed] [Google Scholar]
- 58. Wolinsky JS, Montalban X, Hauser SL, et al. Evaluation of no evidence of progression or active disease (NEPAD) in patients with primary progressive multiple sclerosis in the ORATORIO trial. Ann Neurol. 2018;84(4):527‐536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wolinsky JS, Arnold DL, Brochet B, et al. Long‐term follow‐up from the ORATORIO trial of ocrelizumab for primary progressive multiple sclerosis: a post‐hoc analysis from the ongoing open‐label extension of the randomised, placebo‐controlled, phase 3 trial. Lancet Neurol. 2020;19(12):998‐1009. [DOI] [PubMed] [Google Scholar]
- 60. Hartung H‐P, Gonsette R, König N, et al. Mitoxantrone in progressive multiple sclerosis: a placebo‐controlled, double‐blind, randomised, multicentre trial. Lancet. 2002;360(9350):2018‐2025. [DOI] [PubMed] [Google Scholar]
- 61. Foo E, Russell M, Lily O, Ford H. Mitoxantrone in relapsing‐remitting and rapidly progressive multiple sclerosis: ten‐year clinical outcomes post‐treatment with mitoxantrone. Mult Scler Relat Disord. 2020;44:102330. [DOI] [PubMed] [Google Scholar]
- 62. Cohen JA, Cutter GR, Fischer JS, et al. Benefit of interferon β‐1a on MSFC progression in secondary progressive MS. Neurology. 2002;59(5):679‐687. [DOI] [PubMed] [Google Scholar]
- 63. Secondary Progressive Efficacy Clinical Trial of Recombinant Interferon‐Beta‐1a in MS (SPECTRIMS) Study Group . Randomized controlled trial of interferon‐beta‐1a in secondary progressive MS: clinical results. Neurology. 2001;56(11):1496‐1504. [DOI] [PubMed] [Google Scholar]
- 64. Li DK, Zhao GJ, Paty DW, University of British Columbia MS/MRI Analysis Research Group, SPECTRIMS Study Group . Randomized controlled trial of interferon‐beta‐1a in secondary progressive MS: MRI results. Neurology. 2001;56(11):1505‐1513. [DOI] [PubMed] [Google Scholar]
- 65. Kappos L, European Study Group on interferon β‐1b in Secondary Progressive MS . Placebo‐controlled multicentre randomised trial of interferon β‐1b in treatment of secondary progressive multiple sclerosis. Lancet. 1998;352(9139):1491‐1497. [PubMed] [Google Scholar]
- 66. Panitch H, Miller A, Paty D, Weinshenker B, North American Study Group on Interferon Beta‐1b in Secondary Progressive MS . Interferon beta‐1b in secondary progressive MS: results from a 3‐year controlled study. Neurology. 2004;63(10):1788‐1795. [DOI] [PubMed] [Google Scholar]
- 67. Kappos L, Weinshenker B, Pozzilli C, et al. Interferon beta‐1b in secondary progressive MS: a combined analysis of the two trials. Neurology. 2004;63(10):1779‐1787. [DOI] [PubMed] [Google Scholar]
- 68. Hommes OR, Sørensen PS, Fazekas F, et al. Intravenous immunoglobulin in secondary progressive multiple sclerosis: randomised placebo‐controlled trial. Lancet. 2004;364(9440):1149‐1156. [DOI] [PubMed] [Google Scholar]
- 69. Kapoor R, Ho PR, Campbell N, et al. Effect of natalizumab on disease progression in secondary progressive multiple sclerosis (ASCEND): a phase 3, randomised, double‐blind, placebo‐controlled trial with an open‐label extension. Lancet Neurol. 2018;17(5):405‐415. [DOI] [PubMed] [Google Scholar]
- 70. Sucksdorff M, Tuisku J, Matilainen M, et al. Natalizumab treatment reduces microglial activation in the white matter of the MS brain. Neurol Neuroimmunol Neuroinflamm. 2019;6(4):e574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kappos L, Bar‐Or A, Cree BAC, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double‐blind, randomised, phase 3 study. Lancet. 2018;391(10127):1263‐1273. [DOI] [PubMed] [Google Scholar]
- 72. Tourbah A, Lebrun‐Frenay C, Edan G, et al. MD1003 (high‐dose biotin) for the treatment of progressive multiple sclerosis: a randomised, double‐blind, placebo‐controlled study. Mult Scler. 2016;22(13):1719‐1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Cree BA, Cutter G, Wolinsky JS, et al. Safety and efficacy of MD1003 (high‐dose biotin) in patients with progressive multiple sclerosis (SPI2): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet Neurol. 2020;19(2):988‐997. [DOI] [PubMed] [Google Scholar]
- 74. Sanai SA, Saini V, Benedict RH, et al. Aging and multiple sclerosis. Mult Scler. 2016;22(6):717‐725. [DOI] [PubMed] [Google Scholar]
- 75. Chitnis T, Aaen G, Belman A, et al. Improved relapse recovery in paediatric compared to adult multiple sclerosis. Brain. 2020;143(9):2733‐2741. [DOI] [PubMed] [Google Scholar]
- 76. Malpas CB, Manouchehrinia A, Sharmin S, et al. Early clinical markers of aggressive multiple sclerosis. Brain. 2020;143(5):1400‐1413. [DOI] [PubMed] [Google Scholar]
- 77. Prosperini L, Haggiag S, Tortorella C, Galgani S, Gasperini C. Age‐related adverse events of disease‐modifying treatments for multiple sclerosis: a meta‐regression. Mult Scler. 2020;27:1391‐1402. [DOI] [PubMed] [Google Scholar]
- 78. Iacobaeus E, Arrambide G, Amato MP, et al. Aggressive multiple sclerosis (1): towards a definition of the phenotype. Mult Scler. 2020;26(9):1352458520925369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. De Meo E, Portaccio E, Giorgio A, et al. Identifying the distinct cognitive phenotypes in multiple sclerosis. JAMA Neurol. 2021;78(4):414‐425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Andravizou A, Dardiotis E, Artemiadis A, et al. Brain atrophy in multiple sclerosis: mechanisms, clinical relevance and treatment options. Auto Immun Highlights. 2019;10(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Eijlers AJC, Dekker I, Steenwijk MD, et al. Cortical atrophy accelerates as cognitive decline worsens in multiple sclerosis. Neurology. 2019;93(14):e1348‐e1359. [DOI] [PubMed] [Google Scholar]
- 82. Eshaghi A, Prados F, Brownlee WJ, et al. Deep gray matter volume loss drives disability worsening in multiple sclerosis. Ann Neurol. 2018;83(2):210‐222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Eshaghi A, Young AL, Wijeratne PA, et al. Identifying multiple sclerosis subtypes using unsupervised machine learning and MRI data. Nat Commun. 2021;12(1):2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Koch MW, Mostert J, Greenfield J, Liu WQ, Metz L. Gadolinium enhancement on cranial MRI in multiple sclerosis is age dependent. J Neurol. 2020;267(9):2619‐2624. [DOI] [PubMed] [Google Scholar]
- 85. Elliott C, Belachew S, Wolinsky JS, et al. Chronic white matter lesion activity predicts clinical progression in primary progressive multiple sclerosis. Brain. 2019;142(9):2787‐2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Suthiphosuwan S, Sati P, Absinta M, et al. Paramagnetic rim sign in radiologically isolated syndrome. JAMA Neurol. 2020;77(5):653‐655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Galassi S, Prosperini L, Logoteta A, et al. A lesion topography‐based approach to predict the outcomes of patients with multiple sclerosis treated with interferon beta. Mult Scler Relat Disord. 2016;8:99‐106. [DOI] [PubMed] [Google Scholar]
- 88. Dekker I, Sombekke MH, Balk LJ, et al. Infratentorial and spinal cord lesions: cumulative predictors of long‐term disability? Mult Scler. 2019;26(11):1381‐1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zecca C, Disanto G, Sormani MP, et al. Relevance of asymptomatic spinal MRI lesions in patients with multiple sclerosis. Mult Scler. 2016;22(6):782‐791. [DOI] [PubMed] [Google Scholar]
- 90. Tsagkas C, Magon S, Gaetano L, et al. Spinal cord volume loss: a marker of disease progression in multiple sclerosis. Neurology. 2018;91(4):e349‐e358. [DOI] [PubMed] [Google Scholar]
- 91. Tsagkas C, Naegelin Y, Amann M, et al. Central nervous system atrophy predicts future dynamics of disability progression in a real‐world multiple sclerosis cohort. Eur J Neurol. 2021;28(12):4153‐4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Manouchehrinia A, Stridh P, Khademi M, et al. Plasma neurofilament light levels are associated with risk of disability in multiple sclerosis. Neurology. 2020;94(23):e2457‐e2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Uphaus T, Steffen F, Muthuraman M, et al. NfL predicts relapse‐free progression in a longitudinal multiple sclerosis cohort study. EBioMedicine. 2021;72:103590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Högel H, Rissanen E, Barro C, et al. Serum glial fibrillary acidic protein correlates with multiple sclerosis disease severity. Mult Scler J. 2020;26(2):210‐219. [DOI] [PubMed] [Google Scholar]
- 95. Koch MW, Mostert J, Repovic P, Bowen JD, Uitdehaag B, Cutter G. Reliability of outcome measures in clinical trials in secondary progressive multiple sclerosis. Neurology. 2021;96(1):e111‐e120. [DOI] [PubMed] [Google Scholar]
- 96. Ziemssen T, Tolley C, Bennett B, et al. A mixed methods approach towards understanding key disease characteristics associated with the progression from RRMS to SPMS: physicians' and patients' views. Mult Scler Relat Disord. 2020;38:101861. [DOI] [PubMed] [Google Scholar]
- 97. Cortese R, Collorone S, Ciccarelli O, Toosy AT. Advances in brain imaging in multiple sclerosis. Ther Adv Neurol Disord. 2019;12:1756286419859722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Beynon V, George IC, Elliott C, et al. Chronic lesion activity and disability progression in secondary progressive multiple sclerosis. BMJ Neurol Open. 2022;4(1):e000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Petracca M, Margoni M, Bommarito G, Inglese M. Monitoring progressive multiple sclerosis with novel imaging techniques. Neurol Ther. 2018;7(2):265‐285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Koch MW, Mostert J, Repovic P, et al. MRI brain volume loss, lesion burden, and clinical outcome in secondary progressive multiple sclerosis. Mult Scler. 2022;28(4):561‐572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Oh J, Ontaneda D, Azevedo C, et al. Imaging outcome measures of neuroprotection and repair in MS: a consensus statement from NAIMS. Neurology. 2019;92(11):519‐533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Gingele S, Stangel M. Emerging myelin repair agents in preclinical and early clinical development for the treatment of multiple sclerosis. Expert Opin Investig Drugs. 2020;29(6):583‐594. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.