

Abstract
Chromosomal translocations with gene fusions are uniquely rare events in paraganglioma, mostly involving UBTF::MAML3 gene fusion. Precedent literature suggests that tumors involving MAML3 gene fusion correlate with poor clinical outcomes. Herein, we report a case of metastatic sporadic paraganglioma harboring EWSR1::CREM gene fusion in a 36‐year‐old male, that has not been previously described. The patient presented with large paraspinal mass that was resected the same year. Tumor recurred 3‐years later and on further work‐up, patient was found to have metastases involving both lungs. Histopathologic evaluation of the original primary tumor showed tightly packed irregular nests and cords of cells containing palely eosinophilic cytoplasm. Features considered atypical included: areas of solid growth pattern, coagulative tumor necrosis, focal cellular atypia and angiolymphatic invasion were also identified. By immunohistochemistry, the tumor cells were positive for synaptophysin and chromogranin and negative for keratin. The S100 stain highlights the sustentacular cells and the Ki‐67 proliferation index of 15%. The recurrence specimen was similar but showed increased cellularity, atypia, necrosis, and proliferative activity (Ki‐67 proliferation index of 35%). CT guided biopsy of the right lung lesion was consistent with metastasis. Next generation sequencing identified EWSR1::CREM fusion. The breakpoints were found in chromosome 22: 29683123 for EWSR1 exon 7 (NM_005243.3) and at chromosome 10:35495823 for CREM exon 6 (NM_001267562.1). Fluorescence in situ hybridization for EWSR1 gene rearrangement was positive. In summary, we report a case of metastatic paraganglioma with EWSR1::CREM gene fusion, not previously described in this entity, and expands on the phenotypic diversity within the genetic landscape of EWSR1::CREM gene fusion positive tumors.
Keywords: EWSR1::CREM, metastatic paraganglioma, molecular genetics, sporadic type
1. INTRODUCTION
Paragangliomas are rare non‐epithelial neuroendocrine tumors of sympathetic or parasympathetic origin, that can occur in different locations including head and neck, skull base, and paravertebral region. Head and neck paragangliomas are extremely rare (accounting for 0.6% of all head and neck tumors) and the carotid body is the most common site for occurrence for these lesions. 1 While it has been recognized that the majority of paragangliomas do not metastasize, a subset of these tumors shows recurrent behavior 2 and potential for distant metastasis. 3 Several features including the size and location of tumor, histopathologic findings, proliferation index, levels of plasma methoxytyramine, gene fusions involving MAML3, germline mutations in SDHB and somatic mutations of SETD2/ATRX4 were previously reported to be predictors of aggressive behavior. 4 , 5 , 6 , 7 World Health Organization (WHO) in its most recent classification of paragangliomas and pheocytochromas has maintained a neutral stance about the utility of most scoring systems based on these features but the current conceptual framework defines all paragangliomas as potentially malignant with variable risk for both locally aggressive behavior and distant metastases. 8
Cyclic AMP element modulator (CREM), a member of the CREB family of transcription factors, is a transcription factor that is expressed widely in human tissue and regulates the expression of other genes. 9 The EWS RNA binding protein 1 (EWSR1) regulate a variety of cellular processes including gene expression and RNA processing. EWSR1 gene rearrangements are frequently seen in a growing number of epithelial and soft tissue tumors. 10 On the other hand, CREM gene rearrangements are relatively novel and infrequent, compared to other members of the CREB transcription factors (inclusive of ATF1 and CREB1), and occur commonly in partnership with EWSR1. 9 , 11 They have been reported in a variety of unrelated phenotypical diverse epithelial and mesenchymal tumors with differing prognosis including: hyalinizing clear cell tumors of oral cavity and oropharynx, myxoid mesenchymal tumors, angiomatoid fibrous histiocytoma, abdominal epithelioid neoplasm, spindle/small cell carcinoma, clear cell odontogenic neoplasm, and pulmonary mesenchymal neoplasm 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27
Herein, we report for the first time, a unique case of a metastatic sporadic paraganglioma harboring a unique EWSR1::CREM gene fusion, in a 36 year old male. To the best of our knowledge, this is the first case of metastatic paraganglioma harboring the EWSR1::CREM gene fusion. This report expands the molecular genetics of metastatic paragangliomas and adds to the growing list of phenotypically diverse benign and malignant epithelial and mesenchymal tumors harboring the EWSR1::CREM gene fusion. The biologic consequence and clinical significance of this gene fusion remains to be elucidated.
2. MATERIAL AND METHODS
2.1. Immunohistochemistry
Immunohistochemical stains using the following commercially available antibodies were performed using the Leica Bond III autostainers (Leica Biosystems): Calretenin (Dako, DAK.CALRET.1, 1:600), Chromogranin (Cell Marque, LK2H10, 1:300), Ki67 (Dako, MIB‐1, 1:400), S100 (Dako, Rabbit polyclonal, 1:3000), Synaptophysin (Leica, 27G12, 1:450), CAM 5.2 (BD, CAM5.2, 1:80), AE1/3 (Dako, AE1&AE3, 1:1200), CD56 (Dako, 123C3, 1:100), CD117 (Dako, Rabbit polyclonal, 1:400), EMA (Dako, E29, 1:700), GFAP (Dako, Rabbit polyclonal, 1:10000), HMB45 (Dako, HMB45, 1:100), Melan A (Dako, A103, 1:600), PAX8 (Epitomics, EP298, 1:100), PLAP (Dako, 8A9, 1:25), Progesterone Receptor (Dako, PgR636, 1:600), GATA3 (Biocare, L50‐823, 1:400). All protocols were developed by and performed at the OSU Wexner Medical Center Clinical Laboratory, 410 West 10th Avenue, Doan Hall 310, Columbus, OH 43210. All positive and negative controls showed appropriate staining.
2.2. Next‐generation sequencing
DNA sequencing was performed at Caris Life Sciences, Inc. on representative formalin fixed paraffin embedded tissue sections as described previously. 28 Briefly, DNA was extracted from the tissue enriched for tumor by microdissection and was mechanically sheared by ultrasonicator. DNA libraries were prepared and hybridized to the probes. Amplification was performed and the target captured amplified DNA libraries were sequenced on Illumina HiSeq 4000. A custom designed assay SureSelect XT by Agilent was used to enrich 592 whole‐gene targets. All variants were detected with a >99% confidence based on amplicon coverage and allele frequency. The average sequencing depth of covering was >500 and analytical sensitivity was 5%. The versioned reference identifier used for the transcript ID was Feb.2009 (GRCh37/hg19).
For detection of gene fusions, mRNA was isolated from the formalin fixed paraffin embedded tissue using the Agilent SureSelect XT Low input library preparation reagent kit in conjunction with SureSelect Human All Exon V7 panel and Illumina NovaSeq as described previously. 29 Prior to molecular testing, tumor enrichment was achieved by harvesting targeted tissue using manual microdissection techniques. This assay is designed to detect fusions occurring at known and novel breakpoints within genes. The versioned reference identifier used for the transcript ID was Feb.2009 (GRCh37/hg19).
2.3. Fluorescence in situ hybridization
Interphase fluorescence in situ hybridization (FISH) analysis was performed on a 4‐μm unstained section of representative formalin‐fixed, paraffin‐embedded tissue using a 2‐fluorophore labeled break apart probe set (Abbot Molecular) designed to hybridize EWSR1 translocation breakpoint. Hybridization signals were scored in 100 interphase tumor cells. The cut off level for scoring was 15% for dual color probe system.
3. RESULTS
3.1. Case report
The patient is a 36‐year‐old male with a history of neck pain and a large (5 cm), palpable paraspinal neck mass that was resected in 2016 and confirmed as paraganglioma on histopathological exam. There were no associated episodic headaches, increased sweating, and tachycardia, referable to labile hypertension. Patient presented to the medical oncology clinic 3 years after the initial resection with a chief complaint of a new mass, about 6 cm in greatest dimension, in the neck. A PET scan also detected the presence of lesions in lungs bilaterally. The new tumor was excised, and histopathological exam confirmed the presence of recurrent paraganglioma. Patient was treated with external beam radiation therapy and systemic chemotherapy. A CT guided biopsy was performed for the lesion in right lung, and it was confirmed to be metastatic paraganglioma.
The histopathologic examination of the first (primary) resection was considered diagnostic of paraganglioma. The lesion consisted of tightly packed irregular nests and cords of polygonal cells with pale eosinophilic cytoplasm with focal nuclear atypia (Figure 1A–I). Unique to this case were the presence of atypical features, including solid and infiltrative growth pattern, coagulation tumor necrosis and rare intravascular tumor emboli. By immunohistochemistry, the tumor cells were positive for synaptophysin, chromogranin and negative for calretinin, Cam 5.2, AE1/3, CD56, CD117, EMA (Epithelial Membrane Antigen), GATA3, GFAP, HMB45, Melan A, PAX8, PLAP and Progesterone Receptor. The S100 stain highlighted the sustentacular cells. In keeping with the non‐functional nature of the tumor, plasma levels of free metanephrine and normetanephrine were within normal limits. Immunostaining was also performed for the expression of SDHB and showed normal staining pattern, indicating the absence of germline mutation in the genes of succinate dehydrogenase (SDH) complex involving the SDHB, SDHC and SDHD genes. 30 The Ki‐67 proliferative index was estimated to be 15% by manual quantitation. The microscopic evaluation of the recurrent resection specimen showed similar features except that there were prominent solid and infiltrative growth patterns, punctuated by many confluent areas of coagulation tumor necrosis (Figure 1J–O). By immunohistochemistry, the lesional cells stained similarly as in the primary tumor with the tumor cells positive for synaptophysin, chromogranin, and S100 (in the sustentacular cells) and negative for multiple keratins, confirming the diagnosis. However, the Ki‐67 proliferation index was increased to 35%. Further, the CT guided biopsy of a lung nodule also shows the presence of histologically confirmed metastatic paraganglioma (Figure 1P–S).
FIGURE 1.
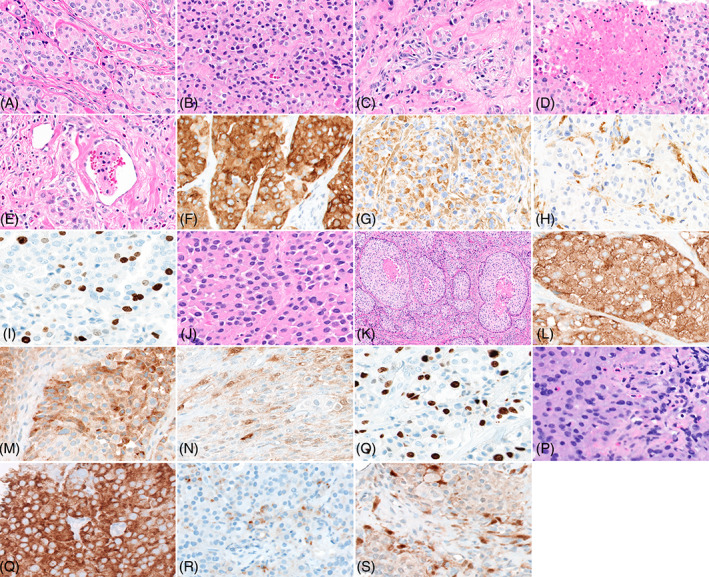
Microscopic and immunohistopathological features of primary, recurrent and metastatic lesion. Panel A shows tumor cells organized in a conventional organoid pattern (40×). Panel B shows area of the lesion with a solid growth pattern (40×). Panel C shows cells with infiltrative growth pattern (40×). Panel D shows area of necrosis within the tumor (40×). Panel E shows area with lymphovascular invasion (40×). Panel F shows tumor cells with strong diffuse positive immunoreactivity to synaptophysin (40×). Panel G shows tumor cells with positive immunoreactivity to chromogranin (40×). Pane H shows sustentacular cells showing immunoreactivity to S100 (40×). Panel I shows reactivity of tumor cells to Ki67 (40×). Panel J shows tumor cells organized in a conventional organoid pattern (40×). Panel K shows area of necrosis (20×). Panel L shows tumor cells showing strong diffuse immunoreactivity to synaptophysin (40×). Panel M shows tumor cells showing diffuse immunoreactivity to chromogranin (40×). Panel N shows immunoreactivity of sustentacular cells to S100 (40×). Panel O shows reactivity of tumor cells to Ki67 (40×). Panel P shows histological features of tumor cells in metastatic lesion in lung (40×). Panel Q shows strong diffuse immunoreactivity to synaptophysin (40×). Panel R shows focal immunoreactivity of tumor cells to chromogranin (40×). Panel S shows immunoreactivity of sustentacular cells to S100 (40×).
Whole transcriptome RNA‐Seq and DNA sequencing studies were performed, and they identified EWSR1::CREM fusion (Figure 2). The breakpoints were found in chromosome 22: 29683123 for EWSR1 exon 7 (NM_005243.3) and at Chromosome 10:35495823 for CREM exon 6 (NM_001267562.1). Previously, breakpoints for this fusion gene have been reported on the exon 7, 8, 9, 10, 11, 13, 14, and 15 for EWSR1 and exon 4, 5, 6, 7, 8, 9, and 13 for CREM (Table 1). The tumor showed low mutational burden and had stable microsatellite profile. No other targetable mutations were identified. Given the rather unexpected and unique molecular genetic profile, we performed fluorescence in situ hybridization for EWSR1 gene rearrangement to corroborate the results of the next generation sequencing. FISH studies showed rearrangement of EWSR1 gene. There was an abnormal pattern of hybridization for the probes spanning the gene at chromosome 22q12 that exceeded the established threshold of 15%. The histological features and immunophenotypic profile, confirmed the diagnosis of paraganglioma. After surgical resection of the secondary neck mass the patient underwent cytotoxic chemotherapy with vincristine, cyclophosphamide and doxorubicin/dacarbazine according to the protocol. 31 The most recent imaging studies performed 5 years after the primary diagnosis demonstrated stable to slightly increased pulmonary parenchymal metastatic nodules by chest CT scan and no evidence of residual/recurrent mass in the posterior left neck by neck CT scan.
FIGURE 2.
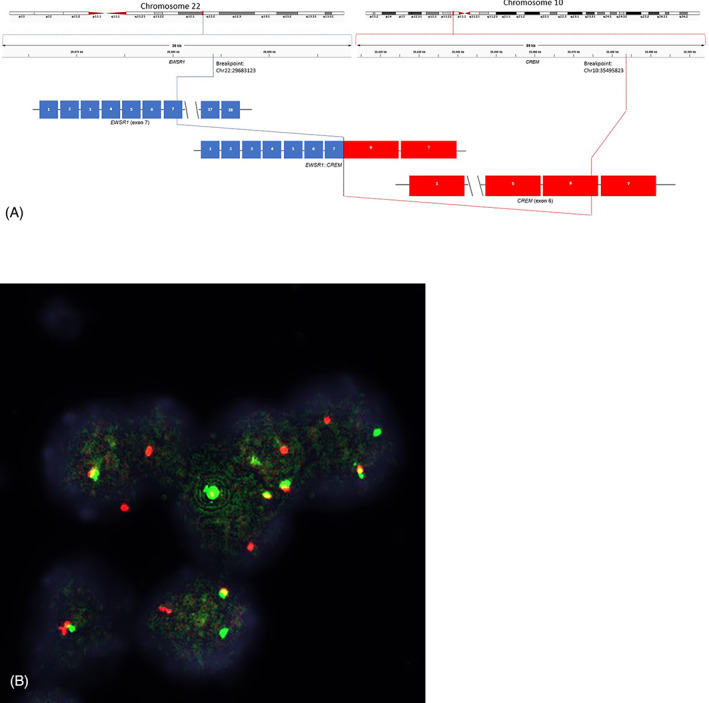
EWSR1::CREM fusion identified by next generation sequencing and confirmed by FISH. Panel A shows schematic for the EWSR1::CREM fusion identified by next generation sequencing. Panel B shows EWSR1 breakapart FISH demonstrating split red and green signal indicating translocation of the gene.
TABLE 1.
Summary of reported tumors including indexed case with EWSR1::CREM gene fusion
| Diagnosis | Site | Morphologic features | Age (years)/sex (M/F) | Number of cases | Genomic breakpoint | Reference |
|---|---|---|---|---|---|---|
| Metastatic paraganglioma | Neck | Polygonal cells with focal nuclear atypia, solid and infiltrative growth pattern, tumor necrosis, intravascular tumor emboli | 36/M | 1 |
EWSR1 exon 7 CREM exon 6 |
Indexed case |
| Myxoid mesenchymal tumor |
Pelvis/distal extremities Cranium Cranium Pelvis/cranium Cranium |
Multinodular growth of epithelioid to spindle shaped cells arranged in nests and cords, fibromyxoid stroma, scattered lymphocytes Spindled/histiocytoid cells with pale eosinophilic cytoplasm Prominent myxoid background Ovoid/round cells with eosinophilic cytoplasm Heterogenous growth pattern with alteration in cell density Myxoid and chondroid stroma Round/ovoid cells with pale cytoplasm Alteration in cell density Myxoid background Monomorphic/mildly atypical cells with oval to spindled nuclei Myxoid matrix Vague nodular architecture |
33/F 55/F 65/F 9/M 20/F 15/F 18/M |
2 1 1 2 1 |
EWSR1 exon 10 CREM exon 7 EWSR1 exon 8 CREM exon 8 and 9 EWSR1 exon 7 CREM exon 7 Not reported EWSR1 exon 14 or 15 CREM exon 7 or intron 6 EWSR1 intron 9 CREM intron 3 |
Lee et al. 2022 12 Kambe et al. 2021 27 White et al. 2019 16 Kao et al. 2017 18 Bale et al. 2017 19 |
| Myxoid angiomatoid fibrous histiocytoma |
Neck Cranium Lung/distal extremities Cranium Cranium |
Syncytial cells in myxoid stroma, multinodular growth and pseudoangiomatoid spaces Polygonal/spindle shaped cells with eosinophilic cytoplasm and vesicular nucleus Small capillaries Myxoid stroma Multinodular myxoid growth, round to spindled cell, prominent lymphoid cuff Round to spindle shaped cells with clear to eosinophilic cytoplasm Alteration in cell density Collagenous to myxoid stroma Atypical histiocytic clear cells with round nuclei, Focal myxoid areas |
43/F 29/M 47/M 50/M 54/M 19/M Recurrence at age of 29 52/M |
1 1 3 1 1 |
EWSR1 exon 13 CREM exon 6 EWSR1 exon 7 CREM exon 7 Not reported EWSR1 exon 7 CREM exon 13 Not reported |
Lee et al. 2022 12 Tan et al. 2021 15 Yoshida et al. 2019 26 Gareton et al. 2018 17 Gilbert et al. 2022 25 |
| Spindled/small cell carcinoma |
Liver Abdomen/chest cavity |
Uniform oval to spindle shaped cells with indistinct cytoplasm Hyalinized stroma Small oval to round spindle cells, delicate collagen, and high mitotic activity |
15/M 15/M 63/M |
1 2 |
EWSR1 exon 11 CREM exon 7 EWSR1 exon 11 CREM exon 7 EWSR1 exon 14 CREM exon 7 |
Shibayama et al. 2022 13 Originally reported in Yoshida et al. 2019 26 Yoshida et al. 2019 26 |
| Clear cell carcinoma |
Buccal mucosa Oropharynx |
Tumor nests consisting of epithelioid and clear cells Partially hyalinized stroma Sheets, nests, and cords of clear cells Hyalinized stroma |
69/F 68/F 75/F 62/M |
1 3 |
Not reported EWSR1 exon 14 CREM exon 5 Not reported for case 2 EWSR1 exon 11 CREM exon 6 |
Hoshino et al. 2021 21 Chapman et al. 2018 20 |
|
Epithelioid Neoplasm |
Kidney |
Epithelioid cells with granular eosinophilic to clear cytoplasm Fibrous stroma |
55/M | 1 | Not reported | Agaimy et al. 2021 22 |
| Clear cell odontogenic carcinoma | Maxilla |
Clear cells and epidermoid cells arranged in clusters Hyalinized stroma |
63/M | 1 |
EWSR1 exon 9 CREM exon 4 |
Breik et al. 2021 24 |
| Clear cell mesenchymal neoplasm | Lungs |
Cuboidal cells with clear cytoplasm Hyalinized stroma |
56/M | 1 |
EWSR1 exon 14 CREM exon 6 |
Komatsu et al. 2020 23 |
| Mesenchymal neoplasm | Cranium | Small round blue cells | 31/M | 1 |
EWSR1 exon 15 CREM exon 5 |
Liu et al. 2020 14 |
| Clear cell sarcoma | Distal extremities | Spindle cells with amphophilic to clear cytoplasm and monomorphic nuclei | 49/F | 1 | Not reported | Yoshida et al. 2019 26 |
4. DISCUSSION
We describe a case of a sporadic metastatic paraganglioma with a unique EWSR1::CREM gene fusion in a 36‐year‐old male patient. To the best of our knowledge, the EWSR1::CREM gene fusion has not been previously described in paraganglioma. Historically, the pathologic diagnosis of metastatic paraganglioma is based on the presence of distant metastasis and there are currently no established histopathologic features or immunophenotypic profile that are considered predictive of the aggressive phenotype. Pertinent to this case is the presence of de novo atypical features at the time of original resection including confluent growth pattern, coagulative necrosis and angiolymphatic tumor emboli. The significance of this finding viz‐a‐viz the development of metastasis is not clear, consistent with precedent literature. 32 , 33
The clinical course of paraganglioma is unpredictable and some cases show aggressive disease potential. 2 , 3 , 4 While a significant portion of these aggressive tumors are associated with germline cancer inheritable genes and well characterized, little is known of the sporadic cases. The majority of the pediatric paragangliomas are associated with certain germline genetic mutations. The mutations are classified into three different clusters based on the mutations in genes regulating pseudohypoxia pathway, WNT and kinase signaling pathway. Most pediatric paraganglioma harbor mutations in the genes regulating pseudohypoxia pathway such as SDHB, SDHD, and VHL. 34 In the case of our patient, no mutations in the known pathways were identified. It is also interesting to note that most of the studies into the genetics of paragangliomas have identified mutations but very few translocations have been reported.
In the metastatic setting, the treatment options are limited with an overall poor prognosis. 35 Metastases are usually seen in regional and distant lymph nodes, lungs, liver, and bone. 36 Although, not formally endorsed by WHO, size and histological features of the tumor, and levels of plasma methoxytyramine are considered to be the indicators of metastatic disease. 4 , 5 , 7 Additionally, a recent comprehensive molecular genomic characterization of paragangliomas appears to suggest that the molecular genetic determinants of aggressive paragangliomas includes: SDHB germline mutations, gene fusions involving MAML3 and somatic mutations involving ATRX. 6 Chromosomal translocations lead to the expression of chimeric proteins that are involved in the tumorigenesis of solid organ malignancies including soft tissue sarcomas, leukemias and lymphomas. However, chromosomal translocations are rare events in paraganglioma. UBTF::MAML3 gene fusion has been reported in sporadic cases of paraganglioma. The fusion positive tumors were not histologically distinctive from the fusion negative tumors. These tumors had an over expression of MAML3, and it was speculated that this may cause an increase in the growth rate of the tumors. The presence of MAML3 fusion gene appeared to correlate with poor clinical outcome in these patients. 6
EWSR1::CREM gene fusions were first described in melanoma cell lines. Knockdown of expression of the fusion product led to a decrease in cell proliferation and invasion of the tumor. The cells also showed features of senescence indicating that the chimeric protein plays an important role in the tumorigenesis. 37 The gene fusion has been detected in many phenotypically diverse epithelial and mesenchymal tumors including hyalinizing clear cell tumors of oropharynx, buccal clear cell carcinoma, abdominal epithelioid neoplasms, pulmonary mesenchymal neoplasm, clear cell odontogenic neoplasm, angiomatoid fibrous histiocytoma, myxoid mesenchymal tumors, and others. 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 Table 1 summarizes reported cases with the EWSR1::CREM gene rearrangement. The prognostic significance of the fusion product is not known in all tumors. Furthermore, the biologic role of this gene fusion in this case is unknown. To our knowledge, no therapeutic options are currently available for the EWSR1::CREM fusion positive tumors. More studies are needed to identify the prognostic and therapeutic potential of this fusion gene. Importantly, this case highlights the phenomenon of molecular pleiotropism that is increasingly reported with the widespread use of next generation sequencing platform in the routine clinical laboratory analysis of solid tumor malignancies. With an increase in the availability of molecular genetic testing, the possibility of identifying novel molecular alterations is going to become more common. This underscores the pivotal role of pathologists in integrating these molecular data in the morphological and immunophenotypic examination of these tumors to provide a rational and complete diagnosis and guide therapy.
In summary, we have presented a case of sporadic metastatic paraganglioma harboring a unique EWSR1::CREM gene fusion. The biologic consequence of this gene fusion in metastatic paraganglioma remains to be determined. Prospective tumor accrual and comprehensive molecular characterization may help uncover molecular genetic subsets of metastatic paraganglioma that may have prognostic and/or therapeutic implications.
AUTHOR CONTRIBUTIONS
Obiajulu Hans Iwenofu conceived of the project. Sehrish Javaid, Ashley Patton, Gabriel Tinoco, Steve Oghumu, and Obiajulu Hans Iwenofu provided essential tools/data. Sehrish Javaid and Obiajulu Hans Iwenofu wrote the article. All authors approved the final manuscript.
CONFLICT OF INTEREST
The authors declare that they do not have any conflicts of interest.
Javaid S, Patton A, Tinoco G, Oghumu S, Iwenofu OH. Metastatic sporadic paraganglioma with EWSR1::CREM gene fusion: A unique molecular profile that expands the phenotypic diversity of the molecular landscape of the EWSR1::CREM gene fusion positive tumors. Genes Chromosomes Cancer. 2023;62(2):85‐92. doi: 10.1002/gcc.23094
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Rana MU, Østhus AA, Heimdal K, Jebsen P, Revheim MER, Osnes TA. Head and neck paragangliomas in Norway, importance of genetics, updated diagnostic workup and treatment. Acta Otolaryngol. 2021;141(3):303‐308. doi: 10.1080/00016489.2020.1845397 [DOI] [PubMed] [Google Scholar]
- 2. Salle SP‐L, Chbat J, Lacroix A, et al. Postoperative recurrences in patients operated for pheochromocytomas and paragangliomas: new data supporting lifelong surveillance. Cancers (Basel). 2022;14(12):2942. doi: 10.3390/CANCERS14122942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Javidiparsijani S, Brickman A, Lin DM, et al. Is regional lymph node metastasis of head and neck paraganglioma a sign of aggressive clinical behavior: a clinical/pathologic review. Ear Nose Throat J. 2021;100(6):447‐453. doi: 10.1177/0145561319863373 [DOI] [PubMed] [Google Scholar]
- 4. Ayala‐Ramirez M, Feng L, Johnson MM, et al. Clinical risk factors for malignancy and overall survival in patients with pheochromocytomas and sympathetic paragangliomas: primary tumor size and primary tumor location as prognostic indicators. J Clin Endocrinol Metab. 2011;96(3):717‐725. doi: 10.1210/JC.2010-1946 [DOI] [PubMed] [Google Scholar]
- 5. Eisenhofer G, Lenders JWM, Siegert G, et al. Plasma methoxytyramine: a novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur J Cancer. 2012;48(11):1739‐1749. doi: 10.1016/J.EJCA.2011.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fishbein L, Leshchiner I, Walter V, et al. Comprehensive molecular characterization of pheochromocytoma and paraganglioma. Cancer Cell. 2017;31(2):181‐193. doi: 10.1016/J.CCELL.2017.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pierre C, Agopiantz M, Brunaud L, et al. COPPS, a composite score integrating pathological features, PS100 and SDHB losses, predicts the risk of metastasis and progression‐free survival in pheochromocytomas/paragangliomas. Virchows Arch. 2019;474(6):721‐734. doi: 10.1007/S00428-019-02553-5 [DOI] [PubMed] [Google Scholar]
- 8. Mete O, Asa SL, Gill AJ, Kimura N, de Krijger RR, Tischler A. Overview of the 2022 WHO classification of paragangliomas and pheochromocytomas. Endocr Pathol. 2022;33(1):90‐114. doi: 10.1007/S12022-022-09704-6 [DOI] [PubMed] [Google Scholar]
- 9. Kaprio H, Heuser VD, Orte K, Tukiainen M, Leivo I, Gardberg M. Expression of transcription factor CREM in human tissues. J Histochem Cytochem. 2021;69(8):495‐509. doi: 10.1369/00221554211032008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Flucke U, van Noesel MM, Siozopoulou V, et al. EWSR1—the most common rearranged gene in soft tissue lesions, which also occurs in different bone lesions: an updated review. Diagnostics (Basel, Switzerland). 2021;11(6):1093. doi: 10.3390/DIAGNOSTICS11061093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thway K, Fisher C. Mesenchymal tumors with EWSR1 gene rearrangements. Surg Pathol Clin. 2019;12(1):165‐190. doi: 10.1016/J.PATH.2018.10.007 [DOI] [PubMed] [Google Scholar]
- 12. Lee PH, Kao YC, Hsieh TH, et al. Myoepithelial and oral intracranial myxoid mesenchymal tumor‐like neoplasms as diagnostic considerations of the ever‐expanding extracranial myxocollagenous tumors harboring FET‐CREB fusions. Pathol Res Pract. 2022;229:153700. doi: 10.1016/J.PRP.2021.153700 [DOI] [PubMed] [Google Scholar]
- 13. Shibayama T, Shimoi T, Mori T, et al. Cytokeratin‐positive malignant tumor in the abdomen with EWSR1/FUS‐CREB fusion: a clinicopathologic study of 8 cases. Am J Surg Pathol. 2022;46(1):134‐146. doi: 10.1097/PAS.0000000000001742 [DOI] [PubMed] [Google Scholar]
- 14. Liu C, Liu Y, Zhao Y, et al. Primary intracranial mesenchymal tumor with EWSR1‐CREM gene fusion: a case report and literature review. World Neurosurg. 2020;142:318‐324. doi: 10.1016/J.WNEU.2020.07.015 [DOI] [PubMed] [Google Scholar]
- 15. Tan NJH, Pratiseyo PD, Wahjoepramono EJ, et al. Intracranial myxoid angiomatoid fibrous histiocytoma with “classic” histology and EWSR1:CREM fusion providing insight for reconciliation with intracranial myxoid mesenchymal tumors. Neuropathology. 2021;41(4):306‐314. doi: 10.1111/NEUP.12737 [DOI] [PubMed] [Google Scholar]
- 16. White MD, McDowell MM, Pearce TM, Bukowinski AJ, Greene S. Intracranial myxoid mesenchymal tumor with rare EWSR1‐CREM translocation. Pediatr Neurosurg. 2019;54(5):347‐353. doi: 10.1159/000501695 [DOI] [PubMed] [Google Scholar]
- 17. Gareton A, Pierron G, Mokhtari K, et al. ESWR1‐CREM fusion in an intracranial myxoid angiomatoid fibrous histiocytoma‐like tumor: a case report and literature review. J Neuropathol Exp Neurol. 2018;77(7):537‐541. doi: 10.1093/JNEN/NLY039 [DOI] [PubMed] [Google Scholar]
- 18. Kao YC, Sung YS, Zhang L, et al. EWSR1 fusions with CREB family transcription factors define a novel myxoid mesenchymal tumor with predilection for intracranial location. Am J Surg Pathol. 2017;41(4):482‐490. doi: 10.1097/PAS.0000000000000788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bale TA, Oviedo A, Kozakewich H, et al. Intracranial myxoid mesenchymal tumors with EWSR1‐CREB family gene fusions: myxoid variant of angiomatoid fibrous histiocytoma or novel entity? Brain Pathol. 2018;28(2):183‐191. doi: 10.1111/BPA.12504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chapman E, Skalova A, Ptakova N, et al. Molecular profiling of hyalinizing clear cell carcinomas revealed a subset of tumors harboring a novel EWSR1‐CREM fusion: report of 3 cases. Am J Surg Pathol. 2018;42(9):1182‐1189. doi: 10.1097/PAS.0000000000001114 [DOI] [PubMed] [Google Scholar]
- 21. Hoshino M, Inoue K, Kaneda T, et al. A case of buccal clear cell carcinoma caused by rare fusion gene: EWSR1‐CREM. Case Rep Dent. 2021;2021:1‐7. doi: 10.1155/2021/5557247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Agaimy A, Stoehr R, Otto M, et al. Intra‐abdominal EWSR1/FUS‐CREM‐rearranged malignant epithelioid neoplasms: two cases of an emerging aggressive entity with emphasis on misleading immunophenotype. Virchows Arch. 2021;480:481‐486. doi: 10.1007/S00428-021-03140-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Komatsu M, Sakai Y, Nishikubo M, et al. EWSR1‐CREM fusion in pulmonary mesenchymal neoplasm showing distinctive clear cell morphology. Pathol Int. 2020;70(12):1020‐1026. doi: 10.1111/PIN.13030 [DOI] [PubMed] [Google Scholar]
- 24. Breik O, Higginson J, Al‐Ajami AK, Mohamed A, Martin T, Amel‐Kashipaz R. Clear cell odontogenic carcinoma: first report of novel EWSR1‐CREM fusion gene in case of long‐term misdiagnosis. Head Neck Pathol. 2021;15(4):1391‐1398. doi: 10.1007/S12105-021-01302-Y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gilbert AR, Yan L, McDougall CM. Broadening the age of incidence of intracranial angiomatoid fibrous histiocytoma with EWSR1‐CREM fusion: a case report. J Neuropathol Exp Neurol. 2020;79(11):1244‐1246. doi: 10.1093/JNEN/NLAA114 [DOI] [PubMed] [Google Scholar]
- 26. Yoshida A, Wakai S, Ryo E, et al. Expanding the phenotypic spectrum of mesenchymal tumors harboring the EWSR1‐CREM fusion. Am J Surg Pathol. 2019;43(12):1622‐1630. doi: 10.1097/PAS.0000000000001331 [DOI] [PubMed] [Google Scholar]
- 27. Kambe A, Kuwamoto S, Shimizu T, et al. A case of intracranial myxoid mesenchymal tumor with EWSR1:CREM fusion in an adult female: extensive immunohistochemical evaluation. Neuropathology. 2021;41(4):315‐323. doi: 10.1111/NEUP.12740 [DOI] [PubMed] [Google Scholar]
- 28. Jonna S, Vanderwalde A, Nieva J, et al. Effect of prior therapy on tumor mutational burden in NSCLC. Transl Lung Cancer Res. 2021;10(3):1231‐1238. doi: 10.21037/TLCR-20-1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ahn JS, Ervin J, Cummings TJ, López GY, Wang SHJ. Papillary glioneuronal tumor with a novel GPR37L1‐PRKCA fusion. J Neuropathol Exp Neurol. 2021;80(10):1004‐1006. doi: 10.1093/JNEN/NLAB055 [DOI] [PubMed] [Google Scholar]
- 30. Papathomas TG, Oudijk L, Persu A, et al. SDHB/SDHA immunohistochemistry in pheochromocytomas and paragangliomas: a multicenter interobserver variation analysis using virtual microscopy: a multinational study of the European Network for the Study of Adrenal Tumors (ENS@T). Mod Pathol. 2015;28(6):807‐821. doi: 10.1038/modpathol.2015.41 [DOI] [PubMed] [Google Scholar]
- 31. Huang H, Abraham J, Hung E, et al. Treatment of malignant pheochromocytoma/paraganglioma with cyclophosphamide, vincristine, and dacarbazine: recommendation from a 22‐year follow‐up of 18 patients. Cancer. 2008;113(8):2020‐2028. doi: 10.1002/CNCR.23812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Torres‐Costa M, Flores B, Torregrosa N, et al. Malignant prediction in paragangliomas: analysis for clinical risk factors. Langenbecks Arch Surg. 2021;406(7):2441‐2448. doi: 10.1007/S00423-021-02222-9 [DOI] [PubMed] [Google Scholar]
- 33. Yamazaki Y, Gao X, Pecori A, et al. Recent advances in histopathological and molecular diagnosis in pheochromocytoma and paraganglioma: challenges for predicting metastasis in individual patients. Front Endocrinol (Lausanne). 2020;11:587769. doi: 10.3389/FENDO.2020.587769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yen K, Lodish M. Pheochromocytomas and paragangliomas. Curr Opin Pediatr. 2021;33(4):430‐435. doi: 10.1097/MOP.0000000000001029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hamidi O. Metastatic pheochromocytoma and paraganglioma: recent advances in prognosis and management. Curr Opin Endocrinol Diabetes Obes. 2019;26(3):146‐154. doi: 10.1097/MED.0000000000000476 [DOI] [PubMed] [Google Scholar]
- 36. Jimenez C. Treatment for patients with malignant pheochromocytomas and paragangliomas: a perspective from the hallmarks of cancer. Front Endocrinol (Lausanne). 2018 May;9:277. doi: 10.3389/FENDO.2018.00277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Giacomini CP, Sun S, Varma S, et al. Breakpoint analysis of transcriptional and genomic profiles uncovers novel gene fusions spanning multiple human cancer types. PLoS Genet. 2013;9(4):e1003464. doi: 10.1371/JOURNAL.PGEN.1003464 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.