

Summary
C3 is the central effector molecule of the complement system, mediating its multiple functions through different binding sites and their corresponding receptors. We will introduce the C3 forms (native C3, C3 [H2O], and intracellular C3), the C3 fragments C3a, C3b, iC3b, and C3dg/C3d, and the C3 expression sites. To highlight the important role that C3 plays in human biological processes, we will give an overview of the diseases linked to C3 deficiency and to uncontrolled C3 activation. Next, we will present a structural description of C3 activation and of the C3 fragments generated by complement regulation. We will proceed by describing the C3a interaction with the anaphylatoxin receptor, followed by the interactions of opsonins (C3b, iC3b, and C3dg/C3d) with complement receptors, divided into two groups: receptors bearing complement regulatory functions and the effector receptors without complement regulatory activity. We outline the molecular architecture of the receptors, their binding sites on the C3 activation fragments, the cells expressing them, the diversity of their functions, and recent advances. With this review, we aim to give an up‐to‐date analysis of the processes triggered by C3 activation fragments on different cell types in health and disease contexts.
Keywords: anaphylatoxin, autoimmunity, cancer, complement C3, inflammation, opsonin
1. INTRODUCTION
The growing interest in recent years has put the complement system, part of the innate immune response, in the spotlight. Many novel functions of the complement system have been discovered, exceeding the limits of its bactericidal activity and complementing the action of antibodies. Biochemical and biophysical studies are the foundations of research in the complement system field. Over the years, accumulating evidence has contributed to the precise knowledge of the molecular mechanisms governing the complement system available to us today. Complement component C3 is considered the “Swiss Army Knife” of innate immunity and host defense and for a good reason. 1 In this review, we will outline the main C3 functions and how they are carried out through interactions with complement receptors.
1.1. C3—What is it and where to find it
C3 is the central component of the complement system, present in the blood in concentrations of more than 1 mg/mL, which makes it one of the most represented proteins in circulation. Native C3 is considered biologically inactive, but its activation fragments have a multitude of biological functions. A plethora of structure–function studies helped to unveil its multiple biologically active sites important in the mediation of complement effector functions, recently reviewed in Geisbrecht et al 2 C3 is mainly expressed by the liver and circulates in plasma, but most of the cell types in the human body express some amount of C3. It is abundant in circulation, tissues, and even intracellularly. In each of these locations, it can be present in different activation states, and it can have different functions (Figure 1).
FIGURE 1.
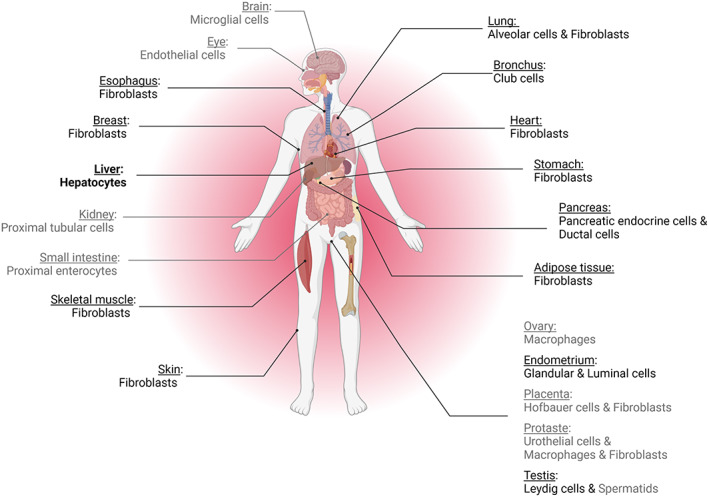
Expression of C3 in different organs and cell types. Gray‐colored items indicate expression lower than 200 transcripts per million (nTPM), in black expression is between 200 and 2500 nTPM, and in bold expression is above 2500 nTPM. Of note, hepatocytes reach 8000 nTPM. Data from protein atlas https://www.proteinatlas.org/
In the circulation and in tissues, C3 is activated by a series of enzymatic reactions upon pathogen infection or cell damage. Each cleavage gives biologically active fragments, indicated by lowercase letters, such as C3a, C3b, iC3b, C3d, and C3d. For the last cleavage fragment, the C3dg form is rapidly transformed to C3d in tissues and on cell surfaces by proteases, and the available tools most often do not allow us to recognize whether C3dg or C3d is present. From this point onward, C3d is used to indicate this last fragment, which in some contexts could be C3dg. The C3 activation fragments serve as inflammatory modulators (the anaphylatoxin C3a) or opsonins (C3b, iC3b, and C3d) and have context‐ and/or receptor‐dependent pro‐ and anti‐inflammatory, destructive, or protective functions. The diversity of the functions is not restricted to the C3 cleavage fragments. The C3 conformation is also a source of diverse biological functions. Native C3 can undergo a conformational change upon binding of a water molecule to generate C3(H2O), a biologically active form resembling C3b in circulation. Intracellular forms of C3 were also recently discovered. This intracellular C3 could either (a) be internalized from the extracellular space in the form of C3(H2O), 3 (b) be expressed by the cell 4 , 5 , 6 from the same start site as the secreted form of C3, or (c) be from an alternative form, generating a shorter, cytoplasmic form lacking disulfide bridges and glycosylation. 7 The intracellular C3 forms regulate cell metabolism, autophagy and contribute to the intracellular detection of cytoinvasive pathogens, as described in detail in this special issue by Blom et al 8 Keeping in mind the diversity of the biologically active forms of C3, its widespread expression and its presence both in the extracellular and intracellular spaces, it is logical to expect that C3 is a key protein for the functioning of the human body.
2. WHY IS C3 SO IMPORTANT—LESSONS FROM C3 DEFICIENCY
The pathological consequences of C3 deficiency in humans are illustrative of the key functional relevance of C3 and its activation fragments. C3 deficiency is extremely rare and results in recurrent pyogenic infections, mainly caused by Streptococcus pneumoniae and Neisseria meningitidis. In isolated cases, C3‐deficient patients could suffer from immune complex (IC)‐related diseases such as systemic lupus erythematosus (SLE)‐like illness and renal diseases, although the deficiency of C3 is not considered a genuine SLE predisposing factor, in contrast to the deficiency of C1q and the other components of the classical pathway (CP). 9 , 10 Mechanistically, C3 deficiencies connected to a lack of opsonization are associated with impaired dendritic cell differentiation, memory B‐cell responses, and regulatory T‐cell development in humans. 11 , 12
Mouse models are instrumental in further understanding the implication of C3 in physiology and in complement‐mediated diseases. C3 knockout in vivo has been used to study several biological or pathological processes in the nervous, circulatory, skeletal, respiratory, digestive, urinary, and visual systems. From these results, the beneficial roles of complement C3, corresponding to the harmful effects of C3 deficiency, and detrimental ones, corresponding to the protective effects of C3 deficiency, can be deduced and will be reviewed in this section. The fact that C3 has been found to play a ubiquitous role underlines once again its importance in multiple contexts.
2.1. Protective role of C3
The complement system, and in particular the CP, has an established role in maintaining homeostasis by removing pathogens and damage‐associated molecular patterns (PAMPSs and DAMPs, respectively) from the circulation in the case of infection but also in the case of tissue injury, autoimmune diseases, and cancer. Confirming its role in protection from infection in humans, in vivo studies in mice established a direct link between C3 deficiency and breakage of the immune defense in bacterial sepsis, acute influenza virus infection, Lyme borreliosis, and fungal infection. 13 , 14 , 15 , 16 Opsonophagocytosis, oxidative stress, and cell lysis are key functions of complement to ensure the rapid elimination of invading bacteria. Indeed, C3 deficiency was shown to increase bacterial load in a model of sepsis induced by cecal ligation and puncture. A decrease in inflammatory mediators was associated with failure to induce complement‐specific functions (phagocytosis, oxidative burst, and cell lysis) 15 (see Section 4). C3 also controls acute influenza virus infection by increasing viral clearance through the regulation of CD4+ and CD8+ T‐cell responses. T‐helper cell‐dependent IgG responses are diminished in C3‐deficient mice due to the failure to target viral antigens to lymphoid organs and to proceed to later stages of complement activation. 14 The Borrelia burgdorferi infection model elucidated that complement contribution is important to control infection dissemination in the early stages of the disease and to prevent the early development of arthritis, while other mechanisms come into play at later stages. 13 Moreover, the presence of C3 was shown to enhance B‐cell responses to many antigens. Fungi are not susceptible to complement‐mediated cell lysis, and the role of C3 in protecting against fungal infection is connected to fungal clearance. Interestingly, complement deficiency is protective when mice are challenged with high doses of fungi. 16
The CP of the complement system has an important role in clearing ICs formed when the host is challenged by an invading pathogen but also in the context of autoimmunity and cancer by facilitating their transport to liver and spleen macrophages. Where circulating ICs are involved in a wide spectrum of pathology, the importance of C3 to prevent their deposition has mainly been evidenced in the kidney. In vivo studies have shown that C3 deficiency leads to the deposition of IgM and IgA ICs in the glomeruli of mice immunized with apoferritin. 17 In a model of lupus nephritis, C3‐induced protection was attributed to IC clearance. 18
Moreover, complement plays a role in tissue healing and repair, as recorded here in the aging central nervous system, in liver injury, in trauma, and in skin psoriatic injury. In the developing brain, complement contributes to synaptic circuit refinement by pruning away excess synaptic connections, a fundamental process for obtaining optimal cognitive ability in adulthood. 19 , 20 , 21 Conversely, complement is also involved in neurodevelopmental and neurodegenerative diseases, and the molecular mechanisms implicated are the subject of active investigation. 22 , 23 , 24 , 25 In Alzheimer's mouse models, C3 deficiency exacerbates the accumulation of amyloid beta plaques and neurodegeneration; therefore, C3 is critical in the clearance of these deposits 26 , 27 (see Section 8.2). Strikingly, recent evidence highlights that constitutive loss of C3 in mice results in spontaneous locomotor deficits during the aging process, with decreased speed and gait ataxia, which can be observed in age‐related locomotor deficits and in the absence of anatomical alterations linked to cerebellar function. 28
The complement system is known to take part in other healing processes in organisms. Specifically, C3 has a fundamental role in liver regeneration and the removal of damaged tissue after toxic liver injury 29 , 30 , 31 ; it also participates in bone development, hematopoietic stem cell homing and engraftment, and amphibians, such as newts and salamanders, limb regeneration. 32 Complement is known to mediate platelet activation, and platelets can activate complement. C3 is essential for physiological hemostasis, and its deficiency results in increased bleeding times, delayed thrombosis, and unstable thrombi. 33 Therefore, functional crosstalk of complement with the coagulation cascade is particularly important upon trauma, where an early clotting response coordinates with complement‐mediated early inflammatory signals that eliminate DAMPs and invading pathogens. 34 In the context of inflammation, C3 was recently found to protect against psoriasis‐like skin inflammation. 35 The absence of C3 exacerbated the inflammatory phenotype by upregulating IFN‐γ+ T‐cell responses, and the phenotype was reversed by caspase inhibition, indicating that C3 exerts its protective role by inhibiting apoptosis. 35
Among the numerous physiological functions of C3 that remain unclear, C3 has been involved in apoptosis and angiogenesis regulation. In a model of retinopathy of prematurity, C3−/− mice displayed increased neovascularization, 36 and in a model of muscle ischemia, angiogenesis was associated with increased neutrophil and macrophage infiltration. 37
C3 has a large number of direct and indirect downstream functions, the molecular details of which are discussed in the coming sections. Before that, to highlight the double‐edged sword of C3 activation, we will give an overview of the deleterious effects of C3 activation.
2.2. Deleterious effects related to C3 activation
While C3 functioning is key in normal physiology, excessive activation of C3 is a hallmark of many complement‐mediated diseases. In this case, protective effects of C3 deficiency are observed in experiments in vivo. The excessive activation of C3, leading to the formation of active fragments C3b and C3a, has been implicated in many acute autoimmune or inflammatory disease models, including acute kidney inflammation, neurotrauma, anti‐phospholipid thrombosis, asthma, and allogeneic transplantation. In the kidney, the iconic organ of complement activation, C3 knockout has been shown to be protective, not only in several models of acute kidney injury, such as ischemia‐reperfusion injury, 38 in which it was associated with reduced neutrophil infiltration and NET formation 39 but also in rhabdomyolysis, where it was associated with inflammatory monocyte infiltration. 40 C3 deficiency decreases the risk of rejection in renal transplantation, even if this deficiency is restricted to the kidney, by modulating T‐cell responses. 41 Finally, C3 deficiency reduced the risk of pyelonephritis in a model of Escherichia coli pyelonephritis. 42 C3 usage was shown to be part of the E. coli mechanism to invade the renal epithelium. Strikingly, despite these protective effects of C3 knockout in ischemic and infectious mouse models in the kidney, in all mouse models of glomerulonephritis, such as lupus and IgA nephropathy, in which complement activation fragments are detected, C3 knockout was protective. This is a key paradox in C3 functions, which can at best be partially explained by the impaired protective IC clearance function of C3 opsonins.
C3 has a detrimental role in the acute assault, such as focal brain ischemia, spinal cord injury, and intracerebral hemorrhage. 43 , 44 , 45 Under these conditions, C3 deficiency protects against inflammatory tissue damage and promotes nerve fiber regeneration after spinal cord injury. In an induced intracerebral hemorrhage, behavioral tests show decreased asymmetry in limb use in C3‐deficient mice, and brain water content measurements indicate lower levels of brain edema. 45 In anti‐phospholipid‐induced thrombosis and fetal loss, C3 deficiency protects against enhanced thrombosis, endothelial cell activation, and fetal injury. 46 , 47 Interestingly, C3 is a biomarker of non‐alcoholic fatty liver disease in rheumatoid arthritis patients, 48 and C3 contributes to steatosis in fatty liver disease in mice. 49 The putative mechanism of action of C3 in this disease has recently been described: C3 upregulates the expression of glycine transfer RNA‐derived fragments, which in turn downregulate the expression of Sirt1, a regulator of lipid metabolic pathways. 50 In the pathogenesis of asthma, C3 deficiency alters the allergic response by reducing the characteristic symptoms of airway hyperresponsiveness and IgE and IgG responses through the downregulation of IL‐4 production, an important cytokine for Th2 function. 51 In pulmonary arterial hypertension, the absence of C3 attenuates prothrombotic and proangiogenic symptoms in a mouse model of the disease. 52 Regulated innate immune responses are critical for the success of allogeneic transplantation. The impact of excessive complement activation, particularly of C3, has been studied in an animal model for graft‐versus‐host disease (GVHD). C3−/− mice showed less mortality and GVHD than wild‐type mice, associated with decreases in donor Th1‐ and Th17‐cell differentiation, which are critical in the development of GVHD. 53
If complement‐mediated responses against self are intrinsic to several inflammatory and autoimmune diseases, the age‐dependent increase in the expression of complement components suggests their implication in age‐related diseases. 54 Indeed, in arthritis and atherosclerosis, inflammatory signals amplified by C3 are detrimental, 55 , 56 , 57 and C3 deficiency ameliorates age‐related changes in mouse kidneys, manifesting as tissue sclerosis and fibrosis. 58 This finding may be linked to a detrimental role of C3 in renal fibrosis, exerted by polarizing macrophages toward a proinflammatory phenotype, increasing proinflammatory cytokine expression, and causing peritubular capillary rarefaction due to decreased angiogenesis. 59 Not only was C3 found to play a role in acute neuropathologies, but C3‐deficient mice are also protected from age‐related hippocampal decline and neurodegeneration, 60 , 61 , 62 , 63 and in experimental autoimmune encephalomyelitis, a mouse model for multiple sclerosis, C3 is necessary for full disease development. 64 The absence of chemotactic activity toward inflammatory cells induced by C3 knockdown is beneficial in alleviating choroidal neovascularization, a common cause of age‐related macular degeneration. 65
Finally, C3 deficiency was found to protect against tumor growth in multiple mouse models. 66 , 67 Cutaneous squamous cell carcinoma was found to induce epidermal hyperplasia and drive tumorigenesis. 68 In a model of carcinogen‐induced sarcoma, C3 and C3a receptors (C3aR) deficiency was associated with reduced tumor growth, reduced accumulation and functional skewing of tumor‐associated macrophages, increased T‐cell activation, and response to anti‐PD‐1 therapy. 69 Moreover, the local production of the proinflammatory anaphylatoxins C3a and C5a is crucial to the tumor response to radiotherapy and concomitant stimulation of tumor‐specific immunity. 70 Many other examples highlight the role of C3 activation fragments in different types of cancers. 71 Overall, C3 contributes to the context‐dependent functions of the complement system in cancer, where the outcome is defined by the site of complement activation, the composition of the tumor microenvironment, and the sensitivity of the tumor cell to complement attack, as recently reviewed by Roumenina et al 72 Pancancer transcriptomic analysis based on the prognostic impact of complement revealed that the coregulated overexpression of complement genes, including C3, confers either poor or favorable prognosis depending on the cancer type. Interestingly, in a subgroup of tumors called the “Protective C3” group, C3 appeared to influence the patient prognosis independently from the other complement proteins. 72
C3−/− mice are indeed susceptible to infections and have deficits in homeostatic processes, but they are protected in a multitude of inflammation‐related disease models. These observations illustrate the “double‐edged sword” of the functioning of the C3 activation fragments—their defensive action could turn offensive if misdirected in a pathological process. Not only is C3 involved in multiple biological processes, but this occurs in physiology, aging, allergy, and chronic and acute disease contexts, highlighting the importance and complexity of C3‐mediated responses and the broad potential for therapeutic intervention. Compstatins are an ever‐growing family of C3‐targeting inhibitors 73 developed to address this ambitious goal. The original compound is a C3 inhibitor that can block all complement pathways and therefore all the C3 fragment‐mediated functions. 74 Compstatins are derivatives of a 13‐residue peptide macrocycle, and they bind C3 between the MG4 and MG5 domains 75 (see Section 3). In 2021, the compstatin pegcetacoplan was approved for the treatment of paroxysmal nocturnal hemoglobinuria (PNH), and several other compstatin‐derived compounds are under investigation for periodontal disease, COVID‐19, age‐related macular degeneration, and neurodegenerative diseases. 73
3. FORMATION OF C3 AND C5 CONVERTASES—THE HEART OF THE COMPLEMENT CASCADE
C3 is the central molecule of the complement system and is the point of convergence of all its activation pathways. Structurally, C3 is part of the A2M protein family, with which it shares the domain architecture. Like all family members, it contains 8 macroglobulin‐like (MG) domains, a linker region (LNK), an anaphylatoxin domain (ANA), a C1r/s, Uegf, B (CUB) domain, and a thioester (TE) domain. One last domain is present at the C‐terminus of complement components C3, C4, and C5 called the C345c domain. Mature C3 is formed by an α‐ and a β‐chain, which are held together by disulfide bridges. The α‐chain encompasses domains MG1 to the first half of MG6 and the LNK, while the second half of MG6, ANA, MG7, CUB, TE, MG8, and C345c domains form the β‐chain (Figure 2, left). 76 One characteristic structural unit of A2M family proteins is the “MG ring”, formed by the MG domains arranged in a ring‐like fashion, visible in the right panel of Figure 2.
FIGURE 2.
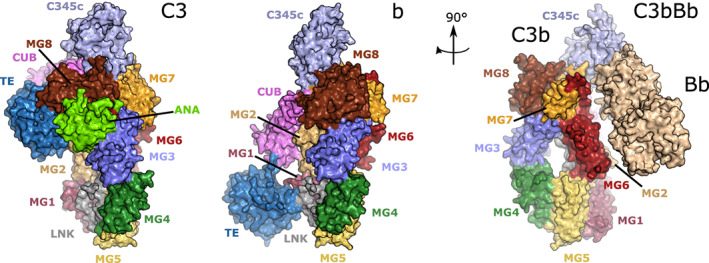
The conformational change occurring upon C3 activation by a C3 convertase. When C3 (left, PDB ID 2A73) is cleaved by a C3 convertase (C4b2a or C3bBb), the anaphylatoxin domain is released to generate C3a in the fluid phase, and C3b which opsonizes the target surface, thanks to an extensive conformational change exposing the thioester in the TE domain, allowing it to react with a nucleophile on the surface (center, PDB ID 2I07). C3b can now form the alternative pathway C3 proconvertase by binding to FB. FB is cleaved by FD to give the alternative pathway C3 convertase C3bBb (right, PDB ID 2 WIN). All figures displaying structures were prepared in PyMOL 2.3.0 https://pymol.org/2/ and edited in Inkscape.
Complement can be activated by three pathways: the classical pathway (CP), lectin pathway (LP), and alternative pathway (AP). 77 C3 is cleaved by the CP and LP C3 convertase C4b2b (updated nomenclature from C4b2a according to the new guidelines 78 ), while in the AP, it is cleaved by the AP C3 convertase C3bBb into the fragments C3a and C3b. Cleavage of C3 at an Arg‐Ser bond releases the C3a anaphylatoxin in the fluid phase and triggers a dramatic conformational change in the nascent C3b molecule. 79 The TE domain is released, exposing the reactive glutamine acyl group of the internal thioester (Figure 2, center). Nucleophilic attack at this acyl group is predominantly by water hydroxyl groups, with C3b remaining in the fluid phase. However, a small proportion of the reactive C3 will undergo nucleophilic attack by surface hydroxyl or amino groups, with consequent covalent attachment to that surface. This process is known as opsonization, and it can either trigger phagocytosis or proceed to the formation of the AP C3 convertase by binding to factor B (FB) and, following activation by FD, generating the AP C3bBb convertase (Figure 2, right). 80 The fact that C3b deposited by the CP can in turn form a C3 convertase, which will amplify C3b deposition on the activator surface, is known as the “amplification loop” of complement activation. AP C3 convertase is very short‐lived and needs to be stabilized by properdin (FP) to have a fully functional impact in vivo. The binding of properdin leads to a 10‐fold increase in the half‐life of AP C3 convertase. 81 , 82
Properdin is the only known positive regulator of the complement system, and it will be described in detail in another contribution to this series. A model for C3 substrate binding to the AP C3 convertase is presented in Figure 3, 83 where it is evident that the C3/C3b MG4 mediates important enzyme–substrate contacts, making it a good target for complement inhibition at the C3 level. In the AP, inhibiting these contacts affects both the C3 substrate and the C3b molecule portion of the convertase enzyme (Figure 3). In the CP and LP, interfering with these interactions prevents the association of the C3 substrate molecule with the convertases (Figure 3, left). Another source of complement C3 activation is spontaneous hydrolysis of the labile thioester group. This nucleophilic attack by water to the C3 thioester is known as the “tick‐over” mechanism, and it is accompanied by a conformational change in C3 resulting in a C3b‐like conformation, generating a molecule called C3(H2O). 84 C3(H2O) is able to bind to FB and, after FB cleavage by FD, to form a fluid phase convertase. The role of this fluid phase convertase is still debated in the complement field. Some suggest that the labile thioester provides a constant low level of complement activation, which has the role of providing protection against invading pathogens and foreign substances such as toxins. However, this view is challenged by the absence of experimental evidence on the existence of a fluid phase convertase. The actual contribution of this pathway to complement activation requires further investigation. 85 , 86 , 87 Another burning question is whether and how a C3 convertase functions in the intracellular space. The intracellular cleavage of C3 and generation of a C3a‐like molecule are now clearly evidenced, 4 , 6 , 88 , 89 , 90 , 91 and its biological relevance for cellular metabolism and functioning is undeniable, but the mechanism remains poorly understood. Experimental evidence suggests that this cleavage is made by cathepsin L in lysosomes 4 or by intracellular FB. 89 An intracellular C3bBb complex cleaving C5 was even detected in macrophages. 92 Whether a genuine C3/C5 convertase, similar to the extracellular convertase, exists intracellularly needs further investigation, as it would require fully folded intracellular C3 and FB. Describing the molecular mechanisms of the generation of intracellular C3a and C5a will be a major breakthrough in the coming years.
FIGURE 3.
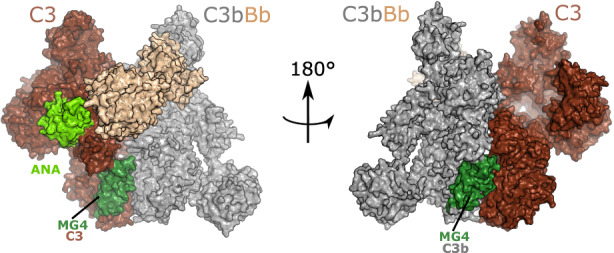
The substrate‐bound alternative pathway C3 convertase. A model of substrate binding by the alternative pathway C3 convertase based on the crystal structure with PDB ID: 3PVM. C3b in the C3 convertase is shown in gray, Bb in roseate. The substrate C3 is shown in brown, with the highlighted ANA domain in lime green, and the MG4 domain in green. The figure underlines the importance of the MG4 domain interface for efficient convertase activity. In fact, residues in MG4 are part of the interface between the substrate C3 and the C3b in the convertase (left), but also between the C3b in the convertase and the substrate C3 (right). To generate the model, C3 was aligned to C5 in the CVF:C5 complex (PDB ID 3PVM), while C3b bound to Bb was aligned to CVF.
4. TERMINAL PATHWAY OF COMPLEMENT—LYTIC CELL DEATH AS A FIRST LINE OF DEFENSE
Sustained C3b deposition at the activator surface results in a switch of substrate specificity of the convertases from C3 to C5. It is not yet known whether the C5 convertases form a trimeric complex by binding of a C3b molecule to the C3 convertases C4b2b and C3bBb or whether the additional C3b molecule at the surface is needed to prime C5 for cleavage by C4b2b/C3bBb. 93 , 94 However, recent data suggest that the conformational activation of C5 (or C5 priming) could be a mechanistic explanation for ongoing terminal pathway activity in the presence of C5 inhibitors. 95 The understanding of the molecular mechanism and structural organization of the C5 convertases is a major challenge to be fully resolved in the upcoming years. The C5 convertases are denoted C4b2bC3b and C3bBbC3b, and they cleave C5 to release the anaphylatoxin C5a and the larger C5b, which is very unstable and can only associate with the membrane if it immediately binds to C6. 96 , 97 The resulting C5b6 complex can then bind successively to C7, C8, and multiple copies of C9. C8 starts membrane penetration, followed by C9, and forms a pore, named the membrane attack complex (MAC), leading to cell lysis. 98 , 99 However, MAC also has other nonlytic, proinflammatory functions, reviewed in ref. [100]. The steps from C5 cleavage to MAC assembly are known as the terminal pathway of the complement system.
5. REGULATION OF C3 ACTIVATION
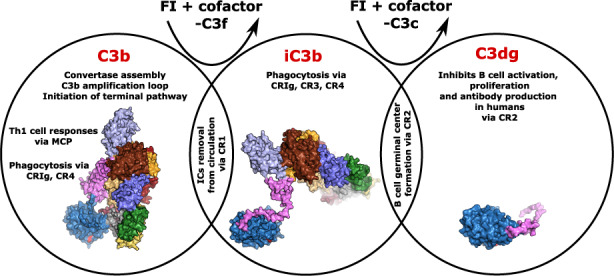
The activation of C3 is dangerous for host cells; therefore, C3 activation is tightly controlled by multiple regulators, part of the regulators of complement activation (RCA) protein family, which for C3 are complement receptor 1 (CR1), factor H (FH), factor H‐like 1 (FHL1), decay‐accelerating factor (DAF), and membrane cofactor protein (MCP). 101 These proteins are characterized by a shared structural unit called a complement control protein (CCP) domain (alternatively called SUSHI or short consensus repeat domain). The repetition of this domain forms the structure of these proteins. The RCA family members that bind to C3b for its degradation are FH, FHL1, CR1, and MCP. 102 FH is the most important regulator of the AP, and its detailed description is presented in another contribution to this series. The RCAs, except DAF, act as cofactors of the protease factor I (FI), in mediating C3b inactivation. Two cleavages by FI in the CUB domain of C3 release the fragment C3f and leave the iC3b fragment bound to the surface. A third and final cleavage releases the C3c fragment and leaves the C3dg fragment bound to the surface, which is further cleaved by proteases to yield C3d (Figure 4). 103 , 104
FIGURE 4.
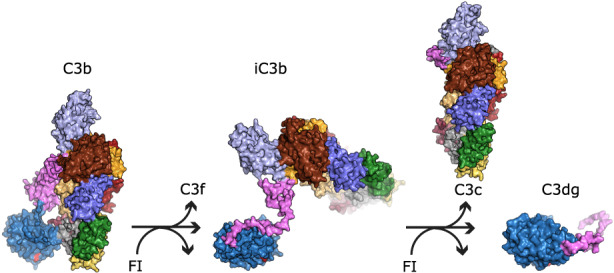
The cleavage steps carried out by FI during C3b regulation. When C3b (left, PDB ID 2I07) is cleaved by FI in the CUB domain (pink), the C3f fragment is released in the fluid phase, and iC3b remains flexibly attached to the target surface (center, PDB ID 7AKK). A last FI cleavage in the CUB domain releases the C3c fragment and leaves C3d bound to the surface (right, PDB ID 7AKK). The thioester glutamine is shown in red, anchoring the TE domain to the activator surface. The location of the remaining iC3b domains is putative, as they are flexibly attached to the TE domain through what remains of the CUB domain (pink).
While part of the binding site of FH and MCP is on the TE domain of C3b, CR1 does not have an affinity for this domain. Hence, FH and MCP can only catalyze FI cleavage up to iC3b formation under physiological conditions, as the third cleavage site is less available for FI cleavage, being between the TE and CUB domains, which are held together by the cofactor MCP or FH. When CR1 is the cofactor for FI, the protease can proceed to the generation of opsonin C3d. 104 CR1 and FH also exert decay acceleration activity, a property shared with membrane‐bound DAF, by binding the convertase and accelerating the decay of the AP C3 convertase. Mutations in CFH, CFI, and C3 are risk factors for the genetic disease atypical hemolytic uremic syndrome (aHUS), and they are involved in the pathogenesis of a group of renal diseases called C3 glomerulopathies (C3G). 105 , 106 , 107 , 108 Autoantibodies against C3 are frequent in patients with lupus nephritis and correlate with disease activity. 109 , 110 , 111 The C3 nephritic factors are autoantibodies recognizing the C3 convertase, but not unbound C3 or C3b, found in C3G, and they impair the decay acceleration activity of the regulators. 112 , 113 The localization for their binding sites is unknown, but it involves neoepitopes formed in the context of the AP C3 and C5 convertases complex.
Detection of the C3 activation fragments in ex vivo models of complement activation and in tissues is important for clinical practice and for understanding disease pathogenesis. Analysis of C3 activation fragments and C5b‐9 on cultured endothelial cells incubated with serum from aHUS patients closely resembles the pathophysiological context of this disease. 114 It has shown elevated deposits from the sera of patients or model conditions of aHUS, malignant hypertension, elevated liver enzymes, low platelet syndrome (HELLP syndrome), sickle cell disease, preeclampsia, lupus nephritis, etc109, 115, 116, 117, 118 In one case, it was used to adjust the therapeutic regimen with a complement‐blocking drug, as the test showed increased deposition of C5b‐9 before the relapse of the disease, encouraging further studies. 119 To make specific diagnoses, tissue staining for C3 activation fragments is critical in clinical practice, especially on kidney biopsies. Anti‐C3c (detecting C3b/iC3b but not C3d) immunostaining is performed as a routine work‐up on kidney biopsies, which detect only ongoing complement activation. In lupus nephritis, C3c versus C3d (stains C3 and the activation fragments C3b, iC3b, and C3d) staining allows the differentiation between active and inactive lupus nephropathy. 120 Interestingly, rhabdomyolysis‐induced acute kidney injury was long considered complement‐independent because the anti‐C3c staining on kidney biopsies was largely negative. A recent study indeed demonstrated negative anti‐C3c but strongly positive anti‐C3d, 40 which can be explained by the rapid cleavage of iC3b to C3c and then to C3d 121 because all biopsies were performed more than 24 hours after the initiation of rhabdomyolysis. This study illustrates the importance of staining for C3d to understand the disease process when complement activation is expected, but the anti‐C3c staining is negative, notably in chronic processes. Finally, tissue staining for C3c or C3d also detects local/intracellular C3 production in fixed and paraffin‐embedded tissues. 122 This is clearly evidenced in renal cancer, where a fraction of the tumor cells produce C3 (and hence stain positive) and deposit C3 activation fragments on their surface due to complement activation. 123 The two patterns could be distinguished and quantified separately, as local production but not surface deposits correlate with poor prognosis in this type of cancer. 124
Here, we have described the C3 cleavage fragments that elicit complement effector functions (Figure 5). In the following sections, we will explore these functions by detailing their interactions with complement receptors.
FIGURE 5.
Different opsonins can trigger specific effector functions. Each circle encloses the effector functions of the represented opsonins, and through which receptors they are implemented. Cell‐type‐specific effector functions were omitted for clarity, and they are presented in tables at the end of each section describing the corresponding receptor.
6. THE C3A:C3AR AXIS: CONTEXT‐DEPENDENT PRO‐ AND ANTI‐INFLAMMATORY ACTIVITY, EXERTED IN THE CIRCULATION, TISSUE MICROENVIRONMENT, AND THE INTRACELLULAR SPACE
The only bioactive fluid phase fragment generated by C3 cleavage is anaphylatoxin C3a. C3a binding to its receptor (C3aR) elicits a response at the activation site involving histamine release from mast cells, smooth muscle contraction, augmented vascular permeability, and mast cell chemokine secretion 125 (Table 1). The biological activity of C3a is regulated by carboxypeptidase‐N, which cleaves off the C‐terminal arginine residue and generates the inactive molecule C3a‐desArg. 126 , 127 C3aR is a transmembrane G‐protein‐coupled receptor distributed in peripheral tissue and the central nervous system and is expressed by all leukocytes. 128 Recently, a mouse atlas of C3aR1 mRNA levels in several organs has allowed a precise mapping of the expressing cells and tissues and a comparison with known human data. 129 , 130 Interestingly, C3aR was found to be stored intracellularly in eosinophils and macrophages, as reported for CD4+ T cells, suggesting that it may function intracellularly in several contexts. 4 Indeed, a recent study showed that it can downregulate mitochondrial metabolism in oxidatively stressed epithelial cells. 131 C3a has been reported to have bactericidal activity exerted by binding to the membrane and inducing cell lysis. 132 With increasing evidence over the years, research has pointed to a dual role of C3a depending on the context, and C3a is currently described as a modulator of inflammation. 133 In particular, C3a can act as an anti‐inflammatory molecule on neutrophils, attenuating their mobilization from the bone marrow to the circulation after ischemia‐reperfusion injury, 134 and it decreases the death rate in sepsis in mice. 135 It mediates the beneficial C3 function of tissue regeneration after liver injury (see Section 2.1) by stimulating cell proliferation, and it contributes to hematopoietic stem cell retention in the bone marrow, homing, and engraftment. 136 , 137 , 138 In the brain, the C3a:C3aR axis has an important role in the recovery after the acute phase of ischemic injury, modulating neurogenesis and axonal and synaptic plasticity. 139 In chronic inflammation, such as autoimmune disease, C3a acts as a proinflammatory mediator. High levels of C3aR expression are found in the glomeruli of lupus nephritis patients. 140 In cancer, C3a can have protumoral effects impacting immune system activation at the tumor site and favoring tumor progression. The C3a:C3aR interaction maintains an immunosuppressive environment in sarcoma and promotes tumor progression by skewing the phenotype of tumor‐associated macrophages, 69 while in lung cancer, C3a:C3aR signaling acts on CD4+ T cells and induces an inhibitory phenotype. 141 In pancreatic ductal adenocarcinoma, C3a:C3aR activates the extracellular‐regulated kinase pathway, inducing epithelial‐to‐mesenchymal transition. 142 Hence, C3a has pleiotropic effects depending on the specific context in which it engages its receptor C3aR. Therapeutic intervention to inhibit or recover its activity should be carefully targeted to the desired location and time point.
TABLE 1.
Cells expressing C3aR and the known functions triggered by C3a binding
| Expressing cell type | Function | References |
|---|---|---|
| Mast cells | Histamine release | [125] |
| Eosinophils, macrophages | Unknown, intracellular? | [129, 130] |
| CD4+ T cells | Important for homeostasis, induction of inhibitory phenotype in lung cancer | [4, 141] |
| Epithelial cells | Metabolism downregulation | [131] |
| Neutrophils | Attenuates mobilization | [134] |
| Hematopoietic stem cells | Retention in the bone marrow, homing, engraftment | [136, 137, 138] |
| Astrocytes, neurons | Modulates neurogenesis, and axonal and synaptic plasticity | [139] |
7. RECEPTORS BEARING COMPLEMENT REGULATORY FUNCTIONS: CR1, MCP, DAF, AND CRIG
7.1. C3b interaction with CR1
C3b is tightly regulated by CR1, which is capable of dissociating C3/C5 convertases and serving as a cofactor for FI to degrade C3b into iC3b and C3d. CR1 is a membrane‐bound complement receptor and a member of the RCA family. Similar to the other members, CR1 has a modular domain architecture formed by 30 CCP domains, a transmembrane domain, and a cytoplasmic tail. 143 The C3b binding interactions are mediated by two regions, each composed of three CCP domains, CCP8‐10 and CCP15‐17. 144 , 145 , 146 CR1 is one of the cofactors binding to C3b and aiding in FI degradation. CR1 also has decay‐accelerating activity, exerted by binding to C3b in the C3 convertase and by dislodging the bound FB (Figure 2, right). CR1 is expressed on many different cell types, namely, monocytes, granulocytes, B and some T lymphocytes, kidney podocytes, and particularly cells of the circulatory system 147 (Table 2). CR1 expressed on erythrocytes represents 90% of the total circulating CR1. 148 In addition to its regulatory role in C3b convertase formation, CR1 is also important for the fixation of ICs and their removal from circulation by transporting them to the liver and spleen. 149 Due to its primary role in IC clearance, CR1 is an important receptor for the maintenance of homeostasis, and CR1 (and CR2; see Section 8.1) is abnormally expressed on B cells of lupus patients. 150 The role of CR1 in Alzheimer's pathogenesis is not yet clear. Studies have reported that impaired C3b regulation by CR1 due to overexpression of a subfunctional isoform is correlated with an increased risk of Alzheimer's disease development 151 , 152 ; however, a recent review highlights that an isoform with the most active sites might be responsible for the increased disease risk. 153
TABLE 2.
Cells expressing CR1 and the known functions triggered by C3b binding
A secondary function of CR1, triggered by high C3b density, is the induction of IL‐1 production and secretion of IL‐1β on monocytes. 154 C3b binding to CR1 has been reported to modulate adaptive immunity, particularly by inhibiting B‐cell differentiation and T‐cell proliferation. 155 , 156 , 157 , 158 , 159 Finally, CR1 also facilitates human T‐cell infection with C3 fragment‐opsonized HIV by generating C3 fragments through its cofactor activity, which can in turn interact with CR2. 160 Due to its multiple functions and implications in disease, CR1‐based therapeutics are being developed to control C3 deposition on tissues and complement activation in the circulation; one example is CSL040. CSL040 is a soluble version of CR1 truncated at residue 1392, and it is promising for preventing complement‐mediated kidney damage. 161 , 162 Another CR1‐based inhibitor consists of a monoclonal antibody targeting C3d covalently linked to CR1 CCP1‐10 domains (C3d mAb‐CR11‐10). CR1 CCP1‐10 are important for decay‐accelerating activity exerted on the CP and AP convertases. This inhibitory fusion protein could allow targeting the complement inhibitory activity of CR1 CCP1‐10 to sites of ongoing complement activation, avoiding systemic complement inhibition and its side effects. 163
7.2. The multiple functions elicited by C3b binding to MCP
C3b can also be degraded by FI in the presence of membrane cofactor protein (MCP or CD46) as a cofactor. MCP is composed of 4 CCP domains, 1‐3 serine/threonine‐rich domains, a transmembrane domain, and a cytoplasmic tail, 164 , 165 and it is a member of the RCA protein family. MCP is expressed on all cells exposed to the circulatory complement system, except erythrocytes (Table 3); MCP binds to C3b deposited by the AP on host cells and promotes its inactivation by FI. 166 , 167 In addition to its regulatory function of C3 deposits, MCP has an important signaling role in CD4+ T cells. 4 Extracellular activation of C3aR and MCP by C3a and C3b, respectively, promotes effector functions, in agreement with the impaired Th1 responses in MCP‐ and C3‐deficient patients. Moreover, the MCP cytoplasmic tail enables Treg cells to remain in an anti‐inflammatory state. MCP in CD8+ T cells transmits important signals for fatty acid metabolism for optimal effector functions. 168 In both CD4+ and CD8+ T cells, MCP plays a role in cell metabolism, although through different signaling events. 169 CD8+ T cells regulate amino acid influx and fatty acid metabolism, while CD4+ T cells tune amino acid influx, oxidative phosphorylation, and glycolysis. Loss of function mutations on the MCP gene leads to the development of aHUSz, 170 , 171 and elevated serum levels of soluble MCP have been reported in one study for SLE patients, 172 indicating how the lost ability to regulate complement by C3b binding at the host cell surface is detrimental for the host.
TABLE 3.
Cells expressing MCP and the known functions triggered by C3b binding
7.3. Decay‐accelerating factor of C3 and C5 convertases
The C3 and C5 convertases can be dissociated by a decay‐accelerating factor (DAF or CD55). DAF is a GPI‐anchored receptor part of the RCA protein family. The ectodomain is composed of four CCP domains arranged in a rod‐like fashion. 173 It is expressed by all blood cells and cells in contact with blood and tissue fluid (Table 4). It exerts its function by binding to C3b in the C3 and C5 convertases, and it induces the decay of the complexes without acting as a cofactor for FI activity. The DAF inhibitory activity of the generation of C3a and C5a fragments by complement convertases is crucial in the response to vascular injury, preventing excessive leukocyte accumulation and thickening of the vascular lining by neovascularization. 174 In addition to its homeostatic regulatory functions, preventing undesired complement activation, DAF is a negative modulator of T‐cell immunity, inhibiting the costimulatory activity exerted by the AP through inhibition of products of AP activation on antigen‐presenting cells and on T cells. 175 New research found that DAF expression is downregulated during germinal center (GC) formation. In this context, augmented C3a:C3aR signaling is necessary for sustaining GC formation; concomitantly, the MAC inhibitor CD59 is upregulated so that B cells are protected from lysis by the terminal pathway of complement. 176 Paralleling its role in normal physiology, DAF is involved in the rare genetic disease PNH, where a mutation in the PIGA gene leads to loss of the GPI anchor, and DAF can no longer protect host cells from complement attack. 177
TABLE 4.
Cells expressing DAF and the known functions triggered by C3b binding
7.4. C3b and iC3b interact with the CRIg receptor on tissue‐resident macrophages
The early complement opsonin C3b has a pivotal role in silent phagocytosis, and the key receptor for this process is the complement receptor of the immunoglobulin superfamily (CRIg or V‐set and Ig domain‐containing 4 [VSIG4]). The N‐terminal C3b/iC3b‐binding domain of CRIg belongs to the IgV family of immunoglobulin‐like domains, 178 , 179 and the binding site is located between the MG3 and MG4 domains of C3b/iC3b. 180 As predicted by the location of its binding site and its structure, CRIg does not act as a cofactor for FI‐mediated C3b degradation; however, it acts as a potent inhibitor of the AP C3 convertase by preventing the C3b molecule in the convertase from binding to the C3 substrate molecule (Figure 3). CRIg is expressed by tissue‐resident macrophages, in particular Kupffer cells in the liver, and other macrophages in the digestive organs, as well as by neutrophils and DCs (Table 5). Subsets of these macrophages also express C3 and/or C1q, but the functional link is unknown. 181 CRIg participates in the phagocytosis of microbes, but it is also important to restrain T‐cell responses, and it is upregulated in mouse autoimmune tissue. 182 Its expression also inhibits macrophage activation by reprogramming mitochondrial pyruvate metabolism and inhibiting reactive oxygen species secretion. 183 The mechanisms for these processes have not yet been elucidated, and whether they are mediated by C3b binding is unknown. The binding of complement fragments triggers the recycling of CRIg on endosomes, depleting the inhibitory activity of the receptor at the surface and promoting inflammation. However, silent phagocytosis not only of pathogens but also of cell debris is important for host homeostasis, as it ensures continuous removal of potential complement activators from circulation. 178 Thus, CRIg is another way in which the innate immune system contributes to host homeostasis and fine‐tunes adaptive immune responses. The binding site on C3b/iC3b of CRIg between MG3 and MG4 is shared by the inhibitory nanobody hC3Nb2. 184 , 185 The nanobody blocks all complement pathways at the C3 cleavage event, indicating how natural receptor binding sites are an inspiration for complement‐targeting molecules and potential therapeutics.
TABLE 5.
Cells expressing CRIg and the known functions triggered by C3b and iC3b binding
8. RECEPTORS MEDIATING COMPLEMENT EFFECTOR FUNCTIONS, WITHOUT COMPLEMENT REGULATORY ACTIVITY: CR2, CR3, AND CR4
8.1. iC3b and C3d interaction with CR2
iC3b and C3d regulate immune cell activation via interaction with complement receptor 2 (CR2, CD21). CR2 has a modular architecture similar to that of CR1; however, it possesses 15 or 16 CCP domains. CR2 mediates the effector functions of iC3b and C3d on B cells, follicular dendritic cells in lymphoid organs, and epithelial cells (Table 6), and the binding site for C3d is at the first two CCP domains. 186 , 187 , 188 In mice, CR1 and CR2 are products of the same gene (Cr2), and alternative splicing leads to the shorter form of the receptor CR2. In humans, they are encoded by separate genes (CR1 and CR2), giving rise to two different proteins with distinct functions. 189 While in mice CR2 acts as a coreceptor for the B‐cell receptor, amplifying the B‐cell response, in human B cells, the costimulatory effect of CR2 is much less pronounced. In mice, the opsonized antigen binds to the B‐cell receptor, while the bound C3d simultaneously binds to CR2 expressed on the same B cell. This coligation lowers the threshold for B‐cell activation by 10 000‐fold. 190 These functions have been extensively studied in mice; however, in humans, C3d‐mediated coengagement of CR2 with the B‐cell receptor has an inhibitory effect. In particular, it suppresses the expression of the activation marker CD69, IL‐6 secretion, proliferation, and antibody production of B cells. 191 Another important function of CR2 is mediating antigen transport to follicular dendritic cells and antigen presentation during GC formation and the generation of high‐affinity antibodies. 192 , 193 CP activation by IgG clustering leads to the decoration of the antigen with iC3b molecules and the formation of ICs. These molecules interact with CR3 (see Section 8.2) on the surface of subcapsular sinus macrophages, which carry ICs to the lymph nodes. Here, the ICs are relayed to B cells through CR2 interaction with C3d and are transported to follicular dendritic cells, where the antigen is retained through CR2:iC3b interaction during GC formation and B‐cell affinity maturation. 194 Decreased CR2 expression leads to the development of SLE and rheumatoid arthritis. 195 , 196 In the context of lupus, CR2 binds to IFNα with the same CCP1‐2 domains used for the iC3b, C3d interaction, 197 and this could be involved in lupus pathogenesis since the receptor would no longer be able to bind opsonized ICs and carry out its function.
TABLE 6.
Cells expressing CR2 and the known functions triggered by C3dg binding
| Expressing cell type | Function | References |
|---|---|---|
| B cells and follicular dendritic cells | Inhibits B cell activation, proliferation, and antibody production in humans, sustains germinal center formation | [190, 191, 192, 193] |
| Tumor cells | Reactivates the immune response to tumor | [198] |
| Marginal zone B cells | MZ B cells acquire DC functions | [199] |
In addition to its function in IC transport, C3d has a novel role in anti‐tumor immunity, mediated by its interaction with CR2, as it amplifies anti‐tumor T‐cell responses. In fact, C3d deposition on irradiated tumor cells, or its recombinant expression inside tumor cells, resulted in the recruitment of CD8+ cytotoxic T cells, depletion of Tregs, and suppression of T‐cell PD‐1 expression in a mouse model. 198 Recent research has demonstrated that the C3d:CR2 interaction plays a role in marginal zone B cells (MZ B cells), which can acquire dendritic cell (DC) functions. Namely, CR2 on MZ B cells interacts with C3d deposited by tick‐over of C3 on MHC II molecules on the DC surface, and MZ B cells acquire the antigen‐presenting ability by trogocytosis. 199 This cooperation between DCs and MZ B cells allows a broader antigen repertoire presentation to CD4+ T cells. From this discussion on the findings related to the CR2 functions, it emerges that careful consideration has to be given to results obtained only in mouse models, and the findings should be reproduced in human‐derived material.
8.2. iC3b and C3d trigger phagocytosis by interacting with their integrin‐like receptors CR3 and CR4
The late opsonins iC3b and C3d have the specialized functions of inducing phagocytosis and clearance of targets through interactions with complement receptors 3 and 4 (CR3, CD11b/CD18, or αMβ2 and CR4, CD11c/CD18, or αXβ2). CR3 and CR4 belong to the β2 family of integrins and hence to a structurally different family of receptors. Integrins are heterodimeric adhesion molecules formed by α and β subunits. They are organized into an extracellular ligand‐binding region, a transmembrane domain, and a cytoplasmic tail. Ligand binding dependent on the chelation of an Mg2+ ion triggers a conformational change that propagates a mechanical signal through the actin cytoskeleton. 200 , 201 In CR3 and CR4, the α subunit is an alpha‐I domain, which has the peculiarity of increased flexibility, allowing binding to more rigid and less accessible ligands on cell surfaces. 202 CR3 and CR4 are expressed on all leukocytes (Table 7), and their structures allow them to carry out specific functions. Generally, CR3 binds preferentially to positively charged ligands, while CR4 binds preferentially to negatively charged ligands. 203 iC3b is the main ligand for CR3 and CR4; however, the two receptors target different domains. 204 , 205 , 206 , 207 CR3 can bind to MG1‐MG2, MG6‐MG7, and the TE domain, while available evidence suggests that CR4 binds iC3b at the MG3‐MG4 interface. The interaction of iC3b with CR3 on macrophages and granulocytes promotes the phagocytosis of ICs. 208 Moreover, CR3 is important for the intracellular degradation of the phagocytized target. 209 An established function of complement and the phagocytic receptor CR3 is at play during brain development and in neurodegenerative diseases. First, in the developing brain, complement contributes to the sculpting of synaptic circuits. Weak synapses, opsonized by iC3b, are eliminated through interaction with CR3 on the surface of microglia, the brain‐resident macrophages. 20 , 210 Second, the iC3b:CR3 receptor interaction is important for the removal of amyloid plaques in brains affected by Alzheimer's disease. 27 Recent findings contributed to further differentiation of the functions of CR3 and CR4. While it was confirmed that the iC3b:CR3 interaction is prevalent for the phagocytic response, the iC3b:CR4 interaction is dominant in the adhesion of monocytes, monocyte‐derived macrophages, and monocyte‐derived dendritic cells to fibrinogen on podosomes, thus facilitating cell migration. 211 , 212 Notably, CR3 and CR4 deficiency results in life‐threatening recurrent infections, as these receptors are important for leukocyte extravasation and migration to the inflammatory site. 213
TABLE 7.
Cells expressing CR3 and CR4 and the known functions triggered by iC3b and C3dg binding
| Interaction | Expressing cell type | Function | References |
|---|---|---|---|
| iC3b:CR3 | Macrophages and granulocytes | Phagocytosis, degradation of phagocytized target | [208, 209] |
| iC3b:CR4 | Monocytes, monocytes derived macrophages and dendritic cells | Adhesion | [212, 213] |
| iC3b:CR3, iC3b:CR4 | Leukocytes | Extravasation and migration to the inflammatory site | [214] |
| iC3b:CR3 | Microglia | Phagocytosis | [20, 210, 211] |
| iC3b:CR3 | T cells | Inhibition of T cell proliferation | [217] |
| C3dg:CR3 | Immune cells in the tumor microenvironment | Activation of immune responses | [218] |
| iC3b:CR3 | Immune cells in the tumor microenvironment | Inhibition of immune responses | [218] |
The iC3b:CR3 interaction contributes to both pathogen removal and maintenance of immune tolerance, 214 as apoptotic cells are also opsonized by iC3b. Their uptake by marginal zone dendritic cells is mediated in part via CR3 and to a lesser extent by CR4, and it causes decreased secretion of proinflammatory cytokines without any effect on the anti‐inflammatory cytokines, thus contributing to the maintenance of peripheral T‐cell tolerance. 215 In vitro studies suggest that the iC3b interaction with CR3 inhibits T‐cell proliferation and IL‐2 release, in line with the hypothesis of a complement‐mediated regulatory function of the iC3b:CR3 interaction on T cells. 216 In the eye, the iC3b:CR3 interaction is key for the development of antigen‐specific tolerance. CR3 stimulation of antigen‐presenting cells results in the sequential production of transforming growth factor‐β2 and interleukin‐10, the latter being essential for the induction of tolerance. 217 At the same time, low‐grade complement activation generates enough C3b to sustain the defense against pathogens in this immune‐privileged site.
8.3. iC3b and C3d as regulators of the immune response and promoters of immune tolerance
The interplay between the opsonins iC3b and C3d with CR2, CR3, and CR4 is key in the balance between triggering the immune response and maintaining immune tolerance. Despite the progress in understanding this process described above, the exact mechanisms at play are still not fully understood and are likely context and organ‐dependent. It has been suggested that iC3b and C3d may have opposite functions in immune regulation in the tumor microenvironment, as they can engage CR3 in two receptor conformations: upon iC3b binding, CR3 adopts the extended trans conformation, while it binds C3d in the closed cis conformation. 218 In this configuration, binding to C3d would occur on the surface of CR3‐expressing cells, activating immune cells. iC3b binding would instead take place on a neighboring cell, extending up to 150 Å distance, thereby inhibiting immune responses. Competition between these two responses is actively taking place in the tumor microenvironment at the synapse between tumor and immune cells. Targeted delivery of C3d to the surface of tumor cells could boost the immune activation induced by C3d binding to CR3. 218 Moreover, C3d associated with tumor cells, or even free C3d, recruits, accelerates, and amplifies anti‐tumor T‐cell responses, allowing reactivation of immunity, up to the point of preventing tumor growth in mouse models. 198 C3d stimulates the increase in tumor‐infiltrating CD8+ T cells by depleting Tregs and suppressing the T‐cell expression of PD‐1 through its interaction with CR2. Therefore, modulating the iC3b/C3d balance in tissue could either help maintain tolerance or stimulate immune responses, a process that, if disrupted, could result in autoimmunity, which is now being explored in anti‐cancer therapy.
9. CONCLUSION
Herein, we have illustrated the central role of complement C3 in the realization of complement effector functions. In particular, C3 can be seen as a basket that is opened upon complement activation, releasing the multitasking mediators of complement functions. Early opsonin C3b contributes to further complement activation and phagocytosis, while anaphylatoxin C3a and the late opsonins iC3b and C3d have multiple immune‐related functions. We have outlined, with structural explanations, how the different fragments carry out specific functions, mainly driven by three synergistic events: (a) release of a bioactive fragment in the fluid phase, (b) conformational changes brought by complement activation that reveal new binding sites and add flexibility, and (c) the exposed binding sites bear differential specificity for the receptors belonging to the structural families of G‐protein‐coupled receptors, RCA, immunoglobulin superfamily, and β2 integrins, thus enabling a multitude of unrelated functions. Furthermore, the expression patterns of the receptors on different cell types add a layer of complexity to the differentiation of the responses triggered by the binding of the C3 fragments.
We can now understand how C3 activation has elegantly regulated context‐dependent functions and how a tip of this balance is a hallmark of many diseases. Thanks to the precise knowledge of C3‐dependent effector functions, an open avenue is available for the development of C3‐targeting therapeutics tailored to the specific disease context.
CONFLICT OF INTEREST
The authors do not declare any conflict of interest to this review.
ACKNOWLEDGEMENTS
A.Z. was supported by the Independent Research Fund Denmark, grant DFF‐1025‐00015B. This work was supported by grants to LTR from: Institut National Du Cancer INCa_16096; Agence Nationale de la Recherche ANR‐21‐CE14‐0066‐02; Les Entreprises contre le Cancer (GEFLUC), Comité de Paris de la Ligue contre le cancer grant RS22/75‐37 and l’Idex Sorbonne Université (Programmes Investissements d’Avenir Émergences 2021‐2022). This work is also supported by The Labex Immuno‐Oncology Excellence Program, INSERM, Université de Paris Cité and Sorbonne Université.
Zarantonello A, Revel M, Grunenwald A, Roumenina LT. C3‐dependent effector functions of complement. Immunol Rev. 2023;313:120‐138. doi: 10.1111/imr.13147
This article is part of a series of reviews covering The Alternative Pathway or Amplification Loop of Complement appearing in Volume 313 of Immunological Reviews.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
REFERENCES
- 1. Ricklin D, Reis ES, Mastellos DC, Gros P, Lambris JD. Complement component C3 – the “Swiss Army Knife” of innate immunity and host defense. Immunol Rev. 2016;274(1):33‐58. doi: 10.1111/imr.12500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Geisbrecht BV, Lambris JD, Gros P. Complement component C3: a structural perspective and potential therapeutic implications. Semin Immunol. 2022;101627. doi: 10.1016/j.smim.2022.101627. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Elvington M, Liszewski MK, Bertram P, Kulkarni HS, Atkinson JP. A C3(H20) recycling pathway is a component of the intracellular complement system. J Clin Invest. 2017;127(3):970‐981. doi: 10.1172/JCI89412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liszewski MK, Kolev M, Le Friec G, et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity. 2013;39(6):1143‐1157. doi: 10.1016/j.immuni.2013.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. King BC, Kulak K, Krus U, et al. Complement component C3 is highly expressed in human pancreatic islets and prevents β cell death via ATG16L1 interaction and autophagy regulation. Cell Metab. 2019;29(1):202‐210.e6. doi: 10.1016/j.cmet.2018.09.009 [DOI] [PubMed] [Google Scholar]
- 6. Friščić J, Böttcher M, Reinwald C, et al. The complement system drives local inflammatory tissue priming by metabolic reprogramming of synovial fibroblasts. Immunity. 2021;54(5):1002‐1021.e10. doi: 10.1016/j.immuni.2021.03.003 [DOI] [PubMed] [Google Scholar]
- 7. Kremlitzka M, Colineau L, Nowacka AA, et al. Alternative translation and retrotranslocation of cytosolic C3 that detects cytoinvasive bacteria. Cell Mol Life Sci. 2022;79(6):291. doi: 10.1007/s00018-022-04308-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. King BC, Blom AM. Intracellular complement: evidence, definitions, controversies, and solutions. Immunol Rev. 2022. doi: 10.1111/imr.13135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cook HT, Botto M. Mechanisms of disease: the complement system and the pathogenesis of systemic lupus erythematosus. Nat Clin Pract Rheumatol. 2006;2(6):330‐337. doi: 10.1038/ncprheum0191 [DOI] [PubMed] [Google Scholar]
- 10. Macedo ACL, Isaac L. Systemic lupus erythematosus and deficiencies of early components of the complement classical pathway. Front Immunol. 2016;7. doi: 10.3389/fimmu.2016.00055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ghannam A, Pernollet M, Fauquert JL, et al. Human C3 deficiency associated with impairments in dendritic cell differentiation, memory B cells, and regulatory T cells. J Immunol. 2008;181(7):5158‐5166. doi: 10.4049/jimmunol.181.7.5158 [DOI] [PubMed] [Google Scholar]
- 12. Ghannam A, Fauquert JL, Thomas C, Kemper C, Drouet C. Human complement C3 deficiency: Th1 induction requires T cell‐derived complement C3a and CD46 activation. Mol Immunol. 2014;58(1):98‐107. doi: 10.1016/j.molimm.2013.11.010 [DOI] [PubMed] [Google Scholar]
- 13. Lawrenz MB, Wooten RM, Zachary JF, et al. Effect of complement component C3 deficiency on experimental Lyme borreliosis in mice. Infect Immun. 2003;71(8):4432‐4440. doi: 10.1128/IAI.71.8.4432-4440.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kopf M, Abel B, Gallimore A, Carroll M, Bachmann MF. Complement component C3 promotes T‐cell priming and lung migration to control acute influenza virus infection. Nat Med. 2002;8(4):373‐378. doi: 10.1038/nm0402-373 [DOI] [PubMed] [Google Scholar]
- 15. Flierl MA, Rittirsch D, Nadeau BA, et al. Functions of the complement components C3 and C5 during sepsis. FASEB J. 2008;22(10):3483‐3490. doi: 10.1096/fj.08-110595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tsoni SV, Kerrigan AM, Marakalala MJ, et al. Complement C3 plays an essential role in the control of opportunistic fungal infections. Infect Immun. 2009;77(9):3679‐3685. doi: 10.1128/IAI.00233-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Quigg RJ, Lim A, Haas M, Alexander JJ, He C, Carroll MC. Immune complex glomerulonephritis in C4‐ and C3‐deficient mice. Kidney Int. 1998;53(2):320‐330. doi: 10.1046/j.1523-1755.1998.00723.x [DOI] [PubMed] [Google Scholar]
- 18. Sekine H, Reilly CM, Molano ID, et al. Complement component C3 is not required for full expression of immune complex glomerulonephritis in MRL/lpr mice. J Immunol. 2001;166(10):6444‐6451. doi: 10.4049/jimmunol.166.10.6444 [DOI] [PubMed] [Google Scholar]
- 19. Stevens B, Allen NJ, Vazquez LE, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131(6):1164‐1178. doi: 10.1016/j.cell.2007.10.036 [DOI] [PubMed] [Google Scholar]
- 20. Schafer DP, Lehrman EK, Kautzman AG, et al. Microglia sculpt postnatal neural circuits in an activity and complement‐dependent manner. Neuron. 2012;74(4):691‐705. doi: 10.1016/j.neuron.2012.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Johnson MB, Stevens B. Pruning hypothesis comes of age. Nature. 2018;554(7693):438‐439. doi: 10.1038/d41586-018-02053-7 [DOI] [PubMed] [Google Scholar]
- 22. Bonifati DM, Kishore U. Role of complement in neurodegeneration and neuroinflammation. Mol Immunol. 2007;44(5):999‐1010. doi: 10.1016/j.molimm.2006.03.007 [DOI] [PubMed] [Google Scholar]
- 23. Lee JD, Coulthard LG, Woodruff TM. Complement dysregulation in the central nervous system during development and disease. Semin Immunol. 2019;45:101340. doi: 10.1016/j.smim.2019.101340 [DOI] [PubMed] [Google Scholar]
- 24. Schartz ND, Tenner AJ. The good, the bad, and the opportunities of the complement system in neurodegenerative disease. J Neuroinflammation. 2020;17(1):354. doi: 10.1186/s12974-020-02024-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zelek WM, Morgan BP. Targeting complement in neurodegeneration: challenges, risks, and strategies. Trends Pharmacol Sci. 2022;43(8):615‐628. doi: 10.1016/j.tips.2022.02.006 [DOI] [PubMed] [Google Scholar]
- 26. Maier M, Peng Y, Jiang L, Seabrook TJ, Carroll MC, Lemere CA. Complement C3 deficiency leads to accelerated amyloid β plaque deposition and neurodegeneration and modulation of the microglia/macrophage phenotype in amyloid precursor protein transgenic mice. J Neurosci. 2008;28(25):6333‐6341. doi: 10.1523/JNEUROSCI.0829-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fu H, Liu B, Frost JL, et al. Complement component C3 and complement receptor type 3 contribute to the phagocytosis and clearance of fibrillar Aβ by microglia. Glia. 2012;60(6):993‐1003. doi: 10.1002/glia.22331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. DeKorver NW, Chaudoin TR, Zhao G, Wang D, Arikkath J, Bonasera SJ. Complement component C3 loss leads to locomotor deficits and altered cerebellar internal granule cell in vitro synaptic protein expression in C57BL/6 mice. Mol Neurobiol. 2021;58(11):5857‐5875. doi: 10.1007/s12035-021-02480-0 [DOI] [PubMed] [Google Scholar]
- 29. Markiewski MM, Mastellos D, Tudoran R, et al. C3a and C3b activation products of the third component of complement (C3) are critical for Normal liver recovery after toxic injury. J Immunol. 2004;173(2):747‐754. doi: 10.4049/jimmunol.173.2.747 [DOI] [PubMed] [Google Scholar]
- 30. Markiewski MM, DeAngelis RA, Strey CW, et al. The regulation of liver cell survival by complement. J Immunol. 2009;182(9):5412‐5418. doi: 10.4049/jimmunol.0804179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Thorgersen EB, Barratt‐Due A, Haugaa H, et al. The role of complement in liver injury, regeneration, and transplantation. Hepatology. 2019;70(2):725‐736. doi: 10.1002/hep.30508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rutkowski MJ, Sughrue ME, Kane AJ, Ahn BJ, Fang S, Parsa AT. The complement cascade as a mediator of tissue growth and regeneration. Inflamm Res. 2010;59(11):897‐905. doi: 10.1007/s00011-010-0220-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gushiken FC, Han H, Li J, Rumbaut RE, Afshar‐Kharghan V. Abnormal platelet function in C3‐deficient mice. J Thromb Haemost. 2009;7(5):865‐870. doi: 10.1111/j.1538-7836.2009.03334.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huber‐Lang M, Kovtun A, Ignatius A. The role of complement in trauma and fracture healing. Semin Immunol. 2013;25(1):73‐78. doi: 10.1016/j.smim.2013.05.006 [DOI] [PubMed] [Google Scholar]
- 35. Zheng QY, Liang SJ, Xu F, et al. Complement component 3 prevents imiquimod‐induced psoriatic skin inflammation by inhibiting apoptosis in mice. Int Immunopharmacol. 2020;85:106692. doi: 10.1016/j.intimp.2020.106692 [DOI] [PubMed] [Google Scholar]
- 36. Langer HF, Chung KJ, Orlova VV, et al. Complement‐mediated inhibition of neovascularization reveals a point of convergence between innate immunity and angiogenesis. Blood. 2010;116(22):4395‐4403. doi: 10.1182/blood-2010-01-261503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Götz P, Braumandl A, Kübler M, et al. C3 deficiency leads to increased angiogenesis and elevated pro‐angiogenic leukocyte recruitment in ischemic muscle tissue. Int J Mol Sci. 2021;22(11):5800. doi: 10.3390/ijms22115800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Park P, Haas M, Cunningham PN, et al. Inhibiting the complement system does not reduce injury in renal ischemia reperfusion. J Am Soc Nephrol. 2001;12(7):1383‐1390. doi: 10.1681/ASN.V1271383 [DOI] [PubMed] [Google Scholar]
- 39. Wu X, You D, Cui J, et al. Reduced neutrophil extracellular trap formation during ischemia reperfusion injury in C3 KO mice: C3 requirement for NETs release. Front Immunol. 2022;13:781273. doi: 10.3389/fimmu.2022.781273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Boudhabhay I, Poillerat V, Grunenwald A, et al. Complement activation is a crucial driver of acute kidney injury in rhabdomyolysis. Kidney Int. 2021;99(3):581‐597. doi: 10.1016/j.kint.2020.09.033 [DOI] [PubMed] [Google Scholar]
- 41. Pratt JR, Basheer SA, Sacks SH. Local synthesis of complement component C3 regulates acute renal transplant rejection. Nat Med. 2002;8(6):582‐587. doi: 10.1038/nm0602-582 [DOI] [PubMed] [Google Scholar]
- 42. Springall T, Sheerin NS, Abe K, Holers VM, Wan H, Sacks SH. Epithelial secretion of C3 promotes colonization of the upper urinary tract by Escherichia coli . Nat Med. 2001;7(7):801‐806. doi: 10.1038/89923 [DOI] [PubMed] [Google Scholar]
- 43. Mocco J, Mack WJ, Ducruet AF, et al. Complement component C3 mediates inflammatory injury following focal cerebral ischemia. Circ Res. 2006;99(2):209‐217. doi: 10.1161/01.RES.0000232544.90675.42 [DOI] [PubMed] [Google Scholar]
- 44. Guo Q, Li S, Liang Y, et al. Effects of C3 deficiency on inflammation and regeneration following spinal cord injury in mice. Neurosci Lett. 2010;485(1):32‐36. doi: 10.1016/j.neulet.2010.08.056 [DOI] [PubMed] [Google Scholar]
- 45. Yang S, Nakamura T, Hua Y, et al. Intracerebral hemorrhage in complement C3‐deficient mice. Acta Neurochir Suppl. 2006;96:227‐231. doi: 10.1007/3-211-30714-1_49 [DOI] [PubMed] [Google Scholar]
- 46. Pierangeli SS, Girardi G, Vega‐Ostertag M, Liu X, Espinola RG, Salmon J. Requirement of activation of complement C3 and C5 for antiphospholipid antibody–mediated thrombophilia. Arthritis Rheum. 2005;52(7):2120‐2124. doi: 10.1002/art.21157 [DOI] [PubMed] [Google Scholar]
- 47. Holers VM, Girardi G, Mo L, et al. Complement C3 activation is required for antiphospholipid antibody‐induced fetal loss. J Exp Med. 2002;195(2):211‐220. doi: 10.1084/jem.200116116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ursini F, Russo E, Mauro D, et al. Complement C3 and fatty liver disease in rheumatoid arthritis patients: a cross‐sectional study. Eur J Clin Invest. 2017;47(10):728‐735. doi: 10.1111/eci.12798 [DOI] [PubMed] [Google Scholar]
- 49. Bykov I, Junnikkala S, Pekna M, Lindros KO, Meri S. Complement C3 contributes to ethanol‐induced liver steatosis in mice. Ann Med. 2006;38(4):280‐286. doi: 10.1080/07853890600664608 [DOI] [PubMed] [Google Scholar]
- 50. Zhong F, Hu Z, Jiang K, et al. Complement C3 activation regulates the production of tRNA‐derived fragments Gly‐tRFs and promotes alcohol‐induced liver injury and steatosis. Cell Res. 2019;29(7):548‐561. doi: 10.1038/s41422-019-0175-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Drouin SM, Corry DB, Kildsgaard J, Wetsel RA. Cutting edge: the absence of C3 demonstrates a role for complement in Th2 effector functions in a murine model of pulmonary allergy. J Immunol. 2001;167(8):4141‐4145. doi: 10.4049/jimmunol.167.8.4141 [DOI] [PubMed] [Google Scholar]
- 52. Bauer EM, Zheng H, Comhair S, Erzurum S, Billiar TR, Bauer PM. Complement C3 deficiency attenuates chronic hypoxia‐induced pulmonary hypertension in mice. PLoS One. 2011;6(12):e28578. doi: 10.1371/journal.pone.0028578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ma Q, Li D, Nurieva R, et al. Reduced graft‐versus‐host disease in C3‐deficient mice is associated with decreased donor Th1/Th17 differentiation. Biol Blood Marrow Transplant. 2012;18(8):1174‐1181. doi: 10.1016/j.bbmt.2012.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gaya Da Costa M, Poppelaars F, Van Kooten C, et al. Age and sex‐associated changes of complement activity and complement levels in a healthy Caucasian population. Front Immunol. 2018;9. doi: 10.3389/fimmu.2018.02664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hietala MA, Jonsson IM, Tarkowski A, Kleinau S, Pekna M. Complement deficiency ameliorates collagen‐induced arthritis in mice. J Immunol. 2002;169(1):454‐459. doi: 10.4049/jimmunol.169.1.454 [DOI] [PubMed] [Google Scholar]
- 56. Buono C, Come CE, Witztum JL, et al. Influence of C3 deficiency on atherosclerosis. Circulation. 2002;105(25):3025‐3031. doi: 10.1161/01.CIR.0000019584.04929.83 [DOI] [PubMed] [Google Scholar]
- 57. Persson L, Borén J, Robertson AKL, Wallenius V, Hansson GK, Pekna M. Lack of complement factor C3, but not factor B, increases hyperlipidemia and atherosclerosis in apolipoprotein E−/− low‐density lipoprotein receptor−/− mice. Arterioscler Thromb Vasc Biol. 2004;24(6):1062‐1067. doi: 10.1161/01.ATV.0000127302.24266.40 [DOI] [PubMed] [Google Scholar]
- 58. Wu X, Lin L, Cui J, Chen Y, Yang L, Wan J. Complement C3 deficiency ameliorates aging related changes in the kidney. Life Sci. 2020;260:118370. doi: 10.1016/j.lfs.2020.118370 [DOI] [PubMed] [Google Scholar]
- 59. Cui J, Wu X, Song Y, Chen Y, Wan J. Complement C3 exacerbates renal interstitial fibrosis by facilitating the M1 macrophage phenotype in a mouse model of unilateral ureteral obstruction. Am J Physiol Renal Physiol. 2019;317(5):F1171‐F1182. doi: 10.1152/ajprenal.00165.2019 [DOI] [PubMed] [Google Scholar]
- 60. Perez‐Alcazar M, Daborg J, Stokowska A, et al. Altered cognitive performance and synaptic function in the hippocampus of mice lacking C3. Exp Neurol. 2014;253:154‐164. doi: 10.1016/j.expneurol.2013.12.013 [DOI] [PubMed] [Google Scholar]
- 61. Shi Q, Colodner KJ, Matousek SB, et al. Complement C3‐deficient mice fail to display age‐related hippocampal decline. J Neurosci. 2015;35(38):13029‐13042. doi: 10.1523/JNEUROSCI.1698-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shi Q, Chowdhury S, Ma R, et al. Complement C3 deficiency protects against neurodegeneration in aged plaque‐rich APP/PS1 mice. Sci Transl Med. 2017;9(392):eaaf6295. doi: 10.1126/scitranslmed.aaf6295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Westacott LJ, Haan N, Evison C, et al. Dissociable effects of complement C3 and C3aR on survival and morphology of adult born hippocampal neurons, pattern separation, and cognitive flexibility in male mice. Brain Behav Immun. 2021;98:136‐150. doi: 10.1016/j.bbi.2021.08.215 [DOI] [PubMed] [Google Scholar]
- 64. Szalai AJ, Hu X, Adams JE, Barnum SR. Complement in experimental autoimmune encephalomyelitis revisited: C3 is required for development of maximal disease. Mol Immunol. 2007;44(12):3132‐3136. doi: 10.1016/j.molimm.2007.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tan X, Fujiu K, Manabe I, et al. Choroidal neovascularization is inhibited via an intraocular decrease of inflammatory cells in mice lacking complement component C3. Sci Rep. 2015;5(1):15702. doi: 10.1038/srep15702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Markiewski MM, DeAngelis RA, Benencia F, et al. Modulation of the antitumor immune response by complement. Nat Immunol. 2008;9(11):1225‐1235. doi: 10.1038/ni.1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Magrini E, Minute L, Dambra M, Garlanda C. Complement activation in cancer: effects on tumor‐associated myeloid cells and immunosuppression. Semin Immunol. 2022;101642. doi: 10.1016/j.smim.2022.101642. In press. [DOI] [PubMed] [Google Scholar]
- 68. Jackson WD, Gulino A, Fossati‐Jimack L, et al. C3 drives inflammatory skin carcinogenesis independently of C5. J Invest Dermatol. 2021;141(2):404‐414.e6. doi: 10.1016/j.jid.2020.06.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Magrini E, Di Marco S, Mapelli SN, et al. Complement activation promoted by the lectin pathway mediates C3aR‐dependent sarcoma progression and immunosuppression. Nat Cancer. 2021;2(2):218‐232. doi: 10.1038/s43018-021-00173-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Surace L, Lysenko V, Fontana AO, et al. Complement is a central mediator of radiotherapy‐induced tumor‐specific immunity and clinical response. Immunity. 2015;42(4):767‐777. doi: 10.1016/j.immuni.2015.03.009 [DOI] [PubMed] [Google Scholar]
- 71. Revel M, Daugan MV, Sautés‐Fridman C, Fridman WH, Roumenina LT. Complement system: promoter or suppressor of cancer progression? Antibodies (Basel). 2020;9(4):E57. doi: 10.3390/antib9040057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Roumenina LT, Daugan MV, Petitprez F, Sautès‐Fridman C, Fridman WH. Context‐dependent roles of complement in cancer. Nat Rev Cancer. 2019;19(12):698‐715. doi: 10.1038/s41568-019-0210-0 [DOI] [PubMed] [Google Scholar]
- 73. Lamers C, Mastellos DC, Ricklin D, Lambris JD. Compstatins: the dawn of clinical C3‐targeted complement inhibition. Trends Pharmacol Sci. 2022;43(8):629‐640. doi: 10.1016/j.tips.2022.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mastellos DC, Yancopoulou D, Kokkinos P, et al. Compstatin: a C3‐targeted complement inhibitor reaching its prime for bedside intervention. Eur J Clin Invest. 2015;45(4):423‐440. doi: 10.1111/eci.12419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Janssen BJC, Halff EF, Lambris JD, Gros P. Structure of Compstatin in complex with complement component C3c reveals a new mechanism of complement inhibition. J Biol Chem. 2007;282(40):29241‐29247. doi: 10.1074/jbc.M704587200 [DOI] [PubMed] [Google Scholar]
- 76. Janssen BJC, Huizinga EG, Raaijmakers HCA, et al. Structures of complement component C3 provide insights into the function and evolution of immunity. Nature. 2005;437(7058):505‐511. doi: 10.1038/nature04005 [DOI] [PubMed] [Google Scholar]
- 77. Merle NS, Church SE, Fremeaux‐Bacchi V, Roumenina LT. Complement system part I – molecular mechanisms of activation and regulation. Front Immunol. 2015;6:262. doi: 10.3389/fimmu.2015.00262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Complement Nomenclature Sub‐committee » IUIS. IUIS . https://iuis.org/committees/nom/complement‐nomenclature‐sub‐committee/. Accessed July 22, 2022.
- 79. Janssen BJC, Christodoulidou A, McCarthy A, Lambris JD, Gros P. Structure of C3b reveals conformational changes that underlie complement activity. Nature. 2006;444(7116):213‐216. doi: 10.1038/nature05172 [DOI] [PubMed] [Google Scholar]
- 80. Rooijakkers SHM, Wu J, Ruyken M, et al. Structural and functional implications of the alternative complement pathway C3 convertase stabilized by a staphylococcal inhibitor. Nat Immunol. 2009;10(7):721‐727. doi: 10.1038/ni.1756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Fearon DT, Austen KF. Properdin: binding to C3b and stabilization of the C3b‐dependent C3 convertase. J Exp Med. 1975;142(4):856‐863. doi: 10.1084/jem.142.4.856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hourcade DE. The role of properdin in the assembly of the alternative pathway C3 convertases of complement. J Biol Chem. 2006;281(4):2128‐2132. doi: 10.1074/jbc.M508928200 [DOI] [PubMed] [Google Scholar]
- 83. Laursen NS, Andersen KR, Braren I, Spillner E, Sottrup‐Jensen L, Andersen GR. Substrate recognition by complement convertases revealed in the C5‐cobra venom factor complex. EMBO J. 2011;30(3):606‐616. doi: 10.1038/emboj.2010.341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Pangburn MK, Müller‐Eberhard HJ. Initiation of the alternative complement pathway due to spontaneous hydrolysis of the thioester of C3. Ann N Y Acad Sci. 1983;421:291‐298. doi: 10.1111/j.1749-6632.1983.tb18116.x [DOI] [PubMed] [Google Scholar]
- 85. Harrison RA. The properdin pathway: an “alternative activation pathway” or a “critical amplification loop” for C3 and C5 activation? Semin Immunopathol. 2018;40(1):15‐35. doi: 10.1007/s00281-017-0661-x [DOI] [PubMed] [Google Scholar]
- 86. Ekdahl KN, Mohlin C, Adler A, et al. Is generation of C3(H2O) necessary for activation of the alternative pathway in real life? Mol Immunol. 2019;114:353‐361. doi: 10.1016/j.molimm.2019.07.032 [DOI] [PubMed] [Google Scholar]
- 87. Fromell K, Adler A, Åman A, et al. Assessment of the role of C3(H2O) in the alternative pathway. Front Immunol. 2020;11. doi: 10.3389/fimmu.2020.00530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Martin M, Leffler J, Smoląg KI, et al. Factor H uptake regulates intracellular C3 activation during apoptosis and decreases the inflammatory potential of nucleosomes. Cell Death Differ. 2016;23(5):903‐911. doi: 10.1038/cdd.2015.164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Yan B, Freiwald T, Chauss D, et al. SARS‐CoV‐2 drives JAK1/2‐dependent local complement hyperactivation. Sci Immunol. 2021;6(58):eabg0833. doi: 10.1126/sciimmunol.abg0833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kemper C, Kolev M. Enzymatic reactions and detection of C3 cleavage fragments. Bio Protoc. 2014;4(16):e1205. doi: 10.21769/BioProtoc.1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Dutta K, Friscic J, Hoffmann MH. Targeting the tissue‐complosome for curbing inflammatory disease. Semin Immunol. 2022;101644. doi: 10.1016/j.smim.2022.101644. In press. [DOI] [PubMed] [Google Scholar]
- 92. Niyonzima N, Rahman J, Kunz N, et al. Mitochondrial C5aR1 activity in macrophages controls IL‐1β production underlying sterile inflammation. Sci Immunol. 2021;6(66):eabf2489. doi: 10.1126/sciimmunol.abf2489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Berends ETM, Gorham RD, Ruyken M, et al. Molecular insights into the surface‐specific arrangement of complement C5 convertase enzymes. BMC Biol. 2015;13(1):93. doi: 10.1186/s12915-015-0203-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Zwarthoff SA, Berends ETM, Mol S, et al. Functional characterization of alternative and classical pathway C3/C5 convertase activity and inhibition using purified models. Front Immunol. 2018;9. doi: 10.3389/fimmu.2018.01691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Mannes M, Dopler A, Zolk O, et al. Complement inhibition at the level of C3 or C5: mechanistic reasons for ongoing terminal pathway activity. Blood. 2021;137(4):443‐455. doi: 10.1182/blood.2020005959 [DOI] [PubMed] [Google Scholar]
- 96. Hadders MA, Bubeck D, Roversi P, et al. Assembly and regulation of the membrane attack complex based on structures of C5b6 and sC5b9. Cell Rep. 2012;1(3):200‐207. doi: 10.1016/j.celrep.2012.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Heesterbeek DA, Bardoel BW, Parsons ES, et al. Bacterial killing by complement requires membrane attack complex formation via surface‐bound C5 convertases. EMBO J. 2019;38(4):e99852. doi: 10.15252/embj.201899852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Serna M, Giles JL, Morgan BP, Bubeck D. Structural basis of complement membrane attack complex formation. Nat Commun. 2016;7(1):10587. doi: 10.1038/ncomms10587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Menny A, Serna M, Boyd CM, et al. CryoEM reveals how the complement membrane attack complex ruptures lipid bilayers. Nat Commun. 2018;9(1):5316. doi: 10.1038/s41467-018-07653-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Xie CB, Jane‐Wit D, Pober JS. Complement membrane attack complex: new roles, mechanisms of action, and therapeutic targets. Am J Pathol. 2020;190(6):1138‐1150. doi: 10.1016/j.ajpath.2020.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Hourcade D, Holers VM, Atkinson JP. The regulators of complement activation (RCA) gene cluster. In: Dixon FJ, ed. Advances in Immunology. Vol 45. Academic Press; 1989:381‐416. doi: 10.1016/S0065-2776(08)60697-5 [DOI] [PubMed] [Google Scholar]
- 102. Forneris F, Wu J, Xue X, et al. Regulators of complement activity mediate inhibitory mechanisms through a common C3b‐binding mode. EMBO J. 2016;35(10):1133‐1149. doi: 10.15252/embj.201593673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Ross G, Newman S, Lambris J, Devery‐Pocius J, Cain J, Lackmann P. Generation of three different fragments of bound C3 with purified factor I or serum. II. Location of binding sites in the C3 fragments for factors B and H, complement receptors, and bovine conglutinin. J Exp Med. 1983;158(2):334‐352. doi: 10.1084/jem.158.2.334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Xue X, Wu J, Ricklin D, et al. Regulator‐dependent mechanisms of C3b processing by factor I allow differentiation of immune responses. Nat Struct Mol Biol. 2017;24(8):643‐651. doi: 10.1038/nsmb.3427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Frémeaux‐Bacchi V, Miller EC, Liszewski MK, et al. Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood. 2008;112(13):4948‐4952. doi: 10.1182/blood-2008-01-133702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Roumenina LT, Frimat M, Miller EC, et al. A prevalent C3 mutation in aHUS patients causes a direct C3 convertase gain of function. Blood. 2012;119(18):4182‐4191. doi: 10.1182/blood-2011-10-383281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Schramm EC, Roumenina LT, Rybkine T, et al. Mapping interactions between complement C3 and regulators using mutations in atypical hemolytic uremic syndrome. Blood. 2015;125(15):2359‐2369. doi: 10.1182/blood-2014-10-609073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Chauvet S, Roumenina LT, Bruneau S, et al. A familial C3GN secondary to defective C3 regulation by complement receptor 1 and complement factor H. J Am Soc Nephrol. 2016;27(6):1665‐1677. doi: 10.1681/ASN.2015040348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Vasilev VV, Noe R, Dragon‐Durey MA, et al. Functional characterization of autoantibodies against complement component C3 in patients with lupus nephritis. J Biol Chem. 2015;290(42):25343‐25355. doi: 10.1074/jbc.M115.647008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Birmingham DJ, Bitter JE, Ndukwe EG, et al. Relationship of circulating anti‐C3b and anti‐C1q IgG to lupus nephritis and its flare. Clin J Am Soc Nephrol. 2016;11(1):47‐53. doi: 10.2215/CJN.03990415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Vasilev VV, Radanova M, Lazarov VJ, Dragon‐Durey MA, Fremeaux‐Bacchi V, Roumenina LT. Autoantibodies against C3b‐functional consequences and disease relevance. Front Immunol. 2019;10:64. doi: 10.3389/fimmu.2019.00064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Marinozzi MC, Chauvet S, Le Quintrec M, et al. C5 nephritic factors drive the biological phenotype of C3 glomerulopathies. Kidney Int. 2017;92(5):1232‐1241. doi: 10.1016/j.kint.2017.04.017 [DOI] [PubMed] [Google Scholar]
- 113. Donadelli R, Pulieri P, Piras R, et al. Unraveling the molecular mechanisms underlying complement dysregulation by nephritic factors in C3G and IC‐MPGN. Front Immunol. 2018;9:2329. doi: 10.3389/fimmu.2018.02329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Meuleman MS, Duval A, Fremeaux‐Bacchi V, Roumenina LT, Chauvet S. Ex vivo test for measuring complement attack on endothelial cells: from research to bedside. Front Immunol. 2022;13. doi: 10.3389/fimmu.2022.860689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Marinozzi MC, Vergoz L, Rybkine T, et al. Complement factor B mutations in atypical hemolytic uremic syndrome—disease‐relevant or benign? J Am Soc Nephrol. 2014;25(9):2053‐2065. doi: 10.1681/ASN.2013070796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Noris M, Galbusera M, Gastoldi S, et al. Dynamics of complement activation in aHUS and how to monitor eculizumab therapy. Blood. 2014;124(11):1715‐1726. doi: 10.1182/blood-2014-02-558296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Palomo M, Blasco M, Molina P, et al. Complement activation and thrombotic microangiopathies. Clin J Am Soc Nephrol. 2019;14(12):1719‐1732. doi: 10.2215/CJN.05830519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Roumenina LT, Chadebech P, Bodivit G, et al. Complement activation in sickle cell disease: dependence on cell density, hemolysis and modulation by hydroxyurea therapy. Am J Hematol. 2020;95(5):456‐464. doi: 10.1002/ajh.25742 [DOI] [PubMed] [Google Scholar]
- 119. Galbusera M, Noris M, Gastoldi S, et al. An ex vivo test of complement activation on endothelium for individualized eculizumab therapy in hemolytic uremic syndrome. Am J Kidney Dis Off J Natl Kidney Found. 2019;74(1):56‐72. doi: 10.1053/j.ajkd.2018.11.012 [DOI] [PubMed] [Google Scholar]
- 120. Wilson HR, Medjeral‐Thomas NR, Gilmore AC, et al. Glomerular membrane attack complex is not a reliable marker of ongoing C5 activation in lupus nephritis. Kidney Int. 2019;95(3):655‐665. doi: 10.1016/j.kint.2018.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Schulze M, Pruchno CJ, Burns M, Baker PJ, Johnson RJ, Couser WG. Glomerular C3c localization indicates ongoing immune deposit formation and complement activation in experimental glomerulonephritis. Am J Pathol. 1993;142(1):179‐187. [PMC free article] [PubMed] [Google Scholar]
- 122. Daugan MV, Revel M, Lacroix L, Sautès‐Fridman C, Fridman WH, Roumenina LT. Complement detection in human tumors by immunohistochemistry and immunofluorescence. Methods Mol Biol. 2021;2227:191‐203. doi: 10.1007/978-1-0716-1016-9_18 [DOI] [PubMed] [Google Scholar]
- 123. Roumenina LT, Daugan MV, Noé R, et al. Tumor cells hijack macrophage‐produced complement C1q to promote tumor growth. Cancer Immunol Res. 2019;7(7):1091‐1105. doi: 10.1158/2326-6066.CIR-18-0891 [DOI] [PubMed] [Google Scholar]
- 124. Daugan MV, Revel M, Thouenon R, et al. Intracellular factor H drives tumor progression independently of the complement Cascade. Cancer Immunol Res. 2021;9(8):909‐925. doi: 10.1158/2326-6066.CIR-20-0787 [DOI] [PubMed] [Google Scholar]
- 125. Hugli TE. Structure and function of C3a anaphylatoxin. In: Lambris JD, ed. The Third Component of Complement. Current Topics in Microbiology and Immunology. Springer; 1990:181‐208. doi: 10.1007/978-3-642-74977-3_10 [DOI] [PubMed] [Google Scholar]
- 126. Matthews KW, Mueller‐Ortiz SL, Wetsel RA. Carboxypeptidase N: a pleiotropic regulator of inflammation. Mol Immunol. 2004;40(11):785‐793. doi: 10.1016/j.molimm.2003.10.002 [DOI] [PubMed] [Google Scholar]
- 127. Bajic G, Yatime L, Klos A, Andersen GR. Human C3a and C3a desArg anaphylatoxins have conserved structures, in contrast to C5a and C5a desArg. Protein Sci Publ Protein Soc. 2013;22(2):204‐212. doi: 10.1002/pro.2200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Settmacher B, Rheinheimer C, Hamacher H, et al. Structure‐function studies of the C3a‐receptor: C‐terminal serine and threonine residues which influence receptor internalization and signaling. Eur J Immunol. 2003;33(4):920‐927. doi: 10.1002/eji.200323293 [DOI] [PubMed] [Google Scholar]
- 129. Quell KM, Karsten CM, Kordowski A, et al. Monitoring C3aR expression using a Floxed tdTomato‐C3aR reporter Knock‐in mouse. J Immunol. 2017;199(2):688‐706. doi: 10.4049/jimmunol.1700318 [DOI] [PubMed] [Google Scholar]
- 130. Laumonnier Y, Karsten CM, Köhl J. Novel insights into the expression pattern of anaphylatoxin receptors in mice and men. Mol Immunol. 2017;89:44‐58. doi: 10.1016/j.molimm.2017.05.019 [DOI] [PubMed] [Google Scholar]
- 131. Ishii M, Beeson G, Beeson C, Rohrer B. Mitochondrial C3a receptor activation in oxidatively stressed epithelial cells reduces mitochondrial respiration and metabolism. Front Immunol. 2021;12. doi: 10.3389/fimmu.2021.628062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Nordahl EA, Rydengård V, Nyberg P, et al. Activation of the complement system generates antibacterial peptides. Proc Natl Acad Sci USA. 2004;101(48):16879‐16884. doi: 10.1073/pnas.0406678101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Coulthard LG, Woodruff TM. Is the complement activation product C3a a proinflammatory molecule? Re‐evaluating the evidence and the myth. J Immunol. 2015;194(8):3542‐3548. doi: 10.4049/jimmunol.1403068 [DOI] [PubMed] [Google Scholar]
- 134. Mike C L W, Faith H B, Jason P L L, et al. The receptor for complement component C3a mediates protection from intestinal ischemia‐reperfusion injuries by inhibiting neutrophil mobilization. Proc Natl Acad Sci USA. 2013;110(23):9439‐9444. doi: 10.1073/pnas.1218815110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Kildsgaard J, Hollmann TJ, Matthews KW, Bian K, Murad F, Wetsel RA. Cutting edge: targeted disruption of the C3a receptor gene demonstrates a novel protective anti‐inflammatory role for C3a in endotoxin‐shock. J Immunol. 2000;165(10):5406‐5409. doi: 10.4049/jimmunol.165.10.5406 [DOI] [PubMed] [Google Scholar]
- 136. Ratajczak J, Reca R, Kucia M, et al. Mobilization studies in mice deficient in either C3 or C3a receptor (C3aR) reveal a novel role for complement in retention of hematopoietic stem/progenitor cells in bone marrow. Blood. 2004;103(6):2071‐2078. doi: 10.1182/blood-2003-06-2099 [DOI] [PubMed] [Google Scholar]
- 137. Reca R, Mastellos D, Majka M, et al. Functional receptor for C3a anaphylatoxin is expressed by normal hematopoietic stem/progenitor cells, and C3a enhances their homing‐related responses to SDF‐1. Blood. 2003;101(10):3784‐3793. doi: 10.1182/blood-2002-10-3233 [DOI] [PubMed] [Google Scholar]
- 138. Lee H, Ratajczak MZ. Innate immunity: a key player in the mobilization of hematopoietic stem/progenitor cells. Arch Immunol Ther Exp (Warsz). 2009;57(4):269‐278. doi: 10.1007/s00005-009-0037-6 [DOI] [PubMed] [Google Scholar]
- 139. Stokowska A, Pekna M. Complement C3a: shaping the plasticity of the Post‐stroke brain. In: Lapchak PA, Zhang JH, eds. Cellular and Molecular Approaches to Regeneration and Repair. Springer series in translational stroke research. Springer International Publishing; 2018:521‐541. doi: 10.1007/978-3-319-66679-2_26 [DOI] [Google Scholar]
- 140. Mizuno M, Blanchin S, Gasque P, Nishikawa K, Matsuo S. High levels of complement C3a receptor in the glomeruli in lupus nephritis. Am J Kidney Dis. 2007;49(5):598‐606. doi: 10.1053/j.ajkd.2007.02.271 [DOI] [PubMed] [Google Scholar]
- 141. Kwak JW, Laskowski J, Li HY, et al. Complement activation via a C3a receptor pathway alters CD4+ T lymphocytes and mediates lung cancer progression. Cancer Res. 2018;78(1):143‐156. doi: 10.1158/0008-5472.CAN-17-0240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Suzuki R, Okubo Y, Takagi T, et al. The complement C3a‐C3a receptor Axis regulates epithelial‐to‐mesenchymal transition by activating the ERK pathway in pancreatic ductal adenocarcinoma. Anticancer Res. 2022;42(3):1207‐1215. doi: 10.21873/anticanres.15587 [DOI] [PubMed] [Google Scholar]
- 143. Krych‐Goldberg M, Atkinson JP. Structure–function relationships of complement receptor type 1. Immunol Rev. 2001;180(1):112‐122. doi: 10.1034/j.1600-065X.2001.1800110.x [DOI] [PubMed] [Google Scholar]
- 144. Krych M, Hourcade D, Atkinson JP. Sites within the complement C3b/C4b receptor important for the specificity of ligand binding. Proc Natl Acad Sci USA. 1991;88(10):4353‐4357. doi: 10.1073/pnas.88.10.4353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Krych‐Goldberg M, Hauhart RE, Subramanian VB, et al. Decay accelerating activity of complement receptor type 1 (CD35). Two active sites are required for dissociating C5 convertases. J Biol Chem. 1999;274(44):31160‐31168. doi: 10.1074/jbc.274.44.31160 [DOI] [PubMed] [Google Scholar]
- 146. Kisserli A, Tabary T, Cohen JHM, Duret V, Mahmoudi R. High‐resolution melting PCR for complement receptor 1 length polymorphism genotyping: an innovative tool for Alzheimer’s disease gene susceptibility assessment. Jove J Vis Exp. 2017;125:e56012. doi: 10.3791/56012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Crehan H, Holton P, Wray S, Pocock J, Guerreiro R, Hardy J. Complement receptor 1 (CR1) and Alzheimer’s disease. Immunobiology. 2012;217(2):244‐250. doi: 10.1016/j.imbio.2011.07.017 [DOI] [PubMed] [Google Scholar]
- 148. Birmingham DJ, Hebert LA. CR1 and CR1‐like: the primate immune adherence receptors. Immunol Rev. 2001;180(1):100‐111. doi: 10.1034/j.1600-065X.2001.1800109.x [DOI] [PubMed] [Google Scholar]
- 149. Satyam A, Hisada R, Bhargava R, Tsokos MG, Tsokos GC. Intertwined pathways of complement activation command the pathogenesis of lupus nephritis. Transl Res. 2022;245:18‐29. doi: 10.1016/j.trsl.2022.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Erdei A, Isaák A, Török K, et al. Expression and role of CR1 and CR2 on B and T lymphocytes under physiological and autoimmune conditions. Mol Immunol. 2009;46(14):2767‐2773. doi: 10.1016/j.molimm.2009.05.181 [DOI] [PubMed] [Google Scholar]
- 151. Lambert JC, Heath S, Even G, et al. Genome‐wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41(10):1094‐1099. doi: 10.1038/ng.439 [DOI] [PubMed] [Google Scholar]
- 152. Hazrati LN, Van Cauwenberghe C, Brooks PL, et al. Genetic association of CR1 with Alzheimer’s disease: a tentative disease mechanism. Neurobiol Aging. 2012;33(12):2949.e5‐2949.e12. doi: 10.1016/j.neurobiolaging.2012.07.001 [DOI] [PubMed] [Google Scholar]
- 153. Torvell M, Carpanini SM, Daskoulidou N, Byrne RAJ, Sims R, Morgan BP. Genetic insights into the impact of complement in Alzheimer’s disease. Genes. 2021;12(12):1990. doi: 10.3390/genes12121990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Bacle F, Haeffner‐Cavaillon N, Laude M, Couturier C, Kazatchkine MD. Induction of IL‐1 release through stimulation of the C3b/C4b complement receptor type one (CR1, CD35) on human monocytes. J Immunol. 1990;144(1):147‐152. [PubMed] [Google Scholar]
- 155. Weiss L, Delfraissy JF, Vazquez A, Wallon C, Galanaud P, Kazatchkine MD. Monoclonal antibodies to the human C3b/C4b receptor (CR1) enhance specific B cell differentiation. J Immunol. 1987;138(9):2988‐2993. [PubMed] [Google Scholar]
- 156. Józsi M, Prechl J, Bajtay Z, Erdei A. Complement receptor type 1 (CD35) mediates inhibitory signals in human B lymphocytes. J Immunol. 2002;168(6):2782‐2788. doi: 10.4049/jimmunol.168.6.2782 [DOI] [PubMed] [Google Scholar]
- 157. Kremlitzka M, Polgár A, Fülöp L, Kiss E, Poór G, Erdei A. Complement receptor type 1 (CR1, CD35) is a potent inhibitor of B‐cell functions in rheumatoid arthritis patients. Int Immunol. 2013;25(1):25‐33. doi: 10.1093/intimm/dxs090 [DOI] [PubMed] [Google Scholar]
- 158. Kremlitzka M, Mácsik‐Valent B, Polgár A, Kiss E, Poór G, Erdei A. Complement receptor type 1 suppresses human B cell functions in SLE patients. J Immunol Res. 2016;2016:e5758192. doi: 10.1155/2016/5758192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Wagner C, Ochmann C, Schoels M, et al. The complement receptor 1, CR1 (CD35), mediates inhibitory signals in human T‐lymphocytes. Mol Immunol. 2006;43(6):643‐651. doi: 10.1016/j.molimm.2005.04.006 [DOI] [PubMed] [Google Scholar]
- 160. Delibrias CC, Kazatchkine MD, Fischer E. Evidence for the role of CR1 (CD35), in addition to CR2 (CD21), in facilitating infection of human T cells with opsonized HIV. Scand J Immunol. 1993;38(2):183‐189. doi: 10.1111/j.1365-3083.1993.tb01711.x [DOI] [PubMed] [Google Scholar]
- 161. Wymann S, Dai Y, Nair AG, et al. A novel soluble complement receptor 1 fragment with enhanced therapeutic potential. J Biol Chem. 2021;296:100200. doi: 10.1074/jbc.RA120.016127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Wymann S, Mischnik M, Leong D, et al. Sialylation‐dependent pharmacokinetics and differential complement pathway inhibition are hallmarks of CR1 activity in vivo. Biochem J. 2022;479(9):1007‐1030. doi: 10.1042/BCJ20220054 [DOI] [PubMed] [Google Scholar]
- 163. Fahnoe KC, Liu F, Morgan JG, et al. Development and optimization of bifunctional fusion proteins to locally modulate complement activation in diseased tissue. Front Immunol. 2022;13. doi: 10.3389/fimmu.2022.869725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Liszewski MK, Post TW, Atkinson JP. Membrane cofactor protein (MCP or CD46): newest member of the regulators of complement activation gene cluster. Annu Rev Immunol. 1991;9:431‐455. doi: 10.1146/annurev.iy.09.040191.002243 [DOI] [PubMed] [Google Scholar]
- 165. Seya T, Hirano A, Matsumoto M, Nomura M, Ueda S. Human membrane cofactor protein (MCP, CD46): multiple isoforms and functions. Int J Biochem Cell Biol. 1999;31(11):1255‐1260. doi: 10.1016/S1357-2725(99)00092-8 [DOI] [PubMed] [Google Scholar]
- 166. Oglesby TJ, Allen CJ, Liszewski MK, White DJ, Atkinson JP. Membrane cofactor protein (CD46) protects cells from complement‐mediated attack by an intrinsic mechanism. J Exp Med. 1992;175(6):1547‐1551. doi: 10.1084/jem.175.6.1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Barilla‐LaBarca ML, Liszewski MK, Lambris JD, Hourcade D, Atkinson JP. Role of membrane cofactor protein (CD46) in regulation of C4b and C3b deposited on cells. J Immunol. 2002;168(12):6298‐6304. doi: 10.4049/jimmunol.168.12.6298 [DOI] [PubMed] [Google Scholar]
- 168. Arbore G, West EE, Rahman J, et al. Complement receptor CD46 co‐stimulates optimal human CD8+ T cell effector function via fatty acid metabolism. Nat Commun. 2018;9(1):4186. doi: 10.1038/s41467-018-06706-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Kolev M, Dimeloe S, Le Friec G, et al. Complement regulates nutrient influx and metabolic reprogramming during Th1 cell responses. Immunity. 2015;42(6):1033‐1047. doi: 10.1016/j.immuni.2015.05.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Fremeaux‐Bacchi V, Moulton EA, Kavanagh D, et al. Genetic and functional analyses of membrane cofactor protein (CD46) mutations in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2006;17(7):2017‐2025. doi: 10.1681/ASN.2005101051 [DOI] [PubMed] [Google Scholar]
- 171. Richards A, Kathryn Liszewski M, Kavanagh D, et al. Implications of the initial mutations in membrane cofactor protein (MCP; CD46) leading to atypical hemolytic uremic syndrome. Mol Immunol. 2007;44(1):111‐122. doi: 10.1016/j.molimm.2006.07.004 [DOI] [PubMed] [Google Scholar]
- 172. Kawano M, Seya T, Koni I, Mabuchi H. Elevated serum levels of soluble membrane cofactor protein (CD46, MCP) in patients with systemic lupus erythematosus (SLE). Clin Exp Immunol. 1999;116(3):542‐546. doi: 10.1046/j.1365-2249.1999.00917.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Lukacik P, Roversi P, White J, et al. Complement regulation at the molecular level: the structure of decay‐accelerating factor. Proc Natl Acad Sci USA. 2004;101(5):1279‐1284. doi: 10.1073/pnas.0307200101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Sakuma M, Morooka T, Wang Y, et al. The intrinsic complement regulator decay‐accelerating factor modulates the biological response to vascular injury. Arterioscler Thromb Vasc Biol. 2010;30(6):1196‐1202. doi: 10.1161/ATVBAHA.110.205559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Heeger PS, Lalli PN, Lin F, et al. Decay‐accelerating factor modulates induction of T cell immunity. J Exp Med. 2005;201(10):1523‐1530. doi: 10.1084/jem.20041967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Cumpelik A, Heja D, Hu Y, et al. Dynamic regulation of B cell complement signaling is integral to germinal center responses. Nat Immunol. 2021;22(6):757‐768. doi: 10.1038/s41590-021-00926-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Brodsky RA. Paroxysmal nocturnal hemoglobinuria. Blood. 2014;124(18):2804‐2811. doi: 10.1182/blood-2014-02-522128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Helmy KY, Katschke KJ, Gorgani NN, et al. CRIg: a macrophage complement receptor required for phagocytosis of circulating pathogens. Cell. 2006;124(5):915‐927. doi: 10.1016/j.cell.2005.12.039 [DOI] [PubMed] [Google Scholar]
- 179. Yuan X, Yang BH, Dong Y, Yamamura A, Fu W. CRIg, a tissue‐resident macrophage specific immune checkpoint molecule, promotes immunological tolerance in NOD mice, via a dual role in effector and regulatory T cells. eLife. 2017;6:e29540. doi: 10.7554/eLife.29540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Wiesmann C, Katschke KJ, Yin J, et al. Structure of C3b in complex with CRIg gives insights into regulation of complement activation. Nature. 2006;444(7116):217‐220. doi: 10.1038/nature05263 [DOI] [PubMed] [Google Scholar]
- 181. Revel M, Sautès‐Fridman C, Fridman WH, Roumenina LT. C1q+ macrophages: passengers or drivers of cancer progression. Trends Cancer. 2022;8(7):517‐526. doi: 10.1016/j.trecan.2022.02.006 [DOI] [PubMed] [Google Scholar]
- 182. Vogt L, Schmitz N, Kurrer MO, et al. VSIG4, a B7 family–related protein, is a negative regulator of T cell activation. J Clin Invest. 2006;116(10):2817‐2826. doi: 10.1172/JCI25673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Li J, Diao B, Guo S, et al. VSIG4 inhibits proinflammatory macrophage activation by reprogramming mitochondrial pyruvate metabolism. Nat Commun. 2017;8(1):1322. doi: 10.1038/s41467-017-01327-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Pedersen H, Jensen RK, Hansen AG, et al. A C3‐specific nanobody that blocks all three activation pathways in the human and murine complement system. J Biol Chem. 2020;295(26):8746‐8758. doi: 10.1074/jbc.RA119.012339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Zarantonello A, Pedersen H, Laursen NS, Andersen GR. Nanobodies provide insight into the molecular mechanisms of the complement Cascade and offer new therapeutic strategies. Biomolecules. 2021;11(2):298. doi: 10.3390/biom11020298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Cooper NR, Bradt BM, Rhim JS, Nemerow GR. CR2 complement receptor. J Invest Dermatol. 1990;94(6, Supplement):s112‐s117. doi: 10.1111/1523-1747.ep12876069 [DOI] [PubMed] [Google Scholar]
- 187. van den Elsen JMH, Isenman DE. A crystal structure of the complex between human complement receptor 2 and its ligand C3d. Science. 2011;332(6029):608‐611. doi: 10.1126/science.1201954 [DOI] [PubMed] [Google Scholar]
- 188. Carroll MC, Isenman DE. Regulation of humoral immunity by complement. Immunity. 2012;37(2):199‐207. doi: 10.1016/j.immuni.2012.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Jacobson AC, Weis JH. Comparative functional evolution of human and mouse CR1 and CR2. J Immunol. 2008;181(5):2953‐2959. doi: 10.4049/jimmunol.181.5.2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Dempsey PW, Allison MED, Akkaraju S, Goodnow CC, Fearon DT. C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science. 1996;271(5247):348‐350. doi: 10.1126/science.271.5247.348 [DOI] [PubMed] [Google Scholar]
- 191. Kovács KG, Mácsik‐Valent B, Matkó J, Bajtay Z, Erdei A. Revisiting the coreceptor function of complement receptor type 2 (CR2, CD21); Coengagement with the B‐cell receptor inhibits the activation, proliferation, and antibody production of human B cells. Front Immunol. 2021;12. doi: 10.3389/fimmu.2021.620427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Fang Y, Xu C, Fu YX, Holers VM, Molina H. Expression of complement receptors 1 and 2 on follicular dendritic cells is necessary for the generation of a strong antigen‐specific IgG response. J Immunol. 1998;160(11):5273‐5279. [PubMed] [Google Scholar]
- 193. Roozendaal R, Carroll MC. Complement receptors CD21 and CD35 in humoral immunity. Immunol Rev. 2007;219(1):157‐166. doi: 10.1111/j.1600-065X.2007.00556.x [DOI] [PubMed] [Google Scholar]
- 194. Phan TG, Green JA, Gray EE, Xu Y, Cyster JG. Immune complex relay by subcapsular sinus macrophages and noncognate B cells drives antibody affinity maturation. Nat Immunol. 2009;10(7):786‐793. doi: 10.1038/ni.1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Nagro CJD, Kolla RV, Rickert RC. A critical role for complement C3d and the B cell coreceptor (CD19/CD21) complex in the initiation of inflammatory arthritis. J Immunol. 2005;175(8):5379‐5389. doi: 10.4049/jimmunol.175.8.5379 [DOI] [PubMed] [Google Scholar]
- 196. Prokopec KE, Rhodiner M, Matt P, Lindqvist U, Kleinau S. Down regulation of Fc and complement receptors on B cells in rheumatoid arthritis. Clin Immunol. 2010;137(3):322‐329. doi: 10.1016/j.clim.2010.08.006 [DOI] [PubMed] [Google Scholar]
- 197. Asokan R, Hua J, Young KA, et al. Characterization of human complement receptor type 2 (CR2/CD21) as a receptor for IFN‐α: a potential role in systemic lupus erythematosus. J Immunol. 2006;177(1):383‐394. doi: 10.4049/jimmunol.177.1.383 [DOI] [PubMed] [Google Scholar]
- 198. Platt JL, Silva I, Balin SJ, et al. C3d regulates immune checkpoint blockade and enhances antitumor immunity. JCI Insight. 2017;2(9):90201. doi: 10.1172/jci.insight.90201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Schriek P, Ching AC, Moily NS, et al. Marginal zone B cells acquire dendritic cell functions by trogocytosis. Science. 2022;375(6581):eabf7470. doi: 10.1126/science.abf7470 [DOI] [PubMed] [Google Scholar]
- 200. Sim RB, Malhotra V, Day AJ, Erdei A. Structure and specificity of complement receptors. Immunol Lett. 1987;14(3):183‐190. doi: 10.1016/0165-2478(87)90099-X [DOI] [PubMed] [Google Scholar]
- 201. Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu Rev Immunol. 2007;25:619‐647. doi: 10.1146/annurev.immunol.25.022106.141618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Xie C, Zhu J, Chen X, Mi L, Nishida N, Springer TA. Structure of an integrin with an alphaI domain, complement receptor type 4. EMBO J. 2010;29(3):666‐679. doi: 10.1038/emboj.2009.367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Vorup‐Jensen T, Jensen RK. Structural immunology of complement receptors 3 and 4. Front Immunol. 2018;9. doi: 10.3389/fimmu.2018.02716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Xu S, Wang J, Wang JH, Springer TA. Distinct recognition of complement iC3b by integrins αXβ2 and αMβ2. Proc Natl Acad Sci USA. 2017;114(13):3403‐3408. doi: 10.1073/pnas.1620881114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Choi J, Buyannemekh D, Nham SU. Moieties of complement iC3b recognized by the I‐domain of integrin αXβ2. Mol Cells. 2020;43(12):1023‐1034. doi: 10.14348/molcells.2020.0197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Jensen RK, Bajic G, Sen M, Springer TA, Vorup‐Jensen T, Andersen GR. Complement receptor 3 forms a compact high‐affinity complex with iC3b. J Immunol. 2021;206(12):3032‐3042. doi: 10.4049/jimmunol.2001208 [DOI] [PubMed] [Google Scholar]
- 207. Fernández FJ, Santos‐López J, Martínez‐Barricarte R, et al. The crystal structure of iC3b‐CR3 αI reveals a modular recognition of the main opsonin iC3b by the CR3 integrin receptor. Nat Commun. 2022;13(1):1955. doi: 10.1038/s41467-022-29580-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Huber H, Polley MJ, Linscott WD, Fudenberg HH, Müller‐Eberhard HJ. Human monocytes: distinct receptor sites for the third component of complement and for immunoglobulin G. Science. 1968;162(3859):1281‐1283. doi: 10.1126/science.162.3859.1281 [DOI] [PubMed] [Google Scholar]
- 209. Rothlein R, Springer TA. Complement receptor type three‐dependent degradation of opsonized erythrocytes by mouse macrophages. J Immunol. 1985;135(4):2668‐2672. [PubMed] [Google Scholar]
- 210. Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci. 2012;35:369‐389. doi: 10.1146/annurev-neuro-061010-113810 [DOI] [PubMed] [Google Scholar]
- 211. Lukácsi S, Nagy‐Baló Z, Erdei A, Sándor N, Bajtay Z. The role of CR3 (CD11b/CD18) and CR4 (CD11c/CD18) in complement‐mediated phagocytosis and podosome formation by human phagocytes. Immunol Lett. 2017;189:64‐72. doi: 10.1016/j.imlet.2017.05.014 [DOI] [PubMed] [Google Scholar]
- 212. Erdei A, Lukácsi S, Mácsik‐Valent B, Nagy‐Baló Z, Kurucz I, Bajtay Z. Non‐identical twins: different faces of CR3 and CR4 in myeloid and lymphoid cells of mice and men. Semin Cell Dev Biol. 2019;85:110‐121. doi: 10.1016/j.semcdb.2017.11.025 [DOI] [PubMed] [Google Scholar]
- 213. Mitroulis I, Alexaki VI, Kourtzelis I, Ziogas A, Hajishengallis G, Chavakis T. Leukocyte integrins: role in leukocyte recruitment and as therapeutic targets in inflammatory disease. Pharmacol Ther. 2015;147:123‐135. doi: 10.1016/j.pharmthera.2014.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Lamers C, Plüss CJ, Ricklin D. The promiscuous profile of complement receptor 3 in ligand binding, immune modulation, and pathophysiology. Front Immunol. 2021;12. doi: 10.3389/fimmu.2021.662164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Morelli AE, Larregina AT, Shufesky WJ, et al. Internalization of circulating apoptotic cells by splenic marginal zone dendritic cells: dependence on complement receptors and effect on cytokine production. Blood. 2003;101(2):611‐620. doi: 10.1182/blood-2002-06-1769 [DOI] [PubMed] [Google Scholar]
- 216. Wagner C, Hänsch GM, Stegmaier S, Denefleh B, Hug F, Schoels M. The complement receptor 3, CR3 (CD11b/CD18), on T lymphocytes: activation‐dependent up‐regulation and regulatory function. Eur J Immunol. 2001;31(4):1173‐1180. doi:10.1002/1521‐4141(200104)31:4<1173::AID‐IMMU1173>3.0.CO;2‐9 [DOI] [PubMed] [Google Scholar]
- 217. Sohn JH, Bora PS, Suk HJ, Molina H, Kaplan HJ, Bora NS. Tolerance is dependent on complement C3 fragment iC3b binding to antigen‐presenting cells. Nat Med. 2003;9(2):206‐212. doi: 10.1038/nm814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Herbert A. Complement controls the immune synapse and tumors control complement. J Immunother Cancer. 2020;8(2):e001712. doi: 10.1136/jitc-2020-001712 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.