

Abstract
From late 2017 to early 2018, clade 2.3.4.4B H5N8 highly pathogenic avian influenza (HPAI) viruses caused mass die‐offs of thousands of coastal seabirds along the southern coastline of South Africa. Terns (Laridae) especially were affected, but high mortalities in critically endangered and threatened species like African Penguins (Spheniscus demersus) caused international concern and, exactly a year later, the disease recurred at a key African Penguin breeding site on Halifax Island, Namibia. Twenty‐five clade 2.3.4.4B H5N8 HPAI viruses from coastal seabirds and a Jackal Buzzard (Buteo rufofuscus) were isolated and/or sequenced in this study. Phylogenetic analyses of the full viral genomes and time to the most recent common ancestor (tMRCA) analyses of the HA, NA, PB1 and PA genes determined that the South African coastal seabird viruses formed a monophyletic group nested within the South African genotype 4 viruses. This sub‐lineage likely originated from a single introduction by terrestrial birds around October 2017. Only the HA and NA sequences were available for the Namibian penguin viruses, but the phylogenetic data confirmed that the South African coastal seabird viruses from 2017 to 2018 were the source and the most closely related South African virus was found in a gull. tMRCA analyses furthermore determined that the progenitors of the five genotypes implicated in the earlier 2017 South African outbreaks in wild birds and poultry were dated at between 2 and 4 months prior to the index cases. tMRCA and phylogenetic data also showed that the novel genotype 6 virus introduced to South Africa in 2018, and later also detected in Nigeria and Poland in 2019, most likely arose in late 2017 in West, Central or East Africa. We propose that it continued to circulate there, and that an unidentified reservoir was the source of both the South African outbreaks in early 2018 and in Nigeria in mid‐2019.
Keywords: avian influenza virus, clade 2.3.4.4B H5N8, cormorants, penguins, terns
1. INTRODUCTION
The fourth inter‐continental wave of Goose/Guangdong (Gs/GD) highly pathogenic avian influenza (HPAI) was caused by clade 2.3.4.4 sub‐group B lineage H5Nx viruses that emerged in June 2016 in the Russian Federation (Lee et al., 2017). As with three prior inter‐continental waves, HPAI viruses spread globally over long distances in asymptomatic infections of migratory wild birds (Fusaro et al., 2019). Thus, between November 2016 and January 2017, outbreaks of clade 2.3.4.4B H5N8 HPAI viruses that originated in Europe, the Middle East or Central and South Asia (Khomenko et al., 2018; Fusaro et al., 2019) were introduced to wild birds and poultry in Egypt, Tunisia and Nigeria, followed by Niger, Cameroon and Uganda before reaching the Democratic Republic of the Congo (DRC) in April 2017 (Abolnik et al., 2019). In May 2017, H5N8 HPAI broke out on a commercial poultry farm near Harare, Zimbabwe, and a month later the first outbreaks were reported about 1200 km further south in commercial poultry flocks at Villers and Standerton, South Africa (SA). These were the first reported cases of Gs/GD‐lineage HPAI H5 viruses south of the tropic of Capricorn, disseminated via the climate‐driven movements of Afro‐tropical waterfowl (Khomenko et al., 2018; Fusaro et al., 2019; Abolnik et al., 2019).
The SA epidemic which started in the southern hemisphere winter of 2017 was characterized by two genetically distinct spatial clusters. The first cluster included the index cases and spanned adjacent regions of the Gauteng, eastern Mpumalanga, North‐West, northern KwaZulu‐Natal and northern Free State provinces, referred to here as the north‐central region of SA (Figures 1 and 2) (Abolnik et al., 2019; Abolnik 2020). This cluster was characterized by high viral genetic diversity with four different genotypes, which were named 1, 2, 3 and 5 for clarity only, detected in dead or moribund wild birds and poultry between June and October 2017 (Abolnik et al., 2019). The second and much larger southern cluster affected the Western and Eastern Cape provinces, with the index case detected in commercial ostriches 7 weeks after the first outbreaks in the north‐central region. The Western Cape province was the most severely affected in the 2017 epidemic and more than 70% of the layer hen population was culled to prevent further spread of the disease. Many wild birds in the province were also affected, including pigeons and doves (Columbidae), Spur‐winged Geese (Plectropterus gambensis), Helmeted Guineafowl (Numida meleagris), Blue Cranes (Anthropoides paradiseus), House Sparrows (Passer domesticus), Peregrine Falcons (Falco peregrinus), Pied Crows (Corvus albus), African Sacred Ibis (Threskiornis aethiopicus), Black‐headed Herons (Ardea melanocephala) and Egyptian Geese (Alopochen aegyptiaca) (Roberts, 2018). All the outbreaks in this southern cluster were caused by a single genotype, designated as 4 for clarity (Abolnik et al., 2019).
FIGURE 1.
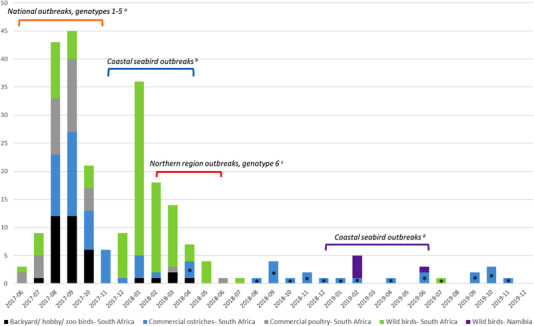
Progression of H5N8 clade 2.3.4.4B HPAI cases in southern Africa based on data reported to the WOAH. Full details of the outbreaks are described in (a) Khomenko et al. (2018) and Abolnik et al. (2019), (b) Khomenko et al. (2018), Roberts et al. (2022) and this study, (c) Abolnik (2020) and (d) Molini et al. (2020). *Cases reported to the WOAH from routine surveillance in commercial ostriches after March 2018 and in hunted Egyptian Geese in July 2019 were based on the detection of H5‐specific antibodies in sera without confirmation of the presence of H5N8 HPAI virus. The last confirmed detection of H5N8 HPAI virus in South Africa was in July 2018, in a dead Blue Crane in the Theewaterskloof District of the Western Cape Province (Roberts, 2020).
FIGURE 2.
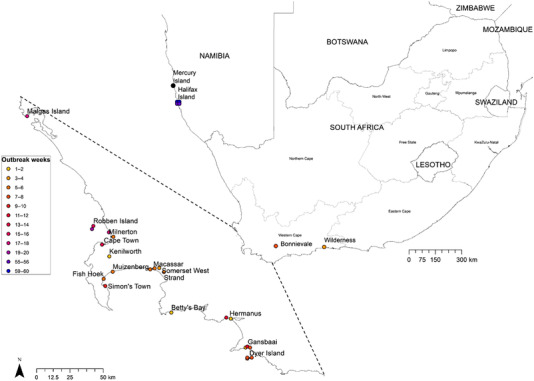
Geographic locations of clade 2.3.4.4B H5N8 HPAI viruses analysed in this study. Locations are indicated in fortnightly increments, according to the progression of the outbreak. Outbreak weeks 1–2 commence with the last week of December 2017.
By the spring of 2017, the outbreaks in poultry and wild terrestrial birds in the Cape provinces and the rest of SA had subsided (Figure 1) (Abolnik et al., 2019), but in the mid‐summer period of December 2017, large numbers of sick and dead Swift (Greater‐crested) Terns (Thalasseus bergii) started being reported along the southwestern coastline, with the cause confirmed by laboratory testing as H5N8 HPAI. The event was reminiscent of the 1961 outbreaks along the same coastline, in which 1300 Common Terns (Sterna hirundo) perished due to an H5N3 HPAI virus of unknown origin (Becker, 1966). This time, however, the spread of H5N8 HPAI to other threatened and critically endangered wild birds like African Penguins (Spheniscus demersus), Cape Gannets (Morus capensis) and African Oystercatchers (Haematopus moquini) was alarming. Between December 2017 and May 2018, an estimated 7415 birds of 15 species, including Swift Terns (forming the majority), African Penguins, Cape Gannets, Common Terns, Sandwich Terns (Thalasseus sandvicensis), Hartlaub's Gulls (Chroicocephalus hartlaubii), Cape Cormorants (Phalacrocorax capensis), Crowned Cormorants (Microcarbo coronatus) and African Oystercatchers, succumbed to suspected or confirmed H5N8 HPAI, at 31 sites along a 1000‐km stretch of the southern coastline of SA from the Olifants River Estuary to Gqeberha (formerly Port Elizabeth) (Khomenko et al., 2018; Roberts, 2018; Roberts et al., 2022). Clinical signs included diarrhoea, corneal oedema, weakness and incoordination, circling and/or the inability to fly, neurological signs like head tremors, poor balance, torticollis, seizures and death (Roberts, 2018).
Concurrently, a new cluster of H5N8 HPAI outbreaks appeared in the north‐central region, commencing in mid‐February 2018 in the Limpopo province and lasting until early June 2018, affecting free‐living backyard poultry, ornamental birds, wild birds, a meat‐type Japanese quail farm and a commercial chicken farm. Phylogenetic analysis determined that these outbreaks were caused by a novel sixth genotype that was possibly a new introduction to SA (Figure 1; Abolnik, 2020). Related viruses were later found in Ogun State, Nigeria from a Guineafowl sampled at a live bird market in July 2019 (Laleye et al., 2022), and in an outbreak in commercial turkeys in Poland at the end of December 2019 (Świętoń et al., 2020).
In mid‐December 2018, exactly 1 year after the SA coastal seabird outbreaks, mortalities started increasing in the protected African Penguin breeding colony on Halifax Island, Namibia. More than 350 penguins perished due to H5N8 HPAI infection by the end of February 2019. The causative viruses were partially sequenced and shown to be phylogenetically related to the 2017 SA terrestrial poultry and wild bird strains, but the SA 2018 coastal seabird sequences were not available at the time for comparison (Molini et al., 2020). In this study, we report on the isolation and sequencing of the SA H5N8 HPAI coastal seabird viruses and their phylogenetic relationships with the Namibian 2019 penguin cases. Furthermore, since the genetic sequences of other viruses related to SA's 2017–2018 outbreaks recently became available (Nigeria and Poland, 2019), and because limited analyses were initially performed on the southern African 2017–2018 outbreak viruses (Abolnik et al., 2019; Abolnik, 2020), here we applied a molecular clock to the previous and new sequence data to gain deeper insights into the origins and timing of the incursions of specific genotypes of clade 2.3.4.4B H5N8 HPAI viruses in southern Africa.
2. MATERIALS AND METHODS
2.1. Sample collection
Seabirds that became moribund or died on beaches or at breeding colonies in SA were collected by members of the public, managing conservation authorities, rehabilitation centres and Veterinary Services (Roberts et al., 2022). For this study, whole bird carcases or tissue samples transported in cool boxes with frozen ice packs were submitted to the Western Cape Provincial Veterinary Laboratory (WCPVL). Some moribund birds were received and were humanely euthanized shortly after arrival. In total, samples from 49 individual birds were received for testing. All were coastal seabirds, apart from a raptor—a single Jackal Buzzard (Buteo rufofuscus). Post‐mortem findings included poor body condition, green diarrhoea, enlarged livers and severe lung congestion. Brain, oropharyngeal and cloacal swab samples were collected using nylon flocked swabs (Copan Diagnostics Inc., Murrieta, CA, USA). Swab samples were also taken from the internal contents (i.e. the mixed allantoic–amniotic fluids within each egg) of four abandoned Swift Tern eggs collected at a breeding site at the Victoria & Alfred Waterfront in Cape Town where large numbers of bird deaths due to HPAI were recorded.
2.2. Virus detection and isolation
Oropharyngeal and cloacal swabs were pooled per bird and tested separately from the brain swabs. Swabs were vortexed in 0.5 ml sterile PBS/Glycerol (50% v/v). Viral RNA was extracted from 150–200 μl of each sample using Nucleospin® RNA Virus kits (Macherey‐Nagel GmbH & Co. KG) or a QIAcube HT® automated extraction system with the Cador® Pathogen 96 QIAcube® HT kit (QIAGEN SA [Pty] Ltd), according to the manufacturers’ instructions. One African Penguin RNA sample (476266) was received from the Assurecloud (Pty) Ltd laboratory in Oudtshoorn. Real‐time RT‐PCR (rRT‐PCR) using VetMAX™ Gold AIV detection kits (Thermo Fisher: Life Technologies) that simultaneously target the M and NP genes were used according to the manufacturer's protocol, to screen all samples for the presence AIV group. For AIV‐positive samples, H5 subtype detection was performed using the primers and probes described by Slomka et al. (2007), and the N8 primers and probes originally described by Hoffmann et al. (2016), subsequently modified by APHA (2016), were used for N8 subtype detection. rRT‐PCR cycle threshold (Ct) values <40 were considered positive.
Where swab samples were rRT‐PCR positive for both H5 and N8, virus isolation was carried out using tissue samples from the corresponding birds. Liver, spleen, lung and/or brain tissue was pooled for virus isolation in Specific Antibody Negative embryonated White Leghorn chicken eggs, inoculated via the allantoic route, according to the method described by the WOAH (2019). Pooled amnio–allantoic fluid was harvested from all eggs on the day on which they died, and from any live eggs remaining at the end of the incubation period. The total incubation period was 7 days after the inoculation. A second blind passage was performed on fluids harvested from eggs that were still alive at the end of the 7‐day incubation period. Haemagglutination tests were performed on all harvested egg amnio–allantoic fluids according to the standard method (WOAH, 2019).
2.3. Genome sequencing
RNA extracted from amnio–allantoic fluid was submitted to the Central Analytical Facility of the University of Stellenbosch for Ion Torrent sequencing as described by Abolnik et al. (2019). Ion Torrent reads were imported into the CLC Genomics Workbench 5.2.1 (QIAGEN CLC bio, Aarhus, Denmark; http://www.clcbio.com) and assembled to the eight segments of an H5N8 genome reference sequence (KY621531–KY621538). The following abbreviations for proteins encoded in the eight genomic segments are referred to in the text and figures: Polymerase Basic 2 (PB2) (segment 1); Polymerase Basic 1 (PB1) and PB1‐F2 (segment 2); Polymerase A (PA) (segment 3); Hemagglutinin (HA) (segment 4); Nucleoprotein (NP) (segment 5); Neuraminidase (NA) (segment 6); Matrix 1 (M1) and M2e (segment 7); Non‐structural 1 (NS1); and Nuclear export protein (NEP) (segment 8). The consensus sequences were exported and annotated using the INCUBI Influenza Virus Sequence Annotation Tool (https://www.ncbi.nlm.nih.gov/genomes/FLU/annotation/) prior to deposition in GenBank (Table S1).
2.4. Phylogenetic and evolutionary analysis
Reference sequences were retrieved by BLAST of the NCBI (https://www.ncbi.nlm.nih.gov/nucore) and GISAID Epiflu (https://www.gisaid.org/) databases, and from a search of GISAID database for H5N8 viruses sampled in Africa and Europe between 1 November 2016 and 31 December 2019. Duplicate sequences were removed and unrelated clade 2.3.4.4 H5N8 viruses in the NCBI database were selected to root the trees.
Nucleotide sequences were aligned in MAFFT v.7 (https://mafft.cbrc.jp/alignment/server/), and the translated amino acid sequences were examined for unique molecular markers in the encoded genes. Maximum likelihood (ML) phylogenetic trees were generated with the Tamura‐Nei model in MEGA‐X (v.10.2.5) (Kumar et al., 2018), with 1000 bootstrap replicates. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Trees were drawn to scale, with branch lengths measured in the number of substitutions per site. To assess whether any reassortment occurred between the coastal seabird viruses, the ML trees were visually inspected. The letters A–H were assigned to the coastal seabird sub‐lineage in segments 1–8, respectively, and within each of these, sub‐lineages supported by bootstrap values of at least 95% were designated with numbers, for example A1, A2 and so on, where no number indicated taxa without statistical support. Results were tabulated to assign sub‐genotypes which were designated 4‐1, 4‐2, 4‐3 and so on, and to assess any reassortment events, which would be indicated by the combinations of numbered letters.
BEAST v.2.6.7 software (Bouckaert et al., 2019) was used to produce dated maximum clade credibility (MCC) trees to determine the time to the most recent common ancestor (tMRCA) of the tree nodes. MCC trees were reconstructed using a Hasegawa–Kishono–Yano (HKY) nucleotide substitution model with a gamma distribution of substitution rates, a Coalescent Bayesian Skyline model and a Relaxed Lognormal clock. Markov chain Monte Carlo chains of between 50 and 80 million iterations were performed and assessed with Tracer v.1.7.2 (Rambaut et al., 2018) to ensure that an effective sample size of >200 was achieved, with statistical uncertainty of the nodes (i.e. tTMRCA) reflected in values of the 95% highest posterior density. MCC trees with common ancestor heights were summarized using TreeAnnotator v.2.6.6, and visualized using FigTree v.1.4.2 (http://tree.bio.ac.uk/software/figtree/).
3. RESULTS
3.1. Virus detection and isolation
Forty‐six swab samples from eight coastal seabird species and a Jackal Buzzard plus four abandoned Swift Tern eggs were screened for H5N8 AIV by rRT‐PCR. All four Swift Tern eggs tested negative for the presence of AIV, but 35 (71.4%) of the coastal seabird swab samples tested positive with Ct values ranging from 10.17 to 35.13. All rRT‐PCR‐positive cases were inoculated into embryonated chicken eggs for virus isolation, but isolation attempts were only successful when the Ct values were below 25.97. Twenty‐seven H5N8 viruses were isolated, of which 24, which represented a broad range in species, sampling locations and dates, plus one RNA sample received from another laboratory (A/African penguin/South Africa/476266/2018) were selected for genome sequencing (Table S1; Figure 2).
3.2. Phylogenetic and reassortment analysis of coastal seabird viruses
The phylogenetic analysis for each of the eight genome segments showed that southern African coastal seabird (and Jackal Buzzard) viruses formed a discrete sub‐lineage within the previously described genotype 4 group of South African 2017 H5N8 outbreak viruses, where a genotype reflects genome constellations that aid in the identification of reassortants (Abolnik et al., 2019). Consequently, there was no evidence of reassortment with any terrestrial bird viruses after the coastal seabird sub‐lineage's emergence (Figures 3 and S1a–h). Three coastal seabird sub‐genotypes were identified in the ML tree data, designated 4‐1 to 4‐3, based on a conservative 95% bootstrap value cut‐off (Table 1). Sub‐genotype 4‐1 represented the majority of taxa, which did not form statistically supported sub‐lineages. Sub‐genotype 4‐2 contained three viruses, the earliest being Common Tern/18010371 at Strand at the end of January 2018, with the closely related Sandwich Tern/18020302 and Cape Cormorant/18020303 viruses sampled 2 weeks later at Dyer Island, 82 km away. Sub‐genotype 4‐3 contained three viruses, Swift Tern/18020273 from Bonnievale in mid‐February was genetically closely related to Swift Tern/18030478 sampled 2 weeks later in Cape Town 188 km away, and sub‐genotype 4‐3 was detected again approximately 2 weeks after that at Robben Island, namely Hartlaubs’ gull/18040224.
FIGURE 3.

Time‐scaled maximum clade credibility tree for the HA gene of H5N8 HPAI viruses from southern Africa from 2017 to 2019, with node bars representing the 95% highest posterior density
TABLE 1.
Reassortment analysis of SA coastal seabird H5N8 virus genomes
| Isolate identification | PB2 | PB1 | PA | HA | NP | NA | M | NS | Sub‐genotype |
|---|---|---|---|---|---|---|---|---|---|
| A/Swift tern/South Africa/18010027/2018 (H5N8) | A | B | C | D | E | F | G | H | 4‐1 |
| A/Swift tern/South Africa/18010028/2018 (H5N8) | A | B | C | D | E | F | G | H | 4‐1 |
| A/tern/South Africa/18010043/2018 (H5N8) | A | B | C | D | E | F | G | H | 4‐1 |
| A/tern/South Africa/18010107/2018 (H5N8) | A | B | C | D | E | F | G | H | 4‐1 |
| A/Jackal buzzard/South Africa/18010106/2018 (H5N8) | A | B | C | D | E | F | G | H | 4‐1 |
| A/Common tern/South Africa/18010259/2018 (H5N8) | A | B | C | D | E | F | G | H | 4‐1 |
| A/Sandwich tern/South Africa/18010369/2018 (H5N8) | A | B | C | D | E | F | G | H | 4‐1 |
| A/Swift tern/South Africa/18010370/2018 (H5N8) | A | B | C | D | E | F | G | H | 4‐1 |
| A/Common tern/South Africa/18010371/2018 (H5N8) | A | B | C | D | E | F1 | G | H | 4‐2 |
| A/African penguin/South Africa/18010422/2018 (H5N8) | A | B | C | D | E | F | G | H | 4‐1 |
| A/African penguin/South Africa/18010423/2018 (H5N8) | A | B | C | D | E | F | G | H | 4‐1 |
| A/tern/South Africa/18010417/2018 (H5N8) | A | B | C | D | E | F | G | H | 4‐1 |
| A/Swift tern/South Africa/18020273/2018 (H5N8) | A1 | B | C | D | E | F | G | H | 4‐3 |
| A/Sandwich tern/South Africa/18020302/2018 (H5N8) | A | B | C | D | E | F1 | G | H | 4‐2 |
| A/Cape cormorant/South Africa/18020303/2018 (H5N8) | A | B | C | D | E | F1 | G | H | 4‐2 |
| A/African penguin/South Africa/18020304/2018 (H5N8) | A | B | C | D | E | F | G | H | 4‐1 |
| A/African penguin/South Africa/18020408/2018 (H5N8) | A | B | C | D | E | F | G | H | 4‐1 |
| A/African penguin/South Africa/476266/2018 (H5N8) | A | B | C | D | E | F | G | H | 4‐1 |
| A/Crowned cormorant/South Africa/18030213/2018 (H5N8) | A | B | C | D | E | F | G | H | 4‐1 |
| A/African oystercatcher/South Africa/18030214/2018 (H5N8) | A | B | C | D | E | F | G | H | 4‐1 |
| A/Swift tern/South Africa/18030478/2018 (H5N8) | A1 | B | C | D | E | F | G | H | 4‐3 |
| A/Hartlaub's gull/South Africa/18040224/2018 (H5N8) | A1 | B | C | D | E | F | G | H | 4‐3 |
| A/Swift tern/South Africa/18040275/2018 (H5N8) | A | B | C | D | E | F | G | H | 4‐1 |
| A/Hartlaub's gull/South Africa/18040367/2018 (H5N8) | A | B | C | D | E | F | G | H | 4‐1 |
| A/African penguin/South Africa/18050256/2018 (H5N8) | A | B | C | D | E | F | G | H | 4‐1 |
Only the HA and NA sequences were available for the Namibian penguin viruses, but the ML and MCC trees clearly indicated that that they shared a most recent common ancestor (MRCA) with the SA coastal seabird viruses. In the HA gene, Hartlaub's gull/18040367, sampled towards the end of the outbreak at Milnerton in Cape Town in April 2018, was the most closely related virus at the root of the Namibian clade, but the bootstrap value of only 61% was relatively low (Figures 3 and S2d).
Analysis of the translated HA protein sequences (not shown) showed that the proteolytic cleavage site (HA0) motif in the coastal seabird viruses from SA and Namibia was PLREKRRKR*GLF, consistent with other clade 2.3.4.4B H5 HPAI viruses. Four unique amino acid mutations were identified in genes of the SA and Namibian coastal seabird viruses, that is N11K and T29S in the HA protein, R95K in the M1 protein and P559A in the PA protein. However, the latter mutation was not found in Hartlaub's gull/18040224, one of the sub‐genotype 4‐3 viruses.
3.3. Estimation of the tMRCA of southern African viruses
To obtain a combined and more accurate estimate to overcome large highest posterior densities (HDPs), which are a result of long branch lengths in the phylogenies, we compared four genes in the tMRCA analysis, namely HA, NA, PB1 and PA (Figures 3 and S2a–c; Table 2). Sub‐lineages in these four genes with BEAST analysis were generally consistent with those in the ML trees. The progenitor of the SA coastal seabird epidemic was dated at October 2017 (the widths of the posterior distributions are given in Table 2) in the HA, PB1 and PA MCC trees, with maximum posterior probability scores of 1. In the NA MCC tree, the coastal seabird progenitor was estimated weeks earlier in September 2017, with a posterior probability of .94. The progenitor to the Namibian penguin outbreaks, for which only HA and NA sequences were available, and a single SA virus Hartlaub's gull/18040367 was dated to March and January 2018, respectively, but the posterior probability of the MRCA node was .83 compared to .26.
TABLE 2.
tMRCA estimates for clade 2.3.4.4B H5N8 HPAI viruses from southern Africa in 2017–2018. The 95% highest posterior density range is in brackets
| Node | HA gene | NA gene | PB1/PB1‐F2 genes | PA/PA‐X genes |
|---|---|---|---|---|
| Namibia coastal seabirds | March 2018 (September 2017 to November 2018) | January 2018 (November 2017 to March 2018) | No sequence available | No sequence available |
| Southern African coastal seabirds | October 2017 (August 2017 to November 2017) | September 2017 (July 2017 to October 2017) | October 2017 (September 2017 to November 2017) | October 2017 (September 2017 to November 2017) |
| Zimbabwe/2017: SA genotypes 2/3 | March 2017 (February 2017 to April 2017) | April 2017 (February 2017 to April 2017) | March 2017 (February 2017 to April 2017) | March 2017 (January 2017 to May 2017) |
| SA genotype 4 | See below | April 2017 (February 2017 to May 2017) | May 2017 (March 2017 to June 2017) | May 2017 (March 2017 to June 2017) |
| SA genotype 1 | See below | April 2017 (February 2017 to May 2017) | April 2017 (February 2017 to June 2017) | April 2017 (November 2016 to May 2017) |
| SA genotype 1/4 | February 2017 (December 2016 to May 2017) | January 2017 (August 2016 to April 2017) | February 2017 (January 2017 to April 2017) | March 2017 (December 2016 to April 2017) |
| SA genotype 3 | April 2017 (January 2017 to June 2017) | May 2017 (February 2017 to June 2017) | June 2017 (May 2017 to July 2017) | June 2017 (April 2017 to June 2017) |
| SA genotype 2/3 | February 2017 (November 2016 to March 2017) | Not applicable a | February 2017 (January 2017 to April 2017) | Not applicable a |
| SA genotype 5 | July 2016 (July 2015 to February 2017) | November 2016 (May 2017 to May 2017) | January 2017 (December 2017 to June 2017) | November 2016 (July 2016 to December 2016) |
| SA genotype 6 | February 2018 (January 2018 to March 2019) | February 2018 (November 2017 to March 2018) | February 2018 (December 2017 to March 2018) | January 2018 (November 2017 to March 2018) |
| SA genotype 6: Nigeria/2019 and Poland/2019 | April 2018 (March 2018 to June 2019) | February 2018 (December 2017 to April 2019) | November 2017 (August 2017 to January 2018) | December 2017 (October 2017 to March 2018) |
| Nigeria/2019: Poland/2019 | January 2019 (October 2018 to June 2019) | September 2018 (March 2018 to January 2019) | Not applicable 1 | September 2018 (May 2018 to January 2019) |
Gene sequences fall in distant clades due to genomic reassortment events
Abbreviation: SA, South Africa.
The following results were obtained for the virus genotypes 1–6 identified in the previous studies (Abolnik et al., 2019; Abolnik, 2020), and the origins of these will now be discussed in detail. One SA genotype 1 virus was implicated in one of two simultaneous poultry index cases, at Villiers in mid‐June 2017, and all four MCC trees dated the genotype 1 MRCA to April 2017 with high posterior probability scores, that is at least 2 months before the strain was detected in SA. The other index case at Standerton was a genotype 2 strain, only detected once in the outbreak, and genotype 3 (n = 2) was identified in wild birds about a month later (Abolnik et al., 2019). The Zimbabwean virus 810/2017 shared an MRCA with genotypes 2 and/or 3 in most of its genome segments, but the SA outbreak strains were not directly derived from it (Abolnik et al., 2019; Figures 3 and S1a–h). The MRCA for the Zimbabwe virus and SA genotypes 2/3 was dated at March 2017 in the HA, PB1 and PA MCC trees (posterior probabilities of .95–1) and at April 2017 in the NA MCC tree (posterior probability of .98); therefore, the consensus places the MRCA at least 2 months prior to the detection of H5N8 in Zimbabwe and 3 months prior to detection in SA.
SA genotype 2 and 3 viruses were closely related in the PB1‐, HA‐, NP‐, NA‐, M‐ and NS‐encoding segments, but not segments 1 and 3 where reassortment with regional low pathogenicity viruses was evident (Abolnik et al., 2019). The MRCA for genotypes 2/3 in the HA and PB1 MCC trees was dated at February 2017 with high posterior probability scores, that is 4 months before either genotype was detected in SA. For genotype 3 individually, the estimated MRCA range was wider between the four MCC trees (April to June 2017), but both the PB1 and PA MCC trees estimated the MRCA at June 2017 with maximum probability scores. Genotype 3 was therefore likely a newer type that emerged just prior to or around the time of its introduction to SA.
Genotypes 4 (n = 41) and 1 (n = 9) shared a common ancestor according to the ML tree analyses, but although they formed statistically supported sub‐lineages for the PB2, PB1, PA, NA and M genes, the distinction was less clear in the HA, NP and NS genes. The HA MCC tree dated the MRCA for genotypes 1/4 to February 2017 (posterior probability of .99), but the RCA for genotype 4 specifically was dated to April 2017 (NA) or May 2017 (PB1 and PA), all with high posterior probability scores. Genotype 4's MRCA was therefore dated by consensus at April to May 2017.
Genotype 5, for which a sole representative was detected in 2017, produced MCC trees with a wide range of estimates from July 2016 to January 2017, but only the PA gene was supported by a strong posterior probability score (1) with the most likely MRCA was thus dated November 2016.
Only two representatives for SA genotype 6 were available for analysis, quail/AI5930 and chicken/499723, and the MCC trees for HA, NA and PB1 all dated the MRCA at February 2018 with the maximum probability scores of 1. The PA MCC tree dated the MRCA a month earlier but with a probability score of .57. The MRCA of the two SA genotype 6 strains and guinea fowl/OG‐G‐11T‐19VIR8424‐7 (Nigeria, early July 2019) and turkey/23 (Poland, late December 2019) was dated with the maximum posterior probability scores at November and December 2017 in the PB1 and PA MCC trees, respectively, whereas the HA MCC tree dated the ancestral virus at April 2018 (posterior probability of .98) and in the NA MCC tree at February 2018 (posterior probability score of .54). Thus, the consensus MRCA of November–December 2017 is most likely. The Nigerian and Polish strains shared an MRCA that was dated at either January 2019 (HA) or September 2018 (NA and PA), all with maximum posterior probability scores. The PB1 MRCA could not be assessed because the Polish virus PB1 gene was derived from reassortment with other European LPAI viruses (Świętoń et al., 2020).
4. DISCUSSION
Clade 2.3.4.4B H5N8 HPAI viruses caused unprecedented and devastating outbreaks in 2017 in the South African poultry industry which waned with the arrival of spring. However, an ecological disaster was triggered in the summer with the mass die‐offs of thousands of coastal seabirds along the southern coastlines of the Western and Eastern Cape provinces. Terns especially were affected, but mortalities in critically endangered and threatened species like African Penguins caused international concern, and exactly a year later the disease recurred at a key African Penguin breeding site on Halifax Island, Namibia. In this study, we detected, isolated and/or sequenced 25 viral genomes of isolates made from SA coastal seabirds and a Jackal Buzzard, and identified all as clade 2.3.4.4B H5N8 HPAI strains. Phylogenetic and tMRCA analyses determined that the SA coastal seabird viruses formed a monophyletic group, nested within the SA genotype 4 group. Genotype 4 was identified previously as the sole genotype associated with the southern cluster terrestrial outbreaks from August to October 2017 in the Eastern and Western Cape provinces (Abolnik et al., 2019). The ML and MCC data pointed towards a single introduction of a genotype 4 virus into the coastal seabird population in October 2017, coinciding with the tail end of the outbreaks in terrestrial birds. It is possible that a bridge species such as Egyptian Geese or Sacred Ibis, which have been observed mingling with seabirds at coastal areas, could have introduced the virus to a seabird colony (Roberts et al., 2022), but gulls also scavenge on carcasses of other birds opportunistically (Khomenko et al., 2018) and may have become infected before transmitting the virus to terns at shared roosting and feeding sites. Kelp Gulls (Larus dominicanus) are commonly recognized as scavengers of carcasses (Reusch et al., 2020) and the lack of virus detection in sick and dead birds of the species, despite some effort (Roberts et al., 2022), may either indicate lack of infection, or subclinical infections and a possible role in HPAI virus transmission. Hartlaub's Gulls, where H5N8 virus was detected, have not often been observed scavenging carcasses (pers. comm.) but may do so nonetheless, as they are known, for example, to scavenge fish regurgitated by cormorants (Ryan, 1987). Hartlaub's Gulls also nest in colonies with other species such as Swift Terns (Gaglio & Sherley, 2014), so close contact and contaminated regurgitated fish are other routes of infection with HPAI viruses. The Jackal Buzzard may also have been infected by scavenging (Hockey, Dean & Ryan, 2005), though may have travelled to the area where it was found because the few carcasses that were reported from the area were only discovered 2 months later (unpublished laboratory data).
There was no obvious evidence of reassortment within the coastal seabird viruses, and no discernible association between sub‐genotypes and specific locations, but our data showed that specific viruses were disseminated by coastal seabirds over almost 200 km within a 2‐week period. None of the unique mutations identified in the HA, M1 and PA proteins of the coastal seabird viruses are known to be associated with any specific phenotype in functional studies, for example enhanced replication or virulence (Suttie et al., 2019), but they may represent viral adaptation to the marine hosts. Alternatively, they reflect new sub‐lineages that were emerging, and in fact shorebirds have been found to support higher viral mutation rates than wild ducks (Kim et al., 2022). The SA coastal seabird epidemic appeared to end by May 2018 with no further mortalities reported, but there was no ongoing active sampling of coastal seabirds due to a lack of resources. The last laboratory‐confirmed detection of H5N8 HPAI virus in SA was in July 2018, in a dead Blue Crane in the Theewaterskloof District of the Western Cape Province (Roberts, 2020).
The H5N8 HPAI virus‐linked mortalities in Namibian penguins exactly a year after the SA event were unexpected. Our analyses show that the SA coastal seabird strains were the source of the Namibian outbreaks and the MRCA for Namibia's viruses was dated at March 2018, consistent with the timing of the SA epidemic in coastal seabirds. The H5N8 virus probably continued to circulate in the coastal shorebird population off the western coast between SA and Namibia after April 2018. In May 2018, about 20 Swift Terns roosting on Mercury Island, roughly 105 km north of Halifax Island, died after showing clinical signs typical of HPAI. The deaths were suspected to have been caused by H5N8 HPAI viruses (Molini et al., 2020) and may have been the precursor to the outbreak on Halifax Island 7 months later. Unfortunately, no samples were collected from the affected Swift Terns for laboratory diagnosis and further analysis. African Penguins may travel considerable distances between colonies, and individual birds from the Eastern Cape province have been known to travel over 1700 km to Ichaboe Island on the Namibian coast (Whittington et al., 2005), but these migrations are uncommon, whereas flights of Cape Cormorants, Kelp Gulls, African Oystercatchers and Swift Terns between breeding sites on the western SA and Namibia coasts may be more common (Underhill et al., 1999; Le Roux 2006; Rao, Hockey, and Montevecchi, 2014). The regional movements of Swift Terns were previously suggested as the vector (Molini et al., 2020), but based on our phylogenetic evidence, gulls are just as likely to have introduced the virus. Interestingly, no other birds that were resident on Halifax Island, for example Kelp Gulls, Crowned Cormorants, Swift Terns, Hartlaub's Gulls and African Oystercatchers, manifested any disease symptoms (Molini et al., 2020), though H5N8 HPAI virus was detected in most of these species in SA a year prior and Swift Terns appeared highly susceptible. Their apparently healthy status suggests that some or all of these species may have acted as immune carriers. Why southern African coastal seabird populations became prone to HPAI in the peak of summer in two consecutive years is unknown, though congregation at breeding sites could explain the peaks in mortality. Swift Tern deaths started before congregation for peak breeding season, but the bulk of the carcasses were chicks counted at two breeding colonies later in the outbreak, in April and May. Peak breeding for African Penguins generally tends to be around winter in SA, but it usually falls in summer in Namibia (Hockey, Dean & Ryan, 2005; Kemper et al., 2007), so higher densities could have played a role there. Cooler and drier conditions during winter throughout most of SA is thought to favour the transmission and survival of H5N8 HPAI virus (Khomenko et al., 2018) and although the Western Cape peninsula has a winter rainfall climate with a dry summer, the 2017/2018 Cape drought may have created more suitable conditions for the virus to survive through winter.
West Africa, a key overwintering destination for Palaearctic‐breeding ducks, has been an important origin of Gs/GD H5Nx viruses for central, eastern and southern African countries, and Nigeria in particular was identified as an important point for virus introduction and an origin for the spread of Gs/GD H5Nx viruses during multiple intercontinental waves (Fusaro et al., 2019). The limited molecular data available show that West Africa was the source of the southern African outbreaks in 2017 (Fusaro et al., 2019), but the paucity of virus sequence data for many countries in this region and elsewhere on the continent in 2016–2017 makes it impossible to pinpoint the exact hosts and routes for H5N8 HPAI viruses reaching SA. It was previously established that that five genotypes were implicated in the 2017 H5N8 SA outbreaks and that the Zimbabwean virus, detected only a month before the SA event started, was closely related but not a direct source (Abolnik et al., 2019). The northern cluster of the two spatially distinct clusters that defined the SA 2017 epizootic was detected earlier and had a higher viral diversity. The affected area is geographically, temporally and genetically consistent as primary site of introduction into the country for ducks arriving from the north. The highly migratory Knob‐billed (Comb) Duck (Sarkidiornis melanotos) with recorded migrations between SA and Tanzania, the DRC, Sudan and Chad (Underhill et al., 1999) is one example of a candidate vector, but the involvement of several other species with shorter ranges is also possible. An anomalous drought affected central Africa during October–December 2016's wet season, but north‐eastern SA experienced abundant rainfall during December 2016 to April 2017, driving the southward movement of ducks in search of food and consequently the virus (Fusaro et al., 2019). The MRCAs of genotypes 1, 2 and 5 of the northern cluster outbreaks were dated at between 2 and 4 months prior to the index cases, with genotype 3 arising either slightly before or shortly after its introduction into SA. Genotype 5 most likely arose even earlier in November 2016, when multiple sub‐lineages of 2.3.4.4B H5N8 viruses were already known to be circulating in Nigeria (Laleye at al., 2022).
The tMRCA of genotype 4, the singular cause of the southern cluster outbreaks and ultimately the coastal seabird outbreaks, was of particular interest in this study because it was initially detected 7 weeks after the northern cluster outbreaks started, in apparently healthy commercial ostriches, during routine surveillance. There have previously been fears that commercial ostriches, farmed mainly in the Western Cape province under extensive conditions, could play a role as a reservoir and source of HPAI infection for other commercial poultry. Our analysis, however, showed that the MRCA of genotype 4 pre‐dated the country's index cases in the north‐central region by at least 1–4 months, suggesting that genotype 4 was introduced to the Western Cape around the same time as the other genotypes appeared in the north‐central region, and it is likely that ostriches only became infected earlier than other poultry in the province because of their direct association with wild birds.
Novel genotype 6 was detected in a new cluster of eight SA outbreaks in the north‐central region, commencing in early February 2018 with the last confirmed case in early June of that year (Figure 1; Abolnik et al., 2019). It was previously postulated that genotype 6 may have been introduced the previous year along with genotypes 1–5, but had circulated undetected in a local reservoir. Alternatively, it could have been a new introduction in early 2018 with a new influx of infected ducks from the West‐Central African hotspot. The preponderance of genetic evidence favoured genotype 6 as a new introduction, and our tMRCA analysis supports this because the MRCA for quail/AI5930 (sampled early April 2018) and chicken/499723 (sampled early June 2018) was dated at February 2018, that is around the start of the outbreaks, and therefore genotype 6 probably did not circulate locally for long prior to its detection in SA. The publication of two additional genotype 6 viruses detected in Nigeria and Poland in July and December 2019 (Świętoń et al., 2020; Laleye et al., 2022) aided us in narrowing the time of origin for genotype 6 in sub‐Saharan Africa. The tMRCA analysis dated the MRCA with the best statistical support for the genotype 6 cluster to late 2017, that is 2–3 months prior to introduction into SA. The SA and Nigerian genotype 6 strains were similar in all eight genome segments; therefore, the reverse movement of virus from SA to Nigeria was postulated (Laleye et al., 2022). This reverse movement of virus would, however, be unprecedented and highly unlikely because SA has shown itself to be an ecological sink for avian influenza viruses of various subtypes that originated in northern hemisphere, but never a source to northern hemisphere countries, including Nigeria, based on the sequencing of hundreds of AIV genome segments in the past 20 years (NCBI database records). Furthermore, the Nigerian and Polish genotype 6 strains shared an MRCA and were phylogenetically more closely related to each other than the SA virus, with >98.8 nucleotide sequence identity in all genes except for segments 2 and 5 where a reassortment with Eurasian LPAI viruses was detected (Świętoń et al., 2020). Our tMRCA analysis dated the Nigerian/Polish virus MRCA to September 2018 (NA and PA) and January 2019 (HA) with maximum posterior probability scores; therefore, the progenitor was likely still in circulation somewhere between late 2018 and early 2019, months after the genotype was last detected in SA. Our alternative theory based on the new tMRCA data is that genotype 6‐type viruses arose in late 2017 in a localized avian population in West, Central or East Africa and continued to circulate there, and that this unidentified reservoir was the origin of both the South African viruses in early 2018 and the Nigerian viruses in mid‐2019. The Nigerian virus is genetically strongly linked to the Polish strain, but reverse virus spread by wild birds from Africa back to Europe has never been documented before (Świętoń et al., 2020). Unreported cross‐border trade of infected live poultry between Nigeria and Eastern Europe countries should also be considered as a possibility, as such practices between Nigeria and countries in the Middle East and South Asia have been recorded (Fusaro et al., 2019).
In conclusion, in 2017–2018 sub‐Saharan African countries suffered heavy losses to commercial poultry production, as well as conservation, due to the Clade 2.3.4.4B H5N8 viruses that originated in the northern hemisphere. In future, surveillance and reporting efforts for HPAI virus infections in wild birds in sub‐Saharan Africa must be expanded to more countries and intensified. Dated viral sequence data are a powerful tool that can be used to unravel the complex movements and interactions of wild bird populations. More studies are required on the continent to evaluate wild hosts and their movements in response to changing climatic conditions (Khomenko et al., 2018; Fusaro et al., 2019) and agricultural practices. The improved data will enable us to establish early warning systems and informed mitigation strategies to safeguard the region's food security and natural heritage.
AUTHOR CONTRIBUTIONS
Celia Abolnik conceptualized the idea of the study, wrote the original draft and acquired funding. Belinda M. Peyrot, Celia Abolnik, Laura C. Roberts and Tasneem Anthony designed methodology. Belinda M. Peyrot, Celia Abolnik and Laura C. Roberts performed formal analysis. Laura C. Roberts and Belinda M. Peyrot reviewed and edited the manuscript. Celia Abolnik, Laura C. Roberts and Tasneem Anthony performed supervision. All authors have read and agreed to the published version of the manuscript.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ETHICS STATEMENT
The study was approved by the Research and Animal Ethics committees of the University of Pretoria.
Supporting information
Supplemental Figure 1. Maximum likelihood trees for the (a) PB2, (b) PB1, (c) PA, (d) HA, (e) NP, (f) NA, (g) M and (h) NS genes of H5N8 viruses from southern Africa in 2017–2019 and related viruses. South African and Namibian viruses are indicated by coloured dots. The sub‐genotype is indicated in square brackets. For the South African coastal seabird viruses a letter was assigned for genotyping purposes (see Table 1).
Supplemental Figure 2. Time‐scaled maximum clade credibility trees showing the ancestral state reconstruction for the (a) NA, (b) PB1 and (b) PA genes of H5N8 viruses from southern Africa from 2017–2019 and related viruses. South African viruses are indicated in orange (genotypes 1–5), red (genotype 6) and blue (sub‐genotypes 4.1‐4.3) with the genotype or sub‐genotype in square brackets. Namibian viruses are indicated in purple, and other international viruses are in black. The numerical values represent the posterior probability of the node.
Table S1 Information
ACKNOWLEDGEMENTS
Funding was provided by the NRF‐DSI SARChI Grant no. N00705/114612. The authors thank A. Coetzee, M. Cupido, K. Shaw, T. Staal, J. Strydom, L. Waller, Country Animal Clinic personnel, African Penguin and Seabird Sanctuary personnel, False Bay Veterinary Clinic, Kloof Veterinary Hospital, D. Roberts, SANCCOB, M. Walton, SANParks (especially West Coast National Park), Provincial Veterinary Services officials (Worcester, Boland and George offices) and TAH Somerset West for collecting and/or submitting samples to the WCPVL for analysis. A. Vorster assisted with Ion Torrent sequencing, and S. Evert and R. Pieterse assisted with molecular tests.
Peyrot, B. M. , Abolnik, C. , Anthony, T. , & Roberts, L. C. (2022). Evolutionary dynamics of the clade 2.3.4.4B H5N8 high‐pathogenicity avian influenza outbreaks in coastal seabirds and other species in southern Africa from 2017 to 2019. Transboundary and Emerging Diseases, 69, 3749–3760. 10.1111/tbed.14744
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are openly available in the National Center for Biotechnology Information sequence database under the accession numbers listed in Table S1.
REFERENCES
- Abolnik, C. , Pieterse, R. , Peyrot, B. M. , Choma, P. , Phiri, T. P. , Ebersohn, K. , van Heerden, C. J. V. , Vorster, A. A. , van der Zel, G. V. , Geertsma, P. J. , Laleye, A. T. , Govindasamy, K. , & Rauff, D. L. (2019). The incursion and spread of highly pathogenic avian influenza H5N8 clade 2.3.4.4 within South Africa. Avian Diseases, 63(sp1), 149–156. [DOI] [PubMed] [Google Scholar]
- Abolnik, C. (2020). Outbreaks of Clade 2.3.4.4 H5N8 highly pathogenic avian influenza in 2018 in the northern regions of South Africa were unrelated to those of 2017. Transboundary and Emerging Diseases, 67(3), 1371–1381. 10.1111/tbed.13448 [DOI] [PubMed] [Google Scholar]
- Becker, W. B. (1966). The isolation and classification of Tern virus: Influenza A‐Tern South Africa–1961. Journal of Hygiene, 64(3), 309–320. 10.1017/s0022172400040596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouckaert, R. , Vaughan, T. G. , Barido‐Sottani, J. , Duchêne, S. , Fourment, M. , Gavryushkina, A. , Heled, J. , Jones, G. , Kühnert, D. , De Maio, N. , Matschiner, M. , Mendes, F. K. , Müller, N. F. , Ogilvie, H. A. , du Plessis, L. , Popinga, A. , Rambaut, A. , Rasmussen, D. , Siveroni, I. , … Drummond, A. J. (2019). BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Computational Biology, 15(4), e1006650. 10.1371/journal.pcbi.1006650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusaro, A. , Zecchin, B. , Vrancken, B. , Abolnik, C. , Ademun, R. , Alassane, A. , Arafa, A. , Awuni, J. A. , Couacy‐Hymann, E. , Coulibaly, M ' B. , Gaidet, N. , Go‐Maro, E. , Joannis, T. , Jumbo, S. D. , Minoungou, G. , Meseko, C. , Souley, M. M. , Ndumu, D. B. , Shittu, I. , … Monne, I. (2019). Disentangling the role of Africa in the global spread of H5 highly pathogenic avian influenza. Nature Communication, 10(1), 5310. 10.1038/s41467-019-13287-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaglio, D. , & Sherley, R. B. (2014). Nasty neighbourhood: Kleptoparasitism and egg predation of Swift Terns by Hartlaub's Gulls. Ornithological Observations, 5, 131–134. [Google Scholar]
- Hall, T. (1999). BioEdit: A user‐friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series, 41, 95–98. [Google Scholar]
- Hoffmann, B. , Hoffmann, D. , Henritzi, D. , Beer, M. , & Harder, T. C. (2016). Riems influenza a typing array (RITA): An RT‐qPCR‐based low density array for subtyping avian and mammalian influenza A viruses. Scientific Reports, 6, 27211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockey, P. A. R. , Dean, W. R. J. , & Ryan, P. G. (Eds.). (2005). Roberts Birds of Southern Africa (7th ed.). Trustees of the John Voelcker Bird Book Fund. [Google Scholar]
- Kemper, J. , Underhill, L. G. , Roux, J. , Bartlett, P. A. , Chesselet, Y. J. , James, J. A. C. , Jones, R. , Uhongora, N.‐N. , & Wepener, S. (2007). Breeding patterns and factors influencing breeding success of African Penguins Spheniscus demersus in Namibia. In Kirkman S. P. (Ed.), Final report of the BCLME (Benguela Current Large Marine Ecosystem) project on top predators as biological indicators of ecosystem change in the BCLME (pp. 89–99). Animal Demography Unit, University of Cape Town. http://www.adu.org.za/docs/bclme11_penguin_breeding%20success.pdf [Google Scholar]
- Khomenko, S. , Abolnik, C. , Roberts, L. , Waller, L. , Shaw, K. , Monne, I. , Taylor, J. , Dhingra, M. , Pittiglio, C. , Mugyeom, M. , Roche, X. , Fredrick, K. , Kamata, K. , Okuthe, S. , Kone, P. , Wiersma, L. , Von Dobschuetz, S. , Soumare, B. , Makonnen, Y. , … Lubroth, J. (2018). 2016–2018 Spread of H5N8 highly pathogenic avian influenza (HPAI) in sub‐Saharan Africa: Epidemiological and ecological observations . FAO. www.fao.org/3/CA1209EN/ca1209en.pdf
- Kim, G. , Shin, H. M. , Kim, H. R. , & Kim, Y. (2022). Effects of host and pathogenicity on mutation rates in avian influenza A viruses. Virus Evolution, 8(1), veac013. 10.1093/ve/veac013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S. , Stecher, G. , Li, M. , Knyaz, C. , & Tamura, K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution, 35, 1547–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laleye, A. T. , Bianco, A. , Shittu, I. , Sulaiman, L. , Fusaro, A. , Inuwa, B. , Oyetunde, J. , Zecchin, B. , Bakam, J. , Pastori, A. , Olawuyi, K. , Schivo, A. , Meseko, C. , Vakuru, C. , Fortin, A. , Monne, I. , & Joannis, T. (2022). Genetic characterization of highly pathogenic avian influenza H5Nx clade 2.3.4.4b reveals independent introductions in Nigeria. Transboundary and Emerging Diseases, 69(2), 423–433. 10.1111/tbed.14000 [DOI] [PubMed] [Google Scholar]
- Lee, D. , Bertran, K. , Kwon, J. H. , & Swayne, D. E. (2017). Evolution, global spread and pathogenicity of highly pathogenic avian influenza H5Nx clade 2.3.4.4. Journal of Veterinary Science, 18(S1), 269–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Roux, J. (2006). The swift tern Sterna bergii in southern Africa: Growth and movement . University of Cape Town. https://open.uct.ac.za/bitstream/handle/11427/4382/thesis_sci_2006_le_roux_j.pdf?sequence=180 [Google Scholar]
- Molini, U. , Aikukutu, G. , Roux, J. P. , Kemper, J. , Ntahonshikira, C. , Marruchella, G. , Khaiseb, S. , Cattoli, G. , & Dundon, W. G. (2020). Avian influenza H5N8 outbreak in African penguins (Spheniscus demersus), Namibia, 2019. Journal of Wildlife Diseases, 56(1), 214–218. [PubMed] [Google Scholar]
- Rambaut, A. , Drummond, A. J. , Xie, D. , Baele, G. , & Suchard, M. A. (2018). Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Systematic Biology, 67(5), 901–904. 10.1093/sysbio/syy032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao, A. S. , Hockey, P. A. R. , & Montevecchi, W. (2014). Coastal dispersal by pre‐breeding African Black Oystercatchers (Haematopus moquini). Marine Ornithology, 42, 105–112. [Google Scholar]
- Reusch, K. , Suárez, N. , Ryan, P. G. , & Pichegru, L. (2020). Foraging movements of breeding Kelp Gulls in South Africa. Movement Ecology, 8, 1–12. 10.1186/s40462-020-00221-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, L. (2018). Highly pathogenic avian influenza (H5N8) in coastal birds . Western Cape Government Veterinary Services. https://www.elsenburg.com/vetepi/epireport_pdf/2018Bundle.pdf
- Roberts, L. (2020). Avian influenza surveillance in the Western Cape ostriches: 2018–2019 . Western Cape Government Veterinary Services. https://www.elsenburg.com/vetepi/epireport_pdf/2020Bundle.pdf
- Roberts, L. C. , Abolnik, C. , Waller, L. J. , Shaw, K. , Ludynia, K. , Roberts, D. G. , Kock, A. A. , Makhado, A. B. , Mokoala, M. , Snyman, A. , & Abernerthy, D. A. (2022). Descriptive epidemiology and management of the highly pathogenic avian influenza (H5N8) epidemic in South African coastal seabirds in 2018. Transboundary and Emerging Diseases, under review. [Google Scholar]
- Ryan, P. G. (1987). The foraging behaviour and breeding seasonality of Hartlaub's Gull Farus hartlaubii . Marine Ornithology, 15, 23–32. [Google Scholar]
- Slomka, M. J. , Pavlidis, T. , Banks, J. , Shell, W. , McNally, A. , Essen, S. , & Brown, I. H. (2007). Validated H5 Eurasian real‐time reverse transcriptase‐polymerase chain reaction and its application in H5N1 outbreaks in 2005–2006. Avian Diseases, 51, 373–377. [DOI] [PubMed] [Google Scholar]
- Świętoń, E. , Fusaro, A. , Shittu, I. , Niemczuk, K. , Zecchin, B. , Joannis, T. , Bonfante, F. , Śmietanka, K. , & Terregino, C. (2020). Sub‐saharan Africa and Eurasia ancestry of reassortant highly pathogenic avian influenza A (H5N8) virus, Europe, December 2019. Emerging Infectious Diseases, 26(7), 1557–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suttie, A. , Deng, Y.‐M. , Greenhill, A. R. , Dussart, P. , Horwood, P. F. , & Karlsson, E. A. (2019). Inventory of molecular markers affecting biological characteristics of avian influenza A viruses. Virus Genes, 55, 739–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UK Animal and Plant Health Agency (APHA) . (2016). N8 avian influenza virus detection by reverse transcription RealTime (RRT)‐PCR (9/12/16) . https://www.izsvenezie.com/reference‐laboratories/avian‐influenza‐newcastle‐disease/diagnostic‐protocols/
- Underhill, L. , Tree, A. , Oschadleus, H. , & Parker, V. (1999). Review of ring recoveries of waterbirds in southern Africa. Avian Demography Unit, University of Cape Town. [Google Scholar]
- Whittington, P. A. , Randall, R. M. , Randall, B. M. , Wolfaart, A. C. , Crawford, R. J. M. , Klages, N. T. W. , Bartlett, P. A. , Chesselet, Y. J. , & Jones, R. (2005). Patterns of movements of the African Penguin in South Africa and Namibia. American Journal of Marine Science, 27(1), 215–229. [Google Scholar]
- World Organization for Animal Health (WOAH) . (2019). Avian influenza (infection with avian influenza viruses) . https://www.woah.org/fileadmin/Home/eng/Health_standards/tahm/3.03.04_AI.pdf
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Maximum likelihood trees for the (a) PB2, (b) PB1, (c) PA, (d) HA, (e) NP, (f) NA, (g) M and (h) NS genes of H5N8 viruses from southern Africa in 2017–2019 and related viruses. South African and Namibian viruses are indicated by coloured dots. The sub‐genotype is indicated in square brackets. For the South African coastal seabird viruses a letter was assigned for genotyping purposes (see Table 1).
Supplemental Figure 2. Time‐scaled maximum clade credibility trees showing the ancestral state reconstruction for the (a) NA, (b) PB1 and (b) PA genes of H5N8 viruses from southern Africa from 2017–2019 and related viruses. South African viruses are indicated in orange (genotypes 1–5), red (genotype 6) and blue (sub‐genotypes 4.1‐4.3) with the genotype or sub‐genotype in square brackets. Namibian viruses are indicated in purple, and other international viruses are in black. The numerical values represent the posterior probability of the node.
Table S1 Information
Data Availability Statement
The data that support the findings of this study are openly available in the National Center for Biotechnology Information sequence database under the accession numbers listed in Table S1.