

Abstract
Retinoic acid is the main active vitamin A derivate and a key regulator of embryonic development. Excess of retinoic acid can disturb palate development in mice leading to cleft palate. WNT signaling is one of the main pathways in palate development. We evaluated the effects of retinoic acid on palate fusion and WNT signaling in in vitro explant cultures. Unfused palates from E13.5 mouse embryos were cultured for 4 days with 0.5 μM, 2 μM or without retinoic acid. Apoptosis, proliferation, WNT signaling and bone formation were analyzed by histology and quantitative PCR. Retinoic acid treatment with 0.5 and 2.0 μM reduced palate fusion from 84% (SD 6.8%) in the controls to 56% (SD 26%) and 16% (SD 19%), respectively. Additionally, 2 μM retinoic acid treatment increased Axin2 expression. Retinoic acid also increased the proliferation marker Pcna as well as the number of Ki‐67‐positive cells in the palate epithelium. At the same time, the WNT inhibitors Dkk1, Dkk3, Wif1 and Sfrp1 were downregulated at least two‐fold. Retinoic acid also down‐regulated Alpl and Col1a2 gene expression. Alkaline phosphatase (ALP) activity was notably reduced in the osteogenic areas of the retinoic acid‐ treated palates. Our data suggest that retinoic acid impairs palate fusion and bone formation by upregulation of WNT signaling.
Keywords: bone formation, palate fusion, retinoic acid, Wnt signaling
INTRODUCTION
Cleft palate is a disruption of the normal orofacial structures caused by a failure in the growth, elevation or fusion of the palatal shelves during embryonic development [1]. Cleft palate etiology is complex and poorly understood, but it is known to involve genetic as well as environmental factors such as vitamin imbalances [2, 3]. Under normal conditions, the palatal shelves grow out vertically from the maxillary prominences, elevate to a horizontal position, adhere forming the medial epithelia seam and then fuse [4]. Fusion may involve the migration, differentiation and/or apoptosis of medial epithelial seam cells [5]. After the fusion of the palatal shelves, bone formation takes place through intramembranous ossification, in which condensed neural crest‐derived mesenchymal cells differentiate directly into osteoblasts [6].
Several signaling pathways have been related to palatogenesis and cleft palate including fibroblast growth factor, sonic hedgehog and wingless‐INT (WNT) [7, 8, 9, 10]. Canonical WNT signaling is activated by the binding of WNT ligands to the Frizzled (FZD) receptors and the low‐density lipoprotein coreceptors‐related protein 5/6 (LRP5, LRP6) [11]. This interaction stabilizes the cytoplasmic β‐catenin and facilitates its translocation into the nucleus, where it binds to lymphoid enhancer‐binding factor 1/T cell‐specific transcription factor (TCF/LEF) [12]. In the absence of a WNT ligand, β‐Catenin is phosphorylated and degraded in the cytoplasm [13]. Canonical WNT signaling is antagonized by several secreted proteins such as the secreted frizzled related proteins (sFRPs), WNT inhibitory protein 1 (WIF1) and the dickkopf family (DKK) [14]. sFRPs and WIF1 bind to WNT ligands and prevent their interaction with the WNT membrane receptors [15, 16]. sFRPs can also bind to the WNT‐binding domain of the FZD receptors [17]. The DKK family of proteins bind to the WNT co‐receptors LRP5 or LRP6, and to Kremen1 and Kremen2. This complex is then endocytosed resulting in the depletion of LRP from the plasma membrane [18].
Genetic studies have associated WNT gene mutations with cleft palate in humans such as a homozygous nonsense mutation in WNT3 and several single nucleotide polymorphism near the WNT6‐WNT10a cluster at the 2q35 region of chromosome 2 [10, 19]. Experiments in mice have also confirmed the role of WNT signaling in cleft palate [20, 21]. For instance, tissue‐specific deletions of Catnb, Tcf4 or Lef1 from the palatal shelves epithelium in mice disrupt medial epithelial seam disappearance resulting in cleft palate [22]. WNT signaling is also involved in bone formation as it promotes the proliferation and differentiation of mesenchymal stem cells (MSC) into the osteogenic lineage [23]. Several in vivo and in vitro studies indicate that WNT signaling can be disrupted by retinoic acid [24, 25, 26].
Retinoic acid is the main active metabolite of vitamin A and is crucial for normal pattern formation during embryonic development [27]. Retinoic acid regulates gene expression by binding to cellular retinoic acid‐binding proteins (CRABPs) that transport retinoic acid to the nucleus, where it binds to the retinoic acid receptors and retinoid X receptors (RARs/RXRs). This complex then binds to retinoic acid response elements (RARE) in the DNA thus activating target gene expression [28]. An overdose of retinoic acid at different embryonic stages induces congenital malformations in both mouse and humans [29, 30, 31]. One of these malformations induced by retinoic acid is cleft palate [30, 32, 33]. Depending on the stage of administration, retinoic acid inhibits palatal shelves growth or fusion [30, 34]. Several studies show contradictory effects of retinoic acid on osteogenic differentiation, depending mainly on the dose and embryonic stage of exposure [35, 36]. For instance, retinoic acid decreases the differentiation of osteoblasts [37]. In contrast, retinoic acid induces bone mineralization in mouse embryo limbs [36]. However, the molecular mechanisms underlying the effects of retinoic acid on palate development are not clearly understood yet. Here, we hypothesized that retinoic acid disrupts palate fusion and bone formation by affecting WNT signaling. Explant cultures of the mouse embryonic palate show that retinoic acid disrupts palate fusion and osteogenic differentiation. This coincides with a downregulation of WNT inhibitors and an upregulation of the WNT marker gene Axin2.
MATERIAL AND METHODS
In vitro palate culture
Seventy‐two palatal shelves were dissected in cold fetal bovine serum (FBS) from the mouse fetuses of nine different mothers at embryonic day 13.5 using a stereomicroscope (Leica MZ16). The wild‐type mice were from an outbred strain Hsd:ICR (CD1). Experiments were approved by the board for animal experiments of Radboud University, Nijmegen (RUDEC 2015‐0080) according to Dutch laws and regulations. The dissected palates were gently put on a 0.8 μm pore MF‐Millipore membrane filter (Millipore) on top of a sterilized stainless‐steel mesh in a six‐well plate (for detailed description, see Figure 1). Generally, three palates were put in one well. Three different media conditions were used. Control: DMEM/F12 medium (Sigma‐Aldrich) containing 5% FBS (GIBCO), 200 mM glutamine, 400 mg/ml ascorbate, 1% penicillin – streptomycin. The other two media contained control medium with all‐trans‐Retinoic acid (Sigma‐Aldrich) in a concentration of 0.5 μM retinoic acid or 2 μM retinoic acid, diluted in dimethyl sulfoxide (Sigma‐Aldrich). Palates were incubated at 37°C with 5% CO2 for 4 days. Medium was changed and photographs were made every 24 h.
FIGURE 1.
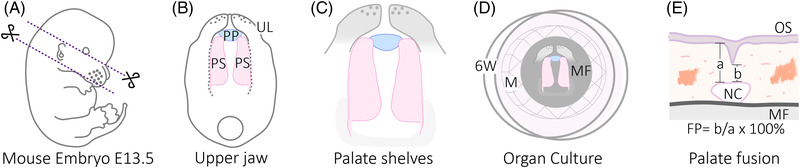
Mouse embryo palate dissection and quantification method for palate fusion. (A) Under the stereomicroscope, mouse embryos were examined to confirm the embryonic stage E13.5 based on Theiler's stages criteria [96]. Each embryo was placed in one drop of cold PBS in a petri dish. To isolate the upper jaw, two transversal cuts were made; one just below the eye level and over the ears and the second from the oral commissures to below the ears. (B) Once the upper jaw is isolated, the oral side is orientated up and the surrounding tissue is carefully removed. PP: primary palate. PS: Palate shelves. UL: Upper lip. (C) In order to maintain the in vivo position of the palatal shelves, some tissue anterior and posterior to the shelves was left. (D) The dissected palates are carefully transferred to a well in a 6‐well plate, containing a stainless mesh with a filter on top and 3.5 ml of medium. 6W: well from a 6‐well plate. M: Stainless‐steel mesh. MF: 8 μM pore MF‐Millipore membrane filter. (E) Diagram showing the quantification of palate fusion on a histological section. OS: Oral side. b: Total thickness of the palate. a: part of the palate fused. NC: Nasal cavity. FP: Fusion percentage
RNA isolation and real‐time quantitative PCR
Three biological replicates, containing at least six pooled palates (from three different mothers) were used for the RNA isolation using the RNeasy MiniKit (Qiagen) according to the manufacturer's protocol. Equal amounts of RNA from each sample (1 μg) was reverse‐transcribed using the iScriptTM Reverse Transcriptase system (Bio‐Rad). Quantitative real‐time PCR reactions were carried out in 25 μl containing 5 μl cDNA (12.5 ng), 4.5 μl RNA‐free water, 2.5 μM forward and reverse primers and 12.5 μl SYBR Green Supermix (Bio‐Rad). The amplifications were performed in a CX96 Real Time System (Bio‐Rad) using the following conditions: initial denaturalization at 95°C for 3 min, followed by 39 cycles performed at 95°C for 15 s and 60°C for 30 s. All data were normalized to the expression of three reference genes (Gapdh, β‐actin and 18 s rRNA). Relative expression was calculated according to the 2−ΔCt method. Primers were obtained from Biolegio and their sequences are summarized in Table 1.
TABLE 1.
Primer sequences
| Gene category | Symbol | Forward Primer (5′−3′) | Reverse Primer (5′−3′) |
|---|---|---|---|
| RA RESPONSIVE | Cyp26b1 | GATCCTACTGGGCGAACACC | GGAGAAGACCTTGCGCTTGT |
| Crabp2 | TGATCTCGACTGCTGGCTTG | TCCCATCGGGTTCCCATAAAG | |
| Rarb | GAAAACGACGACCCAGCAAG | TTACACGTTCGGCACCTTTC | |
| APOPTOSIS MARKER | Trp53 | GGAAGACTCCAGTGGGAACC | CTTCTGTACGGCGGTCTCTC |
| PROLIFERATION MARKER | Pcna | AGAGCATGGACTCGTCTCA | CCAGCACATTTTAGAATTTTGGACA |
| WNT TARGET | Axin2 | GGTTCCGGCTATGTCTTTGC | CAGTGCGTCGCTGGATAACTC |
| WNT INHIBITORS | Dkk3 | GGCCCACAGTCTTCATCAAT | CCAGAGTGGACAGGTGGTCT |
| Dkk1 | CGGGGGATGGATATCCCAGAA | ACGGAGCCTTCTTGTCCTTTG | |
| Kremen1 | TGGGTTTCCATGATCCTTGT | GCATGAGGACGGAGTCTACTG | |
| Sfrp1 | TCTAAGCCCCAAGGTACAACC | GCTTGCACAGAGATGTTCAATG | |
| Sfrp4 | ATCATCCTTGAACGCCACTC | TCGAACACAAGTCCCTCTCA | |
| Wif1 | GCATTCTTTGTTGGGCTTTC | CCATCAGGCTAGAGTGCTCA | |
| BONE DIFFERENTIATION MARKERS | Alpl | CCAGCAAGAAGAAGCCTTTG | AACCCAGACACAAGCATTCC |
| Col1a2 | CCTGGCAAAGACGGACTCAAC | GCTGAAGTCATAACCGCCACTG | |
| Runx2 | CGGACGAGGCAAGAGTTTCA | GGATGAGGAATGCGCCCTAA |
Fusion percentage
After four days in culture, six palates from each concentration group were fixed overnight in 4% paraformaldehyde, embedded in paraffin and sectioned at 5 μm. The sections were stained with hematoxylin and eosin (HE), and photographed with a Zeiss Imager Z1 microscope (Zeiss AxioCam MRc5; Carl Zeiss Microimaging). Measurements were made on every tenth section, of the middle region of each palate using ImageJ software [38]. The fusion percentage (FS) was calculated as the part of the palate that is fuse (b), divided by total thickness of the palate, including any remaining medial epithelial seam (a), as previously reported (Figure 1E) [39].
Immunohistochemistry
Mouse palate shelves cultured in vitro during four days, six palates from each concentration group, were fixed overnight in 4% paraformaldehyde, embedded in paraffin and sectioned at 5 μm. Briefly, the sections were deparaffinized with xylene and rehydrated with a graded series of ethanol. Next, endogenous peroxidase activity was inhibited in 30% H2O2 in phosphate buffered saline (PBS) in the dark at room temperature for 20 min. The sections for DKK3 and KI‐67 staining were boiled with 0.1 M citrate buffer (pH 6.0) in a microwave oven for 10 min and left at room temperature for 20 min to cool down. To reduce non‐specific binding, the samples were incubated in 10% normal donkey serum in PBS for 20 min in the dark. After washing with PBS, the primary antibody against DKK3 and KI‐67 (both from Proteintech) was applied and incubated overnight at 4°C. The sections for WIF1, sFRP4 and AXIN2 received a trypsin treatment for 1 min at 37°C (all from abcam). Then, the primary antibody was applied and labelled with a streptavidin‐biotin immunoperoxidase method using a commercial kit (Vectorlabs). Antibody binding was visualized using diaminobenzidine (DAB) as a chromogene to produce a brown color. Counterstaining was performed with Mayer's hematoxylin. The slides were mounted with DPX mounting medium and photographed with the Zeiss Imager Z1 microscope (Zeiss AxioCam MRc5; Carl Zeiss Microimaging).
Alkaline phosphatase staining
The palate sections were deparaffinized with xylene and rehydrated with a graded series of ethanol. Then, they were washed with MilliQ water and incubated at 37°C with preheated ALP solution of pH 9.5 (100 mM NaCl, 100 mM Tris–HCl, 50 mM MgCl2, 4.5 μl/ml nitroblue tetrazolium and 3.5 μl/ml 5‐bromo‐4‐chloro‐3‐indolyl phosphate) for 60 min and washed again in MilliQ water. Sections were counterstained with natrium acetate 0,1 M (pH 5.1) for 15 min followed by 0.1% Methylgreen in natrium acetate (pH 5.1). Finally, the sections were mounted in Kaisers gelatin and photographed with the Zeiss Imager Z1 microscope (Zeiss AxioCam MRc5; Carl Zeiss Microimaging)
Statistical analysis
The Shapiro‐Wilk test showed that all data were normally distributed. Each culture experiment was performed in triplicate, and the results are presented as mean ± SD. Differences between the groups were evaluated by one‐way ANOVA. Post‐hoc comparisons were made using the Tukey's multiple comparison test. Differences were considered significant if p < 0.05. All statistical tests were performed with Graphpad Prism version 8.2.1.
RESULTS
Retinoic acid effect in palate fusion
Palates shelves were dissected from mouse embryos at E13.5 and cultured with 0.5 μM and 2 μM retinoic acid for up to 4 days. At day 1, the shelves from the control group had already grown and contacted in the midline (Figure 2A). The palatine rugae also started to be visible (data not shown). At day 1 in the 0.5 μM and 2 μM retinoic acid groups, the anterior region of the shelves was not in contact (Figure 2A). From 2 to 4 days in culture, no evident changes occurred in the palate shelves from the control group. However, in the 0.5 μM retinoic acid group a persistent gap was observed in the anterior region. The length of the palate shelves was measured between the yellow dotted lines (Figure 2A, bottom row). The palates treated with 2 μM retinoic acid showed a progressive reduction in length up to 34% after 4 days in culture, compared to the controls.
FIGURE 2.
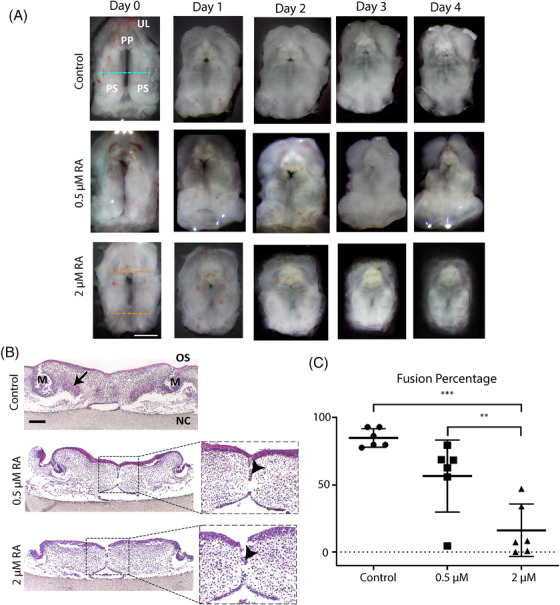
Retinoic acid disrupts palate fusion in mouse palate cultures. Palates were isolated from E13.5 mouse embryos from nine different mothers, cultured oral‐side up for 4 days with 0.5–2 μM or without retinoic acid, fixed, and stained with HE. (A) Representative daily pictures of palates in culture. Green dotted line: middle region. Red dotted line: palate shelves length. PP: primary palate. UL: upper lip. PS: palate shelves. Scale bars: 1 mm. (B) HE staining of frontal sections. Representative pictures of the palates from the middle region, stained with HE. M: molar. OS: oral side. NC: nasal cavity. Arrowheads: medial epithelial seam. Scale bars: 200 μm. (C) Palate shelves fusion percentage. Six palates from each concentration group were used for the measurements. Ten consecutive sections from the middle of each palate were analyzed (* p < 0.05, *** p < 0.001)
Histological sections were evaluated after 4 days in culture. The controls showed limited persistence of medial epithelial seam, molars buds (Figure 2B, M) and mesenchymal condensation (Figure 2B, black arrows). Retinoic acid treatment (0.5 and 2 μM) showed a decreased number of samples with molar buds (data not shown), persistence of the medial epithelial seam (Figure 2B, red arrows) or reduced contact between the palatal shelves (data not shown). Mesenchymal condensations were absent (Figure 2B). Then, to quantify the fusion of the palatal shelves, defined as the disappearance of the medial epithelial seam, the percentage of fusion was measured using image analysis. After 4 days in culture, the control group showed 84% ± 6.8% of fusion (Figure 2C) while in the 0.5 μM retinoic acid group this had decreased to 56 ± 26% (not significant, Figure 2C). The fusion percentage was significantly reduced (16 ± 19%) in the 2 μM retinoic acid group when compared with the control and the 0.5 μM retinoic acid group (p < 0.001 and p < 0.05, respectively; Figure 2C).
Retinoic acid effects on apoptosis, proliferation and WNT signaling
To confirm the functionality of retinoic acid, we determined the expression levels of cytochrome P450 family 26 subfamily B member 1 (Cyp26b1), cellular retinoic acid binding protein 2 (Crabp2) and retinoic acid receptor beta (Rarb) that are involved in retinol‐dependent signaling. As expected, retinoic acid up‐regulated the expression of two retinoic acid‐responsive genes (Figure 3A). Cyp26b1 gene expression showed a 2‐fold increase in the 0.5 μM retinoic acid group (p < 0.05) and a 4‐fold increase in the 2‐μM retinoic acid treated group (p < 0.01). Rarb expression showed a 50‐fold increase in the 2 μM retinoic acid group compared to the controls (p < 0.01). Crabp2 expression only showed a trend towards increased expression.
FIGURE 3.
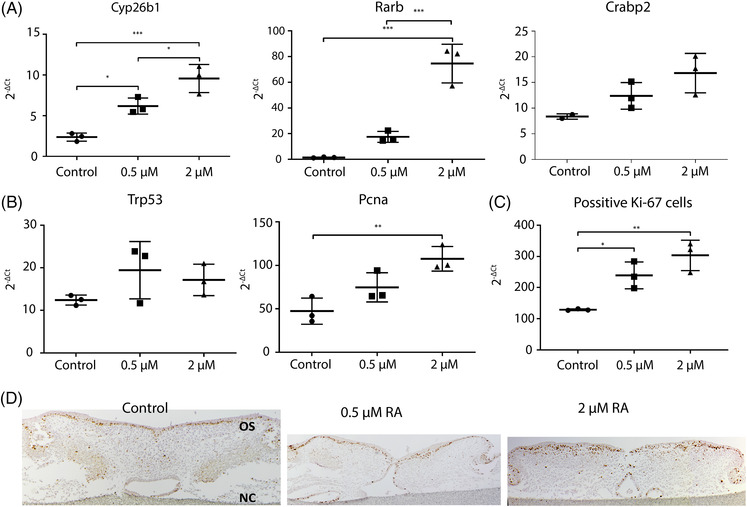
Gene expression analysis. Quantitative real‐time PCR was performed with RNA isolated from mouse palates cultured for 4 days with 0.5–2 μM or without retinoic acid. (A) Expression of retinoic acid‐responsive genes. Data are represented as the mean ± SD (N = 3). * p < 0.05, *** p < 0.001, compared with the controls. (B) Trp53 and Pcna gene expression. Data are represented as the mean ± SD (N = 3). ** p < 0.01. (C) Proliferation marker KI‐67 positive cells. Data are represented as the mean ± SD (N = 3. * p < 0.05, ** p < 0.01, compared with the controls. (D) KI‐67 immunohistochemistry. Representative pictures of the staining. OS: oral side. NC: nasal cavity
We also determined the expression of an apoptosis marker and a proliferation marker (Trp53 and Pcna). After 4 days in culture, no differences were observed in the expression of Trp53 (Figure 3B). Pcna expression showed a 3.2‐fold increased expression only in the palate cultures treated with 2 μM retinoic acid (p < 0.05, Figure 3B). Positive cells for the proliferation marker Ki‐67 were located in the plate epithelium, and the number increased from 129 in the controls, to 239 and 303 in the 0.5 and 2 μM retinoic acid treated groups respectively (p < 0.05, Figure 3C, D).
To evaluate whether the expression of Axin2, a WNT marker gene, was affected by retinoic acid during palate fusion, we analyzed the gene expression of Axin2 (40, 41). Axin2 gene expression did not show significant changes in the 0.5 μM treated group. 2 μM retinoic acid treatment induced a 4.9‐fold increase in Axin2 expression, compared with the control group (p < 0.05; Figure 4A). In the controls, AXIN2 protein expression was located mainly in the oral epithelium and the condensed mesenchyme of the palate (Figure 4B). In the 0.5 μM retinoic acid‐treated group, AXIN2 was almost absent in the mesenchyme but the staining intensity was increased in the oral and nasal epithelium (Figure 4B). In the 2 μM retinoic acid‐treated group, AXIN2 expression was stronger all over the mesenchyme and the oral epithelium (Figure 4B).
FIGURE 4.
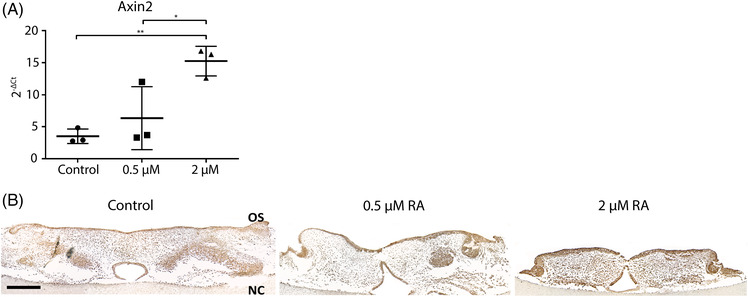
WNT signaling target gene expression. (A) Axin2 gene expression. Data are represented as the mean ± SD (N = 3). * p < 0.05, ** p < 0.01. (B) AXIN2 immunohistochemistry. Representative pictures of the staining. Scale bars: 200 μm. OS: oral side. NC: nasal cavity
WNT inhibitor expression in palate shelves treated with retinoic acid
We have shown earlier that retinoic acid induces the expression of WNT inhibitors during osteogenic differentiation of MC‐3T3 preosteoblasts cultured in vitro (37). To investigate whether the activation of WNT signaling was caused by inhibition of expression of WNT inhibitors, we evaluated the expression of six WNT inhibitors after 4 days of retinoic acid treatment. The expression of dickkopf‐related protein 1 (Dkk1) and dickkopf‐related protein 3 (Dkk3) was significantly down‐regulated compared to the control group (p < 0.001, Figure 5A). The kringle containing transmembrane protein 1 (Kremen1) did not show differences in expression (Figure 5A). The WNT inhibitor factor 1 (Wif1) was down‐regulated in the retinoic acid–treated palates (p < 0.05, Figure 5A). The gene expression of the WNT inhibitor secreted frizzled related protein 1 (Sfrp1) showed a 2‐fold decrease in the palates treated with 0.5 μM retinoic acid (p < 0.01, Figure 5A). Sfrp4 was significantly up‐regulated in the two retinoic acid‐treated groups (p < 0.01, Figure 5A).
FIGURE 5.
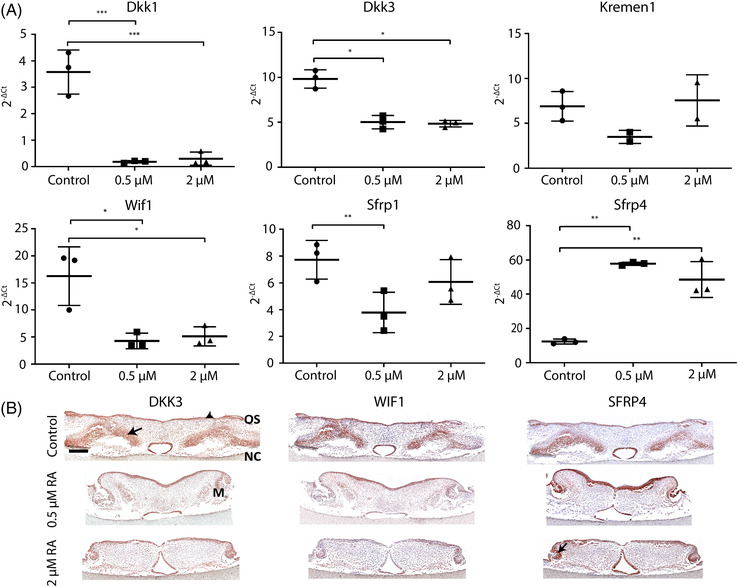
Retinoic acid affects the expression of WNT inhibitors. Palates were isolated from E13.5 mouse embryos from nine different mothers, cultured oral‐side up for 4 days with 0.5–2 μM or without retinoic acid and then processed for qPCR or fixed for histology. (A) Relative WNT inhibitors gene expression. Data are represented as the mean ± SD (N = 3). * p < 0.05, ** p < 0.01, *** p < 0.001. M: molar. OS: oral side. NC: nasal cavity (B) Immunohistochemistry of WNT inhibitors. Representative pictures of the staining of each group. Black arrows: protein expression. Scale bars: 200 μm
To localize the WNT inhibitors DKK3, SFRP4 and Wif1 in the cultured palates we used immunostaining. Palates from the control group showed DKK3 protein expression in the oral and nasal epithelium and in the mesenchymal areas lateral to the middle (Figure 5B, DKK3‐control). However, in the retinoic acid treated groups, mesenchymal expression was almost absent and the signal in the epithelial tissues was reduced (Figure 5B, DKK3).
Palates from the control group, showed WIF1 expression in the nasal and oral epithelium, and in the lateral mesenchymal tissue (Figure 5B, WIF1‐control). In the 0.5 μM retinoic acid‐treated group, oral epithelial WIF1 expression was restricted to the intermolar region, and the mesenchymal expression was strongly reduced. In the 2 μM RA‐treated group, a weak WIF1 expression was present in the lateral parts of the oral epithelium. Palates from the control group, showed a strong SFRP4 protein expression in the nasal epithelium but not in the oral epithelium. The lateral mesenchymal areas were also stained (Figure 5B, SFRP4‐control). Interestingly, the expression in the oral epithelium was greatly increased in the 0.5 μM retinoic acid‐treated palates, which continued into the medial epithelial seam and the nasal epithelium. Additionally, SFRP4 was highly expressed in the molar buds and the surrounding condensed mesenchyme but absent in the lateral mesenchyme. The 2 μM retinoic acid‐treated group showed slightly increased expression in the oral and nasal epithelium, and the molar buds (Figure 5B, SFRP4).
Osteogenic differentiation in palatal shelves treated with retinoic acid
As WNT signaling is related to bone formation, we also analyzed the expression of three marker genes for osteogenic differentiation. The gene expression of the early osteogenic marker alkaline phosphatase (Alpl) was significantly down‐regulated in the palates treated with 0.5 μM and 2 uM retinoic acid (p < 0.01, Figure 6A). The extracellular matrix protein collagen type I alpha 2 (Col1a2) was significantly down‐regulated in the 2 μM retinoic acid‐treated palates (p < 0.05, Figure 6A). Retinoic acid did not significantly inhibit Runx2 expression, but showed a clear trend (p > 0.04, Figure 6A).
FIGURE 6.
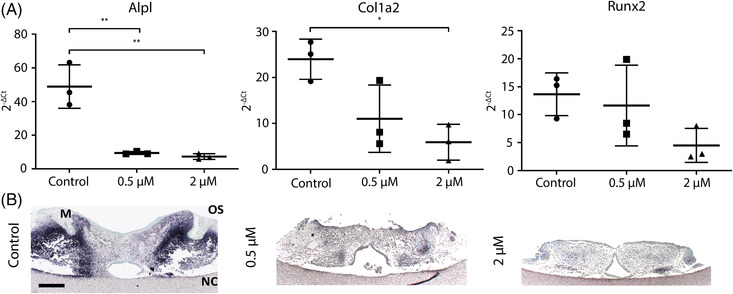
Retinoic acid inhibits the expression of osteogenic markers. After four days in culture RNA from the palates was isolated for qPCR. Additionally, palates were fixed in 4% paraformaldehyde for ALP staining. (A) Relative osteogenic markers gene expression. Data are represented as the mean ± SD (N = 3). * p < 0.05, ** p < 0.01. (B) Alkaline phosphatase staining. Representative pictures of each group. Dark blue staining indicates ALP activity. M: molar tooth bud. OS: oral side. NC: nasal cavity Scale bar: 200 μm
To localize ALP activity, we used an enzymatic staining on histological sections. In the controls, ALP activity was located in the lateral sides of the palatal shelves and around the molar tooth buds. ALP activity was completely absent in the two retinoic acid treated groups except a few small lateral spots in the 0.5 μM retinoic acid‐treated palates (Figure 6B).
DISCUSSION
The complex molecular and cellular regulation of palate development is susceptible to disruptions, which may lead to c left palate [42]. In humans, retinoic acid is known to increase the risk of cleft palate if the serum concentration is outside the normal range of 0.004–0.009 μM [43, 44, 45]. In addition, retinoic acid is known to interact with WNT signaling, which has an essential role during embryogenesis [46, 47]. We hypothesized that retinoic acid disrupts palate development by affecting WNT signaling. Therefore, we cultured palates from mouse embryos (E13.5) to study the effect of retinoic acid on palate fusion and the underlying mechanism.
Our results show that retinoic acid reduces palate fusion in vitro as measured by the reduced disappearance of the medial epithelial seam. Previous studies show similar effects of retinoic acid both in vitro and in vivo, which was related to the inhibition of apoptosis and cell migration in the medial epithelial seam [48, 49, 50]. However, in our data, the expression of the apoptosis marker Trp53 was not affected by retinoic acid. As the medial epithelial seam is only a small part of the total palatal shelves tissue, we might not have picked up an inhibition of Trp53 expression. In addition, we show reduction in the growth of the palatal shelves by retinoic acid. Similarly, studies in pregnant mice exposed to retinoic acid show that the growth of the palatal shelves was reduced because of an inhibition of mesenchymal cell proliferation leading to cleft palate [51, 52, 53, 54]. Conversely, our results show increased Ki‐67 expression by retinoic acid, which seems to be mainly located in the epithelium of the treated palates. Contradictory effects of retinoic acid on cell proliferation have been also reported earlier. While retinoic acid treatment induces proliferation of irradiated murine fetal liver‐derived stromal cells, it also has anti‐proliferative effects on human renal and breast cancer cells [55, 56, 57].
To clarify the mechanism of the retinoic acid effects, we evaluated the activity of WNT signaling. WNT signaling plays an important role during development, controlling both proliferation and differentiation processes [58]. Disruption of WNT signaling during mouse palate development can lead to cleft lip and/or palate [47]. For instance, conditional inactivation of WNT/β‐catenin signaling in the mouse palate epithelium leads to cleft lip and/or palate due to failed palate fusion [59]. It has also been shown that LiCl‐induced WNT activation during palate development inhibits palate fusion inhibition and ossification [60]. Our results show that retinoic acid‐treatment upregulated the expression of the WNT marker gene Axin2. Axin2 is recognized as a good marker gene in vertebrates for WNT signaling as it acts in a negative feedback loop to limit and fine‐tune Wnt signaling [61, 62, 63, 64]. Studies in mouse mesenchymal stem cells and human fetal palatal chondrocytes also showed that retinoic acid stimulated WNT signaling by an increase in the β‐Catenin level during osteogenic induction [65, 66]. However, studies in several types of cancer cells and mouse pre‐osteoblasts have shown that retinoic acid can also down‐regulate WNT signaling [37, 67, 68, 69]. Interestingly, another study reports suppression of Wnt signaling and cleft palate in mouse embryos in vivo by retinoic acid [53]. However, they included tongue tissue in the expression analyses, which might have affected their results as it has been reported that WNT signaling is required to induce proliferation of epithelial cells and differentiation of muscle progenitor cells in the tongue [70]. Our results also showed that the increased AXIN2 expression is mainly localized in the palate epithelium along with an increase in the proliferation marker KI‐67. Enhanced proliferation related to WNT signaling activation has also been reported in mouse and human cardiomyocytes, and human ocular epithelial cells [71, 72]. Our results indicate that increased WNT signaling in retinoic acid‐treated palatal shelves induces proliferation of epithelial cells.
The increased activity of WNT signaling might also explain the persistence of the medial epithelial seam in the retinoic acid‐treated palates. Several studies in the cancer field show that WNT signaling inhibits apoptosis, one of the crucial cellular processes for medial epithelial seam disappearance [73, 74, 75]. For instance, inactivation of the WNT inhibitor DKK1 in human breast cancer cells increases WNT signaling and inhibits apoptosis [75]. In addition, reduced expression of DKK2 in human and mouse breast cancer cells has also been related to inhibition of apoptosis and cell migration [76].
Our results also show that retinoic acid down‐regulates the expression of the WNT inhibitors Dkk1, Dkk3 and Wif1 in the cultured palates. Similar results were found in mouse bone marrow stem cells in which retinoic acid stimulated WNT target gene expression by downregulation of Dkk1 [77]. This was also reported in human neuroblastoma cells and in mouse cerebrovascular development where retinoic acid is required to suppress Dkk and Sfrp expression [78, 79]. Also, during lung development, a retinoic acid‐WNT network seems to maintain lung progenitor cell fate by a retinoic acid‐dependent Dkk1 suppression leading to increased WNT signaling [80]. Together, the data suggest that retinoic acid stimulates WNT signaling through repression of WNT inhibitors. However, further work is needed to clarify the exact mechanism through which retinoic acid downregulates WNT inhibitors in palate development.
Different from the other WNT inhibitors, Sfrp4 gene and protein expression was increased in the palates treated with retinoic acid. This was also shown in pancreatic cancer cells, where retinoic acid induced Sfrp4 expression as well as decreased WNT signaling [81]. Interestingly, SFRPs can also function as WNT enhancers favoring the transport of WNT ligands to the FZD receptor in Xenopus embryos, MDCK cells and Drosophila S2 cells [82, 83]. Similarly, a simultaneous increase in SFRP4 and WNT expression has been shown in mouse and human skin affected by systemic sclerosis [84]. Based on these reported mechanisms of SFRP4, we suggest that it functions as an agonist of WNT signaling after retinoic acid exposure of the palate.
In normal palate development, bone formation starts around the time of fusion in the lateral areas of the palatal shelves [4]. Our results show a reduction in expression of the osteogenic marker Alp by retinoic acid, a trend of reduction in Runx2 and a pronounced decrease in ALP activity in the mesenchyme. Also, in vivo retinoic acid inhibits the development of the palatine and maxillary bones in mouse embryos [85]. In general, increased WNT signaling is related to an increased bone mass in mouse and rat studies [86, 87]. This evidence suggests that retinoic acid reduces WNT signaling and subsequent osteogenesis in palate development. In mesenchymal stem cells, WNT signaling is required for their commitment to the osteoblast lineage, and inhibition of the adipogenic and chondrogenic cell fate [23, 88]. Once commitment is established, canonical WNT signaling is essential for osteoblast precursor proliferation and differentiation [89]. It has also been shown that too low as well as too high serum retinoic acid levels can contribute to poor bone health and skeletal fragility in humans [90]. Moreover, long‐term exposure to retinoic acid impedes osteoblast differentiation and prevents mineralization of mouse pre‐osteoblasts, bone marrow stromal cells and calvarial bone cultures [91, 92, 93]. Additionally, it has been suggested that retinoic acid inhibits the differentiation of osteogenic progenitor cells, leading to a marked reduction in the expression of osteogenic markers (Runx2, Alpl, Sp7) as also shown in our results [94, 95]. We suggest that long‐term retinoic acid exposure inhibits bone formation trough down‐regulation of osteogenic genes.
In summary, this study shows that retinoic acid significantly reduces palate fusion and osteogenic differentiation. Our data suggest that this is correlated to an increased WNT signaling caused by a reduced expression of WNT inhibitors. Further in vivo experiments, for instance using reporter mouse lines, are needed to validate our in vitro findings.
CONFLICTS OF INTEREST
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AUTHOR CONTRIBUTIONS
Conceptualization: Laury Roa, Hans Von den Hoff; Investigation: Laury Roa, Marjon Bloemen. Writing – original draft: Laury Roa, Hans Von den Hoff. Writing‐review and editing: Laury Roa, Hans Von den Hoff, Frank Wagener, Carine Carels. All authors approved the final manuscript.
ACKNOWLEDGEMENTS
This research was supported in part by the Departamento Administrativo de Ciencia Tecnología e Innovación (Colciencias), Colombia.
Roa Fuentes LA, Bloemen M, Carels CEL, Wagener FADTG, Von den Hoff JW. Retinoic acid effects on in vitro palatal fusion and WNT signaling. Eur J Oral Sci. 2022;130:e12899. 10.1111/eos.12899
REFERENCES
- 1. Paiva KBS, Maas CS, Dos Santos PM, Granjeiro JM, Letra A. Extracellular matrix composition and remodeling: current perspectives on secondary palate formation, cleft lip/palate, and palatal reconstruction. Front Cell Dev Biol. 2019;7:340. 10.3389/fcell.2019.00340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dental S, Candotto V, Oberti L, Gabrione F, Greco G, Rossi D, et al. Current concepts on cleft lip and palate etiology. J Biol Regul Homeost Agents. 2019;33(3 Suppl. 1):145‐51. [PubMed] [Google Scholar]
- 3. Jahanbin A, Shadkam E, Miri HH, Shirazi AS, Abtahi M. Maternal folic acid supplementation and the risk of oral clefts in offspring. J Craniofac Surg. 2018;29:e534‐e41. [DOI] [PubMed] [Google Scholar]
- 4. Bush JO, Jiang R. Palatogenesis: morphogenetic and molecular mechanisms of secondary palate development. Development. 2012;139:231‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lan Y, Xu J, Jiang R. Cellular and molecular mechanisms of palatogenesis. Curr Top Dev Biol. 2015;115:59‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nassif A, Senussi I, Meary F, Loiodice S, Hotton D, Robert B, et al. Msx1 role in craniofacial bone morphogenesis. Bone. 2014;66:96‐104. [DOI] [PubMed] [Google Scholar]
- 7. Weng M, Chen Z, Xiao Q, Li R, Chen Z. A review of FGF signaling in palate development. Biomed Pharmacother. 2018;103:240‐7. [DOI] [PubMed] [Google Scholar]
- 8. Xu J, Liu H, Lan Y, Aronow BJ, Kalinichenko VV, Jiang R. A Shh‐Foxf‐Fgf18‐Shh molecular circuit regulating palate development. PLoS Genet. 2016;12:e1005769. 10.1371/journal.pgen.1005769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Paiva KB, Silva‐Valenzuela M, Massironi SM, Ko GM, Siqueira FM, Nunes FD. Differential Shh, Bmp and Wnt gene expressions during craniofacial development in mice. Acta Histochem. 2010;112:508‐17. [DOI] [PubMed] [Google Scholar]
- 10. Reynolds K, Kumari P, Sepulveda Rincon L, Gu R, Ji Y, Kumar S, et al. Wnt signaling in orofacial clefts: crosstalk, pathogenesis and models. Dis Model Mech. 2019;12(2). 10.1242/dmm.037051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nusse R, Clevers H. Wnt/beta‐catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017;169:985‐99. [DOI] [PubMed] [Google Scholar]
- 12. Huber O, Korn R, McLaughlin J, Ohsugi M, Herrmann BG, Kemler R. Nuclear localization of beta‐catenin by interaction with transcription factor LEF‐1. MechDev. 1996;59:3‐10. [DOI] [PubMed] [Google Scholar]
- 13. Pai SG, Carneiro BA, Mota JM, Costa R, Leite CA, Barroso‐Sousa R, et al. Wnt/beta‐catenin pathway: modulating anticancer immune response. J Hematol Oncol. 2017;10:101. 10.1186/s13045-017-0471-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Malinauskas T, Jones EY. Extracellular modulators of Wnt signalling. Curr Opin Struct Biol. 2014;29:77‐84. [DOI] [PubMed] [Google Scholar]
- 15. MacDonald BT, Tamai K, He X. Wnt/beta‐catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bovolenta P, Esteve P, Ruiz JM, Cisneros E, Lopez‐Rios J. Beyond Wnt inhibition: new functions of secreted Frizzled‐related proteins in development and disease. J Cell Sci. 2008;121(Pt 6):737‐46. [DOI] [PubMed] [Google Scholar]
- 17. Taira YMM. Secreted Wnt “inhibitors” are not just inhibitors: regulation of extracellular wnt by secreted frizzled‐related proteins. Dev Growth Differ. 2011;53:911‐23. [DOI] [PubMed] [Google Scholar]
- 18. Verani R, Cappuccio I, Spinsanti P, Gradini R, Caruso A, Magnotti MC, et al. Expression of the Wnt inhibitor Dickkopf‐1 is required for the induction of neural markers in mouse embryonic stem cells differentiating in response to retinoic acid. J Neurochem. 2007;100:242‐50. [DOI] [PubMed] [Google Scholar]
- 19. Beaty TH, Hetmanski JB, Fallin MD, Park JW, Sull JW, McIntosh I, et al. Analysis of candidate genes on chromosome 2 in oral cleft case‐parent trios from three populations. Human Gen. 2006;120:501‐18. [DOI] [PubMed] [Google Scholar]
- 20. Jiang Z, Pan L, Chen X, Chen Z, Xu D. Wnt6 influences the viability of mouse embryonic palatal mesenchymal cells via the beta‐catenin pathway. Exp Ther Med. 2017;14:5339‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lidral DMJMJHAPMTJCAC. Wnt9b is the mutated gene involved in multifactorial nonsyndromic cleft lip with or without cleft palate in A/WySn mice, as confirmed by a genetic complementation test. Birth Defects Res A Clin Mol Teratol. 2006;76:574‐9. [DOI] [PubMed] [Google Scholar]
- 22. WX FH, Wang Y, Li L, Liu C, Yamagami T, Taketo MM, Zhou C, and Chen YP. Epithelial Wnt/β‐catenin signaling regulates palatal shelf fusion through regulation of Tgfβ3 expression. J Dev Biol. 2011;350: 511–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhou H, Mak W, Zheng Y, Dunstan CR, Seibel MJ. Osteoblasts directly control lineage commitment of mesenchymal progenitor cells through Wnt signaling. J BiolChem. 2008;283:1936‐45. [DOI] [PubMed] [Google Scholar]
- 24. Zhu X, Wang W, Zhang X, Bai J, Chen G, Li L, et al. All‐trans retinoic acid‐induced deficiency of the Wnt/beta‐catenin pathway enhances hepatic carcinoma stem cell differentiation. PloS One. 2015;10:e0143255. 10.1371/journal.pone.0143255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xiao W, Jiang W, Shen J, Yin G, Fan Y, Wu D, et al. Retinoic acid ameliorates pancreatic fibrosis and inhibits the activation of pancreatic stellate cells in mice with experimental chronic pancreatitis via suppressing the Wnt/beta‐catenin signaling pathway. PloS One. 2015;10:e0141462. 10.1371/journal.pone.0141462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Osei‐Sarfo K, Gudas LJ. Retinoic acid suppresses the canonical Wnt signaling pathway in embryonic stem cells and activates the noncanonical Wnt signaling pathway. Stem Cells. 2014;32:2061‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Williams AL, Bohnsack BL. What's retinoic acid got to do with it? Retinoic acid regulation of the neural crest in craniofacial and ocular development. Genesis. 2019;57:e23308. 10.1002/dvg.23308 [DOI] [PubMed] [Google Scholar]
- 28. Rhinn M, Dolle P. Retinoic acid signalling during development. Development. 2012;139:843‐58. [DOI] [PubMed] [Google Scholar]
- 29. Zhu Y, Zhu Y, Yin H, Zhou H, Wan X, Zhu J, et al. All‐trans‐retinoic acid induces short forelimb malformation during mouse embryo development by inhibiting chondrocyte maturation rather than by evoking excess cell death. Toxicol Lett. 2012;211:172‐86. [DOI] [PubMed] [Google Scholar]
- 30. Okano J, Suzuki S, Shiota K. Involvement of apoptotic cell death and cell cycle perturbation in retinoic acid‐induced cleft palate in mice. Toxicol Appl Pharmacol. 2007;221:42‐56. [DOI] [PubMed] [Google Scholar]
- 31. Loo CKC, Pearen MA, Pereira TN, Perry‐Keene J, Payton D, Ramm GA. Lung and liver growth and retinoic acid status in human fetuses with congenital diaphragmatic hernia. Early Hum Dev. 2018;116:17‐23. [DOI] [PubMed] [Google Scholar]
- 32. Zhang Y, Dong S, Wang W, Wang J, Wang M, Chen M, et al. Activation of Notch1 inhibits medial edge epithelium apoptosis in all‐trans retinoic acid‐induced cleft palate in mice. Biochem Biophys Res Commun. 2016;477:322‐8. [DOI] [PubMed] [Google Scholar]
- 33. BD A, Harris MW, Birnbaum LS. Etiology of retinoic acid‐induced cleft palate varies with the embryonic stage. Teratology. 1989;40:533‐53. [DOI] [PubMed] [Google Scholar]
- 34. Padmanabhan R, Ahmed I. Retinoic acid‐induced asymmetric craniofacial growth and cleft palate in the TO mouse fetus. Reprod Toxicol. 1997;11:843‐60. [DOI] [PubMed] [Google Scholar]
- 35. Lind T, Sundqvist A, Hu LJ, Pejler G, Andersson G, Jacobson A, et al. Vitamin A is a negative regulator of osteoblast mineralization. PloS One. 2013;8(12). 10.1371/journal.pone.0082388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Masuda E, Shirai K, Maekubo K, Hirai Y. A newly established culture method highlights regulatory roles of retinoic acid on morphogenesis and calcification of mammalian limb cartilage. Biotechniques. 2015;58:318‐24. [DOI] [PubMed] [Google Scholar]
- 37. Roa LA, Bloemen M, Carels CEL, Wagener F, Von den Hoff JW. Retinoic acid disrupts osteogenesis in pre‐osteoblasts by down‐regulating WNT signaling. Int J Biochem Cell Biol. 2019;116:105597. 10.1016/j.biocel.2019.105597 [DOI] [PubMed] [Google Scholar]
- 38. Rueden CT, Schindelin J, Hiner MC, DeZonia BE, Walter AE, Arena ET, et al. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. 2017;18:529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sun D, Vanderburg CR, Odierna GS, Hay ED. TGFbeta3 promotes transformation of chicken palate medial edge epithelium to mesenchyme in vitro. Development. 1998;125:95‐105. [DOI] [PubMed] [Google Scholar]
- 40. Katoh M, Katoh M. WNT signaling pathway and stem cell signaling network. Clin Cancer Res. 2007;13:4042‐5. [DOI] [PubMed] [Google Scholar]
- 41. Rohrs S, Kutzner N, Vlad A, Grunwald T, Ziegler S, Muller O. Chronological expression of Wnt target genes Ccnd1, Myc, Cdkn1a, Tfrc, Plf1 and Ramp3. Cell Biol Int. 2009;33:501‐8. [DOI] [PubMed] [Google Scholar]
- 42. Dixon MJ, Marazita ML, Beaty TH, Murray JC. Cleft lip and palate: understanding genetic and environmental influences. Nat Rev Genet. 2011;12:167‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhou W, Li S. Decreased levels of serum retinoic acid in Chinese children with autism spectrum disorder. Psychiatry Res. 2018;269:469‐73. [DOI] [PubMed] [Google Scholar]
- 44. Manickavasagar B, McArdle AJ, Yadav P, Shaw V, Dixon M, Blomhoff R, et al. Hypervitaminosis A is prevalent in children with CKD and contributes to hypercalcemia. Pediatr Nephrol. 2015;30:317‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Baroni T, Bellucci C, Lilli C, Pezzetti F, Carinci F, Lumare E, et al. Human cleft lip and palate fibroblasts and normal nicotine‐treated fibroblasts show altered in vitro expressions of genes related to molecular signaling pathways and extracellular matrix metabolism. J Cell Physiol. 2010;222:748‐56. [DOI] [PubMed] [Google Scholar]
- 46. Clevers H. Wnt/beta‐catenin signaling in development and disease. Cell. 2006;127(3):469‐80. [DOI] [PubMed] [Google Scholar]
- 47. Liu F, Millar SE. Wnt/beta‐catenin signaling in oral tissue development and disease. J Dent Res. 2010;89:318‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang YM, Dai YJ, Li X, Chen CY, Li WJ, Yu ZL. Inhibition of Smad signaling is implicated in cleft palate induced by all‐trans retinoic acid. Acta Biol Hung. 2011;62:142‐50. [DOI] [PubMed] [Google Scholar]
- 49. Cuervo R, Valencia C, Chandraratna RAS, Covarrubias L. Programmed cell death is required for palate shelf fusion and is regulated by retinoic acid. Dev Biol. 2002;245:145‐56. [DOI] [PubMed] [Google Scholar]
- 50. Zhang YD, Dong SY, Huang HZ. Inhibition of periderm removal in all‐trans retinoic acid‐induced cleft palate in mice. Exp Ther Med. 2017;14:3393‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang Y, Mori T, Iseki K, Hagino S, Takaki H, Takeuchi M, et al. Differential expression of decorin and biglycan genes during palatogenesis in normal and retinoic acid‐treated mice. Dev Dyn. 2003;226:618‐26. [DOI] [PubMed] [Google Scholar]
- 52. Zhang Y, Dong S, Wang J, Wang M, Chen M, Huang H. Involvement of Notch2 in alltrans retinoic acidinduced inhibition of mouse embryonic palate mesenchymal cell proliferation. Mol Med Rep. 2017;16:2538‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hu X, Gao J, Liao Y, Tang S, Lu F. Retinoic acid alters the proliferation and survival of the epithelium and mesenchyme and suppresses Wnt/beta‐catenin signaling in developing cleft palate. Cell Death Dis. 2013;4:e898. 10.1038/cddis.2013.424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu X, Qi J, Tao Y, Zhang H, Yin J, Ji M, et al. Correlation of proliferation, TGF‐beta3 promoter methylation, and Smad signaling in MEPM cells during the development of ATRA‐induced cleft palate. Reprod Toxicol. 2016;61:1‐9. [DOI] [PubMed] [Google Scholar]
- 55. Cheung AM, Tam CK, Chow HC, Verfaillie CM, Liang R, Leung AY. All‐trans retinoic acid induces proliferation of an irradiated stem cell supporting stromal cell line AFT024. Exp Hematol. 2007;35:56‐63. [DOI] [PubMed] [Google Scholar]
- 56. Touma SE, Goldberg JS, Moench P, Guo X, Tickoo SK, Gudas LJ, et al. Retinoic acid and the histone deacetylase inhibitor trichostatin a inhibit the proliferation of human renal cell carcinoma in a xenograft tumor model. Clin Cancer Res. 2005;11:3558‐66. [DOI] [PubMed] [Google Scholar]
- 57. Terao M, Celestini V, Kurosaki M, Vallerga A, Bolis M, Fratelli M, et al. All‐trans retinoic acid perturbs the lipidomic profiles of luminal breast cancer cells characterized by sensitivity to the anti‐proliferative activity of the retinoid. Cancer Res. 2019;79. 10.1158/1538-7445.SABCS18-P2-02-15 31641034 [DOI] [Google Scholar]
- 58. Teo JL, Kahn M. The Wnt signaling pathway in cellular proliferation and differentiation: a tale of two coactivators. Adv Drug Deliv Rev. 2010;62:1149‐55. [DOI] [PubMed] [Google Scholar]
- 59. He F, Xiong W, Wang Y, Li L, Liu C, Yamagami T, et al. Epithelial Wnt/beta‐catenin signaling regulates palatal shelf fusion through regulation of Tgfbeta3 expression. Dev Biol. 2011;350:511‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Meng L, Wang X, Torensma R, Von den Hoff JW, Bian Z. Lithium inhibits palatal fusion and osteogenic differentiation in palatal shelves in vitro. Arch Oral Biol. 2015;60:501‐7. [DOI] [PubMed] [Google Scholar]
- 61. Ramakrishnan A‐B, Cadigan KM. Wnt target genes and where to find them. F1000Res. 2017;6:746. 10.12688/f1000research.11034.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. Wnt/beta‐catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol. 2002;22:1172‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Leung JY, Kolligs FT, Wu R, Zhai Y, Kuick R, Hanash S, et al. Activation of AXIN2 expression by beta‐catenin‐T cell factor. A feedback repressor pathway regulating Wnt signaling. J Biol Chem. 2002;277:21657‐65. [DOI] [PubMed] [Google Scholar]
- 64. Bernkopf DB, Hadjihannas MV, Behrens J. Negative‐feedback regulation of the Wnt pathway by conductin/axin2 involves insensitivity to upstream signalling. J Cell Sci. 2015;128:33‐9. [DOI] [PubMed] [Google Scholar]
- 65. Sozen B, Amadei G, Cox A, Wang R, Na E, Czukiewska S, et al. Self‐assembly of embryonic and two extraembryonic stem cell types into gastrulating embryo‐like structures. Obstet Gynecol Surv. 2019;74:30‐1. [Google Scholar]
- 66. Li N, Xu Y, Zhang H, Gao L, Li J, Wang Y, et al. Excessive retinoic acid impaired proliferation and differentiation of human fetal palatal chondrocytes (hFPCs). Birth Defects Res B Dev Reprod Toxicol. 2014;101:276‐82. [DOI] [PubMed] [Google Scholar]
- 67. Zito G, Naselli F, Saieva L, Raimondo S, Calabrese G, Guzzardo C, et al. Retinoic acid affects lung adenocarcinoma growth by inducing differentiation via GATA6 activation and EGFR and Wnt inhibition. Sci Rep. 2017;7:4770. 10.1038/s41598-017-05047-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zito G, Saotome I, Liu Z, Ferro EG, Sun TY, Nguyen DX, et al. Spontaneous tumour regression in keratoacanthomas is driven by Wnt/retinoic acid signalling cross‐talk. Nat Commun. 2014;5:3543. 10.1038/ncomms4543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mao XM, Li H, Zhang XY, Zhou P, Fu QR, Chen QE, et al. Retinoic acid receptor alpha knockdown suppresses the tumorigenicity of esophageal carcinoma via wnt/beta‐catenin pathway. Dig Dis Sci. 2018;63:3348‐58. [DOI] [PubMed] [Google Scholar]
- 70. Zhu XJ, Yuan X, Wang M, Fang Y, Liu Y, Zhang X, et al. A Wnt/Notch/Pax7 signaling network supports tissue integrity in tongue development. J Biol Chem. 2017;292:9409‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Fan Y, Ho BX, Pang JKS, Pek NMQ, Hor JH, Ng SY, et al. Wnt/beta‐catenin‐mediated signaling re‐activates proliferation of matured cardiomyocytes. Stem Cell Res Ther. 2018;9:338. 10.1186/s13287-018-1086-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nakatsu MN, Ding Z, Ng MY, Truong TT, Yu F, Deng SX. Wnt/beta‐catenin signaling regulates proliferation of human cornea epithelial stem/progenitor cells. Invest Ophthalmol Vis Sci. 2011;52:4734‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Gao L, Chen B, Li J, Yang F, Cen X, Liao Z, et al. Wnt/beta‐catenin signaling pathway inhibits the proliferation and apoptosis of U87 glioma cells via different mechanisms. PloS One. 2017;12:e0181346. 10.1371/journal.pone.0181346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Su N, Wang P, Li Y. Role of Wnt/beta‐catenin pathway in inducing autophagy and apoptosis in multiple myeloma cells. Oncol Lett. 2016;12:4623‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Niu J, Li XM, Wang X, Liang C, Zhang YD, Li HY, et al. DKK1 inhibits breast cancer cell migration and invasion through suppression of β‐catenin/MMP7 signaling pathway. Cancer Cell Int. 2019;19. 10.1186/s12935-019-0883-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Mu J, Hui T, Shao B, Li L, Du Z, Lu L, et al. Dickkopf‐related protein 2 induces G0/G1 arrest and apoptosis through suppressing Wnt/beta‐catenin signaling and is frequently methylated in breast cancer. Oncotarget. 2017;8:39443‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Mirfazeli ES, Arefian E, Nadri S, Rezazadeh Valojerdi R, Kehtari M, Zeynali B. DKK1 expression is suppressed by miR‐9 during induced dopaminergic differentiation of human trabecular meshwork mesenchymal stem cells. Neurosci Lett. 2019;707:134250. 10.1016/j.neulet.2019.05.004 [DOI] [PubMed] [Google Scholar]
- 78. Mishra S, Choe Y, Pleasure SJ, Siegenthaler JA. Cerebrovascular defects in Foxc1 mutants correlate with aberrant WNT and VEGF‐A pathways downstream of retinoic acid from the meninges. Dev Biol. 2016;420:148‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Duffy DJ, Krstic A, Halasz M, Schwarzl T, Konietzny A, Iljin K, et al. Retinoic acid and TGF‐beta signalling cooperate to overcome MYCN‐induced retinoid resistance. Genome Med. 2017;9:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Chen F, Cao Y, Qian J, Shao F, Niederreither K, Cardoso WV. A retinoic acid‐dependent network in the foregut controls formation of the mouse lung primordium. J Clin Invest. 2010;120:2040‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Froeling FE, Feig C, Chelala C, Dobson R, Mein CE, Tuveson DA, et al. Retinoic acid‐induced pancreatic stellate cell quiescence reduces paracrine Wnt‐beta‐catenin signaling to slow tumor progression. Gastroenterology. 2011;141:1486‐97, 97 e1‐14. [DOI] [PubMed] [Google Scholar]
- 82. Mii Y, Taira M. Secreted Frizzled‐related proteins enhance the diffusion of Wnt ligands and expand their signalling range. Development. 2009;136:4083‐8. [DOI] [PubMed] [Google Scholar]
- 83. Uren A, Reichsman F, Anest V, Taylor WG, Muraiso K, Bottaro DP, et al. Secreted frizzled‐related protein‐1 binds directly to Wingless and is a biphasic modulator of Wnt signaling. J Biol Chem. 2000;275:4374‐82. [DOI] [PubMed] [Google Scholar]
- 84. Bayle J, Fitch J, Jacobsen K, Kumar R, Lafyatis R, Lemaire R. Increased expression of Wnt2 and SFRP4 in Tsk mouse skin: role of Wnt signaling in altered dermal fibrillin deposition and systemic sclerosis. J Invest Dermatol. 2008;128:871‐81. [DOI] [PubMed] [Google Scholar]
- 85. Jacobs H, Dennefeld C, Feret B, Viluksela M, Hakansson H, Mark M, et al. Retinoic acid drives aryl hydrocarbon receptor expression and is instrumental to dioxin‐induced toxicity during palate development. Environ Health Perspect. 2011;119:1590‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Krishnan V, Bryant HU, Macdougald OA. Regulation of bone mass by Wnt signaling. J Clin Invest. 2006;116:1202‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Baron R, Rawadi G. Wnt signaling and the regulation of bone mass. Curr Osteoporos Rep. 2007;5:73‐80. [DOI] [PubMed] [Google Scholar]
- 88. Song L, Liu M, Ono N, Bringhurst FR, Kronenberg HM, Guo J. Loss of wnt/beta‐catenin signaling causes cell fate shift of preosteoblasts from osteoblasts to adipocytes. J Bone Miner Res. 2012;27:2344‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med. 2013;19:179‐92. [DOI] [PubMed] [Google Scholar]
- 90. Wu AM, Huang CQ, Lin ZK, Tian NF, Ni WF, Wang XY, et al. The relationship between vitamin a and risk of fracture: meta‐analysis of prospective studies. J Bone Miner Res. 2014;29:2032‐9. [DOI] [PubMed] [Google Scholar]
- 91. Conaway HH, Pirhayati A, Persson E, Pettersson U, Svensson O, Lindholm C, et al. Retinoids stimulate periosteal bone resorption by enhancing the protein RANKL, a response inhibited by monomeric glucocorticoid receptor. J Biol Chem. 2011;286:31425‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bi W, Liu Y, Guo J, Lin Z, Liu J, Zhou M, et al. All‐trans retinoic‐acid inhibits heterodimeric bone morphogenetic protein 2/7‐stimulated osteoclastogenesis, and resorption activity. Cell Biosci. 2018;8:48. 10.1186/s13578-018-0246-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Green AC, Kocovski P, Jovic T, Walia MK, Chandraratna RAS, Martin TJ, et al. Retinoic acid receptor signalling directly regulates osteoblast and adipocyte differentiation from mesenchymal progenitor cells. Exp Cell Res. 2017;350:284‐97. [DOI] [PubMed] [Google Scholar]
- 94. Bond SR, Lau A, Penuela S, Sampaio AV, Underhill TM, Laird DW, et al. Pannexin 3 is a novel target for Runx2, expressed by osteoblasts and mature growth plate chondrocytes. J Bone Miner Res. 2011;26:2911‐22. [DOI] [PubMed] [Google Scholar]
- 95. Li N, Kelsh RN, Croucher P, Roehl HH. Regulation of neural crest cell fate by the retinoic acid and Pparg signalling pathways. Development. 2010;137:389‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Theiler K. The house mouse: atlas of embryonic development. New York: Springer‐Verlag. 1989. [Google Scholar]